

Abstract
Automated relation extraction (RE) from biomedical literature is critical for many downstream text mining applications in both research and real-world settings. However, most existing benchmarking datasets for biomedical RE only focus on relations of a single type (e.g. protein–protein interactions) at the sentence level, greatly limiting the development of RE systems in biomedicine. In this work, we first review commonly used named entity recognition (NER) and RE datasets. Then, we present a first-of-its-kind biomedical relation extraction dataset (BioRED) with multiple entity types (e.g. gene/protein, disease, chemical) and relation pairs (e.g. gene–disease; chemical–chemical) at the document level, on a set of 600 PubMed abstracts. Furthermore, we label each relation as describing either a novel finding or previously known background knowledge, enabling automated algorithms to differentiate between novel and background information. We assess the utility of BioRED by benchmarking several existing state-of-the-art methods, including Bidirectional Encoder Representations from Transformers (BERT)-based models, on the NER and RE tasks. Our results show that while existing approaches can reach high performance on the NER task (F-score of 89.3%), there is much room for improvement for the RE task, especially when extracting novel relations (F-score of 47.7%). Our experiments also demonstrate that such a rich dataset can successfully facilitate the development of more accurate, efficient and robust RE systems for biomedicine.
Availability: The BioRED dataset and annotation guidelines are freely available at https://ftp.ncbi.nlm.nih.gov/pub/lu/BioRED/.
Keywords: relation extraction, named entity recognition, biomedical dataset, biomedical natural language processing
Introduction
Biomedical natural language processing (BioNLP) and text-mining methods/tools make it possible to automatically unlock essential information published in the medical literature, including genetic diseases and their relevant variants [1, 2], chemical-induced diseases [3] and drug response in cancer [4]. However, two crucial and building block steps in the general biomedical information extraction pipeline remain challenging. The first is named entity recognition and linking (NER/NEL), which automatically recognizes the boundary of the entity spans (e.g. ESR1) of a specific biomedical concept (e.g. gene) from the free text and further links the spans to the particular entities with database identifiers (e.g. NCBI Gene ID: 2099). The second is relation extraction (RE), which identifies an entity pair with certain relations.
To facilitate the development and evaluation of NLP and machine learning methods for biomedical NER/NEL and RE, significant efforts have been made on relevant corpora development [5–10]. However, most existing corpora focus only on relations between two entities and within single sentences. For example, Herrero-Zazo et al. [8] developed a drug–drug interaction (DDI) corpus by annotating relations only if both drug names appear in the same single sentence. As a result, multiple individual NER/RE tools need to be created to extract biomedical relations beyond a single type (e.g. extracting both DDI and gene–disease relations).
Additionally, in the biomedical domain, extracting novel findings that represent the fundamental reason why an asserted relation is published as opposed to background or ancillary assertions from the scientific literature is of significant importance. To the best of our knowledge, none of the previous works on (biomedical) relation annotation, however, included such a novelty attribute.
In this work, we first give an overview of NER/NEL/RE datasets and show their strengths and weaknesses. Furthermore, we present a rich biomedical relation extraction dataset (BioRED). We further annotated the relations as either novel findings or previously known background knowledge. We summarize the unique features of the BioRED corpus as follows: (i) BioRED consists of biomedical relations among six commonly described entities (i.e. gene, disease, chemical, variant, species and cell line) in eight different types (e.g. positive correlation). Such a setting supports developing a single general-purpose RE system in biomedicine with reduced resources and improved efficiency. More importantly, several previous studies have shown that training a machine learning algorithm on multiple concepts simultaneously on one dataset, rather than multiple single-entity datasets, can lead to better performance [11–13]. We expect similar outcomes with our dataset for both NER and RE tasks. (ii) The annotated relations can be asserted either within or across sentence boundaries. For example, as shown in Figure 1 (relation R5 in pink), the variant ‘D374Y’ of the PCSK9 gene and the causal relation with the disease ‘autosomal dominant hypercholesterolemia’ are in different sentences. This task, therefore, requires relations to be inferred by machine reading across the entire document. (iii) Finally, our corpus is enriched with novelty annotations. This novel task poses new challenges for (biomedical) RE research and enables the development of NLP systems to distinguish between known facts and novel findings, a greatly needed feature for extracting new knowledge and avoiding duplicate information toward the automatic knowledge construction in biomedicine.
Figure 1.

An example of a relation and the relevant entities displayed on TeamTat (https://www.teamtat.org).
To assess the challenges of BioRED, we performed benchmarking experiments with several state-of-the-art methods, including Bidirectional Encoder Representations from Transformers (BERT)-based models. We find that existing deep-learning systems perform well on the NER task but only modestly on the novel RE task, leaving it an open problem for future NLP research. Furthermore, the detailed analysis of the results confirms the benefit of using such a rich dataset toward creating more accurate, efficient and robust RE systems in biomedicine.
Overviews of NER/NEL/RE datasets
Named entity recognition and named entity linking
Existing NER/NEL datasets cover most of the key biomedical entities, including gene/proteins [14–16], chemicals [17, 18], diseases [9, 19], variants [20–22], species [23, 24] and cell lines [25]. Nonetheless, NER/NEL datasets usually focus on only one concept type; the very few datasets that annotate multiple concept types [26, 27] do not contain relation annotations. Table 1 summarizes some widely used gold standard NER/NEL datasets, including the annotation entity type, corpus size and the task applications.
Table 1.
Overview of gold standard NER/NEL datasets
| Dataset | Text size | Entity type (#mentions) | Task type |
|---|---|---|---|
| JBLPBA [26] | 2404 abstracts | Protein (35 336), DNA (10 589), RNA (1069), cell line (4330) and cell type (8639) | NER |
| NCBI Disease [19] | 793 abstracts | Disease (6892) | NER, NEL |
| CHEMDNER [18] | 10 000 abstracts | Chemical (84 355) | NER |
| BC5CDR [9] | 1500 abstracts | Chemical (15 935), Disease (12 850) | NER, NEL |
| LINNAEUS [24] | 100 PMC full text | Species (4259) | NER, NEL |
| tmVar [20] | 500 abstracts | Variant (1431) | NER, NEL |
| NLM-Gene [14] | 550 abstracts | Gene (15 553) | NER, NEL |
| GNormPlus [12] | 694 abstracts | Gene (9986) | NER, NEL |
Due to the limitation of the entity type in NER datasets, most of the state-of-the-art entity taggers were developed individually for a specific concept. A few studies (e.g. PubTator [28]) integrate multiple entity taggers and apply them to specific collections or even to the entire PubMed/PubMed Central (PMC). In the development process, some challenging issues related to integrating entities from multiple taggers, such as concept ambiguity and variation emerged [29]. Moreover, the same articles need to be processed multiple times by multiple taggers. Huge storage space also is required to store the results of the taggers. In addition, based on clues from previous NER studies [12, 30], we realized that a tagger trained with other concepts performs as well or even better than a tagger trained on only a single concept, especially for highly ambiguous concepts. A gene tagger GNormPlus trained on multiple relevant concepts (gene/family/domain) boosts the performance of a gene/protein significantly. Therefore, a rich NER corpus can help develop a method that can recognize multiple entities simultaneously to reduce the hardware requirement and achieve better performance. Only a very few datasets [5, 27] curate multiple concepts in the text, but no relation is curated in these datasets.
