

Abstract
Background
Oral nirmatrelvir/ritonavir (Paxlovid®) aims to avoid severe COVID‐19 in asymptomatic people or those with mild symptoms, thereby decreasing hospitalization and death. Due to its novelty, there are currently few published study results. It remains to be evaluated for which indications and patient populations the drug is suitable.
Objectives
To assess the efficacy and safety of nirmatrelvir/ritonavir (Paxlovid®) plus standard of care compared to standard of care with or without placebo, or any other intervention for treating COVID‐19 and for preventing SARS‐CoV‐2 infection.
To explore equity aspects in subgroup analyses.
To keep up to date with the evolving evidence base using a living systematic review (LSR) approach and make new relevant studies available to readers in‐between publication of review updates.
Search methods
We searched the Cochrane COVID‐19 Study Register, Scopus, and WHO COVID‐19 Global literature on coronavirus disease database, identifying completed and ongoing studies without language restrictions and incorporating studies up to 11 July 2022.
This is a LSR. We conduct monthly update searches that are being made publicly available on the open science framework (OSF) platform.
Selection criteria
Studies were eligible if they were randomized controlled trials (RCTs) comparing nirmatrelvir/ritonavir plus standard of care with standard of care with or without placebo, or any other intervention for treatment of people with confirmed COVID‐19 diagnosis, irrespective of disease severity or treatment setting, and for prevention of SARS‐CoV‐2 infection.
We screened all studies for research integrity. Studies were ineligible if they had been retracted, or if they were not prospectively registered including appropriate ethics approval.
Data collection and analysis
We followed standard Cochrane methodology and used the Cochrane risk of bias 2 tool. We rated the certainty of evidence using the GRADE approach for the following outcomes: 1. to treat outpatients with mild COVID‐19; 2. to treat inpatients with moderate‐to‐severe COVID‐19: mortality, clinical worsening or improvement, quality of life, (serious) adverse events, and viral clearance; 3. to prevent SARS‐CoV‐2 infection in post‐exposure prophylaxis (PEP); and 4. pre‐exposure prophylaxis (PrEP) scenarios: SARS‐CoV‐2 infection, development of COVID‐19 symptoms, mortality, admission to hospital, quality of life, and (serious) adverse events.
We explored inequity by subgroup analysis for elderly people, socially‐disadvantaged people with comorbidities, populations from LICs and LMICs, and people from different ethnic and racial backgrounds.
Main results
As of 11 July 2022, we included one RCT with 2246 participants in outpatient settings with mild symptomatic COVID‐19 comparing nirmatrelvir/ritonavir plus standard of care with standard of care plus placebo. Trial participants were unvaccinated, without previous confirmed SARS‐CoV‐2 infection, had a symptom onset of no more than five days before randomization, and were at high risk for progression to severe disease. Prohibited prior or concomitant therapies included medications highly dependent on CYP3A4 for clearance and CYP3A4 inducers.
We identified eight ongoing studies.
Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease
For the specific population of unvaccinated, high‐risk patients nirmatrelvir/ritonavir plus standard of care compared to standard of care plus placebo may reduce all‐cause mortality at 28 days (risk ratio (RR) 0.04, 95% confidence interval (CI) 0.00 to 0.68; 1 study, 2224 participants; estimated absolute effect: 11 deaths per 1000 people receiving placebo compared to 0 deaths per 1000 people receiving nirmatrelvir/ritonavir; low‐certainty evidence, and admission to hospital or death within 28 days (RR 0.13, 95% CI 0.07 to 0.27; 1 study, 2224 participants; estimated absolute effect: 61 admissions or deaths per 1000 people receiving placebo compared to eight admissions or deaths per 1000 people receiving nirmatrelvir/ritonavir; low‐certainty evidence).
Nirmatrelvir/ritonavir plus standard of care may reduce serious adverse events during the study period compared to standard of care plus placebo (RR 0.24, 95% CI 0.15 to 0.41; 1 study, 2224 participants; low‐certainty evidence). Nirmatrelvir/ritonavir plus standard of care probably has little or no effect on treatment‐emergent adverse events (RR 0.95, 95% CI 0.82 to 1.10; 1 study, 2224 participants; moderate‐certainty evidence), and probably increases treatment‐related adverse events such as dysgeusia and diarrhoea during the study period compared to standard of care plus placebo (RR 2.06, 95% CI 1.44 to 2.95; 1 study, 2224 participants; moderate‐certainty evidence). Nirmatrelvir/ritonavir plus standard of care probably decreases discontinuation of study drug due to adverse events compared to standard of care plus placebo (RR 0.49, 95% CI 0.30 to 0.80; 1 study, 2224 participants; moderate‐certainty evidence).
No study results were identified for improvement of clinical status, quality of life, and viral clearance.
Subgroup analyses for equity
Most study participants were younger than 65 years (87.1% of the : modified intention to treat (mITT1) population with 2085 participants), of white ethnicity (71.5%), and were from UMICs or HICs (92.1% of study centres). Data on comorbidities were insufficient.
The outcome ‘admission to hospital or death’ was investigated for equity: age (< 65 years versus ≥ 65 years) and ethnicity (Asian versus Black versus White versus others). There was no difference between subgroups of age. The effects favoured treatment with nirmatrelvir/ritonavir for the White ethnic group. Estimated effects in the other ethnic groups included the line of no effect (RR = 1). No subgroups were reported for comorbidity status and World Bank country classification by income level. No subgroups were reported for other outcomes.
Nirmatrelvir/ritonavir for treating COVID‐19 in inpatient settings with moderate to severe disease
No studies available.
Nirmatrelvir/ritonavir for preventing SARS‐CoV‐2 infection (PrEP and PEP)
No studies available.
Authors' conclusions
There is low‐certainty evidence that nirmatrelvir/ritonavir reduces the risk of all‐cause mortality and hospital admission or death based on one trial investigating unvaccinated COVID‐19 participants without previous infection that were at high risk and with symptom onset of no more than five days. There is low‐ to moderate‐certainty evidence that nirmatrelvir/ritonavir is safe in people without prior or concomitant therapies including medications highly dependent on CYP3A4.
Regarding equity aspects, except for ethnicity, no differences in effect size and direction were identified.
No evidence is available on nirmatrelvir/ritonavir to treat hospitalized people with COVID‐19 and to prevent a SARS‐CoV‐2 infection.
We will continually update our search and make search results available on OSF.
Plain language summary
Is the combination nirmatrelvir plus ritonavir effective for treating or preventing COVID‐19?
Key messages
Nirmatrelvir/ritonavir (Paxlovid®) is evaluated for the treatment of coronavirus disease 2019 (COVID‐19).
Nirmatrelvir/ritonavir may lead to fewer deaths and improve patient condition, as assessed by need for hospitalization or death within 28 days.
Data are only available for non‐vaccinated people at increased risk for disease progression receiving treatment within five days of symptom onset.
We found eight ongoing studies. We will update our search every month.
What is nirmatrelvir/ritonavir (Paxlovid®)?
The combination of nirmatrelvir with ritonavir (Paxlovid®) is a new medicine developed to treat infection with the SARS‐CoV‐2 virus and aims to avoid severe COVID‐19 in people without symptoms, or those with mild symptoms. Ritonavir increases the effectiveness of nirmatrelvir, however it can interact with many other drugs which can increase side effects.
What did we want to find out?
We wanted to know if nirmatrelvir/ritonavir reduces death, illness, and length of infection in people with COVID‐19, or if it is useful in prevention of the disease. We included studies comparing the medicine with placebo (dummy treatment), no treatment, usual care, or any other treatments for COVID‐19. We addressed equity and wanted to know whether there are certain groups of people for which nirmatrelvir/ritonavir works best or is less effective. We looked at elderly people, socially disadvantaged people with comorbidities, people from low‐income and lower‐middle‐income countries, and people from different ethnic and racial backgrounds.
We evaluated the effects of nirmatrelvir/ritonavir in people with COVID‐19 regarding:
– people dying;
– whether people's COVID‐19 symptoms got better or worse;
– quality of life;
– unwanted effects of the drug;
– virus elimination.
For prevention, we sought the effect on preventing COVID‐19 and SARS‐CoV‐2 infection.
What did we do? We searched for randomized controlled trials that investigated nirmatrelvir/ritonavir to prevent or treat COVID‐19 in humans. People receiving nirmatrelvir/ritonavir as treatment had to have laboratory‐test confirmed COVID‐19 and be treated in hospital or as outpatients. People receiving nirmatrelvir/ritonavir to prevent an infection had to have a high risk of contacting the disease or had to have a high risk contact with a confirmed COVID‐19 patient.
We compared and summarized the results of the studies and rated our confidence in the evidence, based on common criteria as to how reliable the evidence is.
For all effects, we examined differences with respect to age groups, level of comorbidity, country according to the World Bank country classification by income level, and ethnicity.
What did we find?
We found one study with 2246 participants that investigated nirmatrelvir/ritonavir compared to placebo for the treatment of COVID‐19 in outpatients. The included participants were not vaccinated, without previous confirmed SARS‐CoV‐2 infection, had a symptom onset of no more than five days before start of the treatment, and were at high risk for progression to severe disease due to a comorbidity or risk factor such as current smoking.
We also found eight ongoing studies that have not yet been completed.
Main results
Treating outpatients with COVID‐19
For the specific population of unvaccinated, high‐risk patients, nirmatrelvir/ritonavir may;
‐ lead to fewer deaths; and
‐ improve patients' condition assessed by need for hospitalization or death within 28 days;
‐ reduce serious unwanted events.
For the specific population of unvaccinated, high‐risk patients, nirmatrelvir/ritonavir probably:
‐ has little effect on any unwanted events;
‐ increases any treatment‐related unwanted events (mostly taste disturbance and diarrhoea);
‐ probably decreases discontinuation of study drug due to unwanted events.
Equity aspects
Most study participants were younger than 65 years, of white ethnicity and were from upper‐middle‐ or high‐income countries. There was no difference in effectiveness between younger and older participants. There was a positive effect in all ethnic groups, which was clearest for people of white ethnicity but numbers of participants in the other ethnic groups were low. No subgroups were reported for different levels of comorbidity and World Bank country classification by income level.
No subgroups were reported for other outcomes.
What are the limitations of the evidence?
Our confidence in the evidence is low to moderate because we could only include one study and some events, such as deaths or serious adverse events were rare. The study did not report everything we were interested in, such as quality of life and symptom resolution and had a highly specific patient population of unvaccinated people at high risk of progression to severe COVID‐19.
How up to date is this evidence?
The evidence is up‐to‐date to 11 July 2022.
According to this review's living approach, we will update our search monthly. We are making search results and new relevant studies publicly available.
Summary of findings
Summary of findings 1. Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease.
|
Patient or population: unvaccinated, nonhospitalized people with mild symptomatic disease (WHO scale 2 to 3) at high risk for progression to severe disease Setting: outpatient Intervention: nirmatrelvir/ritonavir (plus standard of care) Comparison: placebo (plus standard of care) | ||||||
| Outcomes |
Anticipated absolute effects* (95% CI) |
Relative effect (95% CI) | N° of participants (studies) | Certainty of the evidence (GRADE) | Comment | |
| Risk with placebo | Risk with nirmatrelvir/ritonavir | |||||
| All‐cause mortality at day 28 | 11 per 1000 |
0 per 1000 |
RR 0.04 (0.00 to 0.68) | 2224 (1 RCT) | ⊕⊕⊝⊝ Lowa |
Nirmatrelvir/ritonavir may reduce all‐cause mortality1 |
| Difference: 11 fewer per 1000 (11 fewer to 4 fewer) | ||||||
| Worsening of clinical status | ||||||
| Admission to hospital or death within 28 days | 61 per 1000 |
8 per 1000 |
RR 0.13 (0.07 to 0.27) | 2224 (1 RCT) | ⊕⊕⊝⊝ Lowb |
Nirmatrelvir/ritonavir may reduce (COVID‐19‐related) hospitalization or death2 |
| Difference: 53 fewer per 1000 (57 fewer to 45 fewer) | ||||||
| Admission to intensive care unit (ICU) or death within 28 days | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported admission to ICU or death |
| Improvement of clinical status | ||||||
| All initial symptoms resolved at 28 days, and up to the longest follow‐up | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported all initial symptoms resolved |
| Time to symptom resolution | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported time to symptom resolution |
| Quality of life up to 28 days and longest follow‐up available | ‐ | ‐ | ‐ | ‐ | ‐ | No studies reported quality of life |
| Serious adverse events during the study period | 66 per 1000 | 16 per 1000 | RR 0.24 (0.15 to 0.41) | 2224 (1 RCT) | ⊕⊕⊝⊝ Lowc |
Nirmatrelvir/ritonavir may reduce SAEs1 |
| Difference: 50 fewer per 1000 (56 fewer to 39 fewer) | ||||||
| Adverse events | ||||||
| Any grade treatment‐emergent adverse events (TEAE) during the study period | 239 per 1000 |
227 per 1000 |
RR 0.95 (0.82 to 1.10) | 2224 (1 RCT) | ⊕⊕⊕⊝ Moderated |
Nirmatrelvir/ritonavir probably has little or no effect on any TEAE1 |
| Difference: 12 fewer per 1000 (43 fewer to 24 more) | ||||||
| Any grade treatment‐related adverse events (TRAE) during the study period | 38 per 1000 |
78 per 1000 |
RR 2.06 (1.44 to 2.95) | 2224 (1 RCT) | ⊕⊕⊕⊝ Moderated |
Nirmatrelvir/ritonavir probably increases any TRAE (mostly attributed to dysgeusia and diarrhea)1 |
| Difference: 40 more per 1000 (17 more to 74 more) | ||||||
| Discontinuation of study drug due to adverse events | 42 per 1000 |
21 per 1000 |
RR 0.49 (0.30 to 0.80) | 2224 (1 RCT) | ⊕⊕⊕⊝ Moderated |
Nirmatrelvir/ritonavir probably decreases discontinuation of study drug due to adverse events1 |
| Difference: 21 fewer per 1000 (29 fewer to 8 fewer) | ||||||
| Viral clearance at 14 days | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported viral clearance |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk on the comparison group and the relative effect of the intervention (and its 95% confidence interval). CI: confidence interval; RCT: randomized controlled trial; RR: risk ratio; SAE: serious adverse event; AE: adverse event; TEAE: treatment emergent adverse event; TRAE: treatment related adverse event | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is the possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
Explanations on ‘certainty in the evidence (GRADE)’
aDowngraded one level for serious risk of bias (inappropriate analysis) and one level for serious imprecision (few events) bDowngraded one level for serious risk of bias (inappropriate analysis) and one level for serious indirectness (COVID‐19 related hospitalization) cDowngraded one level for serious risk of bias (inappropriate analysis) and one level for serious imprecision (due to few SAEs other than hospitalization or death) dDowngraded one level for serious risk of bias (inappropriate analysis)
Explanations on ‘equity considerations’
Most study participants were younger than 65 years, of white ethnicity, and from upper middle‐ or high‐income countries. No subgroup analysis was possible for comorbidity (high risk versus low risk population) as the included study only investigated a high‐risk population.
1No subgroup analyses were reported for age, ethnicity, and World Bank country classification by income level. We are uncertain whether results are applicable to all prespecified subgroups. 2Subgroup analyses were reported for age and ethnicity only. There was no difference between subgroups of age. The effects favoured a treatment with nirmatrelvir/ritonavir for the white ethnic group. Estimated effects of the other ethnic groups included the line of no effect (RR = 1). Numbers of participants in the other ethnic groups were low. No subgroups were reported for World Bank country classification by income level.
Background
Description of the condition
Having been declared the sixth public health emergency of international concern by the World Health Organization (WHO), severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and the resulting outbreak of coronavirus disease 2019 (COVID‐19) has caused a pandemic that has accelerated at an unprecedented scale. As of May 2022, more than two years after the first reported SARS‐CoV‐2 case, there have been over 500 million confirmed cases of COVID‐19, including more than 6 million deaths in 222 countries and territories (WHO 2021a).
COVID‐19 is a rapidly spreading infectious disease caused by SARS‐CoV‐2 (WHO 2020). SARS‐CoV‐2 is a positive‐sense, double‐stranded ribonucleic acid (RNA) virus that belongs to the Coronaviridae family (Kumar 2020).
SARS‐CoV‐2 uses its spike glycoprotein‐S to bind to an angiotensin‐converting enzyme 2 (ACE2) receptor on a host cell surface to initiate the infection process. Angiotensin‐converting enzyme 2 receptors are expressed in lung, heart, kidney, intestine, and endothelium in the human body. The main expression site that is central to the pathophysiology of COVID‐19 is respiratory epithelium of the nasopharynx. SARS‐CoV‐2 genes can then enter the human cell to begin viral replication and shedding. The process of viral replication is mediated by, and depends on, viral proteolytic enzymes (proteases), including main protease (Mpro, also known as 3C‐like protease, 3CLpro) (Amin 2021; Anand 2003). Viral variants mainly present mutational changes in the spike glycoprotein (Harvey 2021). The spike glycoprotein is recognized by the immune system, and is the main target of vaccines against SARS‐CoV‐2 (Salvatori 2020; Walls 2020). In contrast, the Mpro active binding site is highly conserved between different virus variants and less affected by mutations.
Most individuals with COVID‐19 are either asymptomatic or develop mild symptoms not requiring hospitalizations (approximately 80% to 90%), depending on the time of the investigation, the cohort investigated, and the virus variant (Chen 2010; Funk 2021; Wu 2020). A smaller proportion is affected by severe (approximately 11% to 20%) or critical (approximately 1% to 5%) disease with hospitalization and intensive care unit (ICU) admittance due to respiratory failure, septic shock, or multiple organ dysfunction syndrome (Funk 2021; Wu 2020). Risk for severe disease, hospitalization, and mortality is higher for individuals aged 65 years or older, males, smokers, and individuals with certain underlying medical conditions, such as cancer, chronic kidney disease, chronic obstructive pulmonary disease (COPD), moderate‐to‐severe asthma, immunocompromized state, obesity, sickle cell disease, or type 2 diabetes mellitus (Booth 2021; Huang 2020; Karagiannidis 2020; Petrilli 2020; Williamson 2020). Most common symptoms and signs of acute infection include fever, cough, fatigue, and shortness of breath (Grant 2020). Infection with SARS‐CoV‐2 may also lead to long‐term health conditions including persistent fatigue, cognitive dysfunction, and post‐exertional malaise (Huang 2021).
The gold standard for confirming a SARS‐CoV‐2 infection is the reverse transcription polymerase chain reaction (RT‐PCR)‐based detection of viral RNA from a nasopharyngeal swab test, anterior nares swab test (nasal swab), sputum, or tracheal secretion, with a sensitivity ranging from 70% to 98%, depending on pretest probability (Watson 2020). Offering lower sensitivity but greater practicality and accessibility, antigen tests have received increased attention, especially in point‐of‐care diagnostics of COVID‐19 (Dinnes 2021; WHO 2020a).
