

Abstract
Pulmonary alveolar proteinosis (PAP) is a rare pulmonary disease characterised by alveolar accumulation of surfactant. It may result from mutations in surfactant proteins or granulocyte macrophage-colony stimulating factor (GM-CSF) receptor genes, it may be secondary to toxic inhalation or haematological disorders, or it may be auto-immune, with anti-GM-CSF antibodies blocking activation of alveolar macrophages. Auto-immune alveolar proteinosis is the most frequent form of PAP, representing 90% of cases. Although not specific, high-resolution computed tomography shows a characteristic “crazy paving” pattern. In most cases, bronchoalveolar lavage findings establish the diagnosis. Whole lung lavage is the most effective therapy, especially for auto-immune disease. Novel therapies targeting alveolar macrophages (recombinant GM-CSF therapy) or anti-GM-CSF antibodies (rituximab and plasmapheresis) are being investigated. Our knowledge of the pathophysiology of PAP has improved in the past 20 yrs, but therapy for PAP still needs improvement.
Keywords: Granulocyte macrophage-colony stimulating factor, macrophage, pulmonary lavage, rituximab, surfactant
Pulmonary alveolar proteinosis (PAP) is a rare disease characterised by alveolar accumulation of surfactant composed of proteins and lipids due to defective surfactant clearance by alveolar macrophages. Diagnosis of PAP is initiated by computed tomography (CT) scan and confirmed by staining of bronchoalveolar lavage fluid (BALF). Diagnosis of PAP rarely requires lung biopsy. Three main categories of PAP have been defined depending on the aetiology: auto-immune (previously named primary or idiopathic), secondary and genetic. Genetic PAP is seen especially in children, and radio-clinical presentation depends on the mutated gene. Adult forms are mostly auto-immune (with anti-granulocyte macrophage-colony stimulating factor (GM-CSF) antibodies) and/or secondary to toxic inhalation or haematological disorders, without anti-GM-CSF antibodies.
This review summarises our knowledge of the current pathophysiology of PAP and its three forms.
PATHOPHYSIOLOGY
Surfactant
Surfactant consists of a mixture of proteins and lipids (mostly phosphatidylcholine) secreted by type II pneumocytes. The four main surfactant proteins are SP-A, -B, -C and -D and the corresponding genes SFTPA, B, C and D [1]. Surfactant is cleared by type II pneumocytes and alveolar macrophages. Surfactant decreases alveolar surface tension and prevents end-expiratory alveolar collapse. Surfactant is also involved in the alveolar anti-infectious defence. Mutations of surfactant protein genes are responsible for quantitative and qualitative alterations of secreted surfactant, thus leading to ineffective surfactant and abnormal clearance. Non-secreted proteins accumulate in type II pneumocytes and are toxic to the cell. Cellular lesions may be involved in the pathophysiology of interstitial pneumonia [2].
Abnormalities of surfactant clearance
GM-CSF is a key cytokine in PAP pathophysiology. In vitro, GM-CSF, a growth factor for granulocytes and monocytes, stimulates differentiation, proliferation and survival of myeloid cells: monocytes, neutrophils and dendritic cells [3, 4]. GM-CSF knockout (KO) mice show isolated lung lesions reminiscent of PAP seen in humans [4], probably secondary to defective clearance of surfactant by alveolar macrophages. PU.1 is the key transcription factor controlling the expression of GM-CSF in mouse alveolar macrophages. PU.1 KO mice exhibit PAP, and re-expression of PU.1 prevents the development of PAP [5, 6].
The human receptor of GM-CSF (CSFR) has two subunits, α and β. The mutations of CSF2RA and CSF2RB, the coding genes of the α and β subunits, prevent GM-CSF signalling and induce PAP with intra-alveolar surfactant accumulation without interstitial infiltration. In these patients, alveolar and serum concentrations of GM-CSF are elevated and anti-GM-CSF antibodies are absent [7, 8]. In PAP secondary to haematological disorders, alveolar macrophages are thought to be quantitatively or functionally unable to clear the surfactant. A transient defect of expression of the GM-CSF receptor on macrophages has been described in three patients with PAP secondary to acute myeloid leukaemia, with those macrophages being hyporeactive after stimulation with GM-CSF or interleukin (IL)-3 [9]. After therapy for leukaemia, the receptor expression returned to normal [9].
Auto-immune PAP specifically shows a high concentration of neutralising anti-GM-CSF immunoglobulin (Ig)G antibodies [10, 11]. Anti-GM-CSF antibodies bind GM-CSF with high affinity, thus blocking its activity [12]. Because of reduced or absent GM-CSF stimulation, alveolar macrophages are unable to clear the surfactant and are also less efficient for anti-infectious defence [13, 14]. Furthermore, neutrophils and lymphocytes are functionally modified, which explains some of the opportunistic infections occurring during PAP [15]. Anti-GM-CSF antibodies are thought to be pathogenic as, in vitro, anti-GM-CSF antibodies reproduce on myeloid cells from healthy subjects, abnormalities seen on myeloid cells of PAP patients [12], and, in vivo, perfusion of human anti-GM-CSF antibodies in a non-human primate induced PAP indistinguishable from human PAP, with diminished mRNA expression of PU-1 and peroxisome proliferator-activated receptor-γ, as well as blood neutrophil dysfunction [16]. Renaming idiopathic PAP to auto-immune PAP has been suggested.
AUTO-IMMUNE PAP
Epidemiology
Auto-immune PAP represents ∼90% of all PAP cases [17, 18]. The prevalence varies among countries, from four to 40 cases per million, and the incidence is estimated at almost 0.2 cases per million. Three main studies are available to evaluate the epidemiology and evolution of the disease. In 2001, in an exhaustive review of the literature, Seymour and Presneill [13] found descriptions of 410 patients. In France, a 2002 GERMOP retrospective study described 41 patients [19]. Finally, a report of a large cohort of 248 Japanese patients was published in 2008 [18]. Although PAP is auto-immune in 90% of cases, auto-immune PAP is rarely associated with another auto-immune disease. Indeed in the study by Seymour and Presneill [13], only seven (1.7%) out of 410 patients had another auto-immune disease.
In Japan, PAP is distributed equally among the regions, with a 2:1 ratio of males to females. The mean age of patients is 51 yrs, but PAP may occur from newborns to 72 yr olds [19]. About half of the patients (56%) are smokers, and 85% of the male patients are smokers, which may explain the 2:1 sex ratio. Indeed, the sex ratio is 1:1 in nonsmoker patients [13, 18].
Clinical presentation
Symptoms are not specific, and almost one-third of patients are asymptomatic [18]. Dyspnoea is present in 39% of cases and cough, productive or not, in 21% [18, 19]. Chest pain, loss of weight, fatigue and fever are rare. Haemoptysis is rare, of mild volume and should suggest pulmonary infection [19].
Clinical examination results are often normal. Cyanosis or digital clubbing may be present in up to 30% of cases, and auscultation may reveal crackles [19]. Older patients could present the most severe form of PAP.
