

Abstract
Traumatic brain injury (TBI) affects millions yearly, and is increasingly associated with chronic neuropsychiatric symptoms. We assessed the long-term effects of different bilateral frontal controlled cortical impact injury severities (mild, moderate, severe) on the five-choice serial reaction time task, a paradigm with relatively independent measurements of attention, motor impulsivity and motivation. Moderately- and severely-injured animals exhibited impairments across all cognitive domains that were still evident 14 weeks post-injury, while mild-injured animals only demonstrated persistent deficits in impulse control. However, recovery of function varied considerably between subjects such that some showed no impairment (“TBI-resilient”), some demonstrated initial deficits that recovered (“TBI-vulnerable”) and some never recovered (“chronically-impaired”). Three clinically-relevant treatments for impulse-control or TBI, amphetamine, atomoxetine, and amantadine, were assessed for efficacy in treating injury-induced deficits. Susceptibility to TBI affected the response to pharmacological challenge with amphetamine. Whereas sham and TBI-resilient animals showed characteristic impairments in impulse control at higher doses, amphetamine had the opposite effect in chronically-impaired rats, improving task performance. In contrast, atomoxetine and amantadine reduced premature responding but increased omissions, suggesting psychomotor slowing. Analysis of brain tissue revealed that generalized neuroinflammation was associated with impulsivity even when accounting for the degree of brain damage. This is one of the first studies to characterize psychiatric-like symptoms in experimental TBI. Our data highlight the importance of testing pharmacotherapies in TBI models in order to predict efficacy, and suggest that neuroinflammation may represent a treatment target for impulse control problems following injury.
Keywords: controlled cortical impact, impulsivity, amphetamine, prelimbic, cytokine
Introduction
Traumatic brain injury (TBI) affects 2.5 million people annually in the United States alone, placing estimates for the incidence rate between 12–24% across the lifespan (1). Although the majority of injuries are mild, and patients often recover spontaneously (2), an estimated 1–2% of people in the US still live with permanent disabilities from brain injury (3, 4). TBI is recognized as a major environmental risk factor for neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease (5, 6), and a burgeoning literature is also reporting links between TBI and the development of core psychiatric symptoms such as depression, suicidality, attention deficits and impulse control problems (7, 8).
The development of persistent, long-term cognitive deficits is one of the most debilitating consequences of TBI. There are no targeted treatments for TBI-induced psychiatric complications, and it is unclear whether drugs prescribed for impulse control and attention deficits in non-brain-injured populations are efficacious or even appropriate for TBI patients. Notably, drug classes commonly prescribed for impulse control problems, such as dopaminergic and noradrenergic agents, have not been tested in this population. This information vacuum is compounded by a lack of experimental animal studies examining either long-term cognitive outcomes or the impact of such pharmacological treatments on persistent dysfunction. Chronic pathophysiological changes following TBI, particularly those related to neuroinflammation, have been comparatively well-documented, including up-regulation of specific cytokines such as interleukin (IL)-1β and IL-6 (9–11). Although a growing body of evidence indicates similar pathways could be implicated in psychiatric disorders (12, 13), the relationship between such markers and TBI-induced cognitive impairment remains unclear.
One barrier to progress in the field has been the strong level of endogenous recovery in rat models of TBI on commonly used behavioral assessments such as the Morris water maze that measure primarily hippocampus-dependent spatial memory (14), limiting the study of more complex chronic cognitive impairments and necessitating the use of much more severe injuries than typically observed in human TBI populations (15, 16). The bilateral prefrontal controlled cortical impact (CCI) model of TBI, though used less frequently due to the more complex surgery involved, offers considerable advantages in this regard: it not only leads to enduring cognitive deficits, but targets the area of the brain most heavily implicated in psychiatric symptoms such as depression, inattention, and impulsivity, while replicating much of the pathology observed after unilateral CCI (17–19). Additionally, the adoption of more complex, cognitively-demanding behavioral methodologies, such as those used as standard in the field of behavioral pharmacology for assessing models of psychiatric dysfunction, could benefit experimental TBI studies (20). Combining these two approaches may radically improve the detection of chronic TBI-induced cognitive deficits, and generate a model with not only stronger predictive validity to assess therapeutics, but also a model for evaluating more subtle deficits that occur in milder injuries.
In the current study, we therefore evaluated whether a range of TBI severities centered over the frontal cortex affected performance of the five-choice serial reaction time (5CSRT) task in rats, a widely-used rodent paradigm with high translational validity that measures an aspect of attention and impulse control (21). We also determined whether any of three clinically-relevant drug challenges that have therapeutic value for impulse control (amphetamine, atomoxetine) or have been used to treat TBI (amantadine), were effective at remediating post-injury cognitive impairment. Finally, we examined whether the expression of multiple cytokines post mortem were associated with lasting deficits in 5CSRT performance.
Results and Discussion
Additional data and analyses can be found in supporting information.
Effect of TBI on 5CSRT performance:
In the acute phase (days 7–30 post-injury), animals that received a mild, moderate or severe TBI were impaired on numerous behavioral measures assessed by the 5CSRT (Figure 2 and Table S1), and the magnitude of the impairment broadly reflected the severity of impact (accuracy: each group was different from every other group, p’s < 0.005; prematures: each group was different from every other group, p’s < 0.002, except for the Moderate and Severe group, p = 0.644; omissions: each group was different from every other group, p’s < 0.005, except for the Sham and Mild group, p = 0.192; task efficacy index: each group was different from every other group, p’s < 0.002; see Figure S1, Table S1 and supporting information for group differences in total trials, choice and reinforcer collection latencies).
Figure 2.
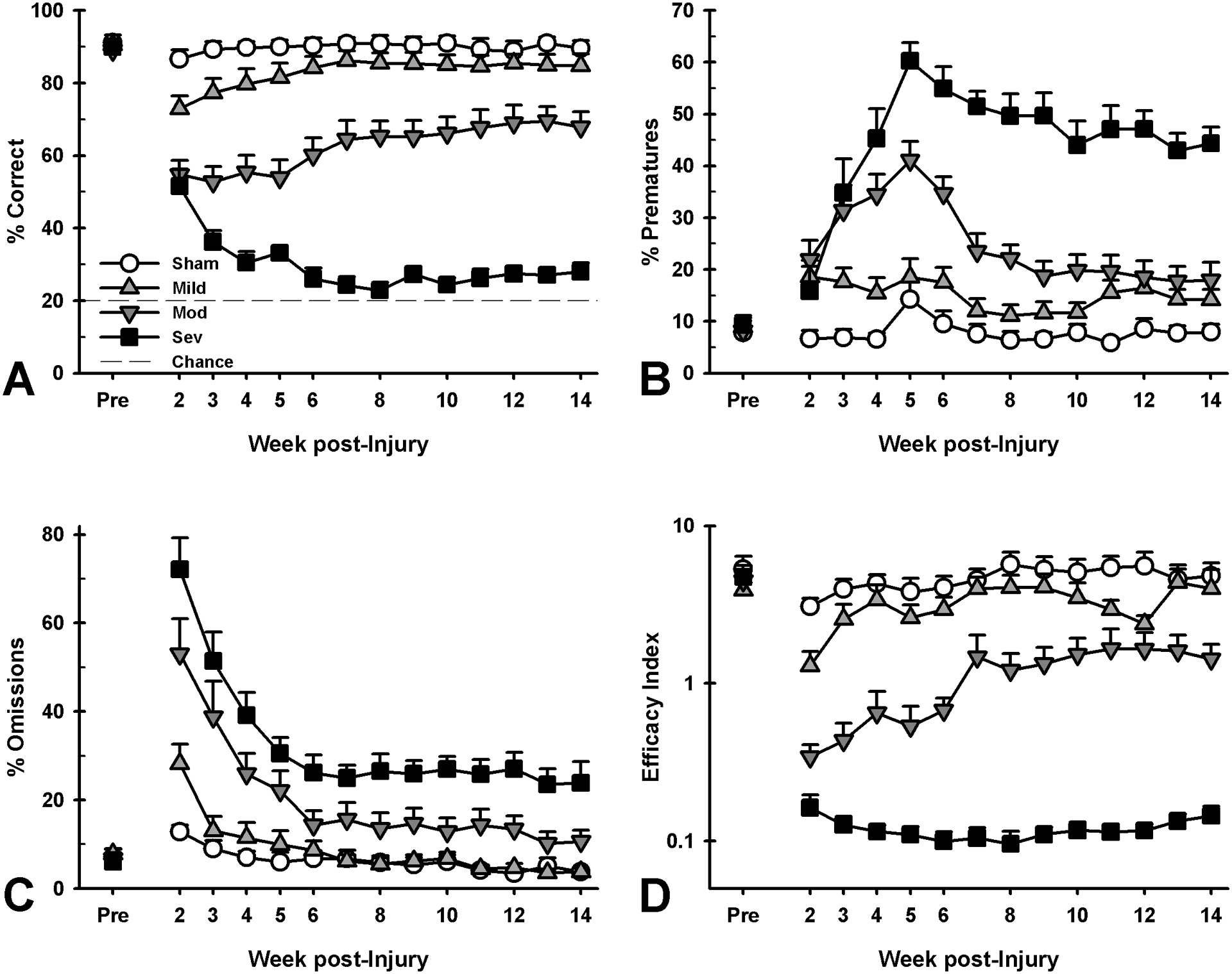
Effects of injury on 5CSRT performance at acute (week 2–5) and chronic (week 5–14) time points. Deficits in all domains were tiered by injury severity. A) Mild-injured rats demonstrated significant acute deficits in attention (p = 0.004) which recovered over time (p = 0.189), while moderate- and severe-injured rats had significant acute (p < 0.001; p < 0.001) and continuing chronic deficits (p < 0.001; p < 0.001). B) Mild-, moderate- and severe-injured rats showed increased impulsive responding in the acute period (p = 0.001; p < 0.001; p < 0.001), which remained elevated throughout chronic testing (p = 0.052; p < 0.001; p < 0.001). C) Mild-injured rats had no significant change in omitted trials (p = 0.192; p = 0.899), yet moderate- and severe-injured rats showed increased omissions at both the acute (p < 0.001; p < 0.001) and chronic (p < 0.040; p < 0.001) time points. D) Mild-injured animals were initially impaired in overall task efficacy (p = 0.001), but recovered during chronic testing (p = 0.115), while moderate- and severe-injured animals demonstrated initial deficits (p < 0.001; p < 0.001) lasting into the chronic period (p < 0.001; p < 0.001). Data shown are mean + SEM.
