

Abstract
Human Leukocyte Antigen (HLA) is a type of molecule residing on the surfaces of most human cells and exerts an essential role in the immune system responding to the invasive items. The T cell antigen receptors may recognize the HLA-peptide complexes on the surfaces of cancer cells and destroy these cancer cells through toxic T lymphocytes. The computational determination of HLA-binding peptides will facilitate the rapid development of cancer immunotherapies. This study hypothesized that the natural language processing-encoded peptide features may be further enriched by another deep neural network. The hypothesis was tested with the Bi-directional Long Short-Term Memory-extracted features from the pretrained Protein Bidirectional Encoder Representations from Transformers-encoded features of the class I HLA (HLA-I)-binding peptides. The experimental data showed that our proposed HLAB feature engineering algorithm outperformed the existing ones in detecting the HLA-I-binding peptides. The extensive evaluation data show that the proposed HLAB algorithm outperforms all the seven existing studies on predicting the peptides binding to the HLA-A*01:01 allele in AUC and achieves the best average AUC values on the six out of the seven k-mers (k=8,9,...,14, respectively represent the prediction task of a polypeptide consisting of k amino acids) except for the 9-mer prediction tasks. The source code and the fine-tuned feature extraction models are available at http://www.healthinformaticslab.org/supp/resources.php.
Keywords: class I HLA-binding peptide prediction, natural language processing, BERT, BiLSTM, feature selection, bioinformatics
Introduction
Peptide is a type of compound formed by the connections of amino acids through peptide bonds and involved in various biological activities [1]. Endogenous peptides are mostly produced by proteolysis within cells and play important biological functions in anti-tumor, immune regulation and endocrine regulation through the interactions with membrane receptors and proteins [2].
T cells in the human immune system may be activated by the target-specific binding of antigenic peptides to the class I and class II Major Histocompatibility Complex (MHC) molecules [3]. Human MHC is also called the Human Leukocyte Antigen (HLA). The main function of the class I HLA (HLA-I) molecules is to present the bound peptides to the T cell antigen receptors on the surfaces of T cells ( [4]. These HLA-I-binding peptides are usually derived from the degraded products of self- or non-self-proteins. The self-produced proteins rarely cause immune responses, while the non-self-peptides can stimulate radical responses of the human immune system. The interaction between the HLA molecules and peptides initiates the subsequent recognition of these foreign peptides by the T cells and controls the size and effectiveness of the immune response. Therefore, a major goal of developing vaccines and immunotherapies is to accurately predict the binding of peptides to the HLA molecules.
There are two main classes of computational studies for the HLA-I-binding peptide prediction problem, i.e. score function and deep learning. (i) The quantitative structure–activity relationship has been widely used to score the class I MHC-/epitope-binding affinity [5]. The traditional sequence scoring functions were also utilized to complement the binding affinity predictions, and various scoring functions were proposed, e.g. amino acid frequency scores [6], WebLogo-based entropy information [7] and Position-specific scoring matrix [8]. Anthem [9] combines five popular scoring functions to encode the peptides and used the wrapper feature selection algorithm to choose a feature subset to train the AODE classification model [10]. (ii) Deep learning exhibits powerful capabilities to extract the latent patterns within the biological sequences. DeepHLApan uses the stacked BiGRU with an attention module to extract the sequential patterns within the peptides [11]. Mei et al. evaluated the performances of 19 HLA-I peptide-binding prediction tools and conducted a comprehensive review of the prediction results from the perspectives of prediction score calculation methods, utilized algorithms and evaluation strategies [12]. This provides a staged progress summary of the existing prediction studies of HLA-I molecules and peptides.
A peptide may be regarded as a life’s language and its contextual information may need a better elaboration way. The natural language processing (NLP) area is rapidly developing recently, and various powerful algorithms have been proposed to extract the latent contextual patterns. Ghosh et al. proposed the contextual version Long Short-Term Memory (LSTM) for the large-scale NLP prediction tasks [13]. Chapman et al. also demonstrated that the contextual features described the clinical text well for the prediction tasks [14]. Rao et al. established the Tasks Assessing Protein Embedding pretrained semi-supervised learning tasks and demonstrated its transferability to the other peptide-based prediction tasks [15].
This study hypothesized that the deep learning-based features may be further encoded by deep neural network for the latent peptide patterns. Bidirectional Encoder Representations from Transformers (BERT) is a popular language representation model [16], and it was further re-tuned with protein sequences such as the Protein Bidirectional Encoder Representations from Transformers (ProtBert) [17]. We tested the hypothesis with the HLA-I-binding prediction framework, HLAB, via the features extracted by the pretrained ProtBert model cascaded with Bi-directional Long Short-Term Memory (BiLSTM). The ProtBert-BiLSTM-extracted features were then enriched by the Uniform Manifold Approximation and Projection (UMAP) [18] algorithm. The features were further refined by feature selection algorithms. The model trained using the optimized features outperformed the existing HLA-I-binding peptide prediction tools or methods.
Materials and Methods
Summary of the dataset
The peptide sequences binding with the HLA-I alleles were retrieved from the study Anthem [9]. Due to the space limitation, the detailed description may be found in the Supplementary Materials and Table 1.
Table 1.
Dataset summarization; overview of the peptides of different lengths used for both the training, validating and independent datasets
| Length | Training | Validating | Testing |
|---|---|---|---|
| 8 | 22 643 | 7 309 | 7 564 |
| 9 | 360 248 | 120 175 | 116 349 |
| 10 | 87 465 | 29 442 | 27 126 |
| 11 | 39 423 | 13 002 | 11 619 |
| 12 | 16 198 | 5 431 | 5 471 |
| 13 | 8 373 | 2 745 | 2 818 |
| 14 | 4 672 | 1 569 | 1 579 |
The pretrained ProtBert model
BERT is a language representation model trained on a very large language corpus [16]. It has achieved the new state-of-the-art results in 11 NLP tasks. BERT’s model architecture is a multi-layer bidirectional transformer encoder. Each layer has 12 or 24 encoder blocks for the BERT-base and BERT-large models, respectively. One layer consists of a multi-head self-attention sub-layer and a fully connected feed-forward sub-layer. A residual connection is deployed around each of the two sub-layers, followed by the layer normalization.
