

Abstract
Pulmonary arterial hypertension (PAH) is a rapidly progressive disease, ultimately leading to right heart failure and death. Accumulating evidence indicates that intervention early in disease progression results in better outcomes than delaying treatment.
In this review we will discuss the assessments and strategies that can be used to monitor disease progression and guide clinical management. Many tools, such as symptoms, functional classification, exercise capacity, haemodynamic measures, findings on cardiac imaging and levels of biomarkers, have shown to be prognostic for survival both at diagnosis and during treatment. However, attempts to define goal thresholds have produced a variety of results.
Several groups have developed risk calculators to estimate individual patients' mortality risk, but the accuracy of these tools across different patient populations remains unknown. What is clear is the importance of regularly assessing a range of parameters and then tailoring treatment goals to each patient. In addition, the use of a multidisciplinary team approach is crucial in order to support patients through all aspects of managing their condition.
There is still an urgent need for prospective collaborative initiatives to assess novel goals and improve treatment strategies that would allow physicians to personalise and optimise clinical management for their patients with PAH.
Keywords: Assessment, disease progression, prognosis, pulmonary hypertension, treatment
Pulmonary arterial hypertension (PAH) is a rapidly progressive disease. Meta-analyses of survival data from randomised controlled trials in PAH have shown that targeted therapies provide a reduction in mortality of ∼40% [1, 2]. However, national pulmonary hypertension (PH) registries show that despite appropriate treatment, patients still have a low expected survival. For example, while 91% of patients were alive 1 yr after diagnosis in the US REVEAL registry [3], longer follow-up in the French Network on Pulmonary Hypertension showed that <60% of patients survived after 3 yrs [4].
Evidence suggests that early intervention may improve outcome. Initial observational data from patients treated with infused epoprostenol indicated that patients with less severe functional impairment and no history of right heart failure at the start of treatment had better outcomes [5, 6]. Subsequently, the randomised controlled EARLY trial investigating bosentan in patients with mildly symptomatic PAH (World Health Organization functional class (WHO FC) II) demonstrated that 6 months of bosentan treatment was associated with haemodynamic improvements and prevention of clinical deterioration [7]. In addition, the ARIES-1 and -2 studies with ambrisentan showed that patients with mildly symptomatic (WHO FC II) PAH had improvement in exercise capacity [8]. Long-term extension data from these studies and BREATHE-1 and -5 (comparing bosentan with placebo in PAH and Eisenmenger's syndrome, respectively) suggest that patients who started treatment with ambrisentan or bosentan, respectively, in the initial placebo-controlled phase appeared to have better outcomes through the extension phase compared with patients who received placebo in the initial 4-month study then switched to active treatment for the extension (although the studies were not powered to show this statistically) [9–12]. Furthermore, meta-analysis of placebo-controlled study data strongly suggests that PAH therapies are effective in improving survival even in studies of short duration (12–16 weeks) [1].
Timely identification of disease progression may prompt physicians to initiate earlier intervention in order to prevent the otherwise inevitable deterioration of right heart function. However, detecting early disease progression is a complex task presenting various challenges [13], mainly because the pathophysiological changes that occur during the initial stages of the disease are generally not detectable. Consequently, the patient becoming aware of symptoms and the changes that are detected in resting haemodynamics are late sequelae of the pathogenic process. Late reporting of symptoms and delayed or misdiagnosis are, therefore, common. In this review, we discuss how physicians can monitor disease progression in order to guide clinical management.
TOOLS AND VARIABLES TO DETECT DISEASE PROGRESSION
Right ventricular (RV) dysfunction is the key driver of PAH disease progression and most tools and variables used to detect clinical deterioration are reflecting RV insult. Several tools are currently used to detect disease progression both at diagnosis and during follow-up of patients with PAH (table 1) [14, 15].
Table 1. Tools and variables for detecting disease progression.
| Clinical assessment |
| Symptoms (NYHA or WHO functional class) |
| Rate of progression |
| PAH aetiology |
| Signs of right ventricular failure |
| Functional (exercise) capacity |
| 6-min walk distance |
| Cardiopulmonary exercise test |
| V′O2,peak |
| V′E/V′CO2 slope |
| Changes in blood pressure |
| Evaluation of right ventricular (dys)function |
| Right heart catheterisation |
| Right atrial pressure |
| Cardiac index |
| Cardiac output |
| Echocardiography |
| Pericardial effusion |
| Tei index (or myocardial performance index) |
| Tricuspid annular plane systolic excursion |
| Right atrial area |
| Left ventricular eccentricity |
| Biomarkers |
| NT-proBNP/BNP |
| Troponin |
| Uric acid |
| Sodium |
| Cardiac magnetic resonance imaging |
| Right ventricular size and function |
| Cardiac output |
| Stroke volume |
NYHA: New York Heart Association; WHO: World Health Organization; PAH: pulmonary arterial hypertension; V′O2,peak: peak oxygen uptake; V′E: minute ventilation; V′CO2: carbon dioxide production; NT-proBNP: N-terminal pro-brain natriuretic protein; BNP: brain natriuretic protein.
