

Abstract
Pulmonary hypertension is a common but complex clinical problem. When suspected in an appropriate clinical setting or detected incidetally, an array of investigative tools are employed with an intent to confirm the diagnosis, define aetiology, evaluate the functional and haemodynamic impairment, define treatment options, monitor the therapy, and establish long-term prognosis. However, no single tool provides comprehensive information that encompasses the aforementioned aims. Therefore, judicious use of these tools is of paramount importance, in order to maximise outcome and cost-effectiveness, while minimising risks and redundancies. Furthermore, a number of promising tools and techniques are emerging rapidly in the arena of pulmonary hypertension. These tools augment our understanding of pathophysiology and natural history of pulmonary hypertension. There is, therefore, increasing need for validating these emerging paradigms in multicentre trials. In this review, we focus on the tools commonly used to evaluate pulmonary arterial hyertension and also define some of the new approaches to pulmonary arterial hypertension.
Keywords: Cardiac magnetic resonance imaging, echocardiography, pulmonary arterial hypertension, right heart catheterisation, right ventricular strain
Pulmonary hypertension (PH) is defined as an increase in mean pulmonary arterial pressure (P̄pa) ≥25 mmHg at rest on right heart catheterisation (RHC) [1]. The clinical classification of PH has evolved through a series of changes since its initial proposal in 1973 at the first international conference on primary PH, endorsed by the World Health Organization [2]. The current clinical classification groups PH into five diagnostic categories [3]: group 1: pulmonary arterial hypertension (PAH); group 2: PH with left-sided heart disease; group 3: PH associated with lung disease and/or hypoxaemia; group 4: PH due to chronic thrombotic and/or embolic disease (CTEPH); and group 5: miscellaneous group.
The clinical evaluation of suspected PH entails investigations that are intended to confirm the diagnosis, define aetiology, evaluate the functional and haemodynamic impairment, define treatment selection and monitor the therapy along with long-term prognostication. In this review, we will focus on the tools commonly used to evaluate PAH and also define some of the new approaches to PAH. Since the diagnosis of PAH involves excluding all other reasonable possibilities, the diagnostic algorithm generally starts with the identification of the more common clinical groups of PH (group 2, left heart disease and group 3, lung diseases), then distinguish group 4, CTEPH, and finally make the diagnosis and recognise the different types in group 1, PAH, and the rarer conditions in group 5. However, an abnormality of one test does not preclude that another abnormality may be contributing or predominant. Contingent tests are recommended to elucidate or confirm results taking into account appropriate clinical context.
TESTS TO CONFIRM THE DIAGNOSIS OF PAH
Right heart catheterisation
RHC is the current reference standard for the diagnosis and assessment of the severity of the haemodynamic impairment in PAH [4, 5]. The utility of RHC depends on the accuracy and completeness of the data obtained. Essential components of RHC are accurate measurements of right atrial pressure (Pra), right ventricular pressure, systolic Ppa (Ppa,syst), diastolic Ppa, P̄pa and pulmonary capillary wedge pressure (Ppcw), along with measurements of oxygen saturation of blood sampled from the superior/inferior vena cava, pulmonary artery and systemic arterial circulation. The influence of intrathoracic pressure on intracardiac pressure measurement is mitigated by recording the measurement at end-expiration when intrathoracic pressure is near zero [6]. Measurements at end-expiration were introduced in the 1980s. Earlier RHC studies averaged pressures over the respiratory cycle. The rationale for measurements at end-expiration is rather that it minimises the influence of lung volume on pulmonary vascular resistance (PVR) [7]. However, in the presence of increased respiratory pressure and volume swings, like in decompensated chronic obstructive pulmonary disease or at high levels of exercise, averaging pressures over the respiratory cycle is preferable. The Fick method remains the gold standard for estimating cardiac output (CO), provided it is used in strictly steady-state conditions, which is rarely the case [8]. In clinical practice, CO based on thermodilution techniques is frequently obtained. Generally, CO measured in triplicate by thermodilution suffices if the values are consistent, provided there is no significant systemic-to-pulmonary shunting, in which case the Fick method is mandatory. Contrary to the general conception, thermodilution CO is not affected by tricuspid regurgitation [9, 10].
Ppcw, a surrogate measurement for left atrial pressure in absence of pulmonary vein obstruction, is required for differentiating group 1 from group 2 categories of PH. To measure the Ppcw, the pulmonary artery catheter tip must be located in a lung segment where pulmonary vein pressure exceeds the alveolar pressure (physiologic zone 3) [11]. In a supine patient during RHC, a major portion of the lung is in zone 3 and, therefore, the catheter tip is naturally directed to this zone. Evidence of distinct A and V waves on Ppcw tracings indicates the catheter tip being in zone 3. Conditions such as hypovolaemia, advanced parenchymal lung disease or positive pressure ventilation will render the alveolar pressure in excess of the pulmonary vein pressure, thus creating zones 1 or 2. In these instances, Ppcw becomes a measure of alveolar pressure rather than left atrial pressure (Pla). The optimal position of the wedging catheter tip can be further ascertained by fluoroscopy and checking the pulmonary vein oxygen saturation, which should be equal to systemic arterial oxygen saturation. If the wedge pressure tracing is suboptimal, a measurement of left ventricle end-diastolic pressure should be obtained by left heart catheterisation.
PVR is obtained by dividing the transpulmonary gradient (P̄pa - Ppcw) by the CO, and is considered a robust diagnostic criterion for PAH because it reflects the influence of transpulmonary gradient and CO and is only elevated if the vascular obstruction occurs within the precapillary pulmonary circulation [5]. PVR distinguishes passive PH (elevated P̄pa, normal PVR) from PH caused by pulmonary vascular disease (elevated P̄pa, elevated PVR). The conventional definition of PAH used in clinical studies includes a P̄pa of ≥25 mmHg at rest in the setting of a normal Ppcw of ≤15 mmHg or less with a PVR >3 Wood units. However, potential exceptions to this definition will involve certain conditions with high flow rates, for instance uncorrected congenital heart disease, sickle cell disease and portopulmonary hypertension. Despite the pivotal role of RHC in evaluation of PAH, the technique has some limitations. Even at experienced centres, RHC procedures are associated with a morbidity of 1.1% and a mortality of 0.055% [12]. The haemodynamic variables obtained at RHC are susceptible to phasic fluctuation due to the respiratory cycle or artifactual fluctuations due to hyperventilation and valsalva manoeuvre. Furthermore, RHC is performed under artificial filling conditions, e.g. at rest and after overnight fasting, which may not correspond to the true pulmonary haemodynamic conditions during patients' daily lives. This technique is also susceptible to common errors in zero levelling, over-damping (insufficient flushing) or under-damping (over-flushing) pressure curves and, in general, the insufficient frequency responses of fluid-filled catheters. The advent of implantable haemodynamic monitors providing continuous haemodynamic data may mitigate some of these limitations and may be useful in monitoring the therapy of PAH, although the intermediate to long-term accuracy of these estimates are not yet defined [13, 14].
Vasodilator challenge
Pulmonary vasodilatation testing is performed to evaluate the reversible component of PAH. It helps identify patients with better prognosis and patients who could potentially benefit from treatment with calcium channel blockers (CCBs) [15–17]. Acute vasodilator testing is most commonly performed using inhaled nitric oxide [18], intravenous epoprostenol [19] or intravenous adenosine [20]. A positive acute response was, until recently, defined as a reduction of both P̄pa and PVR of at least 20% relative to baseline values [21]. However, recent consensus defines a favourable response as a fall in P̄pa ≥10 mmHg to a P̄pa ≤40 mmHg, with an unchanged or increased CO [22]. Using this criterion, only ∼10% of patients with idiopathic PAH will be regarded as acute responders. About 50% of acutely responding idiopathic PAH patients respond long-term to CCBs [17]. While sensitivity of acute vasodilator test is insufficient to identify all patients who may be responsive to long-term CCB therapy, it is sufficiently specific to identify patients who have a high likelihood of benefiting from chronic CCB therapy. Caution should be exercised while performing this test as these agents could lead to an acute increase in right-to-left blood flow and, potentially, overload a poorly compliant left ventricle. Their use in patients with elevated left ventricular filling pressures can cause acute pulmonary oedema [23]. In addition, a minority of patients with PAH will have significant peripheral vasodilatation leading to hypotension (negative responders).
Echocardiography
Doppler echocardiography is the pivotal screening test for noninvasive assessment of suspected PAH and provides several variables which correlate closely with right heart haemodynamics, including Ppa (figs 1 and 2). The Ppa,syst is derived from the tricuspid insufficiency peak gradient (TIPG) using the equation: Ppa,syst = TIPG + estimated Pra. Pra can be estimated based on the diameter and respiratory variation of the inferior vena cava, although often a fixed value of 5 or 10 mmHg is assumed. The TIPG is derived from tricuspid jet velocity (TJV) using the simplified Bernoulli equation: TIPG = 4 × (TJV)2. In 10–25% of patients being evaluated for possible PH, the spectral Doppler profile of tricuspid insufficiency is too weak or insufficient to measure the pressure gradient across the tricuspid valve [24, 25]. The spectral tricuspid regurgitation signal can be augmented by use of contrast echocardiography (e.g. agitated saline or encapsulated microbubble contrast agents), allowing proper measurement of peak TJV. Theoretically, the P̄pa can be derived from Ppa,syst by using the equation: P̄pa = 0.61 × Ppa,syst + 2 mmHg [26]. However, despite strong correlation between TJV and TIPG, the Doppler-derived Ppa,syst may be inaccurate in individual patients. For example, in the ItinerAIR study that comprised of 599 patients with scleroderma, if PH is defined on the basis of Doppler-derived TJV of >3.0 m·s−1 or TJV of 2.5–3.0 m·s−1 with dyspnoea, it corresponded to a false-positive rate of 45% when compared with RHC [27].