Viral transmission is typically inferred from population‐level information. Inherent properties of virus variants of concern, and individual differences in infectiousness among individuals or groups, and differences in local herd immunity make it difficult to contain its spread in the community (WHO 2021a). Currently, the most effective strategy to combat the pandemic is vaccination. COVID‐19 vaccines are effective and can reduce the risk of getting SARS‐CoV‐2 and decrease hospitalization rates (CDC 2021; Juthani 2021). However, vaccination can fail to produce a sufficiently robust immune response, and the response it does elicit can wane over time and be less effective against new variants (Lin 2022). Furthermore, some people cannot get a COVID‐19 vaccine for medical reasons, such as anaphylaxis, or may not develop sufficient immunogenicity following vaccination (NHS 2021). Others are hesitant to get vaccinated due to concerns about vaccine side effects and safety (Altulahi 2021; Wang 2021). The major obstacle in overcoming this pandemic, however, is vaccine inequity in different regions of the world (WHO 2021). Additionally, emerging new virus variants can increase the risk of infection in all countries, including the vaccinated population if vaccines become less effective due to viral immune escape mutations as could be seen with the recent Omicron variant (Ren 2022). Therefore, research on pre‐exposure or post‐exposure prophylaxis of SARS‐CoV‐2 infection and treatment of COVID‐19 is still of high relevance and is being carried out under great pressure worldwide.
Description of the intervention
Pfizer's new drug combination nirmatrelvir/ritonavir (Paxlovid®) aims to avoid severe COVID‐19 in asymptomatic people or those with mild symptoms, thereby decreasing hospitalization and death. Nirmatrelvir/ritonavir is a combination of the SARS‐CoV‐2 protease inhibitor nirmatrelvir, and ritonavir, a CYP3A4 inhibitor used in the treatment of HIV to enhance HIV protease inhibitors. Nirmatrelvir blocks the activity of the SARS‐CoV‐2‐3CLpro protease, an enzyme needed for viral replication. In humans nirmatrelvir is metabolized by the P450 cytochrome enzyme CYP3A4. In order to remain active in the body for longer periods of time, nirmatrelvir is co‐administered with low‐dose ritonavir, to slow down the breakdown of nirmatrelvir (Pfizer 2021). Nirmatrelvir/ritonavir is to be administered orally within five days of symptom onset and taken twice daily for five days. Given the inactivation of CYP3A4 by ritonavir, a common enzyme in drug metabolism, ritonavir interferes with the metabolism of many drugs, can alter their plasma concentrations, and increase drug‐related adverse effects. The applicability of nirmatrelvir/ritonavir may thus be limited in some populations at high risk of severe COVID‐19, such as those with comorbidities requiring medications metabolized using the CYP3A4 enzyme. As 3CLpro, the substrate‐binding site is highly conserved among all coronaviruses and shares no homology with human proteases, a SARS‐CoV‐2‐3CLpro antagonist will be highly specific to SARS‐CoV‐2 and less affected by virus mutations (Dai 2020) compared to antivirals binding to other sites, more prone to mutation.
Therapeutic options for treatment of COVID‐19 in the outpatient setting or for prevention of a SARS‐CoV‐2 infection in close contacts of infected people are still limited. In September 2021, the WHO gave the conditional recommendation to use a combination of neutralizing monoclonal antibodies (mAbs) (casirivimab and imdevimab) in non‐severe COVID‐19 patients at the highest risk of severe disease, and in seronegative patients with severe or critical COVID‐19 (WHO 2021b). A recommendation for sotrovimab, another mAb in high‐risk outpatients followed in January 2022 (WHO 2021b). Accurate clinical prediction guides to establish individual patient risk and benefit from monoclonal antibodies are lacking, and the current usual care for non‐hospitalized COVID‐19 patients varies greatly between countries. Unfortunately, contrary to sotrovimab, the combination of casirivimab and imdevimab has not retained neutralizing activity against the Omicron variant (Takashita 2022). Remdesivir, originally developed to treat hepatitis C, has proven to decrease hospitalization rates in unvaccinated COVID‐19 patients and is currently recommended in several countries and by the WHO for outpatient treatment of infected patients with high risk of disease progression (Gottlieb 2022; NICE 2021; NIH 2021; WHO 2021b). To date only one direct oral antiviral treatment, molnupiravir, has been authorized by the Medicines and Healthcare products Regulatory Agency (MHRA) for infected, non‐hospitalized individuals with at least one risk factor for severe disease (Merck 2021; NCT04575597) and international guidelines are being constantly updated (NICE 2021; WHO 2021b). However, clinical data on molnupiravir regarding efficacy and safety are currently limited. Other strategies to treat COVID‐19 have included re‐purposing existing drugs for an antiviral intention, including ivermectin. However, so far there is no proven effect for ivermectin (Popp 2021a) and, therefore, it should not be used for treatment of COVID‐19 outside well‐designed clinical trials (WHO 2021b). Experimental antivirals being studied include umifenovir (Deng 2020) and favipiravir in combination with molnupiravir (Eloy 2021).
How the intervention might work
Viral non‐structural proteins are important for replication and transcription of SARS‐CoV‐2. The SARS‐CoV‐2‐3CLpro plays a key role in the production of 16 non‐structural proteins of SARS‐CoV‐2. Inhibition of 3CLpro by nirmatrelvir blocks the release of these non‐structural proteins, thereby suppressing further maturation and replication of SARS‐CoV‐2 (Zhang 2021). Boosting with ritonavir, a CYP3A4 inhibitor, is required to increase nirmatrelvir to a concentration that is effective against SARS‐CoV‐2 (Pfizer 2021). There is reason to presume that viral load, infectivity, and disease severity are positively correlated (Fajnzylber 2020; Kawasuji 2020; Liu 2020). Decreasing the viral load by blocking viral replication could thereby prevent disease progression and limit the infectivity of COVID‐19 patients.
Nirmatrelvir (PF‐07321332), the protease inhibitor agent in nirmatrelvir/ritonavir, was developed by modification of an earlier clinical candidate PF‐00835231, originally developed as a potent inhibitor of recombinant SARS‐CoV‐1‐3CLpro during the SARS‐CoV‐1 pandemic in 2002/03. SARS‐CoV‐1‐3CLpro and SARS‐CoV‐2‐3CLpro share 96% sequence homology (Zhang 2020). However, PF‐00835231 needs to be administered intravenously, limiting its application mainly to hospital settings. Stepwise modification led to the new substance nirmatrelvir, with increased oral bioavailability. To date, nirmatrelvir has shown potent inhibition of 3CLpro from all coronavirus types known to infect humans, as well as favourable selectivity profiles against mammalian proteases (Owen 2021).
Lufotrelvir, the phosphate prodrug of PF‐00835231, with a similar mechanism of action to nirmatrelvir but with intravenous administration is currently studied for safety and efficacy in the treatment of hospitalized COVID‐19 patients, including trials in conjunction with remdesivir, as in vitro data showed synergistic effects (de Vries 2020; NCT04501978; NCT04535167).
Why it is important to do this review
Current treatment for hospitalized patients includes supportive care with oxygen in moderate cases, systemic corticosteroids, baricitinib, IL‐6 blockers, and non‐invasive ventilation or invasive mechanical ventilation and extracorporeal membrane oxygenation (ECMO) in severe or critical cases (Agarwal 2020). Overall, data from randomized controlled trials (RCTs) with exception to the aforementioned treatments do not demonstrate a clear, major clinical benefit with most drugs evaluated so far. Therapeutic options for treatment of COVID‐19 in the outpatient setting or for prevention of a SARS‐CoV‐2 infection in close contacts of infected people or in people at risk are still limited. In light of the ongoing potential for evolving virus variants, scarcity of effective treatments, and global vaccination coverage issues, the role of effective oral therapies for patients at high risk of severe disease is of utmost interest for reducing morbidity and mortality secondary to COVID‐19.
The COVID‐19 pandemic has brought social and racial injustice and health inequity in the spotlight of public health. The impact of COVID‐19 disproportionally affects elderly, poor, racial and ethnic minorities, as well as a broad range of vulnerable populations, putting them at increased risk of illness and death (Killerby 2020). Reasons include, but are not limited to, inequitable viral spread in areas of dense population, and limited mitigation capacity due to a higher prevalence of chronic conditions or poor access to high‐quality medical care (Shadmi 2020). Studies of the average effects of interventions, which control for confounding across individual and population‐level characteristics, hide their impact on health equity (Welch 2012). Therefore, special consideration of health equity in studies and meta‐analyses is needed, which can be done by reporting and analysis of population characteristics per outcome on the study‐level.
Prevention of COVID‐19 in people at high risk for developing severe disease requiring hospital level treatment is critical, especially from a global perspective considering limited hospital capacity in low‐income countries (LICs) and lower‐middle‐income countries (LMICs). Antiviral drugs such as nirmatrelvir/ritonavir might therefore be of vital importance in the global fight against SARS‐CoV‐2. It is however yet to be determined whether the fact that nirmatrelvir/ritonavir has to be administered within five days of symptom onset may decrease its applicability due to inadequate healthcare infrastructure and lack of access to public health and medical care in LMICs, in elderly, and in racial and ethnic minority populations.
To allow equity of access across countries, Pfizer has signed a voluntary licence agreement for nirmatrelvir/ritonavir with the Medicines Patent Pool (MPP), a United Nations‐backed public health organization working to increase access to life‐saving medicines for low‐ and middle‐income countries (MPP 2021a; Pfizer 2021). With the MPP having a licence on ritonavir for many years, the agreement will enable MPP to facilitate additional production and distribution of both ritonavir and nirmatrelvir by granting sublicenses to qualified generic medicine manufacturers (MPP 2021b). Pfizer further aims to offer a tiered pricing approach based on the income level of a country, with high‐income countries (HICs) and upper‐middle income countries (UMICs) paying more than LICs, which will pay a not‐for‐profit price (Pfizer 2021).
Pfizer has ongoing trials for nirmatrelvir/ritonavir on clinical outcomes for patients with COVID‐19 at high and standard risk, and for post‐exposure prophylaxis (Pfizer 2021). We expect that many new studies investigating nirmatrelvir/ritonavir will be initiated in hospitals worldwide after Emergency Use Authorization (EUA) by the US Food and Drug Administration (FDA). This review is designed as a living systematic review with continuous monitoring of new and ongoing studies. We aim to keep the evidence based on clinical studies investigating nirmatrelvir/ritonavir for COVID‐19 up to date.
This Cochrane Review will provide a complete evidence profile, based on current Cochrane standards, for nirmatrelvir/ritonavir with regard to efficacy and safety for pre‐ and post‐exposure prophylaxis and treatment of COVID‐19 in out‐ and inpatient settings.
Objectives
To assess the efficacy and safety of nirmatrelvir/ritonavir (Paxlovid®) plus standard of care compared to standard of care with or without placebo, or any other intervention for treating COVID‐19 and for preventing SARS‐CoV‐2 infection.
To explore equity aspects in subgroup analyses.
To keep up to date with the evolving evidence base using a living systematic review (LSR) approach and make new relevant studies available to readers in‐between publication of review updates.
Methods
Criteria for considering studies for this review
Types of studies
Studies were eligible if they were randomized controlled trials (RCTs).
We searched for full‐text journal articles published in PubMed‐indexed and non‐indexed journals, preprint articles, results published in trials registers, clinical study reports (CSRs), and abstract publications. We applied no restrictions on the language of published articles.
We screened all identified studies for research integrity using a tool developed by our group to deal with problematic studies (see Selection of studies).
Types of participants
Treating COVID‐19
Studies were eligible if they included participants with confirmed SARS‐CoV‐2 infection (reverse transcription polymerase chain reaction (RT‐PCR) or antigen testing), regardless of age, gender, ethnicity, serology status, vaccination status, previous SARS‐CoV‐2 infection, and risk factors for developing severe COVID‐19. If studies included participants with a confirmed or suspected COVID‐19 diagnosis, we used only the data for the patient population with confirmed COVID‐19 diagnosis.
COVID‐19 severity was classified according to the WHO clinical progression scale (Marshall 2020) into mild (WHO 1 to 3), moderate (WHO 4 to 5), and severe (WHO 6 to 9).
In cases where data have not been reported separately for people with confirmed or suspected COVID‐19 diagnosis, we excluded the study.
Preventing SARS‐CoV‐2 infection
We synthesized evidence for both, post‐exposure prophylaxis (PEP) and pre‐exposure prophylaxis (PrEP) scenarios. For post‐exposure prophylaxis, we included studies investigating participants who were not infected with SARS‐CoV‐2 at enrolment (negative RT‐PCR), but were at high risk of developing the infection following exposure to infected people or infectious viral particles.
For pre‐exposure prophylaxis, we included studies investigating participants who were not infected with SARS‐CoV‐2 at enrolment (negative RT‐PCR) and were not yet exposed to infected people or infectious viral particles, but are at increased risk of contacting the disease (e.g. healthcare workers).
Participants in both settings were eligible regardless of age, gender, ethnicity, serology status, vaccination status, previous SARS‐CoV‐2 infection, and risk factors for developing severe COVID‐19. Eligible trials must have reported the history of previous SARS‐CoV‐2 infection or serological evidence and the vaccination status of included participants. A history of SARS‐CoV‐2 infection or vaccination was not an exclusion criterion.
Types of interventions
All doses and regimens of nirmatrelvir/ritonavir were eligible for this systematic review. Nirmatrelvir/ritonavir is authorized and approved by the US Food and Drug Administration (FDA) (EUA for Paxlovid®) at a dose of 300 mg (as two 150 mg tablets) of nirmatrelvir with one 100 mg tablet of ritonavir, given twice‐daily for five days.
We compared nirmatrelvir/ritonavir plus standard of care with standard of care with or without placebo, or to any other intervention for treating COVID‐19 and for preventing SARS‐CoV‐2 infection. Co‐interventions (standard of care) must have been comparable between the study arms.
Types of outcome measures
We evaluated core outcomes in accordance with the Core Outcome Measures in Effectiveness Trials (COMET) Initiative for COVID‐19 patients (COMET 2020; Marshall 2020), and additional outcomes that have been prioritized by consumer representatives and the German guideline panel for treatment of people with COVID‐19 (German AWMF Guideline 2021).
We used different outcome sets for the use of nirmatrelvir/ritonavir for treating people with COVID‐19 in the out‐ and inpatient setting, and for preventing SARS‐CoV‐2 infection. If studies were eligible for inclusion regarding study design, population, intervention, and comparator, but no outcomes of interest have been reported, they were not included for meta‐analysis. However, we have summarized reported outcomes for all included studies in the 'Characteristics of included studies' table. We did not exclude studies if they did not report outcomes of interest.
Primary outcomes
Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease
All‐cause mortality at day 28, day 60, time‐to‐event, and up to the longest follow‐up.
-
Worsening of clinical status within 28 days.
Admission to hospital or death.
Admission to intensive care unit (ICU) or death.
-
Improvement of clinical status.
All initial symptoms resolved (asymptomatic) at day 14, day 28, and up to the longest follow‐up.
Time to symptom resolution.
Quality of life, including fatigue and neurological status, assessed with standardized scales (e.g. WHOQOL‐100) at up to 7 days, up to 28 days, and longest follow‐up available.
Serious adverse events (SAEs) during the study period, defined as number of participants with any event.
-
Adverse events (AEs) during the study period, defined as number of participants with any event.
Any grade treatment‐emergent adverse events (TEAEs); adverse events temporally related to the study treatment).
Any grade treatment‐related adverse events (TRAE; adverse events assessed as causally related to the study treatment by the study investigator).
Discontinuation of study drug due to adverse events.
Viral clearance, assessed with RT‐PCR test for SARS‐CoV‐2 at baseline, and 3, 7, and 14 days.
Nirmatrelvir/ritonavir for treating COVID‐19 in inpatient settings with moderate to severe disease
We used a similar outcome set for treating COVID‐19 patients in out‐ and inpatient settings, but with different definitions of the outcomes of ‘Worsening of clinical status’ and ‘Improvement of clinical status’. For inpatient settings we used the following definitions.
-
Worsening of clinical status within 28 days.
Participants with new need for invasive mechanical ventilation or death.
Participants with need for ICU admission or death.
-
Improvement of clinical status within 28 days.
Participants discharged alive.
Participants should be discharged without clinical deterioration or death.
Nirmatrelvir/ritonavir for preventing SARS‐CoV‐2 infection (PrEP and PEP)
We used the same outcome set for PEP and PrEP scenarios, but with different time frames for the outcome assessment. For PEP studies, the relevant period is 14 to 28 days and for PrEP studies, a longer period of up to six months is relevant.
SARS‐CoV‐2 infection (confirmed by RT‐PCR or antigen testing) at 14 days (PEP) and six months (PrEP).
-
Development of clinical COVID‐19 symptoms up to 28 days (PEP) and six months (PrEP); e.g. assessed in accordance with individual items of the WHO scale (Marshall 2020), or any other standardized scale. If the study did not use the standardized WHO scale to assess the status of the participants, we would categorize their status according to the WHO scale with the information provided by the study.
Uninfected (WHO scale 0).
Ambulatory mild disease (WHO scale 1 to 3).
Hospitalized with moderate disease (WHO scale 4 to 5).
Hospitalized with severe disease (WHO scale 7 to 9).
Mortality (WHO scale 10).
All‐cause mortality up to the longest follow‐up.
Admission to hospital or death within 28 days (PEP) and six months (PrEP).
Quality of life assessed with the standardized scale, WHOQOL‐100, up to 28 days (PEP) and six months (PrEP), and at longest follow‐up available.
Serious adverse events during the study period, defined as number of participants with any event.
-
Adverse events during the study period, defined as number of participants with any event.
Any grade treatment‐emergent adverse events (any TEAE; adverse events temporally related to the study treatment).
Any grade treatment‐related adverse events (TRAE; adverse events assessed as causally related to the study treatment by the study investigator).
Discontinuation of study drug due to AEs.
Timing of outcome measurement
We collected information on outcomes from all time points reported in the publications and study reports. If only a few studies contributed data to an outcome, we planned to pool different time points, provided the studies produced valid data and pooling was clinically reasonable. The current review version included one study.
In case of time‐to‐event analysis, e.g. for time to death, we planned to use the longest follow‐up time measured from randomization.
We have reported time points of outcome measurement in the footnotes of the forest plots. We included SAEs and AEs occurring during the study period, includingAEs during active treatment and long‐term AEs. If sufficient data had been available, we planned to group the measurement time points of eligible outcomes into those measured directly after treatment (up to seven days), medium‐term outcomes (up to 14 days), and longer‐term outcomes (28 days or more).
Secondary outcomes
This review specifies no secondary outcomes. All outcomes were treated as a primary outcome set which informed the summary of findings tables.
Search methods for identification of studies
Electronic searches
Our Information Specialist (MIM) conducted systematic searches in the following sources from the inception of each database to 11 July 2022 and placed no restrictions on the language of publication.
-
Cochrane COVID‐19 Study Register (CCSR) (www.covid-19.cochrane.org), comprising:
MEDLINE (PubMed), weekly updates;
Embase, weekly updates;
ClinicalTrials.gov (www.clinicaltrials.gov), daily updates;
World Health Organization International Clinical Trials Registry Platform (ICTRP) (www.who.int/trialsearch), weekly updates;
medRxiv (www.medrxiv.org), weekly updates;
Cochrane Central Register of Controlled Trials (CENTRAL), monthly updates.