The first step is clinical assessment, including symptoms, signs of RV failure, and rate of disease progression. Although the symptoms of PAH are nonspecific, the New York Heart Association (NYHA) functional classification for heart failure [16] has been adapted for PAH as the WHO FC. It is a four-point scale to rate the impact of breathlessness and fatigue according to daily activity. The WHO FC scale ranges from I (no limitation in physical activity) to IV (inability to carry out physical activity without symptoms, or with symptoms present at rest) [15, 17]. A patient presenting with signs of right heart failure (peripheral oedema, ascites and jugular vein distension) should be considered to be in WHO FC IV. Finally, the natural outcome of PAH is influenced by its underlying causes. As an example, patients with PAH associated with systemic sclerosis have a dismal prognosis compared with those with Eisenmenger's syndrome [15].
Functional assessment by NYHA/WHO FC can be completed by the evaluation of exercise capacity using the 6-min walk distance (6MWD) and cardiopulmonary exercise test (CPET). The 6MWD, which remains the primary end-point of most PH clinical trials, has the advantage of being simple to perform and is easily reproducible between clinic visits [18]. Cardiopulmonary exercise testing involves continuous monitoring of gas exchange and ventilation during incremental exercise, and records the following key variables: peak oxygen consumption (V′O2,peak), the slope of increase of minute ventilation relative to carbon dioxide production (V′E/V′CO2 slope) and changes in blood pressure [19].
RV function is defined by the complex interplay between contractility, afterload, compliance and heart rate. There is no such tool that can directly measure the key determinants of RV function. However, it can be approached by invasive haemodynamics, echocardiography, magnetic resonance imaging and biomarker analysis. Right heart catheterisation (RHC) is the gold standard for diagnosing PH, assessing disease severity and determining response to treatment [14, 15], and records a range of variables impacting on prognosis (table 1). Interestingly enough, right atrial pressure, but not pulmonary pressure, has been found to influence outcome. Echocardiography remains a precious tool to approach the right ventricle noninvasively. The tricuspid annular plane systolic excursion (TAPSE) is an indirect marker of RV systolic function and contractility. It is a simple and reproducible variable that is associated with the patient's outcome. The myocardial performance index (also called the Tei index) is another variable with an impact on outcome (table 1). However, both TAPSE and the Tei index are influenced by preload and the degree of tricuspid regurgitation which may limit their use in clinical practice.
Newer imaging techniques are also arousing considerable interest in the assessment of patients with PH [14, 15]. Cardiac magnetic resonance imaging provides a better evaluation of RV size and function compared with echocardiography, allowing noninvasive assessment of cardiac output and stroke volume [20].
Finally, biochemical markers provide another noninvasive tool for assessment and monitoring of RV dysfunction in patients with PH [14, 15]. Increased levels of N-terminal pro-brain natriuretic peptide (NT-proBNP)/BNP, troponin and uric acid have all been shown to correlate with poor prognosis in PH [21–23].
KEY PROGNOSTIC MARKERS
Clinical, functional capacity and haemodynamic assessments provide important prognostic information on the course of PAH which may aid clinical management by prompting initiation or intensification of treatment.
Baseline predictors of survival
Several baseline variables have been shown to have prognostic importance. Despite its apparent subjective nature, WHO FC or NYHA FC remains a powerful predictor of survival [3, 5, 24–27]. For example, in observational studies of patients receiving epoprostenol therapy, survival was significantly longer in patients at NYHA FC III compared with those at NYHA FC IV (fig. 1) [5, 25]. Likewise, in a follow-up of patients receiving subcutaneous treprostinil, worse functional class at baseline was associated with lower survival rates (NYHA FC IV versus NYHA FC III; p<0.001) [27], while a similar association between increased mortality and baseline NYHA FC IV was observed in a 3-yr follow-up of patients receiving oral bosentan [26]. In the US REVEAL registry, patients who were NYHA/WHO FC IV at baseline had a significantly lower 1-yr survival rate than those with better functional class (p<0.05) [3].
Figure 1.