Complementary examinations
Chest radiography
Chest radiography reveals symmetric, bilateral alveolar opacities, without air bronchogram (fig. 1), showing a peri-hilar and basal distribution. Opacities are more rarely asymmetric or with an apical predominance. The pattern resembles acute pulmonary oedema; however, cardiomegaly and pleural effusion are absent [17, 19]. The level of opacities and symptoms are often discrepant [13]. In less severe forms opacities might appear as ground-glass opacities.
Figure 1.

Chest radiograph of a patient with auto-immune pulmonary alveolar proteinosis showing diffuse alveolar opacities with a peri-hilar and basal distribution.
Chest CT scan
A chest CT scan is a major tool in the diagnosis of PAP. Indeed, the pattern observed is highly suggestive of the disease, although not pathognomonic, and should prompt the specific evaluation of BALF. Major abnormalities are ground-glass opacities, septal reticulations and parenchymal consolidation (fig. 2). Reticulations are frequently superimposed on ground-glass opacities, thus forming a “crazy paving” pattern characteristic of PAP. Opacities have a typically geographic distribution, with juxtaposition of healthy and sick zones. The zonal distribution is usually not specific; however, lower zone predominance might be present in 22% of cases [20]. Large focal parenchymal consolidation is rare and should lead to the search for an opportunistic infection [20]. Mediastinal adenopathy and pulmonary nodules are absent [20].
Figure 2.
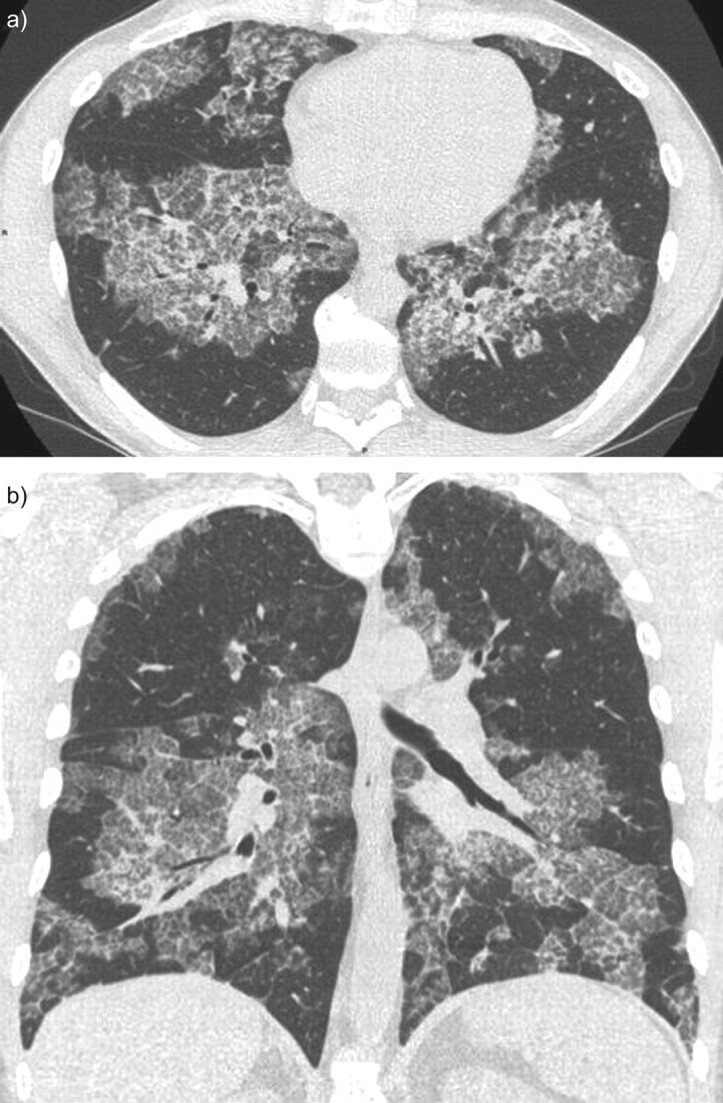
Chest computed tomography scan of a patient with auto-immune pulmonary alveolar proteinosis. Reticulations are superimposed on ground-glass opacities forming a “crazy paving” pattern with a geographic distribution: juxtaposition of healthy and sick zones. a) A horizontal section and b) a coronal section.
Correlation of histology and radiology findings reveals that ground-glass opacities correspond to a lipoproteinaceous alveolar accumulation. Correlations with reticulation are less unequivocal and could correspond to interstitial disease (lipoproteinaceous interstitial accumulation, inflammation or oedema) or lipoproteinaceous alveolar accumulation on the edges of the lobules [21].
A crazy paving pattern is suggestive of, but not specific, to PAP. A crazy paving pattern might be associated with lesional or cardiogenic pulmonary oedema, alveolar haemorrhage, pulmonary infection (mycoplasma, pneumocystis), exogenous lipoid pneumonia or bronchioloalveolar carcinoma [21].
The extent of opacities seen on CT scan seems to be associated with impaired pulmonary function on testing (fig. 2) [22].
BALF
BALF staining is required for the diagnosis of PAP [23]. When performed in a diseased lung area, BALF typically has a milky appearance (fig. 3) but might appear abnormal or normal if performed in a healthy zone with a weak amount of lipoproteinaceous material.
Figure 3.

Bronchoalveolar lavage fluid with a milky appearance in a) normal saline compared to b) pulmonary alveolar proteinosis.
Cytological examination and periodic acid-Schiff (PAS) staining are mandatory for diagnosis. In one study, BAL cellularity was increased (330,000 cells·mL−1) with increased proportion of lymphocytes (mean 57%) [24]. Careful examination reveals large, foamy macrophages containing eosinophilic granules, with extracellular globular hyaline material found homogeneously positive on PAS and negative on Alcian blue staining (fig. 4).
Figure 4.

Periodic acid-Schiff (PAS) staining of bronchoalveolar lavage fluid with pulmonary alveolar proteinosis showing extracellular globular hyaline material homogeneously PAS positive with large, foamy macrophages containing eosinophilic granules. Magnification 400x.
If performed, ultrastructural analysis of BALF reveals numerous lamellar bodies with a structural resemblance to myelin (fig. 5) [13].
Figure 5.
