

Abstract
Pirfenidone is an orally active small molecule that has recently been evaluated in large clinical trials for the treatment of idiopathic pulmonary fibrosis, a fatal disease in which the uncontrolled deposition of extracellular matrix leads to progressive loss of lung function. This review describes the activity of pirfenidone in several well-characterised animal models of fibrosis in the lung, liver, heart and kidney. In these studies, treatment-related reductions in fibrosis are associated with modulation of cytokines and growth factors, with the most commonly reported effect being reduction of transforming growth factor-β. The consistent antifibrotic activity of pirfenidone in a broad array of animal models provides a strong preclinical rationale for the clinical characterisation of pirfenidone in pulmonary fibrosis and, potentially, other conditions with a significant fibrotic component.
Keywords: Animal model, antifibrotic, fibrosis, idiopathic pulmonary fibrosis, pirfenidone
Fibrosis, the dysregulated deposition of extracellular matrix (ECM) with progressive destruction of normal tissue, is a primary or contributing factor in chronic disease states in several organs. Pulmonary fibrosis is associated with numerous diffuse parenchymal lung diseases, of which the most common are idiopathic pulmonary fibrosis (IPF) and sarcoidosis [1]. Cardiac fibrosis is associated with chronic heart failure, atrial fibrillation, and cardiac remodelling following acute myocardial infarction [2, 3]. Renal fibrosis is associated with multiple forms of chronic kidney disease and correlates with impairment of kidney function [4, 5]. Hepatic fibrosis is associated with chronic hepatitis B and C viral infections and nonalcoholic steatohepatitis [6]. Each of these conditions represents a significant unmet medical need, warranting significant research and clinical study into treatment for fibrotic disease.
Pirfenidone (Esbriet®, Pirespa®) is an orally active small molecule comprising a modified phenyl pyridone (fig. 1). The compound exhibits well documented antifibrotic and anti-inflammatory activities in a variety of animal and cell-based models, although its molecular target has not been elucidated. Pirfenidone was initially identified as having anti-inflammatory activity in animal models and evaluated for use as an anti-inflammatory drug [7, 8]. However, the unexpected identification of antifibrotic effects in animals treated with pirfenidone redefined the interest in the compound [9]. Subsequently, pirfenidone has been shown to attenuate fibrosis in numerous animal models, including fibrosis of the lung, liver, heart and kidney.
Figure 1.

Chemical structure of pirfenidone.
The most extensive clinical studies of pirfenidone are for treatment of IPF, a chronic interstitial lung disease characterised by the unregulated deposition of ECM leading to the unremitting destruction of normal lung. Patients diagnosed with IPF typically experience progressive pulmonary insufficiency, and most die of respiratory failure. The estimated median survival upon diagnosis is approximately 3 yrs [1]. The ratio of the estimated prevalence (90,000 individuals) and incidence (30,000 individuals) of IPF in the USA reflects this poor prognosis [10].
IPF is the most common and most life-threatening of the idiopathic interstitial pneumonias (IIPs), a heterogeneous group of diffuse parenchymal lung disorders that are diagnosed on the basis of clinical, radiological and histological features [1]. By definition, IPF is differentiated from other IIPs based on the presence of a pattern of usual interstitial pneumonia in lung histopathology.
Although the pathogenesis of IPF is poorly defined, several hypotheses have been proposed and continue to evolve in response to more detailed characterisations of the disease [11–15]. In the most prevalent theory of IPF pathogenesis, repeated exposure to an as yet undefined stimulus leads to alveolar epithelial cell damage [12–14, 16]. The activated epithelial cell produces a series of cytokines and growth factors (including transforming growth factor (TGF)-β, platelet-derived growth factor (PDGF), fibroblast growth factor (FGF) and tumour necrosis factor (TNF)-α) initiating the migration and proliferation of fibroblasts. In the absence of successful epithelial repair, the wound healing process does not resolve properly and a persistent fibroproliferative state is established. This state is characterised by the presence and proliferation of myofibroblasts (a hallmark cell of IPF), excessive deposition of ECM and loss of basement membrane integrity. Continued epithelial cell injury and initiation at sites throughout the lung gives rise to the pattern of diffuse fibrosis with temporal heterogeneity observed in IPF. Studies examining molecular differences between IPF and control patients support the characterisation of IPF as a disease of active ECM deposition and remodelling; epithelial cell activation; dysregulated expression of cytokines, chemokines, and growth factors; and myofibroblast proliferation [17–21].
This review highlights the preclinical data that demonstrate the antifibrotic activity of pirfenidone in animal models of lung, liver, heart and kidney fibrosis, as well as effects on relevant fibrogenic mediators. Pharmacokinetic data from a subset of animal efficacy studies demonstrate that the antifibrotic effects of pirfenidone are observed at exposures that are achieved clinically, indicating that the effects observed in animal models may have relevance to the clinical pharmacodynamics of pirfenidone. The observation of a robust antifibrotic effect in multiple animal models strongly supports the clinical characterisation of pirfenidone in IPF.
SUMMARY OF CLINICAL TRIALS IN IPF
The earliest clinical study of pirfenidone in IPF, an open-label compassionate use trial in 54 patients with advanced disease, showed that pirfenidone was well tolerated and possibly stabilised lung function, an outcome that warranted further study [22]. A subsequent randomised, double-blind, placebo-controlled trial in 107 Japanese patients with IPF was stopped early following a planned interim analysis at 24 weeks that found an increased incidence of acute IPF exacerbations among patients in the placebo arm [23]. Final analysis of outcomes at week 36 revealed that pirfenidone had a favourable effect on the mean change in vital capacity and the incidence of acute IPF exacerbation. This study was followed by a randomised, double-blind, placebo-controlled phase III trial in 275 Japanese patients with IPF, in which pirfenidone (1,800 mg·day−1) significantly reduced the decline in vital capacity at week 52 (p = 0.04) and improved progression-free survival (p = 0.028) [24]. In contrast to the findings in the phase II trial, no difference was observed between groups in the rate of acute exacerbation and this end-point was not used in the evaluation of clinical efficacy in the subsequent CAPACITY trials (see below).
Pirfenidone was also evaluated in two nearly identical, randomised, double-blind, placebo-controlled, phase III trials (study 004 and study 006) enrolling 435 and 344 patients, respectively, in Australia, Europe and North America (P. Noble and co-workers, Duke University, Durham, NC, USA; manuscript submitted) [25]. In study 004, pirfenidone treatment (2,403 mg·day−1) resulted in a 35% relative reduction in the mean decline in per cent predicted forced vital capacity (FVC) at week 72 (-8.0% versus -12.4% in the pirfenidone and placebo groups, respectively; p = 0.001) and improved progression-free survival time (hazard ratio (HR) 0.64, 95% CI 0.44–0.95; p = 0.023). In study 006, the primary end-point of mean change in per cent predicted FVC at week 72 was not met (-9.0% versus -9.6% in the pirfenidone and placebo groups, respectively; p = 0.501); however, a significant pirfenidone treatment effect was observed at weeks 24 (p<0.001), 36 (p = 0.011), and 48 (p = 0.005) and in the repeated measures analysis of all study time points (p = 0.007). In the pooled analysis, a significant pirfenidone treatment effect was observed for the mean change in per cent predicted FVC (23% relative reduction; -8.5% versus -11.0%; p = 0.005), categorical change in per cent predicted FVC (p = 0.003), mean change in 6-min walk test distance (31% relative difference; -52.8 m versus -76.8 m; p<0.001) and progression-free survival time (HR 0.74, 95% CI 0.57–0.96; p = 0.025). Finally, an independent meta-analysis of progression-free survival time in all three phase III clinical trials of pirfenidone in patients with IPF, including the Japanese study and the multinational CAPACITY studies (n = 1,046), reported a significant overall treatment benefit (HR 0.70, 95% CI 0.56–0.88; p = 0.002) [26].
Smaller exploratory clinical studies have also evaluated the effect of pirfenidone in Hermanksy–Pudlak syndrome [27], focal segmental glomerulosclerosis [4, 28], diabetic nephropathy [29] and advanced liver disease [30]. Results in smaller trials such as these are consistent with a systemic antifibrotic effect and may warrant further investigation.
PHARMACOKINETICS IN HUMANS AND NONCLINICAL SPECIES
Oral pirfenidone is rapidly absorbed and displays high oral bioavailability and broad distribution. In fasted older adults, maximal concentrations (Cmax) in plasma are observed 30 min after an oral dose [31]. Administration with food delays absorption and reduces Cmax but has a comparatively modest effect on overall exposure [31]. The observed reduction in Cmax in the fed state is of potential clinical significance as acute pirfenidone-related side-effects are associated with Cmax in both humans and nonclinical species [31]. Similarly rapid absorption is observed in nonclinical species [32, 33] and a radiometric study in mice demonstrates broad distribution in well perfused tissues including liver, kidney, heart and lung [34].
