

Abstract
We present a novel self-supervised Contrastive LEArning framework for single-cell ribonucleic acid (RNA)-sequencing (CLEAR) data representation and the downstream analysis. Compared with current methods, CLEAR overcomes the heterogeneity of the experimental data with a specifically designed representation learning task and thus can handle batch effects and dropout events simultaneously. It achieves superior performance on a broad range of fundamental tasks, including clustering, visualization, dropout correction, batch effect removal, and pseudo-time inference. The proposed method successfully identifies and illustrates inflammatory-related mechanisms in a COVID-19 disease study with 43 695 single cells from peripheral blood mononuclear cells.
Keywords: scRNA-seq, deep learning, contrastive learning, batch effect removal
INTRODUCTION
Single-cell ribonucleic acid (RNA) sequencing (scRNA-seq) has been a powerful tool for measuring the transcriptome-wide gene expression in individual cells and understanding the heterogeneity among cell populations [1, 2]. It has been facilitating the investigation of several critical biomedical topics, such as cancer [3] and autoimmunity [4]. Despite its promises, the unique properties of the scRNA-seq data, such as extreme sparsity and high variability [5], have posed a number of computational challenges to the community [6, 7]. The key processing step is to obtain a reliable low-dimensional representation for each cell, which can preserve the biological signature of the cell while eliminating technical noise [8, 9].
The existing commonly used methods to perform the above processing are based on different backbone algorithms and assumptions. The earliest methods utilize the traditional dimension reduction algorithms, such as principal component analysis (PCA), followed by k-means or hierarchical clustering to group cells [5, 10–15]. Considering the complexity of the data, researchers have developed multiple kernel-based spectral clustering and feature selection-based clustering methods to learn more robust similarity matrices for cells [16–18]. However, the time and space complexity of such methods impede the broad applications of the methods [5]. In contrast, the graph-based methods and deep learning methods enjoy high speed and scalability [14, 15, 19, 20]. Almost all the recently developed deep learning-based methods are based on autoencoder [5, 9, 21–26] (AE) or variational autoencoder [8, 27, 28] (VAE), which can also incorporate the biostatistical models [29, 30] seamlessly. However, as AE and VAE methods are unsupervised learning methods, it is very difficult to control and decide what the deep learning models will learn, although some very recent studies try to impose constraints and our prior knowledge about the problem onto the low-dimensional space [5, 28]. Researchers have also tried to utilize manual labeling as supervision for training the models, accompanied by transfer learning [23] or meta-learning [31], but such methods encounter scalability issues and have strong assumptions about the homogeneity of different datasets, making them less popular than the above methods. Recently, contrastive learning has attracted researchers’ attention, and it has been used for single-cell multi-omics data integration [32] and analysis [33].
As discussed above, almost all the existing methods are based on unsupervised learning [7], regardless of the specific algorithm. Without accessible supervision, for the deep learning-based methods, it is difficult to guide the training process of the model and explain why a particular transformation is learned, although the model may work well. For example, the functionally similar cells should be close in the transformed space, while distinct cells should be distant [7]; the model should overcome the batch effect and map the cells of the same type but from different experiments into the same region [8]. Unsupervised learning methods may have difficulty in incorporating these requirements explicitly. Here, we propose a novel method, Contrastive LEArning framework for single-cell RNA-sequencing (CLEAR), for integrative scRNA-seq data analysis, based on a new machine learning scenario, self-supervised learning, which can model all the above requirements explicitly. More specifically, we design our method based on self-supervised contrastive learning [34], where we construct the labels from the unlabeled data. For the gene expression profile of each cell, we distort the data slightly by adding noise to the raw data, which mimics the technical noise in the biological experiments. During training, we force the model to produce similar low-dimensional representations for the raw data and the corresponding distorted profile (positive pairs). Meanwhile, we train the model to output distant representations for cells of different types (negative pairs).
CLEAR achieves superior performance on a broad range of fundamental tasks for scRNA-seq data analysis, including clustering, visualization, dropout correction, batch effect removal and pseudo-time inference. As for clustering, CLEAR can outperform the popular tools and recently proposed tools on diverse datasets from different organisms. Applied on a dataset from a COVID-19 disease study with 43 695 single cells from peripheral blood mononuclear cells (PBMCs), CLEAR successfully identifies and illustrates inflammatory-related mechanisms. Further experiments to process a million-scale single-cell dataset demonstrate the scalability and potential of CLEAR to handle the emerging large-scale cell atlases. With the capability of generating effective scRNA-seq data representation while eliminating technical noise, the proposed method can serve as a general computational framework for single-cell data analysis.
RESULTS
Overview of CLEAR
Unlike most existing methods, which are based on unsupervised learning to map the single-cell gene expression profile to the low-dimension space, we develop CLEAR based on self-supervised learning. Notice that we can incorporate our prior knowledge about scRNA-seq data, such as noise and dropout events, into the model training process implicitly and seamlessly when we build the label from the unlabeled data. More specifically, we design CLEAR based on self-supervised contrastive learning [34]. As shown in Figure 1, eventually, we also want to train a deep learning encoder to map the gene expression profile into the low-dimensional space by forcing functionally similar cells close in the transformed space while distinct cells being distant. Here, the model should also be robust to technical noise, such as dropout events. That is, the profiles from the same cell, no matter with or without dropout events, should be mapped into the same place in the low-dimension space. Although it is difficult to estimate the noise level of the real dataset, we can add simulated noise to the data and force the trained model to be robust to the noise. Based on the above idea, we design CLEAR as shown in Figure 1. Given the single-cell gene expression profile, we add different simulated noise, such as Gaussian noise and simulated dropout events, to it (data augmentation), resulting in distorted profiles (augmented data). We also borrow the idea from the genetic algorithm [35] and generate the distorted profile (child) by getting the recombination from the two raw profiles (parents). The raw profile and the corresponding distorted profiles from the same cell are positive pairs, while the profiles from different cells are negative pairs. When training the model, we force the model to produce similar representations for the positive pairs while distinct ones for the negative pairs (contrastive learning). Intuitively, we pull together the representations of functionally close cells in the low-dimensional space while pushing apart the embeddings of the dissimilar ones. CLEAR does not have any assumptions on the data distribution or the encoder architecture. It can eliminate technical noise and generate effective scRNA-seq data representation, which is suitable for a range of downstream applications, such as clustering, batch effect correction and time-trajectory inference, as discussed below.
Figure 1.

Overview of the proposed framework, CLEAR. The proposed method is based on self-supervised contrastive learning. For the gene expression profile of each cell, we distort the data slightly by adding noise to the raw data, which mimics the technical noise in the biological experiments. When training the deep encoder model, we force the model to produce similar low-dimension representations for the raw data and the corresponding distorted profile while distant representations for cells of different types. Intuitively, the deep learning model learns to pull together the representations of similar cells while pushing apart different cells. By considering noise during training, CLEAR can produce effective representations while eliminating technical noise for the scRNA-seq profiles. Such representations have a broad range of applications, including clustering and classification, dropout event and batch effect correction, pseudo-time inference. CLEAR is also scalable to million-scale datasets without any overhead.