Relation extraction
A variety of RE datasets in the general domain have been constructed to promote the development of RE systems [31–33]. Many of the RE datasets focus on extracting relations from a single sentence. Since many relations cross sentence boundaries, moving research from the sentence level to the document level (e.g. DocRED [34], DocOIE [35]) became a popular trend recently. In the biomedical domain, most existing RE datasets [6, 8, 10] focus on sentence-level relations involving a single pair of entities. However, multiple sentences are often required to describe an entire biological process or a relation. We highlight several commonly used biomedical RE datasets in Table 2 (a complete dataset review can be found in Table S1, see Supplementary Data available online at http://bib.oxfordjournals.org/). But only very few datasets contain relations across multiple sentences (e.g. BC5CDR dataset [9]). Most of the datasets [6–10, 36–40], which were widely used for the RE system development [41–45], focus on the single entity pair only (e.g. AIMed [37] to protein–protein interaction). Some of those datasets annotated the relation categories more granular. For example, DDI13 [8] annotated 4 categories (i.e. advise, int, effect and mechanism) of the DDI, ChemProt [10] annotated 5 categories of the chemical–protein interaction and DrugProt [40], an extension of ChemProt, annotated 13 categories. Recently, ChemProt and DDI13 are widely used in evaluating the abilities of biomedical pretrained language models [46–49] on RE tasks.
Table 2.
A summary of biomedical RE and event extraction datasets. The value of ‘-’ means that we could not find the number in their papers or websites. The SEN/DOC Level means whether the relation annotation is annotated in ‘Sentence’, ‘Document’ or ‘Cross-sentence’. ‘Document’ includes abstract, full-text or discharge record. ‘Cross-sentence’ allows two entities within a relation to appear in three surrounding sentences
| Datasets | # Doc./Sent. | # Entities | # Relations | SEN/DOC Levels | Descriptions |
|---|---|---|---|---|---|
| Protein–protein interaction | |||||
| AIMed [37] | 230 abstracts | 4141 genes | 1101 relations | Sentence | The AImed dataset aims to develop and evaluate protein name recognition and protein–protein interaction (PPI) extraction. It contains 750 Medline abstracts, which contain the ‘human’ word, and has 5206 names. Two hundred abstracts previously known to contain protein interactions for PPI extraction were obtained from the Database of Interacting Proteins (DIP) [50] and tagged for both 1101 protein interactions and 4141 protein names. Because negative examples for protein interactions were rare in the 200 abstracts, they manually selected 30 additional abstracts with more than one gene but did not have any gene interactions. |
| BioInfer [6] | 1100 sentences | 4573 proteins | 2662 relations | Sentence | A PPI dataset uses ontologies defining the fine-granted types of entities (like ‘protein family or group’ and ‘protein complex’) and their relationships (like ‘CONTAIN’ and ‘CAUSE’). They developed a corpus of 1100 sentences containing full dependency annotation, dependency types and comprehensive annotation of bio-entities and their relationships. |
| BioCreative II PPI IPS [7] | 1098 full-texts | - | - | Document | The BioCreative II PPI protein interaction pairs subtask (IPS) provides 750 and 356 full texts for training and test sets, respectively. The full text includes corresponding gene mention symbols and PPI pairs. |
| Chemical–protein interaction | |||||
| DrugProt [40] | 5000 abstracts | 65 561 chemicals, 61 775 genes | 24 526 relations | Sentence | The DrugProt dataset aims to promote the development of chemical-gene RE systems, an extension of the ChemProt dataset. It addresses 13 different chemical-gene relations, including regulatory, specific and metabolic relations |
| Chemical–disease interaction | |||||
| BC5CDR [9] | 1500 abstracts | 15 935 chemicals; 12 850 diseases | 3106 relations | Document | BC5CDR consists of 1500 abstracts that chemical and disease mention annotations and their IDs. It annotates chemical-induced disease relation ID pair. There are 1400 abstracts selected from a CTD-Pfizer collaboration-related dataset, and the remaining 100 articles are new curation and are used in the test set. |
| DDI and Drug–ADE(adverse drug effect) interaction | |||||
| ADE [51] | 2972 MEDLINE case report | 5063 drugs; 5776 adverse effects; 231 dosages |
6821 drug-adverse effects; 279 drug-dosage relations | Sentence | The ADE dataset contains drugs and conditions. But the entities do not link to the standard database identifiers. Like most of the relation datasets, ADE annotates the relations (i.e. drug-ADE and drug-dosage relations) at the sentence level. |
| DDI13 [8] | 905 documents | 13 107 drugs | 5028 relations | Sentence | SemEval 2013 DDIExtraction dataset consists of 792 texts selected from the DrugBank database and 233 Medline abstracts. The corpus is annotated with 18 502 pharmacological substances and 5028 DDIs, including both pharmacokinetic (PK) and pharmacodynamic (PD) interactions. |
| n2c2 2018 ADE [52] | 505 summaries | 83 869 entities | 59 810 relations | - | The discharge summaries are from the clinical care database of the MIMIC-III (Medical Information Mart for Intensive Care-III). The summaries are manually selected to contain at least one ADE and annotated with nine concepts and eight relation pairs. The data are split into 303 and 202 for training and test sets, respectively. |
| Variant/gene–disease interaction | |||||
| EMU [21] | 110 abstracts | - | 179 relations | Document | The EMU dataset focuses on finding relationships between mutations and their corresponding disease phenotypes. They use ‘MeSH = mutation’ to select abstracts and use MetaMap [53] to annotate the abstracts that are divided into containing mutations related to prostate cancer (PCa) and breast cancer (BCa). They then use rules and patterns to select subsets of PCa and BCa for annotating. |
| RENET2 [54] | 1000 abstracts, 500 full-texts | - | - | Document | It contains both 1000 abstracts (from RENET [55]) and 500 full texts from PMC open-access subset. For better quality, 500 abstracts of the dataset were refined. The authors used the 500 abstracts to train the RENET2 model and conduct their training data expansion using the other 500 abstracts. They further used the model trained on 1000 abstracts to construct 500 full-text articles. |
| Drug–gene mutation | |||||
| N-ary [56] | - | - | 3462 triples; 137 469 drug–gene relations; 3192 drug–mutation relations; |
Cross-sentence | Authors use distant supervision to construct a cross-sentence drug–gene mutation RE dataset. They use 59 distinct drug–gene mutation triples from the knowledge bases to extract 3462 ternary positive relation triples. The negative instances are generated by randomly sampling the entity pairs/triples without interaction. |
| Event extraction | |||||
| GE09 [57] | 1200 abstracts | - | 13 623 events | Sentence | As the first BioNLP shared task (ST), it aimed to define a bounded, well-defined GENIA event extraction (GE) task, considering both the actual needs and the state-of-the-art in bio-TM technology and to pursue it as a community-wide effort. |
| GE11 [58] | 1210 abstracts, 14 full-text | 21 616 proteins | 18 047 events | Sentence | The BioNLP ST 2011 GE task follows the task definition of the BioNLP ST 2009, which is briefly described in this section. BioNLP ST 2011 took the role of measuring the progress of the community and generalization IE technology to the full papers. |
| CG [59] | 600 abstracts | 21 683 entities | 17 248 events; 917 relations |
Sentence | The BioNLP ST 2013 Cancer Genetics (CG) corpus contains annotations of over 17 000 events in 600 documents. The task addresses entities and events at all levels of biological organization, from the molecular to the whole organism, and involves pathological and physiological processes. |
During the curation of the relations at the sentence level, curators usually do not access the context of the surrounding sentences. Besides, most sentence-level RE datasets do not link the entity names to the concept identifiers (e.g. NCBI Gene ID) in the external resources/databases. Instead, the RE dataset development at the document level relies highly on the concept identifiers. But it is extremely time-consuming, and very limited biomedical datasets annotate the related entities to the concept identifiers. BC5CDR dataset [9] is a widely used dataset with chemical-induced disease relations at the document level. All of the chemicals and diseases are linked to the concept identifiers. However, BC5CDR did not annotate the relations (e.g. treatment) out of the chemical-induced disease category. Peng et al. [56] developed a cross-sentence n-ary relation extraction dataset with the relations among drug, gene and mutation. But the dataset is constructed via distant supervision with the inevitable wrong labeling problem [34] instead of manual curation. Moreover, BioNLP shared task datasets [59–62] provide fine-grained biological event annotations to promote biological activity extraction. In Table 3, we compare BioRED to representative biomedical relation extraction datasets. BioRED covers more types of entity pairs than those datasets.
