

Abstract
Shikimate kinase (EC 2.7.1.71) is a committed enzyme in the seven-step biosynthesis of chorismate, a major precursor of aromatic amino acids and many other aromatic compounds. Genes for all enzymes of the chorismate pathway except shikimate kinase are found in archaeal genomes by sequence homology to their bacterial counterparts. In this study, a conserved archaeal gene (gi|1500322 in Methanococcus jannaschii) was identified as the best candidate for the missing shikimate kinase gene by the analysis of chromosomal clustering of chorismate biosynthetic genes. The encoded hypothetical protein, with no sequence similarity to bacterial and eukaryotic shikimate kinases, is distantly related to homoserine kinases (EC 2.7.1.39) of the GHMP-kinase superfamily. The latter functionality in M. jannaschii is assigned to another gene (gi|1591748), in agreement with sequence similarity and chromosomal clustering analysis. Both archaeal proteins, overexpressed in Escherichia coli and purified to homogeneity, displayed activity of the predicted type, with steady-state kinetic parameters similar to those of the corresponding bacterial kinases: Km,shikimate = 414 ± 33 μM, Km,ATP = 48 ± 4 μM, and kcat = 57 ± 2 s−1 for the predicted shikimate kinase and Km,homoserine = 188 ± 37 μM, Km,ATP = 101 ± 7 μM, and kcat = 28 ± 1 s−1 for the homoserine kinase. No overlapping activity could be detected between shikimate kinase and homoserine kinase, both revealing a >1,000-fold preference for their own specific substrates. The case of archaeal shikimate kinase illustrates the efficacy of techniques based on reconstruction of metabolism from genomic data and analysis of gene clustering on chromosomes in finding missing genes.
Shikimate kinase (EC 2.7.1.71) is the enzyme responsible for converting shikimate to 3-phosphoshikimate, a committed step in the biosynthesis of chorismate (7). The latter is the branching point metabolite and the major precursor of aromatic amino acids, folates, ubiquinones, and many other aromatic compounds.
The chorismate pathway consists of seven enzymatic steps (Fig. 1). It has been extensively studied in Escherichia coli, and the corresponding genes have been identified (for a review, see reference 34). All of the participating enzymes, including shikimate kinase, are conserved in a broad range of organisms, such as bacteria, yeasts, and plants. The chorismate pathway is absent in mammals but seemingly present in some protozoa (27, 35).
FIG. 1.

Pathway reconstruction from genomic data, and chromosomal clustering of chorismate biosynthetic genes. Gene names shown in italic are those of E. coli. Orthologous ORFs found in other genomes are shown by RID numbers from the WIT database. Shaded boxes within a genome row represent proximity on the chromosome.
In the first sequenced archaeal genome of Methanococcus jannaschii (3), genes encoding only four of seven enzymes of chorismate biosynthesis could be identified by sequence similarity with their bacterial and eukaryotic counterparts. Genes for the first two steps and for the shikimate kinase appeared to be missing, but Selkov et al. (37) asserted that the pathway started from 3-dehydroquinate. The growing number of sequenced archaeal genomes and a better understanding of the metabolism of M. jannaschii have strengthened this and other metabolic data (12). Presently, six genes of the pathway can be identified by sequence comparison in Pyrococcus furiosus, Pyrococcus abyssi, Pyrobaculum aerophilum, and Aeropyrum pernix. However, our attempts to identify the gene encoding shikimate kinase using similarity to known versions of the enzyme yielded no plausible candidates in any of the archaeal genomes.