Impairment across multiple domains was still evident in the Moderate and Severe groups during the chronic phase (day 30 – 104 post-injury), whereas deficits had largely resolved in the Mild group with the exception of a strong trend towards increased premature responding (Figure 2 and Table S1; accuracy: each group was different from every other group, p’s < 0.001, except for the Sham and Mild group, p = 0.189; prematures: each group was different from every other group, p’s < 0.014, except for the Sham and Mild group, which approached significance, p = 0.052; omissions: each group was different from every other group, p’s < 0.041, except for the Sham and Mild group, p = 0.899; task efficacy index: each group was different from every other group, p’s < 0.001, except for the Sham and Mild group: p = 0.115; see Figure S1, Table S1 and supporting information for group differences in total trials, choice and collection latencies after moderate and severe, but not mild TBI).
Regardless of injury severity, some rats demonstrated increased susceptibility to the effects of TBI, even in the Mild and Moderate groups (Figure 3A). Rats were reclassified based on their behavioral performance as TBI-Resilient, TBI-Vulnerable, and Chronically-Impaired. Resilient rats had small, transient deficits in attention and task efficacy, while Vulnerable rats demonstrated deficits across all behaviors that recovered over time, but never to baseline levels. However, Chronically Impaired rats were only able to regain minor function with substantial, enduring deficits across all outcome measures. (Figure 3 and Table S2; Resilient: impaired in the acute phase on accuracy and efficacy index, p’s < 0.046, recovered across all variables in the chronic phase, p’s > 0.186; Vulnerable: impaired on all variables in the acute phase, p’s < 0.001, only omissions recovered to baseline level in chronic phase, other p’s < 0.013; Chronically Impaired: impaired on all variables in the acute phase, p’s < 0.001 and the chronic phase, p’s < 0.001; see Figure S2, Table S2 and supporting information for additional group analyses).
Figure 3.
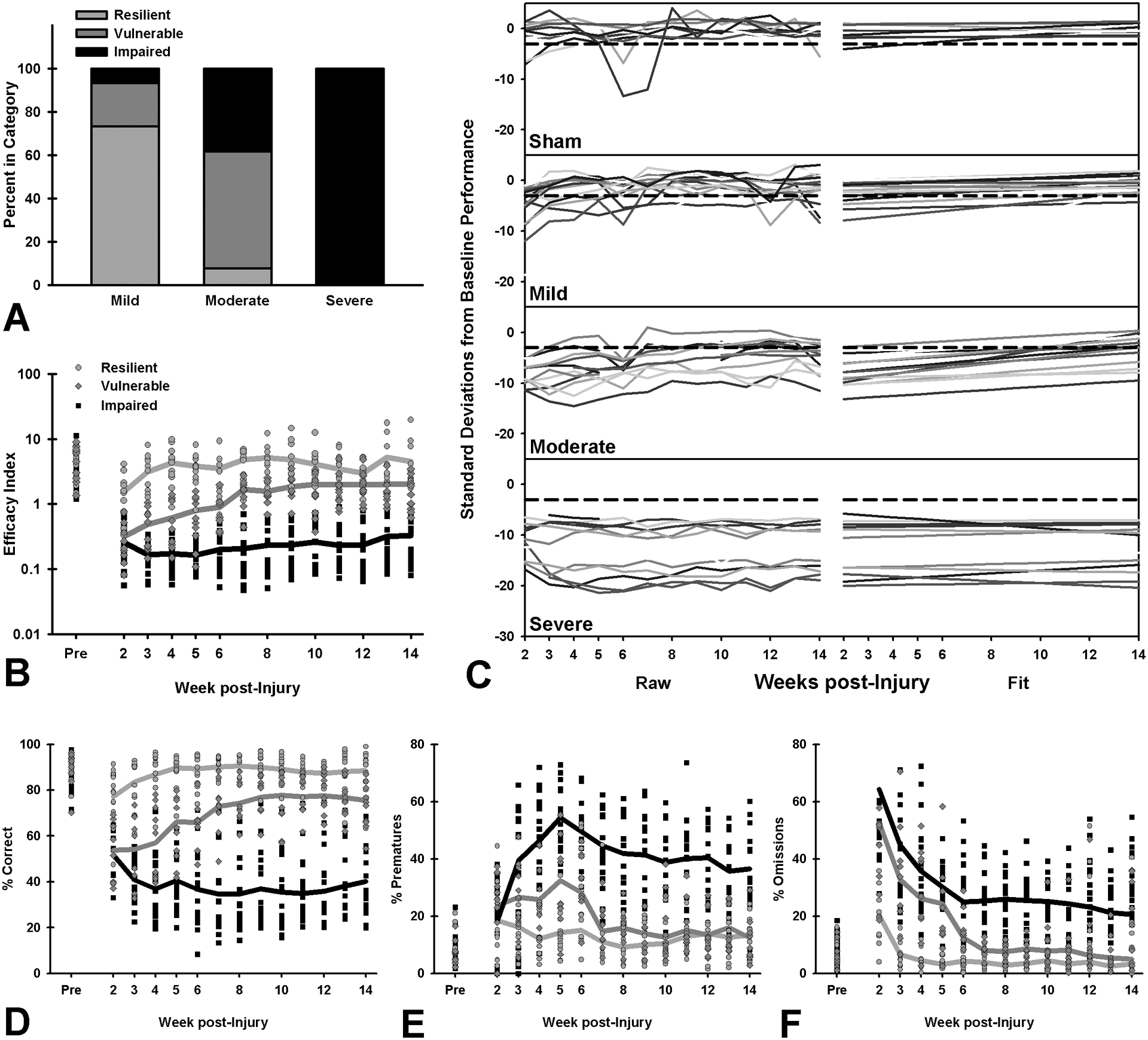
Individual differences in 5CSRT performance and response to brain injury at acute (week 2–5) and chronic (week 5–14) time points. Although sham data is shown for reference in panel C, only injured rats were included in analyses. A) Independent of injury conditions, rats were categorized as “resilient” if they recovered within 5 weeks, “vulnerable” if they recovered by the end of testing (14 weeks), or “chronically impaired” if they never recovered. B) Resilient rats showed acute reductions in task efficacy (p = 0.032) which resolved over time (p = 0.940), while vulnerable rats had continuing deficits despite their recovery (p < 0.001), and chronically impaired rats had unrecovered deficits (p < 0.001). C) The left side of the panel shows raw data for each subject in terms of standard deviations from baseline performance (overall task efficacy), while the right side shows regression fits. Recovery was defined as within 3 standard deviations of (individual) baseline performance (dashed lines). Rats in each injury group show a highly variable response to brain injury. D) Resilient rats demonstrated acute deficits in attention (p = 0.045), which quickly recovered to baseline levels (p = 0.770), while neither vulnerable nor chronically impaired rats fully recovered (p < 0.001; p < 0.001). E) Resilient rats had no change in impulsivity across testing (p = 0.145; p = 0.529), but both vulnerable and chronically impaired rats showed increased impulsivity in the acute period (p < 0.001; p < 0.001), which extended to chronic testing (p = 0.012; p < 0.001). F) Resilient rats showed no change in omitted trials during acute or chronic phases (p = 0.565; p = 0.186), while vulnerable rats showed deficits in the acute (p < 0.001), but not chronic time points (p = 0.445), and chronically impaired rats demonstrated increases throughout testing (p < 0.001). Data shown are individual subjects’ data points and group means.