Baseline New York Heart Association functional class (NYHA FC) predicts survival in patients with pulmonary hypertension using infused epoprostenol therapy. a) 162 patients followed for a median of 36 months. Reproduced from [5] with permission from the publisher. b) 178 patients followed for a median of 26 months. Reproduced from [6] with permission from the publisher.
Exercise capacity, as assessed by 6MWD or CPET, has also been shown to be an independent predictor of survival [3, 4, 26, 28–33]. Several studies have attempted to define a threshold value at which 6MWD is predictive of mortality. For example, a long-term follow-up of 169 patients receiving bosentan therapy identified a baseline 6MWD of <358 m as significantly associated with mortality (p<0.05) [26], while a similar value of <332 m was shown to be predictive of a lower survival rate (p<0.001) in a smaller study of 43 PH patients receiving prostacyclin therapy [29]. A lower threshold was calculated from the US REVEAL registry with 2,716 patients, indicating that a 6MWD of <165 m was significantly associated with mortality risk (HR 1.68; p<0.001) [3]. In a study of 86 patients with idiopathic PAH (referred to as primary PH at that time), the following CPET variables were found to be predictive of survival: V′O2,peak ≤10.4 mL·kg−1·min−1 (p<0.0001) and systolic blood pressure at peak exercise ≤120 mmHg (p<0.0001) [34].
Echocardiography is also important in a risk-assessment strategy, since it provides variables to predict outcome [3, 32, 35–37]. A recent prospective study investigating echocardiographic prognostic factors in PAH identified the following predictors of mortality: mean pulmonary arterial pressure (Ppa) ≥49 mmHg (p=0.012); diastolic Ppa ≥29 mmHg (p=0.006); an abnormal end-diastolic septal curve (p=0.027); inferior vena cava diameter ≥20 mm with respiratory variation of diameter <50% (p=0.018); Tei index ≥0.98 (p=0.035); each 1 mm decrease in TAPSE (p=0.024); and presence of pericardial effusion (p=0.002) [37]. However, this study is in contrast with previous reports suggesting that pulmonary pressures do not influence outcome [3, 5–7, 14, 15, 21, 22, 25, 29]. The presence of pericardial effusion (p<0.003) and indexed right atrial area (p<0.005) were predictors of mortality in a 3-yr follow-up of 81 patients with severe “primary PH” following treatment with prostacyclin plus conventional therapy [32]. Pericardial effusion was also a predictor of mortality from an analysis of the REVEAL registry data (p=0.014) [3]. In a study of 63 patients, for every 1 mm decrease in TAPSE, the unadjusted risk of death increased by 17% (p=0.006) [36]. In a small study involving 26 patients with PH, the Tei index was the single most powerful variable to discriminate patients with PH from healthy patients (being significantly prolonged in PH patients; p<0.001) and was the strongest predictor of clinical status and survival, with each 0.1 unit in the index increasing the risk of death 1.3-fold [35]. Whether or not a single echocardiography-derived variable is superior to another in predicting outcome is unknown. However, in clinical practice, most physicians would rely on the presence of pericardial effusion and TAPSE as determinants of change in the management of PAH.
Several studies have identified the biomarker NT-proBNP (or BNP) as an independent predictor of mortality [3, 22, 30, 38, 39]. NT-proBNP thresholds predicting mortality have been identified as ≥1,400 pg·mL−1 in a 3-yr study of 55 PH patients [39] and ≥1,256 pg·mL−1 in another retrospective analysis of data from 189 PAH patients [38]. BNP levels of ≥150 pg·mL−1 in a study of 60 primary PH patients [22] and >180 pg·mL−1 in the US REVEAL registry of 2,716 PH patients [3] have been shown to be associated with greater mortality risk. A detectable level of cardiac troponin T has also been shown as an independent marker of 2-yr survival rate (p=0.001) in a study of 56 patients with PH [23].
Several haemodynamic measurements have been shown to be independent predictors of mortality in PAH studies. Elevated right artery pressure (Pra) has been consistently identified in several studies to be associated with worse outcome [3, 5, 6, 28, 30, 31, 39–41]. In the REVEAL registry, mean Pra ≥20 mmHg was shown to predict greater mortality risk (p=0.043) [3], while a value of ≥12 mmHg was associated with increased risk of death during long-term follow-up of patients on epoprostenol [6]. Early data from the National Heart Lung and Blood Institute PH registry (1981–1988) showed significantly reduced survival for patients with mean Ppa ≥85 mmHg (p<0.05) [41]. However, this finding has not been confirmed in other analyses, especially in more recent years. Mixed venous oxygen saturation (SV,O2) ≤55% was associated with increased mortality in a retrospective follow-up of patients using subcutaneous treprostinil (p<0.001) [24], as a marker of low cardiac output and increased peripheral oxygen extraction. Greater mortality risk has also been shown for patients with a higher pulmonary vascular resistance (PVR) of >32 Wood units [3] or PVR index of >30 mmHg·L−1·min−1·m−2 [24], with cardiac index and cardiac output also being predictive of long-term survival rates [4, 30, 40, 41]. These studies demonstrate that Pra and cardiac index are strong markers of prognosis, as they reflect the severity of RV dysfunction.