Elnaggar et al. introduced a new NLP model called ProtBert [17], which is obtained via fine-tuning the original BERT model with the protein sequences from the two databases, UniRef100 [19] and BFD [20]. The database UniRef100 is a widely used database of reference protein sequences, and the database BFD merged all the protein sequences available in the database UniProt [21] and the proteins translated from multiple metagenomic sequencing projects. ProtBert increased the number of layers to 30 in order to deliver better performances in the downstream supervised tasks. The authors demonstrated the advantages of the ProtBert model on three tasks, i.e. predicting the secondary structure, subcellular localization and membrane-binding.
This study further tuned the ProtBert model with the HLA-I-binding peptides in the training datasets before extracting the sequence features.
Bi-directional Long Short-Term Memory
LSTM is a subtype of recurrent neural network [22]. Its design characteristics fits the modeling of sequential data like text and time series. The bi-directional version LSTM (BiLSTM) shows a better capability in capturing the text patterns by a combination of forward and backward LSTMs [23]. BiLSTM has been successfully utilized for the predictions of antibacterial and antifungal peptides [24, 25].
Feature dimension reduction and feature selection
The dimensionality of the feature space may be reduced to improve the classification performances of a dataset, including both dimension reduction [26] and feature selection [27] algorithms. It is anticipatable that the removal of irrelevant features will improve the efficiencies of both training and predicting tasks.
The dimension reduction algorithm tries to retain the pairwise distance structure between all the samples (PCA) [28] or prefers the local distances over the global ones (t-SNE) [29]. UMAP transforms the feature space into a new space based on the Riemannian geometry framework and retains more global structure with faster running speed against the t-SNE algorithm [30]. UMAP carries the following advantages compared with the other dimension reduction algorithms. (i) UMAP captures both global and local structures, (ii) UMAP receives less constraints by the sample size of a dataset and (iii) UMAP performs well in a large dataset even with tens of thousands of dimensions. So this study used UMAP to the extracted features for the downstream prediction tasks. We set the number of dimensions after the UMAP dimension reduction to 5~18, and the HLA-I-binding peptide prediction tasks for different HLA-I alleles were optimized to different numbers of dimensions. The final results may be found in the Supplementary Table S2 available online at http://bib.oxfordjournals.org/.
Feature selection has demonstrated its efficacies in reducing the number of the original features and the learned latent features in many studies [6]. There are two main classes of feature selection algorithms, i.e. filters and wrappers [31]. A filter feature selection algorithm evaluates the associations of the individual features with the class labels, while a wrapper evaluates a heuristically selected feature subset for the classification performance. A filter usually runs faster but performs worse than a wrapper. This study used five feature selection algorithms, they are T-test, Wilcoxon rank-sum test (W-test), Random Forest (RF), Recursive Feature Elimination based on Linear Regression method (LR-RFE) and Recursive Feature Elimination based on Support Vector Machine method (SVM-RFE). T-test and W-test rank the features by the ascendent order of the statistical P-values. The other three feature selection algorithms rank the features by their algorithmic default settings. We chose the percent of the top-ranked features as the chosen features to build the classification models.
This study sets three values, 0.55/0.75/0.95, for the feature selection parameter to select features. The experimental data show the necessity of setting this parameter. The detailed parameter choices may be found in Supplementary Table S2 available online at http://bib.oxfordjournals.org/.
The proposed framework HLAB
This study is carried out in the following steps, as illustrated in Figure 1.
Figure 1.

Overview of the proposed framework HLAB. There are the following main modules, including data preprocessing, feature extraction, feature dimension reduction, feature selection and classification.
First, the latent features are extracted from the input sequences. Each input vector is a 49-letter residue sequence, 34 of which are from the HLA and 15 are from the corresponding peptide. The input HLA-I sequence is transformed into a pseudo-sequence by the NetMHCPan algorithm [32]. There is no peptide in the datasets longer than 15 amino acids. So a peptide sequence is encoded as a 15-letter sequence. A peptide shorter than 15 is complemented with the pseudo amino acid ‘X’ to its end so that a 15-letter sequence is loaded into the input vector. The HLA pseudo-sequence is concatenated with the peptide sequence for the next step. The characters ‘[CLS]’ and ‘[SEP]’ are added to the head and end of the entire concatenated vector according to the requirement of the BERT model. The input vector is fed into the ProtBert model and the BiLSTM model for the purpose of feature extraction. The 49D input sequence is encoded as a 1536D high-dimensional feature vector.
Second, the dimension of the feature space is reduced through dimension reduction and feature selection algorithms. We transform the feature space using the UMAP algorithm and then seek out the top-ranked features by their individual associations with the class labels using feature selection algorithms, such as T-test and W-Test.
Third, we establish the classification models using the chosen features on the training datasets and evaluate the trained models on the validating datasets. The prediction model with the best prediction performance on the validating dataset is used for the final prediction of whether the query testing peptides bind to the model-specific HLA-I alleles on the testing dataset.
Based on the parameter settings of BERT [16] and ProtBert [17], we set the learning rate as 5e-5, batch size as 16, dropout rate as 0.1, and the Adam optimizer was used for the model optimization. And the number of hidden unites in the BiLSTM layer is 768. The total number of training epochs is three.
Binary classifiers and their performance metrics
The prediction between each HLA-I allele and its fixed-length binding peptides was a binary classification model, as similar as in [9, 32–38]. The input of the binary classification model is an HLA-peptide pair, and the output of that is 1 or 0, where 1 means the peptide will bind to the HLA allele, and 0 means the peptide will not bind. Seven popular binary classifiers are used to establish the classification models, including logistic regression (LR) [39], support vector machine (SVM) [40], bagging classifier (Bagging) [41], extreme gradient boost (XGBoost) [42], k-nearest neighbor (KNN) [43], decision tree (Dtree) [44] and naive bayes (NB) [45].
