

Abstract
An increasing number of medical services dedicated to the diagnosis, treatment and follow-up of pulmonary hypertension (PH) in children are being established. This has, in turn, increased the need to adapt current guidelines for the treatment of PH to be more relevant to paediatric patients with PH. This article will summarise the data obtained so far from paediatric registries, national cohorts and clinical trials and discuss the best approach for developing a treatment algorithm designed for children with different types of PH. The many unanswered questions, challenges and issues relating to the PH in the paediatric population will also be discussed.
Keywords: Paediatric, pulmonary hypertension, registry, treatment
General awareness of the rare and devastating disease, pulmonary hypertension (PH), has increased significantly in recent years. As a result, many large clinical trials have been completed or are under way, and medical services dedicated to the diagnosis, treatment and follow-up of this disease have been established. A number of these services are exclusively devoted to children with all their different forms of PH. As most guidelines for the treatment of PH are based on data from clinical trials of adults, they are yet to be adapted to the needs of children. We are now at the exciting time when emerging data, as well as clinical experience, can assist in this adaptation.
There is still much to be done; improved referral rates to specialist paediatric centres are required for some of the specific forms of paediatric PH, together with more data from prospective trials. More information is also required about symptoms, diagnosis and outcome of paediatric patients in a real-life setting. These limitations need to be overcome if a specific treatment strategy is to be recommended.
Therefore, the task now ahead for the experts in the field of PH is to amalgamate the findings from paediatric registries, national cohorts and clinical trials, and translate them into meaningful practical advice for paediatricians. This article will summarise the data obtained so far and discuss the best approach for developing a treatment algorithm designed for children with different types of PH. The challenges ahead will also be discussed; there are still many unanswered questions, difficulties and issues relating to the PH in the paediatric population.
DIFFERENCES OF PH IN ADULTS AND CHILDREN
At present, the sparse evidence we have does not provide enough detail on whether there are any substantial differences in the way that PH develops and progresses in adults compared with children. However, data from paediatric cohorts and national registries suggest that there is indeed a difference between adults and children with PH in terms of aetiology; this affects the approach to treatment. For most adult patients, PH is associated with factors such as left-heart disease, whereas the most common forms of PH in children are idiopathic/heritable pulmonary arterial hypertension (IPAH/HPAH), PH due to an underlying congenital heart defect (CHD), and patients with a parenchymal pulmonary disease [1]. Some data relating to the treatment of paediatric PH patients are available; current treatment strategies include treatment with synthetic prostacyclins and their analogues (known as prostanoids), endothelin receptor antagonists (ERAs), and phosphodiesterase type-5 (PDE-5) inhibitors.
PATHOPHYSIOLOGY AND DEVELOPMENT OF PH
PH is a disease characterised by vasoconstriction, and hypertrophy of the vascular smooth muscle cells in the pulmonary arteries, resulting in reduced individual vascular areas and total perfusion areas. Cardiac output is maintained by right ventricular hypertrophy and dilatation, and pulmonary arterial pressure increases. Right heart hypertrophy and dilatation then lead to right heart failure and, eventually, death. Current pulmonary arterial hypertension (PAH)-specific therapies, therefore, primarily re-establish pulmonary vasodilation and, to some extent, reduce smooth muscle proliferation and hypertrophy in the pulmonary arterial vessels. Three main pathways known to be involved in these processes in PH, the prostacyclin pathway, the endothelin pathway and the nitric oxide pathway, are targeted using prostanoids, ERAs and PDE-5 inhibitors, respectively (fig. 1) [2].
Figure 1.

Key pathways involved in the pathophysiology of pulmonary hypertension. Three main pathways are known to be involved in pulmonary vasodilation and, to some extent, smooth muscle proliferation and hypertrophy in the pulmonary arterial vessels: the prostacyclin pathway, the endothelin pathway and the nitric oxide pathway. These pathways are targeted by prostanoids, endothelin receptor antagonists and phosphodiesterase type-5 inhibitors. cAMP: cyclic adenosine monophosphate; cGMP: cyclic guanosine monophosphate. Reproduced from [2] with permission from the publisher.
Translating current therapeutic knowledge to the paediatric population requires that there are no significant differences in the way that these pathways behave within developing tissue in the lungs and heart. It is also important that PH treatments affect vascular remodelling and endothelial dysfunction in a similar way. Indeed, aside from the fact that the heart and lungs are still growing in children, it appears that there is little difference at the molecular level compared to adults. The endothelial dysfunction observed in children is essentially the same as that seen in adults and most histological findings are also similar. While no discernable difference has yet been reported in the expression of endothelin and prostacyclin receptors, and the levels of signalling molecules in normal and diseased tissue, it is probably fair to assume that the processes of growth will affect the mechanisms of disease and treatment effects, either directly or indirectly. For example, children tend to show more medial hypertrophy [3], and it is unclear whether this has an effect on treatment choice. For the time being and in the absence of clear evidence to the contrary, it appears appropriate to view the process of PH progression in children in the same way as in adults and to conclude that similar treatment and strategies can be used for children.