Cardiac magnetic imaging showed that median stroke volume ≤25 mL·m−2 (p=0.010), right ventricular end-diastolic volume ≥84 mL·m−2 (p=0.011) and left ventricular end-diastolic volume ≤40 mL·m−2 (p=0.016) were independent predictors of prognosis in a study involving 64 patients with idiopathic PAH [33].
Other variables and thresholds, such as arterial carbon dioxide tension (Pa,CO2) <4.25 kPa [28], resting systolic blood pressure <110 mmHg, resting heart rate >92 beats·min−1 and diffusing capacity of the lung for carbon monoxide ≤32% have also been shown to be independent predictors of mortality [3].
On-treatment predictors of survival
Response to treatment has been demonstrated to have at least as much prognostic value as disease severity at baseline, with many of the variables discussed previously being predictive of outcome during therapy as well as at diagnosis.
The persistence of severe disease on treatment is unsurprisingly associated with reduced survival time. Patients who remained at NYHA FC III/IV during epoprostenol treatment had a greater risk of death in two follow-up studies [6, 25]. Therefore, a treatment goal of WHO FC I/II has been proposed [30].
Improvement in 6MWD also appears to be of prognostic importance [6, 24, 28, 31]. For example, a 12-week study of treprostinil in PAH reported that survival improved with a >20 m increase from baseline to week 12 [24], and a 12-week study of sildenafil in PAH determined a mean minimally important difference of 41 m [42]. Conversely, a study evaluating factors associated with survival in patients with primary PH showed that the absolute 6MWD value after 3 months of i.v. epoprostenol treatment correlated with survival while its increase from baseline did not (fig. 2) [6].
Figure 2.

Target 6-min walk distance (6MWD) after 3 months of epoprostenol therapy (a) but not change in 6MWD between baseline and 3 months of epoprostenol therapy (b) predicted survival in 178 patients with pulmonary hypertension followed for a median of 26 months. Reproduced from [6] with permission from the publisher.
Several studies have shown that increased levels of NT-proBNP or BNP during treatment were associated with poor outcomes [22, 30, 38, 43]. In a study of 60 PH patients, those with BNP ≥180 pg·mL−1 at 3-months follow-up had a significantly lower survival rate (p<0.05) [22], while in another study, retrospective analysis of 198 patients with PAH revealed that decreases of NT-proBNP of >15% per year were associated with increased survival rate [38]. A treatment goal of NT-proBNP <1,800 ng·L−1 has been proposed [30].
Several invasive haemodynamic variables have prognostic utility during follow-up of PH treatment, such as cardiac index [5, 30, 43], PVR [31, 33], Pra [28], Ppa [5] and SV,O2 [24, 30]. A significant reduction in total pulmonary resistance of 30% relative to baseline after 3 months of epoprostenol treatment was associated with improved outcome [6] and treatment goals of cardiac index ≥2.5 L·min−1·m−2 and SV,O2 of 65% have been proposed [30].
Blood gas analysis in a 12-yr retrospective analysis of 101 patients showed that Pa,CO2 <4.25 kPa was an independent predictor of mortality after 3 months of treatment (p<0.001) [28].
Finally, a cardiac magnetic resonance imaging study has demonstrated that progressive dilatation of the right ventricle and further decreases in stroke volume and left ventricular end-diastolic volume during 1 yr of treatment were independent predictors of treatment failure and mortality in 64 patients with idiopathic PAH [33]. However, these data need to be confirmed in larger studies.
Multidimensional risk assessment
There are conflicting reports on the prognostic value of using a combination of parameters in the management of PH. A recent systematic review of prognostic factors in 54 studies of idiopathic PAH identified 107 factors associated with mortality, of which only 10 were reproducibly predictive across studies: mean Pra (17 studies), functional class (14 studies), cardiac index (13 studies), mean Ppa (10 studies), PVR (10 studies), heart rate (10 studies), 6MWD (eight studies), pericardial effusion severity (five studies), stroke volume index (four studies) and mixed venous arterial oxygen tension or saturations (four studies) [44].