Effects of amphetamine
Rats with TBI showed a differential response to amphetamine administration compared to shams, suggesting damage-dependent changes in monoaminergic systems. As expected, sham controls became more impulsive and less accurate as the dose increased. Mildly-injured rats were similarly more impulsive, but were still accurate, while moderately-injured rats showed no effect of the drug. In contrast, severely-injured rats showed the opposite behavioral response to sham controls, such that these animals appeared less impulsive and more accurate following amphetamine administration (Figure 4 and Table S3; accuracy: Group × Dose interaction, p = 0.009, Sham decreased at 0.6 and 1.0 mg/kg, p’s < 0.013, Mild no change at any dose, p’s > 0.052, Moderate no change at any dose, p’s > 0.151, Severe increased at 1.0 mg/kg, p = 0.002; prematures: Group × Dose interaction, p < 0.001, Sham increased at 0.3, 0.6 and 1.0 mg/kg, p’s < 0.040, Mild increased at 0.3, 0.6 and 1.0 mg/kg, p’s < 0.030, Moderate no change at any dose, p’s > 0.128, Severe decreased at 1.0 mg/kg, p = 0.015). However, there was no significant interaction of group and dose on omissions or the task efficacy index; overall, animals made more omissions and their task efficacy decreased as dose increased. Collectively, these analyses indicate that the beneficial effects of amphetamine observed in severely-injured rats may be mediated by increased omissions, or that there was insufficient power to detect any group-specific effects in general task efficacy (Figure 4 and Table S3; omissions: Dose effect, p < 0.001, increased at 1.0 mg/kg, p = 0.004; task efficacy index: Dose effect, p < 0.001, decreased at 0.3, 0.6, and 1.0 mg/kg, p’s < 0.045; see Figure S4, Table S3 and supporting information for analyses regarding dose-dependent effects of decreasing trials, choice and collection latencies).
Figure 4.
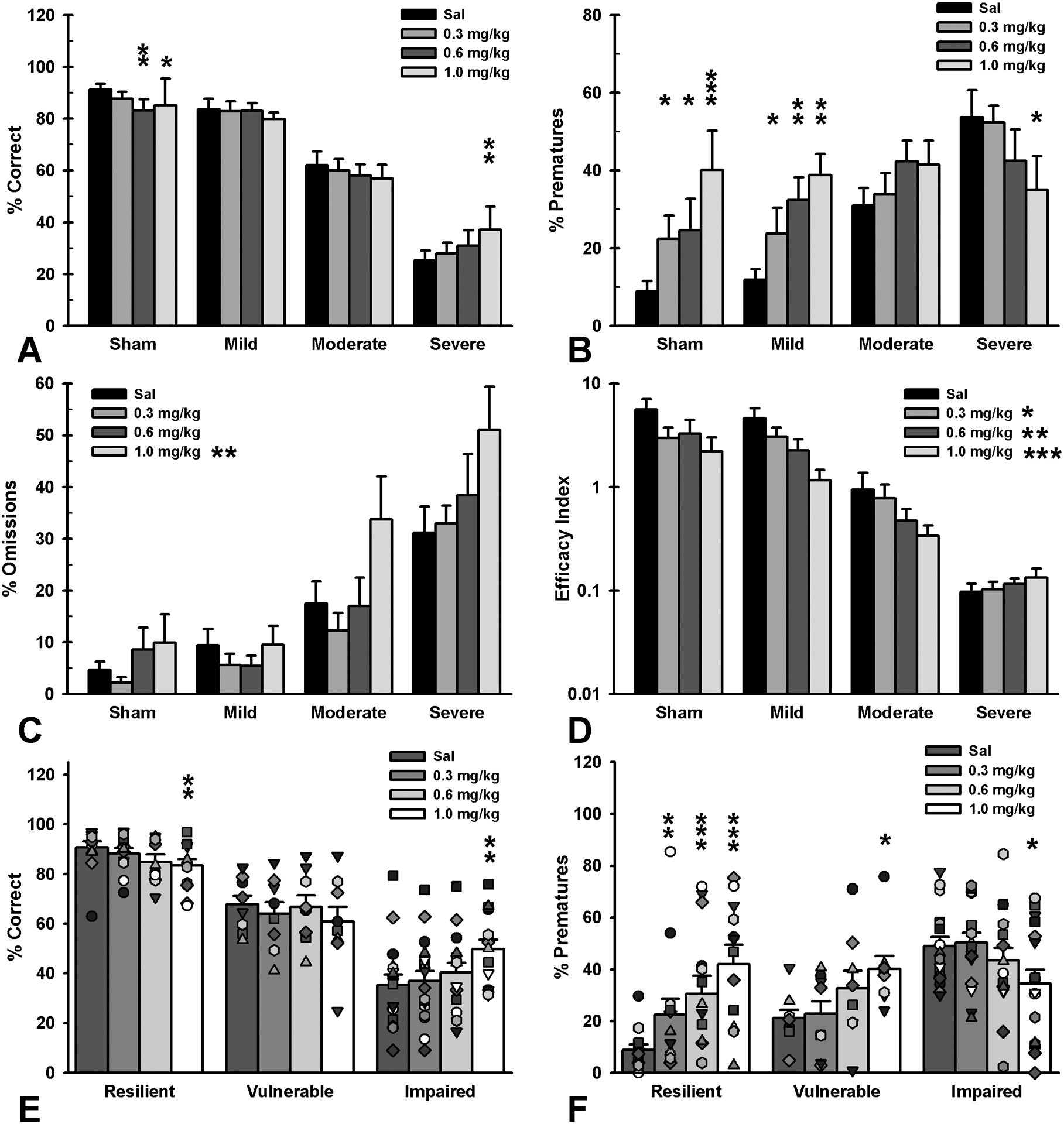
Effects of amphetamine on 5CSRT performance tiered by injury severity, as determined by impact force, vs. injury susceptibility, as determined by trajectory of recovery. A) Severe-injured rats had improved attention at 1.0 mg/kg (p = 0.002), moderate-injured rats showed no change at any dose (p’s > 0.151), mild-injured rats approached impairment at 1.0 mg/kg (p = 0.052) and sham rats were impaired at the 0.6 or 1.0 mg/kg (p = 0.009; p = 0.012). B) Severe-injured rats exhibited reduced impulsivity at 1.0 mg/kg (p = 0.015), moderate-injured rats showed no change across doses (p’s > 0.128), while impulsivity increased in both mild-injured and sham rats at all doses compared to saline (p’s < 0.040). C) Overall, omissions increased at the 1.0 mg/kg dose (p = 0.004). D) In general, rats showed reduced task efficacy at all doses (p’s < 0.045). E) Susceptibility subgroups demonstrated differential effects, with resilient rats showing reduced accuracy at 1.0 mg/kg (p = 0.002), vulnerable rats showing no change at any dose (p’s > 0.137), and chronically impaired rats showing improved function at 1.0 mg/kg (p = 0.002). F) Subgroups also demonstrated similar effects with regards to impulsivity with resilient and vulnerable rats showing increased impulsivity as a function of increasing dose (p’s < 0.021) and chronically impaired rats demonstrating reduced impulsive responding at the 1.0 mg/kg dose (p = 0.012). Data shown are mean + SEM and individual data points in panels E and F, * = p < 0.05, ** = p < 0.01, ***= p < 0.001.
Susceptibility to TBI-induced impairments played a significant role in response to amphetamine. Resilient rats demonstrated decreased response efficacy across all doses, similar to shams, while the Vulnerable group showed a similar, but blunted response and was only significantly impaired at the highest dose. However, the Chronically Impaired group responded in an opposite manner, with increased accuracy, reduced prematures and no decrease in task efficacy, suggesting that amphetamine may be a useful therapeutic in this subset of animals (Figure 4, Figure S2, Table S4; accuracy: Resilient decreased at 1.0 mg/kg, p = 0.002, Vulnerable no effect at any dose, p’s > 0.137, Chronically Impaired increased at 1.0 mg/kg, p = 0.002; prematures: Resilient increased at all doses, p’s < 0.015, Vulnerable increased at 1.0 mg/kg, p = 0.021, Chronically Impaired reduced at 1.0 mg/kg, p = 0.012; omissions: no Group × Dose interaction; task efficacy index: Resilient decreased at all doses, p’s < 0.012, Vulnerable decreased at 1.0 mg/kg, p = 0.009, Chronically Impaired no change at any dose, p’s > 0.338).
Effects of atomoxetine
Unlike amphetamine, there were no contrasting effects of atomoxetine across injury groups. All animals showed a small, but significant drop in accuracy at the lowest dose as well as reduced impulsivity and increased omissions at the highest dose (Figure S5 and Table S5; accuracy: Dose effect, p = 0.020, decreased at 0.1 mg/kg, p = 0.049; prematures: Dose effect, p = 0.043, decreased at 1.0 mg/kg; omissions: Dose effect, p = 0.003, increased at 1.0 mg/kg, p = 0.002; task efficacy index: no Dose effect, p = 0.331; see Figure S5, Table S5 and supporting information for analyses regarding effects of dose decreasing trials). There were no differential responses to atomoxetine based on injury susceptibility classification.