CONSIDERATIONS ON EXAMINING EPIDEMIOLOGY AND CHARACTERISTICS OF PAEDIATRIC PH
When developing clinical programmes for paediatric patients, data obtained from registries and national cohorts are very useful. Epidemiological measures, such as the number of new cases every year (incidence) and the number of cases in a population at a point in time (prevalence), can provide important information when developing management services. Survival data can also be collected, as well as the age at which PH presents itself, the severity of the disease at presentation, and more data on the natural history of PH. These data can help quantify the extent of PH, obtain a more accurate picture of the disease course in children, and identify management strategies currently employed.
A key consideration when planning a treatment strategy for paediatric patients with PH is its aetiology, as this will have an influence on the nature and speed of disease progression, as well as the treatment choice. For example, in the majority of adult patients with PH, it is associated with left-heart disease or lung disease/hypoxaemia [4], for which treatment of the underlying cause of PH is the logical approach [5]. A small percentage of adult patients with PH are classified as having group 1 PH (known as PAH); for this type of PH, the use of PAH-specific therapy alone or in combination with treatment of the underlying cause of the PH is appropriate.
Current treatment guidelines for PH focus on patients with PAH because of the availability of good quality evidence from clinical trials [5]. Within this subgroup of PH, the largest adult patient population consists of those for whom there is no clear cause (IPAH) or PH associated with particular mutations (HPAH) [6, 7]. Other causes for PAH are connective tissue disease, such as scleroderma (PAH-SSc), or congenital heart disease (PAH-CHD). In adults, PAH-SSc is described to progress more rapidly than in those with other aetiologies [8], followed by patients with IPAH/HPAH [9], while the PH tends to be more stable in those with CHD [10]. However, these statements cannot be extrapolated to children without extreme caution, due to the lack of sound evidence. This is especially true for patients with CHD, where PH may be associated with a wide spectrum of functional ability, from patients who show almost normal clinical and exercise tolerance data despite complex lesions, to those who are severely restricted in functional capacity and quality of life [11].
In order to get insight into the distribution of different types of PH in children, information from real-world data is required. Over the past few years, the results of different European registries have been made available, varying greatly in the time period covered (from 5 yrs of follow-up for the Netherlands [12] and the UK [1], to 2 yrs for the Swiss [13] and French [14] registries), the population size they are based on (Swiss [13] versus UK [1]), and the types of PH included (France [14]). Some useful information on prevalence and incidence data can also be derived from single-centre studies, together with information on disease progression and the effects of treatment. The most useful type of information from the real-world is from long-term data provided by larger national networks.
The prevalence of all types of PH in Europe, where awareness of PH is increasing and management of children with CHD is generally state of the art, is estimated between 3.7 [14] and approximately 5.0 [1] cases per million children. Although there are limited data on incidence of paediatric PH, the incidence of IPAH in the UK is estimated to be 0.48 cases per million children, per year [1, 15]. Data also show that the majority of children with PH fall into three categories, which account for at least 70% of all patients: those with IPAH/HPAH, those with PH due to an underlying congenital heart defect, and patients with a parenchymal pulmonary disease [1]; PAH associated with connective tissue disease and portopulmonary hypertension are extremely rare aetiologies (fig. 2). Persistent PH of the newborn is a serious form of PH with its own set of aetiologies, treatment approach and outcomes, which is inherently different to the other forms of PH [16]. It is not discussed in detail in this article or other current guidelines on paediatric PH [5] because it is confined exclusively to the neonatal paediatric age group.
Figure 2.

Prevalence of different aetiologies of pulmonary hypertension (PH) in the paediatric population (results from national registries and paediatric cohorts). ▪: idiopathic/heritable pulmonary arterial hypertension; ▒: PH associated with congenital heart disease (CHD); ▓: PH associated with connective tissue disease; ░: PH associated with lung disease; □: other. Data shown are taken from registries/paediatric cohorts in Switzerland [13], the UK [1], the Netherlands [12] and France [14]. The most common forms of PH in children are idiopathic/heritable pulmonary arterial hypertension, PH associated with CHD and PH associated with lung disease. #: patients with PH associated with CHD were excluded from this cohort; patients shown here were diagnosed with CHD, but this was not thought to be the cause of the PH.
When considering these emerging data, there are at least two very important points to note. First, the different causes of the vast variation of incidence of PH in children between different reports and the inherent bias represented by the recruitment criteria for each registry or collection should be considered. For example, the French paediatric registry specifically excluded children with significant congenital cardiac shunts thought to be responsible for the development of PH [14], whereas these patients comprise between 40% and 50% of patients in other studies [1, 12, 13]. The second point may be even more important, because it is not inherent in the methodology, but in the data themselves. In PH due to CHD, the PH is, notably, not exclusively a result of any “inherent” genetic or idiopathic factor but it is related, in part, to the availability and timeliness of surgical repair in individual countries. Thus, it does not reflect a natural incidence, but one due to medical practice. In other words, PH in children with CHD is relatively low in developed countries due to the advanced medical and surgical care practiced in paediatric cardiology. In other less developed nations, where CHD may not be repaired in early infancy, rates of paediatric PAH-CHD are likely to be higher [17]. It is very difficult, and even more fascinating, to separate true population-based differences in the pattern of PH associated with CHD from this bias. Finally, we should not forget that not only does epidemiology data indirectly reflect the standard of treatment for IPAH and its associated diseases, but also the referral pattern to the specialised paediatric PH centres from our collaborating colleagues.