Scopus.
WHO COVID‐19 Global literature on coronavirus disease (search.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/).
For detailed search strategies, see Appendix 1. As this review is a living systematic review (LSR), we conduct monthly update searches which are being made publicly available on OSF [osf.io/7g49c/; Reis 2022a]. See section ‘Methods for future updates’ on specific LSR methodology.
We do not conduct separate searches of the databases required by the Methodological Expectations of Cochrane Intervention Reviews (MECIR) standards (Higgins 2021), since these databases are being regularly searched for the production of the CCSR.
Searching other resources
We searched other potentially eligible studies or ancillary publications by searching the reference lists of included studies, systematic reviews, and meta‐analyses.
In the event there were no public study results, we planned at the protocol stage to contact the manufacturer (Pfizer Inc.) through their dedicated website (www.pfizer.com/science/clinical_trials/trial_data_and_results/data_requests) to obtain access to individual de‐identified participant data and related study documents, e.g. protocol, statistical analysis plan (SAP), clinical study report (CSR). At the time point of publication of the first review version, the journal publication of one Pfizer study was already available, and no further enquiries were necessary.
Data collection and analysis
Selection of studies
Inclusion criteria
We performed study selection in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2022). Three review authors (Stefanie Reis (SR), Rebecca Kuehn (RK), and Stephanie Weibel (SW)) independently screened titles and abstracts of identified records. We retrieved full‐text articles and independently assessed eligibility of the remaining records against the predefined eligibility criteria. We resolved discrepancies through discussion between the review authors. We included studies irrespective of whether measured outcome data were reported in a 'usable' way. We collated multiple reports of the same study, so that the study, rather than the report, is the unit of interest in the review.
Research integrity screening
Early in this pandemic several studies were identified as unsuitable for public use due to research ethics and integrity concerns and were either retracted, withdrawn, or noted with concern (Bramstedt 2020). Cochrane has published a policy on managing problematic studies and guidance to facilitate research integrity checks in the reviews it publishes, but these checks have not routinely formed part of evidence synthesis processes to date (Cochrane policy ‐ managing problematic studies). Current standard tools for systematic reviews do not systematically consider issues of research integrity. However, there are useful tools available such as the ‘REAPPRAISED’ checklist for evaluation of publication integrity (Grey 2020) or the data extraction sheet from the Cochrane Pregnancy and Childbirth Group that addresses scientific integrity and trustworthiness (Data extraction template 2021). We modified these existing tools and developed a specific tool for studies in this pandemic that we have used for updating the Cochrane Review on ivermectin (Popp 2022; Weibel 2022). This tool along with detailed methodological instructions and critical and important signalling questions to key aspects (domains), is described in the Appendix 2, and elsewhere (Weibel 2022). Briefly, all trials fulfilling the PIC (patient, intervention, and comparator) eligibility criteria were assessed for issues with research integrity, such as retraction notices, prospective trial registration, ethics approval, plausible study authorship, sufficient reporting of methods regarding relevant eligibility criteria (e.g. randomization), and plausibility of study results. Studies were only eligible for the review if they met critical aspects assuring research integrity. Studies were excluded if they were retracted or if they were not prospectively registered in a national or international studies' registry according to the WHO guidelines for clinical trial registration (WHO 2018). All potentially eligible studies with disparities in the reporting of the methods and results were held in ‘awaiting classification’ until the study authors clarified certain questions upon request. The process was documented and decisions were transparently reported.
We documented the study selection process in a PRISMA flow diagram with the total number of studies included, excluded, and ongoing. There are no studies currently awaiting classification. We listed the reasons for exclusion in the 'Characteristics of excluded studies' table.
Data extraction and management
Two review authors (SR and SW) independently extracted data using a standardized data extraction form, including details of the study, participants, intervention, comparator, and outcomes. If necessary, we tried to obtain missing data by contacting the authors of relevant articles. At each step of data extraction, we resolved any discrepancies through discussion between the review authors. In case of discrepancies between different documents of one study (e.g. preprint, journal publication, CSR, registered trial protocol), we planned to contact the authors for clarification.
We extracted the following information, if reported.
General information: author, trial name, title, source, country, language, type of publication/report, and publication date.
Study characteristics: setting and dates, inclusion/exclusion criteria, number of study arms, comparability of groups, length of follow‐up, and funding.
Participant characteristics: number of participants randomized/received intervention/analyzed, COVID‐19 diagnostics, severity of disease, age, gender, race, ethnicity, comorbidities (e.g. diabetes, immunosuppression, obesity), concurrent medication, time since symptom onset, vaccination status (e.g. type of vaccine, number of doses), serology status, and history of SARS‐CoV‐2 infection.
Intervention: dose, frequency, time from symptom onset to treatment initiation, and duration and route of administration.
Control intervention: type of control, dose and frequency, and duration and route of administration.
Outcomes: as specified under Types of outcome measures.
To address health equity, we considered the following population characteristics and report them per outcome on the study‐level in additional tables.
Elderly people (older than 65 years). People of advanced age are at increased risk for severe disease. The intervention (nirmatrelvir/ritonavir) could potentially have greater impact in the elderly.
Persons at social disadvantage due to the number of comorbid health conditions. The intervention (nirmatrelvir/ritonavir) is aimed at persons with at least one risk factor for severe disease. Risk factors include individuals with a comorbid health condition, or multimorbidity, the presence of which is associated with social disadvantage (multimorbidity is associated with a reduction in quality of life, increased disability and premature mortality). The intervention could potentially have greater impact for persons with comorbid health conditions, promoting health equity.
Populations from LICs, LMICs, UMICs, and HICs as defined by the World Bank 2022 (studies were categorized based on the date of first participant enrolment). Differences exist in access to care and the quality of care across LICs, LMICs, UMICs, and HICs. People from LICs and LMICs may not have access to the intervention within five days of onset of symptoms of COVID‐19. Use of diagnostic tools in LICs and LMICs is also limited. In this context, nirmatrelvir/ritonavir could then be seen to have a potentially greater effectiveness for people from UMICs and HICs.
People from different ethnic and racial backgrounds, including minorities. Differences exist in access to care and the quality of care across different ethnic and racial minority groups who may not have access to the intervention within five days of symptom onset of a SARS‐CoV‐2 infection. Nirmatrelvir/ritonavir could therefore be seen as having a lower impact in these population groups
Assessment of risk of bias in included studies
We assessed the risk of bias in the included study using RoB 2 (Higgins 2022a; Sterne 2019). The effect of interest is the effect of assignment at baseline, regardless of whether the interventions were received as intended (the 'intention‐to‐treat effect'). We assessed the risk of bias for all results (outcomes) reported in the included study that we specified as outcomes for the review and that contributed to the review's summary of findings table.
Two review authors (SR, SW) independently assessed the risk of bias of all results. We resolved any disagreements through discussion with a third review author.
The RoB 2 tool considers the following domains:
bias arising from the randomization process;
bias due to deviations from the intended interventions;
bias due to missing outcome data;
bias in measurement of the outcome; and
bias in selection of the reported result.
We assessed the RoB 2 domains using the recommended signalling questions and the following response options:
yes;
probably yes;
probably no;
no; or
no information.
RoB 2 algorithms map responses to signalling questions. We used the proposed algorithm after verification to reach a risk of bias judgement, and assigned one of three levels to each domain:
low risk of bias;
some concerns; or
high risk of bias.
Similarly, we reached an overall risk of bias judgement for a specific outcome by considering all domains resulting in one of the three judgement options described above. Overall low risk of bias of the trial result was assumed when all domains are at low risk; some concerns of bias was assumed when the trial result was judged to raise some concerns in at least one domain for this result, but not at high risk of bias for any domain; overall high risk of bias of the trial result was assumed when the trial was at high risk of bias in at least one domain for this result or when it was judged to have some concerns for multiple domains in a way that substantially lowered confidence in the result (Higgins 2022a).
We used the RoB 2 Excel tool (version beta_v9(6)) to implement RoB 2 (available at www.riskofbias.info/welcome/rob-2-0-tool/current-version-of-rob-2). We stored the full RoB 2 data (e.g. completed Excel tool) in an online repository.
Measures of treatment effect
For dichotomous outcomes, we recorded the number of events and the number of analyzed participants in the intervention and control groups. For any adverse events we counted the number of events as number of participants with (at least) one event. We used the risk ratio (RR) with 95% confidence interval (CI) as the effect measure.
For continuous outcomes, we planned to record the mean, the standard deviation (SD), and the number of analyzed participants in the intervention and control groups. If the standard deviation was not reported, we planned to use standard errors (SEs), CIs, or P values to calculate the standard deviation (SD) with the formulas described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2022b). If studies in future review updates have reported data as median with interquartile range (IQR), we assume that the median is similar to the mean when the distribution of the outcome is similar to the normal distribution (e.g. symmetric IQR). In these cases, the width of the IQR is approximately 1.35 SDs (Higgins 2022b). We planned to use the mean difference (MD) with 95% CI as effect measure. For continuous outcomes measured on different scales (e.g. quality of life), we had planned to perform analyses using the standardized mean difference (SMD). For interpreting SMDs, we would have re‐expressed SMDs in the original units of a particular scale with the most clinical relevance and impact. The current review version did not contain a continuous outcome.
If available in future review updates, we will extract and report hazard ratios (HRs) for time‐to‐event outcomes (e.g. time to death). If HRs are not available, we make every effort to estimate the HR as accurately as possible from available data using the methods proposed by Parmar and Tierney (Parmar 1998; Tierney 2007). If sufficient studies provide HRs, we plan to use HRs rather than RRs or MDs in a meta‐analysis, as they provide more information. The current review version did not contain a time‐to‐event outcome.
We considered effect estimates of dichotomous outcomes with the range of the 95% CIs not crossing 1 (the lines of null effect) and continuous outcomes with the range of the 95% CIs not crossing 0 as statistically significant effect estimates. A statistically significant effect does not necessarily mean that the estimated effect is clinically relevant. Clinical experts assessed the clinical relevance of the effects based on anticipated absolute effects separately, and we reported this transparently.
Unit of analysis issues
The unit of analysis for this review was the randomized participant.
In studies with multiple intervention groups, we planned to combine groups if reasonable (e.g. study arms with different doses of nirmatrelvir/ritonavir). If it was not reasonable to pool the groups, we planned to split the 'shared' comparator group to avoid double‐counting of participants. No study groups were pooled for the current review.
Dealing with missing data
There are many potential sources of missing data in a systematic review or meta‐analysis, which can affect the level of studies, outcomes, summary data, individuals, or study‐level characteristics (Deeks 2022). Incomplete data can introduce bias into the meta‐analysis, if they are not missing at random. We addressed all sources of missing data. Missing studies may be the result of reporting bias, and we addressed this as described in the Assessment of reporting biases section. Missing outcomes and summary data may be the result of selective reporting bias; missing individuals may be the result of attrition from the study or lack of intention‐to‐treat analysis. We addressed these sources of missing data using the RoB 2 tool (Assessment of risk of bias in included studies). If data were incompletely reported, we contacted the study authors to request additional information.
Assessment of heterogeneity
We planned to use the descriptive statistics reported in the 'Characteristics of included studies' table to assess whether the studies within each pairwise comparison were homogeneous enough, with respect to study and intervention details and population baseline characteristics, that the assumption of homogeneity might be plausible. In case of excessive clinical heterogeneity, we planned not to pool the findings of included studies.
We planned to measure statistical heterogeneity using the Chi2 test and the I2 statistic (Deeks 2022), and the 95% prediction interval (PI) for random‐effects meta‐analysis (IntHout 2016). The prediction interval helps in the clinical interpretation of heterogeneity by estimating what true treatment effects can be expected in future settings (IntHout 2016). Calculation of a 95% PI is restricted to meta‐analyses with four or more studies (200 participants or more), since the interval is imprecise when a summary estimate is based on only a few small studies. The current review did not contain meta‐analyses with a sufficient number of studies to investigate heterogeneity. We plan to use the open‐source statistical software R package meta to calculate 95% PIs in review updates (Meta 2022). In future updates, we will declare statistical heterogeneity if the P value is less than 0.1 for the Chi2 statistic, or the I2 statistic is equal to or greater than 40% (40% to 60%: moderate heterogeneity; 50% to 90%: substantial heterogeneity; and 75% to 100%: considerable heterogeneity; Deeks 2022), or the range of the 95% PI reveals a different clinical interpretation of the effect estimate compared to the 95% CI.
Assessment of reporting biases
We tried to identify all research that meets our predefined eligibility criteria. Missing studies can introduce bias to the analysis. We searched for completed non‐published trials in trial registers, contacted authors to seek assurance that results will be made available. We planned to classify these studies as 'awaiting classification' until the results are reported. We also planned to report the number of completed non‐published trials. The current review did not identify completed non‐published trials.
If there were 10 or more relevant studies pooled in a meta‐analysis, we planned to investigate risk of reporting bias (publication bias) in pairwise meta‐analyses using contour‐enhanced funnel plots. If funnel plot asymmetry was suggested by a visual assessment, we planned to perform exploratory analyses (e.g. Rücker's arcsine test for dichotomous data and Egger's linear regression test for continuous data) to further investigate funnel plot asymmetry. A P value of less than 0.1 is considered as the level of statistical significance. We planned to analyze reporting bias using the open‐source statistical software R package meta (Meta 2022). The current review did not contain meta‐analyses with a sufficient number of studies to investigate reporting bias.
Data synthesis
We compared nirmatrelvir/ritonavir plus standard of care with standard of care with or without placebo, or to any active comparator with efficacy. Co‐interventions (standard of care) must have been comparable between the study arms.
We created the following comparisons.
nirmatrelvir/ritonavir plus standard of care versus standard of care (plus/minus placebo); and
nirmatrelvir/ritonavir versus active pharmacological intervention (no studies available for the current review version).
We planned to analyze trials with different objectives of nirmatrelvir/ritonavir use separately, as follows.
Treatment of COVID‐19 in an outpatient setting: participants with confirmed SARS‐CoV‐2 infection.
Treatment of COVID‐19 in an inpatient setting: participants with confirmed SARS‐CoV‐2 infection (no studies available for the current review version).
Prevention of SARS‐CoV‐2 infection (post‐exposure prophylaxis): RT‐PCR negative participants at baseline with a high risk of developing the infection following exposure to infected people or infectious viral particles (no studies available for the current review version).
Prevention of SARS‐CoV‐2 infection (pre‐exposure prophylaxis): RT‐PCR negative participants at baseline not yet exposed to infected people or infectious viral particles but at increased risk of contacting the disease (e.g. healthcare workers) (no studies available for the current review version).
We performed meta‐analyses according to the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2022). Forest plots were used to visualize meta‐analyses.
If clinical and methodological characteristics of individual studies were sufficiently homogeneous, we planned to pool the data in meta‐analyses. When meta‐analyses were feasible, we planned to use the random‐effects model as we assume that the intervention effects are related but might not be the same included studies. For dichotomous outcomes, we performed meta‐analyses using the Mantel‐Haenszel method using a random‐effects model to calculate the summary (combined) intervention effect estimate as a weighted mean of the intervention effects estimated in the individual studies. For continuous outcomes, we planned to use the inverse‐variance method.
We planned to present descriptive statistics only, if we deemed meta‐analysis inappropriate for a certain outcome because of heterogeneity, or because of serious study limitations leading to considerably high risk of bias (e.g competing risk of death not taken into account in outcome measurement). The current review included only one study with different subgroups.
We used RevMan Web for meta‐analysis and to calculate the effect estimate of the only included study (RevMan Web 2020).
Subgroup analysis and investigation of heterogeneity
We reported details of the intervention and population at baseline for the included study in the footnotes of the forest plot.
The current review version included one study, therefore investigation of heterogeneity between studies was not applicable. For future updates, we plan to investigate heterogeneity by visual inspection of the forest plot.
If statistical heterogeneity is present, we plan to investigate heterogeneity by subgroup analysis to calculate risk ratio (RR) or mean difference (MD) in conjunction with the corresponding confidence interval (CI) for each subgroup, if sufficient studies are available.
The following characteristics will be used for subgroup analyses to explore statistical heterogeneity, if reported.
-
Nirmatrelvir/ritonavir used as treatment (in‐ and outpatients):
studies including participants with different severities of condition at baseline (symptomatic versus asymptomatic);
studies including participants with a history of SARS‐CoV‐2 infection/vaccination versus participants with no history of infection/vaccination;
studies with different recruitment periods examining different dominant virus variants circulating at the time of the study (e.g. Alpha versus Beta versus Gamma versus Delta versus Omicron, etc.);
studies that started nirmatrelvir/ritonavir treatment early versus late (more than five days after symptom onset);
studies investigating different doses of nirmatrelvir/ritonavir (low versus recommended versus high). If necessary in future review updates, dosing schemes will be considered and categorized into recommended (300 mg nirmatrelvir/100 mg ritonavir, twice‐daily for 5 days), low (< recommended dose), and high doses (> recommended dose). We planned to analyze different doses in subgroup analysis, if sufficient studies are available. The one included study in the current review did not investigate different doses.
-
Nirmatrelvir/ritonavir used for prevention:
studies including participants with a history of SARS‐CoV‐2 infection/vaccination versus participants with no history of infection/vaccination;
studies investigating different modes of exposure (e.g. working place, nursing home) and burden of exposure (e.g. living in a high‐risk area, high‐risk medical contact) in prevention studies;
studies with different recruitment periods examining different dominant virus variants circulating at the time of the study (e.g. Alpha versus Beta versus Gamma versus Delta versus Omicron, etc.);
studies investigating different doses of nirmatrelvir/ritonavir (low versus recommended versus high).
We investigated health equity considering elderly people, socially disadvantaged people with comorbidities, populations from LICs/LMICs, and people from different ethnic and racial backgrounds using subgroup analysis independent of statistical heterogeneity. We performed the following subgroup analyses for treatment and prevention settings:
studies including different populations regarding age of the population (children versus adults versus older adults (greater than 65 years));
studies including participants with different level of comorbidity (high‐risk versus low‐risk population);
studies including participants from high‐, middle‐, or low‐income country settings and populations according to the World Bank classification (World Bank 2022) (LICs/LMICs versus UMICs/HICs);
studies including different racial and ethnic groups (Asian, Black, White, Hispanic, and minority ethnic groups);
Sensitivity analysis
We planned to conduct sensitivity analyses to test the robustness of the meta‐analyses excluding:
studies with overall high risk of bias;
non‐peer reviewed studies (including preprint articles);
studies reporting data as median instead of mean for continuous outcomes;
studies using no treatment in the comparator arm for patient‐reported outcomes such as symptom resolution.
Summary of findings and assessment of the certainty of the evidence
We presented the main results of the review in summary of findings tables, including a rating of the certainty of evidence based on the GRADE approach. We followed current GRADE guidance as recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2022).
Two review authors (SR, SW) assessed the certainty of evidence, considering risk of bias, inconsistency, imprecision, indirectness, and publication bias. We used the overall RoB 2 assessment and RoB sensitivity analysis to inform the risk of bias judgement underlying the assessment of the certainty of evidence.