Figure 1.

Echocardiographic evaluation of cardiac remodelling in pulmonary arterial hypertension. Short axis and four-chamber views of end diastolic (a, c) and end systolic (b, d) are shown, which demonstrate enlargement of right ventricle and flattening of interventricular septum. e) M-mode demonstrates prominent flattening of interventricular septum. f) Dilatation of inferior vena cava is also noted.
Figure 2.
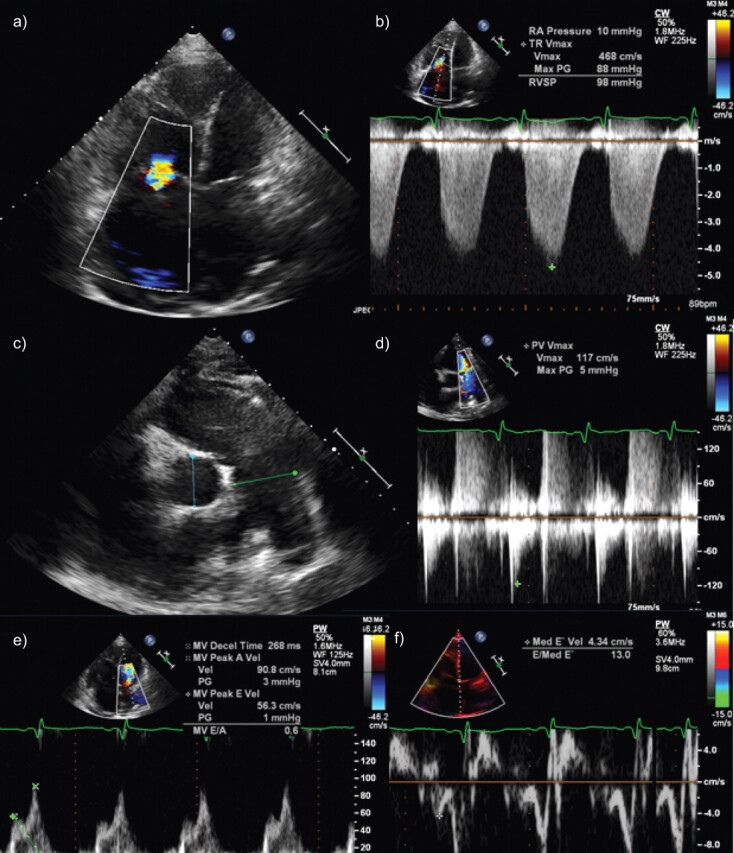
Echocardiographic evaluation of cardiac remodelling in pulmonary arterial hypertension, continued from figure 1. a) Colour Doppler across tricuspid valve reveals flow turbulence due to tricuspid regurgitation. b) Estimated pulmonary artery pressure from regurgitant velocity reveals severely elevated estimated systolic pulmonary artery pressure. c) Dilatation of main pulmonary artery is noted. d) Velocity profile across pulmonic valve is demonstrated where the systolic acceleration time is significantly shortened. e) Mitral inflow pattern revealing reduced E:A ratio. f) Tissue Doppler of interventricular septum reveals impaired tissue relaxation.
Currently, mild PH is arbitrarily defined as TJV of 2.8–3.4 m·s−1, which corresponds to TIPG 31–46 mmHg and to Ppa 36–51 mmHg, if a fixed Pra estimate of 5 mmHg is used [28]. This was endorsed by the 2009 European Society of Cardiology task force guidelines for the diagnosis and treatment of PH (table 1) [4].
Table 1. Definition of pulmonary hypertension (PH).
| Echocardiographic diagnosis: PH unlikely |
| TJV ≤2.8 m·s−1, Ppa,syst ≤36 mmHg, and no additional echocardiographic variables suggestive of PH |
| Echocardiographic diagnosis: PH possible |
| TJV ≤2.8 m·s−1, Ppa,syst ≤36 mmHg, but presence of additional echocardiographic variables suggestive of PH |
| TJV 2.9–3.4 m·s−1, Ppa,syst 37–50 mmHg with/without additional echocardiographic variables suggestive of PH |
| Echocardiographic diagnosis: PH likely |
| TJV >3.4 m·s−1, Ppa,syst >50 mmHg, with/without additional echocardiographic variables suggestive of PH |
TJV: tricuspid jet velocity; Ppa,syst: pulmonary artery systolic pressure. Reproduced from [4], with permission from the publisher.
A major problem of using echocardiography to evaluate the pulmonary circulation as it is practised in many centres is the exclusive reliance on the maximal velocity of tricuspid regurgitation for the calculation of pulmonary artery pressures. Since Ppa is a flow-dependent variable, it is important to couple it with a measurement of CO (Q). This can be measured from the left ventricular outflow tract (LVOT) diameter and velocity integral (VTI) multiplied by cardiac frequency (fC) [29, 30]; Q = (0.785 × (LVOT)2 × VTILVOT) × fC. For the direct calculation of PVR, it is also necessary to know Pla or Ppcw. This can be calculated from the ratio of transmitral Doppler and mitral tissue Doppler E and E' waves [31]; Pla or Ppcw (mmHg) = (1.24 × E/E') + 1.9. P̄pa can be calculated by the equation [32]: P̄pa (mmHg) = 0.61 × Ppa,syst + 2. It is also possible to calculate pulmonary flow at the right ventricular outflow tract (RVOT) by measuring VTIRVOT, but the calculation of volume flow produces a less stable measurement than sampled from the LVOT. VTIRVOT and tricuspid regurgitation flows can be combined to calculate PVR [33] as follows: PVR (in Wood units) = 0.16 + 10 × (TJV/VTIRVOT), or using a simplified equation PVR (in Wood units) = 10 × (TJV/VTIRVOT). If TJV/VTIRVOT <0.2, PVR values are likely to be normal, even in the presence of Doppler evidence of increased Ppa,syst [33]. Another useful independent method to estimate P̄pa is based on the pulsed Doppler measurement of the acceleration time (AT) of pulmonary flow (sampled at the RVOT) [34, 35]: log10 P̄pa = -0.0068 (AT) + 2.1; or P̄pa (mmHg) = 79 - (0.45 × AT). Given the importance of a reliable estimate of Ppa for the diagnosis of PH, and given the uncertainties encountered with the sampling of good quality TJV or pulmonary insufficiency, it is surprising that internal controls based on AT are so uncommonly reported. However, there has been less validation of this method against invasive measurements than reported on the basis of TJV. Accurate estimates of P̄pa are less reliable when AT exceeds 100 ms, the lower limit of normal, and the measurement may need a correction for ejection time at high heart rates [36]. However, there are data suggesting that AT may be more sensitive than tricuspid regurgitation to early or latent pulmonary vasculopathy [37], which may be due to its sensitivity to changes in pulmonary vascular impedance more than PVR. The advantage of the measurement of Ppa on the basis of the AT of pulmonary flow is in a quasi 100% recovery of good quality signals, independently on the level of Ppa. In addition to AT, the shape of the flow wave is of interest, as PH is associated with a deceleration of flow in late or in mid systole (notching). A decreased time to notching has been reported to be of poor prognosis, and to identify proximal obstruction in patients with CTEPH [38]. Finally, as recently emphasised in the guidelines [4], estimations of Ppa or PVR have to be integrated in the context of a complete echocardiographic examination of the heart. An increased TJV in the presence of a completely normal right ventricular size and function does not require further exploration.
Cardiac magnetic resonance
Estimation of right ventricular systolic pressure
Cardiac magnetic resonance imaging (cMRI) (fig. 3) is generally considered a noninvasive reference standard for evaluating biventricular dimensions, geometry and function [40, 41]. The configuration and curvature of the septum is a function of trans-septal pressure gradient [42, 43], whereas transmural pressure gradient determines the left ventricular free wall curvature [44]. Patients with PH may have a substantially reduced trans-septal pressure gradient, which may lead to the frequently observed flattening (or bowing) of the intraventricular septum (IVS) [42]. According to the law of Laplace, wall stress is proportional to the rate of pressure rise (dP) divided by the product of wall thickness and curvature [45]. This relationship may be independently applied to derive wall stress information across the IVS and the free wall. In the absence of asymmetric hypertrophy, the wall stress in the IVS and in the free wall may be assumed to be similar; thus, any change in the dP ratio (i.e. trans-septal pressure gradient divided by effective distending transmural pressure) should affect the curvature ratio RC (ratio of curvature of IVS and curvature of left ventricular free wall) proportionally. Using echocardiography, Resiner et al. [46] proposed an equation to obtain right ventricular systolic pressure (Prv,syst) from systolic blood pressure (SBP): Prv,syst = SBP × [1 - (RC/1.03)]. Using cMRI and RHC as reference measures this equation was validated with a good correlation (r = 0.85) [47].