A very interesting initiative in this context is the foundation of the TOPP (Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension) registry, an international paediatric PH registry drawing data from centres for PH in children from around the world [18]. These data will include details from more than 38 centres in 22 countries and on more than 400–450 cases, and promise to deliver a wealth of urgently needed information on all of the aspects mentioned above, and more.
PRINCIPLES OF TREATMENT IN PH IN ADULTS AND CHILDREN
Current treatment strategies for treating children with PH are analogous to those in adults and are dependent on the characteristics and severity of the disease at presentation. This very complex algorithm can be summarised as follows: if patients show signs of mild to moderate PAH (those in World Health Organization functional class (WHO FC) II or III), together with the appropriate haemodynamic findings, treatment with a single pulmonary vasodilator is the preferred approach. Patients who are acutely vasoreactive can also be treated with calcium channel blockers. If the therapeutic effect is insufficient or waning, or signs of right heart dysfunction become evident, then treatment escalation is recommended. Escalation of treatment includes the use of combination therapy, as well as interventions such as balloon atrial septostomy, in order to prevent low cardiac output status in patients with severely compromised pulmonary blood flow. Other interventions with the same aim, such as the Pott's shunt [19], may be considered if lung transplantation is not available or inappropriate, but may be of high risk and thus are controversial. Eventually, depending on right heart function, lung transplantation alone or a combined heart and lung transplant may be required, although the reality of obtaining organs for this size and age group remains very sobering.
This approach is reflected, and given in all necessary detail, in recent evidence-based, diagnostic and therapeutic guidelines published jointly by three major European medical societies (the European Society of Cardiology, the European Respiratory Society and the International Society of Heart and Lung Transplantation) [5]. These guidelines state that the same treatment paradigm for adults could be considered for use in children, and that the weight of evidence/opinion for treatments used in adults is in favour of efficacy and safety. However, they recognise that the level of evidence for employing these strategies in children is low [5].
Although paediatric PH centres currently use a more case-by-case approach, based on their own experience and interpretation of the available data, adapted for their young patients, some evidence has been accumulated. The UK Service for Pulmonary Hypertension in Children has recently published their retrospective analysis of management for paediatric patients with IPAH [15]. Together with improved survival estimates, these data provide the first quantitative data which might guide treatment decisions (fig. 3). These data support the use of an aggressive approach to treatment in the form of combination therapy which uses two or three classes of drugs, depending on the clinical and haemodynamic stage of PH in children. Management strategies will also depend on what is available for use in different countries and the prevailing philosophy in that country. The management strategy described for the UK uses combination therapy from the initiation of treatment, for example [15], whereas PH centres in France use a step-up approach similar to that advocated in the adult guidelines; some centres may endorse the use of intravenous epoprostenol in infants, while others do not.
Figure 3.

Retrospective analysis of paediatric patients included in the UK National Service for Pulmonary Hypertension in Children. Children were treated with calcium channel blockers (CCBs)±sildenafil, bosentan±sildenafil or prostacyclin ±oral therapy according to vasoreactivity and symptom severity. Decreases in pulmonary vascular resistance index (PVRI) were observed for each treatment regimen. WHO FC: World Health Organization functional class.
There are several questions that are unique for the paediatric patient population, especially considering that children are often growing rapidly and developing while on treatment. Although the latest evidence does suggest that growth and maturity are not impaired in children treated with bosentan (A. Hislop and co-workers, Great Ormond Street Hospital for Sick Children, London, UK; unpublished data) [20], there are still some key issues to consider. A dedicated trial assessing the pharmacokinetics of a paediatric formulation of bosentan [21] surprisingly demonstrated that in young children, maximum plasma availability of this drug was reached at a lower dose than expected. Clearly, similar trials in children are needed for other PH therapies. There is also clear evidence that PH treatments are practical and efficacious to use in babies and young infants [22], and well tolerated in this patient population [23].
AVAILABLE DATA ON PAH-SPECIFIC DRUGS IN THE PAEDIATRIC POPULATION
Historically, the first drugs used in the treatment of paediatric PAH were the prostanoids, as was the case for adults. Intravenous epoprostenol has been used in most of these cases improving survival compared with historical data and increasing exercise tolerance [24–26]. The use of subcutaneous treprostinil is another treatment option for paediatric PH patients, but no specific data on its use in children have been published (table 1). Finally, there are some data from studies investigating inhaled iloprost [28]. As in the adult population, comparing therapy efficiency, in this case between different prostanoids and their modes of application, is not at all trivial and needs careful and sometimes not possible normalisation for disease type, severity, criteria and practicalities of treatment.
