

Abstract
Distinguishing pathogenic variants from non-pathogenic ones remains a major challenge in clinical genetic testing of primary immunodeficiency (PID) patients. Most of the existing mutation pathogenicity prediction tools treat all mutations as homogeneous entities, ignoring the differences in characteristics of different genes, and use the same model for genes in different diseases. In this study, we developed a single nucleotide variant (SNV) pathogenicity prediction tool, Variant Impact Predictor for PIDs (VIPPID; https://mylab.shinyapps.io/VIPPID/), which was tailored for PIDs genes and used a specific model for each of the most prevalent PID known genes. It employed a Conditional Inference Forest model and utilized information of 85 features of SNVs and scores from 20 existing prediction tools. Evaluation of VIPPID showed that it had superior performance (area under the curve = 0.91) over non-specific conventional tools. In addition, we also showed that the gene-specific model outperformed the non-gene-specific models. Our study demonstrated that disease-specific and gene-specific models can improve SNV pathogenicity prediction performance. This observation supports the notion that each feature of mutations in the model can be potentially used, in a new algorithm, to investigate the characteristics and function of the encoded proteins.
Keywords: inborn errors of immunity (IEI), primary immunodeficiency (PID), genetic mutation, variant prediction, machine learning, computational analysis
Introduction
Primary immunodeficiencies (PIDs) refer to a group of >450 inherited diseases characterized by abnormal innate or adaptive immunity, and its prevalence is estimated to be in the excess of 1/1000 in the general population [1]. PIDs lead to increased susceptibility to infections, immune dysregulation and cancers and result in a significant burden to the society as well as the patients’ families. Most PIDs are caused by monogenic mutations [2–4], and genetic testing has been widely used for the diagnosis, treatment and prevention of PIDs. For instance, newborn screening of severe combined immune deficiency (SCID) can lead to life-saving interventions before the occurrence of infections [5]. Accurate identification of disease-causing mutations can also facilitate earlier diagnosis and better management of the disease [6, 7]. Moreover, accurate genetic testing allows better family planning and carrier detection in the patient’s family members [8].
One of the challenges in genetic testing is to distinguish pathogenic variants from non-pathogenic ones. Whole-genome sequencing and whole-exome sequencing usually identify a large number of rare variants, many of which are variants of unknown significance [9]. Out of hundreds of thousands of variants in an exome or genome profile, there is only one or very few which are pathogenic. Separating pathogenic disease-causing mutations from benign ones is a critical and challenging task in genetic testing.
Numerous tools have been developed to predict the pathogenicity of variants, including Sorting Intolerant from Tolerant [10], Polymorphism Phenotyping v2 [11], Combined Annotation-Dependent Depletion [12], MutationTaster [13], MutationAssessor [14] and many others. Most existing tools are using a non-disease-specific approach, thus employing the same model for all diseases. A few existing tools employ a disease-specific approach, for somatic mutations in cancers [15–18], or predict the new pathogenic variants by employing possible association with known pathogenic variants [19, 20]. Genes causing a specific disease or a group of similar diseases tend to be involved in common biological processes, and we hypothesize that they have a higher probability of sharing common characteristics [21]. For instance, many genes associated with Alzheimer’s disease and some other neurodegenerative diseases, including amyotrophic lateral sclerosis, Parkinson’s disease and Huntington’s disease, are all involved in shared pathways with similar protein structures and molecular properties [22]. Moreover, disease-specific variant classifiers have been proved to outperform the state-of-the-art genome-wide tools in inherited cardiac conditions [23] and inherited retinal dystrophies [24]. Therefore, a mutation pathogenicity prediction model tailored for a specific disease or a group of similar diseases would be expected to achieve a better performance also in immune disorders.
The functional impact of non-synonymous single nucleotide variants (SNVs) can be influenced by features of the resulting amino acid substitution, including the change in their size, hydrophobicity, charge, the properties of their context and other molecular properties. Most of the existing tools use a similar model to predict the pathogenicity of mutations in different genes, which can limit the accuracy of the model since different genes can have different characteristics. For instance, one or more types of forces of hydrogen bonding, hydrophobic interactions, ionic bonding and disulfide bridges [25, 26] can be critical in maintaining the three-dimensional (3D) structure of proteins. Thus, the structure of different proteins may rely on different types of forces, and mutations with different properties can exert an influence on their function. The gene-specific thresholds of impact cutoff values may improve the accuracy of existing genome-wide classifiers [27]. Gene-specific mutation pathogenicity prediction models could, therefore, account for the differences in gene properties and its pathogenic variants and have the potential to improve its accuracy by targeted machine learning. Most of the existing tools also select different features and use different algorithms for prediction and thus each of them has its own strengths and limitations. The combination of results from different existing tools has the possibility to overcome the limitation within a single algorithm and the selection of a specific set of features.
