

ABSTRACT
Moxifloxacin is central to treatment of multidrug-resistant tuberculosis. Effects of moxifloxacin on the Mycobacterium tuberculosis redox state were explored to identify strategies for increasing lethality and reducing the prevalence of extensively resistant tuberculosis. A noninvasive redox biosensor and a reactive oxygen species (ROS)-sensitive dye revealed that moxifloxacin induces oxidative stress correlated with M. tuberculosis death. Moxifloxacin lethality was mitigated by supplementing bacterial cultures with an ROS scavenger (thiourea), an iron chelator (bipyridyl), and, after drug removal, an antioxidant enzyme (catalase). Lethality was also reduced by hypoxia and nutrient starvation. Moxifloxacin increased the expression of genes involved in the oxidative stress response, iron-sulfur cluster biogenesis, and DNA repair. Surprisingly, and in contrast with Escherichia coli studies, moxifloxacin decreased expression of genes involved in respiration, suppressed oxygen consumption, increased the NADH/NAD+ ratio, and increased the labile iron pool in M. tuberculosis. Lowering the NADH/NAD+ ratio in M. tuberculosis revealed that NADH-reductive stress facilitates an iron-mediated ROS surge and moxifloxacin lethality. Treatment with N-acetyl cysteine (NAC) accelerated respiration and ROS production, increased moxifloxacin lethality, and lowered the mutant prevention concentration. Moxifloxacin induced redox stress in M. tuberculosis inside macrophages, and cotreatment with NAC potentiated the antimycobacterial efficacy of moxifloxacin during nutrient starvation, inside macrophages, and in mice, where NAC restricted the emergence of resistance. Thus, NADH-reductive stress contributes to moxifloxacin-mediated killing of M. tuberculosis, and the respiration stimulator (NAC) enhances lethality and suppresses the emergence of drug resistance.
KEYWORDS: antimycobacterial, ROS, oxidative stress, moxifloxacin, fluoroquinolone, respiration, N-acetyl cysteine, redox biosensor, NADH, reductive stress, resistance
INTRODUCTION
Antimicrobial resistance is a growing problem for the management of tuberculosis (TB). For example, between 2009 and 2016, the number of global cases of multidrug-resistant tuberculosis (MDR-TB), defined as resistance of Mycobacterium tuberculosis to at least rifampicin and isoniazid, increased annually by over 20% (1). Tuberculosis with additional resistance to a fluoroquinolone and at least one of three injectable agents (kanamycin, amikacin, or capreomycin), termed extensively drug-resistant tuberculosis (XDR-TB), accounted for about 6% of MDR-TB cases in 2018 (2). Alarmingly, the annual XDR-TB cases reported worldwide increased almost 10-fold between 2011 and 2018 (2, 3). Increased prevalence of XDR-TB is not surprising, since the fluoroquinolone-containing combination therapies used to halt progression to XDR-TB utilized early quinolones (ciprofloxacin, ofloxacin, and sparfloxacin) that are only modestly effective anti-TB agents (4).
Efforts to find more active fluoroquinolones led to C-8 methoxy derivatives that exhibit improved ability to kill M. tuberculosis in vitro (5). Two of these compounds, moxifloxacin and gatifloxacin, have been examined as anti-TB agents (6–8). Preclinical studies with moxifloxacin in murine TB models demonstrate effective treatment with reduced relapse frequency as well as treatment-shortening properties (9). Indeed, single-drug clinical studies indicate that the early bactericidal activity of moxifloxacin is similar to that of first-line anti-TB drugs, such as isoniazid and rifampicin. Thus, moxifloxacin could be part of effective multidrug combination treatments to shorten therapy time (10, 11). Shorter treatment could increase treatment compliance, further limiting the progression of MDR- to XDR-TB. However, multiple phase III clinical trials have failed to corroborate the preclinical efficacy data, as moxifloxacin did not shorten treatment time (12–14). One approach for increasing moxifloxacin efficacy is to find ways to increase its lethal action.
The fluoroquinolones have two mechanistically distinct antibacterial effects: (i) they block growth by forming reversible drug-gyrase-DNA complexes that rapidly inhibit DNA synthesis, and (ii) they kill cells (15). Death appears to arise in two ways, one through chromosome fragmentation and the other through accumulation of reactive oxygen species (ROS) (15). The latter appear to dominate when DNA repair is proficient (16, 17). We are particularly interested in fluoroquinolone lethality, because M. tuberculosis possesses a remarkable ability to evade host immune pressures (18). In the absence of an effective immune response, bactericidal drugs are essential for clearing infection quickly. If the redox-based mechanisms of fluoroquinolone lethality seen with Escherichia coli extend to M. tuberculosis, opportunities may exist for increasing ROS and lethality, thereby reducing treatment duration and increasing the cure rate for MDR-TB.
Several methods are available for assessing the contribution of ROS to bactericidal activity. Among these are direct detection of ROS levels using ROS-sensitive dyes, characterization of mutants known to alter ROS levels, and examination of the effects of agents known to suppress ROS accumulation. The present work adds detection of antibiotic-induced changes in redox physiology of M. tuberculosis using a noninvasive, genetic biosensor (Mrx1-roGFP2) (19). The sensor measures the redox potential (EMSH) of a major mycobacterial thiol buffer, mycothiol, thereby assessing oxidative effects independently of radical-sensitive fluorescent dye methods whose interpretation has been debated (20, 21). Mrx1-roGFP2 is well suited for this work, as it has been used to measure EMSH of M. tuberculosis during in vitro growth, infection of macrophages, and exposure to nonquinolone antimicrobials (19, 22–24).
In the present work, we began by asking how well M. tuberculosis fits the paradigm for ROS-mediated killing by antimicrobials as developed from studies using E. coli. We used moxifloxacin as a lethal stressor, because the fluoroquinolones are among the better-understood agents and because moxifloxacin is potentially important for tuberculosis control. M. tuberculosis deviated markedly from E. coli by suppressing respiration rather than increasing it during fluoroquinolone-mediated stress. That raised questions about the source of ROS accumulation, since in E. coli, ROS levels correlate with increased, not decreased, respiration. We found that an increase in NADH (reductive stress) accounted for the increase in ROS and lethality. When we artificially raised respiration, ROS levels increased, and we observed enhanced moxifloxacin lethality in vitro, inside macrophages, and in a murine model of infection. Raising respiration by N-acetyl cysteine (NAC) treatment also reduced the emergence of resistance. Thus, the work provides a mechanistic basis for developing respiration enhancers to increase the lethal action of moxifloxacin and perhaps other anti-TB agents.
RESULTS
Moxifloxacin-mediated lethality associated with oxidative stress during aerobic growth.
To obtain a reference point for M. tuberculosis susceptibility to moxifloxacin, we determined MIC using the resazurin microtiter assay (REMA) (25). The MIC of moxifloxacin ranged from 0.125 μM to 0.5 μM. The MICs were similar for H37Rv and several drug-resistant strains (see Table S1 in the supplemental material; similarity in MICs was also seen for levofloxacin and ciprofloxacin with different M. tuberculosis strains). Thus, strain H37Rv appeared to be representative and appropriate for subsequent experiments.
As an initial probe for moxifloxacin-induced oxidative stress, we examined a transformant of M. tuberculosis strain H37Rv that expresses the redox biosensor Mrx1-roGFP2 from a plasmid (strain Mtb-roGFP2). This biosensor reports the redox state of the mycothiol redox couple (reduced mycothiol [MSH]/oxidized mycothiol [MSSM]) in the bacterial cytosol (Fig. 1A) (19). Exposure of Mtb-roGFP2 to hydrogen peroxide (H2O2) led to rapid (5 min) oxidation of the biosensor in a concentration-dependent manner (Fig. S1). A 2-fold increase in the biosensor ratio is equivalent to the oxidative stress inside M. tuberculosis following treatment with 500 μM H2O2 (Fig. S1).
FIG 1.

Effect of moxifloxacin on biosensor oxidation, ROS level, and bacterial survival. (A) Moxifloxacin increases oxidative stress, which increases the ratio of oxidized (MSSM) to reduced (MSH) mycothiol via mycothiol-dependent peroxiredoxin (Prx) or peroxidase (Mpx). Oxidation of Mrx1-roGFP2 increases fluorescence intensity for excitation at ~400 nm and decreases it for excitation at ~490 nm. (B) Strain M. tuberculosis roGFP2 was exposed to the indicated concentrations of moxifloxacin (1× MIC = 0.5 μM) for 48 h, and the ratiometric response of the biosensor and survival following treatment were determined. (C) Cells were treated as for panel B, and ROS were quantified by flow cytometry using CellROX deep red dye. Cumene hydroperoxide (CHP; 10 mM) served as a positive control. Data represent mean fluorescence intensity of the dye. (D) Exponentially growing M. tuberculosis H37Rv was treated with moxifloxacin at the indicated concentrations for the indicated times; survival was assessed by determining CFU. (E) Time-kill curves for M. tuberculosis treated with 10× MIC of moxifloxacin. Error bars represent standard deviations from the mean. Data represent at least two independent experiments performed in at least duplicate. Statistical significance was calculated against the no-treatment control (****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05).
Increasing moxifloxacin concentration, up to 2.5× MIC (1× MIC = 0.5 μM) during a 48-h incubation, increased the biosensor ratiometric signal by almost 2-fold (Fig. 1B; also, see Fig. S2A and C), indicating elevated levels of MSSM. Above 2.5× MIC, MSSM levels appeared to plateau. Measurement of ROS with CellROX deep red showed a similar increase to 2.5× MIC of moxifloxacin, followed by a plateau of mean fluorescence intensity (Fig. 1C). Moxifloxacin treatment damaged DNA and oxidized lipids (Fig. S3), indicating that drug-induced oxidative stress kills M. tuberculosis by degrading essential biomolecules.
Maximum bacterial killing was observed at 10× MIC, a concentration at which survival decreased by ~2 log10-fold at treatment day 1 and by 6 log10-fold at treatment day 10 (Fig. 1D and E). Survival dropped in a concentration-dependent manner, with a minimum being reached at a higher concentration than observed for maximal oxidative stress (10× MIC) (Fig. 1B to D). The discordance between the biosensor signal plateauing at 2.5× MIC and killing continuing to 10× MIC (Fig. 1B) could be explained by the existence of two lethal mechanisms. One mechanism, chromosome fragmentation (8, 26), likely dominates at high quinolone concentrations, with an ROS-based mechanism dominating at lower concentrations. Indeed, for norfloxacin treatment of E. coli, oxidative effects on killing are seen only at low to moderate drug concentrations (27). As expected, oxidative stress was not observed in M. tuberculosis treated with subinhibitory concentrations of moxifloxacin (Fig. S2A). The increase in biosensor signal was observed as early as 12 h after initiation of moxifloxacin treatment (half the doubling time of untreated control cells); it then increased significantly at 48 h (Fig. S2A and B). Thus, ROS production precedes and contributes to the death of M. tuberculosis. As expected, ROS levels in a moxifloxacin-resistant isolate of M. tuberculosis remained low upon treatment with moxifloxacin (Fig. S4A).
We also compared the ability of a weakly effective fluoroquinolone (ciprofloxacin; minimal bactericidal concentration [MBC] = 2 μM; Table S2), a moderately effective compound (levofloxacin, MBC = 1 μM), and moxifloxacin (MBC = 0.5 μM) to oxidize the biosensor, all at 2.5-fold MBC. Moxifloxacin induced biosensor oxidation after treatment for 12 h and 24 h (Fig. S4B). In contrast, ciprofloxacin did not induce biosensor oxidation, and levofloxacin triggered oxidative stress only after a 24-h treatment (Fig. S4B). Thus, ROS levels do not correlate with MBC, a parameter obtained after a long incubation (see Discussion). The more active quinolones are also more bactericidal at the same fold MIC for several bacterial species, including mycobacteria (5, 28, 29). These data corroborate the link between oxidative stress and moxifloxacin lethality.
High-dose chemotherapy is postulated to slow the emergence of drug resistance if the drug concentration is above the mutant prevention concentration (MPC) (30). However, bacterial strains can paradoxically show elevated survival levels at extremely high drug concentrations. In the case of nalidixic acid at very high concentration, E. coli survival can be 100% (31). Similarly, M. tuberculosis survival increased dramatically at high moxifloxacin concentration (Fig. 1D).
Some aspects of high-concentration survival are discordant when E. coli and M. tuberculosis are compared. For example, with E. coli, this phenotype of high-concentration survival is associated with reduced ROS levels (31); with M. tuberculosis, the biosensor and CellROX signals remained high at high levels of moxifloxacin (Fig. 1B and C). This difference between the organisms, which is unexplained, encouraged further comparisons.
Reduction of moxifloxacin-mediated killing of M. tuberculosis by ROS-mitigating agents.
Work with E. coli supports the idea that ROS contribute to quinolone-mediated killing of bacteria (15). For example, superoxide (O2·−) damages Fe-S clusters and increases the free iron (Fe) pool (32), which may then drive the generation of toxic hydroxyl radical (HO·) via the Fenton reaction (26, 33). To examine the effects of Fe, we assessed ROS levels in M. tuberculosis grown in minimal medium under Fe-deficient and Fe-excess conditions. Fe overload raised ROS levels (Fig. S5). Treatment with thiourea (TU), a thiol-based scavenger of ROS, reversed the Fe-induced ROS increase (Fig. S5). During moxifloxacin treatment of M. tuberculosis, a concentration-dependent increase in free Fe occurs (Fig. S6). Pretreatment with thiourea at the nontoxic concentration of 10 mM (34) increased survival of M. tuberculosis by almost 10-fold during cotreatment with moxifloxacin (1× to 10× MIC) (Fig. 2A and B; Fig. S7). These data support the connection between ROS accumulation and moxifloxacin-mediated killing.
FIG 2.
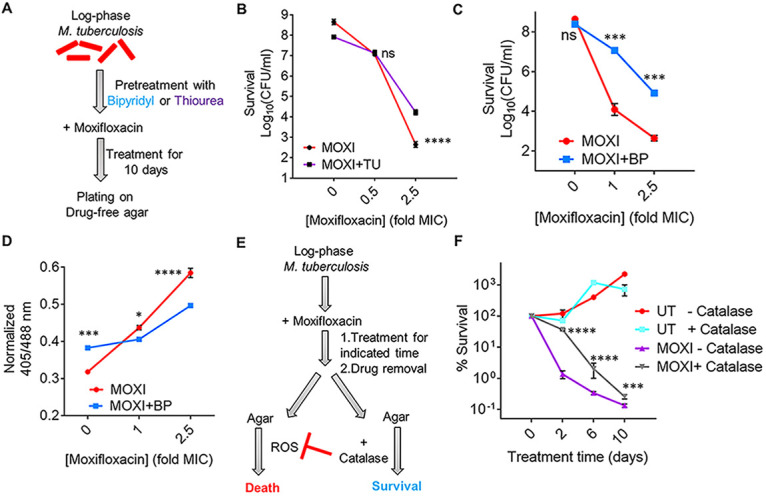
ROS mitigation reduces moxifloxacin-mediated killing of M. tuberculosis. (A) Plan for detecting thiourea (TU) and bipyridyl (BP) effects on moxifloxacin lethality. (B) Exponentially growing M. tuberculosis H37Rv cultures were either left untreated or treated with 10 mM TU for 1 h before addition of the indicated concentrations of moxifloxacin (MOXI; 1× MIC = 0.5 μM) for 10 days followed by determination of CFU. (C) Effect of bipyridyl. M. tuberculosis as for panel B was untreated or treated with 250 μM BP for 15 min prior to addition moxifloxacin as for panel B. (D) The Mrx1-roGFP2 biosensor ratiometric response was determined after 48 h treatment of M. tuberculosis cultures with the indicated concentrations of MOXI alone or with 250 μM BP. (E) M. tuberculosis cultures were treated with moxifloxacin, the drug was removed by washing, and cells were plated on drug-free 7H11 agar with or without catalase followed by CFU determination. (F) M. tuberculosis cultures were treated with 1× MIC of moxifloxacin for the indicated times, washed, and plated with or without catalase (17.5 U/mL of agar). Percentage survival was calculated relative to CFU of cultures at 0 h. Statistical significance was calculated between the drug-alone group and the drug + catalase group. Statistical considerations were as in Fig. 1.