Overall clustering performance
To assess how the representation from CLEAR helps to cluster, we evaluate the proposed method, combined with the 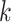 -means clustering algorithm, on 10 published datasets with expert-annotated labels [36–42]. The label information is only available during testing. We compare our model with several state-of-the-art methods that are widely used for scRNA-seq data and belong to different categories, including PCA-based tools (Seurat [10], SC3 [11], CIDR [12], SINCERA [13]), graph-based methods (Seurat [19], scGNN [25]), deep generative models (scVI [8], scDHA [22], scGNN [25], ItClust [23], scRAE [43]), transfer learning approach (ItClust [23]) and two similar works with contrastive learning-based models (contrastive-sc [44], scNAME [45]). Evaluated on 10 datasets covering the life span of humans and mice (Supplementary Method 4), CLEAR achieves substantially better performance in adjusted Rand index (ARI) score and normalized mutual information (NMI) than all the other methods on most datasets (Figure 2A, Supplementary Table 1, Supplementary Figures 1–3, Supplementary Method 3). In particular, on average, CLEAR improves over the second-best method, scDHA, by 4.56% regarding the ARI score. We also perform multiple runs on multiple random seeds to show the stability of CLEAR (Supplementary Table 2), and do the hyperparameter fine-tuning for the baseline methods to achieve a fair comparison (Supplementary Table 3). To better understand the representation produced by each method, we use uniform manifold approximation (UMAP) to project the internal representations into a two-dimensional space and visualize them (Figure 2B, Supplementary Figures 4–12) As shown in the figure, CLEAR learns to embed similar cells within the same clusters while separate dissimilar cells well among different clusters, and produces similar clustering results as the ground truth cell annotation. Furthermore, we evaluated the representations produced by each method with the Leiden clustering algorithms. Starting from a large resolution that the Leiden algorithm would overcluster, we decreased the resolution with a step size equal to 0.01 until it tended to undercluster, as illustrated in Supplementary Figure 13. The river plot at the turning point is shown in Figure 2C. As the PCA-based features are used as the cornerstone for experts in annotating cell types, it is not surprising that Seurat clusters match with experts’ annotation quite well. However, scDHA tends to overcluster oligodendrocyte cells and excitatory cells at the turning point. In contrast, CLEAR better represents the data by achieving nearly perfectly matching performance like Seurat. It suggested that CLEAR and Seurat embeddings have a better internal structure compared with scDHA. Although CLEAR does not access any human supervision on marker genes, it can recover the ground truth directly for this dataset, suggesting that the proposed framework can implicitly capture the data’s biological features. We also performed an ablation study and hyperparameter selection on the data augmentation operations to demonstrate the effect of the novel self-supervised contrastive learning settings, shown in Supplementary Tables 4–6.
-means clustering algorithm, on 10 published datasets with expert-annotated labels [36–42]. The label information is only available during testing. We compare our model with several state-of-the-art methods that are widely used for scRNA-seq data and belong to different categories, including PCA-based tools (Seurat [10], SC3 [11], CIDR [12], SINCERA [13]), graph-based methods (Seurat [19], scGNN [25]), deep generative models (scVI [8], scDHA [22], scGNN [25], ItClust [23], scRAE [43]), transfer learning approach (ItClust [23]) and two similar works with contrastive learning-based models (contrastive-sc [44], scNAME [45]). Evaluated on 10 datasets covering the life span of humans and mice (Supplementary Method 4), CLEAR achieves substantially better performance in adjusted Rand index (ARI) score and normalized mutual information (NMI) than all the other methods on most datasets (Figure 2A, Supplementary Table 1, Supplementary Figures 1–3, Supplementary Method 3). In particular, on average, CLEAR improves over the second-best method, scDHA, by 4.56% regarding the ARI score. We also perform multiple runs on multiple random seeds to show the stability of CLEAR (Supplementary Table 2), and do the hyperparameter fine-tuning for the baseline methods to achieve a fair comparison (Supplementary Table 3). To better understand the representation produced by each method, we use uniform manifold approximation (UMAP) to project the internal representations into a two-dimensional space and visualize them (Figure 2B, Supplementary Figures 4–12) As shown in the figure, CLEAR learns to embed similar cells within the same clusters while separate dissimilar cells well among different clusters, and produces similar clustering results as the ground truth cell annotation. Furthermore, we evaluated the representations produced by each method with the Leiden clustering algorithms. Starting from a large resolution that the Leiden algorithm would overcluster, we decreased the resolution with a step size equal to 0.01 until it tended to undercluster, as illustrated in Supplementary Figure 13. The river plot at the turning point is shown in Figure 2C. As the PCA-based features are used as the cornerstone for experts in annotating cell types, it is not surprising that Seurat clusters match with experts’ annotation quite well. However, scDHA tends to overcluster oligodendrocyte cells and excitatory cells at the turning point. In contrast, CLEAR better represents the data by achieving nearly perfectly matching performance like Seurat. It suggested that CLEAR and Seurat embeddings have a better internal structure compared with scDHA. Although CLEAR does not access any human supervision on marker genes, it can recover the ground truth directly for this dataset, suggesting that the proposed framework can implicitly capture the data’s biological features. We also performed an ablation study and hyperparameter selection on the data augmentation operations to demonstrate the effect of the novel self-supervised contrastive learning settings, shown in Supplementary Tables 4–6.
Figure 2.

The representation from CLEAR benefits clustering and dropout event correction. (A) Clustering performance comparison of different methods on diverse datasets. On average, CLEAR improves over the second-best method, scDHA, by 4.56%, regarding ARI. (B) UMAP visualization of representations produced by CLEAR, scDHA and Seruat on the Hrvatin dataset. (C) River plots of the Hrvatin dataset. CLEAR clustering matches almost perfectly with the expert annotation, without overclustering or underclustering. (D) Clustering performance change of different methods against different artificial dropout percentages in terms of ARI.
Figure 3.

CLEAR corrects batch effects effectively. (A) Upper panel: UMAP visualization showing different methods’ performance on integrating DoubleNeg and pDC cells from two batches. Bottom panel: UMAP visualization showing different methods’ performance on separating four cell types. Notice that CLEAR’s representations also preserve the biological similarity between CD141 cells and CD1C cells. (B) The quantitative performance of different methods on batch effect removal, measured by ARI and ASW.