A binary classifier is evaluated by the following five performance metrics, i.e. area under the receiver operating characteristic (ROC) curve (AUC), sensitivity (Sn), specificity (Sp), accuracy (ACC) and Matthews correlation coefficient (MCC). The measurements TP and FN are the numbers of true positives and false negatives. While the measurements TN and FP represent the numbers of true negatives and false positives. The metrics Sn and Sp are defined as Sn = TP/(TP + FN) and Sp = TN/(TN + FP). The overall accuracy is defined as Acc = (TP + TN)/(TP + FN + TN + FP). The correlation coefficient of the predictions of a binary classifier is defined as MCC = (TP × TN-FP × FN)/sqrt((TP + FP) × (TP + FN) × (TN + FP) × (TN + FN)), where sqrt(x) calculates the squared root of x. AUC is a popular parameter-independent metric to describe a binary classifier.
A classifier is trained using the training dataset and is evaluated on the validating dataset. The model with the best AUC value on the validating dataset is tested on the testing dataset. Unless otherwise specified, the experimental data in the following sections were conducted on the combined validation dataset of all the alleles and all the peptide lengths.
Results and Discussion
Evaluation of model hyperparameters
The deep learning models may perform differently with the different hyperparameter values. We evaluated the two major hyperparameters Epoch and BatchSize shown in Figure 2. A smaller loss value suggested a better model performance. Figure 2A suggested that the model loss did not linearly change with the different epochs, and the loss started to increase after Epoch = 3. So the minimum loss was achieved when Epoch = 3. A similar pattern was also observed for different values of the hyperparameter BatchSize. And the minimum loss 0.0891 was achieved when the BatchSize = 16. The following sections used Epoch = 3 and BatchSize = 16 as the default values.
Figure 2.

Loss evaluation of the model hyperparameters. The changes of the model loss were evaluated for the different values of the hyperparameters (A) Epoch and (B) BatchSize. The horizontal axis gave the value choices of the two hyperparameters and the vertical axis gave the loss values.
Evaluation of the pretrained model weight parameters
The protein evolution and structural information may be distilled into the pretrained ProtBert model through the self-supervised training process, and this section evaluated the contribution of such information to the prediction of the HLA-I-binding peptides. We initialized the ProtBert network with the random weights and denoted this version of ProtBert as the ProtBert_random model. We compared the prediction performances of the pretrained ProtBert and the ProtBert_random models to extract the sequence features and evaluated the prediction performances using the softmax layer for classification. The experimental data, depicted in Figure 3, showed that the pretrained ProtBert model outperformed the ProtBert_random model on all the performance metrics.
Figure 3.
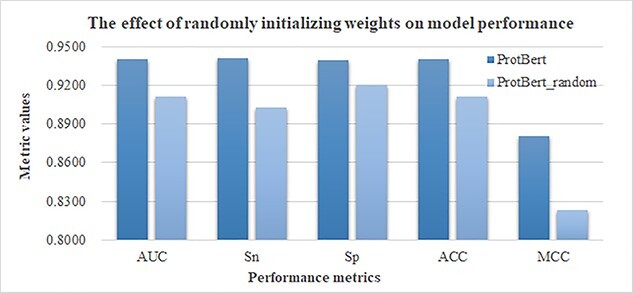
The effect of the pretrained model weight parameters. ProtBert is the model starting with the pretrained model weight parameters, and ProtBert_random is the model initialized with random parameters.
So the pretraining process of the ProtBert model under a large number of the full-length protein sequences is beneficial for the prediction tasks of the HLA-A-binding peptides.
Evaluation of different models based on the self-attention mechanism
This section evaluated the contribution of the self-attention mechanism to the overall prediction performance. The pretrained ProtBert model was based on the Transformer architecture with the self-attention mechanism [17]. The ALBERT model shared the parameters between attention layers in the original BERT model so that the model complexity was significantly reduced [46]. Both ProtBert and ALBERT utilized the self-attention mechanism. The two models were used as the sequence feature encoders and the softmax layer was used for classification. The experimental data, depicted in Figure 4, showed that the ProtBert-based framework outperformed the ALBERT-based model in all the five performance metrics. So both the self-attention mechanism and the fine-tuning on the protein data served as important contributions to the HLA-I-binding peptide prediction tasks.
Figure 4.
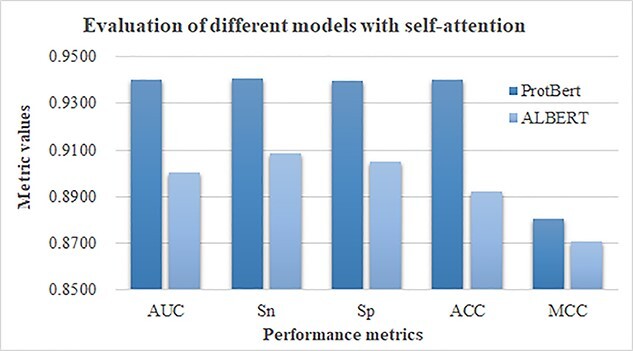
Comparison of the prediction performance of different models based on the self-attention mechanism. The two models ProtBert and ALBERT were evaluated. Both models used the self-attention mechanism. The horizontal axis listed the five investigated performance metrics, i.e. AUC, Sn, Sp, ACC and MCC. The vertical axis gave the metrics values.
Evaluation of the module combinations
Qiao et al. recently proposed a new model BERT-Kcr for the prediction task of the protein lysine crotonylation sites [47]. They loaded the features extracted by the BERT model into different machine learning and deep learning classifiers. The investigated machine learning classifiers included SVM, RF and XGBoost, and the deep learning models included BiLSTM, CNN and fully connected neural network (FCNN). The 10-fold cross-validation experiments showed that the BERT model followed by the BiLSTM network achieved the best AUC value. The authors recommended the extraction of the latent features from the peptide sequences by using the cascaded high-dimensional encoders.