Another test involves chelating free ferrous Fe with bipyridyl, a high-affinity Fe2+ chelator: with E. coli, bipyridyl lowers the lethal action of multiple lethal stressors (33, 35). When we treated M. tuberculosis cultures with a noninhibitory concentration (250 μM) of bipyridyl before moxifloxacin (Fig. 2A), killing was reduced by >100-fold (Fig. 2C), and ROS accumulation was lowered (Fig. 2D). Treatment with bipyridyl or thiourea also protected cells from moxifloxacin-mediated DNA damage and lipid peroxidation (Fig. S3A and B). Collectively, the data indicate that Fe-mediated ROS production is the underlying oxidative stress associated with moxifloxacin lethality.
Work with E. coli also indicates that ROS accumulates even after removal of a lethal stressor (16). When we treated M. tuberculosis with moxifloxacin in liquid medium and then plated cells on antibiotic-free 7H11 agar (see the scheme in Fig. 2E), we found that addition of catalase to the agar reduced bacterial killing (Fig. 2F; peroxide diffuses freely across the cell membrane [36], allowing exogenous catalase to reduce endogenous ROS levels). Replacement of catalase with 2.5% bovine serum albumin, which is not expected to degrade peroxide, failed to protect M. tuberculosis from moxifloxacin-mediated killing (Fig. S8). Finding that an anti-ROS agent suppresses killing after the removal of moxifloxacin indicates that the primary damage (rapid formation of fluoroquinolone-gyrase-DNA complexes) can be insufficient to kill M. tuberculosis. Thus, the poststressor accumulation of ROS and death demonstrated with E. coli occurs with M. tuberculosis.
We note that shorter drug treatment times in 7H9 broth (2 days) prior to catalase treatment on drug-free agar resulted in higher survival levels than longer times (6 to 10 days) (Fig. 2F). However, at later time points (6 to 10 days treatment with moxifloxacin), the protective effect of catalase on bacterial survival decreased (Fig. 2F). A similar phenomenon has been observed with E. coli (16). These data likely reflect ROS acting rapidly to cause accelerated lethality and then having little effect on long-term killing. Overall, studies with ROS-mitigating agents indicate that ROS contribute causally to quinolone-mediated lethality with both E. coli and M. tuberculosis.
Reduction of moxifloxacin lethality by nutrient starvation and hypoxia.
Our data support previous work (37) in which nutrient starvation blocked the lethal action of moxifloxacin (Loebel-cidal concentration [LCC90] > 32 μM) (Fig. S9A and B; Table S2). This phenomenon has also been reported for older quinolones with E. coli (17). With E. coli, these effects have been attributed to a metabolic shift that suppresses respiration, which is thought to be a major source of ROS.
Previous work (37, 38) also shows that hypoxic conditions reduce the lethal action of older fluoroquinolones with M. tuberculosis. We confirmed this observation by showing that moxifloxacin-mediated killing decreases significantly under hypoxic conditions (Fig. S9A and C; Table S2). With the Wayne-cidal concentration (WCC90) assay (37), the moxifloxacin concentration required to kill 90% of hypoxic bacteria after 5 days of drug treatment was ~10 μM (Table S2). This value corresponds to 20× MIC, or 22-fold higher than the 90% lethal dose (LD90) with M. tuberculosis (0.45 μM) cultured under aerobic growth conditions for the same treatment time (Fig. S10).
The hypoxia results are concordant with moxifloxacin lethality being associated with an increase in ROS, an event that should be suppressed by O2 limitation (intracellular generation of ROS may depend largely on the transfer of electrons directly to molecular O2 [39]). However, hypoxic suppression of moxifloxacin lethality could be due to decreased respiration. To test this hypothesis, we stimulated anaerobic respiration by providing nitrate as an alternative electron acceptor (40), since it restores norfloxacin lethality with anaerobic E. coli (20). Surprisingly, nitrate lowered residual anaerobic lethality of moxifloxacin by severalfold (Fig. S9D). Nitrate was even more protective with M. tuberculosis exposed to metronidazole (Fig. S9D), an antimicrobial known to be lethal in the absence of oxygen (41). The surprising protective effect of nitrate, which is considered in more detail in Discussion, encouraged further comparisons with E. coli.
Effect of moxifloxacin on the M. tuberculosis transcriptome.
When we examined the M. tuberculosis transcriptome using published data from a 16-h exposure to 2×, 4×, and 8× MIC of moxifloxacin (42), we found that 359 genes exhibited altered expression (2-fold change across all three treatment conditions). Of these, 219 genes were upregulated, and 140 were downregulated (Data Set S1). Next, we investigated whether deregulated expression of genes after prolonged moxifloxacin exposure (16 h) is largely a secondary effect of induction of other genes. To test this hypothesis, we compared early changes (4-h moxifloxacin exposure) in expression for a set of 28 genes deregulated at 16 h. We saw little difference (Fig. S11A and B). Thus, the patterns we observed at 16 h appear to largely reflect a primary transcription response.
When we classified the differentially expressed genes (DEGs) according to annotated functional categories (43), we found that “Information Pathways and Insertion Sequences and Phages” were 2-fold overrepresented in the moxifloxacin-treated M. tuberculosis transcriptome (Table S3). These data suggest that the bacterium responds to the drug mainly by regulating DNA remodeling, transcription, and translational machinery.
Increased expression was also seen with genes involved in redox homeostasis, such as thioredoxin (trxB1 and trxC), thioredoxin reductase (trxB2), alkyl hydroperoxide reductase (ahpC), the SigH/RshA system, the copper-sensing transcriptional regulator csoR, and the whiB family (whiB4 and whiB7) (Fig. 3A). Indeed, the transcriptome of moxifloxacin-treated M. tuberculosis showed a 67% overlap with the transcriptional signature of H2O2-treated cells (Fig. 3B) (44). Statistical analysis showed a significant overlap between the moxifloxacin transcriptome and the response to oxidative stress (H2O2; P = 5.39e−14) and nitrosative stress (NO; P = 2.13e−8) (Fig. S12; Table S4). Induction of oxidant-responsive genes (sox, sod, and mar) is also seen in norfloxacin-treated E. coli (26). Other conditions, such as hypoxia and acidic pH, showed nonsignificant overlap with the moxifloxacin transcriptome (Fig. S12; Table S4). Collectively, the data are consistent with our working hypothesis that fluoroquinolones stimulate ROS accumulation.
FIG 3.

Whole-genome transcriptome profiling of M. tuberculosis treated with moxifloxacin. (A) Heat map showing gene expression changes due to 16-h treatment of M. tuberculosis with moxifloxacin. DEGs exhibited a 2-fold change across all three treatment conditions (2×, 4×, and 8× MIC of moxifloxacin; 1× MIC = 0.4 μM); the color code for the fold change is at the bottom of the second column (yellow, upregulated genes; turquoise, downregulated genes). Genes are grouped according to function. For genes belonging to bioenergetics processes, the color code for the fold change is at the bottom of the fourth column. *, sdhC and cmtR are deregulated in two treatment conditions (2× and 4×). (B) Venn diagram showing transcriptome overlap between moxifloxacin-mediated (green circle) and H2O2-mediated stress for M. tuberculosis (44); DEGs obtained with treatment with 5 mM H2O2 for 40 min and 5 mM H2O2 for 4 h are shown in beige and blue circles, respectively.
Several genes involved in repairing DNA (recA, -O, -R, and -X, ruvABC, uvrD, ung, dnaE2, xthA, radA, alkA, lexA, nei, and ligB), in DNA metabolism (nrdF2, nrdR, pyrC, and pyrR), and in Fe-S cluster biogenesis (sufR) (45, 46) were also upregulated (Fig. 3A). E. coli treated with norfloxacin shows similar upregulation of DNA damage responses (lexA, uvr, and rec systems and error-prone DNA polymerases IV and V), nucleotide metabolism, and Fe-S cluster biogenesis (iscRUSA) (26). Such results are expected from the known ability of ROS to damage DNA (47, 48).
With M. tuberculosis, moxifloxacin suppressed expression of an Fe-responsive repressor (hupB) and an iron siderophore (mbtF), consistent with drug treatment increasing the intrabacterial pool of labile Fe (Fig. S6A). Additionally, the microarray data suggest that Fe-S clusters in M. tuberculosis are exposed to ROS during treatment with moxifloxacin, resulting in Fe release from damaged clusters and increased expression of Fe-S repair pathways (sufR). In contrast, E. coli upregulates Fe uptake machinery (26). Thus, M. tuberculosis and E. coli may generate ROS in different ways.
In M. tuberculosis, moxifloxacin repressed the expression of energy-efficient respiratory complexes, such as succinate dehydrogenase (rv0248c and sdhC), cytochrome bc1 (qcrB), and type I NADH dehydrogenase (nuo operon) (Fig. 3A). In contrast, the level of the energetically inefficient, non-proton-pumping ndh was increased. These data indicate that M. tuberculosis slows primary respiration and shifts to a lower energy state in response to moxifloxacin. Indeed, several energy-requiring pathways (e.g., cell wall biosynthesis, cofactor biogenesis, cell division, transport, and ESX secretion systems) were downregulated (Fig. 3A and Data Set S1). Moreover, genes coordinating alternate carbon metabolism, such as the gluconeogenesis (pckA) and glyoxylate cycle (icl1) genes, were upregulated (Fig. 3A). Interestingly, Icl1 protects M. tuberculosis from anti-TB drugs (isoniazid, rifampicin, and streptomycin), presumably by counteracting redox imbalance induced by these antibiotics (34). The key idea is that M. tuberculosis enters a quasiquiescent metabolic state in response to stress from moxifloxacin, in contrast to fluoroquinolone effects on E. coli (20, 26).
Moxifloxacin slows respiration.
As a direct test for decelerated respiration, we measured changes in the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) for moxifloxacin-treated M. tuberculosis as readouts for proton-extrusion into the extracellular medium (reflecting glycolysis and tricarboxylic acid [TCA] cycle activity) and for oxidative phosphorylation (OXPHOS), respectively, using a Seahorse XFp analyzer (23, 49). We incubated M. tuberculosis in unbuffered 7H9 medium with glucose in an XF microchamber and exposed the culture to 10× MIC of moxifloxacin and later to the uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP; addition of CCCP stimulates respiration to the maximal level). Changes in OCR and ECAR are reported as percentage of third baseline value. The difference between basal and CCCP-induced OCR estimates the spare (reserve) respiratory capacity (SRC) available to counteract stressful conditions (49, 50).
As expected for a growing, drug-free culture of M. tuberculosis, OCR showed a gradual increase over the duration of the experiment (400 min); OCR increased further upon uncoupling by CCCP (Fig. 4A). In contrast, addition of 10× MIC of moxifloxacin inhibited the time-dependent increase in basal OCR (Fig. 4A), and the level of OCR upon uncoupling by CCCP was markedly lower than in the absence of moxifloxacin (Fig. 4A). In parallel, we measured viability of M. tuberculosis and found that the bacterial culture maintained 100% survival during the entire incubation period and for an additional 4 h (Fig. S13). Thus, the stalled oxygen consumption effect of moxifloxacin cannot be attributed to dead cells.
FIG 4.
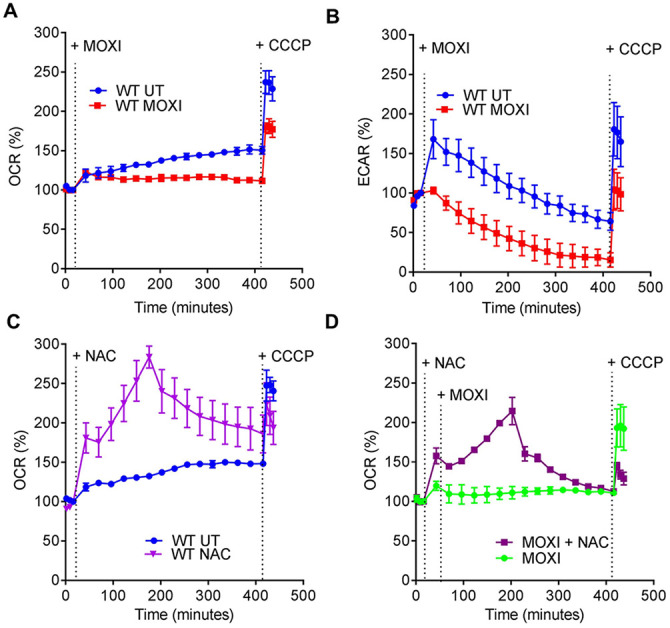
Moxifloxacin-mediated respiration arrest reversed by NAC. (A) OCR (pmol/min). Exponentially growing M. tuberculosis cultures were either left untreated (UT) or treated with 10× MIC of moxifloxacin (MOXI; 5 μM) for the indicated times; black dotted lines indicate when MOXI, NAC (1 mM), or CCCP (10 μM) was added. Determination was done with a Seahorse XFp analyzer. Data are percentages of third baseline values. (B) ECAR (mpH/min) (H+ production or extracellular acidification due to glycolytic and TCA flux). Determination was as for OCR, with data representing percentages of third baseline values. (C and D) NAC (1 mM) addition enhanced OCR of (C) untreated and (D) MOXI-treated cells. Data are representative of two independent experiments performed in triplicate.
The ECAR response increased for 60 min and then gradually dropped for untreated bacteria. This transient, early increase in ECAR was absent in cultures of moxifloxacin-treated M. tuberculosis, which exhibited a greater reduction of ECAR than untreated cultures at late times (Fig. 4B). At the end of the experiment, moxifloxacin-treated cells were completely exhausted of glycolytic capacity (Fig. 4B). The slowing in OCR and ECAR was also seen with treatment at lower concentrations of moxifloxacin (1× and 2.5× MIC) (Fig. S14), while an anti-TB drug (ethambutol) that does not generate ROS failed to significantly affect either OCR or ECAR (Fig. S15).
In summary, moxifloxacin decelerates respiration and carbon catabolism in M. tuberculosis, which likely renders the bacterium metabolically quiescent and therefore less readily killed by moxifloxacin. In support of this hypothesis, a recent study showed that pretreatment with inhibitors of the electron transport chain (ETC), such as the anti-TB drugs bedaquiline (an ATP synthase inhibitor) and Q203 (a cytochrome c oxidase inhibitor), reduces moxifloxacin-mediated killing of M. tuberculosis (51).