CLEAR corrects dropout events and batch effects effectively
Dropout events and batch effects are notorious in scRNA-seq data analysis, which should be handled properly. We next evaluate the robustness of CLEAR when encountering dropout events. We simulate the dropout effects by randomly masking non-zero entries into zero with a hypergeometric distribution. Given the additional artificial dropouts, clustering becomes much more difficult. We test the eight competing approaches together with CLEAR on the Hravtin dataset, containing 48 266 single cells with 25 187 genes and thus 1.2 billion read counts. We set 10%, 30%, 60% and 80% dropout rates for the non-zero entries, respectively (Supplementary Figure 14). High dropout rates provide more difficult conditions for the feature extraction and clustering algorithms. CLEAR achieves the best performance in handling dropout events in terms of clustering, even when 80% of the nonzero entries are masked. Although the extreme dropout events may not happen in the wet lab given the improving sequencing technologies, these artificial dropouts offer a way of evaluating the robustness of the algorithms. The performance of scDHA is similar to that of CLEAR when no dropouts are introduced, but it becomes worse when the dropout rate is 80% (Figure 2D). Except for the extreme scenario where the effectiveness is purely theoretical, CLEAR is also robust to low dropout rates. It suggests the effectiveness of CLEAR under real-world settings.
Although several methods have been proposed to correct batch effects, which are undesirable variability in the scRNA-seq datasets from technical and biological noise, most of them work as separate modules, focusing on one batch effect factor each time, and thus cannot generalize to the large complex atlas projects well, whose batch effects are from multiple factors. CLEAR, however, has the potential to model multiple batch effects in an end-to-end fashion. Here, we assess CLEAR on correcting batch effects. Specifically, we first evaluate CLEAR on the human dendritic cells dataset [46], consisting of batches with shared cell types and biologically similar but unshared cell types. The goal of the batch effect removal algorithms is to integrate common cell types while maintaining separation between highly similar cells in different batches (Methods). As shown in Figure 3A, CLEAR can separate different cell types while mixing up DoubleNeg and pDC cells from different batches. The biological similarity between CD141 cells and CD1C cells is also represented in the figure: the distance between CD141 cell cluster and CD1C cell cluster is closer than the other two clusters. On the other hand, scVI and SIMLR bring DoubleNeg and pDC cells closer but do not mix the batches well. Seurat can mitigate the batch effects in DoubleNeg and pDC cells but split CD141 cells into two clusters. ItCLUST mix up all cells, regardless of batch and cell type, suggesting that it could not handle the dataset. Harmony [47] and iMAP [48] can mix cells from different batches quite well as they are designed for batch effect removal, but the two methods tend to overfit and mix the CD141 with the CD1C cells. Although it is less balanced than CLEAR, it is much better than SIMLR and ItClust. HDMC [49], which is also a contrastive-learning-based method that is specifically designed for batch effect removal tasks, achieves a similar but less balanced performance compared with that of CLEAR.
We further quantify the performance of different methods regarding batch effect removal with two metrics, average silhouette width (ASW) and ARI, on six datasets (Datasets). We further calculate each metric in three aspects: cell type (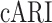 ,
, 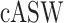 ), batch mixing (
), batch mixing (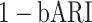 ,
, 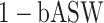 ) and the harmonic mean of the two (
) and the harmonic mean of the two (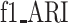 ,
, 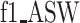 ). As shown in Figure 3B, CLEAR achieves the best balance between cell type identification and batch mixing. Furthermore, CLEAR outperforms all the other baselines under various complex batch effects settings, even though it was not designed to do so (Supplementary Figures 15–20). In particular, on the Tabular Muris Senis cell atlas, which covers the lifespan of a mouse and contains many batches, including cells from several mouses with different identities, ages, genders and from different chips, we used the bladder tissue (Supplementary Figure 15), the diaphragm tissue (Supplementary Figure 16), the limb muscle tissue (Supplementary Figure 18) and the mammary gland tissue (Supplementary Figure 19) as annotated experiments. CLEAR mixes all the cells of the same type from different batches while separating distinct cell types well.
). As shown in Figure 3B, CLEAR achieves the best balance between cell type identification and batch mixing. Furthermore, CLEAR outperforms all the other baselines under various complex batch effects settings, even though it was not designed to do so (Supplementary Figures 15–20). In particular, on the Tabular Muris Senis cell atlas, which covers the lifespan of a mouse and contains many batches, including cells from several mouses with different identities, ages, genders and from different chips, we used the bladder tissue (Supplementary Figure 15), the diaphragm tissue (Supplementary Figure 16), the limb muscle tissue (Supplementary Figure 18) and the mammary gland tissue (Supplementary Figure 19) as annotated experiments. CLEAR mixes all the cells of the same type from different batches while separating distinct cell types well.
Pseudo-time inference
Another thriving topic in scRNA-seq data analysis is pseudo-time inference, also known as trajectory inference. It aims to infer the ordering of cells along a one-dimensional manifold (pseudo-time) from the gene expression profiles. Usually, the inferring algorithms will benefit much from better data representations. Here, we evaluate whether the representation produced by CLEAR can facilitate the downstream pseudo-time inference. We use the CLEAR embeddings and the PAGA [50] algorithm to generate the pseudo-time. We compare it with state-of-the-art methods for inferring pseudo-time, Slingshot [51], SCDHA [22], SCANPY [14] and Monocle3 [52], using two mouse embryo development datasets: Yan [37] and Deng [36].
Figure 4A shows the cells from Yan dataset ordered by pseudo-time (left) and the 2D UMAP embeddings of the cells (right), generated by CLEAR. Ideally, the points in the left figure should fall on a monotonic curve, indicating the monotonically related relationship between the cells and pseudo-time. The time inferred with CLEAR is strongly correlated with the true development stages. In comparison, Monocle3 (Figure 4B) and Slingshot (Figure 4C) mixed the cells from different development stages. We also use the Spearman’s correlation coefficient to quantify the performance. CLEAR achieves the highest value (spcc = 0.971), compared with Monocle3 (spcc = 0.938) and Slingshot (spcc = 0.867). We further illustrate the cell embeddings in the 2D space with UMAP, as shown in the right figure. The smooth lines indicate the time trajectory from CLEAR (Figure 4A), Monocle3 (Figure 4B) and Slingshot (Figure 4C). The trajectories inferred by CLEAR follow the development stages precisely. It starts at the zygote, goes through 2 cells, 4 cells, 8 cells and 16 cells, and finally stops at the blast cells. However, for Monocle3, there is no clear trajectory among the cells. The cells in the early stages tend to mix, while cells in the late stages form another big group.
Figure 4.

CLEAR is helpful for pseudo-time inference. (A) CLEAR’s performance on the pseudo-time inference for the Yan dataset. Left: Cells from the Yan dataset ordered by pseudo-time inferred from CLEAR. Ideally, the points should fall on the diagonal. Right: UMAP visualization of time trajectory inferred from CLEAR. (D) CLEAR’s performance on the pseudo-time inference for the Deng dataset. (B, E) Monocle3’s performance on the pseudo-time inference for the Yan and Deng dataset. (C, F) SCANPY’s performance on the pseudo-time inference for the Yan and Deng dataset.