We planned to create separate summary of findings tables for the use of nirmatrelvir/ritonavir with different intentions (e.g. treatment of people with COVID‐19 in out‐ and inpatient settings, prevention of SARS‐CoV‐2 infection as PEP, and prevention as PrEP), and for different comparisons with regard to the intervention and comparator. The summary of findings tables included the following outcomes.
For use of nirmatrelvir/ritonavir with intention to treat COVID‐19 in an outpatient setting.
All‐cause mortality; all‐cause mortality at longest follow‐up and > 60 days most favourable; if not reported all‐cause mortality day 60, followed by day 28, or time‐to‐event estimate, are reported in the summary of findings table.
Admission to hospital or death within 28 days.
-
Symptom resolution.
All initial symptoms resolved (asymptomatic) at day 14.
Time to symptom resolution.
Quality of life at longest follow‐up available.
Serious adverse events during the study period.
-
Adverse events during the study period.
Any grade treatment‐emergent adverse events (any TEAE).
Any grade treatment‐related adverse events (TRAE).
Viral clearance at 7 days.
For use of nirmatrelvir/ritonavir with intention to treat COVID‐19 in an inpatient setting.
All‐cause mortality; all‐cause mortality at longest follow‐up and > 60 days most favourable; if not reported all‐cause mortality day 60, followed by day 28, or time‐to‐event estimate, are reported in the summary of findings table.
-
Worsening of clinical status within 28 days.
Participants with new need for invasive mechanical ventilation or death.
-
Improvement of clinical status within 28 days.
Participants discharged alive. Participants should be discharged without clinical deterioration or death.
Quality of life at longest follow‐up available.
Serious adverse events during the study period.
-
Adverse events during the study period.
Any grade treatment‐emergent adverse events (any TEAE).
Any grade treatment‐related adverse events (TRAE).
Viral clearance at 7 days.
For use of nirmatrelvir/ritonavir with intention to prevent SARS‐CoV‐2 infection (PEP).
SARS‐CoV‐2 infection (confirmed by RT‐PCR or antigen testing) at 14 days.
Development of clinical COVID‐19 symptoms up to 28 days.
All‐cause mortality up to the longest follow‐up.
Admission to hospital or death within 28 days.
Quality of life at longest follow‐up available.
Serious adverse events during the study period.
-
Adverse events during the study period.
Any grade treatment‐emergent adverse events (any TEAE).
Any grade treatment‐related adverse events (TRAE).
For use of nirmatrelvir/ritonavir with intention to prevent SARS‐CoV‐2 infection (PrEP).
SARS‐CoV‐2 infection (confirmed by RT‐PCR or antigen testing) at six months.
Development of clinical COVID‐19 symptoms up to six months.
All‐cause mortality up to the longest follow‐up.
Admission to hospital or death within six months.
Quality of life at longest follow‐up available.
Serious adverse events during the study period.
-
Adverse events during the study period.
Any grade treatment‐emergent adverse events (any TEAE).
Any grade treatment‐related adverse events (TRAE).
The GRADE assessment result in one of four levels of certainty and these express our confidence in the estimate of effect (Balshem 2011).
High certainty: we are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect.
Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
We addressed equity for all outcomes presented in summary of findings tables. Interpretation of evidence occurred for the specific populations (see Data extraction and management) which are defined as important recipients of the intervention.
Elderly people.
People at social disadvantage due to the number of comorbid health conditions.
People from LICs/LMICs.
People from different ethnic and racial backgrounds, including minorities.
Interpretation considers the questions, whether findings likely to be applicable in those populations, even if they did not make up a large proportion of the participant populations in included studies. We reported inequities in the footnotes of the summary of findings tables.
We used the MAGICapp to create summary of findings tables (MAGICapp), and incorporate the results into RevMan Web manually (RevMan Web 2020).
Methods for future updates ‐ Living systematic review considerations
Our information specialist (MIM) provides us with new search records each month, which two review authors screen, extract, evaluate, and integrate following the guidance for Cochrane living systematic reviews (Cochrane LSR). We maintain an Excel spreadsheet on a monthly basis, which lists the search results and new studies potentially to be included in this review. It is publicly available on the open science framework (OSF) platform [osf.io/7g49c/; Reis 2022a]. Details on this "living method" are available in Metzendorf 2022.
We manually check platform trials for new treatment arms investigating nirmatrelvir/ritonavir.
We wait until the accumulating evidence changes our conclusions of the implications of research and practice before republishing the review. We consider one or more of the following components to inform this decision.
The findings of one or more prioritized outcomes.
The credibility (e.g. GRADE rating) of one or more prioritized outcomes.
New settings, populations, interventions, comparisons, or outcomes studied.
In the case of emerging policy relevance due to global controversies regarding the intervention, we consider republishing an updated review even though our conclusions remain unchanged. We review the review scope and methods approximately monthly, or more frequently if appropriate, in light of potential changes in COVID‐19 research (e.g. when additional comparisons, interventions, subgroups, or outcomes, or new review methods become available).
Results
Description of studies
Results of the search
The literature search up to 11 July 2022 resulted in 217 records. After deduplication in Endnote (EndNote 2013) 177 records remained. During the title and abstract screening 154 records were judged as irrelevant as they did not meet the prespecified inclusion criteria. We proceeded to full‐text screening with 23 reports. We considered published full‐texts in journals or on preprint servers or, if these were unavailable, trials’ register entries. We excluded seven reports with reasons after full‐text assessment. No study is currently awaiting classification. Sixteen reports met our eligibility criteria regarding patient population, intervention, and comparator of which 12 reports belong to ongoing studies (eight studies). Four reports (one study) with results have been identified. No study with results was excluded due to concerns regarding research integrity. Finally, one study was included in our qualitative synthesis. Due to the limited number of available studies, meta‐analysis was not possible. The search process is shown in Figure 1.
1.
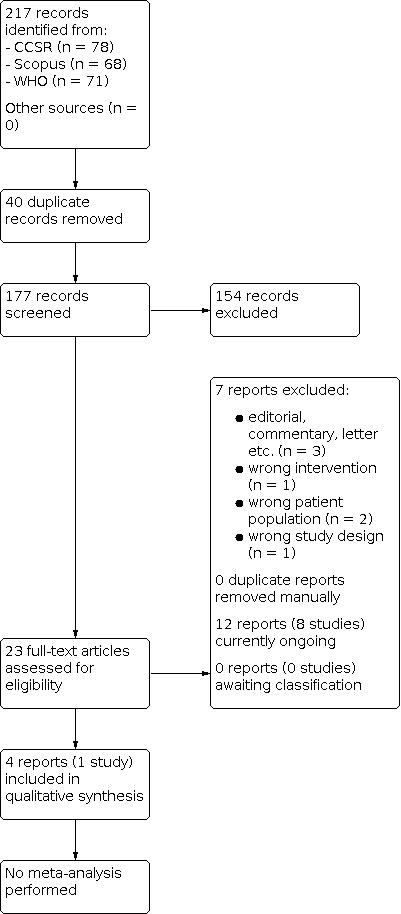
PRISMA diagram
This is a living systematic review (LSR) and we conduct monthly update searches which are being made publicly available on the open science framework (OSF) platform [osf.io/7g49c/; Reis 2022a].
Eligibility screening for research integrity
All eligible studies regarding patient population, intervention and comparator with results were evaluated for issues with research integrity:
One study with results identified by the search in May 2022 was evaluated (EPIC‐HR 2021).
For EPIC‐HR 2021 there were no concerns regarding research integrity. There were no retraction notice or concerns expressed elsewhere, and the trial was prospectively registered with adequate ethics approval. All study authors listed on the main publication are from Pfizer and none of the study investigators from the 343 study centres was among the authors. The method of randomization was sufficiently reported, and the study results were plausible.
The research integrity assessment is described in Appendix 2 and decisions regarding this review's study pool are transparently reported and publicly available (Supplementary File_Nirmatrelvir_Research Integrity).
Included studies
Details of the included study is reported in the Characteristics of included studies.
Design and publication status
We included one multi‐centre randomized controlled trial (RCT) with 2246 randomized adults comparing nirmatrelvir/ritonavir with control (EPIC‐HR 2021). Of 2246 participants, 1120 were randomized to the intervention arm and 1126 to the control arm (EPIC‐HR 2021). The study was a double‐blinded, placebo‐controlled trial and was funded by Pfizer (EPIC‐HR 2021). The study was published as peer‐reviewed journal article (EPIC‐HR 2021).
Setting
The trial took place at 343 sites worldwide (EPIC‐HR 2021). Of those, 27 sites (7.9%) were placed in LICs and LMICs. All other sites (316, 92.1%) were placed in UMICs or HICs. No information was provided regarding the actual number of included participants per site/country. Participants were recruited between July and December 2021.
Participants
The included study investigated nirmatrelvir/ritonavir for treatment of non‐hospitalized COVID‐19 patients (EPIC‐HR 2021). No study investigating nirmatrelvir/ritonavir for treating hospitalized COVID‐19 patients and preventing SARS‐CoV‐2 infection has been included in this review.
We classified participants in EPIC‐HR 2021 as WHO 2 to 3 on the WHO clinical progression scale (Marshall 2020). Participants had confirmed SARS‐CoV‐2 infection with symptom onset of no more than five days before randomization. Mean time between symptom onset and start of treatment was 2.96 days with 66.3% of participants starting treatment within three days (EPIC‐HR 2021). As per exclusion criteria, all participants were unvaccinated against SARS‐CoV‐2. Further exclusion criteria were previous confirmed SARS‐CoV‐2 infection, hospitalization for COVID‐19 or therapy with convalescent COVID‐19 plasma (EPIC‐HR 2021).
Median age was 46 years with an interquartile range(IQR) of 18 to 88 and 51.1% of participants were male (EPIC‐HR 2021). Pregnant or breastfeeding females were excluded from the study and all participants of reproductive age were expected to use a highly effective method of contraception (EPIC‐HR 2021). Age was reported for the modified intention to treat (mITT1) population (2,085 participants; EPIC‐HR 2021) according to groups of ≥ 65 years and < 65 years, with 87.1% younger than 65 years. Regarding race and ethnicity, most participants identified as White (71.5%), 14% as Asian, and 4.9% as Black.
Participants were eligible for inclusion into the trial if they were at high risk for progression to severe disease due to at least one coexisting condition or had other characteristics associated with an increased risk of developing severe illness from COVID‐19 (EPIC‐HR 2021). In the full analysis set, 61.0% of participants had two or more coexisting conditions or characteristics associated with an increased risk of developing severe COVID‐19 (EPIC‐HR 2021). The most common prespecified characteristics and comorbidities associated with an increased risk of developing severe disease were a body mass index (BMI) over 25 kg/m2 in 1807 participants (80.5%), current smoking in 876 participants (39.0%), and hypertension in 739 participants (32.9%) (EPIC‐HR 2021). Frequency of comorbidities were separately reported for the mITT1 population only (subgroup analyses); 79.7% had none or one comorbidity and 20.3% of the 2085 participants in the mITT1 population had two or more comorbidities, most commonly a BMI over 25 kg/m2 (80.0%), hypertension (33.0%), and diabetes (12.1%). The study did not report the number of participants without any comorbidity. Few participants had other baseline comorbidities (e.g. chronic lung disease, cardiovascular disorder, chronic kidney disease, HIV infection, sickle cell disease, neurodevelopmental disorder, and cancer).
Monoclonal antibodies were allowed as concomitant treatment. Prohibited prior or concomitant therapies included medications highly dependent on CYP3A4 for clearance and CYP3A4 inducers (EPIC‐HR 2021).
Interventions and comparators
In the included study, nirmatrelvir/ritonavir was administered orally at a dose of 300 mg/100 mg twice daily for five days and compared with placebo (EPIC‐HR 2021). Mean time between symptom onset and start of treatment was 2.96 days with 66.3% of participants starting treatment within three days (EPIC‐HR 2021). All participants received additional standard of care. Standard of care varied throughout study centres. Prohibited within 28 days prior to dosing of study intervention were any medications or substances known to be strong inducers of CYP3A4. Medications highly dependent on CYP3A4 for clearance and for which elevated plasma concentrations may be associated with serious and/or life‐threatening events were not permitted during the study period. Treatment with COVID‐19 monoclonal antibodies as part of standard of care was allowed in US study centres only.
No studies investigating other comparators were available.
Outcome measures
Primary outcome of the included study was ‘COVID‐19‐related hospitalization or death from any cause through day 28, which we used for the analysis of (any) ‘admission to hospital or death within 28 days’ (EPIC‐HR 2021). Subgroup analyses of this outcome were available for participants < 65 years of age and ≥ 65 years of age as well as for White, Asian, Black/African American and a mixed ethnic group termed "other" (mITT1 population; see EPIC‐HR 2021). Further outcomes eligible for this review included all‐cause mortality at 28 days, any grade treatment‐related and treatment‐emergent adverse events, serious adverse events (SAEs), and discontinuation of study drug. All safety outcomes were assessed at 34 days (EPIC‐HR 2021). No subgroups were reported for any outcome other than the primary outcome of the study (EPIC‐HR 2021). The included study did not report on worsening of clinical status assessed as admission to intensive care unit or death, symptom resolution, time to symptom resolution, quality of life and viral clearance.
Excluded studies
Details of excluded studies are reported in the Characteristics of excluded studies.
We excluded seven reports (six studies) that did not match our inclusion criteria. Two reports (one study) analysed an ineligible study population of healthy participants without exposure to SARS‐CoV‐2 (Singh 2022), one report focused on an intervention other than nirmatrelvir/ritonavir (NCT05305547), and three reports were classified as a commentary (Caceres 2022; Elliott 2022; Wang 2022). One report had a wrong study design (EPIC‐PEDS 2022).
Ongoing studies
Details of ongoing studies are reported in the Characteristics of ongoing studies.
We classified a total of eight studies with12 reports as ongoing. Seven studies investigate nirmatrelvir/ritonavir for treatment of COVID‐19 (ChiCTR2200059390; PANORAMIC 2021; RECOVERY 2020; EPIC‐SR 2021; NCT05321394; NCT05341609;NCT05386433), and one study for post‐exposure prevention (EPIC‐PEP 2021). Of the seven studies investigating nirmatrelvir/ritonavir for treatment of COVID‐19, one study is focusing on an inpatient setting (RECOVERY 2020), while four others investigate nirmatrelvir/ritonavir in outpatient settings (EPIC‐SR 2021; NCT05321394; NCT05341609; PANORAMIC 2021). Two studies did not include information about the study setting in the trial registration (ChiCTR2200059390; NCT05386433). Three studies investigate nirmatrelvir/ritonavir plus standard of care versus standard of care for treatment of COVID‐19 (NCT05386433; PANORAMIC 2021; RECOVERY 2020), two use an additional placebo in the comparator group (EPIC‐SR 2021; EPIC‐PEP 2021), and three studies compare nirmatrelvir/ritonavir with active comparators (ChiCTR2200059390; NCT05321394; NCT05341609). Except for one (NCT05386433), all ongoing studies are currently recruiting. Estimated enrolment numbers ranges from 40 participants to well over 2000 participants. Of the eight ongoing trials, one is conducted in the UK (PANORAMIC 2021), one in Italy (NCT05321394), three in China (ChiCTR2200059390; NCT05386433; NCT05341609),and one in Ghana, India, Indonesia, Nepal, South Africa, Sri Lanka, the UK, and Vietnam (RECOVERY 2020). The other two studies are conducted at over 300 study locations worldwide (EPIC‐SR 2021; EPIC‐PEP 2021). Of the eight ongoing studies, three are funded by pharmaceutical companies (EPIC‐PEP 2021; EPIC‐SR 2021; NCT05341609). Five trials are estimated to be completed in 2022 (ChiCTR2200059390; EPIC‐PEP 2021; EPIC‐SR 2021; NCT05321394; NCT05386433), two in 2023 (NCT05341609; PANORAMIC 2021), and one in 2032 (RECOVERY 2020).
Studies awaiting classification
No study is currently awaiting classification.
Risk of bias in included studies
We assessed methodological quality and risk of bias for the included trial (EPIC‐HR 2021) using the RoB 2 tool recommended in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2022a). The included trial contributed 12 study results to six outcomes. Six study results (whole study population) contributed to all six outcomes and six additional study results (subgroups for age and ethnicity) contributed to one of the six outcomes. To account for different analysis sets used for subgroup analyses in the trial, the outcome ‘hospitalization or death’ has been assessed for the whole population as well as the individual subgroups using ROB 2. The RoB 2 judgements for all study results per outcome and for all domains are available in an interactive risk of bias table (Supplementary File_Nirmatrelvir_Risk of Bias) and are briefly summarized below. The completed RoB 2 tool with responses to all assessed signalling questions is available online at: Supplementary File_Nirmatrelvir_Risk of Bias
Overall risk of bias by study
All 12 study results included from EPIC‐HR 2021 have been assessed as some concerns for the overall risk of bias.
Overall risk of bias by outcome
This section summarises the risk of bias per outcome for all outcomes included in the Table 1.
We have some concern regarding risk of bias for all outcomes included in the Table 1. For the outcomes ‘all‐cause mortality at 28 days’, ‘worsening of clinical status: admission to hospital or death at 28 days’, ‘serious adverse events (SAEs) during the study period’ and ‘any grade treatment‐emergent adverse events during the study period’ the included study been assessed as overall some concern of bias due to use of an inappropriate per‐protocol analysis (EPIC‐HR 2021). For the outcome ‘admission to hospital or death at 28 days’, we applied the respective risk of bias assessments to the subgroup analyses, as well (EPIC‐HR 2021). For the outcomes ‘treatment‐related adverse events during the study period’ and ‘discontinuation of study medication due to adverse events’ the study has been assessed as overall some concern of bias due to use of an inappropriate per‐protocol analysis and lack of prospective outcome registration (EPIC‐HR 2021).
Effects of interventions
See: Table 1
One study investigating nirmatrelvir/ritonavir for treatment of non‐hospitalized COVID‐19 patients has been included in the qualitative synthesis of this review (EPIC‐HR 2021). The included study compared nirmatrelvir/ritonavir plus standard of care with standard of care plus placebo for treatment of mild COVID‐19 in an unvaccinated population at high risk of progression to severe COVID‐19 and without previous SARS‐CoV‐2 infection.
Subgroup analyses between studies to explore heterogeneity and sensitivity analysis to test robustness of the results could not be performed due to insufficient number of studies.
We looked at subpopulations to address health equity (age and ethnicity) for one outcome (‘admission to hospital or death at 28 days’) reported in the one study. No further results for relevant subpopulations have been reported by the study (EPIC‐HR 2021).
The main findings are summarized in Table 1
Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease
All‐cause mortality at day 28
Data on all‐cause mortality at day 28 were reported for 2224 participants in one study (EPIC‐HR 2021). At 28 days none of the participants in the nirmatrelvir/ritonavir and 12 participants in the comparator group had died (Analysis 1.1). Nirmatrelvir/ritonavir plus standard of care may reduce all‐cause mortality at 28 days compared to standard of care plus placebo (RR 0.04, 95% CI 0.00 to 0.68; 1 study, 2224 participants; low‐certainty evidence). We downgraded the certainty of evidence one level for serious risk of bias due to inappropriate analysis and one level for serious imprecision due to few events.