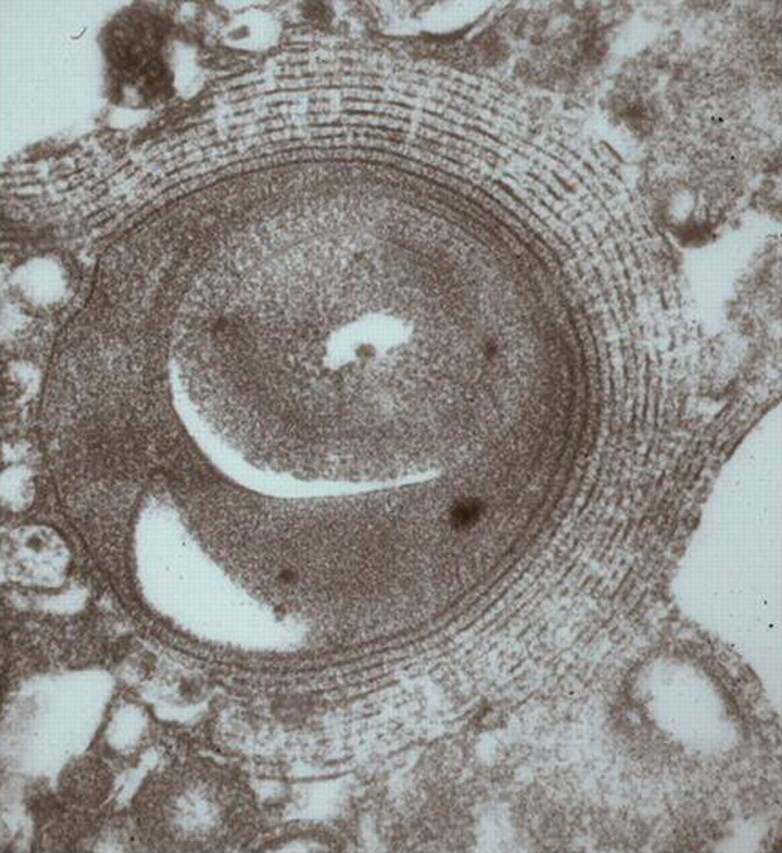
Ultrastructural analysis of bronchoalveolar lavage fluid with pulmonary alveolar proteinosis showing numerous lamellar bodies.
Anti-GM-CSF antibodies
Anti-GM-CSF antibodies are specific to auto-immune PAP. Two methods of dosages are available, an ELISA (the gold standard) and a functional assay which uses the capacity of serum containing anti-GM-CSF antibodies to inhibit the proliferation of the TF1 cell line, a cell line very sensitive to GM-CSF activity [16]. Anti-GM-CSF antibodies may be detected at low concentration in healthy subjects and could participate in the regulation of myeloid cells [25]. Anti-GM-CSF antibodies of IgG, IgA or IgM isotypes have been detected in sera from patients with acute myeloid leukaemia, with their concentration being associated with disease activity [26]. A concentration >19 μg·mL−1 is specific to auto-immune PAP, and a concentration <10 μg·mL−1 has a good negative predictive value [27].
Other biological examinations
Results of routine biological tests are normal. Increased serum lactate dehydrogenase (LDH) may be elevated in half of the cases and then between two and three times the normal range in half of the cases. The serum levels of carcinoembryonic antigen and KL-6 could be higher than those for other diffuse interstitial pneumonia and could be associated with disease severity [18]. However, these markers are not useful for diagnosis and do not help in the therapeutic decision.
The serum levels of the surfactant proteins SP-A, -B and -D are increased and could be associated with disease severity [18]. However, levels of SP-A and -B do not change with therapy [28]. In contrast, the level of SP-D in nonhuman primates was helpful in screening PAP development after injection of anti-GM-CSF antibodies. Determining the level of SP-D could help monitor human disease [16].
Pulmonary function tests and exercise capacity
Pulmonary function and exercise capacity tests are important for therapeutic decisions. Spirometry frequently shows a restrictive pattern but may give normal results in 10–30% of cases. Smoking patients may show an obstructive pattern. The most constant and significant modifications are hypoxaemia and reduced diffusing capacity of the lung for carbon monoxide (DL,CO) (40–50%) with increased alveolar–arterial gradient [17, 19]. Therapy may be introduced in patients with desaturation results on the 6-min walking test.
Open lung biopsy
Special attention to BALF, associated with clinical and CT-scan typical presentation, is often sufficient for diagnosis and open-lung biopsy is not necessary for diagnosis [17]. Transbronchial biopsy may be helpful. In the Japanese cohort of 203 auto-immune PAP, open-lung biopsy was performed for diagnosis in 8% of cases and transbronchial biopsy in 42% of cases [18].
Prognosis and evolution
From spontaneous remission to death, disease evolution is unpredictable. Indeed, among the 39 asymptomatic, untreated patients from the Japanese cohort (17%), 11 (28%) showed spontaneous improvement, three (7%) had worsened disease and symptoms, and 25 (64%) had stable disease. In the French cohort of 41 patients, 10 out of the 15 untreated patients showed spontaneous improvement [18, 19]. However, among 11 patients with spontaneous improvement in the pre-inclusion phase of a therapeutic trial, nine patients secondarily worsened [29]. Since the wide use of therapeutic lavage, 5-yr survival with auto-immune PAP is almost 95% [18].
Approximately 5% of patients with auto-immune PAP, mostly untreated patients, may present with opportunistic infections. Pulmonary infection with Aspergillus, mycobacteria and Nocardia has been reported. Furthermore, pulmonary fibrosis secondary to PAP was reported before the description of anti-GM-CSF antibodies and genetic PAP. Indeed fibrosis secondary to PAP could be genetic PAP and not be associated with anti-GM-CSF antibodies. Non-haematological cancers have been reported, but these associations may be a coincidence [18].
Therapy
The standard of care is symptomatic whole lung lavage. Numerous therapies targeting an enhancement of the surfactant clearance have been investigated either targeting alveolar macrophages with exogenous GM-CSF or aiming at reducing levels of anti-GM-CSF antibodies with plasmapheresis or rituximab.
Whole lung lavage
Numerous techniques have been reported. The first method reported in 1961 is no longer used. Saline was blindly injected through a percutaneous transtracheal endobronchial catheter. The fluid was re-aspirated through the catheter and evacuated by violent coughing [30]. Classical therapeutic bronchoalveolar lavage is performed under general anaesthesia in an operating room or an intensive care unit. The patient is intubated with a double-lumen endotracheal tube and fibre-optic bronchoscopy is performed to confirm the appropriate tube placement. The patient under curarisation is placed in the dorsal or lateral decubitus position, with the lung being lavaged in the uppermost position. The non-lavaged lung is mechanically ventilated. 1 L of warmed (37°C) saline is injected in the lung. Fluid is then collected by gravity after opening the outflow tube. Manual or mechanical chest percussion might be performed to improve drainage. The process is repeated until the fluid becomes less opaque; 15 L of saline are generally necessary. The patient is extubated a few hours later depending on the clinical evolution. The controlateral lung may be lavaged 24–48 h later [19, 31]. The most frequent complications are low oxygen saturation, convulsions, pneumothorax, pleural effusion and fever, which may reveal infection. Retrospective data suggest that whole lung lavage could improve survival [13]. In 85% of cases, patients show symptomatic, radiographic and functional improvement after whole lung lavage: a mean improvement in forced expiratory volume in 1 s of 0.26 L, in vital capacity of 0.5 L, in DL,CO of 4.4 mL·mmHg−1·min−1 and in arterial oxygen tension (Pa,O2) of 20 mmHg, and a mean reduction of alveolar–arterial gradient of 30 mmHg. One procedure is enough for half the patients, which suggests that presence of anti-GM-CSF antibodies is not sufficient to maintain the disease. The other half of the patients will need to repeat the whole lung lavage, on average only one [13].
Successful lobar lavage with fibre-optic bronchoscopy under local anaesthesia for patients with less severe disease has been reported [19, 32, 33].