While it is important to assess a range of different clinical, echocardiographic and haemodynamic parameters that provide important prognostic information, conflicting results may be obtained from different parameters. Some parameters, such as 6MWD, provide indirect information on RV function and may be influenced by other disease and patient characteristics. Disease progression will vary between patients and, therefore, target values and treatment goals should be adjusted to the individual patient. For example, while a 6MWD of >400 m is usually considered acceptable for most patients with PAH, younger patients are often capable of walking >500 m even with severe PH due to greater pre-disease functioning, and so additional CPET and RHC is required to properly assess RV function.
Combinations of prognostic variables have been determined in several studies as being predictive for improved survival. For example, Sitbon et al. [45] defined long-term responders to calcium channel blockers as those with NYHA FC I/II and near normal haemodynamics after at least 1 yr of follow-up, while the same investigators had previously shown that NYHA FC I/II and decreases in total peripheral resistance were predictive of improved survival after 3 months of epoprostenol treatment in patients with primary PH [6].
The European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines provide recommendations on several parameters with established importance for assessing disease severity, stability and prognosis, highlighting the importance of obtaining a panel of data derived from clinical evaluation, exercise tests, biochemical markers, and echocardiographic and haemodynamic assessments (table 2) [15, 46]. Criteria have been developed using these variables to classify patients as “stable and satisfactory” (meeting most criteria in the “better prognosis” column of table 2), “stable and not satisfactory” (some “better prognosis” criteria not fulfilled), and “unstable and deteriorating” (fulfilling most findings listed in the “worse prognosis” column). A similar patient status classification according to risk factor assessment has been developed by Nickel et al. [30], categorising patients as “stable/satisfactory” or “unstable/deteriorating”.
Table 2. Variables with established importance for assessing disease severity, stability and prognosis in pulmonary arterial hypertension.
| Determinants of prognosis | Better prognosis | Worse prognosis |
| Clinical evidence of RV failure | No | Yes |
| Rate of progression of symptoms | Slow | Rapid |
| Syncope | No | Yes |
| WHO-FC | I, II | IV |
| 6MWD | Longer (>500 m)¶ | Shorter (<300 m) |
| Cardiopulmonary exercise testing | Peak O2 consumption >15 mL·min−1·kg−1 | Peak O2 consumption <12 mL·min−1·kg−1 |
| BNP/NT-proBNP plasma levels | Normal or near-normal | Very elevated and rising |
| Echocardiographic findings# | No pericardial effusion TAPSE >2.0 cm | Pericardial effusion TAPSE <1.5 cm |
| Haemodynamics | Pra <8 mmHg and CI ≥2.5 L·min−1·m−2 | Pra >15 mmHg or CI ≤2.0 L·min−1·m−2 |
RV: right ventricular; WHO-FC: World Health Organization functional class; 6MWD: 6-min walk distance; BNP: brain natriuretic peptide; NT-proBNP: N-terminal proBNP; TAPSE: tricuspid annular plane systolic excursion; Pra: right atrial pressure; CI: cardiac index. #: TAPSE and pericardial effusion have been selected because they can be measured in the majority of patients; ¶: depending on age. Reprinted from [46] with permission from the publisher.
Various risk stratification equations have been developed based on prognostic parameters. The National Institute of Health (NIH) equation was based on mean Ppa, Pra and cardiac index values from 191 patients diagnosed between 1981 and 1985 [41]. However, studies prospectively validating this equation have found problems with its specificity and correlation. The observed survival from several subsequent studies was greater than the predicted survival calculated with the NIH equation [5, 26, 31, 40]. In addition, a study analysing survival in a Mexican population of patients with primary PH concluded that the equation demonstrated low specificity [47]. Therefore, the historical NIH equation should no longer be used as a means of predicting outcome in PAH.
Other composite risk factors have been developed from the Pulmonary Hypertension Connection registry based on mean Ppa, Pra and cardiac index [40] and the French PAH registry based on sex, 6MWD and cardiac output at diagnosis [48]. A risk calculator was recently developed using data from the REVEAL registry, based on PAH subgroup, demographics, NYHA/WHO FC, vital signs, 6MWD, BNP, echocardiography, pulmonary function tests and RHC findings [49]. The risk calculator was designed to be applicable at any point during the course of disease based on the patient's most recent evaluation; therefore, it may allow regular re-assessment of risk and differentiation of patients with stable and progressive disease. However, the risk calculator does have several significant limitations. To date, it has only been tested retrospectively. In addition, only baseline values were used in the development of the equation, although studies show that long-term survival is dependent on changes in parameters during treatment. The usefulness of the REVEAL risk calculator in clinical practice is, therefore, questionable and still remains to be validated in the daily management of PAH.