Effects of amantadine
Amantadine had similar effects across both sham and injured animals. In a dose-dependent fashion, it reduced premature responding, increased omitted trials and had no major effect on accuracy, but reduced the task efficacy index, suggesting detrimental effects at higher doses. There was no strong differential response to amantadine in the injured animals, but the Moderate group showed a slight sensitivity with increased omissions at the 20 mg/kg dose (Figure S6 and Table S6; accuracy: Dose effect, p = 0.039, however, no change compared to saline; prematures: Dose effect, p < 0.001, decreased at 20 and 40 mg/kg, p’s < 0.002; omissions: Group × Dose interaction, p = 0.013, all groups increased at 40 mg/kg, p’s < 0.011, Moderate increased at 20 mg/kg, p = 0.002; task efficacy index: Dose effect, p = 0.001, decreased at 40 mg/kg, p = 0.010; see Figure S6, Table S6 and supporting information for analyses of trials, choice and collection latencies indicating psychomotor slowing at high doses). As per atomoxetine, susceptibility to long-term cognitive impairments did not lead to a differential response to amantadine.
Lesion analysis and neuroinflammatory markers
Lesion formation was tiered based on injury severity. Cavitation from the anterior portion of the brain to the striatum was measured, as well as ventricle enlargement across all groups compared to sham (Figure 5A, B; p’s < 0.001). Prefrontal cortical levels of cytokines (IL-1α, IL-1β, IL-2, IL-4, IL6, IL10, IL-12, TNFα, IFNγ) from a subset of animals (N = 25) representing a spectrum of behavioral function were measured using multiplex ELISA. IL-1β and IFNγ fell below detectable levels. ANOVAs revealed that only IL-12 was significantly different across the groups, with increased levels regardless of injury severity (Figure 5F and Table S7; F3,20 = 5.46, p = 0.007, Mild and Moderate increased relative to Sham, p’s < 0.041, Severe approached significance, p = 0.053). A correlation matrix examining the relationships between the measured cytokines, lesion size, attention, and impulsivity showed significant relationships between the various inflammatory markers, as well as significant relationships between cytokines IL-1α, IL-6, IL-10, IL-12 and lesion size, attention and impulsivity (Figure 5C–F; p’s < 0.039; for full correlation matrix see Table S8; for cytokines IL-2, IL-4 and TNFα, see Figure S7).
Figure 5.
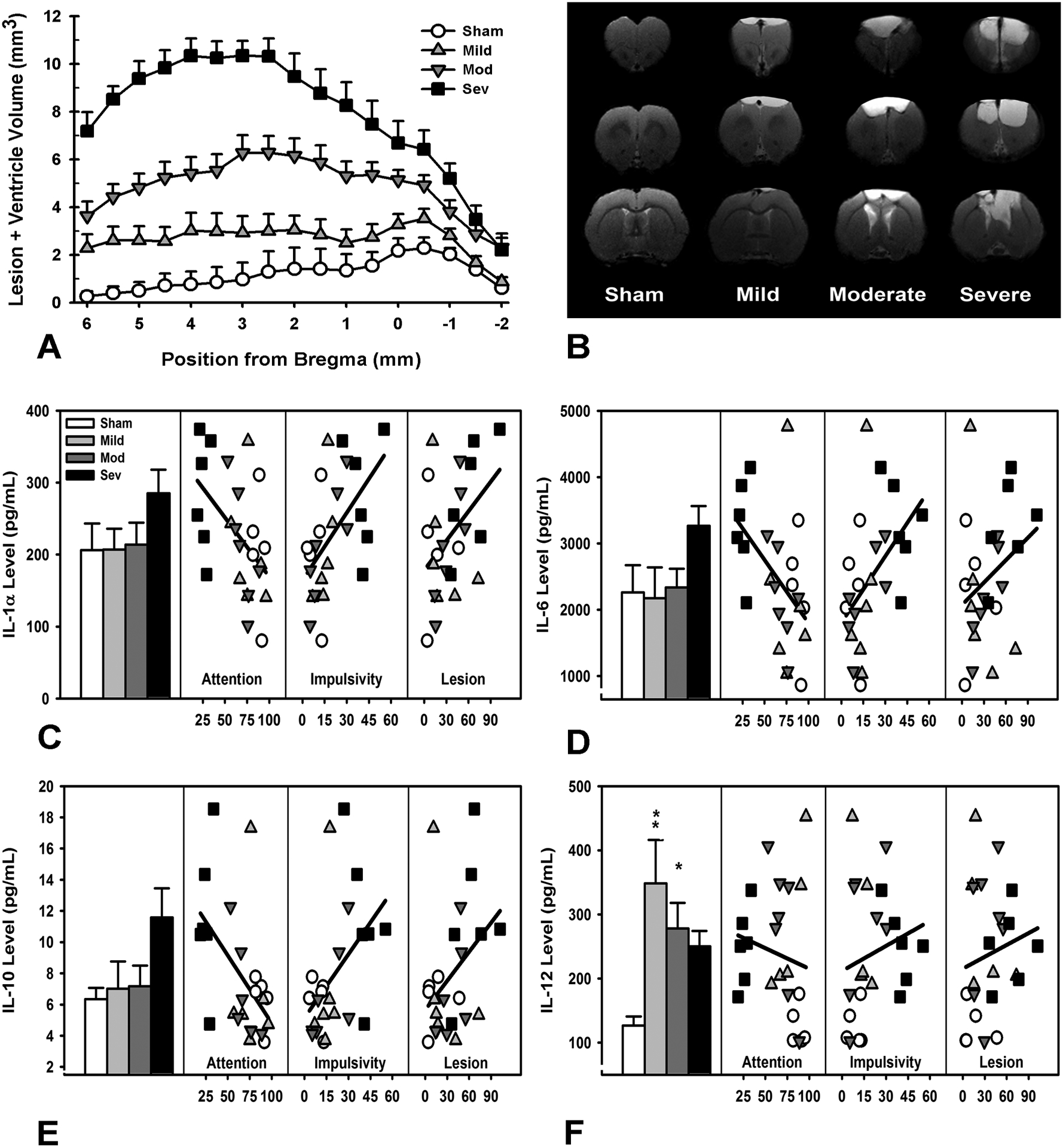
Histological and immune markers and their relationship to functional outcome. A) Lesion cavitation and ventricle size were significantly increased in a severity-dependent manner (p’s < 0.001). B) MRI histoplate demonstrating representative brains from each group. Minor cavitation was evident in mild-injured rats, with increasing damage and ventricular enlargement visible in moderate- and severe-injured rats. C) There were no group differences in IL-1α levels (p = 0.307), however, IL-1α was significantly correlated with attention, impulsivity, and lesion size (p = 0.003; p = 0.001; p = 0.016). D) IL-6 levels were not significantly different across the groups (p = 0.190), however, they were significantly correlated with attention, impulsivity, and lesion size (p < 0.001; p < 0.001; p = 0.008). E) There were no group differences in IL-10 levels (p = 0.172), however, IL-10 was significantly correlated with attention, impulsivity, and lesion size (p’s < 0.001). F) IL-12 levels were significantly increased in mild and moderate TBI groups (p = 0.004; p = 0.040), and approached significance for severe (p = 0.053); IL-12 levels were also significantly correlated with attention, impulsivity, and lesion size (p = 0.027; p = 0.038; p = 0.011). Data shown are mean + SEM in panel A and C-F and raw data points in panels C-F, * = p < 0.05, ** = p < 0.01, ***= p < 0.001.
Given the substantial correlation between cytokines and complexity of cytokine interactions, a PCA was conducted to reduce the data and determine common variance. The PCA revealed three primary components, accounting for 95.97% of the variance in the dataset; PC1 and PC2 both represented generalized neuroinflammation, with relatively strong, equal component loadings (>0.3) for all cytokines, except for IL-12. In contrast, PC3 was heavily dominated by an IL-12 loading (0.93), with weak contributions from other cytokines (Figure S8 and Table S9). The principal components captured injury-specific effects with unequal expression across the groups; the Severe group differed from Sham on PC2, and the Mild and Moderate groups showed lower levels of PC3 (Figure 6A–C, Table S12; PC1 F3,21 = 0.45, p = 0.719; PC2 F3,21 = 3.89, p = 0.024, Severe decreased relative to Sham, p = 0.014; PC3 F3,21 = 7.91, p = 0.001, Moderate and Mild decreased relative to Sham, p’s < 0.025). In order to determine the relative contributions to behavioral dysfunction, the principal components were then analyzed, along with lesion size using multiple regression. Larger lesions were associated with lower performance on attention and task efficacy index measures, while both larger lesion size and increases in PC2 were associated with increased impulsivity (Figure 6 and Table S10; accuracy: lesion, p < 0.001; prematures: lesion & PC2, p’s < 0.037, omissions: no significant predictors; task efficacy index: lesion, p = 0.001). Analysis of TBI susceptibility data revealed that lesion size was predictive of degree of recovery for attention, omissions, and the task efficacy index, but a model with both lesion size and PC1 better accounted for recovery of function in premature responding (Table S10; accuracy: lesion, p = 0.043; prematures: lesion, p = 0.043, PC2, p = 0.056, omissions: lesion, p = 0.108; task efficacy index: lesion, p = 0.020).
Figure 6.