Table 1. Studies of prostanoids in paediatric pulmonary hypertension patients.
| First author [Ref.] | Patients n | Dosage ng·kg−1·min−1 | Description | End-points | Results |
| Yung [24] | 35 | Not specified | Retrospective study of children treated with i.v. epoprostenol | Treatment success, defined as freedom from death, transplantation, or atrial septostomy | Significant improvement in survival |
| Nakayama [25] | 27 | 24.7±6.7 | Single-centre study of paediatric patients receiving i.v. epoprostenol investigating survival | WHO FC | Improvement in survival and 6MWD |
| Plasma brain natriuretic peptide | |||||
| Echocardiography, | |||||
| 6MWD | |||||
| Haemodynamic parameters | |||||
| Lammers [26] | 39 | 32.5±12 | Single-centre study of children with IPAH and PAH-CHD treated with i.v. epoprostenol | Survival | Improved survival, WHO FC, exercise tolerance and ability to thrive |
| WHO FC | |||||
| Echocardiography | |||||
| 6MWD | |||||
| Barst [27] | 31 | 122±36 | Nonresponders to CCBs were treated with i.v. epoprostenol | Survival Haemodynamics | Improved survival, and haemodynamics |
| Ivy [28] | 22 | N/A | Short- and long-term outcome of children with IPAH and PAH-CHD treated with inhaled iloprost | Cardiac catheterisation | Sustained improvement in WHO FC observed in some children |
| Standard lung function testing | |||||
| 6MWT | |||||
| WHO FC | |||||
| Haemodynamic parameters |
Doses are presented as mean±sd. WHO FC: World Health Organization functional class; 6MWD: 6-min walk distance; PAH: pulmonary arterial hypertension; IPAH: idiopathic PAH; CHD: congenital heart disease; 6MWT: 6-min walk test; CCB: calcium channel blocker.
Oral therapy is obviously a more convenient mode of application in children, due to tolerability issues. The first data relating to oral therapy were from a small number of case studies using the PDE-5 inhibitor, sildenafil [29]. Indeed, sildenafil was used in children before any other oral treatments were approved for the treatment of PAH, both because it was widely available as a treatment for its main indication (erectile dysfunction in adults) and also because trials with its precursor, dipyridamole, were already under way. Therefore, the role of sildenafil in PAH therapy was instantly recognised and established. Treatment with sildenafil was associated with improvements in haemodynamics and exercise capacity [30–32], as well as improvement in WHO FC [32]. However, the number of patients enrolled in these studies was relatively small and most data are from single-centre case series (table 2). Perhaps because of its use before any oral treatments were approved for PH, it continues to be prescribed in a highly variable mode, while there is a real need for more data on the effect of sildenafil in children. Fortunately, a number of trials with at least one major randomised controlled clinical trial are currently underway and should result in valuable data very soon.
Table 2. Studies of phosphodiesterase type-5 inhibitors in paediatric pulmonary arterial hypertension (PAH) patients.
| First author [Ref.] | Patients n | Description | Age of patients yrs | End-points | Results |
| Oliveira [30] | 6 | Case series of children with IPAH treated with sildenafil | 10 (3–19) | Haemodynamics6MWDWHO FC | Improved WHO FC and systemic arterial oxygen saturation in all patients |
| Oxygen saturation | Some improvements in haemodynamics and exercise capacity | ||||
| Karatza [31] | 3 | Case series investigating oxyhaemoglobin saturations and exercise tolerance in children treated with sildenafil | 10.4 (6–13) | Haemodynamics6MWDWHO FC | Improved WHO FC and exercise capacity observed in all three patients |
| Humpl [32] | 14 | Open-label study of children with IPAH and PAH-CHD sildenafil | 10.5 (5–18) | 6MWDHaemodynamics | Improvements in haemodynamics and exercise capacity observed |
Data for age is presented as mean (range). IPAH: idiopathic PAH; CHD: congenital heart disease; 6MWD: 6-min walk distance; WHO FC: World Health Organization functional class.
In contrast, because of the well-orchestrated and co-ordinated approach to the clinical trials investigating bosentan leading to its approval as a PAH-specific therapy in adults, more opportunities were available and more attention was given to its efficacy and safety in children. Indeed, bosentan is approved for the treatment of children with PAH in Europe, based on the results of studies in children. This includes data which demonstrate that although the pharmacokinetics of bosentan were similar in healthy adults and children with PH [33], exposure to bosentan in children plateaus out at lower doses relative to patient weight [21]. Treatment with bosentan in paediatric PH patients is associated with improvements in haemodynamics [33–35], improved WHO FC [20, 35] and improved survival compared with historical controls [20, 36]. Table 3 summarises the data from clinical trials investigating bosentan as a treatment for children with PH. As part of the post-marketing surveillance of bosentan, 146 children aged 2–12 yrs with a median exposure to bosentan of 29.1 weeks were analysed and compared to the adult population [23]. This analysis demonstrated that bosentan was well tolerated by children. In particular, the incidence of elevated aminotransferases in children is lower than that observed in adults [23].