Pirfenidone has a terminal half-life of approximately 2.5 h in healthy adults [31]. In humans and nonclinical species, pirfenidone is extensively metabolised. The principal site of metabolism is the 5-methyl position of the pyridone ring, resulting in stepwise formation of 5-hydroxymethyl pirfenidone and a 5-carboxcylic acid metabolite [31, 34]. The 5-carboxcylic acid metabolite accounts for 95% of the recovered dose in healthy older adults [31]. Results from in vitro experiments with cell fractions from human liver and recombinant cytochrome P450 (CYP) enzymes indicate that pirfenidone is primarily metabolised by CYP1A2, although multiple other CYP isoforms contribute to metabolism as well (InterMune, Inc., Brisbane, CA, USA; data on file) [35]. The primary route of excretion in humans and nonclinical species is via urine. In humans, 80% of an orally administered dose is recovered in urine [31]. The rapid metabolism and elimination of pirfenidone is consistent with the use of thrice-daily oral administration in clinical trials.
Efficacy studies in nonclinical species commonly administer pirfenidone continuously as a mixture in feed (most commonly 0.5%). Oral administration of pirfenidone to rats as a 0.4% component of feed results in a relatively constant plasma concentration. The observed concentration is similar to the minimal plasma concentration (Cmin) observed in the aforementioned clinical study 004 (InterMune, Inc.; data on file) [33].
EFFECTS IN MODELS OF PULMONARY FIBROSIS
Bleomycin-induced pulmonary fibrosis
The bleomycin model is a standard but imperfect model of pulmonary fibrosis. Bleomycin administration results in oxidative stress and acute inflammation with the subsequent onset of pulmonary fibrosis [36, 37]. Several histological characteristics of the bleomycin model are similar to those of IPF, including epithelial cell injury with reactive hyperplasia, basement membrane damage, clusters of spindle-shaped mesenchymal cells resembling fibroblastic foci, and interstitial as well as intra-alveolar fibrosis [37]. Although these fibrotic lesions resemble those in IPF, fibrosis in the bleomycin model has several key differences from human disease, including partial reversibility and the rapid onset and progression in response to a well-defined insult. Moreover, several variations of the bleomycin model in preclinical species have notable consequences. Two important differences between variants of the bleomycin model include: 1) the use of single versus multiple bleomycin challenges, where multiple administrations result in a more lasting fibrosis; and 2) intratracheal administration of bleomycin, which results in bronchiocentric fibrosis, versus systemic (intravenous or intraperitoneal) administration, which results in a subpleural fibrosis that is more similar to that observed in IPF [37]. Overall, the bleomycin model continues to be the most common model of pulmonary fibrosis, although the details of the experimental system and supporting data from other models should be considered in evaluating potential antifibrotic activity.
Several studies have demonstrated an effect of pirfenidone in the bleomycin model of pulmonary fibrosis (table 1) [38–47]. Oku et al. [38] examined the effect of oral pirfenidone over the course of a 42-day model initiated by repeated intravenous administration of bleomycin (table 1 and fig. 2a). Administration of pirfenidone via oral gavage (30 or 100 mg·kg−1·day−1 in three divided doses) minimised early lung oedema and lung pathology, and reduced hydroxyproline accumulation when treatment was initiated concurrent with lung damage. These treatment effects were associated with normalised pulmonary expression of pro-inflammatory and fibrogenic proteins. In the same study, high dose prednisolone (3 or 15 mg·kg−1 once daily) also reduced lung oedema and pro-inflammatory proteins but, unlike pirfenidone, did not reduce fibrosis or fibrogenic proteins. The inability of prednisolone to modulate fibrosis in this model is consistent with previous findings in which prednisolone effectively reduced the transient bronchiocentric fibrosis associated with intratracheal bleomycin challenge but did not reduce the more durable peripheral fibrosis associated with systemic bleomycin administration [56–58].
Table 1. Effects of pirfenidone in animal models of lung, heart, kidney, and liver fibrosis.
| System | Reference | In vivo model | Species | Protocol | Total daily dose/route | Fibrosis | Fibrotic markers | Inflammatory markers | Oxidative stress | Functional end-points |
| Lung | Oku et al. [38] | Bleomycin | Mouse | Prophylactic | 10, 30*, 100* mg·kg−1 p.o.# | Decreased | Decreased | Decreased | NA | NA |
| H. Oku; personal communication | Bleomycin | Mouse | Therapeutic | 10, 30*, 100* mg·kg−1 p.o.# | Decreased | NA | NA | NA | NA | |
| Kakugawa et al. [39] | Bleomycin | Mouse | Therapeutic | 400* mg·kg−1 p.o.¶ | Decreased | Decreased | NA | NA | NA | |
| Iyer et al. [40] | Bleomycin | Hamster | Prophylactic | 0.5%* in chow | Decreased | Decreased | NA | Decreased | NA | |
| Iyer et al. [41] | Bleomycin | Hamster | Therapeutic+ | 0.5%* in chow | Decreased | Decreased | NA | Decreased | NA | |
| Iyer et al. [42] | Bleomycin | Hamster | Prophylactic | 0.5%* in chow | Decreased | Decreased | NA | Decreased | NA | |
| Gurujeyalakshmi et al. [43] | Bleomycin | Hamster | Prophylactic | 0.5%* in chow | NA | Decreased | NA | NA | NA | |
| Schelegle et al. [44] | Bleomycin | Hamster | Prophylactic | 0.5%* in chow | Decreased | NA | NA | NA | Improved | |
| Liu et al. [45] | Transplant | Rat | Prophylactic | 0.5%* in chow | Decreased | Decreased | NA | No effect | Improved | |
| Zhou et al. [46] | 1) Transplant | Rat | Prophylactic | 0.5%* in chow | Decreased | Decreased | NA | NA | NA | |
| 2) Transplant | Rat | Therapeutic | 0.5% in chow | No effect | NA | NA | NA | NA | ||
| Hirano et al. [47] | Allergen induced dysfunction | Mouse | Prophylactic | 125, 250, 500* mg·kg−1 s.c.§ | Decreased | Decreased | Decreased | NA | Improved | |
| Heart | Lee et al. [2] | Congestive heart failure | Dog | Prophylactic | 2400* mg p.o.# | Decreased | Decreased | Decreased | NA | Improved |
| Nguyen et al. [3] | MI | Rat | Therapeutic | 1.2%* in chow | Decreased | NA | NA | NA | Improved | |
| Mirkovic et al. [33] | Hypertension | Rat | Therapeutic | 0.4%* in chow | Decreased | NA | NA | NA | Improved | |
| Kidney | Shimizu et al. [48] | 1) Continuous ureteral obstruction | Rat | Prophylactic | 0.6–0.9%* in chow | Decreased | Decreased | NA | NA | NA |
| 2) Continuous ureteral obstruction | Rat | Therapeutic | 0.6–0.9%ƒ in chow | Trend | Decreased | NA | NA | NA | ||
| 3) Temporary ureteral obstruction | Rat | Therapeutic | 0.6–0.9%* in chow | Decreased | NA | NA | NA | Improved | ||
| Takakura et al. [49] | 5/6 nephrectomy | Rat | Therapeutic | 1%* in chow | Decreased | Decreased | NA | NA | Trend | |
| Shimizu et al. [50] | 5/6 nephrectomy | Rat | Therapeutic | 0.6–0.9%* in chow | Decreased | Decreased | NA | NA | Improved | |
| RamachandraRao et al. [51] | Diabetic db/db | Mouse | Therapeutic | 0.5%* in chow | Decreased | Decreased | NA | NA | No effect | |
| Liver | Salazar-Montes et al. [52] | 1) Carbon tetrachloride | Rat | Therapeutic | 200ƒ mg·kg−1 p.o.§ | Trend | Decreased | NA | Decreased | Improved |
| 2) Bile duct ligation | Rat | Therapeutic | 200* mg·kg−1 p.o.§ | Decreased | Decreased | NA | Decreased | No effect | ||
| Garcia et al. [53] | 1) Carbon tetrachloride | Rat | Therapeutic## | 500* mg·kg−1 p.o. | Decreased | Decreased | NA | NA | Improved | |
| 2) Carbon tetrachloride | Rat | Therapeutic¶¶ | 200* mg·kg−1 p.o.§ | Decreased | NA | NA | NA | NA | ||
| 3) Bile duct ligation | Rat | Prophylactic | 200*, 500 mg·kg−1 p.o.§ | Decreased | NA | NA | NA | NA | ||
| Di Sario et al. [54] | Dimethylnitrosamine | Rat | Therapeutic | 0.5%* in chow | Decreased | Decreased | Decreased | NA | Improved | |
| Tada et al. [55] | Dimethylnitrosamine | Rat | Prophylactic | 500* mg·kg−1 p.o.§ | Decreased | Trend | NA | NA | No effect |
NA: not assessed; MI: myocardial infarction. *: statistically significant treatment effect (per reference); #: given in three divided daily doses; ¶: given in two divided daily doses; +: pirfenidone treatment initiated following the second of three bleomycin insults; §: given in one daily dose; ƒ: treatment-associated trend; ##: pirfenidone dosed following cessation of carbon tetrachloride; ¶¶: pirfenidone dosed in final 3 weeks of an 11-week model with continuous carbon tetrachloride.
Figure 2.