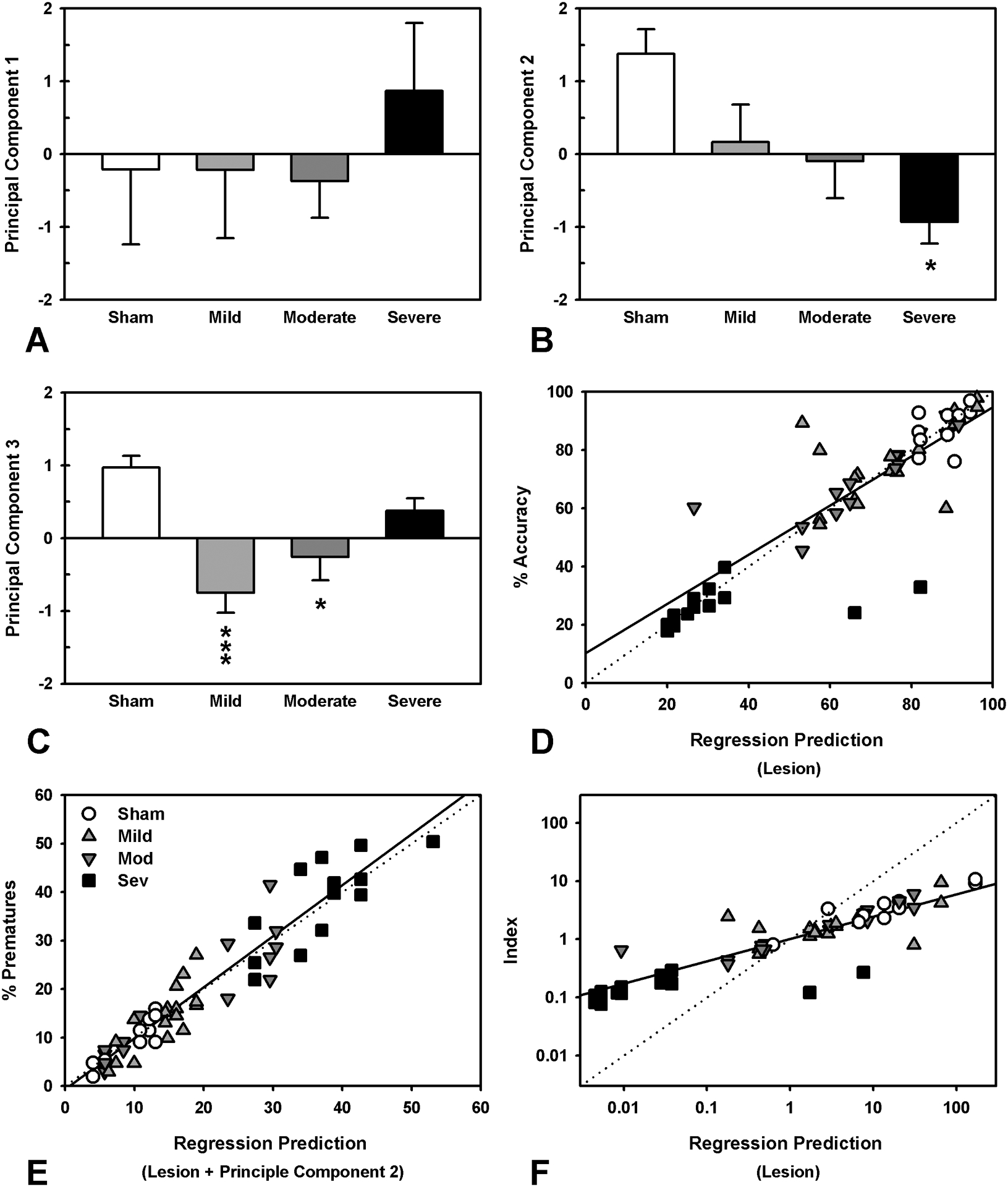
Comparison of neuroinflammation principal components across injury groups and regression analyses of lesion and principal component data and their relationship to behavioral outcomes. A) PC1 did not differ across injury groups (p = 0.719). B) PC2 was significantly lower in the severe TBI group (p = 0.014). C) Both the mild and moderate TBI group had significantly lower levels of PC3 (p < 0.001; p = 0.019). E) Multiple regression revealed that lesion size was most strongly associated with accuracy (p < 0.001). F) A regression with both lesion size (p = 0.030) and principal component 2 (p = 0.036) best accounted for premature responses. G) Multiple regression showed that lesion size (p = 0.001) was significantly associated with task efficacy, although with a much poorer model fit compared to other measures. Data shown are mean + SEM in panels A-C, and raw versus predicted data points in D-F; the dashed line demonstrates perfect prediction, while the solid line represents the actual model, * = p < 0.05, ** = p < 0.01, ***= p < 0.001.
Discussion
In this study, we demonstrated that frontal TBI, at multiple levels of severity, resulted in substantial, persistent deficits in several domains of function, namely attention, impulse control, ability to complete trials, choice and reinforcer collection latencies. In particular, high impulsivity was the most pervasive symptom and persistent elevations in impulsivity were still evident over three months post-injury, even in mildly-injured animals. To our knowledge, this is the first study to replicate impulse control deficits often described in patients (7, 22, 23) using an animal model of TBI, and our data also reproduce the considerable individual variation in recovery trajectory that is observed in human patients. The unique ability of amphetamine to reduce impulsivity and improve attention in chronically impaired animals is of substantial clinical interest given the lack of options for treating this population. Furthermore, the long-term neuroinflammation caused by brain injury was specifically associated with increased impulsivity, even when accounting for gross tissue loss. While long-lasting inflammation has been previously identified throughout the brain (9, 11, 24, 25), the present data identified increased inflammation in the frontal cortex, and suggest that it may be directly involved in the modulation of pro-impulsive behaviors.
The 5CSRT task is highly regarded for its ability to parse unique domains of cognitive function (26). However, the current study necessitated a novel measure, the task efficacy index, in order to capture the extensive nature of the deficits. This measure is derived from the ratio of beneficial actions (correct responses) to detrimental actions (incorrect responses, premature responses, omitted responses). As such, it provides information about how these variables vary together, and was useful not only for the ‘big picture’ of injury, but also in evaluating the overall effects of pharmacological challenges. By combining complex behavioral analyses with MRI scanning to fully characterize the extent of the lesion, as well as multiple markers of cognitive decline, we were able to capture a specific phenotype of chronic impulsive and attentional deficits, and evaluate relevant physiological sequelae of TBI.
One of our most interesting and novel findings is that not all rats responded to injury in the same fashion, despite the use of CCI, arguably the most reproducible method of experimental TBI (27). Although deficits were broadly tied to the severity of the impact, several rats within the mild and moderate injury groups developed chronic deficits that never fully recovered. These differences again reflect similarities to the human condition, in which some patients successfully recover with relatively minor interventions while others go on to develop debilitating neuropsychiatric symptoms (2). Although this level of individual variation in response to experimental TBI is rarely found or reported, capturing this variance in animal models is critical for the identification of factors that confer vulnerability or resilience to TBI-induced cognitive impairment, an issue of considerable relevance to therapeutic development.
A prime example of this can be seen in our own data, in that beneficial effects of amphetamine on cognitive function only fully emerged when animals were stratified by their level of impairment rather than severity of impact: those that were most susceptible to the cognitive sequelae of injury showed improvement, compared to unchanged or impaired performance in more resilient groups. Such a treatment response may indicate that TBI has induced a shift in dopaminergic signaling in severely affected subjects (28, 29) that may be remediated with psychostimulants. However, studies in patients have found mixed results with administration of methylphenidate (30, 31). The large-scale disruptions of neurotransmitter systems following more severe injuries may also explain why a drug such as amphetamine, which has multiple mechanisms of action, may have greater benefits than a more selective compound such as atomoxetine which is a relatively selective noradrenaline reuptake inhibitor (32). Although atomoxetine (1.0 mg/kg) was able to reduce premature responding across all animals, replicating previous reports (33), this was accompanied by increased omissions. Similarly, amantadine, which has been used experimentally in human TBI patients (34) and animal models (35, 36), greatly decreased impulsive responses, but this was again confounded by psychomotor slowing as evidenced by increased omissions, response latencies and an overall decrease in task efficacy. This study is the first use of amantadine as an acute challenge, and it may require multiple doses to achieve efficacy, explaining differences observed here versus previous TBI studies (35, 36). Collectively, such findings highlight the importance of testing therapeutic efficacy in an injury model rather than exclusively in healthy rats, and the need for further research into which drugs might be uniquely effective in TBI patients.
An increased prevalence of pre-morbid behavioral vulnerabilities to TBI is one of the hypotheses proposed to explain why some individuals experience worse recovery following an injury event (37). However, in the context of the current study, we were not able to identify any pre-existing behavioral traits (e.g. increased impulsivity) predicting susceptibility (Figure 3), suggesting that any relationship between prior cognitive function and injury outcome is based on environmental, rather than neurobiological, factors. We did, however, observe changes in markers of neuroinflammation in the frontal cortex of injured rats that were associated with impairments in impulsivity. Multiple regression analyses indicated that, while lesion size was an important component of attentional impairment and overall task efficacy, this failed to account for all of the observed deficits. Most interestingly, while many specific cytokines were not significantly elevated across the groups, changes in generalized neuroinflammation, as identified by principal components analysis (PC2), were strongly associated with chronic impulsivity as well as the degree of recovery in impulse control. This underscores the dual nature of the inflammatory response to injury - both harmful and beneficial - as emphasized by others (38).