Based on this observation, this study constructed the feature extraction layer by different cascaded combinations of the network modules for the prediction tasks of HLA-I-binding peptides. We evaluated the prediction performances of four different end-to-end module combinations, including the ProtBert model alone, the ProtBert cascaded with BiLSTM, the ProtBert model cascaded with the convolutional neural network (CNN) and the ProtBert model with BiLSTM and Attention mechanism. The training set was used for the model training, and the validation set was used for model evaluation. Figure 5 shows that the best module combination was ProtBert + BiLSTM. First, an additional module BiLSTM to ProtBert outperformed the module ProtBert alone with 0. 08% in AUC. If we replaced BiLSTM with CNN, the module combination ProtBert + CNN performed even worse than the ProtBert module alone. The attention layer did not improve the module combination ProtBert + BiLSTM in the performance metric AUC.
Figure 5.
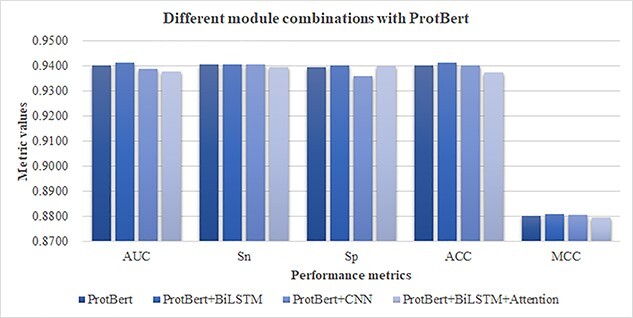
Average performances of the combinations of different HLAB modules on all the HLA-I prediction tasks. The performance metrics are averaged over all the prediction tasks. The performances are calculated on the validating dataset.
Based on the ablation experiments and the observation from the literature, the following sections used the module combination of ProtBert + BiLSTM.
The necessity of feature dimension reduction module
This study investigated a total of 360 different HLA-binding peptide prediction tasks, and the total number of samples was nearly 890 000. The feature extraction step used the ProtBert and BiLSTM cascade modules and generated the feature vector with the dimension 1536. We anticipated that it would be particularly time-consuming to conduct the model training for the large datasets with high-dimensional features. We designed the following experiment to estimate the overall model training time.
We selected the binding prediction task between HLA-A*02:01 and 9-mer peptides as an example dataset. This prediction task has 23 435 samples. We randomly selected 1000, 2000, 3000, …, 10 000 samples as the sub-datasets to train the models without using the feature dimensionality reduction step. The experimental results are shown in Figure 6. According to the model training times for the 10 example datasets, we fit the functional relationship between the size of the dataset and the time of the model training and used a quadratic polynomial function to approximate the nonlinear relationship. The fit function was  , where x and y were the sizes of the dataset and the model training time in hour, respectively.
, where x and y were the sizes of the dataset and the model training time in hour, respectively.
Figure 6.
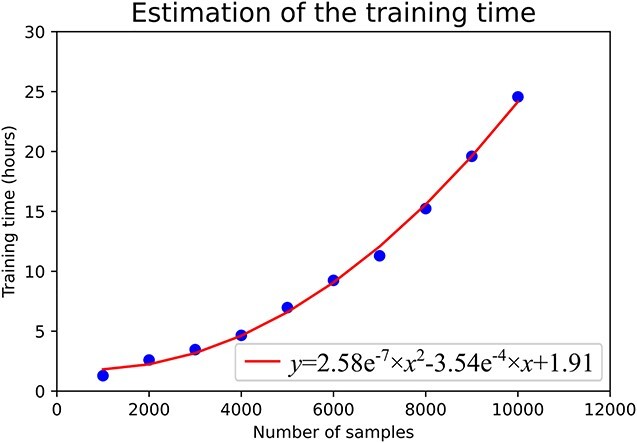
The correlation between the training time and the dataset size. The fit function was illustrated as the curve.
Figure 6 shows that the training time increased very fast as the number of the training sample increased. We used the fit function to estimate that the total time required for the model training for all the tasks was about 1382.89 hours, or about 57 days. So we conducted the feature dimensionality reduction step before the feature selection step to further reduce the model training time.
Performances of different feature selection algorithms and the rates of the selected features
Figure 7 shows the number of times a feature selection algorithm achieving the best prediction performances by collaborating with the seven classifiers on all the HLA-I-binding prediction tasks. Some prediction tasks may have more than one feature selection algorithms achieving the best performance using the same classifier. The detailed data are provided in the Supplementary Table S2 available online at http://bib.oxfordjournals.org/. T-test selects the feature subsets with the best classification performances for >60% of 2121 prediction tasks (1275), while the second-ranked feature selection algorithm RF performs the best on only 26.78% (568) of the prediction tasks. Although T-test performs very well on many of the HLA-I-binding prediction tasks, the remaining 39.89% of the prediction tasks rely on the other feature selection algorithms to find the best feature subsets. So this study evaluates the five feature selection algorithms using the training and validating datasets for each prediction task, and the best feature selection algorithm on the validating dataset is used on the testing dataset.
Figure 7.
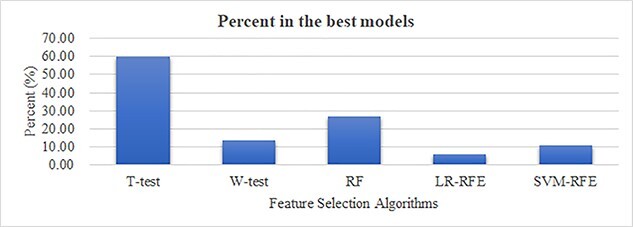
Percent of the times that a feature selection algorithm achieving the best performances by collaborating with seven classifiers for all the HLA-I-binding prediction tasks. The seven classifiers are Dtree, Bagging, XGBoost, NB, SVM, LR and KNN. The five feature selection algorithms, T-test/W-test/RF/LR-RFE/SVM-RFE, are evaluated.