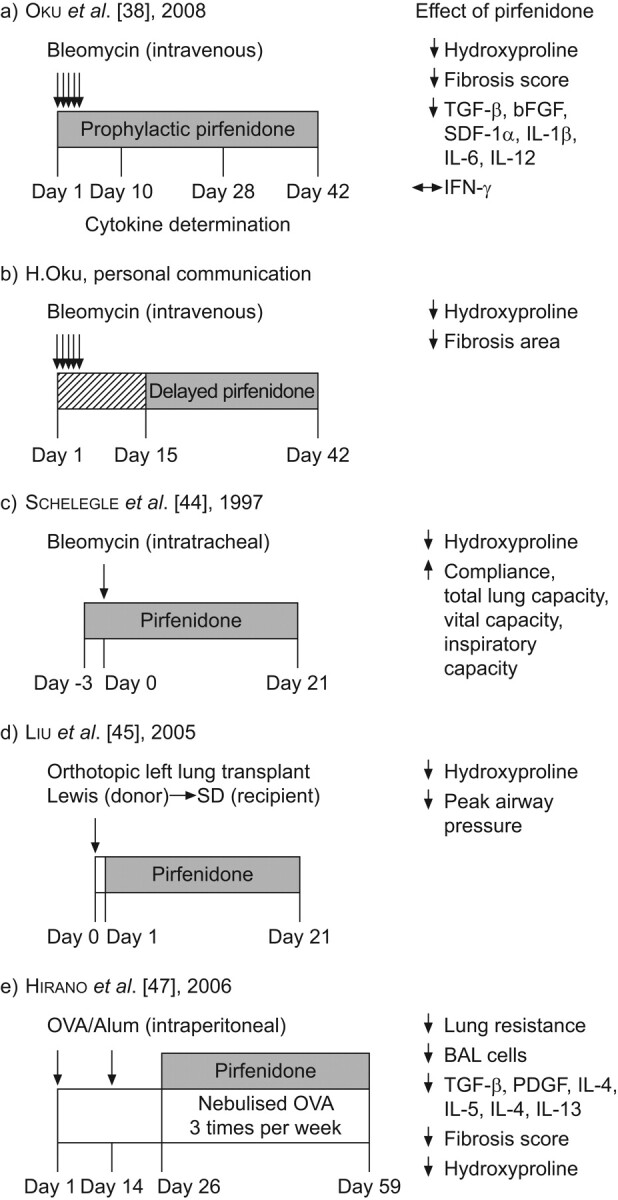
The antifibrotic activity of pirfenidone in preclinical models of pulmonary fibrosis. a) Experimental design and effects of prophylactic pirfenidone treatment on bleomycin-induced pulmonary fibrosis [38]. b) Effects of delayed pirfenidone treatment on bleomycin-induced pulmonary fibrosis (H. Oku, Shionogi and Co. Ltd, Osaka, Japan; personal communication). c) Effects of prophylactic pirfenidone treatment on bleomycin-induced pulmonary fibrosis [44]. d) Experimental design and effects of pirfenidone in an orthotopic lung transplant model [45]. e) The effects of pirfenidone in a mouse model of airway remodelling and hyperresponsiveness induced by sensitisation and challenge with ovalbumin [47]. BAL: bronchoalveolar lavage; bFGF: basic fibroblast growth factor; IL: interleukin; IFN: interferon; OVA: ovalbumin; PDGF: platelet-derived growth factor; SD: Sprague-Dawley; SDF: stromal cell-derived factor; TGF: transforming growth factor.
In order to evaluate the effect of pirfenidone on an established fibrotic process, the authors performed a second study in which pirfenidone administration was initiated at day 15 (table 1 and fig. 2b; H. Oku, Shionogi and Co. Ltd, Osaka, Japan; personal communication). A kinetic characterisation in the model above established that by day 15 inflammation had subsided and fibrosis was established and progressing. Although lung cytokines were not assessed, other technical aspects of the model were unchanged. In this study, administration of 30 or 100 mg·kg−1·day−1 pirfenidone by oral gavage (in three divided doses) alleviated the bleomycin-induced increases in lung hydroxyproline content and pulmonary fibrosis, but did not reduce fibrosis to a level below that already established at the initiation of treatment. Delayed administration of pirfenidone at doses of 10, 30 and 100 mg·kg−1 reduced bleomycin-induced elevations in lung hydroxyproline by 30%, 40% and 60%, respectively. These results are almost indistinguishable from the 30%, 60% and 70% reductions observed with prophylactic treatment [38]. A parallel pharmacokinetic study demonstrated that pirfenidone exposures at both effective doses were lower than those obtained in IPF patients treated with 2,403 mg·day−1, the dose administered in the aforementioned clinical studies 004 and 006. Thus, pirfenidone can markedly reduce the progression of an established bleomycin-induced fibrotic process at clinically relevant exposures.
The antifibrotic effects of pirfenidone were also demonstrated in a series of shorter duration bleomycin models initiated by a single intratracheal administration of bleomycin to hamsters (table 1) [40–44]. In these studies, prophylactic administration of pirfenidone (0.5% in feed throughout the study) showed a consistent reduction in lung collagen content, with a subset of these studies demonstrating additional reductions in prolyl hydrolyase activity (a marker of collagen synthesis) [40–42] and expression of collagen mRNA [42]. Three of these studies demonstrated significant pirfenidone-related reductions in markers of oxidative stress, including lipid peroxidation [40], superoxide dismutase activity [40, 41], myeloperoxidase activity [40, 41] and malondialdehyde equivalents [42]. Importantly, Schelegle et al. [44] demonstrated that prophylactic pirfenidone treatment (0.5% in chow) in a 21 day model reduced bleomycin-induced increases in lung hydroxyproline by ∼70% and preserved functional measures of lung compliance, total lung capacity, vital capacity and inspiratory capacity (fig. 2c).
Transplant-induced pulmonary fibrosis
Pirfenidone is also effective in reducing post-transplant pulmonary fibrosis and dysfunction in preclinical lung allograft models. Chronic lung rejection, which is manifested clinically as obliterative bronchiolitis, is a chronic fibrotic disorder that is a significant cause of death in long-term survivors of lung transplantation. In preclinical lung allograft models, fibrosis develops following injury from rejection and/or infection, replacing lung parenchyma with fibrotic tissue and resulting in pulmonary dysfunction and airway obstruction [45, 46].
Liu et al. [45] examined the effect of a prophylactic pirfenidone regimen (0.5% in feed) in a model of orthotopic left lung transplant between Sprague-Dawley and Lewis rats (table 1 and fig. 2d) [45]. Pirfenidone treatment significantly reduced interstitial and peribronchial fibrosis, preserved alveolar/interstitial structure and reduced allograft-induced increases in collagen content by ∼45%. In addition, pirfenidone ameliorated increases in peak airway pressure in the transplanted lung.
Zhou et al. [46] demonstrated a pirfenidone treatment effect in a murine heterotopic tracheal allograft model (table 1). In this model, subcutaneous tracheal transplantation from a BALB/c donor to a C57BL/6 recipient results in rejection of the transplanted tissue with loss of respiratory epithelium, inflammatory cell infiltration, inflammation, intraluminal granulation and fibrosis with eventual occlusion of the airway [46]. The fibrotic components develop more slowly and were prevalent at day 28. Prophylactic pirfenidone treatment (0.5% in feed) continuing throughout the 28-day study reduced collagen deposition, epithelial cell loss and intraluminal degranulation. Moreover, 64% of the untreated allografts demonstrated complete occlusion of the airway with granulation tissue, compared to only 7% in the pirfenidone treatment group. Therapeutic treatment regimens starting at either day 9 or 16 and continuing through day 28 failed to provide a demonstrable benefit. Collectively, these studies indicate that the antifibrotic effects of pirfenidone in pulmonary models are not unique to fibrosis that results from a chemical insult such as bleomycin.
Allergen-induced airway remodelling
Hirano et al. [47] demonstrated antifibrotic and anti-inflammatory effects of pirfenidone in short- and long-term models of allergen-induced airway dysfunction. Repeated exposure to allergen can induce chronic inflammation and lead to airway remodelling including subepithelial fibrosis, myofibroblast hyperplasia, smooth muscle proliferation and vascular changes. In a short-term animal model of allergen-induced airway dysfunction, a mouse is sensitised to allergen (adjuvant-formulated ovalbumin; OVA) via two intraperitoneal injections (days 0 and 14) followed by repeated challenges with an aerosolised version of the same allergen (days 28, 29 and 30). Pirfenidone treatment on days 26–31 (125, 250 and 500 mg·kg−1 via subcutaneous injection) significantly reduced methacholine-induced airway hyperresponsiveness (AHR; 500 mg·kg−1 dose), with dose-dependent reductions in bronchoalveolar lavage (BAL) cell counts, growth factors and cytokines. Airway remodelling, including subepithelial fibrosis and thickening of the peribronchial smooth muscle layer, was observed in a longer term model in which the initial sensitisation was followed with ∼5 weeks in which animals received nebulised OVA three times per week (days 26–59; table 1 and fig. 2e). In this model, pirfenidone treatment (500 mg·kg−1·day−1 via subcutaneous injection) throughout the period of nebulised OVA challenge showed treatment effects on AHR, cell counts and cytokine expression as in the short-term model. Pirfenidone treatment also markedly reduced thickening of the peribronchiolar smooth muscle layer, reduced fibrosis as assessed by histology, and reduced hydroxyproline accumulation by ∼60%.