ROS derive from increased NADH.
Since moxifloxacin slows respiratory metabolism (Fig. 4A) while increasing ROS levels (Fig. 1B and C), a source of ROS other than respiration must exist. Slowed respiration during anoxia or chemical hypoxia places the ETC in a reduced state, and NADH accumulates. This phenomenon is known as reductive stress (52). Using a redox cycling assay, we detected accumulation of NADH in M. tuberculosis treated with 1×, 2.5×, and 10× MIC of moxifloxacin at 48 h (Fig. 5A). NAD+ was raised at 1× MIC of moxifloxacin, but it showed no significant difference from the untreated control at elevated moxifloxacin concentrations (Fig. 5B); the total NAD/H pool decreased at high drug concentrations (2.5× and 10× MIC; Fig. S16). Thus, the NADH/NAD+ ratio increased as moxifloxacin concentration increased (Fig. 5C), indicating reductive stress.
FIG 5.
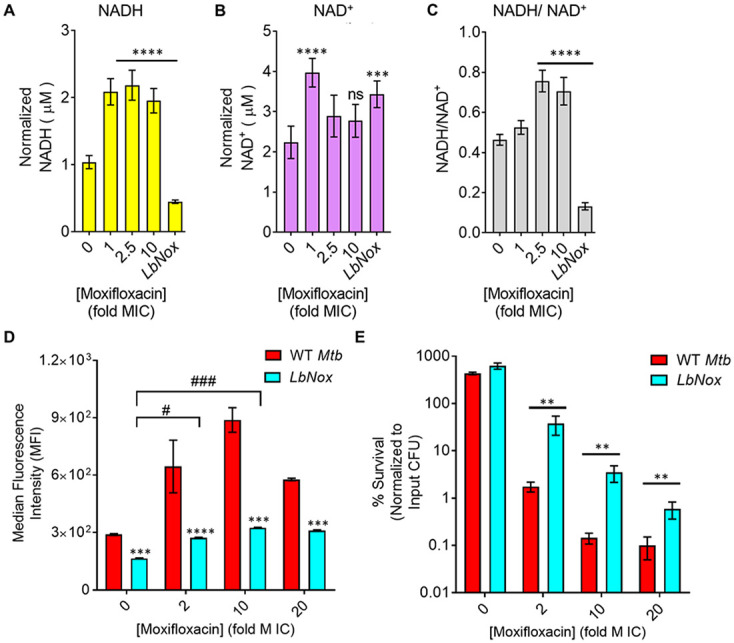
Dissipation of NADH reductive stress diminishes moxifloxacin-induced ROS increase and lethality with M. tuberculosis. (A and B) Detection of (A) NADH and (B) NAD+ levels. M. tuberculosis was treated with moxifloxacin (1× MIC = 0.5 μM) for 2 days, and NADH and NAD+ levels were determined by an alcohol dehydrogenase-based redox cycling assay. (C) NADH/NAD+ ratio. Untreated M. tuberculosis expressing LbNox served as a control. P was determined by unpaired two-tailed Student's t test analyzed relative to the untreated control. (D) ROS response to moxifloxacin. Wild-type M. tuberculosis H37Rv (WT Mtb) and cells expressing LbNox were exposed to the indicated concentrations of moxifloxacin for 48 h, and ROS were quantified by flow cytometry using CellROX deep red. (E) Cultures of exponentially growing wild-type M. tuberculosis (WT Mtb) and LbNox were treated with the indicated concentrations of moxifloxacin for 48 h, and survival was assessed by determining CFU. Statistical considerations were as for Fig. 1 (****, P < 0.0001; *** and ###, P < 0.001; **, P < 0.01; #, P < 0.05; ns, not significant).
We also assessed NADH-reductive stress by expressing a genetically encoded, noninvasive biosensor of the NADH/NAD+ ratio (Peredox) (53) (Fig. S17). A ratiometric increase in Peredox fluorescence indicated an increase in the NADH/NAD+ ratio upon moxifloxacin treatment (1× to 40× MIC) at 48 h. We observed a similar increase in the Peredox ratio upon treatment of M. tuberculosis with bedaquiline, a drug known to increase the NADH/NAD+ ratio (Fig. S17) (53). As with NADH, NADPH accumulated, and the NADPH/NADP+ ratio increased in response to moxifloxacin treatment (Fig. S18). Thus, moxifloxacin triggers NAD(P)H accumulation in M. tuberculosis.
In principle, stalled ETC and excessive reducing equivalents (NADH) can stimulate ROS production in two ways. In one, NADH mobilizes bound iron, thereby increasing the labile iron pool (54). As previously mentioned, we found a drug-concentration-dependent increase in the free iron pool in M. tuberculosis after moxifloxacin treatment (Fig. S6). Additionally, NADH autooxidation keeps iron in a reduced state (Fe2+); thus, NADH can drive the Fenton reaction toward hydroxyl radical generation (54). Second, reduced components of the electron transport chain can directly transfer electrons to molecular oxygen to generate ROS (55, 56). If these ideas are correct, dissipation of the NADH overload, i.e., reductive stress, should lower the labile iron pool, ROS surge, and moxifloxacin lethality.
We lowered the NADH/NAD+ ratio with an M. tuberculosis strain that constitutively expresses Lactobacillus brevis NADH oxidase (Mtb-LbNox) (Fig. 5C; Fig. S17) (57). In comparison to wild-type M. tuberculosis, Mtb-LbNox shows decreased levels of NADH and increased NAD+ without affecting the total pool of NAD+ + NADH (Fig. 5A to C; Fig. S19). As expected, moxifloxacin-treated Mtb-LbNox displayed a significantly reduced free-iron pool (Fig. S6), ROS surge (Fig. 5D), decreased DNA damage (Fig. S6), and a 25- to 30-fold-higher survival rate than wild-type cells (Fig. 5E). Thus, the increased NADH/NAD+ ratio occurring during metabolic quiescence caused by moxifloxacin accounts for the increased labile iron pool, which in turn catalyzes Fenton-mediated production of ROS and cell death.
The growth, OCR, and ECAR of Mtb-LbNox in 7H9 broth were the same as those of wild-type cells (Fig. S20). Furthermore, the MICs of moxifloxacin and other anti-TB drugs (rifampicin, ethambutol, and bedaquiline) for Mtb-LbNox were similar to those for wild-type M. tuberculosis (Tables S1 and S5), indicating that the interaction between drug and its primary target is unaffected by overexpression of LbNox. In contrast, Mtb-LbNox exhibited a 2-fold-lower MIC of isoniazid, which is consistent with the interaction of isoniazid and its primary target (enoyl acyl carrier protein [enoyl-ACP] reductase [InhA]) depending on the NADH/NAD+ ratio in M. tuberculosis (58). Thus, overexpression of LbNox appears to have no effect on growth, metabolism, or respiration of M. tuberculosis that would complicate our interpretation that NADH dissipation protects from moxifloxacin-mediated killing.
N-acetyl cysteine stimulates respiration, oxidative stress, and moxifloxacin lethality.
Previous work showed that stimulating respiration using cysteine and N-acetyl cysteine (NAC) elevates the killing activity of combinations of anti-TB drugs (59, 60). Since M. tuberculosis responded to moxifloxacin by dampening OXPHOS, we expected that countering the dampening with NAC would provide a way to increase ROS further and therefore enhance moxifloxacin lethality. We first measured time-dependent changes in OCR of M. tuberculosis upon exposure to a nontoxic dose of NAC (1 mM). Addition of NAC alone produced a sharp increase in OCR that reached its maximal level by 200 min (Fig. 4C). At later times, OCR gradually declined; it did not increase upon addition of CCCP (Fig. 4C), indicating that NAC-stimulated respiration exhausted the spare respiratory capacity of M. tuberculosis. Pretreatment with NAC reversed moxifloxacin-mediated slowing of respiration (Fig. 4D). As with NAC alone, the OCR of M. tuberculosis, treated with NAC plus moxifloxacin, increased for 200 min and then gradually dropped; CCCP remained ineffective at stimulating OCR (Fig. 4D). These data indicate that NAC-stimulated oxygen consumption by M. tuberculosis overcomes the dampening effect of moxifloxacin on bacterial respiration.
Respiration, stimulated by NAC, augmented ROS accumulation upon moxifloxacin treatment and increased lethality (Fig. 6A). NAC elevated the Mrx1-roGFP2-dependent redox signal for moxifloxacin-treated M. tuberculosis, confirming the increase in oxidative stress (Fig. 6B; Fig. S2C and D). We emphasize that NAC had no effect on moxifloxacin MIC, as measured by REMA and 7H11 agar plate assay (Fig. S21A and B). Thus, the primary interaction between drug and DNA gyrase (cleaved-complex formation) is unaffected by NAC. Supplementation of moxifloxacin at 1× and 5× MIC with 1 mM NAC reduced survival by 21- and 11-fold, respectively (Fig. 6C), levels that are consistent with published results obtained using drug combinations (60).
FIG 6.
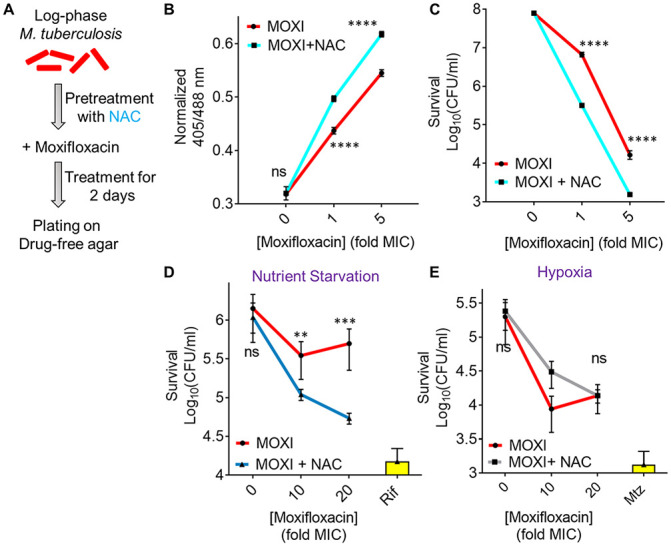
N-acetyl cysteine increases oxidative stress and moxifloxacin-mediated killing of M. tuberculosis. NAC (1 mM) was administered 1 h before addition of moxifloxacin (MOXI; 1× MIC = 0.5 μM) at the indicated concentrations followed by 48 h of incubation. (A) Experimental plan. (B) Oxidative stress, measured by the ratiometric response of the Mrx1-roGFP2 biosensor. (C) Bacterial survival, measured by plating on 7H11 agar. (D) Effect of NAC and moxifloxacin combination with dormant bacilli. M. tuberculosis cultures were starved of nutrients for 14 days and then treated with moxifloxacin for 5 days in the presence or absence of NAC (1 mM) before determination of survival. Rifampicin (Rif; 25 μM) served as a positive control. (E) NAC (1 mM) was added when cultures were placed in Vacutainer tubes followed by the treatment conditions indicated in Fig. S2A and D. Metronidazole (Mtz; 10 mM) served as a positive control. Statistical considerations were as for Fig. 1.
As a complement to the suppression of killing by addition of catalase after drug removal (Fig. 2F), we treated M. tuberculosis cultures with 1× and 5× MIC of moxifloxacin and then plated the cells on 7H11 agar with 1 mM NAC. Postdrug addition of NAC blocked formation of colonies that would otherwise have been observed (Fig. S22). Extended incubation (>4 weeks) resulted in the appearance of small colonies on the NAC-containing plates, which was likely due to instability/oxidation of NAC (half-life [t1/2] ~ 14 h) under aerobic conditions (61–63).
NAC also potentiated moxifloxacin lethality (5- to 7-fold) with nutrient-starved, dormant M. tuberculosis (Fig. 6D). These data agree with recent results which showed that activation of oxidative metabolism and respiration, through addition of l-cysteine to nutrient-starved cultures, reduced the fraction of persister subpopulations and enhanced the in vitro lethal activity of isoniazid and rifampicin (64). As expected, a similar treatment with NAC failed to enhance moxifloxacin lethality under hypoxic conditions (Fig. 6E), consistent with NAC functioning by inducing oxygen consumption and elevating ROS levels.
To address the effect of NAC on the acquisition of moxifloxacin resistance, we measured the mutant prevention concentration (MPC), i.e., the minimal concentration of moxifloxacin at which no moxifloxacin-resistant clone emerges on moxifloxacin-containing 7H11 agar inoculated with ~2.5 × 109 bacteria (30, 65). With a clinical MDR isolate of M. tuberculosis (NHN1664), moxifloxacin exhibited an MPC of 4 μM that was reduced to 2 μM upon cotreatment with either 1 mM or 2 mM NAC (Table S6). For a lower moxifloxacin concentration (1 μM, 2× MIC), the fraction of cells recovered decreased by 10-fold and 100-fold when coplated with 1 mM and 2 mM NAC, respectively (Table S6). Reduction of MPC is expected when lethal activity is increased (66).
We also addressed the possibility that some effects of NAC derived from adducts formed with moxifloxacin. A series of biochemical tests (thin-layer chromatography, nuclear magnetic resonance [NMR], fluorescence assays, and liquid chromatography-mass spectrometry [LC-MS]) revealed no evidence of adduct formation (Fig. S23).
Moxifloxacin-induced redox imbalance during infection of macrophages.
The Mrx1-roGFP2 biosensor previously showed that first-line anti-TB drugs cause an oxidative shift in the EMSH of M. tuberculosis residing inside macrophages (19). When we infected macrophages with M. tuberculosis H37Rv or an MDR clinical isolate (strain NHN1664), each expressing the biosensor Mrx1-roGFP2, M. tuberculosis displayed redox heterogeneity (EMSH-basal, −280 mV; EMSH-oxidized, −240 mV; and EMSH-reduced, −320 mV); the EMSH-reduced subpopulation was predominant (19, 23, 67) (Fig. 7A; Fig. S24). Treatment of M. tuberculosis-infected THP-1 macrophages with various concentrations of moxifloxacin significantly increased the EMSH-oxidized subpopulation (Fig. 7A, fuchsia line; Fig. S24).
FIG 7.

Moxifloxacin-induced oxidative shift in EMSH and killing of M. tuberculosis inside macrophages. THP-1 macrophages, infected with Mtb-roGFP2 (MOI = 1:10), were treated with moxifloxacin (MOXI; 1× MIC = 0.5 μM) immediately after infection and incubated for the indicated times. (A) Approximately 10,000 infected macrophages were analyzed by flow cytometry to quantify changes in the EMSH of M. tuberculosis subpopulations. (B) Bacterial survival kinetics after MOXI treatment of THP-1 macrophages infected with Mtb-roGFP2 (CFU determination). (C) Mtb-roGFP2-infected THP-1 macrophages were treated with MOXI at the indicated concentrations in the presence or absence of NAC (1 mM) immediately after infection and incubated for the indicated times; analysis was as for panel A. (D) THP1 macrophages, infected by Mtb-roGFP2, were treated with NAC (1 mM or 2 mM), MOXI (10 μM), or the combination of NAC plus MOXI at those concentrations. After the indicated incubation times, the bacterial load in the macrophages was determined by plating on drug-free agar. P was determined by two-tailed Student's t test compared to MOXI-alone treatment at each time point. Statistical considerations were as for Fig. 1.