1.1. Analysis.

Comparison 1: Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease , Outcome 1: All‐cause mortality at day 28
Subgroup analysis
We could not perform subgroup analyses for age, ethnicity, comorbidity and World Bank country classification by income level due to missing data (Table 2).
1. Equity assessment .
| Outcome | Study | Age, n (%) | Comorbidities, n (%) | Race/Ethnicity, n (%) | World Bank country classification by income level, n (%) |
| All‐cause mortality at 28 days | EPIC‐HR 2021 | NR | NR | NR | NR |
| Worsening of clinical status: admission to hospital or death at 28 days | EPIC‐HR 2021 | Children: NR < 65 years: 1817 (87%)1 ≥ 65 years: 268 (13%)1 |
NR | Asian: 315 (14%)1 Black: 110 (4.9%)1 Hispanics: NR White: 1607 (71.5%)1 Minorities: NR |
NR |
| Serious adverse events during the study period | EPIC‐HR 2021 | NR | NR | NR | NR |
| Any grade treatment emergent adverse events during the study period | EPIC‐HR 2021 | NR | NR | NR | NR |
| Any grade treatment related adverse events during the study period | EPIC‐HR 2021 | NR | NR | NR | NR |
| Discontinuation of study drug due to adverse events during the study period | EPIC‐HR 2021 | NR | NR | NR | NR |
Abbreviations: NR: not reported;n: number of participants
Worsening of clinical status
Admission to hospital or death within 28 days
Data on worsening of clinical status assessed as admission to hospital or death within 28 days were available for 2224 participants in the included study (EPIC‐HR 2021). The outcome was defined as ‘COVID‐19 related hospital admission or death’ in the study. At 28 days, nine patients in the nirmatrelvir/ritonavir group and 68 in the comparator group had been admitted to hospital or were dead (Analysis 1.2). Nirmatrelvir/ritonavir plus standard of care may reduce admission to hospital or death within 28 days compared to standard of care plus placebo (risk ratio (RR) 0.13, 95% confidence interval (CI) 0.07 to 0.27; 1 study, 2224 participants; low‐certainty evidence). We downgraded the certainty of evidence one level for serious risk of bias due to inappropriate analysis and one level for serious indirectness as the study only assessed COVID‐19‐related hospitalizations.
1.2. Analysis.

Comparison 1: Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease , Outcome 2: Worsening of clinical status: admission to hospital or death at 28 days
Subgroup analysis
Subgroup analyses were available regarding age and ethnicity for the mITT1 population (Table 2). We could not perform subgroup analyses for World Bank country classification by income level due to missing data. No subgroup analysis was possible for comorbidity (high risk versus low risk population) as the included study only investigated COVID‐19 patients at high risk for disease progression which is either due to coexisting conditions (e.g. current smoking) or comorbidities.
Participants under 65 years versus 65 years and older
Of 1817 adult participants younger than 65 years of age, seven in the nirmatrelvir/ritonavir and 46 in the comparator group were admitted to hospital or died (RR 0.15, 95% CI 0.07 to 0.34) (Analysis 1.3). Of 268 participants 65 years or older, one in the nirmatrelvir/ritonavir, and 20 in the comparator group were admitted to hospital or died (RR 0.05, 95% CI 0.01 to 0.38) (Analysis 1.3). The test for subgroup differences indicated no difference between both groups (P = 0.33) and no heterogeneity (I2 = 0%). In both groups, the risk ratio favoured treatment with nirmatrelvir/ritonavir, but the number of the included participants 65 years or older was low.
1.3. Analysis.

Comparison 1: Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease , Outcome 3: Worsening of clinical status: admission to hospital or death at 28 days (subgroup analysis based on age)
Participants from different ethnic groups
Of 1486 participants identifying as White, eight in the nirmatrelvir/ritonavir and 52 in the comparator group were admitted to hospital or died (RR 0.16, 95% CI 0.07 to 0.33) (Analysis 1.4). Of 94 participants identifying as Black or African American, none in the nirmatrelvir/ritonavir and one in the comparator group were admitted to hospital or died (RR 0.29, 95% CI 0.01 to 7.04) (Analysis 1.4). Of 296 participants identifying as Asian, none in the nirmatrelvir/ritonavir and seven in the comparator group were admitted to hospital or died (RR 0.07, 95% CI 0.00 to 1.19) (Analysis 1.4). Of 209 participants categorized as "other" ethnicity, none in the nirmatrelvir/ritonavir and six in the comparator group were admitted to hospital or died (RR 0.08, 95% CI 0.00 to 1.34) (Analysis 1.4). The test for subgroup differences indicated no difference between all pairwise groups (P = 0.88) and no heterogeneity (I2 = 0%). The estimated effect favoured a treatment with nirmatrelvir/ritonavir for the White ethnic group, all estimated effects of the other ethnic groups included the line of no effect (RR = 1). Numbers of participants in the other ethnic groups were low.
1.4. Analysis.

Comparison 1: Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease , Outcome 4: Worsening of clinical status: admission to hospital or death at 28 days (subgroup analysis based on ethnicity)
Admission to intensive care unit or death within 28 days
No study reported data for worsening of clinical status assessed as admission to intensive care unit or death within 28 days.
Improvement of clinical status
All initial symptoms resolved at 28 days and up to the longest follow‐up
The included study did not report data for improvement of clinical status assessed as all initial symptoms resolved at 28 days and up to the longest follow‐up.
Time to symptom resolution
The included study did not report data for improvement of clinical status assessed as time to symptom resolution.
Quality of life up to 28 days and longest follow‐up available
The included study did not report data for quality of life at any time point.
Serious adverse events during the study period
Data on serious adverse events during the study period were available for 2224 participants in one study (EPIC‐HR 2021). Eighteen patients in the nirmatrelvir/ritonavir and 74 in the comparator group experienced serious adverse events during the 34 days observation period (Analysis 1.5). Nirmatrelvir/ritonavir plus standard of care may reduce serious adverse events during the study period compared to standard of care plus placebo (RR 0.24, 95% CI 0.15 to 0.41; 1 study, 2224 participants; low‐certainty evidence). We downgraded the certainty of evidence one level for serious risk of bias due to inappropriate analysis and one level for serious imprecision as there were few serious adverse events other than hospitalization or death.
1.5. Analysis.

Comparison 1: Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease , Outcome 5: Serious adverse events during the study period
Subgroup analysis
We could not perform subgroup analyses for age, ethnicity, comorbidity and World Bank country classification by income level due to missing data (Table 2).
Adverse events
Any grade treatment‐emergent adverse events during the study period
Data on any grade treatment‐emergent adverse events during the study period were available for 2224 participants in one study (EPIC‐HR 2021). In total, 251 patients in the nirmatrelvir/ritonavir and 266 in the placebo comparator group experienced treatment emergent adverse events during the 34 days observation period (Analysis 1.6). Nirmatrelvir/ritonavir plus standard of care probably has little or no effect on treatment‐emergent adverse events during the study period compared to standard of care plus placebo (RR 0.95, 95% CI 0.82 to 1.10; 1 study, 2224 participants; moderate‐certainty evidence). We downgraded the certainty of evidence one level for serious risk of bias due to inappropriate analysis.
1.6. Analysis.

Comparison 1: Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease , Outcome 6: Any grade treatment‐emergent adverse events during the study period
Subgroup analysis
We could not perform subgroup analyses for age, ethnicity, comorbidity and World Bank country classification by income level due to missing data (Table 2).
Any grade treatment‐related adverse events during the study period
Data on any grade treatment‐related adverse events during the study period were available for 2224 participants in one study (EPIC‐HR 2021). 86 patients in the nirmatrelvir/ritonavir and 42 in the comparator group experienced treatment‐related adverse events during the 34 days observation period (Analysis 1.7). Nirmatrelvir/ritonavir plus standard of care probably increases treatment‐related adverse events during the study period compared to standard of care plus placebo (RR 2.06, 95% CI 1.44 to 2.95; 1 study, 2224 participants; moderate‐certainty evidence). Treatment‐related adverse events were mostly attributed to dysgeusia and diarrhoea. We downgraded the certainty of evidence one level for serious risk of bias due to inappropriate analysis.
1.7. Analysis.

Comparison 1: Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease , Outcome 7: Any grade treatment‐related adverse events during the study period
Subgroup analysis
We could not perform subgroup analyses for age, ethnicity, comorbidity and World Bank country classification by income level due to missing data (Table 2)
Discontinuation of study drug due to adverse events
Data on discontinuation of study drug due to adverse events were available for 2224 participants in one study (EPIC‐HR 2021). 23 patients in the nirmatrelvir/ritonavir and 47 in the comparator group discontinued the study drug due to adverse events (Analysis 1.8). Nirmatrelvir/ritonavir plus standard of care probably decreases discontinuation of study drug due to adverse events compared to standard of care plus placebo (RR 0.49, 95% CI 0.30 to 0.80; 1 study, 2224 participants; moderate‐certainty evidence). Treatment related adverse events were mostly attributed to dysgeusia and diarrhoea. We downgraded the certainty of evidence one level for serious risk of bias due to inappropriate analysis.
1.8. Analysis.

Comparison 1: Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease , Outcome 8: Discontinuation of study drug due to adverse events during the study period
Subgroup analysis
We could not perform subgroup analyses for age, ethnicity, comorbidity and World Bank country classification by income level due to missing data (Table 2).
Viral clearance at 14 days
No study reported data on viral clearance at 14 days.
Nirmatrelvir/ritonavir for treating COVID‐19 in inpatient settings with moderate to severe disease
No study with published results investigating nirmatrelvir/ritonavir for treating COVID‐19 in inpatient settings with moderate to severe disease has been included in this review.
Nirmatrelvir/ritonavir for preventing SARS‐CoV‐2 infection (PrEP and PEP)
No study with published results investigating nirmatrelvir/ritonavir for preventing SARS‐CoV‐2 infection has been included in this review.
Discussion
Summary of main results
This Cochrane Review aimed to assess the efficacy and safety of nirmatrelvir/ritonavir for treating and preventing COVID‐19.
Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease
For people with a confirmed diagnosis of COVID‐19, we identified one randomized controlled trial (RCT) with 2246 randomized participants with mild symptomatic COVID‐19 (WHO 2‐3) conducted in the outpatient setting comparing nirmatrelvir/ritonavir plus standard of care to standard of care plus placebo. Trial participants were unvaccinated, without previous confirmed SARS‐CoV‐2 infection, had a symptom onset of no more than five days before randomization, and were at high risk for progression to severe disease.
The study was assessed without concerns for research integrity.
The main findings of this review are summarized in Table 1. Briefly:
Nirmatrelvir/ritonavir may reduce all‐cause mortality at 28 days and clinical worsening assessed as admission to hospital or death within 28 days (low‐certainty evidence).
Nirmatrelvir/ritonavir may reduce serious adverse events during the study period (low‐certainty evidence); there were few serious adverse events other than hospitalization or death.
Nirmatrelvir/ritonavir probably has little or no effect on any treatment‐emergent adverse events during the study period (moderate‐certainty evidence)
Nirmatrelvir/ritonavir probably increases any treatment‐related adverse events during the study period (moderate‐certainty evidence); treatment‐related adverse events were mostly attributed to dysgeusia and diarrhoea.
Nirmatrelvir/ritonavir probably decreases discontinuation of study drug due to adverse events (moderate‐certainty evidence).
We identified no study results reporting on improvement of clinical status, quality of life, and viral clearance. We identified four ongoing trials investigating nirmatrelvir/ritonavir for treatment of COVID‐19 in outpatient settings with mild disease.
Equity
No subgroups were reported for World Bank country classification by income level.
No subgroup analysis was possible for comorbidity (high‐risk versus low‐risk population) as the included study only investigated COVID‐19 patients at high risk for disease progression, which was either due to coexisting conditions (e.g. current smoking) or comorbidities. Reported only for the: modified intention to treat (mITT1) patient population, 79.7% had none or one comorbidity and 20.3% of the participants had two or more comorbidities, most commonly a body mass index (BMI) over 25 kg/m2 (80.0%), hypertension (33.0%), and diabetes (12.1%). The study did not report the number of participants without any comorbidity. Few participants had other baseline comorbidities (e.g. chronic lung disease, cardiovascular disorder, chronic kidney disease, HIV infection, sickle cell disease, neurodevelopmental disorder, and cancer). For the full analysis set, only the combination of coexisting conditions or characteristics associated with an increased risk of developing severe COVID‐19 was reported, with 61.0% of participants having two or more such characteristics or comorbidities. The most common prespecified characteristic associated with an increased risk of developing severe disease was current smoking in 876 participants (39.0%).
Despite the study being conducted in 343 sites worldwide, only 27 sites were placed in LICs and LMICs and all other 316 sites (92.1%) were placed in UMICs or HICs. No separate data were available for participants from LICs, LMICs, UMICs, or HICs.
Regarding equity, we highlight that most study participants in the included study were younger than 65 years and of white ethnicity.
For the outcome ‘admission to hospital or death’ the following subgroups were investigated for equity: age (< 65 years versus ≥ 65 years) and ethnicity (Asian versus Black versus White versus other). For age, the effects favoured a treatment with nirmatrelvir/ritonavir in both groups but the number of included participants in the subgroup of 65 years or older was low. For ethnicity, the effects favoured a treatment with nirmatrelvir/ritonavir for the White ethnic group. Estimated effects of the other ethnic groups included the line of no effect (RR = 1). Numbers of participants in the other ethnic groups were low.
For all other outcomes included in this review no subgroups were reported. As most participants were younger than 65 years and of white ethnicity, we are uncertain whether results are applicable to the other prespecified subgroups.
Nirmatrelvir/ritonavir for treating COVID‐19 in inpatient settings with moderate to severe disease
We did not identify any completed trials investigating nirmatrelvir/ritonavir for treatment of COVID‐19 in inpatient settings but one ongoing trial.
Nirmatrelvir/ritonavir for preventing SARS‐CoV‐2 infection (PrEP and PEP)
We did not identify any completed trials investigating nirmatrelvir/ritonavir for prevention of COVID‐19 but one ongoing trial investigating nirmatrelvir/ritonavir as post‐exposure prophylaxis after exposure to SARS‐CoV‐2.
Overall completeness and applicability of evidence
Originating from only one included study with 2246 randomized participants, the certainty of evidence for the efficacy of nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings is low and for safety aspects low to moderate.
The included study investigated participants with mild symptomatic COVID‐19, corresponding to WHO clinical progression scale 2 to 3. Considering that only unvaccinated participants with high risk for disease progression due to coexisting conditions or characteristics were included in the trial, external validity of the results is limited and results may not be transferable to a broader population of vaccinated participants or those with standard risk for disease progression. As of 20 May 2022 there have been more than 500 million confirmed SARS‐CoV‐2 cases reported to the WHO and more than 12 billion vaccine doses have been administered (WHO 2021a). The influence of the vaccination status as well as previous SARS‐CoV‐2 infections could not be assessed in this review.
With ritonavir being a CYP3A4 inhibitor, nirmatrelvir/ritonavir bears the potential for significant drug‐drug interactions with many medications commonly used, especially in comorbid patients. The exclusion criteria of the study prohibited prior or concomitant therapies including medications highly dependent on CYP3A4 for clearance. Pfizer published an extensive list of potentially significant drug interactions, including contraindicated drugs like HMG‐CoA reductase inhibitors and antiarrhythmics (Pfizer 2022). If known problematic concomitant medications cannot be discontinued or reduced, Pfizer advises against the use of nirmatrelvir/ritonavir, thereby further limiting the transferability of results to a broader high risk population.
Surprisingly only 6% of participants in the study received or were expected to receive COVID‐19 monoclonal antibody treatment, despite the sixth version of the WHO living guideline published 24 September 2021 suggesting "treatment with casirivimab and indevimab for those at highest risk of hospital admission" (WHO 2021b). Since the emergence of the Omicron BA.1 variant, however, casirivimab and indevimab have lost their efficacy (Takashita 2022).
All participants included in this review were enrolled until December 2021 which coincided with the start of the Omicron wave, therefore the findings of this review might not be directly applicable to the treatment situation of patients which are infected with later (sub‐)variants of SARS‐CoV‐2. Different recruitment periods could not be assessed.
According to Pfizer, nirmatrelvir/ritonavir is to be administered within five days of symptom onset. In the included study, mean time between symptom onset and start of treatment was 2.96 days with 66.3% of participants starting treatment within three days which might have increased the effectiveness of treatment. At the same time this limits applicability outside idealized study conditions where time between symptom onset, confirmation of SARS‐CoV‐2 infection and treatment start may well exceed three days due to limited health care access or testing capacities in LICs and LMICs but also disadvantaged groups other than ethnic and racial minorities, for example; refugees, mental or physically disabled patients, and women in HICs.
Reported data of EPIC‐HR 2021 are incomplete and inconsistent. Outcome data on the overall population of 2,246 randomized patients are missing and the Full Analysis Set (FAS) is not reported. Instead, multiple versions of modified intention to treat (mITT) analyses (mITT, mITT1, and mITT2; Table S2; EPIC‐HR 2021) are presented. The primary analysis of the original study protocol from 18 June 2021 was defined as the mITT1 population including participants who were treated ≤ 5 days after COVID‐19 symptom onset. The mITT analysis set which is the main analysis set of the publication was added in the amendment 2 (2 August 2021) to include just those participants who were treated ≤ 3 days after COVID‐19 symptom onset. Any clear explanation why the authors did not report the FAS but provide the mITT as main analyses is missing in the publication. We would have expected the FAS to be presented as the primary analysis set and the mITTs as additional sensitivity analyses. The presented mITT analyses are focusing on smaller populations. In the abstract, the interim and final analysis of the mITT population with 1,379 of all 2,246 randomized participants is presented, with 13 deaths reported. However, according to the results section and Figure 2, only nine patients died in this mITT population, which is inconsistent and confusing.
Finally, there is no detailed information on the clinical characteristics of included participants for the FAS, particularly with regard to the comorbidities as risk factors for disease progression. The distribution of comorbidities can only be extrapolated for the mITT population using the subgroup analyses presented in Figure S2c. The information for all patients (FAS) is missing.
To date, there are no completed studies that investigate nirmatrelvir/ritonavir for the prevention of COVID‐19 or for COVID‐19 treatment in inpatient settings. We found eight ongoing studies of which five are planned to be completed within the next months.
Certainty of the evidence
The certainty of evidence for prioritized outcomes presented in the summary of findings table ranged from low to moderate (Table 1).
We downgraded the certainty of evidence for all outcomes included in our summary of findings table due to risk of bias arising from use of an inappropriate per‐protocol analysis only including participants randomly assigned to study intervention who took ≥ 1 dose of study intervention and had ≥ 1 post‐baseline visits through day 28. Additionally, the outcomes ‘treatment‐related adverse events during the study period’ and ‘discontinuation of study medication due to adverse events’ were not prospectively registered. All‐cause mortality at 28 days and serious adverse events during the study period were further downgraded for serious imprecision due to low number of events. The outcome ‘admission to hospital or death within 28 days’ was additionally downgraded for serious indirectness as the study only assessed COVID‐19 related hospitalizations.