EFFECTS IN MODELS OF CARDIAC FIBROSIS
Cardiac fibrosis is a significant factor in several disorders, including chronic hypertension [33] and congestive heart failure [59], and contributes to morbidity and cardiac dysfunction following myocardial infarction [3]. Areas of fibrotic tissue compromise cardiac function, interfere with coordinated electrical conduction and increase vulnerability to arrhythmias. At a cellular and molecular level, there are similarities between cardiac fibrosis and fibrosis in other organs including persistent myofibroblasts and elevated fibrogenic mediators (TGF-β, PDGF and connective tissue growth factor) [60].
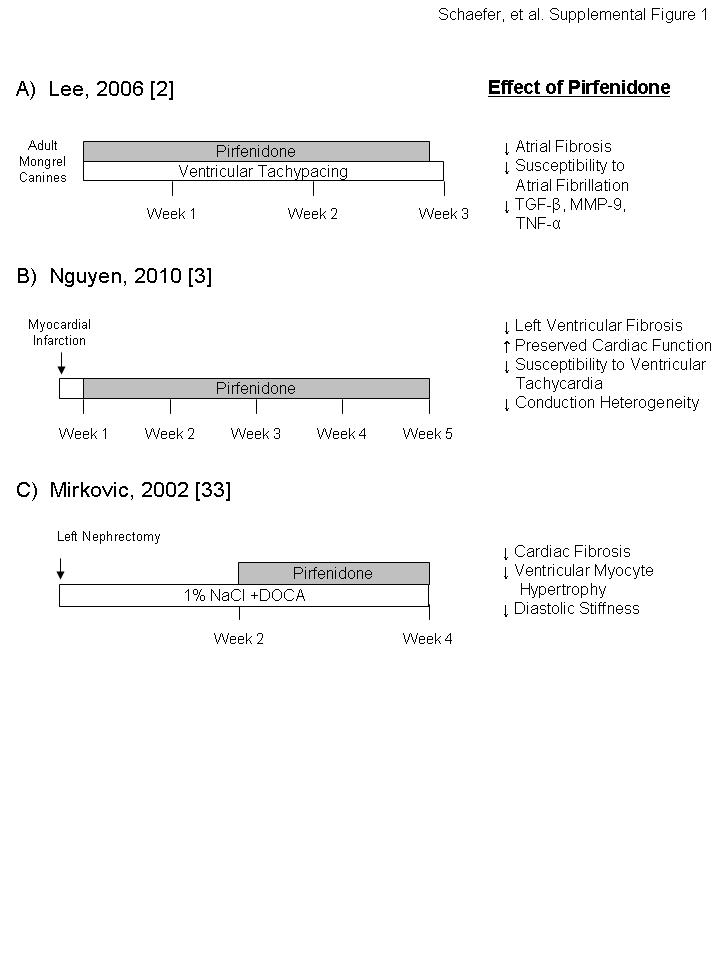
Lee et al. [2] examined the ability of pirfenidone to reduce atrial fibrosis and related susceptibility to atrial fibrillation in a model of heart failure. Congestive heart failure was induced over a 3-week period by ventricular tachypacing in dogs outfitted with a single-chamber pacemaker (table 1 and online supplementary material). Dietary administration of pirfenidone (800 mg three times a day) reduced congestive heart failure-induced left atrial fibrosis by ∼50% and significantly reduced both left atrial conduction heterogeneity and susceptibility to atrial fibrillation.
Persistent fibrosis following myocardial infarction is believed to compromise viable tissue adjacent to the infarct [3]. Finger-like projections of fibrotic tissue into this infarct border zone lead to heterogeneous conduction and predispose patients to ventricular arrhythmias. Nguyen et al. [3] evaluated the ability of pirfenidone to limit fibrosis following myocardial infarction in a rat ischaemic reperfusion model (table 1 and online supplementary material). Pirfenidone treatment (1.2% dietary administration) was initiated 1 week after ischaemic reperfusion and continued for 4 weeks. Pirfenidone treatment resulted in a smaller infarct size with ∼50% less left ventricular fibrosis and less fibrosis extending into adjacent tissue. This finding was associated with a preservation of cardiac function (left ventricular ejection fraction) and reduced susceptibility to ventricular arrhythmias.
Cardiac fibrosis develops in response to chronic hypertension and is associated with increased diastolic stiffness, loss of cardiac function and increased risk of arrhythmias [33]. Mirkovic et al. [33] examined the effect of pirfenidone on cardiac fibrosis in hypertensive rats (table 1 and online supplementary material). Hypertension was initiated by removal of the left kidney with either no further treatment (control rats) or initiation of 1% sodium chloride in drinking water and subcutaneous injections of deoxycorticosterone acetate. Pirfenidone treatment (0.4% in feed) was initiated 2 weeks after surgery and continued through the conclusion of the study at week 4. Pirfenidone treatment attenuated ventricular myocyte hypertrophy and prevented both cardiac fibrosis and increases in diastolic stiffness. Cardiac fibrosis and diastolic stiffness were reduced to levels slightly below those observed in parallel animals at initiation of treatment (week 2), suggesting that pirfenidone may reverse nascent fibrosis in this model. As previously mentioned, a parallel pharmacokinetic study demonstrated that the pirfenidone plasma concentration was relatively constant over a 24-h period and similar to the Cmin observed in IPF patients dosed with 2,403 mg·day−1. Collectively, these studies demonstrate that pirfenidone can exert antifibrotic effects in organs outside of the lung, indicating that the fibrotic pathways targeted by pirfenidone are not tissue specific.
EFFECTS IN MODELS OF RENAL FIBROSIS
Renal fibrosis is the final common pathway for nearly all forms of kidney disease which progress to renal failure [61], and the progression of fibrosis correlates with loss of kidney function [62, 63]. Many of the cellular and molecular mediators of renal fibrosis are common to fibrosis in other organs including the presence of persistent fibroblasts and myofibroblasts, elevated fibrogenic mediators (TGF-β, PDGF, interleukin (IL)-1β and basic (b)FGF), and an imbalance of metalloproteinases and their inhibitors [63]. The effect of pirfenidone in reducing renal fibrosis has been established in a several studies, including models driven by partial nephrectomy [49, 50], unilateral ureteral obstruction (UUO) [48], diabetic nephropathy [51], cyclosporine [64, 65] and vanadate [66]. A limited subset of these studies is summarised below.
One well-characterised model of renal fibrosis uses UUO to induce progressive tubulointerstitial fibrosis. Shimizu et al. [48] examined the effect of pirfenidone in two versions of the UUO model in rats (table 1 and online supplementary material). In one implementation, fibrosis developed with continuous obstruction over a 3-week period. In this model, prophylactic treatment with pirfenidone (0.6–0.9% in feed) yielded a ∼50% reduction in UUO-induced collagen deposition and also reduced expression of collagen and TGF-β mRNAs. Delayed administration of pirfenidone starting 1 week after onset of obstruction and continuing throughout the study showed a trend toward reduced collagen deposition and a significant decrease in TGF-β mRNA expression. In the second implementation of the UUO model, the obstruction was released after 1 week (table 1 and online supplementary material). Elevated hydroxyproline was noted in parallel animals at the time the obstruction was released and fibrosis continued to develop over the subsequent 5 weeks, despite removal of the obstruction. These results suggest that a fibrotic process was established and progressing when the obstruction was released. Pirfenidone treatment (0.6–0.9% in feed) initiated with release of obstruction significantly inhibited further renal fibrosis but did not reduce fibrosis to a level below that already established at the initiation of treatment. At the end of the study, UUO-induced hydroxyproline accumulation was reduced by ∼60%, with evidence of decreased interstitial fibrosis in histological sections. The reversibility of reductions in renal function in this model is inversely correlated to the length of obstruction, which is expected to define the extent of tissue damage. As measured by inulin clearance 5 weeks after release of obstruction, pirfenidone-treated animals showed a partial recovery of renal function whereas untreated animals did not. Together, these experiments show that pirfenidone treatment can prevent renal fibrosis, ameliorate ongoing renal fibrosis and reduce collagen mRNA expression.
In the rat subtotal (5/6) nephrectomy model, surgical resection of two-thirds of one kidney and the entire contralateral kidney results in inflammation and fibrosis in the remnant kidney. Takakura et al. [49] demonstrated that prophylactic pirfenidone treatment (1% in feed) reduced the increase in area staining positive for fibrosis by ∼80% with trends toward improvement of proteinuria as well as creatinine and blood urea nitrogen levels (table 1 and online supplementary material). Shimizu et al. [50] also evaluated the effect of pirfenidone in the 5/6 nephrectomy model. In this study, pirfenidone treatment (0.6–0.9% in feed) prevented ∼60% of collagen accumulation following nephrectomy and also reduced expression of TGF-β and collagen mRNAs (table 1). These effects were associated with improved creatinine clearance. Overall, pirfenidone showed a significant antifibrotic effect in this well-characterised model of chronic kidney disease.
EFFECTS IN MODELS OF HEPATIC FIBROSIS
Hepatic fibrosis occurs in patients with chronic liver disease associated with viral hepatitis, alcoholism and autoimmune liver disease [6, 67]. Hepatic fibrosis is driven by many of the same fibrogenic mediators as in other organs and hepatic stellate cells, which, like myofibroblasts, are hyperactivated with respect to ECM deposition and production of growth factors [6].