Figure 3.

Cardiac magnetic resonance imaging (MRI) evaluation of cardiac remodelling in pulmonary arterial hypertension (PAH). a) Short axis view of cine-MRI frame at systole and b) corresponding tagged MRI images are shown. c) Three-dimensional strain maps from normal (NL) compared with PAH subjects are shown in various phases of the cardiac cycle. Adapted from [39].
Sustained increase in right ventricular afterload in patients with PAH leads to alteration in right ventricle morphology; the change mirrors the severity of P̄pa. Using this concept, ventricular mass index (VMI), which is the ratio between magnetic resonance imaging (MRI)-derived right ventricle mass and left ventricle mass, were obtained in patients with suspected PAH and correlated with echocardiographic and invasive haemodynamic variables [48]. The correlation between VMI and RHC-derived P̄pa was stronger than the correlation using echocardiographically derived P̄pa. Confidence intervals were also narrower for VMI than those for echocardiography. The sensitivity and specificity of VMI as a predictor of P̄pa (using VMI of 0.6 as a diagnostic cut-off) were 84% and 71%, compared with 89% and 57% for echocardiography.
Phase contrast MRI-derived flow parameters
Phase contrast (PC)-MRI technique can be used to derive velocity and flow measurements in the region of interest such as pulmonary arteries. This involves use of specialised software that is routinely available from most major vendors to calculate velocity information for each pixel within the region of interest. Therefore peak and mean velocities are obtained. Integrating them over the cardiac cycle in the main pulmonary artery provides stroke volume measurements. Pulmonary artery strain, or distensibility, which is calculated as per cent change of pulmonary artery cross sectional area compared to end-diastolic area, can be obtained either from cine-MRI sequence or from PC images. Other parameters such as AT and ejection time can also be obtained from PC-MRI dataset.
When these PC-MRI parameters were compared with RHC-derived parameters, average velocity of pulmonary artery flow and minimum pulmonary artery area emerged as useful parameters for the evaluation of PAH (area under receiver operating characteristic curve 0.90 for average velocity and 0.95 for minimum pulmonary artery area) [49]. Use of velocity-encoded cine-MRI has revealed reduced peak systolic velocity and greater post-systolic retrograde flow in the pulmonary arteries of PAH patients, with retrograde flow proportional to pulmonary resistance and inversely proportional to flow volume [50]. In a study of 25 patients with PAH, mean pulmonary artery peak flow velocity, pulmonary artery blood flow and pulmonary artery distensibility were found to be significantly lower than in a matched group of volunteers (p=0.002, p=0.002 and p=0.008, respectively) [51].
Laffon et al. [52] have proposed a computerised algorithm for estimating P̄pa based on cMRI-derived indices (e.g. pulmonary artery cross-sectional area and blood flow velocity) that are weighted using patient-specific biophysical parameters (e.g. height, weight and heart rate). When this algorithm was applied to a series of 31 patients undergoing RHC, the calculated P̄pa based on MRI-derived parameters correlated strongly with catheterisation-derived values (r=0.92). However, in a subsequent study that assessed 44 patients with established PAH, a poor correlation was found between catheterisation-derived P̄pa and five different MRI-derived measures [53], including pulmonary vascular index, AT (defined as time from onset of pulmonary artery forward flow to maximum velocity), the ratio between AT and ejection time, and the algorithm proposed by Laffon et al. [52] discussed previously. The only significant correlation between catheterisation-based P̄pa and MRI-based measures was for ventricular mass index (r=0.56, p<0.001).
Computed tomography scan
Topographic manifestations of PAH on cardiopulmonary computed tomography (CT) are: increase in the ratio of the widest short axis diameter of the right ventricle to the left ventricle on axial CT, flattening and eventual convexity of the IVS with progressive right ventricle enlargement, and reflux of contrast from the right atrium into the inferior vena cava [54]. It has been shown that a ratio of the mean pulmonary artery to ascending aorta size >1 is indicative of PAH [55]. In addition, the coronary sinus is dilated and correlates to pulmonary pressures determined by RHC [56]. CT scanning of the lungs can also provide morphological hints to help rule out other disorders, such as CTEPH and luminal and mural thrombi. Limitations of the use of CT in PAH diagnosis include: CT does not provide pulmonary pressure; the absence of changes in the pulmonary artery does not eliminate the possibility that a patient has PAH; pulmonary artery and ascending aorta diameters may change with body size; dilation of the main pulmonary artery can occur with other pulmonary disorders, such as pulmonary fibrosis; and size estimates of the pulmonary artery may require more than one axial dimension [54].
TESTS TO PROGNOSTICATE PAH
Right heart catheterisation
The prognostic significance of the haemodynamic variables to predict outcomes in PAH was illustrated in the pre-epoprostenol era by the National Institutes of Health registry [57]. On univariate analysis, increased P̄pa (OR 1.16, 95% CI 1.05–1.28), increased P̄ra (OR 1.99, 95% CI 1.47–2.69), and decreased cardiac index (CI) (OR 0.62, 95% CI 0.46–0.82) were associated with increased risk of death. Baseline Pra >20 mmHg carries a 6-month mortality rate of almost 100% without treatment. These variables were also robust predictors in multivariate analysis. Using these three variables, a regression equation was formulated to predict the probability of survival (P) in 1, 2 or 3 yrs (t) after diagnosis: P(t) = {H(t)}A(x,y,z), where H(t) = {0.88 - 0.14t + 0.01t2}, A(x,y,z) = e(0.007325x + 0.0526y - 0.3275z), t = years, x = P̄pa (mmHg), y = P̄ra (mmHg), and z = CI (L·min−1·m−2). This equation was subsequently validated [58] but its utility in the post-epoprostenol era has not been established. Some evidence supports that the baseline haemodynamic variables may confer less prognostic information in epoprostenol-treated patients [59]. The P̄pa may lose its prognostic significance along the natural course of the PAH as the failing right ventricle attenuates the P̄pa [58]. Positive response to acute vasodilator testing portends excellent prognosis with 95% survival at 5 yrs [17, 21]. However, these prognostic indices were derived from patients with idiopathic PAH and may not be directly applicable to PAH related to connective tissue disorders, congenital systemic-to-pulmonary shunts, HIV infection, or portal-pulmonary hypertension. Important information about predicting survival in PAH has been provided by a French registry and the REVEAL registry. Multivariable analysis of 354 consecutive adult patients with idiopathic, familial or anorexigen-associated PAH (56 incident and 298 prevalent cases) in the French registry showed female sex, greater 6-min walk distance and higher CO are jointly significantly associated with improved survival [60]. Analysis of 2,716 patients in the REVEAL registry, which is a multicentre, observational, US-based registry, have demonstrated additional key predictors of survival and helped generate a clinically useful prognostic equation [61].
Echocardiography
While echocardiography is pivotal in the PAH assessment paradigm as an early screening tool, the prognostic information of the echocardiographic variables in PAH is limited due to relatively small series. Those indices with the best prognostic significance identified by multivariate analysis are pericardial effusion [61–63], indexed right atrium area [63], left ventricular eccentricity index [63] and the right ventricular Doppler index [64, 65]. Estimated Ppa,syst derived from TJV has been shown to be non-prognostic [63]. The tricuspid annular plane systolic excursion (TAPSE), which measures the systolic displacement of the tricuspid annulus toward the right ventricle apex, has been shown to correlate closely with right ventricular ejection fraction [66]. In PH (majority PAH), TAPSE <1.5 cm is associated with clinical deterioration and worst prognosis and TAPSE >2.0 cm indicates better prognosis [67]. Similarly, TAPSE of ≥1.8 cm was associated with survival estimates of 94% and 88% at 1 and 2 yrs, respectively, and survival estimates of 60% and 50%, respectively, in subjects with a TAPSE ≤1.8 cm [67].
The myocardial performance index (Tei index or right ventricular Doppler index) is a more physiological index and is calculated as the ratio between the sum of the times of the isovolumic periods and the ejection time for the right ventricle [64]. Normal value for Tei index is 0.28±0.04, and it usually increases in diseases associated with right ventricle dysfunction [64]. The Tei index appears to be predictive of an adverse outcome on univariate analysis and by multivariate regression analysis [64, 65].
Cardiovascular magnetic resonance
cMRI provides morphological and functional parameters with high accuracy, reproducibility and low inter-study variability [68]; these characteristics render it an attractive prognostication and follow-up tool. In a study of 64 patients with idiopathic PAH, baseline right ventricular stroke volume index >25 mL·m−2, right ventricular end-diastolic volume <84 mL·m−2, and left ventricular end-diastolic volume (LVEDV) >40 mL·m−2 were predictive of survival [69]. In a multivariate analysis, only 6-min walk test (6MWT) added incremental prognostic information over these three cMRI variables. In addition, a further decrease in stroke volume, progressive right ventricular dilatation, and further decrease in LVEDV at 1-yr follow-up were the strongest predictors of mortality in this study group [69]. Pulmonary artery distensiblity, as measured by relative cross-sectional area change by cMRI was predictive of survival in a cohort of 86 PAH patients. Those with a relative cross-sectional area change of <16% had a greater mortality than those with a value >16% [70].