Herein, we developed a disease-specific tool to predict the pathogenicity of SNVs for PIDs, Variant Impact Predictor for PIDs (VIPPID), using a gene-specific approach by training sub-models for each of common PID genes. This method can be used to predict the functional consequences of SNVs in PID genes with improved accuracy in comparison with the existing tools. In this study, to predict their pathogenicity, we used the information of 85 features of SNVs, which comprehensively describe the properties of the mutations. In our tool, scores from different existing pathogenicity prediction tools were also incorporated to enable our model to utilize a most comprehensive set of information. To our knowledge, this is the first genomic functional consequence prediction tool that is solely designed for PIDs.
Methods
Selection of the training data set for the prediction model
Data of mutations with known pathogenicity for PIDs were collected from the Resource of Asian Primary Immunodeficiency Diseases (RAPID) database [28], Human Gene Mutation Database (HGMD) [29] and Clinical significance variants (ClinVar) [30] to generate a comprehensive PID genomic variant data set. All single nucleotide variants from the RAPID database were selected. Pathogenic variants or likely disease-causing mutations of 46 common PIDs known genes (29 very frequent and 17 frequent diseases, Table 1) based on our current knowledge were retrieved from HGMD (marked as ‘disease mutation’ or ‘likely disease-causing mutations’) and ClinVar (marked as ‘pathogenic’ or ‘likely pathogenic’ germline mutations). Both lists of mutations collected from HGMD and ClinVar databases were manually curated to exclude those that are associated with conditions unrelated to PIDs. Finally, PID mutations collected from RAPID, HGMD and ClinVar were merged and duplicated entries were removed and subsequently used as the training data set.
Table 1.
The statistic of SNVs used as a training data set for machine learning
| Gene | No. of PID SNVs | No. of non-PID SNVs | Disease | IUIS classification | Inheritance | OMIM |
|---|---|---|---|---|---|---|
| AIRE | 57 | 68 | APECED (APS-1), autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy | Immune dysregulation | AR or AD | 240300 |
| ATM | 149 | 178 | Ataxia-telangiectasia | Syndromic combined immunodeficiencies | AR | 607585 |
| BTK | 317 | 62 | BTK deficiency, X-linked agammaglobulinemia | Predominantly antibody deficiencies | XL | 300300 |
| C3 | 69 | 82 | C3 deficiency (LOF) | Complement defects | AR | 120700 |
| C3 GOF | AD GOF | |||||
| CD40LG | 69 | 46 | CD40 ligand (CD154) deficiency | Combined immunodeficiencies | XL | 308230 |
| CFH | 173 | 207 | Factor H deficiency | Complement defects | AR or AD | 134370 |
| CFTR | 679 | 510 | Cystic fibrosis | Phagocytic defects | AR | 602421 |
| CYBB | 177 | 101 | Macrophage gp91 phox deficiency | Phagocytic defects | XL | 300645 |
| ELANE | 143 | 128 | Elastase deficiency (Severe congential neutropenia 1) | Phagocytic defects | AD | 130130 |
| FANCA | 113 | 135 | Fanconi anemia type A | Syndromic combined immunodeficiencies | AR | 227650 |
| FAS | 59 | 70 | ALPS-FAS | Immune dysregulation | AD or AR | 134637 |
| FOXP3 | 48 | 57 | IPEX, immune dysregulation, polyendocrinopathy, enteropathy X-linked | Immune dysregulation | XL | 300292 |
| IL2RG | 103 | 92 | γc deficiency (common gamma chain SCID, CD132 deficiency) | Combined immunodeficiencies | XL | 308380 |
| ITGB2 | 48 | 57 | Leukocyte adhesion deficiency type 1 | Phagocytic defects | AR | 600065 |
| KMT2D | 137 | 164 | Kabuki syndrome (types 1 and 2) | Syndromic combined immunodeficiencies | AD | 602113 |
| MEFV | 157 | 188 | Familial Mediterranean fever | Autoinflammatory disorders | AR LOF | 249100 |
| AD | 134610 | |||||
| MVK | 120 | 144 | Mevalonate kinase deficiency (Hyper IgD syndrome) | Autoinflammatory disorders | AR | 260920 |
| NLRP3 | 125 | 150 | Muckle–Wells syndrome | Autoinflammatory disorders | AD GOF | 191900 |
| NOD2 | 99 | 118 | Blau syndrome | Autoinflammatory disorders | AD | 186580 |
| PRF1 | 130 | 156 | Perforin deficiency (FHL2) | Immune dysregulation | AR | 170280 |
| RAG1 | 127 | 152 | RAG deficiency | Combined immunodeficiencies | AR | 179615 |
| RAG2 | 49 | 58 | RAG deficiency | Combined immunodeficiencies | AR | 179616 |
| SERPING1 | 199 | 201 | C1 inhibitor deficiency | Complement defects | AD | 606860 |
| STAT1 | 115 | 109 | STAT1 deficiency | Innate immune deficiencies | AD LOF | 614892 |
| AR LOF | 600555 | |||||
| STAT3 | 140 | 135 | AD-HIES STAT3 deficiency (Job syndrome) | Syndromic combined immunodeficiencies | AD LOF | 147060 |
| STAT3 GOF mutation | AD GOF | 102582 | ||||
| TAZ | 70 | 64 | Barth syndrome (3-Methylglutaconic aciduria type II) | Phagocytic defects | XL | 300394 |
| TNFRSF1A | 96 | 115 | TNF receptor-associated periodic syndrome | Autoinflammatory disorders | AD | 142680 |
| UNC13D | 80 | 96 | UNC13D/Munc13–4 deficiency (FHL3) | Immune dysregulation | AR | 608897 |
| WAS | 120 | 112 | Wiskott–Aldrich syndrome (WAS LOF) | Syndromic combined immunodeficiencies | XL | 300392 |
| X-linked neutropenia/myelodysplasia | XL GOF | 300299 | ||||
| Others | 897 | 482 | ||||
| Total | 4865 | 4237 |
IUIS, International Union of Immunological Societies; LOF, loss of function; GOF, gain of function; AD, autosomal dominant; AR, autosomal recessive; XL, X-linked; OMIM, Online Mendelian Inheritance in Man; APS-1: Autoimmune polyglandular syndrome type 1; ALPS-FAS, autoimmune lymphoproliferative syndrome (ALPS) with FAS mutation; FHL2, familial hemophagocytic lymphohistiocytosis type 2; HIES, hyper IgE syndromes; TNF, tumor necrosis factor receptor; and IPEX, immunodysregulation polyendocrinopathy enteropathy X-linked.