Neuroinflammation has been implicated in a number of psychiatric disorders in which impulsivity is prominent, most notably bipolar disorder and suicidality, but also impulsive aggression (13, 39, 40). Although the relationship between brain injury and the emergence of neuropsychiatric symptoms is complex, chronic inflammation may represent a mechanistic link, accounting for a portion of the increased susceptibility following TBI. Further, understanding this mechanism could potentially lead to therapeutics aimed at improving long-term dysfunction by either augmenting or replacing existing pharmacotherapies. Drugs such as lithium and other glycogen synthase kinase-3 inhibitors are already being explored as potential treatments (41, 42), although caution should be exercised as components of the inflammatory response can be beneficial to recovery, as seen in the current study. Of additional interest is the cytokine IL-12, which was highly elevated regardless of injury severity and dominated one principal component that accounted for 11% of the variance in all cytokine activity. Though IL-12 levels were significantly correlated with behavioral function, the IL-12-dominated principal component was not independently associated with functional outcomes in multiple regression analyses. IL-12 has not been shown to remain elevated in previous studies (43, 44), which suggests other sources of production beyond the macrophages that are likely responsible for levels acutely after brain injury, such as astrocytes or microglia (45). It would be useful to determine the time-course underlying prefrontal IL-12 expression following injury in future studies. Although inflammatory changes are likely occurring throughout the brain in response to prefrontal CCI, the 5CSRT is very sensitive to frontal damage, and direct modulation of singular cytokine expression within this region has resulted in behavioral change on other cognitive assessments that rely on intact frontocortical signaling (48). As such, inflammatory changes in the PFC have a high likelihood of contributing to the impulse control deficits observed here.
Brain injury is a complex problem, the solution to which has eluded scientists for several decades. The current study integrated several facets of TBI relevant to the human condition: multimodal cognition, relevant therapeutics, and measurement of long-lasting changes in cytokine levels at the site of impact. By using a clinically-relevant behavioral task, we were able to demonstrate a phenotype of impaired attention and increased impulsivity, which can now be used to answer numerous questions regarding the development of chronic deficits in brain injury. Our data highlight the potential for monoaminergic therapies to alleviate behavioral dysfunction in the most severely impaired, and emphasize the need to evaluate therapeutic agents in special populations. Finally, we have also identified that the neuroinflammatory response is specifically implicated in increased impulsivity post-injury, and that this may explain some of the individual differences in neurocognitive response to TBI. Further work targeting these pathways may yield therapeutic agents that can improve the lives of the millions living with cognitive disabilities due to brain injury.
Materials and Methods
Further details of experimental procedures, including apparatus, surgery, behavioral manipulations, MRI scanning, tissue collection, ELISA, and statistical analyses can be found in the supporting information. The experimental timeline can be found in Figure 1.
Figure 1.
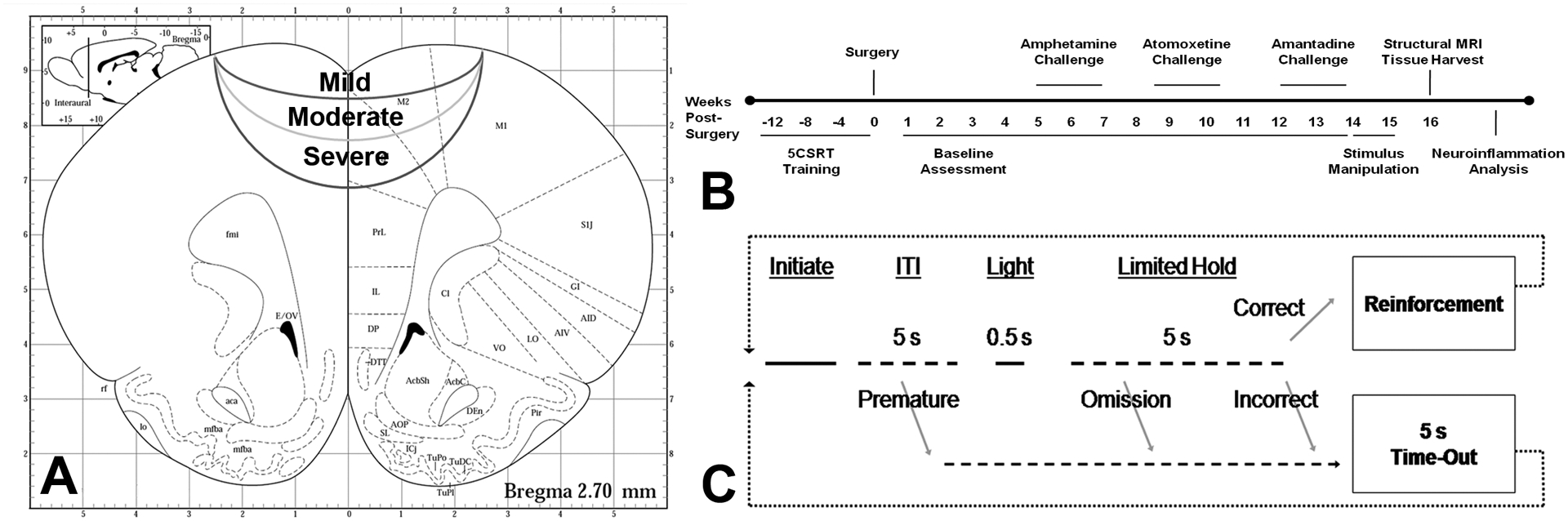
Study overview, including injury location, experimental timeline and five-choice serial reaction time (5CSRT) task description. A) Injury coordinates in stereotaxic space. Injuries were centered on the midline at +3.0 mm from bregma. Severe injuries impacted to a depth of −2.5 mm at 3 m/s, moderate injuries were −1.7 mm at 2 m/s (44% of severe force) and mild injuries were −0.8 mm at 1 m/s (11% of severe force). Adapted from Paxinos and Watson’s The Rat Brain in Stereotaxic Coordinates, 4th ed. B) Experimental timeline showing when training, assessment, pharmacological challenges and end points occurred. C) Task diagram for the 5CSRT task. After initiating a trial by making a nose poke response at the illuminated food tray, rats must wait 5 s for the brief stimulus light to appear at one of the five holes. Once that occurs, a nose poke at the correct hole is reinforced with a 0.45 mg sugar pellet. Incorrect or omitted responses are punished with a 5 s time-out in which the houselight comes on and no pellets may be earned. Responses made prematurely at the 5-hole array, before the stimulus light appears, are also punished with a 5 s time-out. Correct responses provide a measure of attention, and premature responses provide a measure of motor impulsivity. Latencies to make a choice and to collect the reinforcer were also recorded.
Subjects
Subjects were 50 Long-Evans male rats, approximately 2.5 months old at the start of the experiment and 6 months old at surgery. Rats were food restricted to 85% free-feeding weight (14–20g chow daily); water was available ad libitum. Rats were pair-housed on a reverse light cycle in standard cages during training and single-housed following surgery. A plastic hut and paper towel were available as enrichment. Housing and testing were performed in accordance with the Canadian Council on Animal Care and all procedures were approved by the University of British Columbia Animal Care Committee.
Behavioral Training
5CSRT task training followed previous methods (21, 49). Rats were trained to initiate a trial with a nose poke to the food hopper. Then, after a 5 s delay, a brief (0.5 s) stimulus light would be presented in one of the five response holes. A correct response—a nose poke into the illuminated hole—was reinforced by delivery of a sugar pellet, whereas a response in any other hole, or a failure to respond within 5 s, was scored as incorrect or as an omission, respectively, and punished by a 5-sec time-out. Premature responses made before the stimulus light came on provided a measure of motor impulsivity and were also punished with a 5 s timeout. There were a maximum of 100 trials per session and premature responses did not add to the total trial count (see Figure 1).
Surgery
Animals received either a single bilateral frontal CCI surgery or sham procedure as previously described (17, 50, 51). After a 6.0 mm diameter, circular craniotomy was performed, all injuries were induced with a circular, 5 mm diameter, flat-faced tip centered over the medial prefrontal cortex (AP +3.0, ML 0.0 from bregma). Once 5CSRT behavior was deemed statistically stable, animals were divided into groups matched for baseline performance and assigned to one of four surgical conditions (see Figure 1): severe TBI (n = 12): impact depth DV −2.5 mm @ 3 m/s for 0.5 s, as per previous work (50); moderate TBI (n = 13): impact settings 2/3 of severe, DV −1.7 mm @ 2 m/s for 0.5 s (force = 44.4% of severe); mild TBI (n = 15): impact settings 1/3 of severe, DV −0.8 @ 1 m/s for 0.5 s (force = 11.1% of severe); Sham (n = 10) surgeries followed an ‘intact sham’ procedure with no craniotomy, as recently recommended for maximal translational validity (52, 53).