For the Deng dataset, we did a similar analysis (Figure 4D–F). CLEAR and Slingshot correctly reconstruct the time trajectory, where Monocle3 failed to infer the pseudo-time for 4 cells, 8 cells, 16 cells and blast. Although the Spearman’s correlation coefficient of Slingshot is marginally higher than that of CLEAR, it is worth noting that CLEAR preserved better internal structure between 8 cells and 16 cells compared with Slingshot. The results of SCANPY and scDHA are shown in Supplementary Figure 21. Taking all the experimental results together, the cell embeddings from CLEAR can facilitate the downstream algorithms in producing biologically meaningful trajectories.
CLEAR illustrates peripheral immune cells atlas and inflammatory-related mechanisms in COVID-19
To demonstrate the application potential of CLEAR on real-world biological research, we apply it to analyze a newly published COVID-19 dataset [53] (GEO accession number GSE150728), containing 44 721 cells (43 695 cells after quality control) collected from six healthy controls and seven COVID-19 samples. Four of the seven COVID samples are collected from patients with acute respiratory distress syndrome (ARDS) in clinical settings (Figure 5A, Supplementary Table 7). We perform dimensionality reduction by CLEAR and graph-based clustering, identifying 32 clusters and visualizing them via UMAP. We calculate each cluster’s differential expressed genes to annotate cell types manually (Supplementary Table 9). The cell types of monocytes (CD14+ and CD16+), T cells (CD4+ and CD8+), natural killer (NK) cells, B cells, plasmablasts, conventional dendritic cells (DCs), plasmacytoid dendritic cells (pDC), stem cell, eosinophil, neutrophil, platelets, and red blood cells are identified (Figure 5B and D, Supplementary Table 8).
Figure 5.

Peripheral immune cells atlas and inflammatory-related mechanisms in COVID-19 revealed by CLEAR. (A, B) UMAP visualization of the COVID-19 cell atlas (A) colored by COVID status and (B) colored by 13 cell type clusters (n = 43 695 cells). (C) Bar plot showing the relative percentage of different cell types comparing three COVID-19 statuses (HDs, moderate status and severe status). (D) Stacked violin plot overview of the top-important marker genes expression for each cell type. (E) UMAP visualization of the key proinflammatory cytokines expression in both CD14+ and CD16+ monocytes. (F) Heatmap of IFN-stimulated genes and MHC-related genes in CD14+ monocyte. (G, H) Boxplots showing the mean (G) MHC-related score and (H) ISG score in CD14+ monocyte colored by different COVID statuses (HDs—blue, moderate—orange and severe (ARDS)—red). (I, J) GSEA of differential expressed gene (|LogFC| > 0.25) sets between (I) moderate CD14+ monocyte and healthy donor CD14+ monocyte and (J) severe CD14+ monocyte and healthy donor CD14+ monocyte. Red represents upregulated GO biological pathway, and blue represents downregulated GO biological pathway.
To assess the general atlas of immune responses and perturbation during different COVID-19 statuses, we quantify the proportions of immune cell subsets in health donors (HDs), moderate (without ARDS) or severe COVID-19 (with ARDS) individuals (Figure 5C). Consistent with previous reports [53–55], several immune cell subsets vary between healthy donors and COVID-19 samples, and we observe a significant depletion of NK cells, DC, pDC and CD16+ monocytes. We also note an elevated frequency of plasmablasts, especially in patients with ARDS, which indicates that, together with the published clinical observations [56], acute COVID-19 response may be associated with a severe humoral immune response.
Several previous studies have shown that severe COVID-19 has been associated with dysregulated immune responses, which may be induced by the abnormal activation or suppression of inflammatory reaction [57–60]. To reveal inflammatory-related mechanisms in COVID-19, we perform transcription level analysis on monocytes in more granularity. We examine the expression level of the marker genes of the cytokine storm. We choose a set of genes from published papers, including IL1B, IL2, IL6, IL10 and TNF [61, 62], all of which encode cytokines. Consistent with recent research with deeper profiling of immune cells [53, 59], we do not find significant expression of these proinflammatory genes in monocytes (Figure 5E), suggesting that COVID-19 may also present an immune suppression status. Notably, IL2 and IL10 come from different cell populations; IL2 mainly comes from lymphocytes, but IL10 is primarily produced by monocytes, which may indicate that both monocytes and T/B cells are repressed. To further analyze transcription changes driving monocyte response remodeling in COVID-19, we conduct differential expression analysis and cellular pathway analysis by comparing COVID samples with HDs. Given that the dysregulation of CD14+ monocyte plays a more dominant role in COVID-19 progress [63], we especially investigate the transcription profile changes in CD14+ monocytes. An increased IFN-stimulated gene (ISG) set and decreased major histocompatibility complex (MHC) molecules in CD14+ monocyte compared to HDs are observed (Figure 5F, Supplementary Table 10,11). Interestingly, our method also suggests not only a decrease in the MHC II molecules in CD14+ monocyte in COVID patients, but also the MHC I (HLA-C, HLA-E, etc.) molecules decrease in monocytes. This may further tell that SARS-CoV-2 inhibits not only the expression of MHC class II genes but also MHC class I genes. Together, scoring the samples with published MHC-related genes and ISGs respectively also reveal that downregulation of MHC gene expression and upregulation of ISGs are significant in CD14+ monocytes across all the COVID patients (Figure 5G and H, Supplementary Table 12). The dominant effect of the IFN response is consistent with acute viral infection. But the suppression of MHC molecules may hinder the ability to activate lymphocytes and raise an effective anti-viral response. We then apply Gene Ontology (GO) analysis, combined with gene set enrichment analysis (GSEA), to study the biological pathway changes in CD14+ monocytes with different COVID statuses. Significant ISG upregulation in CD14+ monocyte in moderate samples is also reflected in the pathway analysis, such as type I interferon response (Figure 5I), which may indicate a more active interferon level in moderate COVID patients and have the potential to become a clinical blood test marker to monitor COVID progress. Interestingly, we also find a secretion pathway and myeloid leukocyte activation upregulation in severe samples (Figure 5J). This may suggest a dysregulated CD14+ monocytes activation in patients with ARDS. Furthermore, corresponding with our finding in Figure 5F that S100A8 and S100A9 genes are upregulated in severe samples, our method gives a potential target, S100A8/A9, to eliminate immune damage in severe COVID and thus raise patient survival rate.
CLEAR handles million-scale scRNA-seq datasets
With the unprecedented increase in sequencing scale of the recent scRNA-seq experiment platform, the ability to process million-scale single-cell sequencing datasets is increasingly essential. However, many published tools require complicated parameter setting tuning and cause burdens on the users with the split-merge process [9]. CLEAR can perform million-level dataset dimension reduction in parallel while getting rid of the tedious parameter tuning process. To test the scalability of CLEAR, we apply it to a million-level COVID PBMC scRNA-seq dataset (GEO accession number GSE158055), which contains around 1.5 million cells from COVID samples. We use CLEAR with the default parameters to conduct dimension reduction, visualizing the produced representations of the dataset with UMAP. CLEAR identifies 40 clusters in 3 h on a NVIDIA V100 Tensor Core GPU, which are then annotated manually according to each cluster’s top 100 differential expressed genes (Figure 6A). Among them, we find 13 subtypes and then plot selected marker genes for each cell type. Satisfyingly, a significant expression track of these marker genes is obtained for these subtypes (Figure 6B), which could be a solid support to the cell type labeling. Performing sensitive feature extraction while eliminating technical noise on the million-scale dataset, CLEAR is an easy-to-use and well-performed large-scale scRNA-seq data analysis tool, which has the potential to assist the construction and refinement of cell atlases.