Gene abbreviations: AIRE, autoimmune regulator; ATM, ATM serine/threonine kinase; BTK, Bruton tyrosine kinase; C3, Complement Component 3; CD40LG, CD40 ligand; CFH, complement factor H; CFTR, CF transmembrane conductance regulator; CYBB, cytochrome b-245 beta chain; ELANE, elastase, neutrophil expressed; FANCA, FA complementation group A; FAS, Fas cell surface death receptor; FOXP3, forkhead box P3; IL2RG, interleukin 2 receptor subunit gamma; ITGB2, integrin subunit beta 2; KMT2D lysine methyltransferase 2D; MEFV, MEFV innate immuity regulator, pyrin; MVK, mevalonate kinase; NLRP3, NLR family pyrin domain containing 3; NOD2, nucleotide binding oligomerization domain containing 2; PRF1, perforin 1; RAG1, recombination activating 1; RAG2, recombination activating 2; SERPING1, serpin family G member 1; STAT1, signal transducer and activator of transcription 1; STAT3 signal transducer and activator of transcription 3; TAZ tafazzin, phospholipid-lysophospholipid transacylase; TNFRSF1A, TNF receptor superfamily member 1A; UNC13D, unc-13 homolog D; WAS, WASP actin nucleation promoting factor.
To collect non-PID pathogenic mutation set for training, SNVs in PID genes from the exome samples of the Genome Aggregation Database (gnomAD, version r2.1.1) [31] were downloaded and annotated by the Variant Effect Predictor [32]. Among the SNVs from the gnomAD database, only those absent in the collected PID mutation data set and reported as ‘benign’ or ‘uncertain significance’ in the ClinVar database (version 2021-02-21) [33] were selected. Exome data from gnomAD were used because it is the collection of mutations from 125 748 persons, containing a large number of rare mutations, which can reduce the risk of overestimating the performance of the model caused by only using high-frequency mutations as non-PID mutations in the training model. We also filtered out the mutations with an allele frequency ≥ 0.01 in the gnomAD database; we only kept the mutations with an alternative allele count ≥1 and used them as non-PID mutations for training.
Mutation annotation
To acquire information about different features of SNVs, ‘SNVBox’ [34] was used for annotation. The tool provided information on the properties of amino acid substitution, properties of the mutation in an exonic context, evolutionary conservation and properties of mutations in a 3D structure context and properties from the Universal Protein Resource (UniProt) database [35]. In addition, all SNVs in the training data set were also annotated by ‘VarCards’ [36], an integrated annotation engine. Scores from 20 existing genomic variation functional consequences prediction tools were extracted and used as features of SNVs in our VIPPID model. As there are significantly more non-PID pathogenic mutations than PID mutations in most genes, which can cause a class imbalance in machine learning and thus, to balance the two classes, we ensured that the ratio of non-PID to PID mutations was ≤ 1.2, by randomly selected non-PID mutations if they exceed this ratio.
Machine learning and calculation of feature importance
Data were processed with Perl version 5.22 and R version 3.4.4. The VIPPID was trained as a Conditional Inference Forest machine learning model using the R package ‘caret’. Function ‘Boruta’ in R package ‘Boruta’ was used for feature selection with maxRuns = 50 to select candidate features that most likely contribute to the accuracy of the model and remove redundant features. Features were assigned as either ‘Confirmed’, ‘Rejected’ or ‘Tentative’ by ‘Boruta’ in its final decision based on comparison with shadow features, and features assigned as ‘Confirmed’ or ‘Tentative’ were selected and used for model training. Besides, a score to indicate the importance of the feature was also calculated for each feature by ‘Boruta’. Function ‘tuneRF’ in R package ‘randomForest’ was used for model tuning with ‘stepFactor’ = 2.0, ‘improve’ = 1e−5 and ‘ntree’ = 500. The ‘mtry’ parameter, which is the number of predictors sampled for splitting at each node, was selected by choosing the one with the least out-of-bag error, and an upper limit of 20 was set for ‘mtry’ to reduce the risk of overfitting. Then, ‘Conditional Inference Forest’ was trained using function ‘cforest’ in package ‘party’. ‘Conditional Inference Forest’ was used instead of ‘Random Forest’ as the latter has been shown to have selection bias [37], which is in favor of variables that are continuous or have a higher number of categories. To evaluate the performance of the model, the model was trained and tested using nonoverlapping data sets (cross-validation). To reduce potential bias in evaluation, 10 times cross-validation was performed. Receiver operating characteristic (ROC) curves were used for the quantification of the model’s performance and the comparison with other models. R package ‘pROC’ was used for ROC curves generation and area under the curve (AUC) values calculation.