Exercise tolerance
6MWT is a well standardised and reproducible test, which provides quantitative measures of exercise tolerance in terms of distance walked, dyspnoea on exertion (Borg scale) and finger oxygen saturation. Walking distances of <332 m [71] or <250 m [59] and oxygen desaturation >10% [72] indicate impaired prognosis in PAH. In the pivotal epoprostenol idiopathic PAH trial, performance in the unencouraged 6MWT was found to be an independent predictor of survival [73], leading to the acceptance of this test by the Food and Drug Administration and the European Agency for the Evaluation of Medicinal Products as an primary end-point for the evaluation of treatment effects in PAH in clinical trials.
Maximal oxygen consumption determined by progressive exercise testing with cycle ergometry has also been found to be an independent predictor of survival in idiopathic PAH. Following multivariate analysis of clinical, haemodynamic and exercise parameters in 70 of the 86 patients that were able to complete the exercise protocol, peak oxygen uptake (10.4 mL·kg−1·min−1) and peak systolic arterial pressure during exercise (120 mmHg) independently predicted a worse prognosis in idiopathic PAH [74]. In addition, in idiopathic PAH patients treated with epoprostenol, the exercise duration has also been shown to be predictive of survival [75]. While the results of both methods do correlate in PAH, cardiopulmonary exercise test (CPET) failed to confirm improvements observed with 6MWT in randomised controlled trials [76, 77]. This discrepancy has been attributed to lack of standardisation and insufficient expertise in performing CPET [78].
Biomarkers
Both atrial natriuretic peptide and brain natriuretic peptide (BNP) are released from myocardium in response to wall stress and share similar physiological properties inducing vasodilatation and natriuresis. In a PAH study, baseline plasma BNP was an independent predictor of mortality after multivariate analysis, with supramedian level of baseline BNP of ≥150 pg·mL−1 associated with significantly lower survival rate than those with an inframedian level of ≤150 pg·mL−1. After 3 months of target therapy, a supramedian value ≥180 pg·mL−1 was associated with strikingly worse survival [79]. Similarly, N-terminal pro-BNP (NT-proBNP), a high molecular precursor of BNP, is related to alteration in right ventricle morphology and dysfunction as assessed by echocardiography and RHC in PH patients with baseline serum NT-proBNP levels of ≥1,400 pg·mL−1 portending poor long-term prognosis [80]. Detectable cardiac troponin T also confers a poor prognosis, potentially due to the effect of right ventricle strain and ischaemia [81]. Elevated uric acid levels in PAH are also associated with severity of New York Heart Association (NYHA) functional class and impaired CO [82]. Furthermore, increased uric acid levels are independently associated with long-term mortality of patients with PAH [82].
TESTS FOR TREATMENT SELECTION AND MONITORING DISEASE
Conventionally, RHC with appropriate clinical context has been used to diagnose and determine initial treatment selection. However, there is no universally accepted consensus about the timing and frequency of subsequent RHCs. Pragmatically, most centres perform RHC when there is a change in clinical status, when change in treatment is considered, and 3–6 months after initiating new treatments, so as to ensure that the pulmonary haemodynamics are in the desired range. Currently, RHC and echocardiography are the primary tools used in the diagnosis and monitoring of PAH. However, cMRI has already superseded echocardiography as a reference standard for the noninvasive evaluation of right ventricle morphology and function that is integral to PAH management. Increasing evidence suggests that cMRI can be used effectively to guide and monitor PAH therapy [69, 83].
Figure 4 summarises various tools and parameters that are used for prognosticating and treatment selection in PAH. Interestingly, most of the predictors in PAH relate to the function of the right ventricle rather than the severity of the elevation in Ppa.
Figure 4.

Tests for prognostication and treatment selection in pulmonary arterial hypertension (PAH). WHO: World Health Organization; 6MWT: 6-min walk test; CPET: cardiopulmonary exercise testing; V′O2: oxygen uptake; DL,CO: diffusing capacity of the lung for carbon monoxide; NT-proBNP: N-terminal pro-brain natriuretic peptide; BNP: brain natriuretic peptide; RHC: right heart catheterisation; CI: cardiac index; TAPSE: tricuspid annular plane systolic excursion; MRI: magnetic resonance imaging; RVEDVI: right ventricular end-diastolic volume index; LVEDVI: left ventricular end-diastolic volume index; PC SVI: phase-contrast stroke volume index.
FUTURE DIRECTIONS
The right ventricle–pulmonary artery coupling
The haemodynamic variables in patients with PAH obtained from RHC are of prognostic value [57] but survival is more closely related to flow (CO) than to pressure (P̄pa) [59, 75]. Moreover, NYHA functional class and exercise capacity correlates better to CO than P̄pa [71, 73]. Clinical signs of right heart failure are often not clearly related to the progression of PH, as assessed by P̄pa and PVR [84]. These observations suggest that, although RHC provides important clinical information, it neglects a number of important variables that may have important bearing on the outcomes of patients. These include considerations related to pulsatile blood flow, nonuniform patterns of histopathological changes in the pulmonary vasculature, especially in early stages of disease, and complex interaction of right ventricle with the pulmonary axis in PAH.
The prognosis/longevity in PH is highly dependent on the adaptive capacity of the right ventricle [57, 58, 85]. Nearly 25% of the total work required of the right ventricle goes into creating the pulmonary artery pulse pressure. Quantifying ventricular function in isolation without taking into consideration the pulmonary vascular function ignores the important dependency of ventricular output on arterial impedance. This ventricular–vascular coupling can be quantified by using the ventricular pressure–volume loops obtained by RHC and is expressed as ventricular end-systolic elastance (Ees) and arterial effective elastance (Ea) [86]. Ees is an index of ventricular function, Ea is a surrogate of vascular function, and their ratio measures the efficiency of ventricular–vascular haemodynamic interactions [87, 88]. When the ventricle and the vasculature are efficiently coupled, minimal energy is wasted engendering the pulse pressure and maximal energy is transmitted in the mean pressure [89]. In this case, the ventricle operates at a maximum efficiency and submaximal stroke work such that Ees/Ea >1.5. In contrast, for a failing ventricle or high impedance vasculature, energy may be dissipated through a variety of mechanisms. In these cases, Ees/Ea <1 and ventricular–vascular uncoupling occurs [90, 91]. Pressure–volume loops at different levels of preload are required for measuring Ees, which can be difficult to achieve clinically. Single-beat methods have been developed to enable calculation of Ees and Ea from only right ventricle and Ppa waveforms [92]. The single-beat method to measure right ventricle–arterial coupling has been applied at the bedside [93, 94]. However, the application of the method needs expertise and specialised training. It is currently being refined and automated to make it easier to use in clinical practice and to base it on noninvasive measurements, i.e. recalculation of a right ventricle pressure curve from tricuspid regurgitation jet velocity and a relative change in right ventricle volume from pulmonary flow measurements.
To address aspects related to pulmonary impedance, the pulmonary vascular impedance spectrum (PVZ) has been proposed as a comprehensive measure of pulmonary vascular function obtained from spectral analysis of Ppa and flow waves [95]. The PVZ spectrum is expressed as a pressure/flow ratio and a phase angle, both of which are expressed as a function of frequency. Evidence suggests that it is a superior predictor of right ventricular failure in PAH compared to PVR [96]. Although feasibility of performing instantaneous Ppa and flow measurements during a routine RHC combined with Doppler echocardiography in patients with PAH has been shown [97], there is still a paucity of studies utilising PVZ in the PAH patient population, due to the technical difficulties of instantaneous pressure and flow measurements requiring high-fidelity, manometer-tipped catheters and flowmeters [98, 99]. Therefore, the clinical utility of PVZ in assessing responses to therapy and the prognosis of patients with severe PH is unknown.
Right ventricle strain, ventricular interaction, myocardial fibrosis and synchronicity
Strain is defined as deformation of an object compared with its initial shape and strain rate represents the deformation rate. Both MRI and echocardiography can be used to quantify these measurements (fig. 3). Echocardiographic strain and strain rate measurements have described right ventricle function in patients with PH [100] and seem to correlate to invasive haemodynamic variables [101]. Changes in right ventricle and septal systolic strain may be an indicator of early right ventricle dysfunction [102]. Attenuated right ventricle free wall longitudinal strain is associated with a greater degree of disease progression and cor pulmonale within 6 months. Patients with right ventricle free wall strain worse than -12.5% were associated with a greater degree of disease progression within 6 months, a greater requirement for loop diuretics, and/or a greater degree of lower extremity oedema, and it also predicted mortality. After adjusting for age, sex, PH cause and functional class, patients who had a 5% absolute decline in right ventricle free wall strain at 1 yr were associated with a 2.9-fold higher adjusted mortality rate per 5% absolute decline in right ventricle free wall strain at 1 yr [103].