Chronic intoxication of rats with carbon tetrachloride (CCl4) results in elevated liver enzymes and hepatic fibrosis. Salazar-Montes et al. [52] evaluated the efficacy of oral administration of pirfenidone at 200 mg·kg−1 in this model (table 1 and online supplementary material). Pirfenidone treatment decreased liver fibrosis by 40% and significantly decreased collagen I mRNA expression. Markers of liver damage (aspartate aminotransferase, alanine aminotransferase (ALT), total bilirubin and direct bilirubin) were significantly reduced in pirfenidone-treated animals relative to controls, demonstrating a functional benefit of pirfenidone treatment. Pirfenidone treatment also reduced markers of oxidative stress, including nitrites and malondialdehyde in the liver; these changes were accompanied by reductions in expression of superoxide dismutase and catalase mRNAs. Garcia et al. [53] evaluated the effects of pirfenidone in two implementations of the CCl4-induced liver fibrosis model (table 1 and online supplementary material). Pirfenidone administration following discontinuation of CCl4 resulted in 70% reductions in area staining positive for fibrosis and the number of hepatic stellate cells with a ∼40% reduction in total liver hydroxyproline. Reduction in hepatic fibrosis was associated with reduced liver enzymes and molecular markers of fibrosis. In a secondary study, pirfenidone administration with continuous CCl4 insult reduced the area staining positive for fibrosis by 40%. Together, these studies demonstrate the ability of pirfenidone treatment to reduce fibrosis, modulate molecular markers of fibrosis and ameliorate liver damage (as assessed by liver enzyme levels) in the CCl4-induced liver fibrosis model.
Dimethylnitrosamine (DMN)-induced liver fibrosis is another commonly used experimental model in rats, in which chronic administration of DMN results in liver remodelling and cirrhosis. Di Sario et al. [54] evaluated pirfenidone (0.5% in feed; dosed in weeks 3–5 of a 5-week model) and found a 70% reduction in the DMN-induced increase in the area of liver staining positive for fibrosis (table 1 and online supplementary material). This was associated with significant reductions in ALT, necroinflammatory score, hepatic stellate cell accumulation, and expression of TGF-β and procollagen-1 mRNAs. In a second study, Tada et al. [55] demonstrated that pirfenidone (500 mg·kg−1·day−1 via oral gavage) resulted in a 40% decrease in fibrotic area (via histopathology) with significant decreases in liver hydroxyproline content and expression of collagen I mRNA (table 1).
The bile duct ligation model is another widely used model of liver fibrosis. Salazar-Montes et al. [52] treated bile duct ligated rats with pirfenidone (200 mg·kg−1 via oral gavage) and observed a 60% decrease in hepatic fibrosis (area staining with Masson’s trichrome) with decreased expression of collagen I mRNA (table 1 and online supplementary material). Pirfenidone treatment also reduced markers of oxidative stress, including nitrites and malondialdehyde, in liver with concomitant reductions in expression of superoxide dismutase and catalase mRNAs. Garcia et al. [53] found that pirfenidone treatment (200 mg·kg−1 via daily oral gavage) reduced both the area staining positive for fibrosis and total liver hydroxyproline by ∼50% (table 1 and online supplementary material). Overall, these studies show the efficacy of pirfenidone in several widely used experimental models of liver fibrosis.
EFFECTS ON MEDIATORS OF FIBROSIS
Several of the in vivo studies described above examined the ability of pirfenidone to regulate the production of cytokines and growth factors or reduce oxidative stress (table 2). These effects may be direct results of pirfenidone treatment or may reflect secondary, knock-on effects related to modulation of an as of yet uncharacterised primary molecular target. In either case, the modulation of cytokines, growth factors and markers of oxidative stress demonstrates that the antifibrotic effects observed in vivo are associated with regulation of pathways relevant to ongoing fibrosis and provide further support for the antifibrotic effects observed.
Table 2. Cytokines and growth factors modulated by pirfenidone treatment in animal models.
| Cytokine/growth factor | Lung | Heart | Kidney | Liver | |||||||
| Oku et al. [38] | Hirano et al. [47] | Liu et al. [45] | Zhou et al. [46] | Lee et al. [2] | Shimizu et al. [50] | Shimizu et al. [48] | Salazar-Montes et al. [52] | Garcia et al. [53] | Di Sario et al. [54] | Tada et al. [55] | |
| Bleomycin | Allergen-induced AHR | Transplant | Transplant | Chronic heart failure | 5/6 nephrectomy | Ureteral obstruction | CCl4 and bile duct ligation models | CCl4 and bile duct ligation models | DMN | DMN | |
| TGF-β | Protein↓ | Protein↓ | Protein↓ | Protein↓ | Protein↓ | mRNA↓ | mRNA↓ | mRNA↓ | mRNA↓ | mRNA↓ | mRNA↓# |
| PDGF | Protein↓ | ||||||||||
| bFGF | Protein↓ | ||||||||||
| IL-18 | Protein↓ | ||||||||||
| SDF-1α/CXCL12 | Protein↓ | ||||||||||
| IFN-γ | Protein↔ | ||||||||||
| HGF | mRNA↑ | ||||||||||
| MMP-2 | mRNA↓ | Protein↔ | mRNA↓ | ||||||||
| MMP-9 | Protein↓ | ||||||||||
| TIMP-1 | mRNA↓ | mRNA↓ | |||||||||
| TIMP-4 | Protein↑ | ||||||||||
| TNF-α | Protein↓ | Protein↓ | |||||||||
| IL-1β | Protein↓ | ||||||||||
| IL-4 | Protein↓ | ||||||||||
| IL-5 | Protein↓ | ||||||||||
| IL-13 | Protein↓ | ||||||||||
| IL-6 | Protein↓ | ||||||||||
| MCP-1 | Protein↓ | ||||||||||
| IL-12p40 | Protein↓ | ||||||||||
Empty fields denote end-points that were either not assessed or not dysregulated in this model. Unless otherwise noted, all effects were statistically significant per publication. AHR: airway hyperresponsiveness; DMN: dimethylnitrosamine; TGF: transforming growth factor; PDGF: platelet-derived growth factor; bFGF: basic fibroblast growth factor; IL: interleukin; SDF: stromal cell-derived factor; CXCL12: chemokine, CXC motif, ligand 12; IFN: interferon; HGF: hepatocyte growth factor; MMP: matrix metalloproteinase; TIMP: tissue inhibitor of metalloproteinase; TNF: tumour necrosis factor; MCP: monocyte chemoattractant protein. #: statistical significance not assessed.
The most commonly evaluated marker of fibrosis in the studies above is TGF-β. 11 studies evaluated expression of TGF-β; all of these studies showed that TGF-β was upregulated in the fibrotic state and that pirfenidone treatment significantly reduced TGF-β expression (table 2). TGF-β is the best known inducer of fibrosis, and is known to induce the expression of additional fibrogenic mediators, facilitate myofibroblast differentiation and induce epithelial–mesenchymal transition [68]. Overexpression of TGF-β in transgenic animals or through delivery of adenoviral vectors leads to significant fibrosis in the lung and other organs [68]. Moreover, TGF-β is overexpressed in samples taken from patients with fibrotic conditions such as cirrhosis, glomerulosclerosis, cardiomyopathy, scleroderma and IPF [68, 69]. The central role of TGF-β in fibrosis has fostered efforts for therapeutic modulation of its activity by multiple approaches, including the use of neutralising monoclonal antibodies, inhibition of the associated TGF-βRI (ALK5) receptor tyrosine kinase and modulation of downstream signalling cascades [69]. Inhibitors of TGF-β have been shown to reduce fibrosis in nonclinical models, including models of pulmonary fibrosis [68, 69]. The ability of pirfenidone to modulate TGF-β expression suggests that it can downregulate the plethora of downstream fibrosis-associated effects.
Pirfenidone also modulates the activities of additional fibrogenic mediators including growth factors, chemokines and matrix metalloproteinases (MMPs). Effects on growth factors include downregulation of PDGF and bFGF. PDGF is a potent fibroblast mitogen and chemoattractant. Expression of PDGF is upregulated in samples from patients with fibrotic diseases, including IPF, scleroderma and bronchiolitis obliterans [68]. Moreover, experimental introduction of PDGF (as a recombinant protein or via viral vector) results in pulmonary fibrosis, and inhibitors of PDGF receptor tyrosine kinase activity ameliorate fibrosis in TGF-β-, radiation- and bleomycin-induced animal models. bFGF is a mitogen for several cell types, including fibroblasts, myofibroblasts and smooth muscle cells. Expression of bFGF is upregulated in response to bleomycin and paraquat injury in experimental models, and has been shown to be upregulated in the lungs of patients with IPF and scleroderma [68]. In the bleomycin model, pirfenidone downregulated the production of stromal cell-derived factor (SDF)-1α/CXCL12. This chemokine induces fibrocyte trafficking/recruitment and facilitates pulmonary fibrosis in experimental models, and was shown to be elevated in lung and plasma of IPF patients [21, 70, 71].
MMPs are significantly upregulated in IPF and experimental models of pulmonary fibrosis [72]. The role of these proteins in establishing and/or maintaining a fibroproliferative state is complex and incompletely understood. MMPs are known to degrade ECM proteins, but also have potential roles in several additional processes, including the activation or degradation of fibrogenic growth factors, basement membrane disruption and epithelial cell apoptosis. In models of cardiac and renal fibrosis, pirfenidone normalised production of MMP-2 and MMP-9, respectively (table 2). Both of these proteins are upregulated in patients with IPF and have been implicated in disruption of the basement membrane.