We did not consider downgrading for publication bias because the intervention is new and most of the studies are still ongoing.
We identified no study results reporting on improvement of clinical status, quality of life, and viral clearance.
Potential biases in the review process
We are confident that we identified all relevant studies using a broad search and will monitor ongoing studies after the publication of this review. This review is a living systematic review, and we maintain a monthly Excel list of new studies potentially to be included in the next review update. This list is publicly available (osf.io/7g49c/; Reis 2022a).
We followed strictly our protocol and there were only few differences between protocol and review. Following the rationale from Pfizer that nirmatrelvir/ritonavir was developed to manage outpatients with COVID‐19, an outcome set for inpatients was not included in the review protocol avoiding the impression that studies for this population are needed. On 28 March 2022, the RECOVERY trial announced nirmatrelvir/ritonavir to be investigated as a potential treatment for patients hospitalized with COVID‐19 (RECOVERY 2020). We therefore added the outcome set for hospitalized COVID‐19 patients to the review. Further, we have changed the definition of our active comparator. In the protocol we planned to compare nirmatrelvir/ritonavir with active comparisons with proven efficacy only. We decided to extend our definition of an eligible active comparator to any active comparator, including new interventions that would be investigated in future trials that may use nirmatrelvir/ritonavir as comparator.
None of the members of the review author team has any affiliation with any stakeholder group who favours or disapproves of nirmatrelvir/ritonavir or the comparators used in relevant studies.
Agreements and disagreements with other studies or reviews
Due to the limited study results available so far, we found no published meta‐analysis focusing on nirmatrelvir/ritonavir alone. A recent meta‐analysis comparing three oral antivirals molnupiravir, fluvoxamin and nirmatrelvir/ritonavir with over 4000 participants from eight trials found that oral antivirals are effective in reducing the mortality and hospitalization rates in patients with COVID‐19 (Wen 2022). The occurrence of adverse events was not increased. However, no separate subgroup analyses per antiviral treatment were reported.
The WHO living guideline recommends treatment with nirmatrelvir/ritonavir only for patients with non‐severe COVID‐19 at highest risk of hospitalization and advises against treatment in patients with non‐severe COVID‐19 at low risk of hospitalization (Agarwal 2020). The evidence summary was informed by two trials (EPIC‐HR 2021; EPIC‐SR 2021), of which the second one is currently still recruiting participants and to date only published as a press release with results from an interim analysis. Press releases are not eligible for the current review.
Authors' conclusions
Implications for practice.
Based on the current evidence (one trial), there is low‐certainty evidence that nirmatrelvir/ritonavir may reduce all‐cause mortality and hospital admission or death within 28 days. Subgroup analyses regarding equity for admission to hospital or death suggested that there are no differences in efficacy regarding patients' age, but we only have data in a mostly white ethnic population and can therefore not assess benefit in other ethnicities.
There is low‐ to moderate‐certainty evidence that nirmatrelvir/ritonavir is safe in people without prior or concomitant therapies including medications highly dependent on CYP3A4 for clearance and CYP3A4 inducers.
This review only included one trial investigating unvaccinated patients without previous infection that were at high risk of disease progression due to coexisting conditions or other characteristics associated with an increased risk of developing severe illness from COVID‐19. There is currently no evidence for the use of nirmatrelvir/ritonavir in a broader population of vaccinated patients, those with previous SARS‐CoV‐2 infection, or those without increased risk for progression to severe disease. External validity of the results is therefore limited. All participants included in this review were enrolled until December 2021 which coincided with the start of the Omicron wave, therefore the findings of this review might not be directly applicable to the treatment situation of patients which are infected with later (sub‐)variants of SARS‐CoV‐2.
With ritonavir being a CYP3A4 inhibitor, nirmatrelvir/ritonavir bears the potential for significant drug‐drug interactions with many medications commonly used, especially in comorbid patients. The exclusion criteria of the study prohibited prior or concomitant therapies including medications highly dependent on CYP3A4 for clearance. If there are known or anticipated drug interactions with concomitant medications which cannot be discontinued or reduced, Pfizer advises against the use of nirmatrelvir/ritonavir, thereby limiting the transferability of results to a broader high risk population.
Currently, there is no evidence to explore the benefits and harms of nirmatrelvir/ritonavir as treatment in patients with moderate to severe COVID‐19 (hospitalized) or as pre‐/post‐exposure prophylaxis.
Implications for research.
There is a need for evidence for the use of nirmatrelvir/ritonavir as treatment in vaccinated patients, those with previous SARS‐CoV‐2 infection or those without increased risk for progression to severe disease. There is also a need for studies investigating the use of nirmatrelvir/ritonavir to prevent SARS‐CoV‐2 infection. For these scenarios and populations we need high‐quality randomized controlled trials (RCTs).
To address equity, we need further trials investigating:
populations from LICs and LMICs;
people from different ethnic and racial backgrounds, including minorities.
We identified eight ongoing studies investigating nirmatrelvir/ritonavir for treatment or prevention of COVID‐19 which will hopefully increase the certainty of evidence in the future and broaden the applicability of results.
In accordance with the living approach of this review, we are continually updating our search and evaluating new potentially relevant trials for inclusion in this review [osf.io/7g49c/; Reis 2022a].
History
Protocol first published: Issue 4, 2022
Risk of bias
Risk of bias for analysis 1.1 All‐cause mortality at day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the mITT2 population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (mITT2). |
Risk of bias for analysis 1.2 Worsening of clinical status: admission to hospital or death at 28 days.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the mITT2 population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (mITT2). |
Risk of bias for analysis 1.3 Worsening of clinical status: admission to hospital or death at 28 days (subgroup analysis based on age).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.3.1 children | ||||||||||||
| Subgroup 1.3.2 age < 65 years of age | ||||||||||||
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the mITT1 population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (mITT1). |
| Subgroup 1.3.3 age ≥ 65 years of age | ||||||||||||
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the mITT1 population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (mITT1). |
Risk of bias for analysis 1.4 Worsening of clinical status: admission to hospital or death at 28 days (subgroup analysis based on ethnicity).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.4.1 White | ||||||||||||
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the mITT1 population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (mITT1). |
| Subgroup 1.4.2 Black/African American | ||||||||||||
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the mITT1 population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (mITT1). |
| Subgroup 1.4.3 Asian | ||||||||||||
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the mITT1 population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (mITT1). |
| Subgroup 1.4.4 Other | ||||||||||||
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the mITT1 population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (mITT1). |
Risk of bias for analysis 1.5 Serious adverse events during the study period.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the SAS population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (SAS). |
Risk of bias for analysis 1.6 Any grade treatment‐emergent adverse events during the study period.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the SAS population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Low risk of bias | The protocol was prospectively registered and the outcome was reported as registered. | Some concerns | Due to per‐protocol analysis (SAS). |
Risk of bias for analysis 1.7 Any grade treatment‐related adverse events during the study period.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the SAS population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Some concerns | The protocol was prospectively registered and the outcome was not registered. | Some concerns | Due to per‐protocol analysis (SAS) and lack of prospective outcome registration. |
Risk of bias for analysis 1.8 Discontinuation of study drug due to adverse events during the study period.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| EPIC‐HR 2021 | Low risk of bias | Centralized web‐based randomization. There are no baseline differences that would suggest a problem with randomization. | Some concerns | Both participants and those delivering the intervention were not aware of intervention received. The analysis was not appropriate (a per‐protocol analysis was used). | Low risk of bias | Data for this outcome were reported for all participants of the SAS population. | Low risk of bias | Measurement of the outcome was appropriate. Outcome assessors were not aware of the intervention received. | Some concerns | The protocol was prospectively registered and the outcome was not registered. | Some concerns | Due to per‐protocol analysis (SAS) and lack of prospective outcome registration. |
Acknowledgements
Editorial and peer reviewer contributions
The following people conducted the editorial process for this article:
Sign‐off Editors: Professor George Rutherford, and Dr Joseph Pryce (CIDG)
Managing Editor (selected peer reviewers, collated peer‐reviewer comments, provided editorial guidance to authors, edited the article): Dr Deirdre Walshe (CIDG);
-
Copy Editor (copy editing and production):
protocol stage: Luisa M Fernandez Mauleffinch, Cochrane Copy Edit Support;
review stage: Heather Maxwell, Cochrane Copy Edit Support
-
Peer‐reviewers (provided comments and recommended an editorial decision):
protocol stage: Dr Paul Hine, Liverpool, UK (clinical/content peer review); Dr Marty Chaplin, CIDG Statistical Editor (statistical peer review); Dr Vittoria Lutje, CIDG Information Specialist (search peer review); Maria Rosaria Cozzolino, RN MSN, Emergency Department, Barking, Havering and Redbridge University Hospitals Trust, UK (consumer peer review). One additional peer reviewer provided clinical content peer review, but chose not to be publicly acknowledged.
review stage: Dr Harshdeep Harshad Acharya, MD, Internal Medicine Resident Physician, Saint Peter's University Hospital, New Jersey, USA (clinical/content peer review); Dr Nour Essale, MD, Master of International Public Health, UK (clinical/content peer review); Dr Audrin Lenin, Department of Medicine, Christian Medical College, Vellore, India (clinical/content peer review); Dr Kerry Dwan, Cochrane Methods Support Unit (statistical peer review); Dr Vittoria Lutje, CIDG Information Specialist (search peer review); Jenny Negus, PPI Advocate and Cochrane Consumer Reviewer (consumer peer review).
Emma Sydenham (Co‐ordinating Editor, Cochrane Injuries) advised on trial regulatory compliance.
The CIDG editorial base is funded by UK aid from the UK Government for the benefit of low‐ and middle‐income countries (project number 300342‐104). The views expressed do not necessarily reflect the UK Government's official policies.
Rebecca Kuehn is supported by the Research, Evidence and Development Initiative (READ‐It). READ‐It (project number 300342‐104) is funded by UK aid from the UK Government; however, the views expressed do not necessarily reflect the UK Government's official policies.
Appendices
Appendix 1. Search strategies
Cochrane COVID‐19 Study Register (CCSR) Search string: "PF‐07321332" OR "PF 07321332" OR "PF07321332" or paxlovid* or nirmatrelvir*
Study characteristics: 1) "Intervention assignment": “Randomised” OR 2) "Study design": "Parallel/Crossover" OR "Unclear" OR 3) "Study type": "Adaptive/Platform"
Scopus TITLE‐ABS‐KEY (("PF‐07321332" OR "PF 07321332" OR "PF07321332" OR paxlovid* OR nirmatrelvir*) OR (random* OR placebo OR trial OR groups OR "phase 3" OR "phase3" OR p3 OR "pIII"))
WHO COVID‐19 Global literature on coronavirus disease Title, abstract, subject: (("PF‐07321332" OR "PF 07321332" OR "PF07321332" OR paxlovid* OR nirmatrelvir*) AND (random* OR placebo OR trial OR groups OR "phase 3" OR "phase3" OR p3 OR "pIII"))
Appendix 2. Critical and important criteria for the Research Integrity Assessment of RCTs investigating nirmatrelvir/ritonavir
| Domain | Signalling questions to critical and important criteria | Assessment | Decision | |
| 1 | Retraction or expression of concern | Is the study retracted? | Check for post‐publication amendments in the systematic search for studies and on the Retraction Watch Database (http://retractiondatabase.org/RetractionSearch.aspx?) | If study is retracted, exclude the study |
| Is there an expression of concerns published elsewhere? | Check for expressed concerns on the journal‘s homepage or preprint server | If expression of concerns are published, (1) send a request to the authors or the journal editors or wait until resolution of the concerns and (2) hold the study in awaiting classification until clarification | ||
| 2 | Trial registration | Does the study report a trial registry number? | Check in the publication or study report | If study is not prospectively registered, exclude the study |
| Is the study prospectively registered? | Check in the trials register the date of protocol submission and first posted. Prospective registration is defined as registration of a trial before enrolment of the first participant as defined by the WHO. It must be determined whether the registers registered (date first posted) without delay at this point in the pandemic. In case of doubt, check for the date first submitted or the authors must be asked for the submission date. | |||
| Are there any inconsistencies in details such as dates and study methods reported in the publication and in the registration documents? | Compare study dates (enrolment, duration, completion) and methods (study type, allocation, masking) between publication and protocol. | If date of registration is unclear or if prospectively registered, but with inconclusive information, (1) send a request to the authors and (2) hold the study in awaiting classification until clarification | ||
| 3 | Ethics approval | Is an ethics approval reported in the publication? | e.g. the study was authorized by the ethics committee XY located in XY. | If ethics approval or participants’ consent is not adequate, exclude the study. If ethics approval or participants’ consent is unclear or incomplete, (1) send a request to the authors and (2) hold the study in awaiting classification until clarification. |
| Is an ethics approval number reported? | Check in the publication, study report and study protocol | |||
| Is the name and location of the ethics committee reported? | Check in the publication, study report and study protocol | |||
| Does a nationally recognized ethics committee as defined in the country’s clinical trial regulations give the ethics committee approval? | Check the ethics committee on the WHO list of national ethics committees (https://apps.who.int/ethics/nationalcommittees/) and the specific regulations for the country on NIH Clinical Trials Regulation website (https://clinregs.niaid.nih.gov/country/mexico#_top). | |||
| Does the study require written informed consent from participants? | Check in the publication, study report and study protocol | |||
| 4 | Study authorship | Are the authors’ affiliations and countries the study is reported to have taken place in consistent? | Check in the publication, study report and study protocol | If study authorship is unclear, (1) send a request to the authors and (2) hold the study in awaiting classification until clarification. If study authorship is still not plausible after contacting the authors,exclude the study. |
| Are countries specified in different parts of the article or as compared to the trials registry consistent? | Check in the publication, study report and study protocol | |||
| Is the number of authors plausible for the study design (e.g. a single author article reporting a randomized controlled trial is impossible)? | Check in the publication, study report and study protocol | |||
| 5 | Methods reporting | Is the study design (e.g. randomization) reported in sufficient detail? | It has to be clear that the study was truthfully randomized. The method used for the randomization must be described and the process must lead to a random allocation of the participants. The sole designation "randomized study" is not sufficient. | If study design is not reported in sufficient detail, (1) send a request to the authors and (2) hold the study in awaiting classification until clarification. If study turns out to be non‐randomized following author contact, exclude the study. |
| Are baseline details reported in sufficient detail to assess whether randomization worked properly? | Check whether patients characteristics, e.g. risk factors for COVID‐19 (age, gender, comorbidities) and co‐interventions, are reported | |||
| 6 | Results | Is the number of patients recruited within the timeframe with the condition plausible? | Check in the publication. Justify the decision based on clinical experience. The decision should be verifiable. | If study results are not plausible, (1) send a request to the authors and (2) hold the study in awaiting classification until clarification. If, after contacting the author, it turns out that study results are not plausible or fabricated,exclude the study. |
| Is there a realistic response rate or number of participants lost to follow up? In cases with zero losses to follow‐up, is there a plausible explanation (e.g. small number of participants, short‐term follow‐up)? | ||||
| Is the study free from results that could be implausible (e.g. massive risk reduction, unexpected outlier data, unusual frequency of an outcome)? | ||||
| Does the number of participants (e.g. women) in each group coincide with the reported randomization method (e.g. block randomization)? | Check in the publication, study report and study protocol | |||
| Is there no noteworthy overlap in text/data with other published articles by the same or different authors without explanation? | ||||
| Is there no excessive similarity or difference in the characteristics of the study participants between groups? | ||||
| Are there no discrepancies between data reported in figures, tables, and text? | ||||
| Are there no calculation errors (e.g. number of participants, percentages, proportions)? | ||||
Potentially eligible RCTs identified during screening were assessed for research integrity hierarchically considering domain 1 to 6. Retraction, lack of prospective registration, lack of adequate ethical approval with informed written consent, implausible study authorship, lack of truthful randomization, implausible study results were triggers that led to exclusion of a RCT. Concerns in any domain put the study in ‘awaiting classification’ and led to further investigations. If no concerns appeared through all domains or could be clarified, e.g. in correspondence with study authors, the RCT met criteria for inclusion in the review and was processed further. For the next review update, included RCTs and RCTs ‘awaiting classification’ must be reassessed for retraction notices.
Data and analyses
Comparison 1. Nirmatrelvir/ritonavir for treating COVID‐19 in outpatient settings with asymptomatic or mild disease .