In the gene-specific model, a sub-model, which is an independent Conditional Inference Forest model, was trained separately for each of the genes with 45 or more reported pathogenic SNVs (very frequent PID, n = 29, Table 1). In the gene-specific model, mutations from all genes were used for training, but mutations from target genes were assigned with weights = 3.0 and mutations from other genes were assigned with weights of 1.0, by setting the ‘weights’ argument in ‘train’ function in ‘caret’ package. This weighting approach was used as it can make the model learning focus on the target genes but not lose the generality of common PID mutation characteristics. In prediction, SNVs from genes where sub-models were available would be predicted by the sub-model; otherwise, for SNVs from genes without a sub-model, the non-gene-specific model would be used for prediction.
We also built a non-gene-specific model for comparison with our gene-specific model. In the non-gene-specific model, all PID genes were trained together in a common model with identical weights and mutations in all PID genes that shared the same parameters in the model. All mutations in PID were predicted by this model.
R package `umap' and `Rtsne' was used for uniform manifold approximation and projection (UMAP) and t-distributed stochastic neighbor embedding (t-SNE), respectively, and package ‘ggplot2’ was used for visualization of the UMAP and t-SNE results. The median of feature importance values of each gene was calculated and used for UMAP and t-SNE analysis, and the feature importance value of −Inf (minus infinity) was assigned as zero in the analysis.
Verification of machine learning algorithms
To further evaluate the performance of our newly developed model, it was applied on an independent mutation set of 26 reviewed pathogenic or likely-pathogenic variants of known PID pathogenic genes from a large unselected PID cohort of 1318 patients [38] and 39 validated in-house PID pathogenic variants. For sensitivity and specificity calculation, the benign variants were randomly selected using the same method as used in the training set.
Results
Mutation data statistic
A total of 11 677 unique pathogenic variants in 175 genes from the RAPID, HGMD and ClinVar databases were collected, and pathogenicity scores predicted by existing tools and feature information from SNVbox were available for 4865 of them from 146 genes, which were used as PID mutations in the machine learning training data set. While 4237 SNVs from gnomAD from the same gene sets were annotated as ‘benign’ or ‘uncertain significance’ in the ClinVar database and have allele frequency < 0.01, they were used as non-PID mutations in the training data set (Table 1). High-frequency variants were excluded from the non-PID group as the mutations in the PID group were mostly rare mutations, which can reduce the risk of model performance overestimation caused by different distribution of allele frequencies in two mutation groups. Among these 146 genes in the training data set, 29 had over 45 PID SNVs, and they were trained by gene-specific models (Table 1).
Mutation annotation and pathogenicity prediction by other tools
Eighty-five features of SNVs were investigated (Table S1, see Supplementary Data available online at https://academic.oup.com/bib, and Figure S1, see Supplementary Data available online at https://academic.oup.com/bib), and a score to quantify each feature of each SNV was calculated. These features include amino acid substitution features, exonic features which were based on mutations and evolutionary conservation in multi-species alignment, features based on 3D structure of the translated protein, features based on amino acid composition in the surrounding regions, features based on multiple sequences alignment of homologous proteins and features from UniProt databases.
Twenty existing SNV pathogenicity prediction tools (Table S2, see Supplementary Data available online at https://academic.oup.com/bib) were used to predict the effects of SNVs in the training set. Prediction scores from existing tools, along with 85 feature scores, were used for VIPPID model training.
Performance of the model
At first, we used a non-gene specific approach, which treated all PID genes as homogenous entities and trained all the genes together in a single Conditional Inference Forest model using 85 features scores and 20 impact scores from other existing prediction tools.
As different genes have distinct characteristics and different features can influence the impact of mutations on them, it is reasonable to train the model for each gene separately. Therefore, we trained an independent Conditional Inference Forest sub-model separately for each gene that has 45 or more reported pathogenic PIDs in the training data set. For the gene-specific model, mutations from all PID genes were used for training, but mutations from the target genes were assigned higher weights than mutations from other genes. This approach was used as it made the model mainly learn the characteristics of target genes, but did not lose the general characteristic across all mutations, it reduced the risk of overfitting caused by a small training sample size. For all the remaining genes with less than 45 mutations, we trained a common sub-model using mutations from all genes with equal weights. When we predicted the pathogenicity of new SNVs in PID genes, they were predicted by the gene-specific model when it was available.