The shift toward the EMSH-oxidized redox state was seen with moderate concentrations of moxifloxacin (5× MIC) at an early time point (12 h postinfection), before M. tuberculosis survival started to drop (Fig. 7A and B). Thus, moxifloxacin-mediated oxidative stress precedes killing: oxidative stress is not simply a consequence of cell death. Bacterial survival was reduced more by higher moxifloxacin concentrations (10× and 20× MIC) or longer incubation times (24 h and 48 h) (Fig. 7B).
We also assessed the effect of NAC on EMSH and killing of M. tuberculosis inside macrophages. NAC itself is not cytotoxic to macrophages up to at least 5 to 10 mM (59, 68), but at those elevated concentrations, it exhibits antimycobacterial properties (68, 69). Consequently, we selected noncytotoxic, nonantituberculous concentrations of NAC (1 mM and 2 mM) (68) for our test with moxifloxacin. NAC supplementation induced an oxidative shift in the EMSH of M. tuberculosis that exceeded that seen with moxifloxacin alone (Fig. 7C; Fig. S24). NAC alone had no effect on survival of M. tuberculosis inside THP-1 macrophages; however, the NAC plus moxifloxacin combination decreased bacterial burden 5 to 10 times more than moxifloxacin alone in both concentration- and time-dependent manners (Fig. 7D). We note that for either isoniazid or an isoniazid-rifampicin combination, NAC potentiates lethality only at late times (day 6 or 7) (59, 60); NAC acts more rapidly with moxifloxacin, as the killing effect is evident at days 1 and 2 (Fig. 7D). Thus, NAC may not function in the same way with all anti-TB drugs inside macrophages.
Moxifloxacin also lowered the level of an EMSH-reduced subpopulation inside macrophages (Fig. 7A and C, blue line; Fig. S24); this subpopulation was further diminished by NAC supplementation. EMSH-reduced subpopulations are phenotypically tolerant to anti-TB drugs (19, 23) and to moxifloxacin (Fig. S25). Overall, results with infected macrophages demonstrate that accelerating respiration and oxidative metabolism by NAC can enhance moxifloxacin lethality.
Potentiation of moxifloxacin lethality with NAC lowers M. tuberculosis burden in a murine model of infection and restricts the emergence of resistance.
We next asked whether NAC-mediated enhancement of moxifloxacin lethality occurs in vivo. Recent work by Vilchèze and Jacobs (60) directed our choice of murine models: they found little enhancement by NAC on the overall lethality of a combination of moxifloxacin and ethionamide when measured after long incubation using an acute model of murine tuberculosis. Since ROS accelerate killing in vitro without increasing the extent of killing (70), we shortened the drug treatment time. For this pilot test, we infected BALB/c mice with the clinical MDR strain M. tuberculosis NHN1664. Three weeks after aerosol-mediated infection at a low dose (~100 bacilli), mice were treated once daily for 10 days with a low dose of moxifloxacin (50 mg/kg body weight), NAC (500 mg/kg body weight), or moxifloxacin plus NAC at the same doses as for monotreatments (Fig. 8A). After 10 days, moxifloxacin monotherapy reduced the bacillary load by 3-fold and 100-fold in lung and spleen, respectively; NAC alone exhibited no antibacterial activity (Fig. 8B and C). The combination of moxifloxacin plus NAC reduced bacterial burden by another 4- and 12-fold beyond that observed for moxifloxacin alone for lung and spleen, respectively (Fig. 8B and C). Thus, this finding indicates that NAC stimulates the lethal action of moxifloxacin in vivo.
FIG 8.
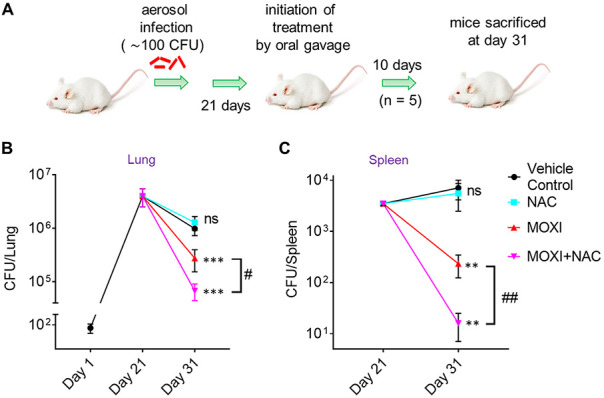
NAC decreases MDR M. tuberculosis survival in mice when combined with moxifloxacin. (A) Experimental protocol. (B and C) Bacterial CFU were enumerated from lungs and spleen at the indicated times. P was determined by unpaired two-tailed Student's t test relative to vehicle control treatment (**, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant). Statistical significance between moxifloxacin (MOXI) alone and MOXI + NAC treatment is also shown (#, P < 0.05; ##, P ≤ 0.01). Error bars represent standard deviations from the mean of bacterial burden in 5 mice per group.
As a proof of concept. we also measured the effect of NAC on the selection of moxifloxacin-resistant mutants in mice as described elsewhere (71, 72). We implanted ~4 × 103 M. tuberculosis NHN1664 organisms in the lungs of BALB/c mice, and at day 14 postinfection, when the mean lung log10 CFU reached ~107, mice received moxifloxacin, NAC, or moxifloxacin plus NAC (Table 1). After 14 days of postinfection treatment, 66% of moxifloxacin-treated mice harbored resistant bacteria, compared to 17% when NAC was also present. The combination of moxifloxacin plus NAC brought the number of mutants down to the level seen with untreated animals (Table 1). Enumeration of total resistant colonies confirmed that while moxifloxacin treatment increases emergence of resistance, NAC supplementation reduced the recovery of moxifloxacin-resistant mutants by 8-fold (Table 1).
TABLE 1.
N-acetyl cysteine decreases recovery of M. tuberculosis-resistant mutants in infected BALB/c mice
| Treatment | No. (%) of mice |
No. of resistant colonies |
||
|---|---|---|---|---|
| Total | Harboring resistant colonies | Total/group | Avg/mousea | |
| UTb | 6 | 1 (17) | 1 | 0.17 |
| NACc | 6 | 2 (33) | 2 | 0.33 |
| MOXId | 6 | 4 (67) | 8 | 1.33 |
| NAC + MOXI | 6 | 1 (17) | 1 | 0.17 |
Total number of resistant colonies in a treatment group divided by total number of mice in that group.
UT, untreated (mice were given water [vehicle control]).
NAC (500 mg/kg body weight) was given once daily for a period of 2 weeks by oral gavage.
Moxifloxacin (MOXI; 50 mg/kg body weight) was given once daily for a period of 2 weeks by oral gavage.
DISCUSSION
M. tuberculosis responds to moxifloxacin through a lethal stress response characterized by suppressed respiration, elevated NADH levels, and ROS accumulation (shown schematically in Fig. 9). Although ROS accumulation is also observed with E. coli during lethal stress, early steps in the M. tuberculosis stress response are distinct. M. tuberculosis enters a metabolically quiescent state characterized by increased NADH levels and NADH/NAD+ ratio plus reduced respiratory and glycolytic rates. This response is opposite to that seen with E. coli (20, 33, 73). Decelerated respiration may be an adaptive strategy against ROS-inducing agents produced by the host. Production of ROS likely derives from an increased NADH/NAD+ ratio, which increases the labile, reduced form of Fe to fuel the Fenton reaction. Interfering with the NADH/NAD+ increase dampened reductive stress and moxifloxacin lethality, suggesting that NADH disposal pathways could be targeted to enhance fluoroquinolone lethality with M. tuberculosis.
FIG 9.

Moxifloxacin-mediated killing of M. tuberculosis involves accumulation of NADH-dependent ROS, which is further enhanced by NAC. (A) Moxifloxacin enters M. tuberculosis and traps gyrase on DNA as reversible, bacteriostatic drug-enzyme-DNA complexes in which the DNA is broken. The bacterium responds by downregulating the expression of genes involved in respiration. The transcriptional changes result in reduced rate of respiration. NADH levels and the ratio of NADH to NAD+ increase. NADH increases the free Fe2+ pool by releasing Fe from ferritin-bound forms and keeps it in a reduced state. ROS damage macromolecules in a self-amplifying process, as indicated by exogenous catalase blocking killing when added after removal of moxifloxacin. (B) Addition of N-acetyl cysteine to cells stimulates respiration and provides more ROS from moxifloxacin-mediated lesions. NAC alone does not induce ROS or trigger death. The additional ROS increases killing by moxifloxacin. Repair of moxifloxacin-mediated lesions, NADH dissipation, Fe sequestration, and ROS detoxification mechanisms contribute to survival. The image was created using BioRender.com.
ROS contribution to lethality.
Studies with E. coli indicate that ROS kill bacteria by damaging macromolecules, i.e., DNA breakage, protein carbonylation, and lipid peroxidation (15). Our data support the general principle that ROS contribute to stress-mediated death of moxifloxacin-treated M. tuberculosis: (i) biosensors show an increase in ROS at lethal drug concentrations; (ii) iron concentrations increase with moxifloxacin treatment; and (iii) two ROS-mitigating agents, thiourea and bipyridyl, reduce oxidative stress and killing. However, the striking increase in survival for E. coli at very high quinolone concentrations is associated with a drop in ROS, while this is not the case with M. tuberculosis; this difference remains unexplained.
Another difference is the lowering of residual fluoroquinolone lethality by nitrate with hypoxic M. tuberculosis (Fig. S9D): the opposite is observed with norfloxacin-treated E. coli. Previous M. tuberculosis work has shown that nitrate protects hypoxic bacilli from killing caused by exposure to reactive nitrogen intermediates (74), acid stress (74), sudden anaerobiosis (75), or inhibition of type II NADH-dehydrogenase (NDH-2) by thioridazine (75). Two phenomena may contribute to nitrate-mediated protection. (i) M. tuberculosis reduces nitrate to nitrite (76), which oxidizes ferrous Fe to the ferric state, thereby displacing Fe from critical Fe-sulfur clusters and inducing bacteriostasis (76). Nitrite also remodels the M. tuberculosis transcriptome similar to several host-imposed stresses that suppress antibiotic-mediated lethality (76) and ensure bacterial survival. (ii) Nitrate, as a terminal electron acceptor, recycles or disposes of excess reducing equivalents that accumulate during anaerobic conditions (77). Discovering strategies or therapeutics to halt such adaptive remodeling (e.g., nitrate reduction during hypoxia) in M. tuberculosis could serve as potential adjunct therapy with moxifloxacin against hypoxic bacilli.
Reductive stress as a source of ROS.
When we dissipated NADH-reductive stress by overexpression of LbNox, the ROS surge was mitigated, and moxifloxacin lethality was lowered. Production of NADH can also be lowered by stimulating the glyoxylate shunt via isocitrate lyase (icl)—deletion of icl increases the lethal action of isoniazid, rifampicin, and streptomycin (34), suggesting that NADH-mediated ROS accumulation may also contribute to the lethal action of these agents. The observed increase in NADPH/NADP+ ratio in response to moxifloxacin exposure could assist M. tuberculosis in mitigating oxidative stress by activating NADPH-dependent antioxidant systems, thioredoxins, and mycothiol. As shown by microarray data, the increase in NADPH/NADP+ ratio could also be a consequence of downregulation of NADPH-requiring anabolic pathways, such as polyketide/lipid biogenesis, amino acid anabolism, and de novo nucleotide synthesis.
In summary, M. tuberculosis appears to tolerate lethal stressors of the host immune system by decelerating respiration coupled with dissipation of NADH-reductive stress. The need to dissipate reductive stress may be of general importance, as this type of stress is also generated and amplified by hypoxia, inhibition of aerobic respiration by NO, and catabolism of fatty acids (78, 79). M. tuberculosis dissipates reductive stress through WhiB3-mediated anabolism of polyketide lipids (78). Other bacterial species also dispose of excess reductant, which they do by (i) using nitrate as a terminal electron acceptor during hypoxia (74), (ii) fueling fermentation under nitrosative stress (80), and (iii) secreting phenazines (redox-active polyketides) that generate NAD+ from NADH (81). Moxifloxacin is the first antimycobacterial agent shown to stimulate NADH-reductive stress.
Lethality enhancement by N-acetyl cysteine.
Vilchèze and Jacobs (60) reported that NAC boosts the lethality of anti-TB drug combinations in vitro. We emphasize that a general, enhancing effect on drug-target interactions by NAC is unlikely, because such effects are usually detected by MIC measurement (15): NAC has no effect on moxifloxacin MIC. This observation reinforces the concept that drug-target interactions are mechanistically distinct from downstream ROS-based killing (protection from killing, which is called tolerance when no change in MIC occurs, is distinct from resistance, which is measured as an increase in MIC).
To simplify interpretation of killing data, we focused on a single drug, moxifloxacin, rather than drug combinations. The previous demonstration that NAC stimulates respiration (60) made it a useful tool for examining killing mechanisms. NAC increased moxifloxacin-mediated ROS accumulation and lethality with cultured M. tuberculosis. Increased killing was initially surprising, because NAC has antioxidant properties expected to reduce moxifloxacin lethality (82). It is likely that the effect of NAC depends on the level of stress, since with other bacterial species, genes (mazF, cpx, and lepA) and treatments (chemical generation of superoxide and H2O2) are known to be protective at low levels of stress but destructive at high ones (genetic deficiencies are protective) (83–86). Indeed, treatment with a sublethal concentration of H2O2 activates the OxyR regulon in E. coli and “primes” the bacterium to counteract subsequent oxidative-stress challenge. At higher concentrations, H2O2 is bactericidal (87). With the high level of stress produced by moxifloxacin, the respiration-stimulating activity of NAC dominates and adds to ROS derived from NADH-reductive stress.
As expected from in vitro studies, NAC increased moxifloxacin-mediated killing of M. tuberculosis inside macrophages and in a murine model of infection. The kinetic effects of ROS explain why our short-term treatment model with infected mice showed stimulation by NAC, while the longer treatment used previously with antimicrobial combinations did not (60).
We addressed the potential of NAC to suppress the emergence of resistance by measuring its effect on MPC. Although MPC is a bacteriostatic parameter (MIC of the least susceptible mutant subpopulation), highly lethal agents lower it, presumably by killing mutants (66). Thus, we were not surprised that stimulation of moxifloxacin lethality by NAC lowered MPC. In a murine infection model, NAC reduced the recovery of resistant mutants by 8-fold.
Although the MPC for moxifloxacin is lower than concentrations achieved in serum by approved doses (88), the clinical situation is likely complex: moxifloxacin shows poor penetration into caseous regions of tubercular granulomas in a rabbit model of experimental tuberculosis (89, 90). Low local moxifloxacin concentrations may promote the emergence of fluoroquinolone resistance (91). Thus, NAC supplementation could increase the probability that moxifloxacin concentration is above the MPC in vivo, but additional work is required to test this idea.