GM-CSF supplemental therapy
GM-CSF (Sargramostim®; Bayer AG, Leverkusen, Germany) may be inhaled or subcutaneously administered [34, 35]. In an open, uncontrolled prospective study 25 patients received 250 μg GM-CSF subcutaneously once a day [36]. If the drug was well tolerated, the posology could be increased to 9 μg·kg−1·day−1. After 9 months, if the treatment was well tolerated, the dose could be increased up to 18 μg·kg−1·day−1. 12 (48%) patients showed improvement with therapy. Side-effects were considered minor and included injection-site oedema, erythema, malaise and shortness of breath. In the GM-CSF responder group improvement was slower than after whole lung lavage. Improvement of Pa,O2 could be of the same magnitude with both therapies, with an increase of 12–19 mmHg with whole lung lavage [37, 38] and a mean increase of 23 mmHg with GM-CSF therapy in GM-CSF-responder patients, and 9.7 mmHg for all GM-CSF patients [34]. Factors associated with response to subcutaneous GM-CSF therapy were hypereosinophilia under therapy, longer delay since diagnosis, higher vital capacity, normal serum LDH concentration and increased serum SP-B concentration [34].
GM-CSF could be more effective when inhaled. Indeed, a retrospective study reported on 12 patients treated with inhaled GM-CSF, 250 μg·12 h−1 in 2 mL of saline; 11 (91%) patients showed improvement [39]. Three other patients described in case reports showed improvement with inhaled GM-CSF [40, 41]. Results of a Japanese prospective study were recently published [29]. The study design involved an induction period and a maintenance period. In the induction period of six 2-week cycles, patients inhaled 125 μg GM-CSF twice a day from day 1 to day 8; a new cycle began on day 14. In the maintenance period of six 2-week cycles, patients received 125 μg GM-CSF once a day from day 1 to day 4; a new cycle began on day 12. Of the 35 treated patients, 24 (68%) showed improvement, 17 in the induction period and seven more in the maintenance period. Serum anti-GM-CSF antibody concentrations did not change with therapy. Two patients experienced severe adverse events that may not have been attributable to GM-CSF due to pulmonary infection and pulmonary tuberculosis.
GM-CSF therapy is now considered an alternative to whole lung lavage. However, Sargramostim® (Bayer) was withdrawn from several national markets and may require specific authorisation from national health authorities according to each country. No clinical or biological marker exists to predict response to GM-CSF and to select patients that could benefit from GM-CSF therapy. Indeed, concentration of anti-GM-CSF antibody and evolution under therapy do not seem to be associated with response [13, 34, 36].
Rituximab and plasmapheresis
Immunosuppressive therapy, particularly corticosteroids, does not seem to be effective in PAP [13] and could increase the risk of pulmonary infection. Theoretically, plasmapheresis should be effective to decrease the concentration of anti-GM-CSF antibodies and improve the disease as in Goodpasture disease [42]. Only two cases have been reported in the literature: a decrease of anti-GM-CSF antibodies was measured in both cases, and one patient showed a striking improvement and the other a mild improvement [43, 44].
Rituximab, a monoclonal antibody directed against the CD20 antigen of B-lymphocytes, could ameliorate PAP by decreasing anti-GM-CSF antibody concentration. Preliminary results of a prospective monocentric open-label study were presented at the 2010 American Thoracic Society meeting [45]; eight out of the 10 treated patients showed improvement with rituximab without modification of serum anti-GM-CSF antibody concentration. Borie et al. [46] described a patient who refused whole lung lavage and exhibited clinical, functional and radiographic improvement 6 months after rituximab therapy. Another group described a patient who was resistant to whole lung lavage but showed marked improvement with rituximab therapy [47]. Rituximab was well tolerated in all patients.
Although the exact place of these treatments is not well defined at present, rituximab therapy could be an alternative for whole lung lavage resistant disease.
SECONDARY PAP
Secondary PAP includes PAP secondary to cancer and particularly haematological diseases and secondary to toxic inhalation.
Immune deficiency
PAP has been rarely associated with immune deficiency, including severe combined immunodeficiency, agammaglobulinaemia [48] or organ transplantation [13]. A few cases of connective tissue diseases have been reported: dermatomyositis [49], rheumatoid arthritis [50] and Behcet's disease [51, 52]. One study suggested that pulmonary pneumocystosis secondary to HIV infection could induce PAP [53]. However, this association has not been reported again. Most patients seem to improve with whole lung lavage.
Cancer
The association of PAP and haematological disorders is well established, mostly myelodysplastic syndromes and acute myeloid leukaemia [52]. PAP could explain up to 10% of pulmonary manifestations during these diseases [54]. Less frequently, PAP has been associated with acute lymphoid leukaemia [55], lymphoma [56] and myeloma [57]. In haematological diseases, alveolar macrophages could be numerically or functionally unable to clear the surfactant.
PAP is generally diagnosed during the evolution of haematological disease and may occur in the absence of detectable tumour after bone marrow transplantation [58]. The diagnosis of PAP and haematological disease may occur simultaneously; in that case, the diagnosis of the haematological disease is generally easy [52, 59].
The classical presentation is respiratory insufficiency with fever in 24% of the cases, particularly in patients with prolonged neutropenia secondary to chemotherapy [54]. Chest radiographs shows diffuse interstitial opacities [54]. Chest CT scans may show the typical crazy paving pattern. CT scans may also be less suggestive by showing ground-glass opacities mainly in the lower zone without subpleural sparing or consolidations (fig. 6) [56, 60]. Fibre-optic bronchoscopy is diagnostic. Interestingly, BALF analysis may reveal PAP and an associated opportunistic infection. Pulmonary infection with Nocardia, Pneumocystis, Acinetobacter, Aspergillus, Cladosporium, or Mycobacerium tuberculosis or non-tuberculosis mycobacteria have been associated with PAP [56, 61]. Positive PAS staining of intra- and extracellular material is sufficient for the diagnosis of PAP and may negate a lung biopsy or autopsy [54, 62]. Indeed, in numerous case reports a diagnosis of PAP is obtained only by autopsy [57].
Figure 6.