Table 3.
Comparison of the BioRED corpus with representative relation extraction datasets
| <D,G> | <D,C> | <D,V> | <C,C> | <C,G> | <G,G> | <V,C> | <V,V> | |
|---|---|---|---|---|---|---|---|---|
| RENET2 | ✓ | |||||||
| BC5CDR | ✓ | |||||||
| EMU | ✓ | |||||||
| DDI13 | ✓ | |||||||
| DrugProt | ✓ | |||||||
| AIMed | ✓ | |||||||
| GE11 | ✓ | |||||||
| N-ary | ✓ | ✓ | ||||||
| CG | ✓ | ✓ | ✓ | ✓ | ✓ | |||
| BioRED | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
D = Disease, G = Gene, C = Chemical and V = Variant.
Methods
Annotation definition/scope
We first analyzed a set of public PubMed search queries by tagging different entities and relations. This data-driven approach allowed us to determine a set of key entities and relations of interest that should be most representative, and therefore the focus of this work. Some entities are closely related biologically and are thus used interchangeably in this work. For instance, protein, mRNA and some other gene products typically share the same names and symbols. Thus, we merged them to a single gene class, and similarly merged symptoms and syndromes to a single disease class. In the end, we have six concept types: (i) Gene: for genes, proteins, mRNA and other gene products; (ii) Chemical: for chemicals and drugs; (iii) Disease: for diseases, symptoms and some disease-related phenotypes; (iv) Variant: for genomic/protein variants (including substitutions, deletions, insertions and others); (v) Species: for species in the hierarchical taxonomy of organisms and (vi) CellLine: for cell lines. Due to the critical problems of term variation and ambiguity, entity linking (also called entity normalization) is also required. We linked the entity spans to specific identifiers in an appropriate database or controlled vocabulary for each entity type (e.g. NCBI Gene ID for genes).
Between any of two different entity types, we further observed eight popular associations that are frequently discussed in the literature: <D,G > for <Disease, Gene>; <D,C > for <Disease, Chemical>; <G,C > for <Gene, Chemical>; <G,G > for <Gene, Gene>; <D,V > for <Disease, Variant>; <C,V > for <Chemical, Variant>; <C,C > for <Chemical, Chemical> and < V,V > for <Variant, Variant>. For relations between more than two entities, we simplified the relation to multiple relation pairs. For example, we simplified the chemicals co-treat disease relation (‘bortezomib and dexamethasone co-treat multiple myeloma’) to three relations: <bortezomib, multiple myeloma, treatment>, <dexamethasone, multiple myeloma, treatment> and < bortezomib, dexamethasone, cotreatment> (treatment is categorized in the Negative_Correlation). Other associations between two concepts are either implicit (e.g. variants frequently located within a gene) or rarely discussed. Accordingly, in this work we focus on annotating those eight concept pairs, as shown in solid lines in Figure 2A. To further characterize relations between entity pairs, we used eight biologically meaningful and nondirectional relation types (e.g. positive correlation; negative correlation) in our corpus as shown in Figure 2B. The details of the relation types are described in our annotation guidelines.
Figure 2.

Relations annotated in BioRED corpus. (A) Categorized relations between concepts. The patterns of the lines between the concepts present the categories: (—) Popular associations: The concept pairs are frequently discussed in the biomedical literature. (══) Implied associations, e.g. the name of a gene can imply the corresponding species. (-----) Rarely discussed associations: Some other relation types are rarely discussed in the biomedical text (and this is why the concept of Cell Line is not listed here). (B) The mapping between the concept pairs and the relation types. The frame widths of the concept pairs/relation types and the bold lines between the two sides proportionally represent the frequencies.
Annotation process
To be consistent with previous annotation efforts, we randomly sampled articles from several existing datasets (i.e. NCBI Disease [19], NLM-Gene [14], GNormPlus [12], BC5CDR [9] and tmVar [20, 60]). A small set of PubMed articles were first used to develop our annotation guidelines and familiarize our annotators with both the task and TeamTat [61], a web-based annotation tool equipped to manage team annotation projects efficiently. Following previous practice in biomedical corpus development, we developed our annotation guidelines and selected PubMed articles consistently with previous studies. Furthermore, to accelerate entity annotation, we used previous annotations combined with automated preannotations (i.e. PubTator [28]), which can then be edited based on human judgment. Unlike entity annotation, each relation is annotated from scratch by hand with an appropriate relation type, except the chemical-induced-disease relations that were previously annotated in BC5CDR.
Every article in the corpus was first annotated by three annotators with a background in biomedical informatics to prevent erroneous and incomplete annotations (especially relations) due to manual annotation fatigue. If an entity or a relation cannot be agreed upon by the three annotators, this annotation was then reviewed by another senior annotator with a background in molecular biology. For each relation, two additional biologists assessed whether it is a novel finding versus background information and made the annotation accordingly. We annotated the entire set of 600 abstracts in 30 batches of 20 articles each. For each batch, it takes approximately 2 h per annotator to annotate entities, 8 h for relations and 6 h for assigning novel versus background label. The details of the data sampling and annotation rules are described in our annotation guidelines.