Behavioral assessment
After seven days recovery, 5CSRT testing resumed. Rats were assessed for TBI-related deficits until all groups showed statistically stable performance (20–25 consecutive sessions of testing, approximately 4 weeks post-injury), before pharmacological challenges began. Baseline testing continued between days of drug administration and during washout weeks. Testing continued to 15 weeks post-injury.
Pharmacological challenges
Doses of each drug were administered according to a Latin square design (8 sequences, counterbalanced) with one day of washout (no behavior) and one day of baseline performance between each dose, and one week of baseline sessions between each drug. All drugs were prepared fresh daily, dissolved in 0.9% sterile saline and administered at a volume of 1 mL/kg, i.p. The assessed drugs were amphetamine (0.0, 0.3, 0.6 and 1.0 mg/kg doses, 10 min prior to testing, sourced from Sigma; (54)), atomoxetine (0.0, 0.1, 0.3 and 1.0 mg/kg doses, 15 min prior to testing, sourced from Tocris; (55)), and amantadine (0, 10, 20, 40 mg/kg doses, 15 min prior to testing, sourced from Sigma, dosing based on (35)).
Structural MRI scanning & lesion quantification
Following behavioral assessment (15 weeks post-injury), rats underwent structural MRI scanning in a 7T MRI. Remaining brain volume, ventricle and lesion size were quantified using T2-weighted image slices taken in 0.5 mm increments from +6.0 to −2.0 mm from bregma using ImageJ (NIH, Bethesda). The slice areas were multiplied by their thickness (0.5 mm) and summed across the entire range in order to generate a volumetric measurement as per the Cavalieri method (56).
Post mortem analysis of cytokine levels
Samples from the orbitofrontal and medial prefrontal cortex were collected at 15 weeks post-injury and analyzed for IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL12, TNFα, and IFNγ via multiplex ELISA (Quansys Q-plex, Logan, UT).
Injury susceptibility determination
Due to considerable variation in post-injury performance, particularly in the mild and moderate groups, all injured rats were also re-categorized in terms of susceptibility to TBI-induced cognitive impairment as either Resilient (<5 weeks to recover), Vulnerable (5–14 weeks to recover) or Chronically Impaired (never recovered) and data reanalyzed. Recovery was designated as performance within 3 standard deviations of individual baseline task efficacy index, which corresponded to the average variability in the sham animal population.
Data analysis
As per previous 5CSRT studies, the following behavioral variables were analyzed: trials completed, percent accuracy [correct / (correct + incorrect)*100], percent premature responses [(prematures / initiated trials)*100], percent omissions [(omissions / trials completed)*100], latencies to make a correct response and to collect the reinforcer. We also computed an additional task efficacy index [correct / (incorrect + omissions + prematures)] to capture the cross-variable nature of the deficits induced by TBI.
Repeated measures data (behavioral outcomes, pharmacological challenges, cytokine/lesion regressions) were analyzed with linear mixed effects regression; univariate data (cytokine levels) were analyzed using ANOVA and a Tukey posthoc test where appropriate; and relationships between variables were analyzed with correlations (cytokines and steady-state behavioral data) and principal components analysis (PCA; cytokine levels). All data were analyzed using R statistical software (http://www.r-project.org/) with the lme4, lmerTest and stats libraries. A p-value equal to or less than 0.05 was considered significant. For more details on data analyses, see supporting information.
Supplementary Material
Acknowledgements
Special thanks to the UBC MRI Research Centre for their assistance in scanning of animals and analysis of MRI data, in particular, Andrew Yung, Barry Bohnet and Dr. Piotr Kozlowski. Thanks to laboratory members who provided feedback during this project.
Funding
This work was supported by the Canadian Institutes for Health Research, the Michael Smith Foundation for Health Research, and the National Institute of Neurological Disorders and Stroke (R21NS087458).
Footnotes
Supporting Information
Supporting information including methodological details, additional analyses, and statistical details is available at the ACS Chemical Neuroscience website.
REFERENCES
- (1).Diaz-Arrastia R, and Kenney K (2014) Epidemiology of traumatic brain injury, in Traumatic Brain Injury (Vos P, and Diaz-Arrastia R, Eds.) pp 183–191, Wiley-Blackwell, Oxford, UK. [Google Scholar]
- (2).Rosenbaum SB, and Lipton ML (2012) Embracing chaos: The scope and importance of clinical and pathological heterogeneity in mTBI. Brain Imaging and Behavior 6, 255–282. [DOI] [PubMed] [Google Scholar]
- (3).Thurman DJ, Alverson C, Dunn KA, Guerrero J, and Sniezek JE (1999) Traumatic brain injury in the United States: A public health perspective. Journal of Head Trauma Rehabilitation 14, 602–615. [DOI] [PubMed] [Google Scholar]
- (4).Zaloshnja E, Miller T, Langlois JA, and Selassie AW (2008) Prevalence of Long-term disability from traumatic brain injury in the civilian population of the United States, 2005. The Journal of Head Trauma Rehabilitation 23, 394–400. [DOI] [PubMed] [Google Scholar]
- (5).Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, and Breitner JCS (2000) Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 55, 1158–1166. [DOI] [PubMed] [Google Scholar]
- (6).Semchuk KM, Love EJ, and Lee RG (1993) Parkinson’s disease: A test of the multifactorial etiologic hypothesis. Neurology 43, 1173–1180. [DOI] [PubMed] [Google Scholar]
- (7).Reeves RR, and Panguluri RL (2011) Neuropsychiatric complications of traumatic brain injury. Journal of Psychosocial Nursing and Mental Health Services 49, 42–50. [DOI] [PubMed] [Google Scholar]
- (8).Vaishnavi S, Rao V, and Fann JR (2009) Neuropsychiatric problems after traumatic brain injury: Unraveling the silent epidemic. Psychosomatics 50, 198–205. [DOI] [PubMed] [Google Scholar]
- (9).Kasturi BS, and Stein DG (2009) Traumatic brain injury causes long-term reduction in serum growth hormone and persistent astrocytosis in the cortico-hypothalamo-pituitary axis of adult male rats. Journal of Neurotrauma 26, 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zou H, Brayer SW, Hurwitz M, Niyonkuru C, Fowler LE, and Wagner AK (2013) Neuroprotective, neuroplastic, and neurobehavioral effects of daily treatment with levetiracetam in experimental traumatic brain injury. Neurorehabilitation and Neural Repair 27, 878–888. [DOI] [PubMed] [Google Scholar]
- (11).Holmin S, and Mathiesen T (1999) Long-term intracerebral inflammatory response after experimental focal brain injury in rat. NeuroReport 10, 1889–1891. [DOI] [PubMed] [Google Scholar]
- (12).Najjar S, Pearlman DM, Alper K, Najjar A, and Devinsky O (2013) Neuroinflammation and psychiatric illness. Journal of Neuroinflammation 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Courtet P, Giner L, Seneque M, Guillaume S, Olie E, and Ducasse D (2015) Neuroinflammation in suicide: Toward a comprehensive model. The World Journal of Biological Psychiatry epub, ahead of print, 1–23. [DOI] [PubMed] [Google Scholar]
- (14).Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, and DeKosky ST (1999) One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. Journal of Neurotrauma 16, 109–122. [DOI] [PubMed] [Google Scholar]
- (15).Pierce JES, Smith DH, Trojanowski JQ, and McIntosh TK (1998) Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience 87, 359–369. [DOI] [PubMed] [Google Scholar]
- (16).Shear DA, Lu X-CM, Bombard MC, Pedersen R, Chen Z, Davis A, and Tortella FC (2010) Longitudinal characterization of motor and cognitive deficits in a model of penetrating ballistic-like brain injury. Journal of Neurotrauma 27, 1911–1923. [DOI] [PubMed] [Google Scholar]
- (17).Hoffman SW, Fülöp Z, and Stein DG (1994) Bilateral frontal cortical contusion in rats: Behavioral and anatomic consequences. Journal of Neurotrauma 11, 417–431. [DOI] [PubMed] [Google Scholar]
- (18).Hoane MR, Akstulewicz SL, and Toppen J (2003) Treatment with vitamin B3 improves functional recovery and reduces GFAP expression following traumatic brain injury in rats. Journal of Neurotrauma 20, 1189–1199. [DOI] [PubMed] [Google Scholar]
- (19).Goss CW, Hoffman SW, and Stein DG (2003) Behavioral effects and anatomic correlates after brain injury: a progesterone dose-response study. Pharmacology Biochemistry and Behavior 76, 231–242. [DOI] [PubMed] [Google Scholar]
- (20).van der Staay FJ (2006) Animal models of behavioral dysfunctions: Basic concepts and classifications, and an evaluation strategy. Brain Research Reviews 52, 131–159. [DOI] [PubMed] [Google Scholar]