NAC is a potential candidate as a lethality enhancer, because it is relatively safe for humans (it has been used to alleviate drug-induced toxicity in TB patients [92, 93]). Furthermore, NAC by itself exhibits antimycobacterial activity in macrophages, mice, and guinea pigs (68, 94), and a small clinical trial in which NAC was administered to TB patients showed improved host health, immunological response, and early sputum conversion (95, 96). The present work provides a mechanistic foundation for refining NAC-based enhancement of antituberculosis agents. Assessing the general clinical significance of ROS enhancement is also complex, because ROS accelerate killing without lowering bacterial survival upon long incubations. This phenomenon has been observed with Staphylococcus aureus (70), E. coli (16), and M. tuberculosis (Fig. 2F; Fig. S4B). With M. tuberculosis-infected mice, NAC stimulates reduction of bacterial load at short but not long incubation times (60). Thus, an appropriate dosing interval must be determined, initially guided by in vitro pharmacodynamic studies (71, 72, 97, 98). Relevant tissue concentrations of NAC and moxifloxacin for various doses over time must also be determined before clinical relevance can be assessed. Therefore, the present report provides a strong foundation for detailed follow-up work to determine the clinical relevance of NAC as an adjunct therapy for moxifloxacin.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
M. tuberculosis H37Rv and the drug-resistant clinical isolates JAL 1934, JAL 2287, JAL 2261, and BND 320 were kind gifts from Kanury V. S. Rao (International Centre for Genetic Engineering and Biotechnology, Delhi, India). The MDR strain NHN1664, originally isolated in China, was obtained from BEI Resources, NIAID, NIH. Strain Mtb-roGFP2 was generated by transforming M. tuberculosis strain H37Rv with an E. coli-mycobacterial shuttle vector, pMV762 (99, 100). Plasmid pMV762 contains the Mrx1-roGFP2 biosensor construct under the control of the M. tuberculosis hsp60 promoter and a hygromycin resistance gene as a selection marker. Hygromycin (catalog no. 0219417091; MP Biomedical, Santa Ana, CA) was used at a final concentration of 50 μg/mL. A plasmid (pGMCgS-0×-Ptb38-LbNOX-FLAG-SD1) containing an LbNox construct was a kind gift from Dirk Schnappinger and Sabine Ehrt (Weill Cornell Medicine, New York, NY, USA). Plasmid was electroporated into wild-type M. tuberculosis H37Rv to create the Mtb-LbNox strain. Streptomycin was used at a final concentration of 25 μg/mL. Plasmid pMV762-Peredox-mcherry was a kind gift from Ashwani Kumar (Council of Scientific and Industrial Research, Institute of Microbial Technology, Chandigarh, India).
All strains, including the laboratory M. tuberculosis strain H37Rv and M. tuberculosis expressing the Mrx1-roGFP2 redox biosensor, were grown in 7H9 broth supplemented with 0.2% glycerol, 0.05% Tween 80, and ADS (0.5% albumin, 0.2% dextrose, and 0.085% NaCl) with shaking at 180 rpm in a rotary shaker incubator (Lab Therm LT-X; Kuhner, Basel, Switzerland) or on 7H11 agar supplemented with ADS or OADC (ADS plus 0.05% oleic acid and 0.004% catalase) (catalog no. C9322; Sigma-Aldrich, St. Louis, MO) or ADC (ADS plus 0.004% catalase) at 37°C. Bacterial strains expressing Mrx1-roGFP2 were grown in medium containing hygromycin.
Determination of MIC under aerobic growth conditions.
MIC was determined by a resazurin microtiter assay (REMA) using 96-well flat-bottom plates (25). M. tuberculosis strains were cultured in 7H9+ADS medium (19) and grown to exponential phase (optical density at 600 nm [OD600] = 0.4 to 0.8). Approximately 1 × 105 bacteria per well were added in a total volume of 200 μL of 7H9+ADS medium. Wells lacking M. tuberculosis served as controls. Additional controls consisted of wells containing cells without drug treatment (growth control). After 5 days of incubation at 37°C in the presence of fluoroquinolone (or NAC), 30 μL of 0.02% resazurin (catalog no. R7017; Sigma-Aldrich, St. Louis, MO) was added, and plates were incubated for an additional 24 h. Fluorescence intensity was measured using a SpectraMax M3 plate reader (Molecular Devices, San Jose, CA) in bottom-reading mode with excitation at 530 nm and emission at 590 nm. Percent inhibition was calculated from the relative fluorescence units compared with an untreated control culture; the MIC was taken as the lowest drug concentration that resulted in at least 90% reduction in fluorescence compared to the untreated growth control.
Determination of bacterial survival.
To determine the number of viable bacilli, aliquots were removed from cultures, cells were concentrated by centrifugation (4,200 × g, 5 min) to remove drug or treatment compounds, and they were resuspended in an equal volume of medium. Tenfold dilutions were prepared, and 20 μL aliquots were spotted on drug-free 7H11 agar plates containing 10% ADC or ADS. Plates were incubated for 3 to 4 weeks at 37°C for CFU enumeration.
Concentration-kill curves were obtained by treating exponentially growing cultures of M. tuberculosis (OD600 of 0.3, or ~5 × 107 cells/mL) with various concentrations of moxifloxacin. Tubes were incubated with shaking at 180 rpm for 10 days at 37°C. Aliquots were taken at various intervals, serially diluted, and plated on drug-free agar for CFU enumeration.
The LD90 (lethal dose) of moxifloxacin under in vitro aerobic-culture conditions was measured by treating 10-mL cultures in 50-mL centrifuge tubes under aerobic conditions (shaking at 180 rpm and 37°C), followed by CFU measurement. The concentration that resulted in a 90% reduction in CFU on treatment day 5, compared with the input control, was defined as the LD90.
To determine the effect of an Fe chelator on moxifloxacin-mediated lethality, 250 μM (a noninhibitory concentration) of bipyridyl (catalog no. D216305; Sigma-Aldrich, St. Louis, MO) was added to bacterial cultures 15 min prior to moxifloxacin addition; bipyridyl was present throughout the experiment (i.e., 10 days).
For experiments with thiourea or N-acetyl cysteine (NAC), thiourea (10 mM) or NAC (1 mM) was added 1 h prior to moxifloxacin and was maintained for 2 days or 10 days. Aliquots were taken at indicated times, serially diluted, and plated for CFU enumeration on drug-free agar.
Poststressor bactericidal activity of moxifloxacin.
M. tuberculosis cultures were treated with moxifloxacin for the indicated durations, the drug was removed by washing, and cells were plated on drug-free 7H11 agar with or without 1 mM NAC or with 2.5% bovine serum albumin (BSA) or catalase (17.5 U/mL of agar), followed by visual CFU determination.
Measurement of EMSH using the Mrx1-roGFP2 redox biosensor.
For intramycobacterial EMSH determination during in vitro growth or under ex vivo macrophage infection conditions, strain Mtb-roGFP2 was grown in 7H9 broth or in THP-1 macrophages, respectively. Cultures were then treated with moxifloxacin, and at various times, they were treated with 10 mM N-ethylmaleimide (NEM; catalog no. E3876; Sigma-Aldrich, St. Louis, MO) for 5 min at room temperature followed by fixation with 4% paraformaldehyde (PFA; catalog no. GRM3660; Himedia, Mumbai, India) for 1 h at room temperature. Bacilli and infected macrophages were analyzed using a FACSVerse flow cytometer (BD Biosciences, San Jose, CA). Intramycobacterial EMSH was determined using the Nernst equation as described previously (19). EMSH is defined as the standard reduction potential of the MSH/MSSM redox couple (reduced mycothiol to oxidized mycothiol).
The biosensor response was measured by analyzing the fluorescence ratio at a fixed emission (510 nm) after excitation at 405 and 488 nm. Data were analyzed using the BD FACSuite software. These ratiometric data were normalized to measurements with cells treated with 10 mM cumene hydroperoxide (catalog no. 247502; Sigma-Aldrich, St. Louis, MO), which reports maximal oxidation of the biosensor, and 20 mM dithiothreitol (Sigma-Aldrich, catalog no. D9779, St. Louis, MO), which reports maximal reduction. Ten thousand events per sample were analyzed.
ROS measurement using CellROX deep red.
Cultures of exponentially growing M. tuberculosis at an initial OD600 of 0.3 were treated with various concentrations of moxifloxacin for 48 h. Sterile 50-mL polypropylene centrifuge tubes (catalog no. P10404; Abdos, Roorkee, India), containing 10 mL of culture, were incubated in a shaker incubator (180 rpm, 37°C). Then, cells were harvested by centrifugation (4,200 × g for 5 min) and resuspended in 100 μL of growth medium. Per the manufacturer’s instructions, CellROX deep red (catalog no. C10422; Invitrogen, Waltham, MA) was added to a final concentration of 5 μM, and cells were agitated on a rocker (Biobee Tech, Bangalore, India) for 30 min. After incubation, cells were washed to remove residual dye by centrifugation (4,200 × g for 5 min). Cells were resuspended in 300 μL phosphate-buffered saline (pH 7.4) and then fixed by addition of 4% PFA for 1 h at room temperature. Fluorescence was measured at a fixed emission (670 nm) after excitation with a red laser (640 nm) using a BD FACSVerse flow cytometer (BD Biosciences, San Jose, CA) with 10,000 events per sample. No autofluorescence was observed.
Determination of DNA damage by TUNEL assay.
DNA damage was measured by using an in situ cell death detection kit with tetramethylrhodamine (TMR) red (catalog no. 12156792910; Roche Molecular Biochemicals, Indianapolis, IN), which is based on TUNEL (TdT-mediated dUTP-X nick end labeling) assay (101, 102). Equal numbers of cells (based on OD600) were taken at various times of treatment with moxifloxacin, washed once by centrifugation, and fixed in 2% PFA. PFA was removed by washing cells, followed by resuspension in 2% sodium dodecyl sulfate (SDS) and a second wash by centrifugation. DNA double-strand breaks were labeled in 100 μL of TUNEL reaction mix for 3 to 4 h. Cells incubated with label solution only (no terminal transferase) were used as negative controls. Fluorescence was measured at a fixed emission (585 nm) after excitation with green-yellow laser (561 nm) using a BD FACSAria flow cytometer (BD Biosciences, San Jose, CA). Ten thousand events were acquired per sample.
Lipid peroxidation assay.
Cultures were grown to mid-log phase (OD600 = 0.6 to 0.8). Lipid hydroperoxides were quantified from cell pellets after centrifugation (4,000 × g, 5 min), using FOX2 reagent (24). Briefly, cell pellets were resuspended in 1:2 chloroform-methanol and mixed by vortexing. Next, chloroform and water were added as a 1:1 mixture. The samples were then centrifuged to separate the water and organic phases. The organic phase was collected and washed twice with water. Two hundred microliters of the organic phase was incubated with 1 mL of FOX2 reagent in the dark for 1 h at 22°C. Lipid hydroperoxides were measured spectrophotometrically at 560 nm and normalized to culture turbidity (OD600).
Macrophage preparation and infection by M. tuberculosis.
The human monocytic cell line THP-1 was grown in RPMI 1640 medium (Cell Clone, Manassas, VA) supplemented with 10% heat-treated (55°C) fetal bovine serum (catalog no. 092916754; MP Biomedical, Santa Ana, CA). A total of 3 × 105 cells/well was seeded into a 24-well cell-culture plate. THP-1 monocytes were stimulated to differentiate into macrophage-like cells by treatment with 20 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich Co., St. Louis, MO) for 18 to 20 h at 37°C and incubated further for 48 h to allow differentiation (19). The resulting macrophage-like cells were infected with Mtb-roGFP2 or MDR M. tuberculosis NHN1664 expressing Mrx1-roGFP2 at a multiplicity of infection (MOI) of 10 and incubated for 4 h at 37°C in 5% CO2. After infection, extracellular bacteria were removed by washing three times (by centrifugation) with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.4 mM KH2PO4, pH 7.4). Moxifloxacin (catalog no. PHR1542; Sigma-Aldrich, St. Louis, MO), with or without NAC (catalog no. A7250; Sigma-Aldrich, St. Louis, MO), was added to infected cells that were incubated for various times. For CFU determination, infected cells were lysed in 7H9 medium containing 0.06% SDS; dilutions were prepared using 7H9 medium, and aliquots were plated on 7H11+OADC agar plates (19). Plates were incubated at 37°C for 3 to 4 weeks before colonies were counted.
Nutrient starvation.
Starvation was achieved as previously described (37). Briefly, cultures of M. tuberculosis H37Rv were grown to exponential phase in roller-culture bottles (catalog no. 430518; Corning, Corning, NY) containing Middlebrook 7H9 medium, supplemented with ADS and 0.05% Tween 80, at 37°C with rolling at 6 rpm in a roller incubator (120 Vac Roll-In incubator; Wheaton, Millville, NJ). Cultures grown to an OD600 of ~0.2 were harvested by centrifugation (4,000 × g for 5 min) followed by two washes with PBS (pH 7.4) supplemented with 0.025% Tween 80 (PBST). Bacterial cells were diluted to a final OD600 of 0.1 in PBS. Fifty milliliters of this suspension was transferred into a roller culture bottle and incubated for 14 days to achieve starvation conditions.
Hypoxia.
For determination of moxifloxacin lethality under hypoxic conditions, bacterial cultures (OD600 = 0.1) were placed in Vacutainer tubes (catalog no. 367812; Becton Dickinson, Franklin Lakes, NJ) followed by incubation for 14 to 21 days at 37°C (103). A high cell density (OD600 = 0.1) was used for rapid achievement of hypoxia, which was observed as decolorization of methylene blue (final concentration was 1.5 μg/mL) in the culture medium. When hypoxia was established, moxifloxacin was added to cultures anaerobically. Metronidazole at 10 mM and isoniazid at 10 μM were used as positive and negative controls, respectively. Drugs were injected in volumes of 100 μL in phosphate-buffered saline following passage of argon through the drug solution to remove residual oxygen. Hypoxic cultures were treated with drugs for 5 days, similar to the incubation time for MIC determination with aerobically growing cells. After treatment, Vacutainer tubes were unsealed, and endpoint bacterial survival was determined by plating on drug-free agar, incubating for 3 to 4 weeks at 37°C, and visually enumerating colonies.
Effect of nitrate on survival of hypoxic bacteria.
Survival during nitrate-dependent respiration was achieved by supplementing M. tuberculosis cultures with 5 mM sodium nitrate. Nitrate was added when cultures were placed in Vacutainer tubes before hypoxia. All other conditions were as described above for hypoxia.
Microarray data analysis.
Gene expression data from GSE71200 (NCBI Gene Expression Omnibus [GEO]) was used for analyzing the response of M. tuberculosis to moxifloxacin (42). The data were expressed as a two-channel microarray with control and drug-exposed M. tuberculosis stained with Cy3 and Cy5 dyes, respectively. We used GSM1829746, GSM1829747, and GSM1829748, which consist of published data for M. tuberculosis exposed to 2×, 4×, or 8× MIC (1× MIC = 0.4 μM) of moxifloxacin for 16 h. Normalized expression data for GSE71200 were downloaded from GEO (104, 105), and probe IDs were mapped to the respective gene IDs. The expression levels for genes having multiple probes were averaged, and genes lacking data were removed from further analysis. Differentially expressed genes (DEGs) were defined as genes that were upregulated or downregulated by at least 2-fold in all 3 moxifloxacin treatment conditions.