Data characteristics
The BioRED corpus contains a total of 20 419 entity mentions, corresponding to 3869 unique concept identifiers. We annotated 6503 relations in total. The proportion of novel relations among all annotated relations in the corpus is 69%. Table 4 shows the numbers of the entities (mentions and identifiers) and relations in the training, development and test sets.
Table 4.
Number of entity (mention and identifier) and relation annotations in the BioRED corpus, the IAA and the distribution between the training, development and test sets. The parenthesized numbers are the unique entities linked with concept identifiers.
| Annotation s | Training | Dev | Tests | Total | IAA | |
|---|---|---|---|---|---|---|
| Document | 400 | 100 | 100 | 600 | - | |
| Entity (ID) | All | 13 351 (2708) | 3533 (956) | 3535 (982) | 20 419 (3869) | 97.01% |
| Gene | 4430 (1141) | 1087 (368) | 1180 (399) | 6697 (1643) | 97.35% | |
| Disease | 3646 (576) | 982 (244) | 917 (244) | 5545 (778) | 96.06% | |
| Chemical | 2853 (486) | 822 (184) | 754 (170) | 4429 (651) | 96.12% | |
| Variant | 890 (420) | 250 (135) | 241 (137) | 1381 (678) | 97.79% | |
| Species | 1429 (37) | 370 (13) | 393 (11) | 2192 (47) | 99.43% | |
| Cell Line | 103 (48) | 22 (12) | 50 (21) | 175 (72) | 99.68% | |
| Relation | 4178 | 1162 | 1163 | 6503 | 77.91% | |
| Relation pair with novelty findings | 2838 | 835 | 859 | 4532 | 85.01% | |
In addition, we computed the inter-annotator-agreement (IAA) for the entity, relation and novelty annotations, where we achieved 97.01, 77.91 and 85.01%, respectively. Figure 3 depicts the distribution of the different concept pairs in the relations.
Figure 3.
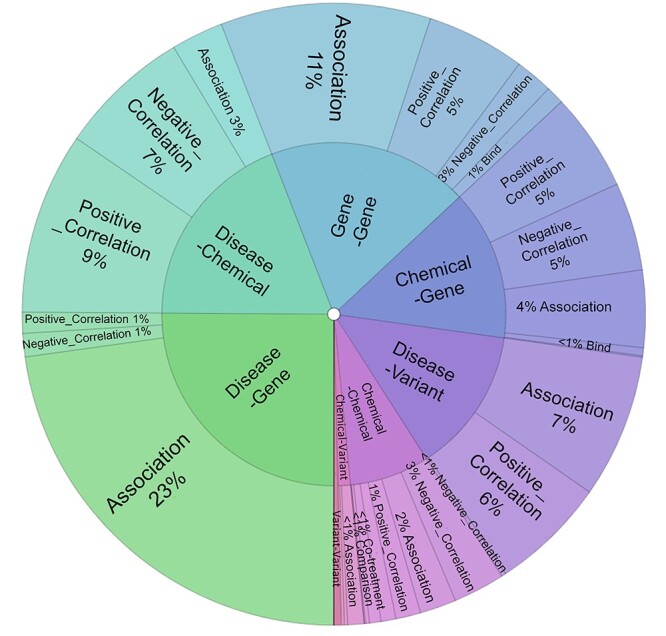
The distribution of concept pairs and relation types in the BioRED corpus.
We also analyzed dataset statistics per document. The average document length consists of 11.9 sentences or 304 tokens. Furthermore, 34 entity spans (3.8 unique entity identifiers) and 10.8 relations are annotated per document. Among the relation types, 52% are associations, 27% are positive correlations, 17% are negative correlations and 2% are involved in the triple relations (e.g. two chemicals co-treat a disease).
Benchmarking methods
To assess the utility and challenges of the BioRED corpus, we conducted experiments to show the performance of leading RE models. For the NER task, each mention span was considered separately. We evaluate three state-of-the-art NER models on the corpus including bidirectional long short-term memory-conditional random field (BiLSTM-CRF), BioBERT-CRF and PubMedBERT-CRF. The input documents are first to split into multiple sentences and encoded into a hidden state vector sequence by BiLSTM [62], BioBERT [49] and PubMedBERT [47], respectively. The models predicted the label corresponding to each of the input tokens in the sequence, then computed the network score using a fully connected layer and decode the best path of the tags in all possible paths by using CRF [63]. Here, we used the BIO (Begin, Inside, Outside) tagging scheme for the CRF layer.
We chose two BERT-based models, BERT-GT [64] and PubMedBERT [47], for evaluating the performance of current RE systems on the BioRED corpus. The first model is BERT-GT, which defines a graph transformer by integrating a neighbor–attention mechanism into the BERT architecture to avoid the effect of the noise from the longer text. BERT-GT was specifically designed for document-level relation extraction tasks and utilizes the entire sentence or passage to calculate the attention of the current token, which brings significant improvement to the original BERT model. PubMedBERT is a trained biomedical language model based on transformer architecture. It is currently a state-of-the-art text-mining method, which applies the biomedical domain knowledge (biomedical text and vocabulary) for the BERT pretrained language model. In the benchmarking, we used the text classification framework for the RE model development.
For both NER and RE evaluations, the training and development sets were first used for model development and parameter optimization before a trained model is evaluated on the test set. Benchmark implementation details are provided in Supplementary Materials A.1. Standard precision, recall and F-score metrics are used. To allow approximate entity matching, we also applied relaxed versions of the F-score to evaluate NER. In this case, as long as the boundary of the predicted entity overlaps with the gold standard span, it is considered as a successful prediction.
Results
NER results on the test set
Table 5 shows the evaluation of NER on the test set. The first run is evaluated by strict metrics. The concept type and boundary of the entity should exactly match the entity in the text. The second run is evaluated by relaxed metrics: The entity boundary should overlap, and the same entity type is required. Unlike BiLSTM-CRF, the BERT-based methods contain well pretrained language models for extracting richer features, hence achieving better performance overall. Further, PubMedBERT performs even better than BioBERT on genes, variants and cell lines. BioBERT uses the original BERT model’s vocabulary generated from general domain text, which causes a lack of understanding of the biomedical entities. On the contrary, PubMedBERT generates the vocabulary from scratch using biomedical text, and it achieves the highest F-score (89.3% in strict metrics). Among these entity types, the PubMedBERT-CRF achieves the highest performance of 97% in F1 score to species entity recognition as less term ambiguity and variation issues are found in species names.
Table 5.