Figure 6.

COVID-19 PBMC cell atlas based on million-scale scRNA-seq dataset. (A) UMAP embedding of PBMCs from all samples (n = 1.46 million cells) colored by manually added cell types. (B) Dot plot showing percent expression and average expression of the selected marker genes for each cell type.
DISCUSSION
scRNA-seq has become a powerful and essential tool in biological research. With the accumulated data and the emerging cell atlases, the demand for practical computational tools to process and analyze such data has never been fully satisfied. Here, we introduce such a framework, CLEAR, based on self-supervised contrastive learning. By introducing noise during training and forcing the model to pull together the representation of functionally similar cells while pushing apart dissimilar cells with a carefully designed task, we manage to train the model to produce effective representations for the single-cell profile.
The major difference of CLEAR compared with other contrastive-learning-based methods is the novel augmentation functions. While all previous methods focused on simple augmentations masking or shuffling, the inductive bias introduced through simple augmentations may hurt model generalization ability [64]. Our augmentations consist of modules derived from the genetic algorithm, which have been proven to be effective for data exploration. Instead of mapping cells into the biased embedding space invariant to masking or shuffling, CLEAR learns to construct a general embedding space that captures the invariant to the biologically meaningful signals. Through comprehensive experiments, CLEAR shows its ability as a real problem-solving tool.
Although self-supervised contrastive learning may help us learn a robust representation of the single-cell data with augmented data, it may also distort the structure of the space in such a way that differences among individual cells from the same clusters are not measurable anymore. Besides, improper augmentation steps will lead to unstable and bad embedding features. We noticed that an ideal encoder would discriminate between cells using multiple distinguishing features like gene co-expression patterns instead of simple suppressed features, and the encoder perform stable across multiple runs. It is less likely that cells from the same cluster are separated, and the clusters vary among multiple runs, given the ideal encoder. Inspired by this, we performed extensive experiments and adjusted the difficulty of the instance discrimination task, which is proven to be effective for the encoder to grab multiple features in some computer vision tasks [65, 66]. In the future, we will investigate proposing a powerful contrastive objective function considering the global features, better augmentation procedure and incorporating our prior knowledge about the data distribution into the learning framework, which helps us eliminate the noise while preserving the meaningful biological difference between different cells. In the future, CLEAR can be further developed from both the biological aspect and the machine learning aspect. CLEAR is a very flexible framework to perform data integration, regardless of the single-omics, multi-dataset integration (cell atlases construction) or multi-omics integration (e.g. the integration of scRNA-seq and scATAC-seq data). In terms of the machine learning technical details, more advanced methods to handle data imbalance, perform data augmentation and incorporate prior knowledge, such as partially labeled data, should be developed. We believe that our framework, CLEAR, will become a powerful alternative approach for single-cell data analysis.
METHODS
The CLEAR framework
The key idea of CLEAR is to learn effective cell representations, considering noise in the data, and to pull together the representation of functionally similar cells, while pushing apart dissimilar cells. We achieved the goal with self-supervised contrastive learning. Given the single-cell gene expression profile, we added different simulated noise, such as Gaussian noise and simulated dropout events, to it (data augmentation), resulting in distorted profiles (augmented data). The two distorted profiles from the same cell are considered positive pairs, while the profiles from different cells are negative pairs. When training the model, we forced the model to produce similar representations for the positive pairs while distinct for the negative pairs (contrastive learning). More specifically, by discriminating the positive pairs from a large number of negatives, CLEAR learns a locally smooth nonlinear mapping function 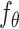 that pulls together multiple distortions of a cell in the embedding space and pushes away the other samples. In the transformed space, cells with similar expression patterns form clusters, which are likely to be cells of the same cell types. The function
that pulls together multiple distortions of a cell in the embedding space and pushes away the other samples. In the transformed space, cells with similar expression patterns form clusters, which are likely to be cells of the same cell types. The function 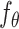 is parameterized by a deep neural network, whose parameters can be optimized in an end-to-end manner. The detailed workflows are as follows.
is parameterized by a deep neural network, whose parameters can be optimized in an end-to-end manner. The detailed workflows are as follows.
(i) Data augmentation. We first performed data augmentation to generate training pairs. Each cell will have two augmented versions, and thus a minibatch of 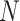 cells is augmented to
cells is augmented to 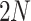 cells. This step will be discussed in detail in Supplementary Method 6.
cells. This step will be discussed in detail in Supplementary Method 6.
(ii) Constructing negative labels with data from multiple minibatches. For data in one minibatch, we can consider the two data points generated from the same gene expression profile as a positive pair while the other combinations as negatives. To make the locally smooth function 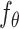 have a global effect, we used negatives from other minibatches. We achieved that by maintaining a queue with data from multiple minibatches. When the current minibatch is enqueued, the oldest minibatch will be dequeued. Within the queue, a specific distorted profile only has one positive pair match, while all the other profiles are negatives for it.
have a global effect, we used negatives from other minibatches. We achieved that by maintaining a queue with data from multiple minibatches. When the current minibatch is enqueued, the oldest minibatch will be dequeued. Within the queue, a specific distorted profile only has one positive pair match, while all the other profiles are negatives for it.
(iii) Loss function. Let 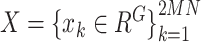 be the queue consisting of a number of gene expression profiles, where
be the queue consisting of a number of gene expression profiles, where 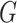 denotes the number of genes;
denotes the number of genes; 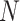 stands for the batch size;
stands for the batch size; 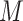 stands for the number of batches stored in the queue. In one batch,
stands for the number of batches stored in the queue. In one batch, 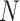 samples are augmented into
samples are augmented into 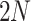 samples. Consequently, the queue consisting of
samples. Consequently, the queue consisting of 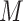 minibatches contains
minibatches contains 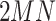 augmented samples.
augmented samples. 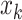 denotes the
denotes the 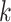 -th (distorted) cell embeddings in the queue. For a pair of positive samples
-th (distorted) cell embeddings in the queue. For a pair of positive samples 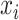 and
and 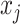 (derived from one original sample), the other
(derived from one original sample), the other 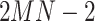 samples are treated as negatives. To distinguish the positive pair from the negatives, we use the following pairwise contrastive InfoNCE loss:
samples are treated as negatives. To distinguish the positive pair from the negatives, we use the following pairwise contrastive InfoNCE loss:
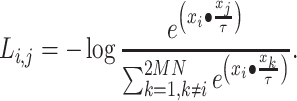 |
(1) |
Note that 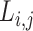 is asymmetrical. Suppose we put all the pairs in an order, such that
is asymmetrical. Suppose we put all the pairs in an order, such that 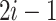 and
and 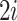 denote the paired augmentations, then the summed-up loss is:
denote the paired augmentations, then the summed-up loss is:
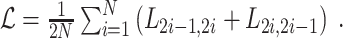 |
(2) |
(iv). Momentum update. As suggested by He et al. [67, 68], a rapidly changing encoder network will reduce the representations’ consistency, resulting in poor performance. To deal with the problem, we utilize two encoders: a slow-evolving key encoder 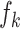 and a fast-evolving query encoder
and a fast-evolving query encoder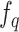 . Denoting the parameters of
. Denoting the parameters of 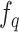 as
as 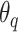 and those of
and those of 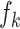 as
as 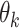 , we updated the query encoder by the normal backpropagation. For the key encoder, we updated it with momentum, which helps the model update in a consistent direction. Each time the key encoder is updated with a much smaller step: by taking a linear combination of the previous key encoder parameters and the newly computed query encoder parameters, we kept the information from previous steps.