We addressed this problem using an approach based on the analysis of gene clustering on the chromosome (31). Briefly, this analysis utilizes the observation that functionally related genes in prokaryotes, such as those that are active in the same metabolic pathway, tend to cluster along the chromosome. By comparing the chromosomal clustering in multiple bacterial genomes, conjectures relating to the functions of previously uncharacterized genes can be formulated. In this study, an open reading frame (ORF) of M. jannaschii (RMJ07785) encoding a hypothetical protein, which is conserved in most of the archaeal genomes, was identified as the best candidate for the missing shikimate kinase gene. (ORF [and corresponding protein] identifiers cited in this report are from the WIT genomic database [http://igweb.integratedgenomics.com/IGwit/].) This protein has no sequence similarity with bacterial and eukaryotic shikimate kinases but is instead distantly related to homoserine kinases. The latter functionality in M. jannaschii is assigned to another protein (RMJ01903), in agreement with both sequence similarity and chromosomal clustering analysis. Homoserine kinase, an enzyme involved in threonine biosynthesis, is a member of the GHMP-kinase superfamily, which initially included four families of enzymes specifically phosphorylating galactose, homoserine, mevalonate, and phosphomevalonate (2). Shikimate kinase activity has never been detected within the fold characteristic of the GHMP-kinase superfamily. Moreover, all previously identified shikimate kinases belong to a structurally unrelated NMP-kinase superfamily, as recently confirmed by X-ray crystallography (19).
Here we report the expression, purification, and characterization of the two putative GHMP-kinases from M. jannaschii, RMJ07885 and RMJ01903. Through verification of the anticipated substrate specificity, we have shown that archaea express a novel shikimate kinase family which, in contrast to its bacterial and eukaryotic counterpart, belongs to the GHMP-kinase superfamily. Our results demonstrate for the first time that shikimate can be phosphorylated by two structurally unrelated enzymes.
MATERIALS AND METHODS
Strains, plasmids, and other reagents.
E. coli strains DH5α, BL21, and BL21/DE3 (Gibco-BRL, Rockville, Md.) were used for cloning and expression. For expression of all genes in E. coli, a pET-derived vector containing the T7 promoter, His6 tag, and TEV-protease cleavage site (such as described elsewhere 30) or a similar vector with the trp promoter (pPROEX-HTa; Gibco-BRL) was used. Genomic DNA of M. jannaschii was a kind gift from Claudia Reich, University of Illinois at Champaign-Urbana. Enzymes for DNA manipulations were from New England Biolabs (Beverly, Mass.) and MBI Fermentas (Vilnius, Lithuania). For PCR, Pfu polymerase (Stratagene, La Jolla, Calif.) was used. Plasmid purification kits and Ni-nitrilotriacetic acid resin were from Qiagen (Valencia, Calif.). Oligonucleotides for PCR and sequencing were from Sigma-Genosys (Woodlands, Tex.). All other chemicals, including the assay components shikimate, galactose, homoserine, mevalonate, NADH, ATP, phosphoenolpyruvate, lactate dehydrogenase, and pyruvate kinase, were from Sigma-Aldrich (St. Louis, Mo.).
Genome analysis.
Genomes cited in this study are listed in Table 1. Our approach to identification of candidates for missing genes is based on comparative genome analysis using the WIT platform (a genomic database and set of tools for functional annotation and metabolic reconstruction).
TABLE 1.