Table 3. Studies of endothelin receptor antagonists in paediatric pulmonary arterial hypertension (PAH) patients.
| First author [Ref.] | Patients n | Description | Age yrs | End-points | Results |
| Barst [33] | 19 | Pharmacokinetics and efficacy of bosentan investigated | NS (3–15) | PharmacokineticsHaemodynamics | Pharmacokinetics of bosentan in children with PAH comparable to healthy adults |
| 6MWD | Improvement in haemodynamics | ||||
| WHO FC | WHO FC improved or stabilised | ||||
| Beghetti [21] | 36 | Pharmacokinetic study of novel paediatric formulation of bosentan | 7.0 (2–11) | Pharmacokinetics | An exposure plateau appears to be reached in children at the dose of 2mg·kg−1 |
| Gilbert [34] | 7 | Uncontrolled study of bosentan in patients with PAH-CHD | 3.8# (1.5–6.4) | Clinical parametersEchocardiography | Significant reduction in right ventricular systolic pressure |
| Haemodynamics | |||||
| Simpson [36] | 7 | Open-label study of children with IPAH treated with bosentan, compared with historical controls | NS | Survival | Improved survival and delayed disease progression |
| Addition of epoprostenol due to disease progression | |||||
| Rosenzweig [35] | 86 | Long-term retrospective study of patients treated with bosentan over median exposure 14 months | 11 (0.8–18) | WHO FC | 46% improved WHO FC |
| Haemodynamics | Improved haemodynamics | ||||
| Survival | |||||
| Maiya [20] | 40 | Long-term retrospective study of children with IPAH and PAH-CHD treated with bosentan | 8.3 (0.6–16) | WHO FCStabilisation of the | Children with IPAH stabilised (with addition of epoprostenol) |
| clinical condition6MWD | WHO FC improved significantly for patients with PAH-CHD | ||||
| van Loon [37] | 10 | Long-term response to bosentan in children compared to adults | 11.8 (4.7–17.3) | WHO FC | Improved 6MWD |
| Transcutaneous oxygen saturation | Survival not significantly different between adults and children | ||||
| Heart rate and blood pressure | Persistence of treatment effect lower in children | ||||
| 6MWD | Improvement in 6MWD at 1 yr but then declined | ||||
| Survival | WHO FC improvement persisted over 1.5 yrs | ||||
| Persistence of treatment effect |
Data for age presented as mean (range), unless otherwise indicated. #: median (range); CHD: congenital heart disease; IPAH: idiopathic PAH; NS: not specified; 6MWD: 6-min walk distance; WHO FC: World Health Organization functional class.
Fortunately, with these oral drugs there are two very effective treatments which continue to show a wide therapeutic breadth and excellent tolerability profile in children. However, as pulmonary hypertension remains a potentially very aggressive disease especially in children, clearly more data and also other novel substance are of vital interest.
LIMITATIONS AND CHALLENGES
The awareness of physicians concerning the nature of PH has improved significantly in recent years. However, there are still many limitations and challenges that need to be overcome before the learning curve for paediatric PH reaches the more reassuring plateau that it is approaching for adults.
One of the most important issues that need to be addressed remains related to increased awareness. Because knowledge of PH is reasonably widespread throughout well-educated medical communities, children with suspected PH are frequently started on treatment by diagnosing and concerned paediatric cardiologists, or respirologists with special PH interest, without proper or timely referral to a dedicated PH centre. The epidemiologically important result of this is that these patients are not registered within national patient cohort studies until they are referred to specialised, tertiary PH centres. Often, there is a significant time delay after initial diagnosis and a clear deterioration in the patient's condition. Thus, vital information which could help the care and treatment of children with PH in the future is lost; this is exemplified by the widespread use of long-term sildenafil treatment in infants, school children and adolescents.
Another substantial obstacle is the dearth of prospective studies specifically investigating the efficacy and safety of PH-specific drugs in the paediatric population. Although we already have some valuable evidence that can form the basis of recommendations, more data are required for detailed guidelines. This is especially the case for data from randomised clinical trials. The logistics of running this type of trial in this disease is limited by small patient numbers available but there are also other limitations, such as clear ethical and practical restraints on the design of the trials. Running placebo-controlled trials of monotherapy in children is clearly not ethical in such a progressive and devastating disease, although trials of combination therapy which use placebo in addition to other background treatments may be acceptable.