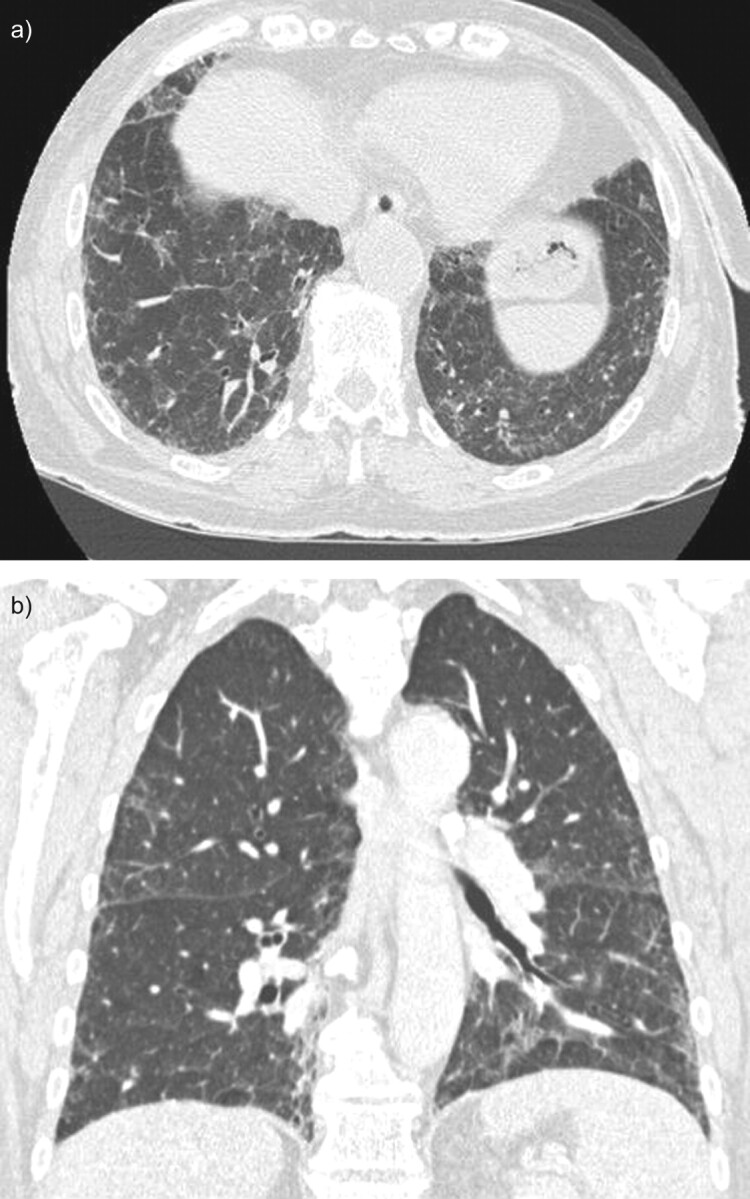
Chest computed tomography (CT) scan of a patient with pulmonary alveolar proteinosis secondary to myelodysplastic syndrome. The CT scan shows ground-glass opacities and reticulations mainly in the lower zone with patchy, slightly peripheral distribution. a) A horizontal section and b) coronal section.
The prognosis is poor and the median time of survival is 16 months. In a Japanese study the causes of death were haematological disease (33%), infection (25%), respiratory insufficiency (25%) and unspecified bleeding (13%). No reported series has specifically evaluated haematological therapies and whole lung lavage. Only two out of the 14 patients who benefited from whole lung lavage showed improvement. Four patients showed improvement after transplantation (allogenic bone marrow n = 3, lung n = 1) [6, 63].
Haematological therapy alone, chemotherapy and/or bone-marrow transplantation can cure PAP, particularly in cases of acute leukaemia [54]. Some patients with respiratory insufficiency may show symptomatic improvement with whole lung lavage [64]. Furthermore, a patient with myeloproliferative syndrome with myelodysplasia showed decreased white blood cell count after whole lung lavage [59]. Regardless, the haematological disease must be controlled [59, 63]. Double symptomatic and aetiological therapy is probably justified, and numerous cases of recovery have been reported after this double therapy [65].
PAP may be associated with non-haematological cancers, most frequently lung cancers (squamous cell cancer n = 2, adenocarcinoma n = 2) and more rarely mesothelioma (n = 1), melanoma (n = 1) with lung metastasis and glioblastoma (n = 1) [66]. For at least one of these cases, anti-GM-CSF antibodies were detected. This association could be a coincidence in view of the frequency of lung cancer.
Inhalation of toxic particles
Numerous case reports or short series describe PAP after inhalation of mineral particles (silica, talc, cement, kaolin), metal particles (aluminium, titanium, indium), or more rarely organic particles (fibres of cellulose) [13, 17, 67]. PAP may occur after massive inhalation of silica and is then called acute silicoproteinosis [13]. Clinical presentation is close to that of auto-immune PAP.
Numerous animal models demonstrated the development of PAP after inhalation of nickel, silica, titanium, quartz, fibreglass, indium or aluminium powder [17, 68, 69]. In a French study reported by Briens et al. [19], 39% of patients reported notable professional exposure to particles potentially responsible for PAP: silica, paint, wood powder, zirconium, epoxy resin, polyvinyl chloride (PVC), cereal powder, cement, copper and welding smoke. Furthermore, in the Japanese cohort of 248 patients with auto-immune PAP, 23% were considered exposed to toxic inhalation [18]. More recently, anti-GM-CSF antibodies were detected in a patient with PAP secondary to indium inhalation [67]. This suggests that some of the secondary PAP may be associated with anti-GM-CSF antibodies and that toxic inhalation could be a trigger for an auto-immune disease [70].
GENETIC PAP
Genetic PAP includes mutations of SFTPB, SFTPC, ATP-binding cassette 3 (ABCA3), NK2 homeobox 1 (NKX2-1) and GM-CSF receptor α or β subunit, as well as lysurinic protein intolerance.
Mutations of SFTPB, SFTPC, ABCA3 and NKX2-1
The term PAP has been widely used in patients with surfactant mutation associated disorders [71]. Indeed, both entities share similar clinical characteristics which probably explain the confusion [72]. The radiological and histological presentations are nonetheless different. In both children and adults, diffuse ground-glass opacities and lung cysts are the common radiological features observed in surfactant protein disorders whereas septal reticulations and parenchymal consolidation, usually described in auto-immune PAP, are less frequent [73, 74]. The histopathological findings in infants with mutations in the SFTPB, SFTPC, ABCA3 or NKX2-1 genes are remarkably similar, demonstrating varying degrees of interstitial thickening and a remodelling of the alveolar epithelium with type II cell hyperplasia, as well as alveolar accumulation of eosinophilic, lipoproteinaceous granular material [73–76]. These features contrast with the relative integrity of the alveolar structure completely filled with granular, PAS-positive material observed in children with genetic defect of GM-CSF receptor [7].
Along with the preventive measures, corticosteroids are the preferred therapy for SFTPC, ABCA3 or NKX2-1 associated disorders [75, 77, 78]. In the absence of studies, the choice of azithromycin and/or hydroxychloroquine in association with steroid remains highly dependent on the habits and experiences of the different centres. Lung transplantation may be considered [73, 74, 79, 80].
Genetic defects in the GM-CSF receptor
The GM-CSF receptor is composed of the binding α chain (CD116), coded by CSF2RA, and the common β chain (CD131), coded by CSF2RB, which is also used by IL-3 and IL-5 receptors.
Mutations of CSF2RA have only been described in children, and a series of eight patients aged 1.5–9 yrs was recently reported [7]. Mutations of CSF2RA could correspond to 6% of all PAP [7]. Transmission is autosomal recessive, but some mutations have varying and incomplete penetrance and disease was found in two asymptomatic children (aged 5 and 8 yrs) after the diagnosis of a familial index case. Some mutations could be responsible for adult-onset PAP [81]. Except for a lower age at disease onset, patients with mutation of CSF2RA present PAP close to auto-immune PAP. Indeed, unlike mutations of surfactant proteins, interstitial cell infiltration is absent. However, alveolar and serum concentration of GM-CSF is increased and anti-GM-CSF antibodies are absent.
Mutation of CSF2RB was suggested in three patients presenting neonatal PAP and was recently confirmed in a 36-yr-old female and a 9-yr-old girl [8, 82, 83]. Both showed disease close to auto-immune PAP without detectable anti-GM-CSF antibodies but with high concentration of GM-CSF [8].