In addition, the performance of VIPPID and other existing tools or algorithms was evaluated and compared. Among the 20 existing SNVs effect prediction tools, VEST3 showed the best performance for PID documented mutations, and it had an AUC value of 0.84 in ROC in 10 times cross-validations. It was followed by REVEL, which had an AUC value of 0.82. Our non-gene specific modeling provided in VIPPID achieved an AUC value of 0.89, showing superior accuracy over all existing tools. When the model was trained in a gene-specific manner, it achieved a superior accuracy of an AUC of 0.91 (Figure 1A).
Figure 1.

(A) Receiver operating characteristic curve for predictions from VIPPID model and existing SNV effect prediction tools and (B) model performance (AUC) comparison for VIPPID and other classifiers in the independent data set. Gene-specific VIPPID model (solid red line) and non-gene-specific VIPPID model (solid blue line) had superior accuracy compared with the existing prediction tools (non-solid lines). The numbers in parentheses were AUC values.
To further evaluate the performance of our model, we applied it on a completely independent mutation set. The prediction also showed the superior performance of VIPPID over existing tools (Figure 1B).
Important features of SNVs in PID genes
The function of different genes can be impacted by mutations with distinct types of features, and the machine learning algorithm calculated a score for each feature. Features with high importance are those that can better separate pathogenic variants from non-pathogenic variants. Meanwhile, they are also more likely to be features that have a high impact on the function of the gene products.
We used a feature selection algorithm based on the ‘Random Forest model’ to select features of SNVs that are most important for each gene. Twenty-four to 30 features were assigned as ‘Confirmed’ or ‘Tentative’ features by the algorithm, which were selected as important features and used in different sub-models. These important features included multi-46-way alignment positional conservation (MGAPHC), entropy (MGAEntropy, MGARelEntropy, ExonSnpDensity and ExonConservation), and their counterpart with hidden Markov model (HMM) and conservation scores (HMMPHC, HMMEntropy and HMMRelEntropy) (Figure 2 and Figure S2, see Supplementary Data available online at https://academic.oup.com/bib). This implied that a high evolutionary constraint and construction similarity were applied to a functional critical set of immune-related genes.
Figure 2.
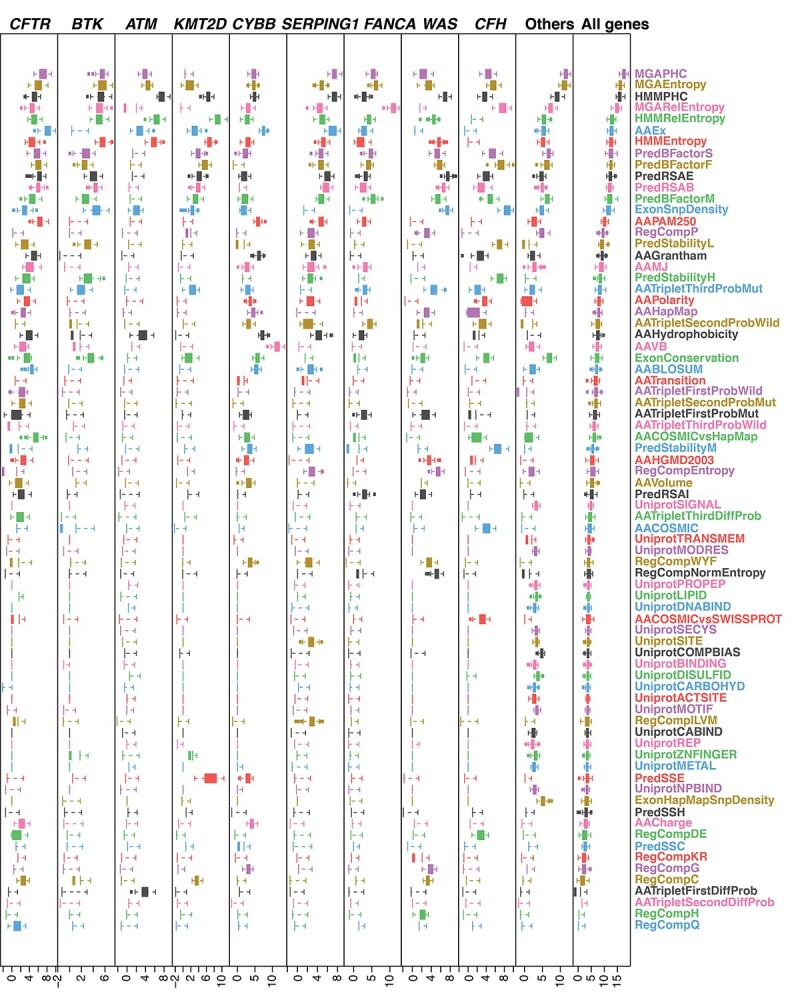
Boxplot of feature importance of SNVs in top 9 frequently mutated PID genes. Importance of features in the top 9 common PID genes (left columns in each figure) and in the remaining other PID genes (the 9th or 10th column in each figure) in the gene-specific model, features were ordered by their importance in all PID genes in the non-gene-specific model (last column). A different gene has shown a unique profile of feature importance.