Diastolic functions comprise ventricular relaxation and filling [64, 104, 105]. The ventricular relaxation can be quantified by isovolumic relaxation time (IVRT), the ventricular filling can be quantified by the peak filling rate and filling pattern. When diastolic function in PAH was evaluated using these indices, IVRT was found to correlate well with right ventricle mass and PVR, as well as disease severity as assessed by Pra, NT-proBNP level, CI and right ventricular ejection fraction, and IVRT was sensitive to afterload reduction with sildenafil [106].
The left ventricle is intimately connected to the right ventricle via the interventricular septum. Moreover, both the right and left ventricle are enclosed within a common pericardial sac occupying the same thoracic cavity. Therefore, changes in the structure and function of the right ventricle also affect the left ventricle. The MRI delayed contrast enhancement technique is used to demonstrate myocardial fibrosis with high spatial resolution. Myocardial delayed enhancement is frequently noted in right ventricular septal insertion points in PAH [107, 108]. Recent work using echocardiographic techniques in PH patients indicates that not only the right ventricle free wall and interventricular septal longitudinal shortening but also the circumferential shortening of the left ventricular lateral wall and interventricular septum and left ventricle systolic torsion are reduced [109]. With RV pressure overload, the septum flattens or even bulges to the left in early diastole. This paradoxical septal motion was classically believed to result from negative interventricular pressure gradient between left and right ventricles during diastole by studies using M-mode echocardiography and haemodynamics [110], and subsequently confirmed by cMRI [111]. Tagged MRI study indicates that this paradoxical septal motion is a result of higher pressure in the right than in the left ventricle leading to interventricular asynchrony due to a prolonged right ventricle contraction. This ventricular asynchrony impedes left ventricle diastolic filling, causing a lowered LVEDV [112, 113]. Right intraventricular dyssynchrony is also more prevalent in patients with PAH compared with controls as measured by two-dimensional strain echocardiography [114] and Doppler echocardiography [115].
CONCLUSIONS
Use of various tools has provided important insights in the natural history of PAH (table 2). No single tool provides comprehensive information regarding disease progression, stability and prognosis. There is on-going progress in better approaches for the evaluation of PAH. It is important to use existing and emerging techniques judiciously with an intent to validate some of the emerging paradigms in multicentre trials [129].
Table 2. Effect of pulmonary arterial hypertension (PAH) therapies on remodelling of the right heart, as assessed by noninvasive tests.
| Therapy | Patients | Conventional end-points | Surrogate end-points |
| cMRI | |||
| Epoprostenol [83] | 11 PAH | ↑ 6MWD | ↑ RVSV |
| ↓ PVR | ↑ CO | ||
| ↔ RVEDV | |||
| ↔ RV mass | |||
| ↔ P̄pa | |||
| Bosentan [116] | 16 PAH | ↑ 6MWD | ↔ RVEF |
| ↑ CI | ↔ RVEDV | ||
| ↓ PVR | ↔ RVSV | ||
| Bosentan/sildenafil [117] | 26 PAH (IPAH and CTD-APAH) | ↑ 6MWD | ↓ RV mass |
| ↑ CI | ↓ LVSEI | ||
| Bosentan and sildenafil [118] | 9 IPAH | ↑ 6MWD | ↑ RVEF |
| 3 CTD-APAH | ↑ CI | ↓ RV mass | |
| 2 CHD-APAH | ↔ RVEDV | ||
| 1 HIV-APAH | ↔ LVEDV | ||
| ↓ RVEDV/LVEDV ratio | |||
| ↔ RVSV index | |||
| Ambisertan [119] | 11 IPAH | ↑ 6MWD | ↑ RVEF |
| 1 drug-induced PAH | ↓ PVR | ↔ RVEDV | |
| ↔ RV mass | |||
| Sildenafil [120] | 4 IPAH | ↑ 6MWD | ↓ RV mass |
| 1 CHD-APAH | ↓ PVR | ||
| Echocardiography | |||
| Epoprostenol [121] | 9 IPAH | ↓ NYHA FC | ↓ RV dilatation |
| 1 CTD-APAH | ↓ TRV | ||
| 2 CHD-APAH | ↓ Ppa,syst/PVVTI | ||
| 4 PPH | ↔ RVMPI | ||
| 1 CTEPH | |||
| 2 sarcoid | |||
| 1 IPF | |||
| Bosentan [122] | 48 IPAH | ↑ 6MWD | ↑ CI |
| 7 CTD-APAH | ↓ NYHA FC | ↑ LVED filling velocity | |
| 1 other PAH | ↑ LVED area | ||
| ↓ LVSEI | |||
| ↓ RVES area | |||
| ↓ RV:LV diastolic areas | |||
| ↓ Pericardial effusion score | |||
| Bosentan [123] | 13 CTD-APAH | ↑ 6MWD | ↓ RVSP |
| ↔ P̄pa | |||
| Sildenafil [124] | 11 PAH | ↔ NYHA FC | ↔ RVSP |
| 1 CTEPH | ↔ 6MWD | ↔ Tei index | |
| 1 PPH | |||
| Epoprostenol/treprostinil s.c./inhaled iloprost plus sildenafil [125] | 13 IPAH | ↓ NYHA FC | ↓ RVES diameter |
| 6 toxic oil PAH | ↓ Right heart failure | ↓ LVDEI | |
| 1 HIV-APAH | ↑ 6MWD | ||
| Epoprostenol [126] | 41 IPAH | NA | ↓ RVED area |
| ↓ LVDEI | |||
| ↓ LVSEI | |||
| ↔ Pericardial effusion score | |||
| ↓ Maximal TRV | |||
| Beraprost/inhaled iloprost/iloprost i.v. plus bosentan [127] | 10 IPAH | ↑ 6MWD | ↓ Tei index |
| 6 other PAH | ↑ 6MWD | ||
| Biomarkers | |||
| Bosentan [128] | 18 CTD-APAH | ↔ WHO class III | ↓ NO |
| ↑ 6MWD | ↓ sCD40L | ||
| ↑ P-selectin | |||
| Bosentan and sildenafil [118] | 9 IPAH | ↑ 6MWD | ↓ NT-proBNP |
| 3 CTD-APAH | |||
| 2 CHD-APAH | |||
| 1 HIV-APAH | |||
| Bosentan/sildenafil [117] | 26 PAH (IPAH and CTD-APAH) | ↑ 6MWD | ↓ BNP |
| ↑ CI | |||
cMRI: cardiac magnetic resonance imaging; IPAH: idiopathic PAH/primary pulmonary hypertension; CTD-APAH: PAH associated with connective tissue disease; CHD-APAH: PAH associated with congenital heart disease; PPH: portopulmonary hypertension; CTEPH: chronic thromboembolic pulmonary hypertension; IPF: idiopathic pulmonary fibrosis; 6MWD: 6-min walk distance; PVR: pulmonary vascular resistance; CI: cardiac index; NYHA FC: New York Heart Association functional class; NA: not applicable; WHO: World Health Organization; RV: right ventricular; RVSV: RV stroke volume; CO: cardiac output; RVEDV: RV end-diastolic volume; P̄pa: mean pulmonary artery pressure; RVEF: RV ejection fraction; LV: left ventricular; LVSEI: LV systolic eccentricity index; LVEDV: LV end-diastolic volume; TRV: tricuspid regurgitation jet velocity; Ppa,syst: pulmonary artery systolic pressure; PVVTI: pulmonary valve velocity time integral; RVMPI: RV myocardial performance index; LVED: LV end-diastolic; RVES: RV end-systolic; RVSP: RV systolic pressure; LVDEI: LV diastolic eccentricity index; RVED: RV end-diastolic; NO: nitric oxide; sCD40L: human soluble CD40 ligand; BNP: brain natriuretic peptide; NT-proBNP: N-terminal pro-BNP.
Footnotes
Provenance
Publication of this peer-reviewed article was supported by Actelion Pharmaceuticals Ltd, Switzerland (principal sponsor, European Respiratory Review issue 122).
Support Statement
This work is supported in part by NIH-NHLBI 5R01HL104018 (H.Gupta).
Statement of Interest
H. Gupta has acted as a scientific consultant to Actelion. R. Naeije has received reimbursements for attending symposiums, speaker fees and fees for consulting from Actelion, Pfizer, Bayer Healthcare, United Therapeutics and LungRX, a fee for organising education from Pfizer, funds for research from Actelion and Pfizer, and funds for a member of staff from Pfizer.