Genomes cited in this study
| Genome | Website | Reference |
|---|---|---|
| Methanococcus jannaschii | http://www.tigr.org/ | 3 |
| Methanobacterium thermoautotrophicum | http://www.biosci.ohio-state.edu/ | 38 |
| Archaeoglobus fulgidus | http://www.tigr.org/ | 18 |
| Pyrococcus horikoshii | http://www.bio.nite.go.jp | 171 |
| Aeropyrum pernix | http://www.bio.nite.go.jp/ | 16 |
| Escherichia coli | http://gib.genes.nig.ac.jp/ | 1 |
| Bacillus subtilis | http://genolist.pasteur.fr/SubtiList/ | 20 |
| Treponema pallidum | http://www.tigr.org/ | 8 |
| Chlamydia trachomatis | http://chlamydia-www.berkeley.edu:4231/ | 39 |
| Thermatoga maritima | http://www.tigr.org/ | 29 |
| Saccharomyces cerevisiae | http://genome-www.stanford.edu/ | 28 |
| Pyrococcus abyssi | http://www.genoscope.cns.fr/ | |
| Pyrococcus furiosus | http://www.genome.utah.edu/ | |
| Clostridium acetobutylicum | http://www.cric.com/ | |
| Corynebacterium diphtheriae | http://www.sanger.ac.uk/ | |
| Streptococcus pneumoniae | http://www.tigr.org/ | |
| Pyrobaculum aerophilum | http://informa.bio.caltech.edu/ |
We use the term “missing gene” to denote a particular enzyme in a pathway (sometimes referred to as “missing enzyme” [4]) for which a corresponding gene has not been cloned or identified otherwise. This term may refer to all organisms or to a subset of organisms. An example of the latter case is the shikimate kinase gene originally discovered in E. coli (7, 23). Shikimate kinase orthologs can be unambiguously identified by sequence comparison in bacterial and some eukaryotic genomes but not in archaeal genomes.
The technique for inferring functional coupling between genes based on their chromosomal arrangement was introduced earlier (31). This technique is implemented in a set of tools within the WIT program. The approach is based on the well-established notion that genes including proteins with related functions (e.g., enzymes of the same metabolic pathway) tend to cluster on the chromosome, at least in prokaryotes. This concept is widely used in WIT, e.g., for resolving ambiguities in functional assignment of paralogs (32). The same principles can be applied to identify candidates for missing genes in metabolic pathways.
The most likely candidates for a sought functional role are presumed to occur among hypothetical proteins (unassigned or ambiguously assigned ORFs) that are clustered on the chromosome with known genes from the same pathway. Using WIT, one can build up evidence of functional coupling between a set of genes. The system has accumulated instances in which a pair of genes that are close in one genome correspond to a pair of genes that are close in another genome. More precisely, the system has tabulated instances of pairs of close bidirectional best hits (PCBBHs) (31). One can formulate a process using this form of evidence to methodically build a case that a gene encoding a hypothetical protein is actually a missing gene. This process can be illustrated by building a spreadsheet (such as the one shown in Fig. 1) where each column corresponds to a function (enzyme), and each row corresponds to an organism, in the following manner. (i) Pick all assigned genes from the pathway of interest within a selected set of organisms (genomes). This set will contain a subset of genomes with a sought “missing gene.” (ii) Add all hypothetical proteins for which the PCBBH evidence with respect to any component of the original spreadsheet is greater than some specified evidence threshold. (iii) Fill in the added columns of hypothetical proteins with bidirectional best hits from all other genomes in the set. Use colors or patterns to depict groups of clustered genes (those that are close on the chromosome) within each organism. (iv) Apply user-defined criteria to score and select the best candidates for experimental verification. Initially genes encoding all added hypothetical proteins are considered to be candidates for a missing gene. Some examples of the useful criteria are relative strength of clustering (as indicated by the number of PCBBHs and the phylogenetic diversity of the organisms for which PCBBHs exist, presence of homologs of the candidate gene in most of the genomes where the sought gene is missing, absence of homologs of the candidate gene in most of the genomes where either the sought enzyme was previously identified in the nonorthologous form or the entire pathway is absent, and motifs or patterns in a sequence of a candidate gene relevant for a sought enzymatic function (e.g., nucleotide-binding motif).
This technique may produce a widely varying number of candidates, depending on the strength of clustering and other user-defined criteria. In the case of the archaeal shikimate kinase, as well as in some other cases when functionally related genes tend to form large operons, overwhelming evidence reveals only one very strong candidate for a sought functional role.
PCR amplification and cloning.