, we updated the query encoder by the normal backpropagation. For the key encoder, we updated it with momentum, which helps the model update in a consistent direction. Each time the key encoder is updated with a much smaller step: by taking a linear combination of the previous key encoder parameters and the newly computed query encoder parameters, we kept the information from previous steps.
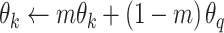 |
(3) |
Here 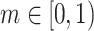 is a momentum coefficient. A large
is a momentum coefficient. A large 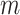 makes the key encoder updates slowly, while small m forces the key encoder to become much like the query encoder. The momentum update makes the encoder network evolve smoothly.
makes the key encoder updates slowly, while small m forces the key encoder to become much like the query encoder. The momentum update makes the encoder network evolve smoothly.
(v). Inference. After we trained the model, the query encoder network 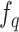 is the final productive network, which outputs the representation (a 128-d vector) of a single cell gene expression profile. After obtaining the representations of all the cells in a dataset, we clustered the cells with the common clustering algorithms (e.g. k-means algorithm, Louvain algorithm and Leiden algorithm). Finally, cell types are assigned to the discovered clusters based on the differential expression genes in the cluster.
is the final productive network, which outputs the representation (a 128-d vector) of a single cell gene expression profile. After obtaining the representations of all the cells in a dataset, we clustered the cells with the common clustering algorithms (e.g. k-means algorithm, Louvain algorithm and Leiden algorithm). Finally, cell types are assigned to the discovered clusters based on the differential expression genes in the cluster.
Architecture and hyperparameters
The encoder neural network in CLEAR consists of two fully connected layers. The query encoder and the key encoder share the same architecture. The first layer has 1024 nodes, while the second layer has 128 nodes. The 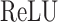 function, defined as
function, defined as 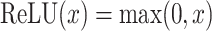 , is used as the nonlinear activation function after the linear transformation. We used Adam optimizer with the learning rate as 1 and the cosine learning schedule. We trained the paired neural networks for 200 epochs. Temperature,
, is used as the nonlinear activation function after the linear transformation. We used Adam optimizer with the learning rate as 1 and the cosine learning schedule. We trained the paired neural networks for 200 epochs. Temperature, 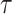 , in the CLEAR’s objective function, is set to be 0.2. The momentum coefficient
, in the CLEAR’s objective function, is set to be 0.2. The momentum coefficient 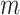 is 0.999. The hyperparameters are determined using grid search with cross-validation.
is 0.999. The hyperparameters are determined using grid search with cross-validation.
Data augmentation
Data augmentation is critical to the success of self-supervised contrastive learning. We use the following ways of data augmentation, considering noise during real experiments. Note that the augmentations are performed in a specific order (as shown below). Not all the steps will be certainly conducted, with each step having a probability of being chosen or dropped.
(i) Random mask. We randomly replaced some gene expression values with zero in the profile of the target cell. The mask percentage is 0.2, while the probability of executing the step is 0.5. Notice that this synthetic noise is similar to the dropout events in the single-cell sequencing experiments.
(ii) Gaussian noise. We adopted the idea from limma [69], used the linear regression model to model the scRNA-seq data, with a Gaussian distribution to model the noise, and then generated augmentations from the Gaussian distribution parameters. We randomly modified some gene expression values in the target cell profile by adding numbers drawn from the Gaussian distribution. To speed up the entire pipeline and prevent overfitting, we did not fit an independent regression model per gene; instead, we set a predefined Gaussian distribution according to the experimental result. The noise percentage is 0.8. The mean of Gaussian distribution is 0, while the SD is 0.2. The probability of executing this step is 0.5.
(iii) Random swap. For a gene expression profile, we randomly chose an even number of gene expression values and constructed pairs from the subset and then swapped the gene expression values inside each pair. The total percentage that performs swapping is 0.1. The probability of execution is 0.5.
(iv) Crossover with another cell. Inspired by the genetic algorithm, we used the crossover operation to generate efficient distorted profiles. We randomly chose another cell in the dataset as the crossover source and then selected some genes from the target gene expression profile, swapping the gene expression value between the two cells. 25% of the gene expression data in one cell will be exchanged with the other cell. The probability of executing this step is 0.5. This exchanging step will not influence the next batch or the next training epoch. Typically, this step could help batch mixing as it pushes the distorted swapped profiles with the raw profiles together. With a small crossover percentage (25%), the model learns the dominant features between the generations while ignoring the side effect features.
(v) Crossover with many cells. We randomly chose several cells in the dataset as the crossover source and some genes from the target gene expression profile and swapped the expression values between the source cell and the target cells. The 25% of the gene expression data in the cell will be exchanged with the selected cells. The probability of execution is 0.5. This step would not influence the next batch or the next training epoch.
Key Points
CLEAR is a self-supervised contrastive learning-based approach that produces effective representations for the scRNA-seq data.
By creating positive pairs through data augmentation that simulates different noise (batch effect, technical noise, dropout), the novel contrastive loss will encourage cells with same cell type to be clustered together despite the noise in the data.
CLEAR outperforms existing methods on popular tasks such as clustering, batch effect removal and dropout correction, and trajectory inference.
The representations of CLEAR enable the downstream analysis. We apply CLEAR on a COVID-19 dataset for identification of differential expressed genes between patients under different severity of COVID for potential target genes in therapeutics.
Supplementary Material
Acknowledgements
We are thankful to all members of the SFB group for kind discussions.
Author Biographies
Wenkai Han is a PhD candidate in computer science at King Abdullah University of Science and Technology (KAUST). His research interest focuses on computational biology and machine learning.
Yuqi Cheng is a PhD student in computational science and engineering at Georgia Institute of Technology. He received his master's degree at Weill Cornell Medicine. His research interest includes bioinformatics and scRNA-seq data analysis.
Jiayang Chen is a visiting student in computer science and engineering at The Chinese University of Hong Kong (CUHK). He is interested in deep learning and its application.
Huawen Zhong is a PhD candidate in bioscience at King Abdullah University of Science and Technology (KAUST). Her major research interests include bioinformatics and deep learning techniques.