REFERENCES
- 1.Badesch DB, Champion HC, Gomez Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009; 54: Suppl. 1, S55–S66. [DOI] [PubMed] [Google Scholar]
- 2.Hatano S, Strasser T. Primary Pulmonary Hypertension. Geneva, World Health Organization, 1975 [Google Scholar]
- 3.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009; 54: Suppl. 1, S43–S54. [DOI] [PubMed] [Google Scholar]
- 4.Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30: 2493–2537. [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009; 53: 1573–1619. [DOI] [PubMed] [Google Scholar]
- 6.Sharkey S. A Guide to Interpretation of Hemodynamic Data in the Coronary Care Unit. 1st Edn. Philadelphia, Lippincott Williams and Wilkins, 1997 [Google Scholar]
- 7.Howell JBL, Permutt S, Proctor DF, et al. Effect of inflation of the lung on different parts of pulmonary vascular bed. J Appl Physiol 1961; 16: 71–76. [DOI] [PubMed] [Google Scholar]
- 8.Fishman AP, McClement J, Himmelstein A, et al. Effects of acute anoxia on the circulation and respiration in patients with chronic pulmonary disease studied during the steady state. J Clin Invest 1952; 31: 770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buffington CW, Nystrom EU. Neither the accuracy nor the precision of thermal dilution cardiac output measurements is altered by acute tricuspid regurgitation in pigs. Anesth Analg 2004; 98: 884–890. [DOI] [PubMed] [Google Scholar]
- 10.Hoeper MM, Maier R, Tongers J, et al. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. Am J Respir Crit Care Med 1999; 160: 535–541. [DOI] [PubMed] [Google Scholar]
- 11.Levitzky M. Pulmonary Physiology. 4th Edn. New York, McGraw Hill, Inc, 1995 [Google Scholar]
- 12.Hoeper MM, Lee SH, Voswinckel R, et al. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J Am Coll Cardiol 2006; 48: 2546–2552. [DOI] [PubMed] [Google Scholar]
- 13.Frantz RP, Benza RL, Kjellström B, et al. Continuous hemodynamic monitoring in patients with pulmonary arterial hypertension. J Heart Lung Transplant 2008; 27: 780–788. [DOI] [PubMed] [Google Scholar]
- 14.Karamanoglu M, McGoon M, Frantz RP, et al. Right ventricular pressure waveform and wave reflection analysis in patients with pulmonary arterial hypertension. Chest 2007; 132: 37–43. [DOI] [PubMed] [Google Scholar]
- 15.Montani D, Savale L, Natali D, et al. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur Heart J 2010; 31: 1898–1907. [DOI] [PubMed] [Google Scholar]
- 16.Morales-Blanhir J, Santos S, de Jover L, et al. Clinical value of vasodilator test with inhaled nitric oxide for predicting long-term response to oral vasodilators in pulmonary hypertension. Respir Med 2004; 98: 225–234. [DOI] [PubMed] [Google Scholar]
- 17.Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111: 3105–3111. [DOI] [PubMed] [Google Scholar]
- 18.Pepke-Zaba J, Higenbottam TW, Dinh-Xuan AT, et al. Inhaled nitric oxide as a cause of selective pulmonary vasodilatation in pulmonary hypertension. Lancet 1991; 338: 1173–1174. [DOI] [PubMed] [Google Scholar]
- 19.Rubin LJ, Groves BM, Reeves JT, et al. Prostacyclin-induced acute pulmonary vasodilation in primary pulmonary hypertension. Circulation 1982; 66: 334–338. [DOI] [PubMed] [Google Scholar]
- 20.Schrader BJ, Inbar S, Kaufmann L, et al. Comparison of the effects of adenosine and nifedipine in pulmonary hypertension. J Am Coll Cardiol 1992; 19: 1060–1064. [DOI] [PubMed] [Google Scholar]
- 21.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992; 327: 76–81. [DOI] [PubMed] [Google Scholar]
- 22.Badesch DB, Abman SH, Simonneau G, et al. Medical therapy for pulmonary arterial hypertension. Chest 2007; 131: 1917–1928. [DOI] [PubMed] [Google Scholar]
- 23.Loh E, Stamler JS, Hare JM, et al. Cardiovascular effects of inhaled nitric oxide in patients with left ventricular dysfunction. Circulation 1994; 90: 2780–2785. [DOI] [PubMed] [Google Scholar]
- 24.Currie P, Seward J, Chan K, et al. Continuous wave Doppler determination of right ventricular pressure: a simultaneous Doppler-catheterization study in 127 patients. J Am Coll Cardiol 1986; 6: 750–756. [DOI] [PubMed] [Google Scholar]
- 25.Hinderliter AL, Willis PW, Long WA, et al. Frequency and severity of tricuspid regurgitation determined by Doppler echocardiography in primary pulmonary hypertension. Am J Cardiol 2003; 91: 1033–1037. [DOI] [PubMed] [Google Scholar]
- 26.Fisher MR, Forfia PR, Chamera E, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med 2009; 179: 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52: 3792–3800. [DOI] [PubMed] [Google Scholar]
- 28.Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: Suppl. 1, S40–S47. [DOI] [PubMed] [Google Scholar]
- 29.Huntsman LL, Stewart DK, Barnes SR, et al. Noninvasive Doppler determination of cardiac output in man. Clinical validation. Circulation 1983; 67: 593–602. [DOI] [PubMed] [Google Scholar]
- 30.Christie J, Sheldahl LM, Tristani FE, et al. Determination of stroke volume and cardiac output during exercise: comparison of two-dimensional and Doppler echocardiography, Fick oximetry, and thermodilution. Circulation 1987; 76: 539–547. [DOI] [PubMed] [Google Scholar]
- 31.Nagueh SF, Middleton KJ, Kopelen HA, et al. Doppler tissue imaging: a noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol 1997; 30: 1527–1533. [DOI] [PubMed] [Google Scholar]
- 32.Chemla D, Castelain V, Humbert M, et al. New formula for predicting mean pulmonary artery pressure using systolic pulmonary artery pressure. Chest 2004; 126: 1313–1317. [DOI] [PubMed] [Google Scholar]
- 33.Abbas AE, Fortuin FD, Schiller NB, et al. A simple method for noninvasive estimation of pulmonary vascular resistance. J Am Coll Cardiol 2003; 41: 1021–1027. [DOI] [PubMed] [Google Scholar]
- 34.Kitabatake A, Inoue M, Asao M, et al. Noninvasive evaluation of pulmonary hypertension by a pulsed Doppler technique. Circulation 1983; 68: 302–309. [DOI] [PubMed] [Google Scholar]
- 35.Chan K-L, Currie PJ, Seward JB, et al. Comparison of three Doppler ultrasound methods in the prediction of pulmonary artery pressure. J Am Coll Cardiol 1987; 9: 549–554. [DOI] [PubMed] [Google Scholar]
- 36.Hatle L, Angleson B. Doppler Ultrasound in Cardiology: Physical Principles and Clinical Applications. 2nd Edn. Philadelphia, Lea and Febiger, 1985 [Google Scholar]
- 37.Huez S, Roufosse F, Vachiéry JL, et al. Isolated right ventricular dysfunction in systemic sclerosis: latent pulmonary hypertension? Eur Respir J 2007; 30: 928–936. [DOI] [PubMed] [Google Scholar]
- 38.Hardziyenka M, Reesink HJ, Bouma BJ, et al. A novel echocardiographic predictor of in-hospital mortality and mid-term haemodynamic improvement after pulmonary endarterectomy for chronic thrombo-embolic pulmonary hypertension. Eur Heart J 2007; 28: 842–849. [DOI] [PubMed] [Google Scholar]
- 39.Venkatesh BA, Schiros CG, Gupta H, et al. Three-dimensional plus time biventricular strain from tagged MR images by phase-unwrapped harmonic phase. J Magn Reson Imaging 2011; [Epub ahead of print DOI: 10.1002/jmri.22665]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Constantine G, Shan K, Flamm SD, et al. Role of MRI in clinical cardiology. Lancet 2004; 363: 2162–2171. [DOI] [PubMed] [Google Scholar]
- 41.Pennell DJ, Sechtem UP, Higgins CB, et al. Clinical indications for cardiovascular magnetic resonance (CMR): consensus panel report. Eur Heart J 2004; 25: 1940–1965. [DOI] [PubMed] [Google Scholar]
- 42.Jessup M, Sutton MSJ, Weber KT, et al. The effect of chronic pulmonary hypertension on left ventricular size, function., and interventricular septal motion. Am Heart J 1987; 113: 1114–1122. [DOI] [PubMed] [Google Scholar]
- 43.Lima JA, Guzman PA, Yin FC, et al. Septal geometry in the unloaded living human heart. Circulation 1986; 74: 463–468. [DOI] [PubMed] [Google Scholar]
- 44.Belenkie I, Smith E, Tyberg J. Ventricular interaction: from bench to bedside. Ann Med 2001; 33: 236–241. [DOI] [PubMed] [Google Scholar]
- 45.Yin FC. Ventricular wall stress. Circ Res 1981; 49: 829–842. [DOI] [PubMed] [Google Scholar]
- 46.Reisner S, Azzam Z, Halmann M, et al. Septal/free wall curvature ratio: a noninvasive index of pulmonary arterial pressure. J Am Soc Echocardiogr 1994; 7: 27–35. [DOI] [PubMed] [Google Scholar]