PH management in clinical practice
Recommendations for follow-up strategies have been published in the ESC/ERS guidelines [15]. Clinical assessment, WHO FC, electrocardiography, 6MWD, CPET, BNP/NT-proBNP and RHC assessments should be performed at baseline and at 3–4 months after initiation or changes in therapy and in case of clinical worsening. The guidelines also recommend that regular follow-up is performed every 3–6 months in stable patients.
It is important to develop goal-orientated strategies with criteria relevant to all patients with PH that are also flexible enough to accommodate individual patients presenting with different parameters of disease progression. Guideline recommendations may also need to be adapted based on resources available in a given clinical setting. At the pulmonary hypertension clinic at Erasme University (Brussels, Belgium) we have developed a management strategy based on that of Hoeper et al. [50] but adapted to individual cases using our own clinical experience (fig. 3).
Figure 3.
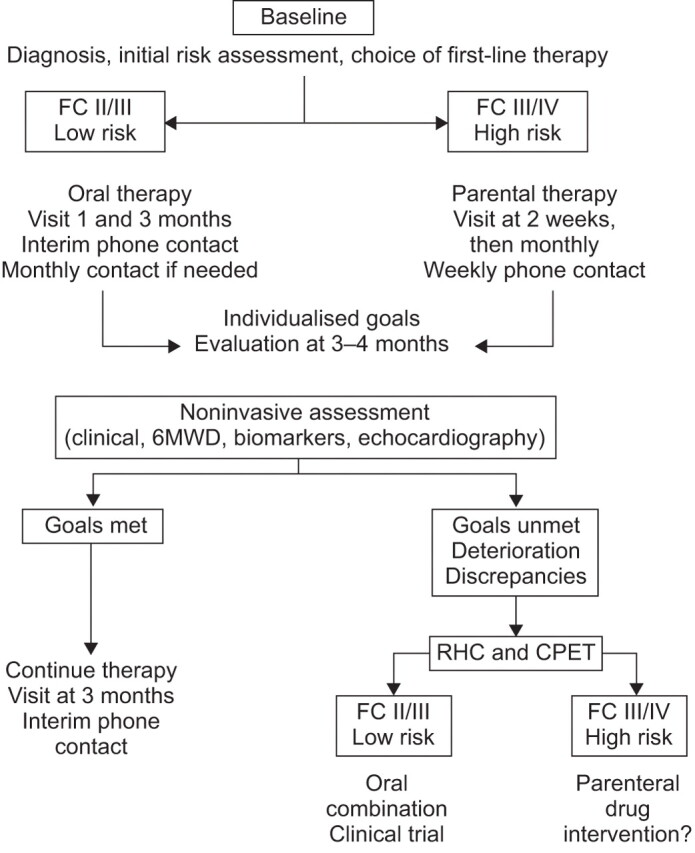
Goal-oriented strategy developed at the pulmonary hypertension clinic at Erasme University (Brussels, Belgium). FC: functional class; 6MWD: 6-min walk distance; RHC: right heart catheterisation; CPET: cardiopulmonary exercise testing.
However, several questions remain with regard to treatment goals. Are treatment goals aggressive enough, and are patients actually achieving these goals? Is it appropriate to use average values before diagnosis as a reference point for goals, as these may not represent optimal functioning for an individual patient? While patients invariably show some improvement after treatment, how can we assess whether an individual patient has achieved the best function possible?
When assessing disease progression, the use of a multidisciplinary team approach is crucial in order to support patients medically, psychologically and socially and to encourage patients to be pro-active in their disease monitoring or management. Open patient discussions regarding treatment plan, monitoring, disease progression and prognosis are invaluable in helping patients to understand and adhere to treatment strategies. Nurses provide a crucial link between the patient and treating physician(s), providing contact for patients in the event of clinical worsening, educating patients on disease, treatment and impact on daily life, and providing emotional support.
CONCLUSIONS
PAH is a progressive disorder requiring early intervention for optimal clinical outcome. Various parameters have been identified as having prognostic value in PH and regular monitoring with a panel of relevant prognostic parameters allows for timely treatment escalation. Risk stratification is important to prevent late intervention which leads to progressive remodelling and right heart failure in the absence of treatment. However, there is still an urgent need for prospective collaborative initiatives to assess novel goals and improve treatment strategies. Goal-orientated strategies help to provide pre-defined, structured ways to assess disease progression and standardisation of treatment between physicians and treatment centres. A global assessment of disease, combining assessment parameters with prognostic significance, would allow physicians to personalise and optimise clinical management for their patients with PAH.