Two ORFs from M. jannaschii were PCR amplified using the following primers: for a predicted shikimate kinase RMJ07785 (gi|1500322), gggtcATGaAAGGAAAAGCCTATGCATTAGCATCTG (5′ primer) and ggggtcgacTTAGTAAATAGAAGCTCCATCATTGTTTGGTTTAG (3′ primer); for a predicted homoserine kinase RMJ01903 (gi|1591748), gggtcATGAAAGTTAGAGTGAAAGCTCCCTGCAC (5′ primer) ggggtcgacTTAAACAACTTCAACTCCTTTACCAACTTCTGTTC (3′ primer). Introduced restriction sites (BspHI for the 5′ end and SalI for the 3′ end) are in boldface; nucleotides not present in the original sequence are in lowercase. Only one mutation, Glu-2 (GAA)→Lys (aAA), was introduced into RMJ07785. PCR amplification was performed using M. jannaschii genomic DNA and Pfu polymerase according to the manufacturer's protocol. PCR fragments were cloned into the expression vectors, which were cleaved by NcoI and SalI. Selected clones were verified by DNA sequence analysis. No mutations compared to the original DNA sequence were observed.
Expression and purification.
Both proteins were expressed as N-terminal fusions with a His6 tag and a TEV-protease cleavage site. Cells were grown to an optical density at 600 nm of 0.8 to 1.0 at 37°C (in 50 ml [for analytical purposes] and in 6 liters [for preparative purification] of Luria-Bertani medium). Isopropyl-β-d-thiogalactopyranoside was added to 0.8 mM, and harvesting was performed after ∼12 h of shaking at 20°C. Expression analysis and protein purification were performed using standard techniques. Briefly, harvested cells were resuspended in A buffer (20 mM HEPES [pH 7] containing 100 mM NaCl, 0.03% Brij 35, and 2 mM β-mercaptoethanol supplemented with 2 mM phenylmethylsulfonyl fluoride and a protease inhibitor cocktail (Sigma-Aldrich). Lysozyme was added to 1 mg/ml. After 20 min of incubation on ice, the cell suspension was frozen in liquid nitrogen. After thawing and sonication, cell debris was removed by centrifugation at 20,000 rpm for 2 h. Tris-HCl buffer (pH 8) was added to the supernatant (50 mM, final concentration), and the supernatant was loaded onto a Ni-nitrilotriacetic acid agarose column. A gradient elution with imidazole (0 to 200 mM in buffer A) was performed using an AKTA fast protein liquid chromatography system (Pharmacia, Uppsala, Sweden). Fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, pooled, concentrated to a final volume of 2 ml in the presence of 1 mM dithiothreitol and 1 mM EDTA, and loaded onto a HiLoad Superdex 200 16/60 column (Pharmacia). Gel filtration was performed in HEPES buffer (pH 7.5) containing 100 mM NaCl, 0.5 mM EDTA, and 1 mM dithiothreitol. Fractions containing active protein were pooled and concentrated to >10 mg/ml; aliquots were frozen in liquid nitrogen and stored at −80°C. In preliminary experiments, we showed that such treatment did not affect the kinase activity.
Enzymatic properties.
The colorimetric coupled assay for homoserine kinase activity was performed as described elsewhere (14), using a Beckman DU-640 to monitor the change in absorbance at 340 nm in a six-cuvette assembly thermostated at 37°C. The 500 μl of mixture contained 100 mM HEPES (pH 8.0), 20 mM KCl, 10 mM MgCl2, 5 mM ATP, 2 mM phosphoenolpyruvate, 0.3 mM NADH, 5 U of lactate dehydrogenase, 2.5 U of pyruvate kinase, 10 mM homoserine, and 0.1 to 10 μg of the enzyme being analyzed. An extinction coefficient of NADH equal to 6.22 mM−1 cm−1 was used for rate calculations. One unit of enzyme was defined as a quantity capable of converting 1 μmol of NADH to NAD+ per min. The same protocol was adopted to measure kinase activity with alternative substrates (10 mM shikimate, mevalonate, and galactose).