Whole lung lavage may be effective [84]. GM-CSF therapy does not seem to be effective [82, 84, 85].
Lysinuric protein intolerance
Lysinuric protein intolerance is an autosomal-recessive disease caused by mutation of SLC7A7 contributing to defective transport of cationic amino acid at the membrane of epithelial cells in the intestine and kidney. The disease seems to be more frequent in Japan, Italy and Finland, where it was first described. In Japan, the estimated prevalence is one out of every 57,000 births. The clinical presentation is characterised by failure to thrive and gastrointestinal symptoms. The most frequent chronic manifestations are related to renal and pancreatic insufficiency.
Lysinuric protein intolerance is diagnosed by the presence of excessive amounts of dibasic amino acids (arginine, lysine, ornithine) in the urine, particularly after protein ingestion, and/or mutation of SLC7A7 [86, 87]. The treatment is based on a low protein diet and oral supplementation with citrulline.
Pulmonary manifestations are variable and range from subclinical interstitial lung disease to respiratory insufficiency. PAP is frequently present. Indeed, expression of SLC7A7 is a target of GM-CSF that could explain the reduced activities of alveolar macrophages and peripheral blood monocytes in patients with lysinuric protein intolerance [88]. Whole lung lavage and nebulised GM-CSF therapy seem to be effective [88]; however, PAP may lead to death and relapse after lung transplantation [89, 90].
CONCLUSIONS
Our knowledge of the pathophysiology of PAP has greatly improved in the past 20 yrs, although the mechanisms leading to auto-immunity against GM-CSF are poorly understood. Therapy of PAP is also in progress and the relative place of whole lung lavage, GM-CSF supplemental therapy and antibody targeted treatments should be better defined.
Footnotes
Provenance
Submitted article, peer reviewed.
Statement of Interest
None declared.
REFERENCES
- 1.Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med 2010; 61: 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harari S, Caminati A. Update on diffuse parenchymal lung disease. Eur Respir Rev 2010; 19: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dranoff G, Crawford AD, Sadelain M, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994; 264: 713–716. [DOI] [PubMed] [Google Scholar]
- 4.Stanley E, Lieschke GJ, Grail D, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA 1994; 91: 5592–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shibata Y, Berclaz PY, Chroneos ZC, et al. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 2001; 15: 557–567. [DOI] [PubMed] [Google Scholar]
- 6.Jouneau S, Uzunhan Y. La lipoprotéinose alvéolaire en 2008 [Alveolar lipoproteinosis in 2008]. Rev Mal Respir 2008; 25: 114–120. [Google Scholar]
- 7.Suzuki T, Sakagami T, Young LR, et al. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. Am J Respir Crit Care Med 2010; 182: 1292–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka T, Motoi N, Tsuchihashi Y, et al. Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB.. J Med Genet 2010; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 9.Dirksen U, Hattenhorst U, Schneider P, et al. Defective expression of granulocyte-macrophage colony-stimulating factor/interleukin-3/interleukin-5 receptor common beta chain in children with acute myeloid leukemia associated with respiratory failure. Blood 1998; 92: 1097–1103. [PubMed] [Google Scholar]
- 10.Kitamura T, Tanaka N, Watanabe J, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med 1999; 190: 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitamura T, Uchida K, Tanaka N, et al. Serological diagnosis of idiopathic pulmonary alveolar proteinosis. Am J Respir Crit Care Med 2000; 162: 658–662. [DOI] [PubMed] [Google Scholar]
- 12.Uchida K, Nakata K, Trapnell BC, et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood 2004; 103: 1089–1098. [DOI] [PubMed] [Google Scholar]
- 13.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med 2002; 166: 215–235. [DOI] [PubMed] [Google Scholar]
- 14.Greenhill SR, Kotton DN. Pulmonary alveolar proteinosis: a bench-to-bedside story of granulocyte-macrophage colony-stimulating factor dysfunction. Chest 2009; 136: 571–577. [DOI] [PubMed] [Google Scholar]
- 15.Uchida K, Beck DC, Yamamoto T, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med 2007; 356: 567–579. [DOI] [PubMed] [Google Scholar]
- 16.Sakagami T, Beck D, Uchida K, et al. Patient-derived GM-CSF autoantibodies reproduce pulmonary alveolar proteinosis in non-human primates.. Am J Respir Crit Care Med 2010; 182: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delaval P, Brinchault G, Corre R, et al. Lipoprotéinose alvéolaire pulmonaire [Pulmonary alveolar phospholipoproteinosis]. Rev Pneumol Clin 2005; 61: 186–192. [DOI] [PubMed] [Google Scholar]
- 18.Inoue Y, Trapnell BC, Tazawa R, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med 2008; 177: 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Briens E, Delaval P, Mairesse MP, et al. Lipoprotéinose alvéolaire pulmonaire [Pulmonary alveolar proteinosis]. Rev Mal Respir 2002; 19: 166–182. [PubMed] [Google Scholar]
- 20.Holbert JM, Costello P, Li W, et al. CT features of pulmonary alveolar proteinosis. AJR Am J Roentgenol 2001; 176: 1287–1294. [DOI] [PubMed] [Google Scholar]
- 21.Johkoh T, Itoh H, Muller NL, et al. Crazy-paving appearance at thin-section CT: spectrum of disease and pathologic findings. Radiology 1999; 211: 155–160. [DOI] [PubMed] [Google Scholar]
- 22.Lee KN, Levin DL, Webb WR, et al. Pulmonary alveolar proteinosis: high-resolution CT, chest radiographic, and functional correlations. Chest 1997; 111: 989–995. [DOI] [PubMed] [Google Scholar]
- 23.Wells AU. The clinical utility of bronchoalveolar lavage in diffuse parenchymal lung disease. Eur Respir Rev 2010; 19: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milleron BJ, Costabel U, Teschler H, et al. Bronchoalveolar lavage cell data in alveolar proteinosis. Am Rev Respir Dis 1991; 144: 1330–1332. [DOI] [PubMed] [Google Scholar]