For overlap analysis of differentially expressed genes in moxifloxacin-exposed bacteria compared with other stress-induced conditions (log2 fold change of >1 or <−1; P values of <0.01 are considered to indicate differentially expressed genes in stress conditions), the GeneOverlap (v1.22.0) package from R (v3.6.3) was used (106, 107). It uses Fisher's exact test to find statistical significance by calculating the P value and the odds ratio for the overlap (a P value of <0.05 and an odds ratio of >1 were taken as the significance thresholds).
OCR and ECAR measurements.
To measure basal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), log-phase M. tuberculosis cultures (OD600 = 0.6 to 0.8) were briefly (1 day) incubated in 7H9 medium containing the nonmetabolizable detergent tyloxapol (catalog no.157162; MP Biomedical, Santa Ana, CA) and lacking ADS or a carbon source. These cultures were then passed 10 times through a 26-gauge syringe needle followed by centrifugation at 100 × g for 1 to 2 min to remove clumps of bacterial cells. The resulting single-cell suspensions of bacteria at 2 × 106 cells/well were placed in the bottom of wells of a Cell-Tak (catalog no. 354240; Corning, Corning, NY)-coated XF culture plate (Agilent/Seahorse Biosciences, Santa Clara, CA). Measurements were performed using a Seahorse XFp analyzer (Agilent/Seahorse Biosciences, Santa Clara, CA) with cells in unbuffered 7H9 growth medium (pH 7.35 lacking monopotassium phosphate and disodium phosphate) containing glucose (2 mg/mL) as a carbon source. OCR and ECAR measurements were recorded for ~21 min (3 initial baseline readings) before addition of moxifloxacin (1×, 2.5× or 10× MIC; 1× MIC = 0.5 μM), which was delivered automatically through the drug ports of the sensor cartridge (Wave Software, Agilent Technologies, Santa Clara, CA). NAC or CCCP was similarly added through drug ports at times indicated in the figures. OCR and ECAR were measured for an additional 6 h in the absence or presence of moxifloxacin and/or NAC. Changes in OCR and ECAR readings triggered by moxifloxacin were calculated as a percentage of the third baseline reading for OCR and ECAR taken prior to drug injection.
ROS measurement under Fe depletion conditions.
Iron starvation was achieved as described previously (108). Briefly, log-phase M. tuberculosis cultures were grown in minimal medium (0.5% [wt/vol] asparagine, 0.5% (wt/vol) KH2PO4, 2% glycerol, 0.05% Tween 80, 10% ADS, 0.5 mg/L of sterile ZnCl2, 0.1 mg/L of MnSO4, and 40 mg/L of MgSO4) to early stationary phase (OD600 of ~1). To remove metal ions, the minimal medium was treated with 5% Chelex-100 (catalog no. 142-2842; Bio-Rad, Hercules, CA) for 24 h with gentle agitation. This Fe-depleted culture was diluted further to an OD600 of 0.1 in the same medium and allowed to grow to early stationary phase to deplete stored Fe. The Fe-depleted cells were further treated with 50 μg/mL of freshly prepared deferoxamine mesylate (catalog no. D9533; Sigma-Aldrich, St. Louis, MO)-containing minimal medium for 6 days. The cells were washed and diluted in minimal medium containing 80 μM FeCl3 or 80 μM FeCl3 + 10 mM thiourea for an additional 4 days. ROS was quantified by CellROX deep red dye using flow cytometry as described in the section “ROS measurement using CellROX deep red.”
Cellular Fe estimation.
Cell-free Fe levels were measured using the ferrozine-based assay as described previously (59, 109). Briefly, M. tuberculosis cultures were grown to an OD600 of 0.3 to 0.4 followed by treatment with 0×, 1×, 2.5×, or 10× MIC of moxifloxacin. After 48 h of treatment, cells were harvested and washed twice with ice-cold PBS. The cell pellets were resuspended in 1 mL of 50 mM NaOH and lysed using a bead beater. The cell lysate sample (300 μL) was mixed with 10 mM HCl (300 μL) followed by addition of Fe detection reagent (6.5 mM ferrozine, 6.5 mM neocuproine, 1 M ascorbic acid, and 2.5 M ammonium acetate in water) (90 μL). The reaction mix was incubated at 37°C for 30 min, and then absorbance at 562 nm was measured. The cellular Fe concentration was determined by plotting the absorbance values against a standard curve of FeCl3 concentration gradient and normalized to protein content. Protein concentration was determined using the Pierce bicinchoninic acid (BCA) protein assay kit (catalog no. 23225; Thermo Scientific, Rockford, IL).
Estimation of NAD+, NADP+, NADH, and NADPH.
M. tuberculosis H37Rv was grown to an OD600 of ~0.35 and treated with 1×, 2.5×, or 10× MIC of moxifloxacin for 48 h. Pyridine nucleotide levels were determined by a redox-cycling assay (78, 110). Protein concentration of NAD+ or NADH extracts was determined using the Pierce BCA protein assay kit (catalog no. 23225; Thermo Scientific, Rockford, IL) to normalize NAD/(P)H and NAD/(P)+ concentrations.
Analysis of mixtures containing moxifloxacin with and without NAC by TLC.
Thin-layer chromatography (TLC) was performed using silica gel 60 GF254 precoated aluminum-backed plates (0.25-mm thickness; Merck, Darmstadt, Germany), and visualization was accomplished by irradiation with UV light at 254 nm. Stock solutions of moxifloxacin (10 mM) and NAC (0.2 M, 2 M, and 20 M) were prepared independently in dimethyl sulfoxide (DMSO) and phosphate buffer (PB; pH 7.4; 10 mM), respectively. In a typical incubation, moxifloxacin (100 μL at a final concentration of 2 mM) was independently reacted with various concentrations of NAC (5 μL; final concentration, 2 mM, 20 mM, or 200 mM) in 395 μL PB (pH 7.4; 10 mM). In a control experiment, moxifloxacin (100 μL; final concentration, 2 mM) was added to 400 μL PB (pH 7.4; 10 mM). The mixtures were incubated at 37°C on an Eppendorf ThermoMixer Comfort (800 rpm). The reactions were monitored by spotting aliquots (5 μL) from the incubation mixtures onto the TLC plate at designated times. The solvent system used was methanol (MeOH) and CHCl3 (1:9).
Analysis of mixtures containing moxifloxacin plus and minus NAC by NMR.
Deuterated PB (10 mM) was prepared by dissolving monobasic potassium phosphate (KH2PO4; 4 mg) and dibasic potassium phosphate (K2HPO4; 12 mg) in deuterated water (D2O), and the pH was adjusted to 7.4 using 40% (wt/wt) sodium deuteroxide solution in D2O (catalog no. 151882; Sigma-Aldrich, St. Louis, MO). Stock solutions of moxifloxacin (10 mM) and NAC (0.2 M) were prepared in DMSO and deuterated PB (pH 7.4; 10 mM), respectively. In a typical reaction, moxifloxacin (200 μL; 2 mM final concentration) was incubated with NAC (10 μL; 2 mM final concentration) in 790 μL deuterated PB (pH 7.4; 10 mM). In a control experiment, moxifloxacin (200 μL; 2 mM final concentration) was added to 800 μL deuterated PB (pH 7.4; 10 mM). The mixtures were incubated at 37°C in an Eppendorf ThermoMixer Comfort (800 rpm). An aliquot (0.5 mL) of the incubation mixture was taken after 1 h of incubation, and 1H NMR and 19F NMR spectra were recorded. 1H NMR was acquired with 64 scans on a JEOL 400 MHz spectrometer using D2O (Sigma-Aldrich) as an internal standard. Chemical shifts (δ) were reported in parts per million downfield from D2O (δ = 4.79 ppm) for 1H NMR. 19F spectra were recorded on a JEOL spectrometer (376 MHz) using an external reference (α,α,α-trifluorotoluene; δF = −63.72 ppm).
Fluorescence-based detection of intracellular levels of moxifloxacin in wild-type M. smegmatis.
Stock solutions of moxifloxacin (0.05 mM) and NAC (200 mM) were prepared in DMSO and phosphate buffer (pH 7.4, 10 mM), respectively. Wild-type M. smegmatis cultures were grown in Middlebrook 7H9 broth supplemented with glycerol (0.2%) and Tween 80 (0.1%). Exponential-phase cultures of wild-type M. smegmatis, grown to OD600 of 0.3 (985 μL), were transferred to 1.5 mL Eppendorf tubes and either left untreated or treated with NAC (5 μL, final concentration 1 mM) for 1 h prior to addition of 2× MIC of moxifloxacin (10 μL, final concentration 0.5 μM). Treated cells were incubated with shaking at 180 rpm in a rotary shaker incubator at 37°C for 48 h. The cell suspension was then centrifuged at 9,391 × g at 4°C for 15 min, and the pellet was washed twice with 1× PBS and resuspended in 1 mL PBS in a microcentrifuge tube. The cells were lysed by sonication using a 130-W ultrasonic processor (Vx 130W) by stepping a microtip with a 4-min pulse-on time (with 3-s on and 3-s off pulse; 60% amplitude; 20-kHz frequency) under ice-cold conditions. An aliquot (100 μL) of whole-cell lysate was withdrawn from the samples and dispensed into a 96-well microtiter plate. Fluorescence ascribed to moxifloxacin (λex = 289 nm and λem = 488 nm), recovered in NAC-untreated or -pretreated M. smegmatis lysates, was recorded using an Ensight multimode plate reader (PerkinElmer, India). Readings were collected from the top with 25 flashes per well and with a focus height adjusted to 9.5 mm.
Intracellular levels of moxifloxacin in M. smegmatis using LC-MS.
Stock solutions of moxifloxacin (0.05 mM) and NAC (200 mM) were prepared as described above. Whole-cell lysates of wild-type M. smegmatis treated with moxifloxacin (0.5 μM, 2× MIC) with or without NAC (1 mM) were prepared as described for the fluorescence-based method employed for the detection of moxifloxacin. An aliquot (100 μL) of whole-cell lysate was withdrawn from the incubation mixtures, diluted with methyl alcohol (100 μL), and centrifuged at 9,390 × g at 4°C for 15 min. The supernatant fluids (50 μL) were removed, diluted with methyl alcohol (50 μL), and analyzed by LC-MS. All measurements were performed using the positive ion mode with high-resolution, multiple-reaction monitoring (MRM-HR) analysis with a Sciex X500R quadrupole time-of-flight (QTOF) mass spectrometer fitted with an Exion ultrahigh-performance LC (UHPLC) system having a Kinetex 2.6-mm hydrophilic interaction liquid chromatography (HILIC) column (100-Å particle size, 150-mm length, and 3-mm internal diameter; Phenomenex, Intek Chromasol Pvt. Ltd., India). Nitrogen was used as the nebulizer gas, with nebulizer pressure set at 50 lb/in2. MS was calibrated in the positive mode, and samples were analyzed with the following parameters: mode, electrospray ionization (ESI); ion source gas 1 = 40 lb/in2; ion source gas 2 = 50 lb/in2; curtain gas = 30; CAD (collisionally activated dissociation) gas = 7; spray voltage = 5,500 V; and temperature = 500°C. The MRM-HR mass spectrometry parameters were as follows: moxifloxacin (Q1, M+H+) = 402.18, moxifloxacin-NAC adduct (Q2, M+H+) = 565.21, declustering potential = 80 V, collision energy = 20 V, collision exit potential = 5 V, and accumulation time = 0.24 s. The LC runs were for 30 min with a gradient of 100% solvent A (0.1% HCOOH in water) for 5 min and a linear gradient of solvent B (acetonitrile, 0% to 100%) for 25 min followed by 100% solvent A for 5 min, all at a flow rate of 0.5 mL/min.
Determination of MPC.
MPC of moxifloxacin with M. tuberculosis NHN1664 was determined by methods described previously (65, 88). Cultures of MDR strain M. tuberculosis NHN1664 were grown to OD600 0.6 to 0.7. Approximately 2.5 × 109 bacilli were plated on 7H11 agar containing either moxifloxacin (2×, 4×, or 8× MIC) alone or in combination with NAC at either 1 mM or 2 mM. Resistant colonies were enumerated after incubation at 37°C for 8 weeks. The drug-resistant phenotype was confirmed by replating on drug-containing 7H11-agar plates. MPC was determined as the lowest concentration of drug that prevented bacterial colony formation when >2 × 109 bacteria were plated on drug-containing 7H11 plates. Mutation frequency with moxifloxacin was calculated as the number of mutants appearing on drug-containing plates divided by the viable input bacteria.
Determination of drug-tolerant M. tuberculosis population ex vivo.
Murine bone marrow-derived macrophages (BMDMs) were infected with Mrx1-roGFP2-expressing M. tuberculosis H37Rv at a multiplicity of infection of 10. After extracellular bacteria were washed off, infected macrophages were left untreated for 24 h for the emergence of the drug-tolerant population. At 24 h postinfection, macrophages harboring EMSH-reduced and EMSH-basal M. tuberculosis were sorted using a BD FACSAria fusion flow cytometer. The sorted, infected BMDMs were seeded into 96-well plates followed by treatment with 3× MIC of moxifloxacin (1× MIC = 0.5 μM) for 48 h. After treatment, cells were lysed with 0.05% sodium dodecyl sulfate (SDS) in 7H9 medium, serially diluted, and plated on 7H11-OADC agar plates. Plates were incubated at 37°C for 3 to 4 weeks before CFU were enumerated.
In vivo drug efficacy.
All animal studies were executed as per guidelines prescribed by the Committee for the Purpose of Control and Supervision of Experiments on Animals, Government of India, with approval from the Institutional Animal Ethical Committee and Biosafety Level-3 Committee. Six- to 8-week-old female pathogen-free BALB/c mice were infected via a low-dose aerosol exposure to the M. tuberculosis MDR strain NHN1664 using a Madison chamber aerosol generation instrument. The short-course mouse model of infection was performed as described previously (111). At day 1 postinfection, three mice were sacrificed to confirm implantation of ~100 CFU of bacteria per mouse. Mice were randomly divided into 4 groups (n = 5 per group). Feed and water were given ad libitum. Treatment with moxifloxacin and/or NAC started 21 days postinfection and continued for 10 days; untreated control mice received water rather than the two test compounds. Treated mice were administered moxifloxacin (50 mg/kg of body weight), NAC (500 mg/kg of body weight), and a combination of moxifloxacin and NAC at 50 mg/kg and 500 mg/kg of body weight, respectively, by oral gavage, once daily. Five infected mice were sacrificed at the start of treatment as pretreatment controls. After treatment, mice were sacrificed, and the lungs and spleens were harvested for measurement of bacterial burden. CFU were determined by plating appropriate serial dilutions on 7H11 (supplemented with OADC) agar plates and counting visible colonies after 3 to 4 weeks of incubation at 37°C. Data were normalized to whole organs.
Detection of moxifloxacin-resistant mutants in murine model of infection.