Pirfenidone treatment modulated a series of inflammatory cytokines that have proposed roles in the initiation and maintenance of a fibrotic process. These include TNF-α, which promotes cell recruitment, fibroblast proliferation, epithelial cell hyperplasia and airway epithelial cell apoptosis [11]. Overexpression of TNF-α in rodent lungs leads to inflammation and mild fibrosis [73]. Pirfenidone treatment also downregulated expression of IL-1β, a cytokine that induces fibroblasts to produce fibrogenic mediators such as PDGF and TGF-β [68]. Overexpression of IL-1β in rodent lungs leads to marked inflammation, tissue damage and chronic fibrosis [73]. The fibrogenic T-helper type 2 cytokines IL-4 and IL-13 are downregulated by pirfenidone treatment. IL-4 promotes ECM deposition and overexpression of IL-13 in mice results in increased expression of TGF-β and pulmonary fibrosis [13].
Pirfenidone reduced markers of oxidative stress in models of pulmonary and hepatic fibrosis. Elevated markers of oxidative stress have been identified in patients with fibrotic disorders of the liver, kidney and lung [74]. Reactive oxygen species are implicated in epithelial cell damage and apoptosis, are induced by TGF-β, and are mediators of TGF-β-induced myofibroblast proliferation, ECM production and contractility [74–76]. Evidence of increased oxidative stress has been demonstrated in serum, BAL and exhaled breath condensate of IPF patients [75], and genes associated with oxidative stress are upregulated in lung tissue from patients with progressive IPF relative to those with more stable disease [18, 19]. A clinical study comparing N-acetylcysteine (NAC) in combination with prednisone and azathioprine to prednisone and azathioprine alone in 155 IPF patients showed that the addition of NAC slowed deterioration of vital capacity and carbon monoxide diffusion capacity [77]. The potential role of antioxidant therapy in IPF is being examined further in a study comparing placebo to NAC or a combination of prednisone, azathioprine and NAC [78].
Overall, the broad effects of pirfenidone across several classes of fibrogenic mediators are consistent with the robust antifibrotic effects observed in vivo.
EFFECTS IN CELL-BASED ASSAYS
In addition to the modulation of fibrotic mediators in animal models, multiple activities of pirfenidone that are potentially relevant to its antifibrotic effects have been reported in cell-based assays. The in vitro antifibrotic activities of pirfenidone (table 3) fall into three general categories: reduction of fibroblast and myofibroblast proliferation [79, 84–86], inhibition of extracellular matrix synthesis/deposition [79–83, 86], and reductions in fibrotic markers [81, 82, 84, 85]. Each of these activities is relevant to the attenuation of chronic fibrosis, is consistent with the effects reported for pirfenidone in animal models, and may be relevant to the use of pirfenidone in the treatment of IPF.
Table 3. Antifibrotic effects in cell-based assays.
| Reference | System | Fibroblast proliferation | ECM production | Fibrotic markers |
| Di Sario et al. [79] | Primary rat hepatic stellate cells | Inhibited PDGF-stimulated proliferation | Reduced TGF-β induced collagen I (protein and mRNA) | |
| Ozes and Blatt [80] | Human lung fibroblasts (HFL-1) | Reduced TGF-β induced collagen (protein) | ||
| Sulfab et al. [81] | Human unbilical vein endothelial cells (HUVEC) | Reduced TGF-β stimulated fibronectin, collagen IV, and thrombospondin-2 proteins# | Reduced TGF-β stimulated ET-1, IGFBP-3 and TGF-β isoforms 1-3 | |
| Nakayama et al. [82] | Normal human lung fibroblasts (NHLF) | Reduced TGF-β induced collagen I expression (protein and mRNA) | Reduced TGF-β induced expression of the collagen chaperone HSP47 (protein and mRNA) | |
| Zhang et al. [83] | Human retinal pigment epithelial (RPE) cells | Reduced TGF-β induced fibronectin (protein) production | ||
| Hewitson et al. [84] | Rat renal fibroblast isolated after ureteral obstruction (a mixture of fibroblasts and myofibroblasts) | Reduced fibroblast proliferation | Collagen protein production unaffected (slight reduction) | Reduced α-SMA and CTGF protein expression |
| Lin et al. [85] | Human tenon’s fibroblasts (isolated from eye and expanded) | Reduced serum-stimulated fibroblast proliferation. Increased population of G1-phase cells and decreased S-phase. | Reduced TGF-β1, -β2, and -β3 mRNA and protein expression; reduced fibroblast migration and collagen matrix contraction | |
| Lee et al. [86] | Human myometrial and leiomyoma smooth muscle cells (benign uterine fibroid tumour cells) | Reduced serum-stimulated fibroblast proliferation | Reduced collagen I and III mRNA expression |
Blank boxes denote end-points that were not assessed. All results statistically significant (per reference) with the exception of one treatment related trend (#). ECM: extracellular matrix; PDGF: platelet derived growth factor; TGF: transforming growth factor; ET: endothelin; IGFBP: insulin-like growth factor binding protein; HSP: heat shock protein; SMA: smooth muscle actin; CTGF: connective tissue growth factor; G1-phase: gap 1 phase; S-phase: synthesis phase.
The anti-inflammatory effects of pirfenidone have also been established in cell-based assays (table 4) [87–89]. The most commonly reported effect of pirfenidone in these studies is reduction of TNF-α production. Other affected cytokines include IL-1β, IL-6, and macrophage inflammatory protein-1β. Pirfenidone treatment also increased lipopolysaccharide (LPS)-stimulated production of the anti-inflammatory cytokine IL-10. The effects on cytokines observed in these cell-based assays overlap with those observed in the animal models described above and are further supported by additional LPS-challenge models in vivo [89, 90].
Table 4. Anti-inflammatory effects in cell-based assays.
| Reference | System | Inflammatory markers |
| Grattendick et al. [87] | Human mononuclear cell line (THP-1) | Reduced LPS-induced secreted TNF-α |
| Reduced LPS-induced cell-associated TNF-α | ||
| Phillips et al. [88] | Human peripheral blood mononuclear cells (PBMC) | Decreased LPS-induced TNF-α, IL-1β, IFN-γ and GM-CSF; increased LPS-stimulated IL-10 |
| Reduced constitutive expression of IL-8, IL-6, and MIP-1β | ||
| Nakazato et al. [89] | Mouse macrophage cell line (RAW264.7) | Reduced LPS-induced secreted TNF-α |
| Reduced LPS-induced cell-associated TNF-α |
LPS: lipopolysaccharide; TNF: tumour necrosis factor; IL: interleukin; IFN: interferon; GM-CSF: granulocyte-macrophage colony-stimulating factor; MIP: macrophage inflammatory protein.
A limited number of studies have examined the antioxidant activity of pirfenidone in vitro. Pirfenidone has been reported to reduce the formation of reactive oxygen species by activated neutrophils and macrophages as well as TGF-β-stimulated murine mesangial cells [51, 91, 92]. Pirfenidone also reduced lipid peroxidation in microsomes and biochemical assays [92, 93]. Biochemical experiments to define the mechanism of antioxidant effect have produced equivocal results. Two reports suggest that pirfenidone quenches the hydroxyl radical generated by Fenton chemistry (the reaction of hydrogen peroxide with ferrous iron). However, there are conflicting reports regarding the ability of pirfenidone to complex iron and potentially prevent the formation of hydroxyl radical rather than act as a radical quencher [91, 93]. With respect to the ability to quench superoxide anion, three reports suggest that this is either not an effect of pirfenidone [93], a weak effect [92], or an effect only in complex with iron [91].
The ability of pirfenidone to modulate multiple cell-based activities in vitro, particularly fibroblast proliferation, ECM deposition and the production of fibrogenic proteins, is consistent with the systemic antifibrotic effects observed in vivo. However, the relative contribution of effects observed in vitro to pharmacodynamic effects observed in vivo is not well defined and several activities may play a role. Figure 3 provides a summary of the broad range of final common pathways that are modulated by pirfenidone treatment in animal models and/or cell-based assays. It is likely that a combination of the observed antifibrotic, anti-inflammatory and antioxidant effects is responsible for the reduction in fibrosis observed across a broad range of animal models [35].
Figure 3.

Summary of activities observed in animal models and cell-based assays. ECM: extracellular matrix.
CONCLUSIONS
Pirfenidone is an orally bioavailable small molecule that has been extensively characterised for the treatment of IPF. Clinical investigation of pirfenidone in IPF was initiated based on the broad antifibrotic activity observed in preclinical animal models. This activity is evident in numerous models of fibrosis in the lung, liver, heart and kidney. Several studies demonstrate a relationship between the reduction in fibrosis and improvements in related functional end-points, including improved lung function, improved heart function, reduced risk of cardiac arrhythmia, improved renal clearance and reduction in biochemical markers of liver damage. In these studies, pirfenidone treatment modulated expression of several growth factors and cytokines that are thought to be relevant to fibrosis, including TGF-β, PDGF, SDF-1α/CXCL12, and TNF-α. Supporting data from cell-based assays extend these findings and demonstrate the ability to reduce fibroblast proliferation and modulate ECM deposition. Overall, the preclinical profile of pirfenidone demonstrates systemic antifibrotic effects that support its clinical evaluation in fibrotic diseases.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors gratefully acknowledge H. Oku (Shionogi and Co. Ltd, Osaka, Japan) for providing unpublished bleomycin model data and U. Costabel (Ruhrlandklinik and Medical Faculty, University of Duisburg/Essen, Essen, Germany) for a critical review of the manuscript. The authors also acknowledge A. Cohen, K. Glasscock, S. Lim and F. Weber (InterMune Inc., Brisbane, CA, USA) for review and assistance in preparation of the manuscript.