We evaluated the impact of the different rates of the selected features in the feature selection step on the model performance, as shown in Figure 8. We selected four prediction tasks as the datasets for this experiment. In order to ensure the distinguishability and integrity of the features after the feature selection step, five values, 0.15/0.35/0.55/0.75/0.95, were evaluated for how they impacted the prediction performances. The experimental results, depicted in Figure 8, showed that the AUC values of the values 0.15 and 0.35 achieved significantly lower AUC values than the other three values 0.55, 0.75 and 0.95 for all the four prediction tasks. Furthermore, the four prediction tasks reached the best AUC values using different rates of the selected features among the three choices 0.55/0.75/0.95. So this study used the best choice of the three rates of the selected features, 0.55/0.75/0.95, for each prediction task.
Figure 8.

Evaluation of the different rates of the selected features for the four subtasks. The evaluation experiments were conducted for the prediction tasks of (A) HLA-A*02:01 and 8-mer peptides, (B) HLA-A*03:01 and 8-mer peptides, (C) HLA-B*27:09 and 13-mer peptides and (D) HLA-B*35:01and 13-mer peptides. The horizontal axis gave the five evaluated values, including 0.15, 0.35, 0.55, 0.75 and 0.95. The vertical axis gave the calculated AUC values of the experiments.
Classifiers perform differently on different datasets
The seven classifiers demonstrate different prediction performances on the seven prediction tasks of the allele HLA-A*01:01, as shown in Figure 9A. The detailed data may be found in the Supplementary Table S3 available online at http://bib.oxfordjournals.org/. All the classifiers achieve reasonable prediction AUC values ≥ 0.9264 for the seven prediction tasks of the allele HLA-A*01:01, as shown in Supplementary Table S3 available online at http://bib.oxfordjournals.org/. The four classifiers, Bagging/KNN/Dtree/NB, tend to be ranked the lowest among the seven classifiers, as shown in Figure 9A. The remaining three classifiers, LR/SVM/XGBoost, show comparable prediction AUC values. All the three classifiers are ranked the best on some of the seven datasets. Since no classifier achieves the best AUC value on all the datasets, the following sections deliver the prediction values on the testing datasets using the best models of the finally chosen features evaluated on the validating datasets.
Figure 9.
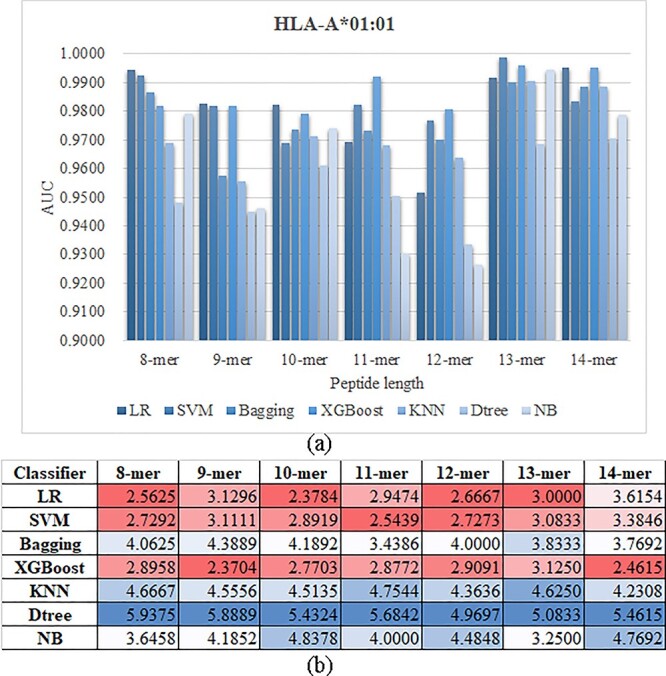
The performance metric AUC values of the seven classifiers on the HLA-I-binding prediction tasks. (A) The rankings of the seven classifiers on the seven k-mers (k = 8, 9, …, 14) of the allele HLA-A*01:01. (B) The average rankings of the seven classifiers on all the HLA-I alleles. The heatmap colors red and blue represent the maximum and minimum values of each column, respectively.
The seven classifiers are further evaluated on all the HLA-I alleles, as shown in Figure 9B. The classifier LR achieves the best average AUC ranks on four (8/10/12/13) out of the seven k-mers of the HLA-I-binding prediction tasks. While the classifier XGBoost achieves the best AUC ranks on only two k-mers (9 and 14). But the average AUC ranks of the two classifiers LR and XGBoost on all the k-mers of all the HLA-I alleles are 2.9000 and 2.7728, respectively. Another classifier SVM achieves the overall average AUC rank 2.9245, which is slightly worse than those (2.9000 and 2.7728) of the two classifiers LR and XGBoost.
So the following sections of this study deliver the best models for the seven k-mers of the HLA-I alleles using different feature selection and classification algorithms.
Performance evaluation of the FCNN
We compared the model performances of the FCNN and the machine learning classifiers on the engineered features in this study, as shown in Figure 10. Five prediction datasets were chosen for this comparison experiment. Figure 10 suggests that the fully connected layer for the deep neural network usually performed very good prediction performances, while the supervised machine learning classifiers may deliver better prediction results on the same set of the extracted features by the deep neural networks. So this study used the classifier algorithms for the binary classification tasks.
Figure 10.