Six- to 8-week-old female, pathogen-free BALB/c mice were infected via a high-dose aerosol exposure to the M. tuberculosis MDR strain NHN1664 using a Madison chamber aerosol generation instrument. At day 1 postinfection, three mice were sacrificed to determine implantation of >103 CFU of bacteria per mouse. Mice were then randomly divided into 4 treatment groups, with 6 mice each in the vehicle control group, the NAC-only group, and the groups receiving either moxifloxacin or a combination of NAC and moxifloxacin. Feed and water were given ad libitum. Treatment with moxifloxacin and/or NAC started 14 days postinfection and continued for 14 days; untreated control mice received water rather than the two test compounds. Treated mice were administered moxifloxacin (50 mg/kg of body weight), NAC (500 mg/kg of body weight), and a combination of moxifloxacin and NAC at 50 mg/kg and 500 mg/kg of body weight, respectively, by oral gavage, once daily. Five infected mice were sacrificed at the start of treatment as pretreatment controls. After treatment, mice were sacrificed, and the lungs and spleens were harvested together. Moxifloxacin-resistant mutants were selected by plating the undiluted homogenates (lung and spleen) on five 7H11 plates containing 0.5 μM moxifloxacin (4× MIC). Plates were incubated for 3 to 4 weeks at 37°C.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism version 8.4.3 software (GraphPad, San Diego, CA). Mean and standard deviation values were plotted as indicated in the figure legends. A P value of less than 0.05 was considered significant. Statistical significance was determined by unpaired two-tailed Student's t test; either one-way or two-way analysis of variance (ANOVA) was performed where comparison of multiple groups was made.
Data availability.
Further information, resources, constructs, and reagents are available on request to A.S.
ACKNOWLEDGMENTS
We thank the following for critical comments on the manuscript: Lanbo Shi, Bo Shopsin, and Xilin Zhao.
This work was supported by Wellcome Trust-Department of Biotechnology (DBT) India Alliance grant IA/S/16/2/502700 (A.S.) and in part by DBT grants BT/PR13522/COE/34/27/2015, BT/PR29098/Med/29/1324/2018, and BT/HRD/NBA/39/07/2018-19 (A.S.), DBT-IISc Partnership Program grant 22-0905-0006-05-987 436, and the Infosys Foundation. A.S. is a senior fellow of Wellcome Trust-DBT India Alliance. S. Singh was supported by a National Postdoctoral Fellowship from the Department of Science and Technology (DST), India vide file number PDF/2018/2941. S. Shee was supported by a fellowship award from Indian Institute of Science (IISc).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We declare that no competing interests exist.
Footnotes
Supplemental material is available online only.
Contributor Information
Karl Drlica, Email: drlicaka@njms.rutgers.edu.
Amit Singh, Email: asingh@iisc.ac.in.
REFERENCES
- 1.Lange C, Chesov D, Heyckendorf J, Leung CC, Udwadia Z, Dheda K. 2018. Drug-resistant tuberculosis: an update on disease burden, diagnosis and treatment. Respirology 23:656–673. 10.1111/resp.13304. [DOI] [PubMed] [Google Scholar]
- 2.WHO. 2019. Global tuberculosis report 2019. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.WHO. 2013. Global tuberculosis report 2013. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 4.Shandil RK, Jayaram R, Kaur P, Gaonkar S, Suresh BL, Mahesh BN, Jayashree R, Nandi V, Bharath S, Balasubramanian V. 2007. Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against Mycobacterium tuberculosis: evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob Agents Chemother 51:576–582. 10.1128/AAC.00414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong Y, Xu C, Zhao X, Domagala J, Drlica K. 1998. Fluoroquinolone action against mycobacteria: effects of C-8 substituents on growth, survival, and resistance. Antimicrob Agents Chemother 42:2978–2984. 10.1128/AAC.42.11.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruan Q, Liu Q, Sun F, Shao L, Jin J, Yu S, Ai J, Zhang B, Zhang W. 2016. Moxifloxacin and gatifloxacin for initial therapy of tuberculosis: a meta-analysis of randomized clinical trials. Emerg Microbes Infect 5:e12. 10.1038/emi.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodríguez JC, Ruiz M, Climent A, Royo G. 2001. In vitro activity of four fluoroquinolones against Mycobacterium tuberculosis. Int J Antimicrob Agents 17:229–231. 10.1016/s0924-8579(00)00337-x. [DOI] [PubMed] [Google Scholar]
- 8.Malik M, Zhao X, Drlica K. 2006. Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Mol Microbiol 61:810–825. 10.1111/j.1365-2958.2006.05275.x. [DOI] [PubMed] [Google Scholar]
- 9.Nuermberger EL, Yoshimatsu T, Tyagi S, O'Brien RJ, Vernon AN, Chaisson RE, Bishai WR, Grosset JH. 2004. Moxifloxacin-containing regimen greatly reduces time to culture conversion in murine tuberculosis. Am J Respir Crit Care Med 169:421–426. 10.1164/rccm.200310-1380OC. [DOI] [PubMed] [Google Scholar]
- 10.Pletz MWR, De Roux A, Roth A, Neumann K-H, Mauch H, Lode H. 2004. Early bactericidal activity of moxifloxacin in treatment of pulmonary tuberculosis: a prospective, randomized study. Antimicrob Agents Chemother 48:780–782. 10.1128/AAC.48.3.780-782.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorman SE, Nahid P, Kurbatova EV, Phillips PPJ, Bryant K, Dooley KE, Engle M, Goldberg SV, Phan HTT, Hakim J, Johnson JL, Lourens M, Martinson NA, Muzanyi G, Narunsky K, Nerette S, Nguyen NV, Pham TH, Pierre S, Purfield AE, Samaneka W, Savic RM, Sanne I, Scott NA, Shenje J, Sizemore E, Vernon A, Waja Z, Weiner M, Swindells S, Chaisson RE, Tuberculosis Trials Consortium . 2021. Four-month rifapentine regimens with or without moxifloxacin for tuberculosis. N Engl J Med 384:1705–1718. 10.1056/NEJMoa2033400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PPJ, Nunn AJ, REMoxTB Consortium . 2014. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med 371:1577–1587. 10.1056/NEJMoa1407426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jindani A, Harrison TS, Nunn AJ, Phillips PPJ, Churchyard GJ, Charalambous S, Hatherill M, Geldenhuys H, McIlleron HM, Zvada SP, Mungofa S, Shah NA, Zizhou S, Magweta L, Shepherd J, Nyirenda S, van Dijk JH, Clouting HE, Coleman D, Bateson ALE, McHugh TD, Butcher PD, Mitchison DA, RIFAQUIN Trial Team . 2014. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med 371:1599–1608. 10.1056/NEJMoa1314210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jawahar MS, Banurekha VV, Paramasivan CN, Rahman F, Ramachandran R, Venkatesan P, Balasubramanian R, Selvakumar N, Ponnuraja C, Iliayas AS, Gangadevi NP, Raman B, Baskaran D, Kumar SR, Kumar MM, Mohan V, Ganapathy S, Kumar V, Shanmugam G, Charles N, Sakthivel MR, Jagannath K, Chandrasekar C, Parthasarathy RT, Narayanan PR. 2013. Randomized clinical trial of thrice-weekly 4-month moxifloxacin or gatifloxacin containing regimens in the treatment of new sputum positive pulmonary tuberculosis patients. PLoS One 8:e67030. 10.1371/journal.pone.0067030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drlica K, Zhao X. 2021. Bacterial death from treatment with fluoroquinolones and other lethal stressors. Expert Rev Anti Infect Ther 19:601–618. 10.1080/14787210.2021.1840353. [DOI] [PubMed] [Google Scholar]
- 16.Hong Y, Zeng J, Wang X, Drlica K, Zhao X. 2019. Post-stress bacterial cell death mediated by reactive oxygen species. Proc Natl Acad Sci USA 116:10064–10071. 10.1073/pnas.1901730116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong Y, Li Q, Gao Q, Xie J, Huang H, Drlica K, Zhao X. 2020. Reactive oxygen species play a dominant role in all pathways of rapid quinolone-mediated killing. J Antimicrob Chemother 75:576–585. 10.1093/jac/dkz485. [DOI] [PubMed] [Google Scholar]
- 18.Ehrt S, Schnappinger D. 2009. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol 11:1170–1178. 10.1111/j.1462-5822.2009.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhaskar A, Chawla M, Mehta M, Parikh P, Chandra P, Bhave D, Kumar D, Carroll KS, Singh A. 2014. Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog 10:e1003902. 10.1371/journal.ppat.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CTY, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ. 2014. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci USA 111:E2100–E2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Imlay JA. 2013. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213. 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mishra S, Shukla P, Bhaskar A, Anand K, Baloni P, Jha RK, Mohan A, Rajmani RS, Nagaraja V, Chandra N, Singh A. 2017. Efficacy of β-lactam/β-lactamase inhibitor combination is linked to WhiB4-mediated changes in redox physiology of Mycobacterium tuberculosis. Elife 6:e25624. 10.7554/eLife.25624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishra R, Kohli S, Malhotra N, Bandyopadhyay P, Mehta M, Munshi M, Adiga V, Ahuja VK, Shandil RK, Rajmani RS, Seshasayee ASN, Singh A. 2019. Targeting redox heterogeneity to counteract drug tolerance in replicating Mycobacterium tuberculosis. Sci Transl Med 11:eaaw6635. 10.1126/scitranslmed.aaw6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nambi S, Long JE, Mishra BB, Baker R, Murphy KC, Olive AJ, Nguyen HP, Shaffer SA, Sassetti CM. 2015. The oxidative stress network of Mycobacterium tuberculosis reveals coordination between radical detoxification systems. Cell Host Microbe 17:829–837. 10.1016/j.chom.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Padiadpu J, Baloni P, Anand K, Munshi M, Thakur C, Mohan A, Singh A, Chandra N. 2016. Identifying and tackling emergent vulnerability in drug-resistant mycobacteria. ACS Infect Dis 2:592–607. 10.1021/acsinfecdis.6b00004. [DOI] [PubMed] [Google Scholar]
- 26.Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. 2007. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol Syst Biol 3:91. 10.1038/msb4100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malik M, Hussain S, Drlica K. 2007. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob Agents Chemother 51:28–34. 10.1128/AAC.00739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X, Xu C, Domagala J, Drlica K. 1997. DNA topoisomerase targets of the fluoroquinolones: a strategy for avoiding bacterial resistance. Proc Natl Acad Sci USA 94:13991–13996. 10.1073/pnas.94.25.13991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao X, Wang JY, Xu C, Dong Y, Zhou J, Domagala J, Drlica K. 1998. Killing of Staphylococcus aureus by C-8-methoxy fluoroquinolones. Antimicrob Agents Chemother 42:956–958. 10.1128/AAC.42.4.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drlica K, Zhao X. 2007. Mutant selection window hypothesis updated. Clin Infect Dis 44:681–688. 10.1086/511642. [DOI] [PubMed] [Google Scholar]
- 31.Luan G, Hong Y, Drlica K, Zhao X. 2018. Suppression of reactive oxygen species accumulation accounts for paradoxical bacterial survival at high quinolone concentration. Antimicrob Agents Chemother 62:e01622-17. 10.1128/AAC.01622-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keyer K, Imlay JA. 1996. Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci USA 93:13635–13640. 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 34.Nandakumar M, Nathan C, Rhee KY. 2014. Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat Comm 5:4306. 10.1038/ncomms5306. [DOI] [PubMed] [Google Scholar]
- 35.Hong Y, Li L, Luan G, Drlica K, Zhao X. 2017. Contribution of reactive oxygen species to thymineless death in Escherichia coli. Nat Microbiol 2:1667–1675. 10.1038/s41564-017-0037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seaver LC, Imlay JA. 2001. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol 183:7182–7189. 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gengenbacher M, Rao SP, Pethe K, Dick T. 2010. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology (Reading) 156:81–87. 10.1099/mic.0.033084-0. [DOI] [PubMed] [Google Scholar]
- 38.Wayne LG, Hayes LG. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 64:2062–2069. 10.1128/iai.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wayne LG, Hayes LG. 1998. Nitrate reduction as a marker for hypoxic shiftdown of Mycobacterium tuberculosis. Tuber Lung Dis 79:127–132. 10.1054/tuld.1998.0015. [DOI] [PubMed] [Google Scholar]
- 41.Wayne LG, Sramek HA. 1994. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob Agents Chemother 38:2054–2058. 10.1128/AAC.38.9.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma S, Minch KJ, Rustad TR, Hobbs S, Zhou SL, Sherman DR, Price ND. 2015. Integrated modeling of gene regulatory and metabolic networks in Mycobacterium tuberculosis. PLoS Comput Biol 11:e1004543. 10.1371/journal.pcbi.1004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kapopoulou A, Lew JM, Cole ST. 2011. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb) 91:8–13. 10.1016/j.tube.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Voskuil M, Bartek I, Visconti K, Schoolnik G. 2011. The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front Microbiol 2:105. 10.3389/fmicb.2011.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anand K, Tripathi A, Shukla K, Malhotra N, Jamithireddy AK, Jha RK, Chaudhury SN, Rajmani RS, Ramesh A, Nagaraja V, Gopal B, Nagaraju G, Narain Seshayee AS, Singh A. 2021. Mycobacterium tuberculosis SufR responds to nitric oxide via its 4Fe–4S cluster and regulates Fe–S cluster biogenesis for persistence in mice. Redox Biol 46:102062. 10.1016/j.redox.2021.102062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das M, Dewan A, Shee S, Singh A. 2021. The multifaceted bacterial cysteine desulfurases: from metabolism to pathogenesis. Antioxidants (Basel) 10:997. 10.3390/antiox10070997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319. 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dwyer DJ, Collins JJ, Walker GC. 2015. Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol 55:313–332. 10.1146/annurev-pharmtox-010814-124712. [DOI] [PubMed] [Google Scholar]
- 49.Lamprecht DA, Finin PM, Rahman MA, Cumming BM, Russell SL, Jonnala SR, Adamson JH, Steyn AJC. 2016. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat Comm 7:12393. 10.1038/ncomms12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saini V, Cumming BM, Guidry L, Lamprecht DA, Adamson JH, Reddy VP, Chinta KC, Mazorodze JH, Glasgow JN, Richard-Greenblatt M, Gomez-Velasco A, Bach H, Av-Gay Y, Eoh H, Rhee K, Steyn AJ. 2016. Ergothioneine maintains redox and bioenergetic homeostasis essential for drug susceptibility and virulence of Mycobacterium tuberculosis. Cell Rep 14:572–585. 10.1016/j.celrep.2015.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee BS, Kalia NP, Jin XEF, Hasenoehrl EJ, Berney M, Pethe K. 2019. Inhibitors of energy metabolism interfere with antibiotic-induced death in mycobacteria. J Biol Chem 294:1936–1943. 10.1074/jbc.RA118.005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mavi PS, Singh S, Kumar A. 2020. Reductive stress: new insights in physiology and drug tolerance of mycobacterium. Antioxid Redox Signal 32:1348–1366. 10.1089/ars.2019.7867. [DOI] [PubMed] [Google Scholar]