Performance of NER models on test set. All numbers are F-scores.
| Metrics | Methods | All | Gene | Disease | Chemical | Species | CellLine | Variant |
|---|---|---|---|---|---|---|---|---|
| Strict | BiLSTM-CRF | 87.1 | 87.3 | 83.3 | 88.2 | 96.3 | 80.9 | 82.9 |
| BioBERT-CRF | 88.7 | 89.5 | 84.8 | 89.7 | 96.7 | 83.5 | 83.9 | |
| PubMedBERT-CRF | 89.3 | 92.4 | 83.5 | 88.6 | 97.0 | 90.5 | 87.3 | |
| Relaxed | BiLSTM-CRF | 92.4 | 92.3 | 92.2 | 91.9 | 96.8 | 85.4 | 93.6 |
| BioBERT-CRF | 93.4 | 93.8 | 93.6 | 91.3 | 97.0 | 90.1 | 92.3 | |
| PubMedBERT-CRF | 93.5 | 94.7 | 92.6 | 91.1 | 97.0 | 92.6 | 94.5 |
RE results on the test set
We also evaluated performance on the RE task by different benchmark schemas: (i) entity pair: to extract the pair of concept identifiers within the relation, (ii) entity pair + relation type: to recognize the specific relation type for the extracted pairs and (iii) entity pair + relation type + novelty: to further label the novelty for the extracted pairs. In this task, the gold-standard concepts in the articles are given. We applied BERT-GT and PubMedBERT to recognize the relations and the novelty in the test set.
As shown in Table 6, the overall performance of PubMedBERT is higher than that of BERT-GT in all schemas. Because the numbers of relations in <D,V>, <C,V> and <V,V> are low, their performance is not comparable to that of other concept pairs, especially <V,V> (the F-score is 0% for two models). In the first schema, BERT-GT and PubMedBERT can achieve performance above 72% for the F-scores, which is expected and promising in the document-level RE task. To predict the relation types (e.g. positive correlation) other than entity pairs, however, is still quite challenging. The best performance on the second schema is only 58.9%, as the number of instances in many relation types is insufficient. The performances of different relation types of our best model using PubMedBert are provided in Supplementary Materials A.2. The performance on the third schema dropped to 47.7%. In some cases, the statements of the relations in abstracts are usually concise, and the details of the related mechanism can only be found in the full text.
Table 6.
Performance on RE task for the first schema: extracting the entity pairs within a relation, second schema: extracting the entity pairs and the relation type and the third schema: further labeling the novelty for the extracted pairs. All numbers are F-scores. The <G,D > is the concept pair of the gene (G) and the disease (D). The columns of those entity pairs present the RE performance in F-scores.
| Eval Schema | Methods | All | <G,D> | <G,G> | <G,C> | <D,V> | <C,D> | <C,V> | <C,C> |
|---|---|---|---|---|---|---|---|---|---|
| Entity pair | BERT-GT | 72.1 | 63.8 | 78.5 | 77.7 | 69.8 | 76.2 | 58.8 | 74.9 |
| PubMedBERT | 72.9 | 67.2 | 78.1 | 78.3 | 67.9 | 76.5 | 58.1 | 78.0 | |
| +Relation type | BERT-GT | 56.5 | 54.8 | 63.5 | 60.2 | 42.5 | 67.0 | 11.8 | 52.9 |
| PubMedBERT | 58.9 | 56.6 | 66.4 | 59.9 | 50.8 | 65.8 | 25.8 | 54.4 | |
| +Novelty | BERT-GT | 44.5 | 37.5 | 47.3 | 55.0 | 36.9 | 51.9 | 11.8 | 48.5 |
| PubMedBERT | 47.7 | 40.6 | 54.7 | 54.8 | 42.8 | 51.6 | 12.9 | 50.3 |
G = gene, D = disease, V = variant and C = chemical.
Benefits of multiple entity recognition and relation extraction
To test the hypothesis that our corpus can result in a single model with better performance, we trained multiple separate NER and RE models, each with an individual concept (e.g. gene) or relation (e.g. gene–gene) for comparison. We used PubMedBERT for this evaluation since it achieved the best performances in both the NER and RE tasks. As shown in Table 7, both models trained on all entities or relations generally perform better than the models trained on most of the entities or relations, while the improvement for RE is generally larger. The performance on NER and RE tasks is both obviously higher in the single model. Especially for entities and relations (e.g. cell lines and chemical–chemical relations) with insufficient amounts, the model trained on multiple concepts/relations can obtain larger improvements. The experiment demonstrated that training NER/RE models with more relevant concepts or relations not only can reduce resource usage but also can achieve better performance.
Table 7.
The comparison of the models trained on all entities/relations to the models trained on individual entity/relation. The <G,D > is the relation of the gene (G) and the disease (D). All models are evaluated by strict metrics.
| Entities /Relations | Types | All entities or relations | Single entity or relation | ||||
|---|---|---|---|---|---|---|---|
| Precision | Recall | F-score | Precision | Recall | F-score | ||
| Entity | G | 92.2 | 92.5 | 92.4 | 90.8 | 91.0 | 90.9 |
| D | 80.7 | 86.5 | 83.5 | 83.2 | 85.7 | 84.4 | |
| C | 87.9 | 89.3 | 88.6 | 87.3 | 92.4 | 89.8 | |
| V | 88.8 | 85.9 | 87.3 | 84.7 | 87.1 | 85.9 | |
| S | 95.8 | 98.2 | 97.0 | 95.2 | 96.4 | 95.8 | |
| CL | 95.6 | 86.0 | 90.5 | 77.1 | 74.0 | 75.5 | |
| Relation | <G,D> | 63.6 | 71.2 | 67.2 | 75.8 | 62.7 | 68.7 |
| <G,G> | 81.5 | 75.0 | 78.1 | 57.3 | 80.0 | 66.8 | |
| <G,C> | 74.1 | 83.1 | 78.3 | 66.7 | 68.9 | 67.8 | |
| <D,V> | 71.2 | 64.9 | 67.9 | 76.5 | 51.5 | 61.5 | |
| <C,D> | 73.3 | 79.9 | 76.5 | 78.2 | 85.2 | 81.5 | |
| <C,V> | 60.0 | 56.3 | 58.1 | 53.3 | 50.0 | 51.6 | |
| <C,C> | 75.3 | 80.9 | 78.0 | 64.2 | 72.3 | 68.0 | |
G = gene, D = disease, C = chemical, V = variant, S = species and CL = cell line.