Other features that were also important for the function of proteins included the probability of whether the wild-type backbone residue is stiff or flexible, and its accessibility residue is buried or exposed (PredBFactorF, PredRSAE, PredRSAB, PredBFactorM and PredBFactorS). This underscores the importance of the impact of mutations in a 3D structural context. Scores describing the probability of amino acid substitution from different matrices (AAPAM250, AAEx, AAMJ and AAGrantham) also provided useful information for the pathogenicity prediction of SNVs associated with PID.
We also looked into the pattern of feature importance of SNVs in PID gene by applying UMAP and t-SNE. Both showed that SNVs in pathogenic genes that belong to the same phenotypic classification of inborn errors of immunity (IEI) somewhat tend to cluster together, such as SNVs in TAZ, CYBB, ELANE, CFTR and ITGB2 that are associated with Phagocytic defects and SNVs in PRF1, FAS, FOXP3, AIRE and UNC13D that caused immune dysregulation are exhibit analogous characteristics. However, SNVs in MVF showed the most distinct pattern from other genes (MEFV, TNFRSF1A, NOD2 and NLRP3) that caused autoinflammatory disorders (Figure 3 and Figure S3, see Supplementary Data available online at https://academic.oup.com/bib). This may indicate the relationship between important properties of these proteins and their functional similarities.
Figure 3.
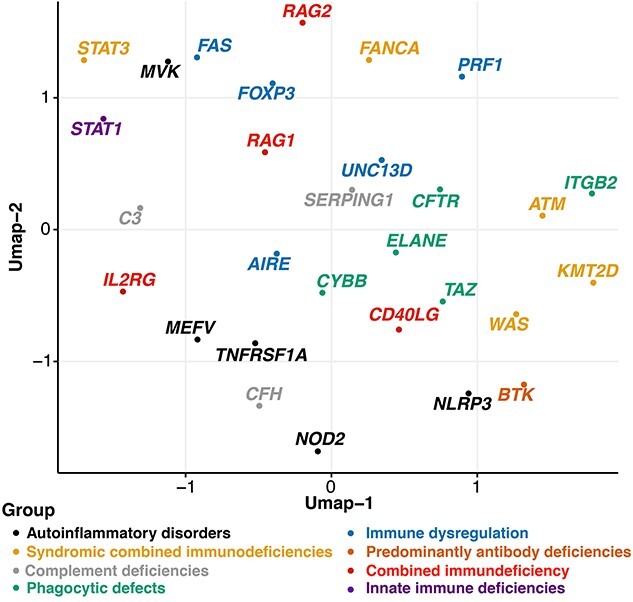
UMAP plot depicting the feature importance of SNVs in 29 PID genes with more than 45 distinct pathogenic mutations. SNVs in pathogenic genes that belong to the same phenotypic classification of IEI somewhat tend to cluster together.
When looking into individual features, interestingly, amino acid hydrophobicity plays an important role in ATM, CFTR, CYBB, FANCA, FOXP3, NOD2, RAG1, SERPING1, TAZ and TNFRSF1A, but exert only a minor impact on the pathogenicity of SNVs in other genes.
Discussion
Many existing tools attempt to predict the functional consequence of non-synonymous SNVs, while very few of them are designed to predict mutations related to one specific disease or one specific group of diseases. In this work, we developed a tool to distinguish PIDs related mutations from non-disease-causing mutations and showed its superior performance over existing tools. One of the reasons for its superiority can be that a disease-specific approach may be better tailored for different etiologies of the diseases. Another reason is its ability to utilize information from 85 different features of SNVs and combining results of other SNVs pathogenicity prediction tools. Although these existing tools are not designed to be disease-specific, they use different algorithms and possess individual strengths and limitations. Machine learning is powerful at selecting information that is most relevant to the classification task [39, 40]. Therefore, it could select features that are most relevant to immune-related genes with similar functions and structure from various sources of information to make a better prediction.
One of the significant advantages of our tool is that it has one sub-model tailored for each common PID gene. In this study, we present that features in different genes have distinct contributions to its pathogenicity, gene-specific and a non-gene-specific approach could result in utilizing a very different set of features for prediction (Figure 2). We presume that one explanation is that their influence was compensated by other features that can better represent the pathogenicity of the SNVs. Although in the independent validation set, VIPPID showed somewhat weaker performance compared with the performance at the cross-validations stage; we also noticed that the performance of other tools was also reduced. This may be caused by the differences in the pathogenicity distribution of the two variant sets, as the disease variants used in the cross-validations were discovered earlier, while the variants in the independent validation set were mostly discovered recently, this means that the former may have clearer disease association and higher variant pathogenicity which made discovery easier. However, we should not exclude the possibility of overfitting of VIPPID and existing tools, as existing tools may also use some of those variants in their training data set.
We propose that, in general, a unified model that assigns a feature of equal importance in all genes cannot yield the best predictive performance. With the rapid development of sequencing technology, numerous pathogenic and non-pathogenic variants are being discovered, and with these data, building a model in a gene-specific manner should be one of the future directions for mutation pathogenicity prediction tool development. We would benefit from this approach since different genes may have very distinct properties in sequence, structure and function. In addition, models tailored for their uniqueness can achieve an improved performance.