Zhihang Hu is a PhD candidate in computer science and engineering at the Chinese University of Hong Kong (CUHK). His research interests include bioinformatics and artificial intelligence.
Siyuan Chen is a PhD candidate in computer science at King Abdullah University of Science and Technology (KAUST). His research interests include the intersection between computer science and biology.
Licheng Zong is a PhD candidate in computer science and engineering at The Chinese University of Hong Kong (CUHK). His research interests include the intersection between deep learning and bioinformatics.
Liang Hong is a PhD candidate in computer science and engineering at The Chinese University of Hong Kong (CUHK). His research interests include machine learning, data analysis and its applications in bioinformatics.
Ting-Fung Chan is a professor at School of Life Science, the Chinese University of Hong Kong (CUHK). His research group focuses on RNomics and bioinformatics in biological processes and diseases.
Irwin King is the chairman and professor of Department of Computer Science and Engineering, The Chinese University of Hong Kong (CUHK). His research group focuses on machine learning, social computing, AI, web intelligence, data mining and multimedia information processing.
Xin Gao is a professor of Computer Science at King Abdullah University of Science and Technology (KAUST), Saudi Arabia. He is the Associate Director of the Computational Bioscience Research Center (CBRC) and Deputy Director of the Smart Health Intiative (SHI) at KAUST. His research focuses at the intersection between AI and biology.
Yu Li is an assistant professor at the Department of Computer Science and Engineering, The Chinese University of Hong Kong (CUHK). His research group focuses on the intersection between machine learning, health care and bioinformatics and developing novel machine learning methods to resolve the computational problems in biology and health care, especially the structured learning problems.
Contributor Information
Wenkai Han, Computer, Electrical and Mathematical Sciences and Engineering Division (CEMSE), Computational Bioscience Research Center (CBRC), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955, Saudi Arabia.
Yuqi Cheng, Department of Computer Science and Engineering (CSE), The Chinese University of Hong Kong (CUHK), Hong Kong SAR, China; Weill Cornell Graduate School of Medical Sciences, Weill Cornell Medicine, New York, NY, 10065, USA.
Jiayang Chen, Department of Computer Science and Engineering (CSE), The Chinese University of Hong Kong (CUHK), Hong Kong SAR, China.
Huawen Zhong, Biological and Environmental Sciences & Engineering Division (BESE), Red Sea Research Center (RSRC), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955, Saudi Arabia.
Zhihang Hu, Department of Computer Science and Engineering (CSE), The Chinese University of Hong Kong (CUHK), Hong Kong SAR, China.
Siyuan Chen, Computer, Electrical and Mathematical Sciences and Engineering Division (CEMSE), Computational Bioscience Research Center (CBRC), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955, Saudi Arabia.
Licheng Zong, Department of Computer Science and Engineering (CSE), The Chinese University of Hong Kong (CUHK), Hong Kong SAR, China.
Liang Hong, Department of Computer Science and Engineering (CSE), The Chinese University of Hong Kong (CUHK), Hong Kong SAR, China.
Ting-Fung Chan, School of Life Sciences and State Key Laboratory of Agrobiotechnology, The Chinese University of Hong Kong, Hong Kong SAR, China.
Irwin King, Department of Computer Science and Engineering (CSE), The Chinese University of Hong Kong (CUHK), Hong Kong SAR, China.
Xin Gao, Computer, Electrical and Mathematical Sciences and Engineering Division (CEMSE), Computational Bioscience Research Center (CBRC), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955, Saudi Arabia; BioMap, Beijing, China.
Yu Li, Department of Computer Science and Engineering (CSE), The Chinese University of Hong Kong (CUHK), Hong Kong SAR, China; The CUHK Shenzhen Research Institute, Hi-Tech Park, Nanshan, Shenzhen, 518057, China.
Data availability
We used 10 datasets for evaluating the performance of clustering and dropouts, one dataset for benchmarking the batch effects removal. Two COVID-PBMC datasets for case study. The detailed information and the links to the publicly available sources of the 13 datasets can be found in the Supplementary Data part. An open-source implementation of CLEAR is available at GitHub: https://github.com/ml4bio/CLEAR, under the MIT license.
Authors’ contributions
Y.L. and W.H. conceived and designed the approach. W.H., J.C., Y.C., H.Z. and Z.H. performed research, conducted analyses, contributed the experiments, developed the metrics and create figures. S.C. and L.Z. drew Figure 1. Y.L. and X.G. supervised the research and the entire project. Y.L., W.H., Y.C. and I.K. contributed to the manuscript. All authors reviewed the manuscript.
Funding
King Abdullah University of Science and Technology FCC/1/1976-44-01, FCC/1/1976-45-01, REI/1/4742-01-01, REI/1/5202-01-01, and REI/1/4940-01-01; Chinese University of Hong Kong (4937025, 4937026, 5501517 and 5501329).
References
- 1. Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet 2013;14:618–30. [DOI] [PubMed] [Google Scholar]
- 2. Shalek AK, Satija R, Shuga J, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014;510:363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maynard A, McCoach CE, Rotow JK, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell 2020;182:1232, e1222–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galen P, Hovestadt V, Wadsworth Ii MH, et al. Single-cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell 2019;176:1265, e1224–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tian T, Zhang J, Lin X, et al. Model-based deep embedding for constrained clustering analysis of single cell RNA-seq data. Nat Commun 2021;12:1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet 2015;16:133–45. [DOI] [PubMed] [Google Scholar]
- 7. Kiselev VY, Andrews TS, Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet 2019;20:273–82. [DOI] [PubMed] [Google Scholar]
- 8. Lopez R, Regier J, Cole MB, et al. Deep generative modeling for single-cell transcriptomics. Nat Methods 2018;15:1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deng Y, Bao F, Dai QH, et al. Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Nat Methods 2019;16:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Satija R, Farrell JA, Gennert D, et al. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 2015;33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kiselev VY, Kirschner K, Schaub MT, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods 2017;14:483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin PJ, Troup M, Ho JWK. CIDR: ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol 2017;18:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo M, Wang H, Potter SS, et al. SINCERA: a pipeline for single-cell RNA-Seq profiling analysis. PLoS Comput Biol 2015;11:e1004575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 2018;19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levine JH, Simonds EF, Bendall SC, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 2015;162:184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang B, Zhu J, Pierson E, et al. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat Methods 2017;14:414–6. [DOI] [PubMed] [Google Scholar]
- 17. Park S, Zhao HY. Spectral clustering based on learning similarity matrix. Bioinformatics 2018;34:2069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vans E, Patil A, Sharma A. FEATS: feature selection-based clustering of single-cell RNA-seq data. Brief Bioinform 2020;22:bbaa306. [DOI] [PubMed] [Google Scholar]
- 19. Butler A, Hoffman P, Smibert P, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dijk DV, Sharma R, Nainys J, et al. Recovering gene interactions from single-cell data using data diffusion. Cell 2018;174:716–729.e727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eraslan G, Simon LM, Mircea M, et al. Single-cell RNA-seq denoising using a deep count autoencoder. Nat Commun 2019;10:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tran D, Nguyen H, Tran B, et al. Fast and precise single-cell data analysis using a hierarchical autoencoder. Nat Commun 2021;12:1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu J, Li X, Hu G, et al. Iterative transfer learning with neural network for clustering and cell type classification in single-cell RNA-seq analysis. Nat Mach Intell 2020;2:607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang J, Agarwal D, Huang M, et al. Data denoising with transfer learning in single-cell transcriptomics. Nat Methods 2019;16:875–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang JX, Ma AJ, Chang YZ, et al. scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses. Nat Commun 2021;12:1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li XJ, Wang K, Lyu YF, et al. Deep learning enables accurate clustering with batch effect removal in single-cell RNA-seq analysis. Nat Commun 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ding JR, Condon A, Shah SP. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat Commun 2018;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ding J, Regev A. Deep generative model embedding of single-cell RNA-Seq profiles on hyperspheres and hyperbolic spaces. Nat Commun 2021;12:2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pierson E, Yau C. ZIFA: dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol 2015;16:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Risso D, Perraudeau F, Gribkova S, et al. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat Commun 2018;9:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brbic M, Zitnik M, Wang S, et al. MARS: discovering novel cell types across heterogeneous single-cell experiments. Nat Methods 2020;17:1200–6. [DOI] [PubMed] [Google Scholar]
- 32. Xu Y, Das P, McCord RP. SMILE: mutual information learning for integration of single-cell omics data. Bioinformatics 2022;38:476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ciortan M, Defrance M. Contrastive self-supervised clustering of scRNA-seq data. BMC Bioinform 2021;22:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen T, Kornblith S, M Net al. A simple framework for contrastive learning of visual representations. In: International conference on machine learning. 2020, (pp. 1597–1607). PMLR.