- 47.Dellegrottaglie S, Sanz J, Poon M, et al. Pulmonary hypertension: accuracy of detection with left ventricular septal-to–free wall curvature ratio measured at cardiac MRI. Radiology 2007; 243: 63–69. [DOI] [PubMed] [Google Scholar]
- 48.Saba TS, Foster J, Cockburn M, et al. Ventricular mass index using magnetic resonance imaging accurately estimates pulmonary artery pressure. Eur Respir J 2002; 20: 1519–1524. [DOI] [PubMed] [Google Scholar]
- 49.Sanz J, Kuschnir P, Rius T, et al. Pulmonary arterial hypertension: noninvasive detection with phase-contrast MR imaging. Radiology 2007; 243: 70–79. [DOI] [PubMed] [Google Scholar]
- 50.Kondo C, Caputo GR, Masui T, et al. Pulmonary hypertension: pulmonary flow quantification and flow profile analysis with velocity-encoded cine MR imaging. Radiology 1992; 183: 751–758. [DOI] [PubMed] [Google Scholar]
- 51.Ley S, Mereles D, Puderbach M, et al. Value of MR phase-contrast flow measurements for functional assessment of pulmonary arterial hypertension. Eur Radiol 2007; 17: 1892–1897. [DOI] [PubMed] [Google Scholar]
- 52.Laffon E, Vallet C, Bernard V, et al. A computed method for noninvasive MRI assessment of pulmonary arterial hypertension. J Appl Physiol 2004; 96: 463–468. [DOI] [PubMed] [Google Scholar]
- 53.Roeleveld RJ, Marcus JT, Boonstra A, et al. A comparison of noninvasive MRI-based methods of estimating pulmonary artery pressure in pulmonary hypertension. J Magn Reson Imaging 2005; 22: 67–72. [DOI] [PubMed] [Google Scholar]
- 54.Devaraj A, Hansell DM. Computed tomography signs of pulmonary hypertension: old and new observations. Clin Radiol 2009; 64: 751–760. [DOI] [PubMed] [Google Scholar]
- 55.Ng C, Wells A, Padley S. A CT sign of chronic pulmonary arterial hypertension: the ratio of main pulmonary artery to aortic diameter. J Thorac Imaging 1999; 14: 270–278. [DOI] [PubMed] [Google Scholar]
- 56.Isaacs D, Hazany S, Gamst A, et al. Evaluation of the coronary sinus on chest computed tomography in patients with and without pulmonary artery hypertension. J Comput Assist Tomogr 2009; 33: 513–516. [DOI] [PubMed] [Google Scholar]
- 57.D'Alonzo G, Barst R, Ayres S, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991; 115: 343–349. [DOI] [PubMed] [Google Scholar]
- 58.Sandoval J, Bauerle O, Palomar A, et al. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation 1994; 89: 1733–1744. [DOI] [PubMed] [Google Scholar]
- 59.Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002; 40: 780–788. [DOI] [PubMed] [Google Scholar]
- 60.Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010; 122: 156–163. [DOI] [PubMed] [Google Scholar]
- 61.Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension. Circulation 2010; 122: 164–172. [DOI] [PubMed] [Google Scholar]
- 62.Eysmann SB, Palevsky HI, Reichek N, et al. Two-dimensional and Doppler-echocardiographic and cardiac catheterization correlates of survival in primary pulmonary hypertension. Circulation 1989; 80: 353–360. [DOI] [PubMed] [Google Scholar]
- 63.Raymond RJ, Hinderliter AL, Willis PW, et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol 2002; 39: 1214–1219. [DOI] [PubMed] [Google Scholar]
- 64.Tei C, Dujardin K, Hodge D, et al. Doppler echocardiographic index for assessment of global right ventricular function. J Am Soc Echocardiogr 1996; 9: 838–847. [DOI] [PubMed] [Google Scholar]
- 65.Yeo TC, Dujardin KS, Tei C, et al. Value of a Doppler-derived index combining systolic and diastolic time intervals in predicting outcome in primary pulmonary hypertension. Am J Cardiol 1998; 81: 1157–1161. [DOI] [PubMed] [Google Scholar]
- 66.Kaul S, Tei C, Hopkins J, et al. Assessment of right ventricular function using two-dimensional echocardiography. Am Heart J 1984; 107: 526–531. [DOI] [PubMed] [Google Scholar]
- 67.Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 2006; 174: 1034–1041. [DOI] [PubMed] [Google Scholar]
- 68.Grothues F, Moon JC, Bellenger NG, et al. Interstudy reproducibility of right ventricular volumes, function, and mass with cardiovascular magnetic resonance. Am Heart J 2004; 147: 218–223. [DOI] [PubMed] [Google Scholar]
- 69.van Wolferen SA, Marcus JT, Boonstra A, et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007; 28: 1250–1257. [DOI] [PubMed] [Google Scholar]
- 70.Gan CT, Lankhaar JW, Westerhof N, et al. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest 2007; 132: 1906–1912. [DOI] [PubMed] [Google Scholar]
- 71.Miyamoto S, Nagaya N, Satoh T, et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med 2000; 161: 487–492. [DOI] [PubMed] [Google Scholar]
- 72.Paciocco G, Martinez FJ, Bossone E, et al. Oxygen desaturation on the six-minute walk test and mortality in untreated primary pulmonary hypertension. Eur Respir J 2001; 17: 647–652. [DOI] [PubMed] [Google Scholar]
- 73.Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996; 334: 296–301. [DOI] [PubMed] [Google Scholar]
- 74.Wensel R, Opitz CF, Anker SD, et al. Assessment of survival in patients with primary pulmonary hypertension. Circulation 2002; 106: 319–324. [DOI] [PubMed] [Google Scholar]
- 75.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension. Circulation 2002; 106: 1477–1482. [DOI] [PubMed] [Google Scholar]
- 76.Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004; 169: 441–447. [DOI] [PubMed] [Google Scholar]
- 77.Barst RJ, McGoon M, McLaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2003; 41: 2119–2125. [DOI] [PubMed] [Google Scholar]
- 78.Oudiz RJ, Barst RJ, Hansen JE, et al. Cardiopulmonary exercise testing and six-minute walk correlations in pulmonary arterial hypertension. Am J Cardiol 2006; 97: 123–126. [DOI] [PubMed] [Google Scholar]
- 79.Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation 2000; 102: 865–870. [DOI] [PubMed] [Google Scholar]
- 80.Fijalkowska A, Kurzyna M, Torbicki A, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 2006; 129: 1313–1321. [DOI] [PubMed] [Google Scholar]
- 81.Torbicki A, Kurzyna M, Kuca P, et al. Detectable Serum Cardiac Troponin T as a marker of poor prognosis among patients with chronic precapillary pulmonary hypertension. Circulation 2003; 108: 844–848. [DOI] [PubMed] [Google Scholar]
- 82.Nagaya N, Uematsu M, Satoh T, et al. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med 1999; 160: 487–492. [DOI] [PubMed] [Google Scholar]
- 83.Roeleveld RJ, Vonk-Noordegraaf A, Marcus JT, et al. Effects of epoprostenol on right ventricular hypertrophy and dilatation in pulmonary hypertension. Chest 2004; 125: 572–579. [DOI] [PubMed] [Google Scholar]
- 84.Sniderman A, Fitchett D. Vasodilators and pulmonary arterial hypertension: the paradox of therapeutic success and clinical failure. Int J Cardiol 1988; 20: 173–181. [DOI] [PubMed] [Google Scholar]
- 85.McLaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J 2005; 25: 244–249. [DOI] [PubMed] [Google Scholar]
- 86.Sagawa K, Maughan L, Suga H, et al. Cardiac Contraction and the Pressure–Volume Relationship. New York, Oxford University Press, 1988 [Google Scholar]
- 87.Asanoi H, Sasayama S, Kameyama T. Ventriculoarterial coupling in normal and failing heart in humans. Circ Res 1989; 65: 483–493. [DOI] [PubMed] [Google Scholar]
- 88.Burkhoff D, Sagawa K. Ventricular efficiency predicted by an analytical model. Am J Physiol 1986; 250: R1021–R1027. [DOI] [PubMed] [Google Scholar]
- 89.O'Rourke M, Avolio A, Nichols W. Left Ventricular–Systemic Arterial Coupling in Humans and Strategies to Improve Coupling in Disease States. New York: Springer-Verlag, 1987 [Google Scholar]
- 90.Fourie PR, Coetzee AR, Bolliger CT. Pulmonary artery compliance: its role in right ventricular–arterial coupling. Cardiovasc Res 1992; 26: 839–844. [DOI] [PubMed] [Google Scholar]
- 91.Kass D, Kelly R. Ventriculo-arterial coupling: concepts, assumptions, and applications. Ann Biomed Eng 1992; 20: 41–62. [DOI] [PubMed] [Google Scholar]
- 92.Brimioulle S, Wauthy P, Ewalenko P, et al. Single-beat estimation of right ventricular end-systolic pressure-volume relationship. Am J Physiol 2003; 284: H1625–H1630. [DOI] [PubMed] [Google Scholar]
- 93.Kuehne T, Yilmaz S, Steendijk P, et al. Magnetic resonance imaging analysis of right ventricular pressure-volume loops. Circulation 2004; 110: 2010–2016. [DOI] [PubMed] [Google Scholar]