By studying the importance of each feature in a gene, it is possible to gain a better insight into the characteristics of the gene as well as the encoded protein. For instance, we found marked importance of hydrophobicity of the SNVs in RAG1, and this may be due to their ability to destabilize the hydrophobic core of the protein, which is essential to maintain its 3D structure. The RAG1–RAG2 interface is also enriched with hydrophobic interactions [41], and the change in hydrophobicity of their amino acids can probably have a dramatic impact on the formation of this important complex. This algorithm may provide a new approach to investigate the characteristics of a gene and its mutations. With the increasing availability of a vast amount of genomic variation data, the newly established approach will be capable to take full advantage of the machine learning technologies. As a result, our understanding of the proteins’ characteristics can be improved by revealing the quantitative contribution of each attribute of variation to the function of the protein. Furthermore, the proposed approach also provides knowledge for the application of various other data mining techniques.
In our studies, a total of 85 features were used to describe SNVs. However, we believe that if we can find more features to describe mutations precisely, it can make this approach even more powerful and more insights would thus be gained.
It should also be noted that our model is trained with PID variants specifically, so the prediction of variants for other diseases is not likely to lead to optimal results. To apply our model to other diseases, retraining with the corresponding variants, and possibly tuning some parameters, will be needed. In addition, as it has been found in previous studies, in some patients, a genetic diagnosis could not be made or phenotypic heterogeneity could not be explained by known genetic variants, which indicates the possible existence of modifying variants or variants with small effects. Our model is based on the training set of two groups, PID-pathogenic and non-pathogenic genes, which contain few variants with a mild clinical effect, so the prediction of a small effect or modifying variants should be compromised. Limited by the size of the training data set and the similarity of characteristics between PID variants and variants causing other diseases, training a model to distinguish PID variants from variants of other diseases is still challenging. However, with the continuous discovery of more PID mutations and the increase of the training data size, building such model may be possible.
Conclusion
We developed an SNVs pathogenicity prediction tool explicitly designed for PIDs and built in a gene-specific approach, which utilized information of 85 features of SNVs and scores from 20 existing prediction tools. Our evaluation showed that its performance on IEI pathogenic variants was superior to the existing tools. In addition, we also suggest that the disease- and gene-specific approach can result in a more tailored model and can be extended to the pathogenicity prediction of other diseases. Investigating the importance of different features of genomic mutations for their pathogenicity will provide a new approach to gain new insight into the characteristics of the studied genes.
Authors’ contributions
M.F., Z.S. and L.H. designed the research; M.F. performed data analysis and wrote the paper; Z.S. designed and wrote the software; L.H., Z.S., H.A., Y.I. and X.J. revised the paper.
Key Points
We develop a single nucleotide variant (SNV) pathogenicity prediction tool Variant Impact Predictor for PID (VIPPID), tailing for genes associated with inborn errors of immunity (IEI).
VIPPID employs gene-specific random forest model and utilizes information of 85 features of SNVs and scores from 20 existing prediction tools.
VIPPID exhibits an excellent performance for IEI pathogenic variants compared with other existing prediction tools, most of which were trained to predict genes of different diseases with the same model.
Supplementary Material
Author Biographies
Mingyan Fang is an associate professor at BGI-Shenzhen and BGI- Singapore. She received a PhD degree in Medical Science from Karolinska Institutet, Sweden. Her main interest lies in human disease research using multi-omics approaches.
Zheng Su is a PhD student at the School of Biotechnology and Biomolecular Sciences, The University of New South Wales. His interests mainly focus on human disease genomic research, multi-omics integration analysis, database construction, computational intelligence and bioinformatics.
Hassan Abolhassani is an assistant professor at the Department of Biosciences and Nutrition, Karolinska Institutet. His research interest is to investigate the pathogenesis of primary antibody deficiency and the immune pathways involved.
Yuval Itan is an assistant professor at the Charles Bronfman Institute for Personalized Medicine, and the Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai. His research interest focuses on investigating human disease genomics for enhancing precision medicine.
Xin Jin is a professor at BGI-Shenzhen and BGI- Singapore. His research interests mainly focused on big data research of genomics and developing new clinical applications based on cell-free nucleic acids.
Lennart Hammarström is a professor at the Department of Biosciences and Nutrition, Karolinska Institutet. He is also a professor at BGI-Shenzhen. His research interests focus on immunogenetics and immunotherapy, trying to understand the molecular basis of a variety of primary immunodeficiencies (PIDs) and develop novel forms of passive immunity that can be used to treat PIDs patients.
Funding
National Natural Science Foundation of China (grant no. 31800765); National Key Research and Development Program of China (grant no. 2020YFC2002902); Jeffrey Modell Foundation; grants provided by the Stockholm County Council (ALF project).
Data availability statement
R scripts used for model training and testing are available on GitHub: https://github.com/myfang2021/VIPPID. And a public server for utilizing it at (https://mylab.shinyapps.io/VIPPID/).