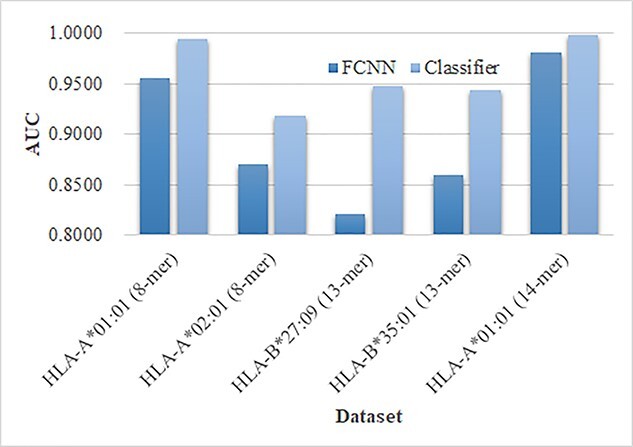
The performance comparison between the FCNN and the machine learning classifiers. The FCNN used the fully connected layer to generate the predictions. The machine learning classifiers (Classifier) generated the predictions using the engineered features in this study. The horizontal axis listed the datasets and the vertical axis gave the AUC values.
Performance comparison on the testing dataset of HLA-A*01:01
Multiple studies have been published for predicting the peptides binding the HLA-I alleles, which are compared with the proposed HLAB framework in this study, as shown in Figure 11. These studies are Anthem [9], MixMHCpred2.0.2 [33], NetMHCpan4.1 [32], NetMHCcons1.1 [34], NetMHCstabpan1.0 [35], ACME [36], MHCSeqNet [37] and DeepSeqPan [38]. A fair comparison is carried out on the testing dataset using the HLAB models with the best performances on the validating datasets for the specific predicting tasks. Five performance metrics are evaluated, i.e. AUC, Sn, Sp, ACC and MCC.
Figure 11.

Performance comparison with the existing studies on predicting the peptides binding to the HLA-A*01:01 allele. (A) A comparison is first carried out for the five performance metrics, AUC/Sn/Sp/ACC/MCC, on predicting the 8-mer peptides binding to the HLA-A*01:01 allele. The vertical axis gives the values of the five performance metrics. (B) The AUC values are compared between the proposed HLAB with the existing studies for predicting the k-mer peptides binding to the HLA-A*01:01 allele, where k = 8, 9, …, 14. The study MHCSeqNet was not evaluated on the 13-mer and 14-mer peptides in the original study.
First, the proposed HLAB outperforms all the seven studies on predicting the 8-mer peptides binding to the HLA-A*01:01 allele, as shown in Figure 11A. HLAB improves the seven algorithms by at least 0.0230 in AUC and by at least 0.0560 in ACC.
Second, the AUC values of all the seven peptide lengths are evaluated for these eight prediction algorithms, as shown in Figure 11B. The algorithm NetMHCpan-4.1 achieves the best prediction AUC values on four peptide lengths, while the proposed HLAB algorithm achieves the best AUC on three. The averaged rank (2.4286) of NetMHCpan-4.1 is slightly better than that (2.8571) of the proposed algorithm HLAB. But if we calculate the average AUC value, HLAB achieves 0.9891, which is better than that (0.9869) of NetMHCpan-4.1. So the proposed algorithm HLAB outperforms all the existing studies on predicting the peptides binding to the HLA-A*01:01 allele in the performance metric AUC.
The ROC curve has been widely used to illustrate how a binary classification model performs [48, 49]. We visualized the ROC curves for six prediction tasks, including (i) HLA-A*01:01 and 8-mer peptide, (ii) HLA-A*01:01 and 9-mer peptide, (iii) HLA-A*01:01 and 10-mer peptide, (iv) HLA-A*01:01 and 11-mer peptide, (v) HLA-A*01:01 and 12-mer peptide and (vi) HLA-A*01:01 and 13-mer peptide, as shown in Figure 12. The areas under the ROC curves for all the six prediction tasks were at least 0.98. The ROC curves and the AUC values suggested that the proposed HLAB algorithm achieved good prediction performances for the HLA-I-binding peptides of different lengths.
Figure 12.

ROC curves of HLAB for the HLA-I peptide binding prediction on the independent datasets. The illustrative datasets were (A) HLA-A*01:01(8-mer), (B) HLA-A*01:01 (9-mer), (C) HLA-A*01:01(10-mer), (D) HLA-A*01:01(11-mer), (E) HLA-A*01:01 (12-mer) and (F) HLA-A*01:01 (13-mer).
Performance comparison on the testing dataset of all the HLA-I alleles
The performances of HLAB on predicting the peptides binding to all the HLA-I alleles are summarized in Figure 13, and the detailed data may be found in the Supplementary Table S4 available online at http://bib.oxfordjournals.org/. Figure 13 shows that the proposed framework HLAB proposed achieved the best average AUC value on the six out of the seven k-mers (except for the 9-mer prediction tasks). HLAB improved the prediction tasks of 13-mer and 14-mer by at least 0.0663 in the average AUC values. HLAB achieved a slightly worse average AUC (0.9769) than that (0.9826) of Anthem for the prediction tasks of 9-mers.
Figure 13.

Performance comparison with the existing studies on the testing datasets of all the HLA-I alleles. For each k-mer (k = 8, 9, …, 14), the average AUC of multiple binary classification tasks is calculated. The average AUC are compared between the proposed HLAB and the existing studies.
Conclusions
This study proposed the feature extraction algorithm HLAB for the HLA-I-binding peptide prediction problem. The experimental data demonstrated the necessity of the cascaded peptide encoding by two NLP networks, ProtBert and BiLSTM. The extracted features may be further refined by feature selection algorithms for different prediction tasks.
The unsupervised cascaded ProtBert + BiLSTM model may be pretrained for the other protein sequence prediction tasks in the future studies. For example, the prediction of the class II HLA-binding peptides may utilize the framework in this study. The potential challenge is that the class II HLA-binding peptides have a wider range of lengths, and the publicly available dataset has fewer samples [50, 51]. Therefore, it is necessary to further explore how the proposed framework may be tuned to achieve the best performances for predicting the class II HLA-binding peptides. And, it is also feasible to apply the unsupervised cascaded ProtBert + BiLSTM model to the prediction tasks of the post-translationally modified peptides [52–54].
Key Points
Contextual information in peptides may be encoded by NLP models.