- (21).Carli M, Robbins TW, Evenden JL, and Everitt BJ (1983) Effects of lesions to ascending noradrenergic neurones on performance of a 5-choice serial reaction task in rats; implications for theories of dorsal noradrenergic bundle function based on selective attention and arousal. Behavioural Brain Research 9, 361–380. [DOI] [PubMed] [Google Scholar]
- (22).Greve KW, Sherwin E, Stanford MS, Mathias C, Love J, and Ramzinski P (2001) Personality and neurocognitive correlates of impulsive aggression in long-term survivors of severe traumatic brain injury. Brain Injury 15, 255–262. [DOI] [PubMed] [Google Scholar]
- (23).Rochat L, Beni C, Billieux J, Azouvi P, Annoni J-M, and Van der Linden M (2010) Assessment of impulsivity after moderate to severe traumatic brain injury. Neuropsychological Rehabilitation 20, 778–797. [DOI] [PubMed] [Google Scholar]
- (24).Morganti JM, Jopson TD, Liu S, Riparip L-K, Guandique CK, Gupta N, Ferguson AR, and Rosi S (2016) CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. The Journal of Neuroscience 35, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Piao C-S, Stoica BA, Wu J, Sabirzhanov B, Zhao Z, Cabatbat R, Loane DJ, and Faden AI (2013) Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiology of Disease 54, 252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Robbins TW (2002) The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology 163, 362–380. [DOI] [PubMed] [Google Scholar]
- (27).Brody DL, MacDonald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, Kim E, Schwetye KE, Holtzman DM, and Bayly PV (2007) Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. Journal of Neurotrauma 24, 657–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bales JW, Wagner AK, Kline AE, and Dixon CE (2009) Persistent cognitive dysfunction after traumatic brain injury: A dopamine hypothesis. Neuroscience & Biobehavioral Reviews 33, 981–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wagner AK, Sokoloski JE, Ren D, Chen X, Khan AS, Zafonte RD, Michael AC, and Dixon CE (2005) Controlled cortical impact injury affects dopaminergic transmission in the rat striatum. Journal of Neurochemistry 95, 457–465. [DOI] [PubMed] [Google Scholar]
- (30).Plenger PM, Dixon CE, Castillo RM, Frankowski RF, Yablon SA, and Levin HS (1996) Subacute methylphenidate treatment for moderate to moderately severe traumatic brain injury: A preliminary double-blind placebo-controlled study. Archives of Physical Medicine and Rehabilitation 77, 536–540. [DOI] [PubMed] [Google Scholar]
- (31).Speech TJ, Rao SM, Osmon DC, and Sperry LT (1993) A double-blind controlled study of methylphenidate treatment in closed head injury. Brain Injury 7, 333–338. [DOI] [PubMed] [Google Scholar]
- (32).Yu G, Li G-F, and Markowitz JS (2016) Atomoxetine: A review of its pharmacokinetics and pharmacogenomics relative to drug disposition. Journal of Child and Adolescent Psychopharmacology 26, 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Navarra R, Graf R, Huang Y, Logue S, Comery T, Hughes Z, and Day M (2008) Effects of atomoxetine and methylphenidate on attention and impulsivity in the 5-choice serial reaction time test. Progress in Neuro-Psychopharmacology and Biological Psychiatry 32, 34–41. [DOI] [PubMed] [Google Scholar]
- (34).Giacino JT, Whyte J, Bagiella E, Kalmar K, Childs N, Khademi A, Eifert B, Long D, Katz DI, and Cho S (2012) Placebo-controlled trial of amantadine for severe traumatic brain injury. New England Journal of Medicine 366, 819–826. [DOI] [PubMed] [Google Scholar]
- (35).Dixon CE, Kraus MF, Kline AE, Ma X, Yan HQ, Griffith RG, Wolfson BM, and Marion DW (1998) Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restorative neurology and neuroscience 14, 285–294. [PubMed] [Google Scholar]
- (36).Wang T, Huang X-J, Van KC, Went GT, Nguyen JT, and Lyeth BG (2014) Amantadine improves cognitive outcome and increases neuronal survival after fluid percussion traumatic brain injury in rats. Journal of Neurotrauma 31, 370–377. [DOI] [PubMed] [Google Scholar]
- (37).Davidson J, Cusimano MD, and Bendena WG (2014) Post-traumatic brain injury genetic susceptibility to outcome. The Neuroscientist 21, 424–441. [DOI] [PubMed] [Google Scholar]
- (38).Lenzlinger PM, Morganti-Kossmann M-C, Laurer HL, and McIntosh TK (2001) The duality of the inflammatory response to traumatic brain injury. Molecular Neurobiology 24, 169–181. [DOI] [PubMed] [Google Scholar]
- (39).Coccaro EF, Lee R, and Coussons-Read M (2014) Elevated plasma inflammatory markers in individuals with intermittent explosive disorder and correlation with aggression in humans. JAMA Psychiatry 71, 158–165. [DOI] [PubMed] [Google Scholar]
- (40).Rosenblat JD, Brietzke E, Mansur RB, Maruschak NA, Lee Y, and McIntyre RS (2015) Inflammation as a neurobiological substrate of cognitive impairment in bipolar disorder: Evidence, pathophysiology and treatment implications. Journal of Affective Disorders 188, 149–159. [DOI] [PubMed] [Google Scholar]
- (41).Shim s. s., and Stuzmann GB (2016) Inhibition of glycogen synthase kinase-3: An emerging target in the treatment of traumatic brain injury. Journal of Neurotrauma epub, ahead of print. [DOI] [PubMed] [Google Scholar]
- (42).Leeds PR, Yu F, Wang Z, Chiu C-T, Zhang Y, Leng Y, Linares GR, and Chuang D-M (2014) A new avenue for lithium: Intervention in traumatic brain injury. ACS Chemical Neuroscience 5, 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gatson JW, Liu M-M, Abdelfattah K, Wigginton JG, Smith S, Wolf S, and Minei JP (2013) Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. Journal of Trauma and Acute Care Surgery 74, 470–475. [DOI] [PubMed] [Google Scholar]
- (44).Yan H, Zhang HW, Wu QL, Zhang GB, Liu K, Zhi DS, Hu ZB, and Zeng XW (2012) Increased leakage of brain antigens after traumatic brain injury and effect of immune tolerance induced by cells on traumatic brain injury. Chinese Medical Journal 125, 1618–1626. [PubMed] [Google Scholar]
- (45).Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, and Ruderisch N (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zhu X-C, Tan L, Jiang T, Tan M-S, Zhang W, and Yu J-T (2014) Association of IL-12A and IL-12B polymorphisms with Alzheimer’s disease susceptibility in a Han Chinese population. Journal of Neuroimmunology 274, 180–184. [DOI] [PubMed] [Google Scholar]
- (47).vom Berg J, Prokop S, Miller KR, Obst J, Kalin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, and Heppner FL (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nature Medicine 18, 1812–1819. [DOI] [PubMed] [Google Scholar]
- (48).Donegan JJ, Girotti M, Weinberg MS, and Morilak DA (2014) A novel role for brain interleukin-6: Facilitation of cognitive flexibility in rat orbitofrontal cortex. The Journal of Neuroscience 34, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Winstanley CA, Zeeb FD, Bedard A, Fu K, Lai B, Steele C, and Wong AC (2010) Dopaminergic modulation of the orbitofrontal cortex affects attention, motivation and impulsive responding in rats performing the five-choice serial reaction time task. Behavioural Brain Research 210, 263–272. [DOI] [PubMed] [Google Scholar]
- (50).Vonder Haar C, Maass WR, Jacobs EA, and Hoane MR (2014) Deficits in discrimination following experimental frontal brain injury are mediated by motivation and can be improved by nicotinamide administration. Journal of Neurotrauma 31, 1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Vonder Haar C, and Winstanley CA (2016) Minor functional deficits in basic response patterns for reinforcement following frontal traumatic brain injury in rats. Journal of Neurotrauma epub, ahead of print. [DOI] [PubMed] [Google Scholar]
- (52).Martens KM, Vonder Haar C, Hutsell BA, and Hoane MR (2012) A discrimination task used as a novel method of testing decision-making behavior following traumatic brain injury Journal of Neurotrauma 29, 2505–2512. [DOI] [PubMed] [Google Scholar]
- (53).Cole JT, Yarnell A, Kean WS, Gold E, Lewis B, Ren M, McMullen DC, Jacobowitz DM, Pollard HB, O’Neil JT, Grunberg NE, Dalgard CL, Frank JA, and Watson WD (2011) Craniotomy: True sham for traumatic brain injury, or a sham of a sham? Journal of Neurotrauma 28, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Comeau WL, Winstanley CA, and Weinberg J (2014) Prenatal alcohol exposure and adolescent stress – unmasking persistent attentional deficits in rats. European Journal of Neuroscience 40, 3078–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Sun H, Cocker PJ, Zeeb FD, and Winstanley CA (2012) Chronic atomoxetine treatment during adolescence decreases impulsive choice, but not impulsive action, in adult rats and alters markers of synaptic plasticity in the orbitofrontal cortex. Psychopharmacology 219, 285–301. [DOI] [PubMed] [Google Scholar]
- (56).Coggeshall RE (1992) A consideration of neural counting methods. Trends in Neurosciences 15, 9–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.