References
- 1. Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol 2007;27(5):497–502. [DOI] [PubMed] [Google Scholar]
- 2. Lim MS, Elenitoba-Johnson KS. The molecular pathology of primary immunodeficiencies. J Mol Diagn 2004;6(2):59–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tangye SG, al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol 2020;40(1):24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fang M, Su Z, Abolhassani H, et al. T cell repertoire abnormality in immunodeficiency patients with DNA repair and methylation defects. J Clin Immunol 2022;42(2):375–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. King JR, Hammarstrom L. Newborn screening for primary immunodeficiency diseases: history, current and future practice. J Clin Immunol 2018;38(1):56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abolhassani H, Aghamohammadi A, Fang M, et al. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet Med 2019;21(1):243–51. [DOI] [PubMed] [Google Scholar]
- 7. Fang M, Abolhassani H, Pan-Hammarström Q, et al. Compound heterozygous mutations of IL2-inducible T cell kinase in a Swedish patient: the importance of early genetic diagnosis. J Clin Immunol 2019;39(2):131–4. [DOI] [PubMed] [Google Scholar]
- 8. Heimall JR, Hagin D, Hajjar J, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol 2018;38(3):320–9. [DOI] [PubMed] [Google Scholar]
- 9. Findlay GM, Daza RM, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018;562(7726):217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4(7):1073–81. [DOI] [PubMed] [Google Scholar]
- 11. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7(4):248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rentzsch P, Witten D, Cooper GM, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47(D1):D886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11(4):361–2. [DOI] [PubMed] [Google Scholar]
- 14. Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol 2007;8(11):R232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaminker JS, Zhang Y, Watanabe C, et al. CanPredict: a computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res 2007;35(Web Server):W595–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carter H, Chen S, Isik L, et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res 2009;69(16):6660–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011;39(17):e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gonzalez-Perez A, Deu-Pons J, Lopez-Bigas N. Improving the prediction of the functional impact of cancer mutations by baseline tolerance transformation. Genome Med 2012;4(11):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smedley D, Jacobsen JOB, Jäger M, et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protoc 2015;10(12):2004–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zemojtel T, Köhler S, Mackenroth L, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med 2014;6(252):252ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang P, Cobat A, Lee YS, et al. A computational approach for detecting physiological homogeneity in the midst of genetic heterogeneity. Am J Hum Genet 2021;108(6):1012–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arneson D, Zhang Y, Yang X, et al. Shared mechanisms among neurodegenerative diseases: from genetic factors to gene networks. J Genet 2018;97(3):795–806. [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang X, Walsh R, Whiffin N, et al. Disease-specific variant pathogenicity prediction significantly improves variant interpretation in inherited cardiac conditions. Genet Med 2021;23(1):69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iancu IF, Avila-Fernandez A, Arteche A, et al. Prioritizing variants of uncertain significance for reclassification using a rule-based algorithm in inherited retinal dystrophies. NPJ Genom Med 2021;6(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andersen NH. Protein structure, stability, and folding. methods in molecular biology. Volume 168 Edited by Kenneth P. Murphy (University of Iowa College of Medicine). Humana Press: Totowa, New Jersey. 2001. ix + 252 pp. $89.50. ISBN 0-89603-682-0. J Am Chem Soc 2001;123(51):12933–4. [Google Scholar]
- 26. Woolley P. Protein stability and folding: Theory and practice. FEBS Lett 1996;379(2):196–6. [Google Scholar]
- 27. Itan Y, Shang L, Boisson B, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods 2016;13(2):109–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keerthikumar S, Raju R, Kandasamy K, et al. RAPID: resource of asian primary immunodeficiency diseases. Nucleic Acids Res 2009;37(Database):D863–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stenson PD, Mort M, Ball EV, et al. The human gene mutation database: 2008 update. Genome Med 2009;1(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46(D1):D1062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536(7616):285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol 2016;17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42(D1):D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong WC, Kim D, Carter H, et al. CHASM and SNVBox: toolkit for detecting biologically important single nucleotide mutations in cancer. Bioinformatics 2011;27(15):2147–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. UniProt Consortium, T . UniProt: the universal protein knowledgebase. Nucleic Acids Res 2018;46(5):2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li J, Shi L, Zhang K, et al. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res 2018;46(D1):D1039–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strobl C, Boulesteix AL, Zeileis A, et al. Bias in random forest variable importance measures: illustrations, sources and a solution. BMC Bioinform 2007;8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thaventhiran JED, Lango Allen H, Burren OS, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 2020;583(7814):90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huynh-Thu VA, Saeys Y, Wehenkel L, et al. Statistical interpretation of machine learning-based feature importance scores for biomarker discovery. Bioinformatics 2012;28(13):1766–74. [DOI] [PubMed] [Google Scholar]
- 40. Dutta D, Paul D, Ghosh P. Analysing feature importances for diabetes prediction using machine learning. Proc IEEE 9th Annu Inf Technol Electron Mobile Commun Conf (IEMCON). 2018; pp. 924–8.
- 41. Kim MS, Lapkouski M, Yang W, et al. Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature 2015;518(7540):507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
R scripts used for model training and testing are available on GitHub: https://github.com/myfang2021/VIPPID. And a public server for utilizing it at (https://mylab.shinyapps.io/VIPPID/).