Appropriate end-points also need to be properly defined and characterised for paediatric studies, as those used in adult studies are difficult to validate in children. This should begin with a more objective determination of WHO FC, which may be trivial for adolescents but is less so for younger children, who are not capable of performing a formal cardiopulmonary exercise test, and infants. For example, the measurement of exercise capacity using 6-min walk distance (6MWD), the primary end-point used in most previous trials, is difficult and very debatable despite established normal values for different ages of children [38]. Although it remains a useful tool for evaluating each child during treatment, it is not possible to perform any 6-min walk tests reliably in children aged <5–6 yrs. The distance walked at age 6 yrs will be very different from what is possible for an older child even when normalised for age. The variability of the 6MWD in children is very high and can easily offset treatment effect; as such, its utility is limited as an end-point. Laboratory markers, such as brain natriuretic peptide [39], could be utilised as surrogate clinical assessments, but have the disadvantage of needing a blood sample: another fact which is trivial in the adult world but which can be a real obstacle in effective long-term care in children. Well-defined and characterised parameters from echocardiograms may be close to ideal as end-points. Although these noninvasive end-points show promise for the future, they have not yet been validated and would require dedicated studies aimed at assessing them. Similarly, although magnetic resonance imaging provides exquisite data on cardiac function and flows, it lacks pressure data, which are necessary for a valid calculation of pulmonary vascular resistance and still can only be obtained by cardiac catheterisation. Even invasively obtained gold standard, end-point measurements from right heart catheterisation are very sensitive to respiratory support, specifically changes of plasma CO2 levels and anaesthetic management in general, and may be flawed when not performed in a careful and standardised way.
Finally, specific data are required for paediatric patients with PAH-CHD, as data on survival and treatment typically have described adult patients with IPAH. Data on children with IPAH are due to emerge very soon [15], and might still reflect the optimised practice of PH treatment in children more than the true targeted treatment effects on survival of patients with paediatric PH. In contrast to IPAH, and as discussed previously, the complex group of patients with PH associated with CHD does not originate and progress in the same way as patients with IPAH. There are a wide range of physiologies of PH associated with CHD, ranging from candidates for Fontan and Glenn operations who have low pulmonary vascular resistance which is yet not low enough to operate at acceptable risk, to the intricacies of left-heart PH and the different types of the Eisenmenger syndrome. For each of these patients, treatment need to be individually considered, and a careful choice of end-points is essential for clinical trials which includes such patients [40].
In conclusion, it is currently an exciting time in the field of paediatric PH. Sufficient data are becoming available for us to begin to draw conclusions on how children may best be treated and managed with currently available therapies. The first treatment algorithms specifically designed for children (fig. 4) [41], and the data to support them (fig. 3) [15], are now being published. However, although much information has already been gained from large cohorts and national registries on the epidemiology of paediatric PH and current treatment strategies, there is still much to be learnt. More information is required about symptoms, diagnosis and outcome of paediatric patients in a real-life setting in order to design and recommend specific and appropriate treatment strategies for children.
Figure 4.
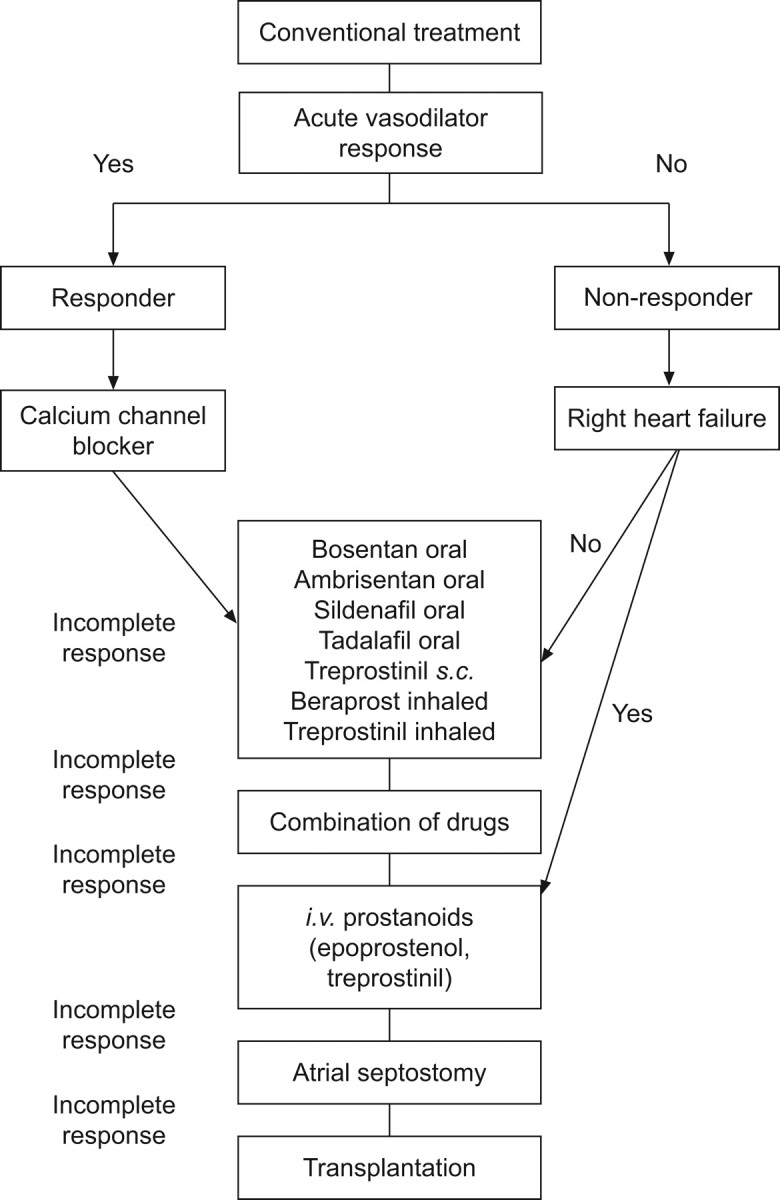
An example of a treatment algorithm specifically designed for paediatrics, in which recommended treatment depends on a variety of factors, including acute vasoreactivity, signs of right heart failure and response to treatment. This algorithm is supported by the treatment algorithm used by the UK National Service for Pulmonary Hypertension in Children, with corresponding clinical and haemodynamic data given in approximate intervals (see fig. 3). s.c: subcutaneously; i.v.: intravenous. Reproduced from [41] with permission from the publisher.