Acknowledgments
The authors received editorial assistance from Adelphi Communications (Bollington, UK), funded by Bayer Pharma AG (Berlin, Germany).
Footnotes
Provenance
Publication of this peer-reviewed article was supported by Bayer Pharma AG, Germany (article sponsor, European Respiratory Review issue 123).
Statement of Interest
J-L. Vachiéry has received fees for speaking from Actelion, GlaxoSmithKline, Pfizer, United Therapeutics, and Bayer-Shering, and fees for consultancy from Actelion, GlaxoSmithKline, Pfizer, and United Therapeutics. He has also received a research grant from GlaxoSmithKline.
REFERENCES
- 1.Galiè N, Manes A, Negro L, et al. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 2009; 30: 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryerson CJ, Nayar S, Swiston JR, et al. Pharmacotherapy in pulmonary arterial hypertension: a systematic review and meta-analysis. Respir Res 2010; 11: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010; 122: 164–172. [DOI] [PubMed] [Google Scholar]
- 4.Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010; 122: 156–163. [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002; 106: 1477–1482. [DOI] [PubMed] [Google Scholar]
- 6.Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002; 40: 780–788. [DOI] [PubMed] [Google Scholar]
- 7.Galiè N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008; 371: 2093–2100. [DOI] [PubMed] [Google Scholar]
- 8.Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008; 117: 3010–3019. [DOI] [PubMed] [Google Scholar]
- 9.Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346: 896–903. [DOI] [PubMed] [Google Scholar]
- 10.Badesch DB, Barst RJ, Galie N, et al. Abstract 2765: Maintenance of improvement in 6-minute walk distance with long term bosentan treatment: results of the BREATHE-1 open-label extension study. Circulation 2006; 114: IL_578. [Google Scholar]
- 11.Gatzoulis MA, Beghetti M, Galie N, et al. Longer-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension study. Int J Cardiol 2008; 127: 27–32. [DOI] [PubMed] [Google Scholar]
- 12.Oudiz RJ, Galiè N, Olschewski H, et al. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol 2009; 54: 1971–1981. [DOI] [PubMed] [Google Scholar]
- 13.Lau EM, Manes A, Celermajer DS, et al. Early detection of pulmonary vascular disease in pulmonary arterial hypertension: time to move forward. Eur Heart J 2011; 32: 2489–2498. [DOI] [PubMed] [Google Scholar]
- 14.McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009; 119: 2250–2294. [DOI] [PubMed] [Google Scholar]
- 15.Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2009; 34: 1219–1263. [DOI] [PubMed] [Google Scholar]
- 16.American Heart Association. Classes of heart failure. www.heart.org/HEARTORG/Conditions/HeartFailure/AboutHeartFailure/Classes-of-Heart-Failure_UCM_306328_Article.jsp Date last updated: August 5, 2011. Date last accessed: November 24, 2011.
- 17.McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004; 126: Suppl. 1, 14S–34S. [DOI] [PubMed] [Google Scholar]
- 18.ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002; 166: 111–117. [DOI] [PubMed] [Google Scholar]
- 19.Oudiz RJ, Barst RJ, Hansen JE, et al. Cardiopulmonary exercise testing and six-minute walk correlations in pulmonary arterial hypertension. Am J Cardiol 2006; 97: 123–126. [DOI] [PubMed] [Google Scholar]
- 20.Torbicki A. Cardiac magnetic resonance in pulmonary arterial hypertension: a step in the right direction. Eur Heart J 2007; 28: 1187–1189. [DOI] [PubMed] [Google Scholar]
- 21.Nagaya N, Uematsu M, Satoh T, et al. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med 1999; 160: 487–492. [DOI] [PubMed] [Google Scholar]
- 22.Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation 2000; 102: 865–870. [DOI] [PubMed] [Google Scholar]
- 23.Torbicki A, Kurzyna M, Kuca P, et al. Detectable serum cardiac troponin T as a marker of poor prognosis among patients with chronic precapillary pulmonary hypertension. Circulation 2003; 108: 844–848. [DOI] [PubMed] [Google Scholar]
- 24.Benza RL, Gomberg-Maitland M, Naeije R, et al. Prognostic factors associated with increased survival in patients with pulmonary arterial hypertension treated with subcutaneous treprostinil in randomized, placebo-controlled trials. J Heart Lung Transplant 2011; 30: 982–989. [DOI] [PubMed] [Google Scholar]