Discussion
The relaxed NER results in Table 5 for overall entity type are over 92% for all methods, suggesting the maturity of current tools for this task. If considering the performance of each concept individually, the recognition of genes, species and cell lines can reach higher performance (over 90% in strict F-score) since the names are often simpler and less ambiguous than other concepts. The best model for genomic variants achieves an F-score of 87.3% in strict metrics and 94.5% in relaxed metrics, which suggests that the majority of the errors are due to incorrect span boundaries. Most variants are not described in accordance with standard nomenclature (e.g. ‘ACG— > AAG substitution in codon 420’), thus it is difficult to exactly identify the boundaries. Similar to genomic variants, diseases are difficult to be identified due to term variability and most errors are caused by mismatched boundaries. For example, our method recognized a part (‘papilledema’) of a disease mentioned (‘bilateral papilledema’) in the text. Disease names also present greater diversity than other concepts: 55.4% of the disease names in the test set are not present in the training/development sets. Chemical names are extremely ambiguous with other concepts: half of the errors for chemicals are incorrectly labeled as other concept types (e.g. gene), since some chemicals are interchangeable with other concepts, such as proteins and drugs. Moreover, we merged the annotations matched by the dictionary to the results of the PubMedBERT-CRF model. However, the performance of the dictionary method heavily depends on the difficulties of the term variation and ambiguity issues. Especially, there are many ambiguous terms in the dictionary, such as ‘B1’, ‘Beta’ and ‘98–4.9’ in Cellosaurus. Although the F1 score of the dictionary cannot compete with the machine learning method, merging the results from both methods can improve the recall of all the concepts (see details in Supplementary Materials A.3).
Experimental results in Table 6 show that the RE task remains challenging in biomedicine, especially for the new task of extracting novel findings. In our observation, there are three types of errors in novelty identification. First, some abstracts do not indicate which concept pairs represent novel findings, and instead, provide more details in the full text. Such cases confused both the human annotators and the computer algorithms. Second, when the mechanism of interaction between two relevant entities is unknown, and the study aims to investigate it but the hypothesized mechanism is shown to be false. Third, the authors frequently mention relevant background knowledge within their conclusion. As an example, ‘We conclude that Rg1 may significantly improve the spatial learning capacity impaired by chronic morphine administration and restore the morphine-inhibited LTP. This effect is NMDA receptor-dependent.’ in the conclusion of the PMID:18308784, the Rg1 responded to morphine as background knowledge. But it is mentioned together with the novelty knowledge pair <Rg1, NMDA receptor>. In this case, our method misclassified the pair < Rg1, morphine> as novel. We also conducted an experiment to evaluate the effect of section information for novelty detection. The experimental results show that the structured section information (e.g. TITLE, PURPOSE, METHODS, RESULTS, … ) can be useful for novelty classification by boosting the best F1 score from 47.7% to 48.9% (see details in Supplementary Materials A.4). However, this result was obtained on a subset of 191 abstracts with structured section information due to limited availability.
The results in Table 7 demonstrate that training NER/RE models on one rich dataset with multiple concepts/relations simultaneously can not only make the trained model simpler and more efficient, but also more accurate. More importantly, we notice that for the entities and relations with a lower number of training instances (e.g. cell lines and chemical–chemical relations), simultaneous prediction is especially beneficial for improving performance. Additionally, merging entity results from different models often poses some challenges, such as ambiguity or overlapping boundaries between different concepts.
Conclusion
In the past, biomedical RE datasets were typically built for a single entity type or relation. To enable the development of RE tools that can accurately recognize multiple concepts and their relations in biomedical texts, we have developed BioRED, a high-quality RE corpus, with one-of-a-kind novelty annotations. Similar to other commonly used biomedical datasets, e.g., BC5CDR [9], we expect BioRED to serve as a benchmark for not only biomedical-specific NLP tools but also for the development of RE methods in the general domain. Additionally, the novelty annotation in BioRED proposes a new NLP task that is critical for information extraction in practical applications. Recently, the dataset was successfully used by the NIH LitCoin NLP Challenge (https://ncats.nih.gov/funding/challenges/litcoin) and a total of over 200 teams participated in the Challenge.
This work has implications for several real-world use cases in medical information retrieval, data curation and knowledge discovery. Semantic search has been commonly practiced in the general domain but much less so in biomedicine. For instance, several existing studies retrieve articles based on the co-occurrence of two entities [65–68] or rank search results by co-occurrence frequency. Our work could accelerate the development of semantic search engines in medicine. Based on the extracted relations within documents, search engines can semantically identify articles by two entities with relations (e.g. 5-FU-induced cardiotoxicity) or by expanding the user queries from an entity (e.g. 5-FU) to the combination of the entity and other relevant entities (e.g. cardiotoxicity, diarrhea).
While BioRED is a novel and high-quality dataset, it has a few limitations. First, we are only able to include 600 abstracts in the BioRED corpus due to the prohibitive cost of manual annotation and limited resources. Nonetheless, our experiments show that except for a few concept pairs and relation types (e.g. variant–variant relations) that occur infrequently in the literature, its current size is appropriate for building RE models. Our experimental results in Table 7 also show that in some cases, the performance of entity class with a small number of training instances (e.g. Cell Line) can be significantly boosted when training together with other entities in one corpus. Second, the current corpus is developed on PubMed abstracts, as opposed to the full text. While full text contains more information, data access remains challenging in real-world settings. More investigation is warranted on this topic in the future.
Key Points
First review on publicly available biomedical named entity recognition and relation extraction (RE) datasets.
We present a first-of-its-kind biomedical relation extraction dataset (BioRED) with multiple entity types and relation pairs at the document level.
The novelty RE task is proposed to differentiate between a novel finding or previously known background knowledge.
Several cutting-edge deep learning models are evaluated on BioRED, and results show that there is much room for improvement for the RE task.
Supplementary Material
Acknowledgments
The authors are grateful to Drs Tyler F. Beck and Christine Colvis, Scientific Program Officer at the NCATS and their entire research team for help with our dataset. The authors would like to thank Rancho BioSciences and specifically, Mica Smith, Thomas Allen Ford-Hutchinson and Brad Farrell for their contribution with data curation.
Author Biographies
Ling Luo, PhD, is a postdoctoral fellow at the National Center for Biotechnology Information, working on biomedical text mining.
Po-Ting Lai, PhD, is a postdoctoral fellow at the National Center for Biotechnology Information, working on biomedical text mining.
Chih-Hsuan Wei, PhD, is a research scientist at the National Center for Biotechnology Information, working on biomedical text mining.
Cecilia Arighi, PhD, is a research associate professor at the University of Delaware. Her research includes improving coverage and access to literature and annotations in UniProt via text mining, integration from external sources and community crowdsourcing.
Zhiyong Lu, PhD, FACMI is a senior investigator and Deputy Director for Literature Search at NCBI/NLM where he leads text mining and machine learning research and directs overall R&D efforts to improve information access in literature databases such as PubMed and LitCovid.
Contributor Information
Ling Luo, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, USA.
Po-Ting Lai, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, USA.
Chih-Hsuan Wei, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, USA.
Cecilia N Arighi, University of Delaware, Newark, DE 19716, USA.
Zhiyong Lu, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, USA.
Funding
National Institutes of Health (NIH) intramural research program; National Library of Medicine; NIH (grant 2U24HG007822-08 to C.N.A.).