- 53.Bhat SA, Iqbal IK, Kumar A. 2016. Imaging the NADH:NAD+ homeostasis for understanding the metabolic response of mycobacterium to physiologically relevant stresses. Front Cell Infect Microbiol 6:145. 10.3389/fcimb.2016.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jaeschke H, Kleinwaechter C, Wendel A. 1992. NADH-dependent reductive stress and ferritin-bound iron in allyl alcohol-induced lipid peroxidation in vivo: the protective effect of vitamin E. Chem Biol Interact 81:57–68. 10.1016/0009-2797(92)90026-h. [DOI] [PubMed] [Google Scholar]
- 55.Kareyeva AV, Grivennikova VG, Vinogradov AD. 2012. Mitochondrial hydrogen peroxide production as determined by the pyridine nucleotide pool and its redox state. Biochim Biophys Acta 1817:1879–1885. 10.1016/j.bbabio.2012.03.033. [DOI] [PubMed] [Google Scholar]
- 56.Vinogradov AD, Grivennikova VG. 2016. Oxidation of NADH and ROS production by respiratory complex I. Biochim Biophys Acta 1857:863–871. 10.1016/j.bbabio.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 57.Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, Mootha VK. 2016. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352:231–235. 10.1126/science.aad4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vilchèze C, Weisbrod TR, Chen B, Kremer L, Hazbón MH, Wang F, Alland D, Sacchettini JC, Jacobs WR, Jr.. 2005. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother 49:708–720. 10.1128/AAC.49.2.708-720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vilchèze C, Hartman T, Weinrick B, Jain P, Weisbrod TR, Leung LW, Freundlich JS, Jacobs WR. 2017. Enhanced respiration prevents drug tolerance and drug resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 114:4495–4500. 10.1073/pnas.1704376114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vilchèze C, Jacobs WR. 2021. The promises and limitations of N- acetylcysteine as a potentiator of first-line and second-line tuberculosis drugs. Antimicrob Agents Chemother 65:e01703-20. 10.1128/AAC.01703-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Held KD, Biaglow JE. 1994. Mechanisms for the oxygen radical-mediated toxicity of various thiol-containing compounds in cultured mammalian cells. Radiat Res 139:15–23. 10.2307/3578727. [DOI] [PubMed] [Google Scholar]
- 62.Jaworska M, Szulińska G, Wilk M, Tautt J. 1999. Capillary electrophoretic separation of N-acetylcysteine and its impurities as a method for quality control of pharmaceuticals. J Chromatogr A 853:479–485. 10.1016/s0021-9673(99)00727-x. [DOI] [PubMed] [Google Scholar]
- 63.Sommer I, Schwebel H, Adamo V, Bonnabry P, Bouchoud L, Sadeghipour F. 2020. Stability of N-acetylcysteine (NAC) in standardized pediatric parenteral nutrition and evaluation of N,N-diacetylcystine (DAC) formation. Nutrients 12:1849. 10.3390/nu12061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Srinivas V, Arrieta-Ortiz ML, Kaur A, Peterson EJR, Baliga NS. 2020. PerSort facilitates characterization and elimination of persister subpopulation in mycobacteria. mSystems 5:e01127-20. 10.1128/mSystems.01127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singh S, Kalia NP, Joshi P, Kumar A, Sharma PR, Kumar A, Bharate SB, Khan IA. 2017. Boeravinone B, a novel dual inhibitor of NorA bacterial efflux pump of Staphylococcus aureus and human p-glycoprotein, reduces the biofilm formation and intracellular invasion of bacteria. Front Microbiol 8:1868. 10.3389/fmicb.2017.01868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cui J, Liu Y, Wang R, Tong W, Drlica K, Zhao X. 2006. The mutant selection window in rabbits infected with Staphylococcus aureus. J Infect Dis 194:1601–1608. 10.1086/508752. [DOI] [PubMed] [Google Scholar]
- 67.Mishra R, Yadav V, Guha M, Singh A. 2021. Heterogeneous host–pathogen encounters coordinate antibiotic resilience in Mycobacterium tuberculosis. Trends Microbiol 29:606–620. 10.1016/j.tim.2020.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amaral EP, Conceição EL, Costa DL, Rocha MS, Marinho JM, Cordeiro-Santos M, D'Império-Lima MR, Barbosa T, Sher A, Andrade BB. 2016. N-acetyl-cysteine exhibits potent anti-mycobacterial activity in addition to its known anti-oxidative functions. BMC Microbiol 16:251. 10.1186/s12866-016-0872-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao R, Teskey G, Islamoglu H, Abrahem R, Munjal S, Gyurjian K, Zhong L, Venketaraman V. 2018. Characterizing the effects of glutathione as an immunoadjuvant in the treatment of tuberculosis. Antimicrob Agents Chemother 62:e01132-18. 10.1128/AAC.01132-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Y, Liu X, Qu Y, Wang X, Li L, Zhao X. 2012. Inhibitors of reactive oxygen species accumulation delay and/or reduce the lethality of several antistaphylococcal agents. Antimicrob Agents Chemother 56:6048–6050. 10.1128/AAC.00754-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Almeida D, Nuermberger E, Tyagi S, Bishai WR, Grosset J. 2007. In vivo validation of the mutant selection window hypothesis with moxifloxacin in a murine model of tuberculosis. Antimicrob Agents Chemother 51:4261–4266. 10.1128/AAC.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ginsburg AS, Sun R, Calamita H, Scott CP, Bishai WR, Grosset JH. 2005. Emergence of fluoroquinolone resistance in Mycobacterium tuberculosis during continuously dosed moxifloxacin monotherapy in a mouse model. Antimicrob Agents Chemother 49:3977–3979. 10.1128/AAC.49.9.3977-3979.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lobritz MA, Belenky P, Porter CBM, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, Collins JJ. 2015. Antibiotic efficacy is linked to bacterial cellular respiration. Proc Natl Acad Sci USA 112:8173–8180. 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tan MP, Sequeira P, Lin WW, Phong WY, Cliff P, Ng SH, Lee BH, Camacho L, Schnappinger D, Ehrt S, Dick T, Pethe K, Alonso S. 2010. Nitrate respiration protects hypoxic Mycobacterium tuberculosis against acid and reactive nitrogen species stresses. PLoS One 5:e13356. 10.1371/journal.pone.0013356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sohaskey CD. 2008. Nitrate enhances the survival of Mycobacterium tuberculosis during inhibition of respiration. J Bacteriol 190:2981–2986. 10.1128/JB.01857-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cunningham-Bussel A, Zhang T, Nathan CF. 2013. Nitrite produced by Mycobacterium tuberculosis in human macrophages in physiologic oxygen impacts bacterial ATP consumption and gene expression. Proc Natl Acad Sci USA 110:E4256–E4265. 10.1073/pnas.1316894110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shapleigh JP. 2009. Dissimilatory and assimilatory nitrate reduction in the purple photosynthetic bacteria, p 623–642. In Hunter CN, Daldal F, Thurnauer MC, Beatty JT (ed), The purple phototrophic bacteria. Advances in photosynthesis and respiration, vol 28. Springer, Dordrecht, The Netherlands. [Google Scholar]
- 78.Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJC. 2009. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog 5:e1000545. 10.1371/journal.ppat.1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik G. 2003. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med 198:705–713. 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Richardson AR, Libby SJ, Fang FC. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672–1676. 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 81.Price-Whelan A, Dietrich LEP, Newman DK. 2007. Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J Bacteriol 189:6372–6381. 10.1128/JB.00505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ezeriņa D, Takano Y, Hanaoka K, Urano Y, Dick TP. 2018. N-acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular H2S and sulfane sulfur production. Cell Chem Biol 25:447–459.E4. 10.1016/j.chembiol.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rodríguez-Rojas A, Kim JJ, Johnston PR, Makarova O, Eravci M, Weise C, Hengge R, Rolff J. 2020. Non-lethal exposure to H2O2 boosts bacterial survival and evolvability against oxidative stress. PLoS Genet 16:e1008649. 10.1371/journal.pgen.1008649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mosel M, Li L, Drlica K, Zhao X. 2013. Superoxide-mediated protection of Escherichia coli from antimicrobials. Antimicrob Agents Chemother 57:5755–5759. 10.1128/AAC.00754-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li L, Hong Y, Luan G, Mosel M, Malik M, Drlica K, Zhao X. 2014. Ribosomal elongation factor 4 promotes cell death associated with lethal stress. mBio 5:e01708-14. 10.1128/mBio.01708-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dorsey-Oresto A, Lu T, Mosel M, Wang X, Salz T, Drlica K, Zhao X. 2013. YihE kinase is a central regulator of programmed cell death in bacteria. Cell Rep 3:528–537. 10.1016/j.celrep.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Imlay JA, Linn S. 1986. Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J Bacteriol 166:519–527. 10.1128/jb.166.2.519-527.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dong Y, Zhao X, Kreiswirth BN, Drlica K. 2000. Mutant prevention concentration as a measure of antibiotic potency: studies with clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 44:2581–2584. 10.1128/AAC.44.9.2581-2584.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O'Brien P, Chen C, Kaya F, Weiner DM, Chen P-Y, Song T, Lee M, Shim TS, Cho JS, Kim W, Cho SN, Olivier KN, Barry CE, Dartois V. 2015. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med 21:1223–1227. 10.1038/nm.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sarathy J, Blanc L, Alvarez-Cabrera N, O’Brien P, Dias-Freedman I, Mina M, Zimmerman M, Kaya F, Ho Liang H-P, Prideaux B, Dietzold J, Salgame P, Savic RM, Linderman J, Kirschner D, Pienaar E, Dartois V. 2019. Fluoroquinolone efficacy against tuberculosis is driven by penetration into lesions and activity against resident bacterial populations. Antimicrob Agents Chemother 63:e02516-18. 10.1128/AAC.02516-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davies Forsman L, Niward K, Kuhlin J, Zheng X, Zheng R, Ke R, Hong C, Werngren J, Paues J, Simonsson USH, Eliasson E, Hoffner S, Xu B, Alffenaar J-W, Schön T, Hu Y, Bruchfeld J. 2021. Suboptimal moxifloxacin and levofloxacin drug exposure during treatment of patients with multidrug-resistant tuberculosis: results from a prospective study in China. Eur Respir J 57:2003463. 10.1183/13993003.03463-2020. [DOI] [PubMed] [Google Scholar]
- 92.Baniasadi S, Eftekhari P, Tabarsi P, Fahimi F, Raoufy MR, Masjedi MR, Velayati AA. 2010. Protective effect of N-acetylcysteine on antituberculosis drug-induced hepatotoxicity. Eur J Gastroenterol Hepatol 22:1235–1238. 10.1097/MEG.0b013e32833aa11b. [DOI] [PubMed] [Google Scholar]
- 93.Kranzer K, Elamin WF, Cox H, Seddon JA, Ford N, Drobniewski F. 2015. A systematic review and meta-analysis of the efficacy and safety of N-acetylcysteine in preventing aminoglycoside-induced ototoxicity: implications for the treatment of multidrug-resistant TB. Thorax 70:1070–1077. 10.1136/thoraxjnl-2015-207245. [DOI] [PubMed] [Google Scholar]
- 94.Palanisamy GS, Kirk NM, Ackart DF, Shanley CA, Orme IM, Basaraba RJ. 2011. Evidence for oxidative stress and defective antioxidant response in guinea pigs with tuberculosis. PLoS One 6:e26254. 10.1371/journal.pone.0026254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mahakalkar SM, Nagrale D, Gaur S, Urade C, Murhar B, Turankar A. 2017. N-acetylcysteine as an add-on to Directly Observed Therapy Short-I therapy in fresh pulmonary tuberculosis patients: a randomized, placebo-controlled, double-blinded study. Perspect Clin Res 8:132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nagrale D, Mahakalkar S, Gaur S. 2013. Supplementation of N-acetylcysteine as an adjuvant in treatment of newly diagnosed pulmonary tuberculosis patients: a prospective, randomized double blind, placebo controlled study. Eur Respir J 42:P2833. [Google Scholar]
- 97.Firsov AA, Vostrov SN, Lubenko IY, Drlica K, Portnoy YA, Zinner SH. 2003. In vitro pharmacodynamic evaluation of the mutant selection window hypothesis using four fluoroquinolones against Staphylococcus aureus. Antimicrob Agents Chemother 47:1604–1613. 10.1128/AAC.47.5.1604-1613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gumbo T, Louie A, Deziel MR, Parsons LM, Salfinger M, Drusano GL. 2004. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis 190:1642–1651. 10.1086/424849. [DOI] [PubMed] [Google Scholar]
- 99.Steyn AJC, Joseph J, Bloom BR. 2003. Interaction of the sensor module of Mycobacterium tuberculosis H37Rv KdpD with members of the Lpr family. Mol Microbiol 47:1075–1089. 10.1046/j.1365-2958.2003.03356.x. [DOI] [PubMed] [Google Scholar]
- 100.Singh A, Mai D, Kumar A, Steyn AJC. 2006. Dissecting virulence pathways of Mycobacterium tuberculosis through protein–protein association. Proc Natl Acad Sci USA 103:11346–11351. 10.1073/pnas.0602817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fan XY, Tang BK, Xu YY, Han AX, Shi KX, Wu YK, Ye Y, Wei ML, Niu C, Wong KW, Zhao GP, Lyu LD. 2018. Oxidation of dCTP contributes to antibiotic lethality in stationary-phase mycobacteria. Proc Natl Acad Sci USA 115:2210–2215. 10.1073/pnas.1719627115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vilchèze C, Hartman T, Weinrick B, Jacobs WR. 2013. Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nat Comm 4:1881. 10.1038/ncomms2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Taneja NK, Tyagi JS. 2007. Resazurin reduction assays for screening of anti-tubercular compounds against dormant and actively growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J Antimicrob Chemother 60:288–293. 10.1093/jac/dkm207. [DOI] [PubMed] [Google Scholar]
- 104.Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. 2013. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41:D991–D995. 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li S, Sinai M. 2019. GeneOverlap: test and visualize gene overlaps. 10.18129/B9.bioc.GeneOverlap. [DOI]
- 107.R Core Team. 2018. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. [Google Scholar]
- 108.Kurthkoti K, Amin H, Marakalala MJ, Ghanny S, Subbian S, Sakatos A, Livny J, Fortune SM, Berney M, Rodriguez GM. 2017. The capacity of Mycobacterium tuberculosis to survive iron starvation might enable it to persist in iron-deprived microenvironments of human granulomas. mBio 8:e01092-17. 10.1128/mBio.01092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fish WW. 1988. Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol 158:357–364. 10.1016/0076-6879(88)58067-9. [DOI] [PubMed] [Google Scholar]
- 110.Chawla M, Parikh P, Saxena A, Munshi M, Mehta M, Mai D, Srivastava AK, Narasimhulu KV, Redding KE, Vashi N, Kumar D, Steyn AJC, Singh A. 2012. Mycobacterium tuberculosis WhiB4 regulates oxidative stress response to modulate survival and dissemination in vivo. Mol Microbiol 85:1148–1165. 10.1111/j.1365-2958.2012.08165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lenaerts AJ, Gruppo V, Brooks JV, Orme IM. 2003. Rapid in vivo screening of experimental drugs for tuberculosis using gamma interferon gene-disrupted mice. Antimicrob Agents Chemother 47:783–785. 10.1128/AAC.47.2.783-785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 to S6, Fig. S1 to S25, and legend of Data Set S1. Download aac.00592-22-s0001.pdf, PDF file, 1.6 MB (1.6MB, pdf)
Data Set S1. Download aac.00592-22-s0002.xlsx, XLSX file, 0.4 MB (464KB, xlsx)
Data Availability Statement
Further information, resources, constructs, and reagents are available on request to A.S.