- 25.Kuhn KP, Byrne DW, Arbogast PG, et al. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med 2003; 167: 580–586. [DOI] [PubMed] [Google Scholar]
- 26.McLaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J 2005; 25: 244–249. [DOI] [PubMed] [Google Scholar]
- 27.Barst RJ, Galie N, Naeije R, et al. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J 2006; 28: 1195–1203. [DOI] [PubMed] [Google Scholar]
- 28.Hoeper MM, Pletz MW, Golpon H, et al. Prognostic value of blood gas analyses in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2007; 29: 944–950. [DOI] [PubMed] [Google Scholar]
- 29.Miyamoto S, Nagaya N, Satoh T, et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med 2000; 161: 487–492. [DOI] [PubMed] [Google Scholar]
- 30.Nickel N, Golpon H, Greer M, et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2011; [Epub ahead of print DOI: 10.1183/09031936.00092311]. [DOI] [PubMed] [Google Scholar]
- 31.Provencher S, Sitbon O, Humbert M, et al. Long-term outcome with first-line bosentan therapy in idiopathic pulmonary arterial hypertension. Eur Heart J 2006; 27: 589–595. [DOI] [PubMed] [Google Scholar]
- 32.Raymond RJ, Hinderliter AL, Willis PW, et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol 2002; 39: 1214–1219. [DOI] [PubMed] [Google Scholar]
- 33.van Wolferen SA, Marcus JT, Boonstra A, et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007; 28: 1250–1257. [DOI] [PubMed] [Google Scholar]
- 34.Wensel R, Opitz CF, Anker SD, et al. Assessment of survival in patients with primary pulmonary hypertension: importance of cardiopulmonary exercise testing. Circulation 2002; 106: 319–324. [DOI] [PubMed] [Google Scholar]
- 35.Tei C, Dujardin KS, Hodge DO, et al. Doppler echocardiographic index for assessment of global right ventricular function. J Am Soc Echocardiogr 1996; 9: 838–847. [DOI] [PubMed] [Google Scholar]
- 36.Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 2006; 174: 1034–1041. [DOI] [PubMed] [Google Scholar]
- 37.Brierre G, Blot-Souletie N, Degano B, et al. New echocardiographic prognostic factors for mortality in pulmonary arterial hypertension. Eur J Echocardiogr 2010; 11: 516–522. [DOI] [PubMed] [Google Scholar]
- 38.Mauritz GJ, Rizopoulos D, Groepenhoff H, et al. Usefulness of serial N-terminal pro-B-type natriuretic peptide measurements for determining prognosis in patients with pulmonary arterial hypertension. Am J Cardiol 2011; 108: 1645–1650. [DOI] [PubMed] [Google Scholar]
- 39.Fijalkowska A, Kurzyna M, Torbicki A, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 2006; 129: 1313–1321. [DOI] [PubMed] [Google Scholar]
- 40.Thenappan T, Shah SJ, Rich S, et al. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J 2010; 35: 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991; 115: 343–349. [DOI] [PubMed] [Google Scholar]
- 42.Gilbert C, Brown MC, Cappelleri JC, et al. Estimating a minimally important difference in pulmonary arterial hypertension following treatment with sildenafil. Chest 2009; 135: 137–142. [DOI] [PubMed] [Google Scholar]
- 43.Miura Y, Fukumoto Y, Sugimura K, et al. Identification of new prognostic factors of pulmonary hypertension. Circ J 2010; 74: 1965–1971. [DOI] [PubMed] [Google Scholar]
- 44.Swiston JR, Johnson SR, Granton JT. Factors that prognosticate mortality in idiopathic pulmonary arterial hypertension: a systematic review of the literature. Respir Med 2010; 104: 1588–1607. [DOI] [PubMed] [Google Scholar]
- 45.Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111: 3105–3111. [DOI] [PubMed] [Google Scholar]
- 46.Mclaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation 2006; 114: 1417–1431. [DOI] [PubMed] [Google Scholar]
- 47.Sandoval J, Bauerle O, Palomar A, et al. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation 1994; 89: 1733–1744. [DOI] [PubMed] [Google Scholar]
- 48.Humbert M, Sitbon O, Yaici A, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J 2010; 36: 549–555. [DOI] [PubMed] [Google Scholar]
- 49.Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL risk score calculator in newly diagnosed patients with pulmonary arterial hypertension. Chest 2011. [Epub ahead of print DOI: 10.1378/chest.11–0676]. [DOI] [PubMed] [Google Scholar]
- 50.Hoeper M, Markevych I, Spiekerkoetter E, et al. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J 2005; 26: 858–863. [DOI] [PubMed] [Google Scholar]