References
- 1. Singhal A, Simmons M, Lu Z. Text mining genotype-phenotype relationships from biomedical literature for database curation and precision medicine. PLoS Comput Biol 2016;12(11):e1005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee K, Lee S, Park S, et al. . BRONCO: Biomedical entity relation oncology corpus for extracting gene-variant-disease-drug relations. Database 2016;2016:baw043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wei C-H, Peng Y, Leaman R, et al. . Assessing the state of the art in biomedical relation extraction: overview of the BioCreative V chemical-disease relation (CDR) task. Database 2016;2016:baw032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baptista D, Ferreira PG, Rocha M. Deep learning for drug response prediction in cancer. Brief Bioinform 2021;22(1):360–79. [DOI] [PubMed] [Google Scholar]
- 5. Kim J-D, Ohta T, Tateisi Y, et al. . GENIA corpus—a semantically annotated corpus for bio-textmining. Bioinformatics 2003;19(suppl_1):i180–2. [DOI] [PubMed] [Google Scholar]
- 6. Pyysalo S, Ginter F, Heimonen J, et al. . BioInfer: a corpus for information extraction in the biomedical domain. BMC Bioinform 2007;8(1):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krallinger M, Leitner F, Rodriguez-Penagos C, et al. . Overview of the protein-protein interaction annotation extraction task of BioCreative II. Genome Biol 2008;9(2):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herrero-Zazo M, Segura-Bedmar I, Martínez P, et al. . The DDI corpus: An annotated corpus with pharmacological substances and drug–drug interactions. J Biomed Inform 2013;46(5):914–20. [DOI] [PubMed] [Google Scholar]
- 9. Li J, Sun Y, Johnson RJ, et al. . BioCreative V CDR task corpus: a resource for chemical disease relation extraction. Database 2016;2016:baw068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krallinger M, Rabal O, Akhondi SA, et al. . Overview of the BioCreative VI chemical-protein interaction Track . In: Proceedings of the sixth BioCreative Challenge Evaluation Workshop, Bethesda, MD USA: BioCreative, 2017, 142–147.
- 11. Wang X, Lyu J, Dong L, et al. . Multitask learning for biomedical named entity recognition with cross-sharing structure. BMC Bioinformat 2019;20(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wei C-H, Kao H-Y, Lu Z. GNormPlus: an integrative approach for tagging genes, gene families, and protein domains. Biomed Res Int 2015;2015:918710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akdemir A, Shibuya T. Analyzing the effect of multi-task learning for biomedical named entity recognition. arXiv preprint arXiv:2011.00425. 2020.
- 14. Islamaj Doğan R, Wei C-H, Cissel D, et al. . NLM-Gene, a richly annotated gold standard dataset for gene entities that addresses ambiguity and multi-species gene recognition. J Biomed Inform 2021;118:103779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morgan AA, Lu Z, Wang X, et al. . Overview of BioCreative II gene normalization. Genome Biol 2008;9(2):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirschman L, Colosimo M, Morgan A, et al. . Overview of BioCreAtIvE task 1B: normalized gene lists. BMC Bioinformat 2005;6(1):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Islamaj Doğan R, Leaman R, Kim S, et al. . NLM-Chem, a new resource for chemical entity recognition in PubMed full text literature. Sci Data 2021;8(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krallinger M, Rabal O, Leitner F, et al. . The CHEMDNER corpus of chemicals and drugs and its annotation principles. J Chem 2015;7(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Islamaj Doğan R, Leaman R, Lu Z. NCBI disease corpus: a resource for disease name recognition and concept normalization. J Biomed Inform 2014;47:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei C-H, Harris BR, Kao H-Y, et al. . tmVar: a text mining approach for extracting sequence variants in biomedical literature. Bioinformatics 2013;29(11):1433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doughty E, Kertesz-Farkas A, Bodenreider O, et al. . Toward an automatic method for extracting cancer-and other disease-related point mutations from the biomedical literature. Bioinformatics 2011;27(3):408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caporaso JG, BaumgartnerWA, Jr, Randolph DA, et al. . MutationFinder: a high-performance system for extracting point mutation mentions from text. Bioinformatics 2007;23(14):1862–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pafilis E, Frankild SP, Fanini L, et al. . The SPECIES and ORGANISMS resources for fast and accurate identification of taxonomic names in text. PLoS One 2013;8(6):e65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gerner M, Nenadic G, Bergman CM. LINNAEUS: a species name identification system for biomedical literature. BMC Bioinform 2010;11(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arighi C, Hirschman L, Lemberger T, et al. . Bio-ID track overview. In: BioCreative VI Challenge Evaluation Workshop, Bethesda, MD USA: BioCreative, 2017, 14–19.
- 26. Kim J-D, Ohta T, Tsuruoka Y, et al. . Introduction to the bio-entity recognition task at JNLPBA. In: Proceedings of the International Joint Workshop on Natural Language Processing in Biomedicine and Its Applications. Geneva, Switzerland: COLING, 2004, 70–75. [Google Scholar]
- 27. Bada M, Eckert M, Evans D, et al. . Concept annotation in the CRAFT corpus. BMC Bioinform 2012;13(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wei C-H, Allot A, Leaman R, et al. . PubTator central: automated concept annotation for biomedical full text articles. Nucleic Acids Res 2019;47(W1):W587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wei C-H, Lee K, Leaman R, et al. . Biomedical mention disambiguation using a deep learning approach . In: Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, New York, United States: Association for Computing Machinery, 2019, 307–313.
- 30. Leaman R, Lu Z. TaggerOne: joint named entity recognition and normalization with semi-Markov Models. Bioinformatics 2016;32(18):2839–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hendrickx I, Kim SN, Kozareva Z, et al. . Semeval-2010 task 8: Multi-way classification of semantic relations between pairs of nominals . In: Proceedings of the 5th International Workshop on Semantic Evaluation, ACL 2010, Uppsala, Sweden: Association for Computational Linguistics, 2019, 33–38.
- 32. Zhang Y, Zhong V, Chen D, et al. . Position-aware attention and supervised data improve slot filling . In: Proceedings of the 2017 Conference on Empirical Methods in Natural Language Processing, Copenhagen, Denmark: Association for Computational Linguistics, 2017, 35–45.
- 33. Walker C, Strassel S, Medero J, et al. . ACE 2005 multilingual training corpus. In: Linguistic Data Consortium, Philadelphia: Linguistic Data Consortium, 2006.
- 34. Yao Y, Ye D, Li P, et al. . DocRED: a large-scale document-level relation extraction dataset. In: Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics, Florence, Italy: Association for Computational Linguistics, 2019, 764–777.
- 35. Dong K, Zhao Y, Sun A, et al. . DocOIE: a document-level context-aware dataset for OpenIE. In: Findings of the Association for Computational Linguistics: ACL-IJCNLP 2021, Association for Computational Linguistics, 2021, 2377–2389.