- 25.Uchida K, Nakata K, Suzuki T, et al. Granulocyte/macrophage-colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood 2009; 113: 2547–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sergeeva A, Ono Y, Rios R, et al. High titer autoantibodies to GM-CSF in patients with AML, CML and MDS are associated with active disease. Leukemia 2008; 22: 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uchida K, Nakata K, Suzuki T, et al. Granulocyte/macrophage colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy individuals.. Blood 2009; 113: 2547–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seymour JF, Doyle IR, Nakata K, et al. Relationship of anti-GM-CSF antibody concentration, surfactant protein A and B levels, and serum LDH to pulmonary parameters and response to GM-CSF therapy in patients with idiopathic alveolar proteinosis. Thorax 2003; 58: 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tazawa R, Trapnell BC, Inoue Y, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med 2010; 181: 1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramirez J, Campbell GD. Pulmonary alveolar proteinosis. Endobronchial treatment. Ann Intern Med 1965; 63: 429–441. [DOI] [PubMed] [Google Scholar]
- 31.Michaud G, Reddy C, Ernst A. Whole-lung lavage for pulmonary alveolar proteinosis. Chest 2009; 136: 1678–1681. [DOI] [PubMed] [Google Scholar]
- 32.Cheng SL, Chang HT, Lau HP, et al. Pulmonary alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest 2002; 122: 1480–1485. [DOI] [PubMed] [Google Scholar]
- 33.Brach BB, Harrell JH, Moser KM. Alveolar proteinosis. Lobar lavage by fiberoptic bronchoscopic technique. Chest 1976; 69: 224–227. [DOI] [PubMed] [Google Scholar]
- 34.Seymour JF, Presneill JJ, Schoch OD, et al. Therapeutic efficacy of granulocyte-macrophage colony-stimulating factor in patients with idiopathic acquired alveolar proteinosis. Am J Respir Crit Care Med 2001; 163: 524–531. [DOI] [PubMed] [Google Scholar]
- 35.Kavuru MS, Sullivan EJ, Piccin R, et al. Exogenous granulocyte-macrophage colony-stimulating factor administration for pulmonary alveolar proteinosis. Am J Respir Crit Care Med 2000; 161: 1143–1148. [DOI] [PubMed] [Google Scholar]
- 36.Venkateshiah SB, Yan TD, Bonfield TL, et al. An open-label trial of granulocyte macrophage colony stimulating factor therapy for moderate symptomatic pulmonary alveolar proteinosis. Chest 2006; 130: 227–237. [DOI] [PubMed] [Google Scholar]
- 37.Prakash UB, Barham SS, Carpenter HA, et al. Pulmonary alveolar phospholipoproteinosis: experience with 34 cases and a review. Mayo Clin Proc 1987; 62: 499–518. [DOI] [PubMed] [Google Scholar]
- 38.Rogers RM, Levin DC, Gray BA, et al. Physiologic effects of bronchopulmonary lavage in alveolar proteinosis. Am Rev Respir Dis 1978; 118: 255–264. [DOI] [PubMed] [Google Scholar]
- 39.Wylam ME, Ten R, Prakash UB, et al. Aerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis. Eur Respir J 2006; 27: 585–593. [DOI] [PubMed] [Google Scholar]
- 40.Robinson TE, Trapnell BC, Goris ML, et al. Quantitative analysis of longitudinal response to aerosolized granulocyte-macrophage colony-stimulating factor in two adolescents with autoimmune pulmonary alveolar proteinosis. Chest 2009; 135: 842–848. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez Portal JA, Rodriguez Becerra E, Sanchez Garrido A. Proteinosis alveolar. Respuesta al tratamiento con factor estimulante de colonias de granulocitos y macrófagos por vía inhalada [Response to inhaled granulocyte-macrophage colony-stimulating factor in a patient with alveolar proteinosis]. Arch Bronconeumol 2009; 45: 150–152. [DOI] [PubMed] [Google Scholar]
- 42.Kluth DC, Rees AJ. Anti-glomerular basement membrane disease. J Am Soc Nephrol 1999; 10: 2446–2453. [DOI] [PubMed] [Google Scholar]
- 43.Kavuru MS, Bonfield TL, Thomassen MJ. Plasmapheresis, GM-CSF, and alveolar proteinosis.. Am J Respir Crit Care Med 2003;167: 1036. [DOI] [PubMed] [Google Scholar]
- 44.Luisetti M, Rodi G, Perotti C, et al. Plasmapheresis for treatment of pulmonary alveolar proteinosis. Eur Respir J 2009; 33: 1220–1222. [DOI] [PubMed] [Google Scholar]
- 45.Marshall I, Malur A, Arce S, et al. Rituximab and pulmonary alveolar proteinosis: what have we learned so far? Am J Respir Crit Care Med 2010; 181: A3994. [Google Scholar]
- 46.Borie R, Debray MP, Laine C, et al. Rituximab therapy in autoimmune pulmonary alveolar proteinosis. Eur Respir J 2009; 33: 1503–1506. [DOI] [PubMed] [Google Scholar]
- 47.Amital A, Dux S, Shitrit D, et al. Therapeutic effectiveness of rituximab in a patient with unresponsive autoimmune pulmonary alveolar proteinosis. Thorax 2010; 65: 1025–1026. [DOI] [PubMed] [Google Scholar]
- 48.Patiroglu T, Akyildiz B, Patiroglu TE, et al. Recurrent pulmonary alveolar proteinosis secondary to agammaglobulinemia. Pediatr Pulmonol 2008; 43: 710–713. [DOI] [PubMed] [Google Scholar]
- 49.Samuels MP, Warner JO. Pulmonary alveolar lipoproteinosis complicating juvenile dermatomyositis. Thorax 1988; 43: 939–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wardwell NR, Jr, Miller R, Ware LB. Pulmonary alveolar proteinosis associated with a disease-modifying antirheumatoid arthritis drug. Respirology 2006; 11: 663–665. [DOI] [PubMed] [Google Scholar]
- 51.Uchiyama M, Nagao T, Hattori A, et al. Pulmonary alveolar proteinosis in a patient with Behcet's disease. Respirology 2009; 14: 305–308. [DOI] [PubMed] [Google Scholar]
- 52.Ishii H, Tazawa R, Kaneko C, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. Eur Respir J 2011; 37: 465–468. [DOI] [PubMed] [Google Scholar]
- 53.Tran Van Nhieu J, Vojtek AM, Bernaudin JF, et al. Pulmonary alveolar proteinosis associated with Pneumocystis carinii. Ultrastructural identification in bronchoalveolar lavage in AIDS and immunocompromised non-AIDS patients. Chest 1990; 98: 801–805. [DOI] [PubMed] [Google Scholar]
- 54.Cordonnier C, Fleury-Feith J, Escudier E, et al. Secondary alveolar proteinosis is a reversible cause of respiratory failure in leukemic patients. Am J Respir Crit Care Med 1994; 149: 788–794. [DOI] [PubMed] [Google Scholar]
- 55.Pamuk GE, Turgut B, Vural O, et al. Pulmonary alveolar proteinosis in a patient with acute lymphoid leukemia regression after G-CSF therapy. Leuk Lymphoma 2003; 44: 871–874. [DOI] [PubMed] [Google Scholar]
- 56.Chung JH, Pipavath SJ, Myerson DH, et al. Secondary pulmonary alveolar proteinosis: a confusing and potentially serious complication of hematologic malignancy. J Thorac Imaging 2009; 24: 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vella FS, Simone B, Giannelli G, et al. Case of multiple myeloma mimicking an infectious disease with fever, intrahepatic cholestasis, renal failure, and pulmonary insufficiency. Am J Hematol 2003; 72: 38–42. [DOI] [PubMed] [Google Scholar]
- 58.Tomonari A, Shirafuji N, Iseki T, et al. Acquired pulmonary alveolar proteinosis after umbilical cord blood transplantation for acute myeloid leukemia. Am J Hematol 2002; 70: 154–157. [DOI] [PubMed] [Google Scholar]