Acknowledgments
We received editorial assistance from T. Newton at Elements Communications Ltd (Westerham, UK), supported by Actelion Pharmaceuticals Ltd (Allschwil, Switzerland).
Footnotes
Provenance
Publication of this peer-reviewed article was supported by Actelion Pharmaceuticals Ltd, Switzerland (unrestricted grant, European Respiratory Review issue 118).
Statement of Interest
I. Schulze-Neick has received consultancy fees and honoraria for talking and organising education, and has served on advisory boards/consulting for both Actelion and Pfizer, who have also supported research and provided funding for members of staff. M. Beghetti has served on advisory boards/consulting for Pfizer, Actelion Pharmaceuticals, Bayer Schering, Pfizer, GlaxoSmithKline, INO Therapeutics, Eli Lilly and Mondobiotech, and has received lecture fees from Actelion Pharmaceuticals, Pfizer and Bayer Schering.
REFERENCES
- 1.Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension: the UK Pulmonary Hypertension Service for Children 2001–2006. Heart 2009; 95: 312–317. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351: 1425–1436. [DOI] [PubMed] [Google Scholar]
- 3.Haworth SG. Pulmonary endothelium in the perinatal period. Pharmacol Rep 2006; 58:Suppl., 153–164. [PubMed] [Google Scholar]
- 4.Gabbay E, Yeow W, Playford D. Pulmonary arterial hypertension (PAH) is an uncommon cause of pulmonary hypertension (PH) in an unselected population: the Armadale echocardiography study. Am J Resp Crit Care Med 2007; 175: A713. [Google Scholar]
- 5.Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30: 2493–2537. [DOI] [PubMed] [Google Scholar]
- 6.Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 2010; 137: 376–387. [DOI] [PubMed] [Google Scholar]
- 7.Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006; 173: 1023–1030. [DOI] [PubMed] [Google Scholar]
- 8.Launay D, Sitbon O, Le PJ, et al. Long-term outcome of systemic sclerosis-associated pulmonary arterial hypertension treated with bosentan as first-line monotherapy followed or not by the addition of prostanoids or sildenafil. Rheumatology (Oxford) 2010; 49: 490–500. [DOI] [PubMed] [Google Scholar]
- 9.Thenappan T, Shah SJ, Rich S, et al. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J 2010; 35: 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimopoulos K, Inuzuka R, Goletto S, et al. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation 2010; 121: 20–25. [DOI] [PubMed] [Google Scholar]
- 11.Haworth SG, Beghetti M. Assessment of endpoints in the pediatric population: congenital heart disease and idiopathic pulmonary arterial hypertension. Curr Opin Pulm Med 2010; 16:Suppl. 1, S35–S41. [DOI] [PubMed] [Google Scholar]
- 12.van Loon RL, Roofthooft MT, van Osch-Gevers M, et al. Clinical characterization of pediatric pulmonary hypertension: complex presentation and diagnosis. J Pediatr 2009; 155: 176–182. [DOI] [PubMed] [Google Scholar]
- 13.Fasnacht MS, Tolsa JF, Beghetti M. The Swiss registry for pulmonary arterial hypertension: the paediatric experience. Swiss Med Wkly 2007; 137: 510–513. [DOI] [PubMed] [Google Scholar]
- 14.Fraisse A, Jais X, Schleich JM, et al. Characteristics and prospective 2-year follow-up of children with pulmonary arterial hypertension in France. Arch Cardiovasc Dis 2010; 103: 66–74. [DOI] [PubMed] [Google Scholar]
- 15.Moledina S, Hislop AA, Foster H, et al. Childhood idiopathic pulmonary arterial hypertension: a national cohort study. Heart 2010; 96: 1401–1406. [DOI] [PubMed] [Google Scholar]
- 16.Konduri GG. New approaches for persistent pulmonary hypertension of newborn. Clin Perinatol 2004; 31: 591–611. [DOI] [PubMed] [Google Scholar]
- 17.Lopes AA, Bandeira AP, Flores PC, et al. Pulmonary hypertension in Latin America: pulmonary vascular disease: the global perspective. Chest 2010; 137:Suppl. 6, 78S–84S. [DOI] [PubMed] [Google Scholar]
- 18.Beghetti M, Berger RMF, Schulze-Neick I, et al. Tracking outcomes and practice in pediatric pulmonary hypertension: the first multinational registry in pediatric pulmonary hypertension. www.peph-association.org/publications/topp_ers_2008_08_16.pdf. 2008.