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality at day 28 | 1 | 2224 | Risk Ratio (M‐H, Random, 95% CI) | 0.04 [0.00, 0.68] |
| 1.2 Worsening of clinical status: admission to hospital or death at 28 days | 1 | 2224 | Risk Ratio (M‐H, Random, 95% CI) | 0.13 [0.07, 0.27] |
| 1.3 Worsening of clinical status: admission to hospital or death at 28 days (subgroup analysis based on age) | 1 | 2085 | Risk Ratio (M‐H, Random, 95% CI) | 0.13 [0.06, 0.27] |
| 1.3.1 children | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.3.2 age < 65 years of age | 1 | 1817 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.07, 0.34] |
| 1.3.3 age ≥ 65 years of age | 1 | 268 | Risk Ratio (M‐H, Random, 95% CI) | 0.05 [0.01, 0.38] |
| 1.4 Worsening of clinical status: admission to hospital or death at 28 days (subgroup analysis based on ethnicity) | 1 | 2085 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.07, 0.29] |
| 1.4.1 White | 1 | 1486 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.07, 0.33] |
| 1.4.2 Black/African American | 1 | 94 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.01, 7.04] |
| 1.4.3 Asian | 1 | 296 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.00, 1.19] |
| 1.4.4 Other | 1 | 209 | Risk Ratio (M‐H, Random, 95% CI) | 0.08 [0.00, 1.34] |
| 1.5 Serious adverse events during the study period | 1 | 2224 | Risk Ratio (M‐H, Random, 95% CI) | 0.24 [0.15, 0.41] |
| 1.6 Any grade treatment‐emergent adverse events during the study period | 1 | 2224 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.82, 1.10] |
| 1.7 Any grade treatment‐related adverse events during the study period | 1 | 2224 | Risk Ratio (M‐H, Random, 95% CI) | 2.06 [1.44, 2.95] |
| 1.8 Discontinuation of study drug due to adverse events during the study period | 1 | 2224 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.30, 0.80] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
EPIC‐HR 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes | Primary study outcome (as defined by the study)
Relevant review outcomes reported
Additional study outcomes reported
|
|
| Notes | Date of publication: 16 Febuary 2022 Sponsor/funding: Pfizer Information on ethics votum: trial sites in the countries HU, ES, CZ and BG provided information on ethics approval in their trial registry entries and obtained the necessary permissions. |
|
mITT: modified intention to treat;RT‐PCR: reverse transcription polymerase chain reaction; SAEs: serious adverse events; TEAEs: treatment‐emergent adverse events; TRAEs: treatment‐related adverse events; WHO: World Health Organization.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Caceres 2022 | Editorial, commentary, letter etc. |
| Elliott 2022 | Editorial, commentary, letter etc. |
| EPIC‐PEDS 2022 | Ineligible study design |
| NCT05305547 | Ineligible intervention: study drug S‐217622 |
| Singh 2022 | Ineligible patient population: healthy adults |
| Wang 2022 | Editorial, commentary, letter etc. |
Characteristics of ongoing studies [ordered by study ID]
ChiCTR2200059390.
| Study name | A randomized controlled study on the efficacy and safety of Huashi Baidu granule in the treatment of novel coronavirus pneumonia (covid‐19) with high risk factors |
| Methods | Trial design: open‐label RCT with parallel assignment Type of record: trial register entry Sample size: 300 Setting: NA Country: China Language: English Number of centres: 1 Study purpose (treatment, prevention): treatment Trial registration number: ChiCTR2200059390 Date of registration: 28 April 2022 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention
Details of control
|
| Outcomes | Primary study outcome:
Relevant review outcomes planned:
Additional study outcomes:
|
| Starting date | May 2022 |
| Contact information | Yuanweian weian_1980@163.com 528 Zhangheng Road, Pudong New Area, Shanghai |
| Notes | Recruitment status: NA Prospective completion date: December 2022 Date last update posted: 16 May 2022 Sponsor/funding: Shuguang Hospital Affiliated to Shanghai University of traditional Chinese Medicine, Shanghai Municipal Health Commission |
EPIC‐PEP 2021.
| Study name | A study of a potential oral treatment to prevent COVID‐19 in adults who are exposed to household member(s) with a confirmed symptomatic COVID‐19 infection |
| Methods | Trial design: double‐blind RCT with 3 parallel arms Type of record: trial register entry Sample size: 2880 Setting: outpatient Country: UK Language: English Number of centres: 358 Study purpose (treatment, prevention): prevention Trial registration number: NCT05047601 Date of registration: 17 September 2021 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention
Details of control group
|
| Outcomes | Primary study outcome:
Relevant review outcomes planned:
Additional study outcomes:
|
| Starting date | 17 September 2021 |
| Contact information | Pfizer CT.gov Call Center 1‐800‐718‐1021 ClinicalTrials.gov_Inquiries@pfizer.com |
| Notes | Recruitment status: recruiting Prospective completion date: 18 April 2022 Date last update posted: 18 April 2022 Sponsor/funding: Pfizer |
EPIC‐SR 2021.
| Study name | Evaluation of protease inhibition for COVID‐19 in standard‐risk patients (EPIC‐SR) |
| Methods | Trial design: double‐blind RCT with 2 parallel arms Type of publication: trial registry entry Setting: outpatients Recruitment dates: NR Country: worldwide Language: English Number of centres: 372 study locations Study purpose (treatment, prevention): treatment Trial registration number: NCT05011513 Date of registration: 18 August 2021 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention:
Treatment details of control group:
Concomitant therapy: NA |
| Outcomes | Primary study outcome:
Relevant review outcomes planned:
Additional study outcomes:
|
| Starting date | 25 August 2021 |
| Contact information | Pfizer CT.gov Call Center1‐800‐718‐1021 ClinicalTrials.gov_Enquiries@pfizer.com |
| Notes | Recruitment status: recruiting Prospective completion date: 30 November 2022 Date last update posted: 13 April 2022 Sponsor/funding: Pfizer |
NCT05321394.
| Study name | Adaptive, randomized, non‐inferiority trial on the use of monoclonal antibodies or antivirals in outpatients with mild or moderate COVID‐19 |
| Methods | Trial design: open‐label RCT with parallel assignment Type of record: trial register entry Sample size: 1095 Setting: outpatient Country: Italy Language: English Number of centres: 19 Study purpose (treatment, prevention): treatment Trial registration number: NCT05321394 Date of registration: 11 April 2022 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention
Details of control
|
| Outcomes | Primary study outcome:
Relevant review outcomes planned:
Additional study outcomes:
|
| Starting date | 7 March 2022 |
| Contact information | Professor Evelina Tacconelli evelina.tacconelli@univr.it |
| Notes | Recruitment status: recruiting Prospective completion date: 30 October 2022 Date last update posted: 12 April 2022 Sponsor/funding: Azienda Ospedaliera Universitaria Integrata Verona |
NCT05341609.
| Study name | A multicentre, single‐blind, randomized, controlled study to evaluate the efficacy and safety of JT001 (VV116) compared with Paxlovid for the early treatment of COVID‐19 in participants with mild to moderate COVID‐19 |
| Methods | Trial design: single‐blind RCT, parallel assignment Type of record: trial register entry Sample size: 864 Setting: outpatients Country: China Language: English Number of centres: 7 Study purpose (treatment, prevention): treatment Trial registration number: NCT05341609 Date of registration: April 22,2022 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention:
Details of control group
|
| Outcomes | Primary study outcomes:
Relevant review outcomes planned:
Additional study outcomes:
|
| Starting date | 4 April 2022 |
| Contact information | Huaqing Duan huaqing.duan@vigonvita.cn |
| Notes | Recruitment status: recruiting Prospective completion date: 30 April 2023 Date last update posted: 29 April 2022 Sponsor/funding: Vigonvita Life Sciences |
NCT05386433.
| Study name | Paxlovid in the treatment of COVID‐19 patients with uremia |
| Methods | Trial design: open‐label RCT Type of record: trial register entry Sample size: 40 Setting: NA Country: China Language: English Number of centres: 1 Study purpose (treatment, prevention): treatment Trial registration number: NCT05386433 Date of registration: 23 May 2022 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention
Details of control:
|
| Outcomes | Primary study outcome:
Relevant review outcomes planned:
Additional study outcomes:
|
| Starting date | June 2022 |
| Contact information | Jieming QU, PhD, Ruijin Hospital |
| Notes | Recruitment status: not yet recruiting Prospective completion date: August 2022 Date last update posted: 23 May 2022 Sponsor/funding: Ruijin Hospital |
PANORAMIC 2021.
| Study name | A clinical trial investigating novel treatments for COVID‐19 in the community (PANORAMIC Trial) |
| Methods | Trial design: double‐blind RCT platform trial Type of record: trial register entry Sample size: 5319 Setting: outpatients Country: United Kingdom, England, Northern Ireland, Scotland, Wales Language: English Number of centres: NR Study purpose (treatment, prevention): treatment Trial registration number: ISRCTN30448031 Date of registration: 28 October 2021 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention
Treatment details of control group
Concomitant therapy: NA |
| Outcomes | Primary study outcome
Relevant review outcomes planned
Additional study outcomes
|
| Starting date | Overall trial start date 1 September 2021 |
| Contact information | Prof. Christopher Butler Nuffield Department of Primary Care Health Sciences University of Oxford Radcliffe Observatory Quarter Woodstock Road Oxford OX2 6GG United Kingdom +44 (0)1865 289670 panoramic@phc.ox.ac.uk |
| Notes | Recruitment status: recruiting Prospective completion date: 30 September 2023 Date last update posted: NR Sponsor/funding: Department of Health and Social Care, National Institute for Health Research (NIHR) (UK). |
RECOVERY 2020.
| Study name | Randomised evaluation of COVID‐19 therapy (RECOVERY) |
| Methods | Trial design: platform trial Type of record: trial register entry Sample size: 50,000 Setting: inpatients Country: Ghana, India, Indonesia, Nepal, South Africa, Sri Lanka, United Kingdom, Vietnam Language: English Number of centres: 195 Study purpose (treatment, prevention): treatment Trial registration number: NCT04381936 Date of registration: 11 May 2020 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention (Part L)
Treatment details of control group
Concomitant therapy: NA |
| Outcomes | Primary study outcomes:
Relevant review outcomes planned:
Additional study outcomes :
|
| Starting date | Overall trial start date 19 March 2020 |
| Contact information | Richard Haynes +44 (0)1865 743743 recoverytrial@ndph.ox.ac.uk |
| Notes | Recruitment status: recruiting Prospective completion date: NR Date last update posted: 7 April 2022 Sponsor/funding:
|
ALT: alanine aminotransferase; AST: Aspartate transferase; BMI: body mass index; ;CT: computed tomography; GFR: glomerular filtration; ICU: intensive care unit; rate;IV: intravenous; RCT: randomized controlled trial; RT‐PCR: reverse transcription polymerase chain reactionSAEs: serious adverse events; ULN: upper limit of normal
Differences between protocol and review
Following the rational from Pfizer that nirmatrelvir/ritonavir is developed to manage outpatients with COVID‐19, an outcome set for inpatients was not included in the protocol avoiding the impression that studies for this population are needed. However, we do not know whether nirmatrelvir/ritonavir might be a valuable antiviral option for COVID‐19 patients at high risk who are hospitalized early after infection. Therefore, we have planned if we identify inpatient studies we include them and use the outcome set for hospitalized COVID‐19 patients published elsewhere (Popp 2021b). On 28 March 2022, the RECOVERY trial announced that Paxlovid® to be investigated as a potential treatment for patients hospitalized with COVID‐19. We added the outcome set for hospitalized COVID‐19 patients to the review.
We have changed the definition of our active comparator. In the protocol we planned to compare nirmatrelvir/ritonavir to active comparisons with proven efficacy only. We decided to extend our definition of an eligible active comparator to any active comparator, including new interventions that would be investigated in future trials that may use nirmatrelvir/ritonavir as comparator.
Contributions of authors
Stefanie Reis (SR): conception of the review; design of the review; search and selection of studies for inclusion in the review; collection of data for the review; assessment of research integrity and risk of bias in included studies; analysis of data; assessment of the certainty in the body of evidence; interpretation of data, and writing of the review.
Maria‐Inti Metzendorf (MIM): search strategy design, conduct of living search and writing of the review.
Rebecca Kuehn (RK): conception of the review; design of the review; search and selection of studies for inclusion in the review; interpretation of data; writing the review.
Maria Popp (MP): conception of the review; design of the review; interpretation of data; proofreading of the review.
Ildikó Gágyor (IG): conception of the review; design of the review; interpretation of data; proofreading of the review.
Peter Kranke (PK): conception of the review; design of the review; interpretation of data; proofreading of the review.
Patrick Meybohm (PM): conception of the review; design of the review; interpretation of data; proofreading of the review.
Nicole Skoetz (NS): conception of the review; design of the review; interpretation of data; proofreading of the review.
Stephanie Weibel (SW): conception of the review; design of the review; co‐ordination of the review; search and selection of studies for inclusion in the review; collection of data for the review; assessment of research integrity and risk of bias in included studies; analysis of data; assessment of the certainty in the body of evidence; interpretation of data, and writing of the review.
Sources of support
Internal sources
-
Department of Anaesthesiology, Intensive Care, Emergency and Pain Medicine, University Hospital Würzburg, Germany
Departmental funding
Liverpool School of Tropical Medicine, UK
External sources
-
Foreign, Commonwealth, and Development Office (FCDO), UK
Project number 300342‐104
Declarations of interest
Stefanie Reis (SR) has no known conflicts of interest to declare.
Maria‐Inti Metzendorf (MIM) has no known conflicts of interest to declare.
Rebecca Kuehn (RK) has no known conflicts of interest to declare.
Maria Popp (MP) has no known conflicts of interest to declare.
Ildikó Gágyor (IG) has no known conflicts of interest to declare.
Peter Kranke (PK) has no known conflicts of interest to declare.
Patrick Meybohm (PM) has no known conflicts of interest to declare.
Nicole Skoetz (NS) has no known conflicts of interest to declare.
Stephanie Weibel (SW) has no known conflicts of interest to declare.
New
References
References to studies included in this review
EPIC‐HR 2021 {published data only}
- Hammond J, Leister-Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, et al, EPIC-HR Investigators. Oral nirmatrelvir for high-risk, nonhospitalized adults with COVID-19. New England Journal of Medicine 2022;386:1397-408. [DOI: 10.1056/NEJMoa2118542] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norisuke K. jRCT2031210267: A study of PF-07321332/ritonavir in nonhospitalized high risk adult participants with COVID-19. jrct.niph.go.jp/en-latest-detail/jRCT2031210267 2021 (first received April 11 2022).
- Pfizer. 2021-002895-38: A phase 2/3 efficacy and safety study of PF-07321332 /ritonavir in nonhospitalized high risk adult participants with COVID 19. clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2021-002895-38 2021 (first received April 11 2022).
- Pfizer. NCT04960202: EPIC-HR: study of oral PF-07321332/ritonavir compared with placebo in nonhospitalized high risk adults with COVID-19. clinicaltrials.gov/ct2/show/NCT04960202 2021 (first received April 11 2022).
References to studies excluded from this review
Caceres 2022 {published data only}
- Caceres J, Maki DG, Hennekens CH. Cautious optimism and considerations for health providers and patients: oral antiviral for early treatment of high-risk patients and vaccines for prevention of COVID-19. American Journal of Medicine 2022;135(7):808-9. [DOI: 10.1016/j.amjmed.2022.02.041] [DOI] [PMC free article] [PubMed] [Google Scholar]
Elliott 2022 {published data only}
- Elliott W, Chan J. Nirmatrelvir and ritonavir tablets (Paxlovid). Internal Medicine Alert 2022;44(1):Issue date: January 15, 2022. [Google Scholar]
EPIC‐PEDS 2022 {published data only}
- Pfizer Inc. A phase 2/3 safety, pharmacokinetics, and efficacy study of nirmatrelvir/ritonavir in pediatric, nonhospitalized symptomatic participantswWith COVID-19 who are at risk of progression to severe disease. clinicaltrials.gov/ct2/show/NCT05261139 2022 (accessed May 15 2022).
NCT05305547 {published data only}
- Shionogi Clinical Trials Administrator. NCT05305547. A study to compare S-217622 with placebo in non-hospitalized high-risk participants with COVID-19. clinicaltrials.gov/ct2/show/NCT05305547 2022 (accessed April 11 2022).
Singh 2022 {published data only}
- Singh RS, Toussi SS, Hackman F, Chan PL, Rao R, Allen R, et al. Innovative randomized phase 1 study and dosing regimen selection to accelerate and inform pivotal COVID-19 trial of nirmatrelvir [online ahead of print]. Clinical Pharmacology & Therapeutics 2022. [DOI: 10.1002/cpt.2603] [DOI] [PMC free article] [PubMed]
- Singh Rsp, Toussi SS, Hackman F, Chan PL, Rao R, Allen R, et al. Innovative randomized phase 1 study and dosing regimen selection to accelerate and inform pivotal COVID-19 trial of nirmatrelvir. medRxiv 2022;2022.02.08:22270649. [DOI: 10.1101/2022.02.08.22270649] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2022 {published data only}
- Wang Z, Yang L. In the age of omicron variant: Paxlovid raises new hopes of COVID-19 recovery. Journal of Medical Virology 2022;94(5):1766-7. [DOI: 10.1002/jmv.27540] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
ChiCTR2200059390 {published data only}
- ChiCTR2200059390. A randomized controlled study on the efficacy and safety of Huashi Baidu granule in the treatment of novel coronavirus pneumonia (covid-19) with high risk factors. chictr.org.cn/showproj.aspx?proj=169088 (first received 28 April 2022).
EPIC‐PEP 2021 {published data only}
- Norisuke K. jRCT2031210349: A study of a potential oral treatment to prevent COVID-19 in adults who are exposed to household member(s) with a confirmed symptomatic COVID-19 infection. jrct.niph.go.jp/en-latest-detail/jRCT2031210349 2021.
- Pfizer. NCT05047601: A post-exposure prophylaxis study of PF-07321332/ritonavir in adult household contacts of an individual with symptomatic COVID-19. clinicaltrials.gov/ct2/show/NCT05047601 2021.
- Pfizer Inc. EUCTR2021-002894-24-ES: A phase 2/3 postexposure prophylaxis study of PF-07321332/ritonavir in household contacts of a patient with COVID-19. clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2021-002894-24 2021.
EPIC‐SR 2021 {published data only}
- Norisuke K. jRCT2031210274: A study of PF-07321332/ritonavir in non-hospitalized low-risk adult participants with COVID-19. jrct.niph.go.jp/en-latest-detail/jRCT2031210274 2021.
- Pfizer. NCT05011513: A study of PF-07321332/ritonavir in non-hospitalized low-risk adult participants with COVID-19. clinicaltrials.gov/ct2/show/NCT05011513 2021.
- Pfizer Inc. EUCTR2021-002857-28-HU: A phase 2/3 efficacy and safety study of PF-07321332/ritonavir in nonhospitalized low-risk adult participants with COVID-19. clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2021-002857-28 2021.
NCT05321394 {published data only}
- Adaptive, randomized, non-inferiority trial on the use of monoclonal antibodies or antivirals in outpatients with mild or moderate COVID-19. ClinicalTrials.gov Identifier: NCT05321394.
NCT05341609 {published data only}
- A multicenter, single-blind, randomized, controlled study to evaluate the efficacy and safety of JT001 (VV116) compared with Paxlovid for the early treatment of COVID-19 in participants with mild to moderate COVID-19. ClinicalTrials.gov Identifier: NCT05341609.
NCT05386433 {published data only}
- NCT05386433. Paxlovid in the treatment of COVID-19 patients with uremia. clinicaltrials.gov/ct2/show/NCT05386433 (first received 23 May 2022).
PANORAMIC 2021 {published data only}
- ISRCTN30448031. A clinical trial investigating novel treatments for COVID-19 in the community. isrctn.com/ISRCTN30448031 (first received 28 October 2021).
RECOVERY 2020 {published data only}
- University of Oxford. Randomised evaluation of COVID-19 therapy (RECOVERY). clinicaltrials.gov/ct2/show/NCT04381936 (first received 11 May 2020).
Additional references
Agarwal 2020
- Agarwal A, Rochwerg B, Lamontagne F, Siemieniuk RA, Agoritsas T, Askie L, et al. A living WHO guideline on drugs for covid-19. BMJ 2020/09/04;370:m3379. [DOI: 10.1136/bmj.m3379] [DOI] [PubMed] [Google Scholar]
Altulahi 2021
- Altulahi N, AlNujaim S, Alabdulqader A, Alkharashi A, AlMalki A, AlSiari F, et al. Willingness, beliefs, and barriers regarding the COVID-19 vaccine in Saudi Arabia: a multiregional cross-sectional study. BMC Family Practice 2021;22(1):247. [DOI: 10.1186/s12875-021-01606-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Amin 2021
- Amin SA, Banerjee S, Ghosh K, Gayen S, Jha T. Protease targeted COVID-19 drug discovery and its challenges: insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorganic & Medicinal Chemistry 2021;29:115860. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Anand 2003
- Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003;300(5626):1763-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Balshem 2011
- Balshem H, Helfand M, Schünemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines – 3: rating the quality of evidence. Journal of Clinical Epidemiology 2011;64(4):401-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Booth 2021
- Booth A, Reed AB, Ponzo S, Yassaee A, Aral M, Plans D, et al. Population risk factors for severe disease and mortality in COVID-19: a global systematic review and meta-analysis. PLOS One 2021;16(3):e0247461. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bramstedt 2020
- Bramstedt KA. The carnage of substandard research during the COVID-19 pandemic: a call for quality. Journal of Medical Ethics 2020;46:803-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
CDC 2021
- Centers for Disease Control and Prevention (CDC). Benefits of getting a COVID-19 vaccine. cdc.gov/coronavirus/2019-ncov/vaccines/vaccine-benefits.html (accessed 14 December 2021).