Footnotes
This article has supplementary material which is available from http//err.ersjournals.com
Provenance
Publication of this peer-reviewed article was supported by InterMune Inc., USA (article sponsor, European Respiratory Review issue 120).
Statement of Interest
All authors are employee shareholders of InterMune, Inc., which recently received European marketing authorisation for Esbriet® (pirfenidone) in the treatment of adult patients with mild to moderate idiopathic pulmonary fibrosis. The article describes unpublished data from the laboratory of H. Oku, an employee of Shionogi and Co. Ltd, which markets pirfenidone for the treatment of idiopathic pulmonary fibrosis in Japan.
REFERENCES
- 1.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304. [DOI] [PubMed] [Google Scholar]
- 2.Lee KW, Everett TH, Rahmutula D, et al. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation 2006; 114: 1703–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen DT, Ding C, Wilson E, et al. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm 2010; 7: 1438–1445. [DOI] [PubMed] [Google Scholar]
- 4.Cho ME, Kopp JB. Pirfenidone: an anti-fibrotic therapy for progressive kidney disease. Expert Opin Investig Drugs 2010; 19: 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelley CJ, Neilson EG. Tubulointerstitial diseases.. In: Brenner BM, ed. The Kidney. 5th Edn. Philadelphia, WB Saunders and Company, 1996; pp. 1655–1679. [Google Scholar]
- 6.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008; 134: 1655–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gadekar SM. N-Substituted Pyridone and General Method for Preparing Pyridones. United States patent, number 3,839,346 (patent); 1974. [Google Scholar]
- 8.Lasky J. Pirfenidone. IDrugs 2004; 7: 166–172. [PubMed] [Google Scholar]
- 9.Margolin SB. Composition for Raparation and Prevention of Fibrotic Lesions. United States patent, number 5,716,632 (patent); 1990. [Google Scholar]
- 10.Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006; 174: 810–816. [DOI] [PubMed] [Google Scholar]
- 11.Noble PW, Homer RJ. Idiopathic pulmonary fibrosis, new insights into pathogenesis. Clin Chest Med 2004; 25: 749–758. [DOI] [PubMed] [Google Scholar]
- 12.du Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov 2010; 9: 129–140. [DOI] [PubMed] [Google Scholar]
- 13.Strieter RM, Mehrad B. New mechanisms of pulmonary fibrosis. Chest 2009; 136: 1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001; 134: 136–151. [DOI] [PubMed] [Google Scholar]
- 15.Vancheri C, Failla M, Crimi N, et al. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J 2010; 35: 496–504. [DOI] [PubMed] [Google Scholar]
- 16.Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc 2006; 3: 364–372. [DOI] [PubMed] [Google Scholar]
- 17.Zuo F, Kaminski N, Eugui E, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA 2002; 99: 6292–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boon K, Bailey NW, Yang J, et al. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS One 2009; 4: e5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selman M, Carrillo G, Estrada A, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One 2007; 2: e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selman M, Pardo A, Barrera L, et al. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med 2006; 173: 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang IV, Burch LH, Steele MP, et al. Gene expression profiling of familial and sporadic interstitial pneumonia. Am J Respir Crit Care Med 2007; 175: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raghu G, Johnson WC, Lockhart D, et al. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: results of a prospective, open-label phase II study. Am J Respir Crit Care Med 1999; 159: 1061–1069. [DOI] [PubMed] [Google Scholar]
- 23.Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2005; 171: 1040–1047. [DOI] [PubMed] [Google Scholar]
- 24.Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2009; 35: 821–829. [DOI] [PubMed] [Google Scholar]
- 25.Noble P, Albera C, Bradford W, et al. The CAPACITY (CAP) trials: randomized, double-blind, placebo-controlled, phase III trials of pirfenidone (PFD) in patients with idiopathic pulmonary fibrosis (IPF). Am J Respir Crit Care Med 2009; 179: A1129. [Google Scholar]
- 26.Spagnolo P, Del Giovane C, Luppi F, et al. Non-steroid agents for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2010; 9: CD003134. [DOI] [PubMed] [Google Scholar]
- 27.Gahl WA, Brantly M, Troendle J, et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky–Pudlak syndrome. Mol Genet Metab 2002; 76: 234–242. [DOI] [PubMed] [Google Scholar]
- 28.Cho ME, Smith DC, Branton MH, et al. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2007; 2: 906–913. [DOI] [PubMed] [Google Scholar]
- 29.Sharma K, Kopp J, Fervenza F, et al. Improvement of renal function with pirfenidone in patients with diabetic nephropathy. NDT Plus 2009; 2: Suppl. 2, ii1514. [Google Scholar]
- 30.Armendariz-Borunda J, Islas-Carbajal MC, Meza-Garcia E, et al. A pilot study in patients with established advanced liver fibrosis using pirfenidone. Gut 2006; 55: 1663–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubino CM, Bhavnani SM, Ambrose PG, et al. Effect of food and antacids on the pharmacokinetics of pirfenidone in older healthy adults. Pulm Pharmacol Ther 2009; 22: 279–285. [DOI] [PubMed] [Google Scholar]
- 32.Bruss ML, Margolin SB, Giri SN. Pharmacokinetics of orally administered pirfenidone in male and female beagles. J Vet Pharmacol Ther 2004; 27: 361–367. [DOI] [PubMed] [Google Scholar]
- 33.Mirkovic S, Seymour AM, Fenning A, et al. Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA-salt hypertensive rats. Br J Pharmacol 2002; 135: 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giri SN, Wang Q, Xie Y, et al. Pharmacokinetics and metabolism of a novel antifibrotic drug pirfenidone, in mice following intravenous administration. Biopharm Drug Dispos 2002; 23: 203–211. [DOI] [PubMed] [Google Scholar]
- 35.Azuma A. Pirfenidone: antifibrotic agent for idiopathic pulmonary fibrosis. Expert Rev Respir Med 2010; 4: 301–310. [DOI] [PubMed] [Google Scholar]
- 36.Moeller A, Ask K, Warburton D, et al. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol 2008; 40: 362–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chua F, Gauldie J, Laurent GJ. Pulmonary fibrosis: searching for model answers. Am J Respir Cell Mol Biol 2005; 33: 9–13. [DOI] [PubMed] [Google Scholar]
- 38.Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol 2008; 590: 400–408. [DOI] [PubMed] [Google Scholar]
- 39.Kakugawa T, Mukae H, Hayashi T, et al. Pirfenidone attenuates expression of HSP47 in murine bleomycin-induced pulmonary fibrosis. Eur Respir J 2004; 24: 57–65. [DOI] [PubMed] [Google Scholar]
- 40.Iyer SN, Wild JS, Schiedt MJ, et al. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med 1995; 125: 779–785. [PubMed] [Google Scholar]
- 41.Iyer SN, Margolin S, Hyde DM, et al. Lung fibrosis is ameliorated by pirfenidone fed in diet after the second dose in a three-dose bleomycin-hamster model. Exp Lung Res 1998; 24: 119–132. [DOI] [PubMed] [Google Scholar]
- 42.Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on procollagen gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther 1999; 289: 211–218. [PubMed] [Google Scholar]
- 43.Gurujeyalakshmi G, Hollinger MA, Giri SN. Pirfenidone inhibits PDGF isoforms in bleomycin hamster model of lung fibrosis at the translational level. Am J Physiol Lung Cell Mol Physiol 1999; 276: L311–L318. [DOI] [PubMed] [Google Scholar]
- 44.Schelegle ES, Mansoor JK, Giri S. Pirfenidone attenuates bleomycin-induced changes in pulmonary functions in hamsters. Proc Soc Exp Biol Med 1997; 216: 392–397. [DOI] [PubMed] [Google Scholar]
- 45.Liu H, Drew P, Gaugler AC, et al. Pirfenidone inhibits lung allograft fibrosis through l-arginine-arginase pathway. Am J Transplant 2005; 5: 1256–1263. [DOI] [PubMed] [Google Scholar]
- 46.Zhou H, Latham CW, Zander DS, et al. Pirfenidone inhibits obliterative airway disease in mouse tracheal allografts. J Heart Lung Transplant 2005; 24: 1577–1585. [DOI] [PubMed] [Google Scholar]
- 47.Hirano A, Kanehiro A, Ono K, et al. Pirfenidone modulates airway responsiveness, inflammation, and remodeling after repeated challenge. Am J Respir Cell Mol Biol 2006; 35: 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimizu T, Kuroda T, Hata S, et al. Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int 1998; 54: 99–109. [DOI] [PubMed] [Google Scholar]