BERT and BiLSTM are two popular NLP models to encode peptide sequences.
The ProtBert-encoded peptide features may be further enriched by BiLSTM.
The ProtBert-BiLSTM cascade framework efficiently encodes the HLA-I-binding peptides.
Feature selection is also important to improve the prediction of HLA-I-binding peptides.
Supplementary Material
Acknowledgements
We really appreciate the anonymous reviewers’ constructive remarks and useful suggestions that have substantially improved the manuscript!
Author Biographies
Yaqi Zhang is a postgraduate student at the School of Biology & Engineering, Guizhou Medical University, and the College of Computer Science and Technology, Jilin University, whose research interests include bioinformatics algorithm design and development.
Gancheng Zhu is a postgraduate student at the College of Computer Science and Technology, Jilin University, whose research interests include bioinformatics algorithm design and development.
Kewei Li is a postgraduate student at the College of Computer Science and Technology, Jilin University, whose research interests include bioinformatics algorithm design and development.
Fei Li is a PhD student at the College of Computer Science and Technology, Jilin University. His research interests include biomedical big data modeling.
Lan Huang is a professor at the College of Computer Science and Technology, Jilin University, Changchun, Jilin, China. Her research interests include bioinformatics and systems biology.
Meiyu Duan is a PhD student at the College of Computer Science and Technology, Jilin University. Her research interests include biomedical big data modeling.
Fengfeng Zhou is a professor at the School of Biology & Engineering, Guizhou Medical University, and the College of Computer Science and Technology, and Key Laboratory of Symbolic Computation and Knowledge Engineering of Ministry of Education, Jilin University. His research interests include biomedical big data.
Funding
Senior and Junior Technological Innovation Team (20210509055RQ); National Natural Science Foundation of China (62072212 and U19A2061); Jilin Provincial Key Laboratory of Big Data Intelligent Computing (20180622002JC); Fundamental Research Funds for the Central Universities (JLU).
Data availability
The Python implementation and the fine-tuned models of HLAB are freely available at http://www.healthinformaticslab.org/supp/resources.php.
References
- 1. Rudinger J. Characteristics of the amino acids as components of a peptide hormone sequence. In: Peptide Hormones. Palgrave, London: Springer, 1976, 1–7. [Google Scholar]
- 2. Guerrero A, Dallas DC, Contreras S, et al. Mechanistic peptidomics: factors that dictate specificity in the formation of endogenous peptides in human milk. Mol Cell Proteomics 2014;13:3343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol 2013;31:443–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Labrecque N, Whitfield LS, Obst R, et al. How much TCR does a T cell need? Immunity 2001;15:71–82. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y, Zhou P, Lin Y, et al. Quantitative prediction of class I MHC/epitope binding affinity using QSAR modeling derived from amino acid structural information. Comb Chem High Throughput Screen 2015;18:75–82. [DOI] [PubMed] [Google Scholar]
- 6. Chen Z, Zhao P, Li F, et al. iFeature: a Python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018;34:2499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crooks GE, Hon G, Chandonia JM, et al. WebLogo: a sequence logo generator. Genome Res 2004;14:1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang S, Bai Z, Han J, et al. Improving the prediction of HLA class I-binding peptides using a supertype-based method. J Immunol Methods 2014;405:109–20. [DOI] [PubMed] [Google Scholar]
- 9. Mei S, Li F, Xiang D, et al. Anthem: a user customised tool for fast and accurate prediction of binding between peptides and HLA class I molecules. Brief Bioinform 2021;22:bbaa415. [DOI] [PubMed] [Google Scholar]
- 10. Webb GI, Boughton JR, Wang Z. Not so naive Bayes: aggregating one-dependence estimators. Mach Learn 2005;58:5–24. [Google Scholar]
- 11. Wu J, Wang W, Zhang J, et al. DeepHLApan: a deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity. Front Immunol 2019;10:2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mei S, Li F, André L, et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief Bioinform 2020;4:1119–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghosh S, Vinyals O, Strope B, et al. Contextual lstm (clstm) models for large scale nlp tasks. arXiv preprint arXiv:1602.06291 2016.
- 14. Chapman W, Dowling J, Chu D. ConText: an algorithm for identifying contextual features from clinical text. In: Biological, Translational, and Clinical Language Processing, Association for Computational Linguistics (ACL) Publisher, Prague, 2007, 81–8.
- 15. Rao R, Bhattacharya N, Thomas N, et al. Evaluating protein transfer learning with TAPE. Adv Neural Inf Process Syst 2019;32:9689. [PMC free article] [PubMed] [Google Scholar]
- 16. Devlin J, Chang MW, Lee K, et al. BERT: pre-training of deep bidirectional transformers for language understanding. arXiv preprint arXiv:1810.04805 2018.
- 17. Elnaggar A, Heinzinger M, Dallago C, et al. ProtTrans: towards cracking the language of Life’s code through self-supervised deep learning and high performance computing. arXiv preprint arXiv:2007.062252020.
- 18. Mcinnes L, Healy J. UMAP: uniform manifold approximation and projection for dimension reduction. J Open Source Softw 2018;3:861. [Google Scholar]
- 19. Suzek BE, Wang Y, Huang H, et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015;31:926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martin S, Johannes S. Clustering huge protein sequence sets in linear time. Nat Commun 2018;9:2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. UniProt, Consortium . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2018;47:D506–D515.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Q, Chen J, Wang Y, et al. DeepTorrent: a deep learning-based approach for predicting DNA N4-methylcytosine sites. Brief Bioinform 2021;22:bbaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hasegawa D, Kaneko N, Shirakawa Set al. Evaluation of speech-to-gesture generation using bi-directional LSTM network. In: Proceedings of the 18th International Conference on Intelligent Virtual Agents. Sydney NSW Australia: Association for Computing Machinery, New York, NY, United States, 2018, 79–86.