- 36. Ding J, Berleant D, Nettleton D, et al. . Mining MEDLINE: abstracts, sentences, or phrases? In: Biocomputing 2002. Kauai, Hawaii, USA: World Scientific, 2001, 326–37. [DOI] [PubMed] [Google Scholar]
- 37. Bunescu R, Ge R, Kate RJ, et al. . Comparative experiments on learning information extractors for proteins and their interactions. Artif Intell Med 2005;33(2):139–55. [DOI] [PubMed] [Google Scholar]
- 38. Nédellec C. Learning language in logic-genic interaction extraction challenge. In: 4. Learning Language in Logic Workshop (LLL05). Born, Germany: ACM-Association for Computing Machinery, 2005. [Google Scholar]
- 39. Fundel K, Küffner R, Zimmer R. RelEx—relation extraction using dependency parse trees. Bioinformatics 2007;23(3):365–71. [DOI] [PubMed] [Google Scholar]
- 40. Miranda A, Mehryary F, Luoma J, et al. . Overview of DrugProt BioCreative VII track: quality evaluation and large scale text mining of drug-gene/protein relations. In: Proceedings of the BioCreative VII Challenge Evaluation Workshop, BioCreative, 2021, 11–21.
- 41. Airola A, Pyysalo S, Björne J, et al. . All-paths graph kernel for protein-protein interaction extraction with evaluation of cross-corpus learning. BMC Bioinform 2008;9(11):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peng Y, Rios A, Kavuluru R, et al. . Extracting chemical–protein relations with ensembles of SVM and deep learning models. Database 2018;2018:bay073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yadav S, Ekbal A, Saha S, et al. . Feature assisted stacked attentive shortest dependency path based Bi-LSTM model for protein–protein interaction. Knowledge-Base Syst 2019;166:18–29. [Google Scholar]
- 44. Luo L, Yang Z, Cao M, et al. . A neural network-based joint learning approach for biomedical entity and relation extraction from biomedical literature. J Biomed Inform 2020;103:103384. [DOI] [PubMed] [Google Scholar]
- 45. Li Y, Chen Y, Qin Y, et al. . Protein-protein interaction relation extraction based on multigranularity semantic fusion. J Biomed Inform 2021;123:103931. [DOI] [PubMed] [Google Scholar]
- 47. Raj Kanakarajan K, Kundumani B, Sankarasubbu M. BioELECTRA: pretrained biomedical text encoder using discriminators. In: Proceedings of the 20th Workshop on Biomedical Language Processing, Association for Computational Linguistics, 2021, 143–154.
- 47. Gu Y, Tinn R, Cheng H, et al. . Domain-specific language model pretraining for biomedical natural language processing. ACM Trans Comput Healthc 2021;3(1):1–23. [Google Scholar]
- 48. Alrowili S, Vijay-Shanker K. BioM-transformers: building large biomedical language models with BERT, ALBERT and ELECTRA. In: Proceedings of the 20th Workshop on Biomedical Language Processing. Association for Computational Linguistics, 2021, 221–227. [Google Scholar]
- 49. Lee J, Yoon W, Kim S, et al. . BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2020;36(4):1234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xenarios I, Fernandez E, Salwinski L, et al. . DIP: the database of interacting proteins: 2001 update. Nucleic Acids Res 2001;29(1):239–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gurulingappa H, Rajput AM, Roberts A, et al. . Development of a benchmark corpus to support the automatic extraction of drug-related adverse effects from medical case reports. Journal of Biomedical Informatics 2012;45(5):885–92. [DOI] [PubMed] [Google Scholar]
- 52. Henry S, Buchan K, Filannino M, et al. . 2018 n2c2 shared task on adverse drug events and medication extraction in electronic health records. J Am Med Inform Assoc 2019;27(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aronson AR. Effective mapping of biomedical text to the UMLS Metathesaurus: the MetaMap program. In: Proceedings of the AMIA Symposium. Washington, DC, USA: American Medical Informatics Association, 2001, 17–21. [PMC free article] [PubMed] [Google Scholar]
- 54. Su J, Wu Y, Ting H-F, et al. . RENET2: high-performance full-text gene–disease relation extraction with iterative training data expansion. NAR Genomics. Bioinformatics 2021;3(3):lqab062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu Y, Luo R, Leung H, et al. . Renet: A deep learning approach for extracting gene-disease associations from literature . In: International Conference on Research in Computational Molecular Biology. Washington, DC, USA: Springer, 2019, 272–284. [Google Scholar]
- 56. Peng N, Poon H, Quirk C, et al. . Cross-sentence n-ary relation extraction with graph lstms. Trans Assoc Comput Linguist 2017;5:101–15. [Google Scholar]
- 57. Kim J-D, Ohta T, Pyysalo S, et al. . Overview of BioNLP’09 shared task on event extraction . In: Proceedings of the BioNLP 2009 Workshop Companion Volume for Shared Task, Boulder, Colorado: Association for Computational Linguistics, 2009, 1–9.
- 58. Kim J-D, Wang Y, Takagi T, et al. . Overview of genia event task in bionlp shared task 2011 . In: Proceedings of BioNLP shared task 2011 workshop, Portland, Oregon, USA: Association for Computational Linguistics, 2011, 7–15.
- 59. Pyysalo S, Ohta T, Rak R, et al. . Overview of the cancer genetics and pathway curation tasks of bionlp shared task 2013. BMC Bioinformat 2015;16(10):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wei C-H, Phan L, Feltz J, et al. . tmVar 2.0: integrating genomic variant information from literature with dbSNP and ClinVar for precision medicine. Bioinformatics 2018;34(1):80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Islamaj Doğan R, Kwon D, Kim S, et al. . TeamTat: a collaborative text annotation tool. Nucleic Acids Res 2020;48(W1):W5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput 1997;9(8):1735–80. [DOI] [PubMed] [Google Scholar]
- 63. Lafferty J, McCallum A, Pereira FC. Conditional random fields: probabilistic models for segmenting and labeling sequence data. In: Proceedings of the Eighteenth International Conference on Machine Learning. San Francisco, CA, United States: Association for Computing Machinery, 2001, 282–289.
- 64. Lai P-T, Lu Z. BERT-GT: cross-sentence n-ary relation extraction with BERT and Graph Transformer. Bioinformatics 2020;36(24):5678–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Allot A, Peng Y, Wei C-H, et al. . LitVar: a semantic search engine for linking genomic variant data in PubMed and PMC. Nucleic Acids Res 2018;46(W1):W530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thomas P, Starlinger J, Vowinkel A, et al. . GeneView: a comprehensive semantic search engine for PubMed. Nucleic Acids Res 2012;40(W1):W585–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dörpinghaus J, Klein J, Darms J, et al. . SCAIView-A Semantic Search Engine for Biomedical Research Utilizing a Microservice Architecture. In: SEMANTICS Posters&Demos, Vienna, Austria: CEUR-WS, 2018.
- 68. Pang X, Bou-Dargham MJ, Liu Y, et al. Accelerating cancer research using big data with BioKDE platform. In: Proceedings of the American Association for Cancer Research Annual Meeting, Chicago, IL. Philadelphia (PA): AACR, 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.