- 49.Takakura K, Fujimori A, Chikanishi T, et al. Renoprotective properties of pirfenidone in subtotally nephrectomized rats. Eur J Pharmacol 2009; 629: 118–124. [DOI] [PubMed] [Google Scholar]
- 50.Shimizu T, Fukagawa M, Kuroda T, et al. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int Suppl 1997; 63: S239–S243. [PubMed] [Google Scholar]
- 51.RamachandraRao SP, Zhu Y, Ravasi T, et al. Pirfenidone is renoprotective in diabetic kidney disease. J Am Soc Nephrol 2009; 20: 1765–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salazar-Montes A, Ruiz-Corro L, Lopez-Reyes A, et al. Potent antioxidant role of pirfenidone in experimental cirrhosis. Eur J Pharmacol 2008; 595: 69–77. [DOI] [PubMed] [Google Scholar]
- 53.Garcia L, Hernandez I, Sandoval A, et al. Pirfenidone effectively reverses experimental liver fibrosis. J Hepatol 2002; 37: 797–805. [DOI] [PubMed] [Google Scholar]
- 54.Di Sario A, Bendia E, Macarri G, et al. The anti-fibrotic effect of pirfenidone in rat liver fibrosis is mediated by downregulation of procollagen alpha-I, TIMP-1 and MMP-2. Dig Liver Dis 2004; 36: 744–751. [DOI] [PubMed] [Google Scholar]
- 55.Tada S, Nakamuta M, Enjoji M, et al. Pirfenidone inhibits dimethylnitrosamine-induced hepatic fibrosis in rats. Clin Exp Pharmacol Physiol 2001; 28: 522–527. [DOI] [PubMed] [Google Scholar]
- 56.Chaudhary NI, Schnapp A, Park JE. Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am J Respir Crit Care Med 2006; 173: 769–776. [DOI] [PubMed] [Google Scholar]
- 57.Tamagawa K, Taooka Y, Maeda A, et al. Inhibitory effects of a lecithinized superoxide dismutase on bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med 2000; 161: 1279–1284. [DOI] [PubMed] [Google Scholar]
- 58.Phan SH, Thrall RS, Williams C. Bleomycin-induced pulmonary fibrosis. Effects of steroid on lung collagen metabolism. Am Rev Respir Dis 1981; 124: 428–434. [DOI] [PubMed] [Google Scholar]
- 59.Everett TH, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm 2007; 4: S24–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res 2010; 106: 1675–1680. [DOI] [PubMed] [Google Scholar]
- 61.Ito Y, Aten J, Bende RJ, et al. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 1998; 53: 853–861. [DOI] [PubMed] [Google Scholar]
- 62.Cohen EP. Fibrosis causes progressive kidney failure. Med Hypotheses 1995; 45: 459–462. [DOI] [PubMed] [Google Scholar]
- 63.Eddy AA. Molecular insights into renal interstitial fibrosis. J Am Soc Nephrol 1996; 7: 2495–2508. [DOI] [PubMed] [Google Scholar]
- 64.Brook NR, Waller JR, Bicknell GR, et al. The novel antifibrotic agent pirfenidone attenuates the profibrotic environment generated by calcineurin inhibitors in the rat salt-depletion model. Transplant Proc 2005; 37: 130–133. [DOI] [PubMed] [Google Scholar]
- 65.Brook NR, Waller JR, Bicknell GR, et al. The experimental agent pirfenidone reduces pro-fibrotic gene expression in a model of tacrolimus-induced nephrotoxicity. J Surg Res 2005; 125: 137–143. [DOI] [PubMed] [Google Scholar]
- 66.Al-Bayati MA, Xie Y, Mohr FC, et al. Effect of pirfenidone against vanadate-induced kidney fibrosis in rats. Biochem Pharmacol 2002; 64: 517–525. [DOI] [PubMed] [Google Scholar]
- 67.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000; 275: 2247–2250. [DOI] [PubMed] [Google Scholar]
- 68.Kolb M, Xing Z, Gauldie J. Growth factors.. In: Barnes P, Drazen J, Rennard S, et al., eds. Asthma and COPD. London, Academic Press, 2002; pp. 283–289 [Google Scholar]
- 69.Prud’homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest 2007; 87: 1077–1091. [DOI] [PubMed] [Google Scholar]
- 70.Phillips RJ, Burdick MD, Hong K, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 2004; 114: 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andersson-Sjoland A, de Alba CG, Nihlberg K, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol 2008; 40: 2129–2140. [DOI] [PubMed] [Google Scholar]
- 72.Lagente V, Boichot E, Pardo A, et al. Role of matrix metalloproteases in pulmonary fibrosis. In: Parnham MJ, ed. Matrix Metalloproteinases in Tissue Remodelling and Inflammation. Basle, Birkhäuser Basel, 2008; pp. 39–55 [Google Scholar]
- 73.Kelly M, Kolb M, Bonniaud P, et al. Re-evaluation of fibrogenic cytokines in lung fibrosis. Curr Pharm Des 2003; 9: 39–49. [DOI] [PubMed] [Google Scholar]
- 74.Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Radic Biol Med 2010; 48: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bargagli E, Olivieri C, Bennett D, et al. Oxidative stress in the pathogenesis of diffuse lung diseases: a review. Respir Med 2009; 103: 1245–1256. [DOI] [PubMed] [Google Scholar]
- 76.Hecker L, Vittal R, Jones T, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 2009; 15: 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Demedts M, Behr J, Buhl R, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2005; 353: 2229–2242. [DOI] [PubMed] [Google Scholar]
- 78.Klingsberg RC, Mutsaers SE, Lasky JA. Current clinical trials for the treatment of idiopathic pulmonary fibrosis. Respirology 2010; 15: 19–31. [DOI] [PubMed] [Google Scholar]
- 79.Di Sario A, Bendia E, Svegliati Baroni G, et al. Effect of pirfenidone on rat hepatic stellate cell proliferation and collagen production. J Hepatol 2002; 37: 584–591. [DOI] [PubMed] [Google Scholar]
- 80.Ozes ON, Blatt LM. Development of a high throughput collagen assay using a cellular model of idiopathic pulmonary fibrosis. Chest 2006; 130: 230S. [Google Scholar]
- 81.Sulfab M, Blatt LM, Ozes ON. The effects of pirfenidone and interferon-inducible T-cell alpha chemoattractant on transforming growth factor-β1-mediated synthesis of extracellular matrix proteins in endothelial cells. Am J Respir Crit Care Med 2007; 175: A730. [Google Scholar]
- 82.Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-β1-stimulated human lung fibroblasts. Life Sci 2008; 82: 210–217. [DOI] [PubMed] [Google Scholar]
- 83.Zhang S, Shiels IA, Ambler JS, et al. Pirfenidone reduces fibronectin synthesis by cultured human retinal pigment epithelial cells. Aust NZ J Ophthalmol 1998; 26: S74–S76. [DOI] [PubMed] [Google Scholar]
- 84.Hewitson TD, Kelynack KJ, Tait MG, et al. Pirfenidone reduces in vitro rat renal fibroblast activation and mitogenesis. J Nephrol 2001; 14: 453–460. [PubMed] [Google Scholar]
- 85.Lin X, Yu M, Wu K, et al. Effects of pirfenidone on proliferation, migration, and collagen contraction of human Tenon’s fibroblasts in vitro. Invest Ophthalmol Vis Sci 2009; 50: 3763–3770. [DOI] [PubMed] [Google Scholar]
- 86.Lee B-S, Margolin S, Nowak R. Pirfenidone: a novel pharmacological agent that inhibits leiomyoma cell proliferation and collagen production. J Clin Endocrinol Metabol 1998; 83: 219–223. [DOI] [PubMed] [Google Scholar]
- 87.Grattendick KJ, Nakashima JM, Feng L, et al. Effects of three anti-TNF-α drugs: etanercept, infliximab and pirfenidone on release of TNF-α in medium and TNF-α associated with the cell in vitro. Int Immunopharmacol 2008; 8: 679–687. [DOI] [PubMed] [Google Scholar]
- 88.Phillips R, Wang T, Blatt LM, et al. Pirfenidone mediates differential effects on lipopolysaccharide-induced cytokine expression in human peripheral mononuclear cells. Chest 2005; 128: 169S. [Google Scholar]
- 89.Nakazato H, Oku H, Yamane S, et al. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-α at the translational level. Eur J Pharmacol 2002; 446: 177–185. [DOI] [PubMed] [Google Scholar]
- 90.Oku H, Nakazato H, Horikawa T, et al. Pirfenidone suppresses tumor necrosis factor-α, enhances interleukin-10 and protects mice from endotoxic shock. Eur J Pharmacol 2002; 446: 167–176. [DOI] [PubMed] [Google Scholar]
- 91.Mitani Y, Sato K, Muramoto Y, et al. Superoxide scavenging activity of pirfenidone-iron complex. Biochem Biophys Res Commun 2008; 372: 19–23. [DOI] [PubMed] [Google Scholar]
- 92.Giri SN, Leonard S, Shi X, et al. Effects of pirfenidone on the generation of reactive oxygen species in vitro. J Environ Pathol Toxicol Oncol 1999; 18: 169–177. [PubMed] [Google Scholar]
- 93.Misra HP, Rabideau C. Pirfenidone inhibits NADPH-dependent microsomal lipid peroxidation and scavenges hydroxyl radicals. Mol Cell Biochem 2000; 204: 119–126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.