- 24. Singh V, Shrivastava S, Kumar Singh S, et al. StaBle-ABPpred: a stacked ensemble predictor based on biLSTM and attention mechanism for accelerated discovery of antibacterial peptides. Brief Bioinform 2021;24:bbab439. [DOI] [PubMed] [Google Scholar]
- 25. Sharma R, Shrivastava S, Kumar Singh S, et al. Deep-AFPpred: identifying novel antifungal peptides using pretrained embeddings from seq2vec with 1DCNN-BiLSTM. Brief Bioinform 2021;23:bbab422. [DOI] [PubMed] [Google Scholar]
- 26. Chen Z, Pang M, Zhao Z, et al. Feature selection may improve deep neural networks for the bioinformatics problems. Bioinformatics 2020;36:1542–52. [DOI] [PubMed] [Google Scholar]
- 27. Chatterjee S, Biswas S, Majee A, et al. Breast cancer detection from thermal images using a Grunwald-Letnikov-aided dragonfly algorithm-based deep feature selection method. Comput Biol Med 2021;141:105027. [DOI] [PubMed] [Google Scholar]
- 28. Hotellings H. Analysis of a complex of statistical variables into principal components. Br J Educ Psychol 1932;24:417–520. [Google Scholar]
- 29. Laurens VDM, Hinton G. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–605. [Google Scholar]
- 30. McInnes L, Healy J, Melville J. Umap: uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.034262018.
- 31. Veneroni C, Acciarito A, Lombardi E, et al. Artificial intelligence for quality control of oscillometry measures. Comput Biol Med 2021;138:104871. [DOI] [PubMed] [Google Scholar]
- 32. Birkir R, Bruno A, Sinu P, et al. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res 2020;48:W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bassani-Sternberg M, Chong C, Guillaume P, et al. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput Biol 2017;13:e1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karosiene E, Lundegaard C, Lund O, et al. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 2012;64:177–86. [DOI] [PubMed] [Google Scholar]
- 35. Rasmussen, Michael, Fenoy, et al. Pan-specific prediction of peptide-MHC class I complex stability, a correlate of T cell immunogenicity. J Immunol 2016;197:1517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu Y, Wang Z, Hu H, et al. ACME: pan-specific peptide–MHC class I binding prediction through attention-based deep neural networks. Bioinformatics 2019;23:4946–4954. [DOI] [PubMed] [Google Scholar]
- 37. Phloyphisut P, Pornputtapong N, Sriswasdi S, et al. MHCSeqNet: a deep neural network model for universal MHC binding prediction. BMC Bioinform 2019;20:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Z, Cui Y, Xiong Z, et al. DeepSeqPan, a novel deep convolutional neural network model for pan-specific class I HLA-peptide binding affinity prediction. Sci Rep 2019;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong C, Qiao Y, Shang C, et al. Non-contact screening system based for COVID-19 on XGBoost and logistic regression. Comput Biol Med 2021;141:105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang W, Han R, Zhang M, et al. A network-based method for brain disease gene prediction by integrating brain connectome and molecular network. Brief Bioinform 2021;23:bbab459. [DOI] [PubMed] [Google Scholar]
- 41. Hu J. An approach to EEG-based gender recognition using entropy measurement methods. Knowl Based Syst 2018;140:134–41. [Google Scholar]
- 42. Prabha A, Yadav J, Rani A, et al. Design of intelligent diabetes mellitus detection system using hybrid feature selection based XGBoost classifier. Comput Biol Med 2021;136:104664. [DOI] [PubMed] [Google Scholar]
- 43. Wolff J, Backofen R, Gruning B. Robust and efficient single-cell Hi-C clustering with approximate k-nearest neighbor graphs. Bioinformatics 2021;37:4006–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ghiasi MM, Zendehboudi S. Application of decision tree-based ensemble learning in the classification of breast cancer. Comput Biol Med 2021;128:104089. [DOI] [PubMed] [Google Scholar]
- 45. Shen Y, Li Y, Zheng HT, et al. Enhancing ontology-driven diagnostic reasoning with a symptom-dependency-aware naive Bayes classifier. BMC Bioinform 2019;20:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lan Z, Chen M, Goodman S, et al. ALBERT: A Lite BERT for self-supervised learning of language representations. arXiv preprint arXiv:1909.11942 2019.
- 47. Qiao Y, Zhu X, Gong H. BERT-Kcr: prediction of lysine crotonylation sites by a transfer learning method with pre-trained BERT models. Bioinformatics 2021;38:648–54. [DOI] [PubMed] [Google Scholar]
- 48. Bell DR, Chen SH. Toward guided mutagenesis: Gaussian process regression predicts MHC class II antigen mutant binding. J Chem Inf Model 2021;61:4857–67. [DOI] [PubMed] [Google Scholar]
- 49. Sosnina EA, Sosnin S, Nikitina AA, et al. Recommender systems in antiviral drug discovery. ACS Omega 2020;5:15039–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gopalakrishnan V, Aayush G, Srinivasaraghavan G, et al. MHCAttnNet: predicting MHC-peptide bindings for MHC alleles classes I and II using an attention-based deep neural model. Bioinformatics 2020;36:i399–i406. Supplement_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Junet V, Daura X. CNN-PepPred: an open-source tool to create convolutional NN models for the discovery of patterns in peptide sets—application to peptide–MHC class II binding prediction. Bioinformatics 2021;37:4567–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li F, Fan C, Marquez-Lago TT, et al. PRISMOID: a comprehensive 3D structure database for post-translational modifications and mutations with functional impact. Brief Bioinform 2019;21:1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li Q, Fisher K, Meng W, et al. GMSimpute: a generalized two-step Lasso approach to impute missing values in label-free mass spectrum analysis. Bioinformatics 2019;36:257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang C, Tan X, Tang D, et al. GPS-Uber: a hybrid-learning framework for prediction of general and E3-specific lysine ubiquitination sites. Brief Bioinform 2022;23:bbab574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Python implementation and the fine-tuned models of HLAB are freely available at http://www.healthinformaticslab.org/supp/resources.php.