- 59.Pollack SM, Gutierrez G, Ascensao J. Pulmonary alveolar proteinosis with myeloproliferative syndrome with myelodysplasia: bronchoalveolar lavage reduces white blood cell count. Am J Hematol 2006; 81: 634–638. [DOI] [PubMed] [Google Scholar]
- 60.Ishii H, Trapnell BC, Tazawa R, et al. Comparative study of high-resolution CT findings between autoimmune and secondary pulmonary alveolar proteinosis. Chest 2009; 136: 1348–1355. [DOI] [PubMed] [Google Scholar]
- 61.Goldschmidt N, Nusair S, Gural A, et al. Disseminated Mycobacterium kansasii infection with pulmonary alveolar proteinosis in a patient with chronic myelogenous leukemia. Am J Hematol 2003; 74: 221–223. [DOI] [PubMed] [Google Scholar]
- 62.Dai MS, Lee SC, Ho CL, et al. Impact of open lung biopsy for undiagnosed pulmonary infiltrates in patients with hematological malignancies. Am J Hematol 2001; 68: 87–90. [DOI] [PubMed] [Google Scholar]
- 63.Ishii H, Nakata K, Inoue Y, et al. Clinical course of GM−CSF autoantibody negative pulmonary alveolar proteinosis (NAPAP): efficacy of lavage therapy. Am J Respir Crit Care Med 2009; 179: A3035. [Google Scholar]
- 64.Numata A, Matsuishi E, Koyanagi K, et al. Successful therapy with whole-lung lavage and autologous peripheral blood stem cell transplantation for pulmonary alveolar proteinosis complicating acute myelogenous leukemia. Am J Hematol 2006; 81: 107–109. [DOI] [PubMed] [Google Scholar]
- 65.Tabata S, Shimoji S, Murase K, et al. Successful allogeneic bone marrow transplantation for myelodysplastic syndrome complicated by severe pulmonary alveolar proteinosis. Int J Hematol 2009; 90: 407–412. [DOI] [PubMed] [Google Scholar]
- 66.Su KC, Lay SL, Perng RP, et al. Lung cancer may develop subsequently or coincidently with pulmonary alveolar proteinosis. Lung Cancer 2007; 58: 144–148. [DOI] [PubMed] [Google Scholar]
- 67.Cummings KJ, Donat WE, Ettensohn DB, et al. Pulmonary alveolar proteinosis in workers at an indium processing facility. Am J Respir Crit Care Med 2010; 181: 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lison D, Laloy J, Corazzari I, et al. Sintered indium-tin-oxide (ITO) particles: a new pneumotoxic entity. Toxicol Sci 2009; 108: 472–481. [DOI] [PubMed] [Google Scholar]
- 69.Oller AR, Kirkpatrick DT, Radovsky A, et al. Inhalation carcinogenicity study with nickel metal powder in Wistar rats. Toxicol Appl Pharmacol 2008; 233: 262–275. [DOI] [PubMed] [Google Scholar]
- 70.Costabel U, Nakata K. Pulmonary alveolar proteinosis associated with dust inhalation: not secondary but autoimmune? Am J Respir Crit Care Med 2010; 181: 427–428. [DOI] [PubMed] [Google Scholar]
- 71.Tredano M, Griese M, Brasch F, et al. Mutation of SFTPC in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease. Am J Med Genet A 2004; 126A: 18–26. [DOI] [PubMed] [Google Scholar]
- 72.Deutsch GH, Young LR, Deterding RR, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med 2007; 176: 1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mechri M, Epaud R, Emond S, et al. Surfactant protein C gene (SFTPC) mutation-associated lung disease: high-resolution computed tomography (HRCT) findings and its relation to histological analysis. Pediatr Pulmonol 2010; 45: 1021–1029. [DOI] [PubMed] [Google Scholar]
- 74.van Moorsel CH, van Oosterhout MF, Barlo NP, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med 2010; 182: 1419–1425. [DOI] [PubMed] [Google Scholar]
- 75.Guillot L, Carre A, Szinnai G, et al. NKX2-1 mutations leading to surfactant protein promoter dysregulation cause interstitial lung disease in “Brain-Lung-Thyroid Syndrome”. Hum Mutat 2010; 31: E1146–E1162. [DOI] [PubMed] [Google Scholar]
- 76.Young LR, Nogee LM, Barnett B, et al. Usual interstitial pneumonia in an adolescent with ABCA3 mutations. Chest 2008; 134: 192–195. [DOI] [PubMed] [Google Scholar]
- 77.Thouvenin G, Abou Taam R, Flamein F, et al. Characteristics of disorders associated with genetic mutations of surfactant protein C. Arch Dis Child 2010; 95: 449–454. [DOI] [PubMed] [Google Scholar]
- 78.Doan ML, Guillerman RP, Dishop MK, et al. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax 2008; 63: 366–373. [DOI] [PubMed] [Google Scholar]
- 79.Hamvas A, Nogee LM, Mallory GB, Jr, et al. Lung transplantation for treatment of infants with surfactant protein B deficiency. J Pediatr 1997; 130: 231–239. [DOI] [PubMed] [Google Scholar]
- 80.Nogee LM, Dunbar AE, 3rd, Wert SE, et al. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 2001; 344: 573–579. [DOI] [PubMed] [Google Scholar]
- 81.Motoi N, Tanaka T, Tsuchihashi Y, et al. A case of acquired pulmonary alveolar proteinosis with defective signaling through common beta chain of GM-CSF receptor. Am J Respir Crit Care Med 2010; 181: A2995. [Google Scholar]
- 82.Dirksen U, Nishinakamura R, Groneck P, et al. Human pulmonary alveolar proteinosis associated with a defect in GM-CSF/IL-3/IL-5 receptor common beta chain expression. J Clin Invest 1997; 100: 2211–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suzuki T, Maranda B, Sakagami T, et al. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur Respir J 2011; 37: 201–204. [DOI] [PubMed] [Google Scholar]
- 84.Suzuki T, Sakagami T, Rubin BK, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med 2008; 205: 2703–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martinez-Moczygemba M, Doan ML, Elidemir O, et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med 2008; 205: 2711–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sperandeo MP, Andria G, Sebastio G. Lysinuric protein intolerance: update and extended mutation analysis of the SLC7A7 gene. Hum Mutat 2008; 29: 14–21. [DOI] [PubMed] [Google Scholar]
- 87.Broer S. Lysinuric protein intolerance: one gene, many problems. Am J Physiol Cell Physiol 2007; 293: C540–541. [DOI] [PubMed] [Google Scholar]
- 88.Barilli A, Rotoli BM, Visigalli R, et al. In lysinuric protein intolerance system y+L activity is defective in monocytes and in GM-CSF-differentiated macrophages. Orphanet J Rare Dis 2010; 5: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Santamaria F, Parenti G, Guidi G, et al. Early detection of lung involvement in lysinuric protein intolerance: role of high-resolution computed tomography and radioisotopic methods. Am J Respir Crit Care Med 1996; 153: 731–735. [DOI] [PubMed] [Google Scholar]
- 90.Santamaria F, Brancaccio G, Parenti G, et al. Recurrent fatal pulmonary alveolar proteinosis after heart-lung transplantation in a child with lysinuric protein intolerance. J Pediatr 2004; 145: 268–272. [DOI] [PubMed] [Google Scholar]