- 94.Wauthy P, Naeije R, Brimioulle S. Left and right ventriculo-arterial coupling in a patient with congenitally corrected transposition. Cardiol Young 2005; 15: 647–649. [DOI] [PubMed] [Google Scholar]
- 95.Nichols W, O'Rourke M. McDonald's Blood Flow in Arteries. 3rd Edn. London, Edward Arnold, 1990 [Google Scholar]
- 96.Grant BJ, Lieber BB. Clinical significance of pulmonary arterial input impedance. Eur Respir J 1996; 9: 2196–2199. [DOI] [PubMed] [Google Scholar]
- 97.Huez S, Brimioulle S, Naeije R, et al. Feasibility of routine pulmonary arterial impedance measurements in pulmonary hypertension. Chest 2004; 125: 2121–2128. [DOI] [PubMed] [Google Scholar]
- 98.Haneda T, Nakajima T, Shirato K, et al. Effects of oxygen breathing on pulmonary vascular input impedance in patients with pulmonary hypertension. Chest 1983; 83: 520–527. [DOI] [PubMed] [Google Scholar]
- 99.Laskey WK, Ferrari VA, Palevsky HI, et al. Pulmonary artery hemodynamics in primary pulmonary hypertension. J Am Coll Cardiol 1993; 21: 406–412. [DOI] [PubMed] [Google Scholar]
- 100.Huez S, Vachiéry J-L, Unger P, et al. Tissue Doppler imaging evaluation of cardiac adaptation to severe pulmonary hypertension. Am J Cardiol 2007; 100: 1473–1478. [DOI] [PubMed] [Google Scholar]
- 101.Dambrauskaite V, Delcroix M, Claus P, et al. Regional right ventricular dysfunction in chronic pulmonary hypertension. J Am Soc Echocardiogr 2007; 20: 1172–1180. [DOI] [PubMed] [Google Scholar]
- 102.Kittipovanonth M, Bellavia D, Chandrasekaran K, et al. Doppler myocardial imaging for early detection of right ventricular dysfunction in patients with pulmonary hypertension. J Am Soc Echocardiogr 2008; 21: 1035–1041. [DOI] [PubMed] [Google Scholar]
- 103.Sachdev A, Villarraga HR, Frantz RP, et al. Right ventricular strain for prediction of survival in patients with pulmonary arterial hypertension. Chest 2011; 139: 1299–1309. [DOI] [PubMed] [Google Scholar]
- 104.Leeuwenburgh BPJ, Steendijk P, Helbing WA, et al. Indexes of diastolic RV function: load dependence and changes after chronic RV pressure overload in lambs. Am J Physiol 2002; 282: H1350–H1358. [DOI] [PubMed] [Google Scholar]
- 105.Stojnic B, Brecker S, Xiao H, et al. Left ventricular filling characteristics in pulmonary hypertension: a new mode of ventricular interaction. Br Heart J 1992; 68: 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gan CT, Holverda S, Marcus JT, et al. Right ventricular diastolic dysfunction and the acute effects of sildenafil in pulmonary hypertension patients. Chest 2007; 132: 11–17. [DOI] [PubMed] [Google Scholar]
- 107.Blyth KG, Groenning BA, Martin TN, et al. Contrast enhanced-cardiovascular magnetic resonance imaging in patients with pulmonary hypertension. Eur Heart J 2005; 26: 1993–1999. [DOI] [PubMed] [Google Scholar]
- 108.McCann GP, Gan CT, Beek AM, et al. Extent of MRI delayed enhancement of myocardial mass is related to right ventricular dysfunction in pulmonary artery hypertension. Am J Roentgenol 2007; 188: 349–355. [DOI] [PubMed] [Google Scholar]
- 109.Puwanant S, Park M, Popović ZB, et al. Ventricular geometry, strain, and rotational mechanics in pulmonary hypertension. Circulation 2010; 121: 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tanaka H, Tei C, Nakao S, et al. Diastolic bulging of the interventricular septum toward the left ventricle. An echocardiographic manifestation of negative interventricular pressure gradient between left and right ventricles during diastole. Circulation 1980; 62: 558–563. [DOI] [PubMed] [Google Scholar]
- 111.Marcus JT, Vonk Noordegraaf A, Roeleveld RJ, et al. Impaired left ventricular filling due to right ventricular pressure overload in primary pulmonary hypertension. Chest 2001; 119: 1761–1765. [DOI] [PubMed] [Google Scholar]
- 112.Marcus JT, Gan CT-J, Zwanenburg JJM, et al. Interventricular mechanical asynchrony in pulmonary arterial hypertension: left-to-right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol 2008; 51: 750–757. [DOI] [PubMed] [Google Scholar]
- 113.Vonk-Noordegraaf A, Marcus JT, Gan CT, et al. Interventricular mechanical asynchrony due to right ventricular pressure overload in pulmonary hypertension plays an important role in impaired left ventricular filling. Chest 2005; 128: Suppl. 6, 628S–630S. [DOI] [PubMed] [Google Scholar]
- 114.Kalogeropoulos AP, Georgiopoulou VV, Howell S, et al. Evaluation of right intraventricular dyssynchrony by two-dimensional strain echocardiography in patients with pulmonary arterial hypertension. J Am Soc Echocardiogr 2008; 21: 1028–1034. [DOI] [PubMed] [Google Scholar]
- 115.Schwartz DJ, Kop WJ, Park MH, et al. Evidence for early right ventricular and septal mechanical activation (interventricular dyssynchrony) in pulmonary hypertension. Am J Cardiol 2008; 102: 1273–1277. [DOI] [PubMed] [Google Scholar]
- 116.Chin KM, Kingman M, de Lemos JA, et al. Changes in right ventricular structure and function assessed using cardiac magnetic resonance imaging in bosentan-treated patients with pulmonary arterial hypertension. Am J Cardiol 2008; 101: 1669–1672. [DOI] [PubMed] [Google Scholar]
- 117.Wilkins MR, Paul GA, Strange JW, et al. Sildenafil versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) Study. Am J Respir Crit Care Med 2005; 171: 1292–1297. [DOI] [PubMed] [Google Scholar]
- 118.van Wolferen SA, Boonstra A, Marcus JT, et al. Right ventricular reverse remodelling after sildenafil in pulmonary arterial hypertension. Heart 2006; 92: 1860–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Blalock SE, Matulevicius S, Mitchell LC, et al. Long-term outcomes with ambrisentan monotherapy in pulmonary arterial hypertension. J Card Fail 2010; 16: 121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Michelakis ED, Tymchak W, Noga M, et al. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation 2003; 108: 2066–2069. [DOI] [PubMed] [Google Scholar]
- 121.Nath J, DeMarco T, Hourigan L, et al. Correlation between right ventricular indices and clinical improvement in epoprostenol treated pulmonary hypertension patients. Echocardiography 2005; 22: 374–379. [DOI] [PubMed] [Google Scholar]
- 122.Galiè N, Hinderliter AL, Torbicki A, et al. Effects of the oral endothelin-receptor antagonist bosentan on echocardiographic and Doppler measures in patients with pulmonary arterial hypertension. J Am Coll Cardiol 2003; 41: 1380–1386. [DOI] [PubMed] [Google Scholar]
- 123.Cozzi F, Montisci R, Marotta H, et al. Bosentan therapy of pulmonary arterial hypertension in connective tissue diseases. Eur J Clin Invest 2006; 36: 49–53. [DOI] [PubMed] [Google Scholar]
- 124.Bhatia S, Frantz RP, Severson CJ, et al. Immediate and long-term hemodynamic and clinical effects of sildenafil in patients with pulmonary arterial hypertension receiving vasodilator therapy. Mayo Clinic Proceedings 2003; 78: 1207–1213. [DOI] [PubMed] [Google Scholar]
- 125.Ruiz MJ, Escribano P, Delgado JF, et al. Efficacy of sildenafil as a rescue therapy for patients with severe pulmonary arterial hypertension and given long-term treatment with prostanoids: 2-year experience. J Heart Lung Transplant 2006; 25: 1353–1357. [DOI] [PubMed] [Google Scholar]
- 126.Hinderliter AL, Willis PW, Barst RJ, et al. Effects of long-term infusion of prostacyclin (epoprostenol) on echocardiographic measures of right ventricular structure and function in primary pulmonary hypertension. Circulation 1997; 95: 1479–1486. [DOI] [PubMed] [Google Scholar]
- 127.Seyfarth H, Jr, Pankau H, Hammerschmidt S, et al. Bosentan improves exercise tolerance and Tei index in patients with pulmonary hypertension and prostanoid therapy. Chest 2005; 128: 709–713. [DOI] [PubMed] [Google Scholar]
- 128.Cella G, Vianello F, Cozzi F, et al. Effect of bosentan on plasma markers of endothelial cell activity in patients with secondary pulmonary hypertension related to connective tissue diseases. J Rheumatol 2009; 36: 760–767. [DOI] [PubMed] [Google Scholar]
- 129.ClinicalTrials.gov. Combination Therapy in Pulmonary Arterial Hypertension (COMPASS 3). NCT00433329. http://clinicaltrials.gov/ct2/show/NCT00433329 Date last updated: January 11, 2011. Date last accessed: September 5, 2011..