- 35. Eiben AE, Raué PE, Ruttkay Z. Genetic algorithms with multi-parent recombination. In: Parallel Problem Solving from Nature — PPSN III. Berlin, Heidelberg: Springer, 1994, 78–87. [Google Scholar]
- 36. Deng Q, Ramsköld D, Reinius B, et al. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014;343:193–6. [DOI] [PubMed] [Google Scholar]
- 37. Yan L, Yang M, Guo H, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol 2013;20:1131–9. [DOI] [PubMed] [Google Scholar]
- 38. Pollen AA, Nowakowski TJ, Shuga J, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol 2014;32:1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kolodziejczyk AA, Kim JK, Tsang JC, et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 2015;17:471–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muraro MJ, Dharmadhikari G, Grün D, et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst 2016;3:385, e383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hrvatin S, Hochbaum DR, Nagy MA, et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neurosci 2018;21:120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Consortium TM . A single cell transcriptomic atlas characterizes aging tissues in the mouse. Nature 2020;583:590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mondal AK, Asnani H, Singla P, et al. scRAE: deterministic regularized autoencoders with flexible priors for clustering single-cell gene expression data. IEEE/ACM Transactions on Computational Biology and Bioinformatics 2021. [DOI] [PubMed]
- 44. Ciortan M, MJBB D. Contrastive self-supervised clustering of scRNA-seq data. BMC bioinformatics 2021;22:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wan H, Chen L, Deng MJB. scNAME: neighborhood contrastive clustering with ancillary mask estimation for scRNA-seq data. Bioinformatics 2022;38:1575–83. [DOI] [PubMed] [Google Scholar]
- 46. Tran HTN, Ang KS, Chevrier M, et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol 2020;21:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 2019;16:1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang D, Hou S, Zhang L, et al. iMAP: integration of multiple single-cell datasets by adversarial paired transfer networks. Genome Biol 2021;22:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X, Wang J, Zhang H, et al. HDMC: a novel deep learning-based framework for removing batch effects in single-cell RNA-seq data. Bioinformatics 2021;38:1295–303. [DOI] [PubMed] [Google Scholar]
- 50. Wolf FA, Hamey FK, Plass M, et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol 2019;20:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Street K, Risso D, Fletcher RB, et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 2018;19:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilk AJ, Rustagi A, Zhao NQ, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 2020;26:1070–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuri-Cervantes L, Pampena MB, Meng W, et al. Immunologic perturbations in severe COVID-19/SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar]
- 55. Kuri-Cervantes L, Pampena MB, Meng W, et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol 2020;5:eabd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao J, Yuan Q, Wang H, et al. Antibody responses to SARS-CoV-2 in patients with novel coronavirus disease 2019. Clin Infect Dis 2020;71:2027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Choudhary S, Sharma K, Silakari O. The interplay between inflammatory pathways and COVID-19: a critical review on pathogenesis and therapeutic options. Microb Pathog 2021;150:104673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol 2021;93:250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schulte-Schrepping J, Reusch N, Paclik D, et al. Suppressive myeloid cells are a hallmark of severe COVID-19. medRxiv 2020; 2020.2006.2003.20119818. [Google Scholar]
- 60. Unterman A, Sumida TS, Nouri N, et al. Single-cell omics reveals dyssynchrony of the innate and adaptive immune system in progressive COVID-19. medRxiv 2020; 2020.2007.2016.20153437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guo C, Li B, Ma H, et al. Single-cell analysis of two severe COVID-19 patients reveals a monocyte-associated and tocilizumab-responding cytokine storm. Nat Commun 2020;11:3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ragab D, Salah Eldin H, Taeimah M, et al. The COVID-19 cytokine storm; what we know so far. Front Immunol 2020;11:1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schulte-Schrepping J, Reusch N, Paclik D, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 2020;182:1419, e1423–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xiao T, Wang X, Efros AA, et al. What should not be contrastive in contrastive learning. arXiv preprint arXiv:2008.05659 (2020).
- 65. Robinson J, Chuang C-Y, Sra S, et al. Contrastive learning with hard negative samples. arXiv preprint arXiv 2010; 04592 2020. [Google Scholar]
- 66. Kalantidis Y, Sariyildiz MB, Pion N, et al. Hard negative mixing for contrastive learning. arXiv preprint arXiv 2010; 01028 2020. [Google Scholar]
- 67. Chen X, Fan H, Girshick R, et al. Improved baselines with momentum contrastive learning. arXiv preprint arXiv 2003; 04297 2020. [Google Scholar]
- 68. He K, Fan H, Wu Yet al. Momentum contrast for unsupervised visual representation learning. In: Proceedings of the IEEE/CVF conference on computer vision and pattern recognition. Seattle, WA, USA: IEEE, 2020, p. 9729–38.
- 69. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We used 10 datasets for evaluating the performance of clustering and dropouts, one dataset for benchmarking the batch effects removal. Two COVID-PBMC datasets for case study. The detailed information and the links to the publicly available sources of the 13 datasets can be found in the Supplementary Data part. An open-source implementation of CLEAR is available at GitHub: https://github.com/ml4bio/CLEAR, under the MIT license.