- 19.Blanc J, Vouhé P, Bonnet D. Potts shunt in patients with pulmonary hypertension. N Engl J Med. 2004; 350: 623. [DOI] [PubMed] [Google Scholar]
- 20.Maiya S, Hislop AA, Flynn Y, et al. Response to bosentan in children with pulmonary hypertension. Heart 2006; 92: 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beghetti M, Haworth SG, Bonnet D, et al. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: the FUTURE-1 study. Br J Clin Pharmacol 2009; 68: 948–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tissot C, Beghetti M. Advances in therapies for pediatric pulmonary arterial hypertension. Expert Rev Respir Med 2009; 3: 265–282. [DOI] [PubMed] [Google Scholar]
- 23.Beghetti M, Hoeper MM, Kiely DG, et al. Safety experience with bosentan in 146 children 2–11 years old with pulmonary arterial hypertension: results from the European Postmarketing Surveillance program. Pediatr Res 2008; 64: 200–204. [DOI] [PubMed] [Google Scholar]
- 24.Yung D, Widlitz AC, Rosenzweig EB, et al. Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation 2004; 110: 660–665. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama T, Shimada H, Takatsuki S, et al. Efficacy and limitations of continuous intravenous epoprostenol therapy for idiopathic pulmonary arterial hypertension in Japanese children. Circ J 2007; 71: 1785–1790. [DOI] [PubMed] [Google Scholar]
- 26.Lammers AE, Hislop AA, Flynn Y, et al. Epoprostenol treatment in children with severe pulmonary hypertension. Heart 2007; 93: 739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barst RJ, Maislin G, Fishman AP. Vasodilator therapy for primary pulmonary hypertension in children. Circulation 1999; 99: 1197–1208. [DOI] [PubMed] [Google Scholar]
- 28.Ivy DD, Doran AK, Smith KJ, et al. Short- and long-term effects of inhaled iloprost therapy in children with pulmonary arterial hypertension. J Am Coll Cardiol 2008; 51: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abrams D, Schulze-Neick I, Magee AG. Sildenafil as a selective pulmonary vasodilator in childhood primary pulmonary hypertension. Heart 2000; 84: E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliveira EC, Amaral CF. [Sildenafil in the management of idiopathic pulmonary arterial hypertension in children and adolescents]. J Pediatr (Rio J) 2005; 81: 390–394. [DOI] [PubMed] [Google Scholar]
- 31.Karatza AA, Bush A, Magee AG. Safety and efficacy of sildenafil therapy in children with pulmonary hypertension. Int J Cardiol 2005; 100: 267–273. [DOI] [PubMed] [Google Scholar]
- 32.Humpl T, Reyes JT, Holtby H, et al. Beneficial effect of oral sildenafil therapy on childhood pulmonary arterial hypertension: twelve-month clinical trial of a single-drug, open-label, pilot study. Circulation 2005; 111: 3274–3280. [DOI] [PubMed] [Google Scholar]
- 33.Barst RJ, Ivy D, Dingemanse J, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther 2003; 73: 372–382. [DOI] [PubMed] [Google Scholar]
- 34.Gilbert N, Luther YC, Miera O, et al. Initial experience with bosentan (tracleer) as treatment for pulmonary arterial hypertension (PAH) due to congenital heart disease in infants and young children. Z Kardiol 2005; 94: 570–574. [DOI] [PubMed] [Google Scholar]
- 35.Rosenzweig EB, Ivy DD, Widlitz A, et al. Effects of long-term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol 2005; 46: 697–704. [DOI] [PubMed] [Google Scholar]
- 36.Simpson CM, Penny DJ, Cochrane AD, et al. Preliminary experience with bosentan as initial therapy in childhood idiopathic pulmonary arterial hypertension. J Heart Lung Transplant 2006; 25: 469–473. [DOI] [PubMed] [Google Scholar]
- 37.Lammers AE, Hislop AA, Flynn Y, et al. The six-minute walk test: normal values for children of 4–11 years of age. Arch Dis Child 2008; 93: 464–468. [DOI] [PubMed] [Google Scholar]
- 38.Bernus A, Wagner BD, Accurso F, et al. Brain natriuretic peptide levels in managing pediatric patients with pulmonary arterial hypertension. Chest 2009; 135: 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schulze-Neick I, Beghetti M. Classifying pulmonary hypertension in the setting of the congenitally malformed heart: cleaning up a dog's dinner. Cardiol Young 2008; 18: 22–25. [DOI] [PubMed] [Google Scholar]
- 40.Tissot C, Ivy DD, Beghetti M. Medical therapy for pediatric pulmonary arterial hypertension. J Pediatr. 2010; 157: 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Loon RL, Hoendermis ES, Duffels MG, et al. Long-term effect of bosentan in adults versus children with pulmonary arterial hypertension associated with systemic-to-pulmonary shunt: does the beneficial effect persist? Am Heart J 2007; 154: 776–782. [DOI] [PubMed] [Google Scholar]