Chen 2010
- Chen S, Jonas F, Shen C, Hilgenfeld R. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein Cell 2010;1(1):59-74. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cochrane policy ‐ managing problematic studies
- Cochrane Database of Systematic Reviews: editorial policies. Cochrane policy on managing potentially problematic studies. cochranelibrary.com/cdsr/editorial-policies#problematic-studies (assessed 04 April 2022).
COMET 2020
- Core Outcome Measures in Effectiveness Trials (COMET) Initiative. Core outcome set developers’ response to COVID-19. comet-initiative.org/Studies/Details/1538 (accessed 13 June 2021).
Dai 2020
- Dai W, Zhang B, Jiang X-M, Su H, Li J, Zhao Y, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020;368(6497):1331-5. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Data extraction template 2021
- Cochrane Pregnancy and Childbirth Group. pcg_data_extraction_form_v_2.2_-_13_july_2021_1.docx. pregnancy.cochrane.org/author-resources-new-reviews (accessed 9 December 2021).
Deeks 2022
- Deeks JJ, Higgins JP, Altman DG. Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Deng 2020
- Deng L, Li C, Zeng Q, Liu X, Li X, Zhang H, et al. Arbidol combined with LPV/r versus LPV/r alone against corona virus disease 2019: a retrospective cohort study. Journal of Infection 2020;81(1):e1-5. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
de Vries 2020
- Vries M, Mohamed AS, Prescott RA, Valero-Jimenez AM, Desvignes L, O’Connor R, et al. Comparative study of a 3CL pro inhibitor and remdesivir against both major SARS-CoV-2 clades in human airway models. bioRxiv 2020 ;95(10):1819-20. [DOI: 10.1101/2020.08.28.272880] [DOI] [Google Scholar]
Dinnes 2021
- Dinnes J, Deeks JJ, Berhane S, Taylor M, Adriano A, Davenport C, et al . Rapid, point-of-care antigen and molecular-based tests for diagnosis of SARS-CoV-2 infection. Cochrane Database of Systematic Reviews 2021, Issue 3. Art. No: CD013705. [DOI: 10.1002/14651858.CD013705] [DOI] [PMC free article] [PubMed] [Google Scholar]
Eloy 2021
- Eloy P, Le Grand R, Malvy D, Guedj J. Combined treatment of molnupiravir and favipiravir against SARS-CoV-2 infection: One + zero equals two? eBioMedicine 2021;74:103663. [DOI: 10.1016/j.ebiom.2021.103663] [DOI] [PMC free article] [PubMed] [Google Scholar]
EndNote 2013 [Computer program]
- EndNote. Version EndNote 20. Philadelphia, PA: Clarivate, 2013.
EUA for Paxlovid
- US Food and Drug Administration (FDA). Fact sheet for healthcare providers: Emergency Use Authorization for Paxlovid. www.fda.gov/media/155050/download (accessed 14 February 2022).
Fajnzylber 2020
- Fajnzylber J, Regan J, Coxen K, Corry H, Wong C, Rosenthal A, et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nature Communications 2020;11(1):5493. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Funk 2021
- Funk T, Pharris A, Spiteri G, Bundle N, Melidou A, Carr M, et al. Characteristics of SARS-CoV-2 variants of concern B.1.1.7, B.1.351 or P.1: data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Euro Surveillance 2021;26(16):2100348. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
German AWMF Guideline 2021
- Blankenfeld H, Kaduszkiewicz H, Kochen MM, Pömsl J. [SARS-CoV-2/Covid-19 Informationen und Praxishilfen für niedergelassene Hausärztinnen und Hausärzte]. awmf.org/leitlinien/detail/ll/053-054.html (accessed 14 February 2022).
Gottlieb 2022
- Gottlieb RL, Vaca CE, Paredes R, Mera J, Webb BJ, Perez G, et al. Early remdesivir to prevent progression to severe Covid-19 in outpatients. New England Journal of Medicine 2022;386(4):305-15. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Grant 2020
- Grant MC, Geoghegan L, Arbyn M, Mohammed Z, McGuinness L, Clarke EL, et al. The prevalence of symptoms in 24,410 adults infected by the novel coronavirus (SARS-CoV-2; COVID-19): a systematic review and meta-analysis of 148 studies from 9 countries. PLOS One 2020;15(6):e0234765. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Grey 2020
- Grey A, Bolland MJ, Avenell A, Klein AA, Gunsalus CK. Check for publication integrity before misconduct. Nature 2020;577(7789):167-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Harvey 2021
- Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nature Reviews Microbiology 2021;19(7):409-24. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2021
- Higgins JP, Lasserson T, Chandler J, Tovey D, Thomas J, Flemyng E, et al. Methodological Expectations of Cochrane Intervention Reviews (MECIR). community.cochrane.org/mecir-manual/ (accessed 12 December 2021).
Higgins 2022a
- Higgins JP, Savović J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JPT Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Higgins 2022b
- Higgins JP, Li T, Deeks JJ, editor(s). Chapter 6: Choosing effect measures and computing estimates of effect. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Huang 2020
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395(10223):497-506. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Huang 2021
- Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet 2021;397(10270):220-32. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
IntHout 2016
- IntHout J, Ioannidis JP, Rovers MM, Goeman JJ. Plea for routinely presenting prediction intervals in meta-analysis. BMJ Open 2016;12(6):e010247. [DOI: 10.1136/bmjopen-2015-010247] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Juthani 2021
- Juthani PV, Gupta A, Borges KA, Price CC, Lee AI, Won CH, et al. Hospitalisation among vaccine breakthrough COVID-19 infections. Lancet Infectious Diseases 2021;21(11):1485-6. [DOI: 10.1016/S1473-3099(21)00558-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Karagiannidis 2020
- Karagiannidis C, Mostert C, Hentschker C, Voshaar T, Malzahn J, Schillinger G, et al. Case characteristics, resource use, and outcomes of 10021 patients with COVID-19 admitted to 920 German hospitals: an observational study. Lancet Respiratory Medicine 2020;8(9):853-62. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kawasuji 2020
- Kawasuji H, Takegoshi Y, Kaneda M, Ueno A, Miyajima Y, Kawago K, et al. Transmissibility of COVID-19 depends on the viral load around onset in adult and symptomatic patients. PLOS One 2020;15(12):e0243597. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Killerby 2020
- Killerby ME, Link-Gelles R, Haight SC, Schrodt CA, England L, Gomes DJ, et al. Characteristics associated with hospitalization among patients with COVID-19 - Metropolitan Atlanta, Georgia, March–April 2020. Morbidity and Mortality Weekly Report 2020;69:790-4. [DOI: 10.15585/mmwr.mm6925e1] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kumar 2020
- Kumar M, Al Khodor S. Pathophysiology and treatment strategies for COVID-19. Journal of Translational Medicine 2020;18(1):353. [DOI: 10.1186/s12967-020-02520-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 2022
- Lefebvre C, Glanville J, Briscoe S, Featherstone R, Littlewood A, Marshall C, et al. Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Lin 2022
- Lin DY, Gu Y, Wheeler B, Young H, Holloway S, Sunny SK, et al. Effectiveness of Covid-19 vaccines over a 9-month period in North Carolina. New England Journal of Medicine 2022;386(10):933-41. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2020
- Liu Y, Yan L, Wan L, Xiang T, Le A, Liu J, et al. Viral dynamics in mild and severe cases of COVID-19. Lancet 2020;20(6):656-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Marshall 2020
- Marshall JC, Murthy S, Diaz J, Adhikari NK, Angus DC, Arabi YM, et al. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infectious Diseases 2020;20(8):e192-7. [DOI: 10.1016/S1473-3099(20)30483-7] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Merck 2021
- Merck. Merck and Ridgeback’s investigational oral antiviral molnupiravir reduced the risk of hospitalization or death by approximately 50 percent compared to placebo for patients with mild or moderate Covid-19 in positive interim analysis of phase 3 study. merck.com/news/merck-and-ridgebacks-investigational-oral-antiviral-molnupiravir-reduced-the-risk-of-hospitalization-or-death-by-approximately-50-percent-compared-to-placebo-for-patients-with-mild-or-moderat/ (accessed 1 October 2021).
Meta 2022 [Computer program]
- Meta: General Package for Meta-Analysis. Schwarzer G, Version 5.2-0. The R Project, 2022. Available at cran.r-project.org/package=meta.
Metzendorf 2022
- Metzendorf MI, Weibel S, Reis S, McDonald S. A pragmatic and open science-based solution to a current problem in the reporting of Living Systematic Reviews – the Cochrane Review on nirmatrelvir/ritonavir (Paxlovid®). Manuscript submitted to publication, 2 June 2022.
MPP 2021a
- Medicines Patent Pool (MPP). Medicines Patent Pool. medicinespatentpool.org/who-we-are/about-us (accessed 1 December 2021).
MPP 2021b
- Medicines Patent Pool (MPP). Nirmatrelvir - License agreement; November 2021. Available at medicinespatentpool.org/licence-post/pf-07321332.
NCT04501978
- NCT04501978. ACTIV-3: Therapeutics for Inpatients with COVID-19 (TICO) [A multicenter, adaptive, randomized, blinded controlled trial of the safety and efficacy of investigational therapeutics for hospitalized patients with COVID-19]. clinicaltrials.gov/ct2/show/NCT04501978 (first received 6 August 2020).
NCT04535167
- NCT04535167. First-in-human study to evaluate safety, tolerability, and pharmacokinetics following single ascending and multiple ascending doses of PF-07304814 in hospitalized participants with COVID-19 [A phase 1B, 2-part, double-blind, placebo-controlled, sponsor-open study, to evaluate the safety, tolerability and pharmacokinetics of single ascending (24-hour, part 1) and multiple ascending (120-hour, part 2) intravenous infusions of PF-07304814 in hospitalized participants with COVID-19]. clinicaltrials.gov/ct2/show/NCT04535167 (first received 1 September 2020).
NCT04575597
- NCT04575597. Efficacy and safety of molnupiravir (MK-4482) in non-hospitalized adult participants with covid-19 (MK-4482-002) [A phase 2/3, randomized, placebo-controlled, double-blind clinical study to evaluate the efficacy, safety, and pharmacokinetics of MK-4482 in non-hospitalized adults with COVID-19]. clinicaltrials.gov/ct2/show/NCT04575597 (first received 5 October 2020).
NHS 2021
- National Health Service (NHS) UK. Who cannot have vaccines. nhs.uk/conditions/vaccinations/why-vaccination-is-safe-and-important/ (accessed 14 December 2021).
NICE 2021
- National Institute for Health and Care Excellence (NICE). NG191: COVID-19 rapid guideline: managing COVID-19. nice.org.uk/guidance/ng191 (accessed 14 December 2021).
NIH 2021
- National Institutes of Health (NIH). COVID-19 treatment guidelines panel. Coronavirus disease 2019 (COVID-19) treatment guidelines. covid19treatmentguidelines.nih.gov/ (accessed 14 December 2021).
Owen 2021
- Owen DR, Allerton CM, Anderson AS, Aschenbrenner L, Avery M, Berritt S, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021;374(6575):1586-93. [PMID: ] [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815-34. [DOI: ] [DOI] [PubMed] [Google Scholar]
Petrilli 2020
- Petrilli CM, Jones SA, Yang J, Rajagopalan H, O'Donnell L, Chernyak Y, et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 2020;369:m1966. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pfizer 2021
- Pfizer Inc. Pfizer’s novel COVID-19 oral antiviral treatment candidate reduced risk of hospitalization or death by 89% in interim analysis of phase 2/3 EPIC-HR study. pfizer.com/news/press-release/press-release-detail/pfizers-novel-covid-19-oral-antiviral-treatment-candidate (accessed 23 November 2021).
Pfizer 2022
- Pfizer. Potentially Significant Drug Interactions, including Contraindicated DrugsPAXLOVIDTM (nirmatrelvir tablets; ritonavir tablets). https://pfizermedical.pfizerpro.com/api/vc/en/medical/assets/5773bb0d-ab2b-40bb-8988-be557bd70923/Potentially%20Significant%20Drug%20Interactions.pdf (accessed 20 May 2022) 2022;1.1:1-9.
Popp 2021a
- Popp M, Stegemann M, Metzendorf M-I, Gould S, Kranke P, Meybohm P, et al. Ivermectin for preventing and treating COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 7. Art. No: CD015017. [DOI: 10.1002/14651858.CD015017.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Popp 2021b
- Popp M, Stegemann M, Riemer M, Metzendorf M-I, Romero CS, Mikollajewska A, et al. Antibiotics for the treatment of COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 10. Art. No: CD015025. [DOI: 10.1002/14651858.CD015025] [DOI] [PMC free article] [PubMed] [Google Scholar]
Popp 2022
- Popp M, Reis S, Schießer S, Hausinger R Ilona, Stegemann M, et al. Ivermectin for preventing and treating COVID-19. Cochrane Database of Systematic Reviews 2022, Issue 6. Art. No: CD015017. [DOI: 10.1002/14651858.CD015017.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reis 2022a
- Reis S, Metzendorf M, Kuehn R, Popp M, Gagyor I, Kranke P, et al. Living evidence base for Cochrane Review "Nirmatrelvir combined with ritonavir for preventing and treating COVID‐19". osf.io/7g49c/ (created 30 May 2022). [DOI: 10.17605/OSF.IO/7G49C] [DOI] [PMC free article] [PubMed]
Ren 2022
- Ren SY, Wang WB, Gao RD, Zhou AM. Omicron variant (B.1.1.529) of SARS-CoV-2: mutation, infectivity, transmission, and vaccine resistance. World Journal of Clinical Cases 2022;10(1):1-11. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan Web 2020 [Computer program]
- Review Manager Web (RevMan Web). Version 1.22.0. The Cochrane Collaboration, 2020. Available at revman.cochrane.org.
Salvatori 2020
- Salvatori G, Luberto L, Maffei M, Aurisicchio L, Roscilli G, Palombo F, et al. SARS-CoV-2 SPIKE PROTEIN: an optimal immunological target for vaccines. Journal of Translational Medicine 2020;18(1):222. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2022
- Schünemann HJ, Higgins JPT Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Shadmi 2020
- Shadmi E, Chen Y, Dourado I, Faran-Perach I, Furler J, Hangoma P, et al. Health equity and COVID-19: global perspectives. International Journal for Equity in Health 2020;19(1):104. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JA, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898. [DOI: 10.1136/bmj.l4898] [PMID: ] [DOI] [PubMed] [Google Scholar]
Supplementary File_Nirmatrelvir_Research Integrity
- Weibel S, Reis S. Supplementary File_Nirmatrelvir_Research Integrity Assessment (Version 1). Zenodo 2022. [DOI: 10.5281/zenodo.7074190] [DOI]
Supplementary File_Nirmatrelvir_Risk of Bias
- Weibel S, Reis S. Supplementary File_Nirmatrelvir_Risk of Bias Excel Tool (Version 1). Zenodo 2022. [DOI: 10.5281/zenodo.7074194] [DOI]
Takashita 2022
- Takashita E, Kinoshita N, Yamayoshi S, Sakai-Tagawa Y, Fujisaki S, Ito M, et al. Efficacy of antibodies and antiviral drugs against Covid-19 Omicron variant. New England Journal of Medicine 2022;386(10):995-8. [DOI: 10.1056/NEJMc2119407] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8:16. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Walls 2020
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;183(6):1735. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2021
- Wang C, Han B, Zhao T, Liu H, Liu B, Chen L, et al. Vaccination willingness, vaccine hesitancy, and estimated coverage at the first round of COVID-19 vaccination in China: a national cross-sectional study. Vaccine 2021;39(21):2833-42. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Watson 2020
- Watson J, Whiting PF, Brush JE. Interpreting a Covid-19 test result. BMJ 2020;369:m1808. [DOI: 10.1136/bmj.m1808] [DOI] [PubMed] [Google Scholar]
Weibel 2022
- Weibel S, Popp M, Reis S, Skoetz N, Garner P, Sydenham E. Identifying and managing problematic trials: a Research Integrity Assessment (RIA) tool for randomized controlled trials in evidence synthesis. www.medrxiv.org/content/10.1101/2022.05.31.22275756v1 (first received 05 June 2022). [DOI: 10.1101/2022.05.31.22275756] [DOI] [PMC free article] [PubMed]
Welch 2012
- Welch V, Petticrew M, Tugwell P, Moher D, O'Neill J, Waters E, et al. PRISMA-Equity 2012 Extension: reporting guidelines for systematic reviews with a focus on health equity. PLOS Medicine 2012;9(10):e1001333. [DOI: 10.1371/journal.pmed.1001333] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wen 2022
- Wen W, Chen C, Tang J, Wang C, Zhou M, Cheng Y, et al. Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID-19: a meta-analysis. Annals of Medicine 2022;54(1):516-23. [DOI: 10.1080/07853890.2022.2034936] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2018
- World Health Organization (WHO). International standards for clinical trial registries – 2nd edition. Geneva: World Health Organization; 2018. Licence: CC BY-NC-SA 3.0 IGO. Available at who.int/ictrp/International_Standards_for_Clinical_Trial_Registration_2018.pdf.
WHO 2020
- World Health Organization (WHO). Report of the WHO-China joint mission on coronavirus disease 2019 (COVID-19). who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report (accessed 24 November 2021).
WHO 2020a
- World Health Organization (WHO). Advice on the use of point-of-care immunodiagnostic tests for COVID-19: scientific brief. apps.who.int/iris/handle/10665/331713 (accessed 8 April 2020).
WHO 2021
- World Health Organization (WHO). Vaccine equity. who.int/campaigns/vaccine-equity (accessed 14 December 2021).
WHO 2021a
- World Health Organization (WHO). WHO coronavirus disease (COVID-19) dashboard. covid19.who.int/table (accessed 23 May 2022).
WHO 2021b
- World Health Organization (WHO). Therapeutics and COVID-19: living guideline . app.magicapp.org/#/guideline/nBkO1E/rec/E850m0 24 September 2021 (accessed 23 May 2022);version 6:15-23.
Williamson 2020
- Williamson EJ, Walker AJ, Bhaskaran K, Bacon S, Bates C, Morton CE, et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020;584(7821):430-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
World Bank 2022
- World Bank. The world by income and region. datatopics.worldbank.org/world-development-indicators/the-world-by-income-and-region.html (accessed 15 February 2022).
Wu 2020
- Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020;323(13):1239-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
Zhang 2020
- Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020;368(6489):409-12. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2021
- Zhang Y, Tang LV. Overview of targets and potential drugs of SARS-CoV-2 according to the viral replication. Journal of Proteome Research 2021;20(1):49-59. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Reis 2022
- Reis S, Popp M, Kuehn R, Metzendorf M-I, Gagyor I, Kranke P, et al. Nirmatrelvir combined with ritonavir for preventing and treating COVID‐19. Cochrane Database of Systematic Reviews 2022, Issue 4. Art. No: CD015395. [DOI: 10.1002/14651858.CD015395] [DOI] [PMC free article] [PubMed] [Google Scholar]