Steady-state kinetic parameters for homoserine kinase and shikimate kinase were determined using the same assay adapted for 96-well plates (250 μl per well) and time-resolved absorbance readings in a Tecan (Tecan-US, Durham, N.C.) Spectrafluor Plus thermostated at 37°C with a 340-nm filter. ATP concentration was varied in the range of 14 to 350 μM. The reaction was started by adding a last component (0.1 to 5 mM homoserine or shikimate) with 96-tip automated pipetting station Quadra-96 (Tomtec, Hamden, Conn.). The initial rates were determined using Magellan (version 2.22) software (Tecan-US). Global nonlinear fitting of initial rates (V) versus both substrate concentrations (A is ATP; B is homoserine or shikimate) was performed using Sigma-Plot 2000 (Jandel Scientific) and the most general equation for a steady-state bireactant model (36),
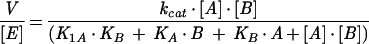 |
1 |
where [E] is the enzyme concentration, KA and KB are Michaelis constants for corresponding substrates, and K1A is the dissociation constant for an ATP-enzyme complex. In the case of shikimate kinase, no acceptable fit could be obtained with this model. Parallel lines were observed on the double-reciprocal plot, indicating that a ping-pong model is a better approximation of this system, and a corresponding equation was used for fitting the data (36):
 |
2 |
However, without additional experiments it is impossible to distinguish between a true ping-pong mechanism and a sequential mechanism with a relatively low Kd of the first adduct (36).
RESULTS
Reconstruction of the chorismate pathway and prediction of the archaeal shikimate kinase.
Figure 1 illustrates the reconstruction of the chorismate biosynthetic pathway from genomic data as it is known for E. coli (Fig. 2). Presented in Fig. 1 is a limited set of representative genomes for illustrative purposes (more than 50 complete and partial microbial genomes were analyzed). The chorismate pathway in its entirety is remarkably conserved over a broad range of organisms, and homologs of all seven genes can be easily identified by sequence comparison. In most prokaryotes these genes are either all present or all missing. The latter case is illustrated by representatives of bacterial (Treponema pallidum) and archaeal (Pyrococcus horikoshii) genomes. P. horikoshii contains no genes associated with the biosynthesis of aromatic amino acids (24), and at least one aromatic amino acid, tryptophan, is required for its growth (11).
FIG. 2.

Chorismate biosynthesis pathway in E. coli (modified from reference 34). Enzyme names and corresponding E. coli genes are displayed.
In M. jannaschii, as well as in M. thermoautotrophicum and Archaeoglobus fulgidus, genes for steps 1, 2, and 5 are missing. The most likely interpretation of the absence of 7P-2-dehydro-3-deoxy-d-arabinoheptulosonate synthase (step 1) and 3-dehydroquinate synthase (step 2) is that these organisms use a completely different pathway for the formation of 3-dehydroquinate. This explanation is in agreement with labeling studies of Methanococcus maripaludis showing that erythrose-4-phosphate is not a precursor for chorismate (41).
On the other hand, one would expect shikimate kinase activity to be present in all archaea which contain enzymes for the two steps preceding and two steps following the phosphorylation of shikimate. Therefore, a missing archaeal shikimate kinase gene most likely represents a case of so-called nonorthologous gene displacement, meaning that the same functional role is performed by a structurally unrelated protein. The same assertion was presented by Makarova et al. (25). The increasing number of sequenced genomes is revealing many examples of nonorthologous gene displacement, as recently reviewed (9).
Comparative analysis of multiple microbial genomes using the WIT platform revealed a large number of PCBBHs among all of the genes listed in Fig. 1. In many genomes, these genes occur within large operon-like clusters, such as in Thermatoga maritima. Chromosomal clustering is illustrated in Fig. 1 and graphically presented by the alignment of selected contigs in Fig. 3. No clustering of chorismate biosynthetic genes is observed in M. jannaschii and other methanogenic archaea, while P. abyssi, P. furiosus, and A. pernix display remarkable clustering of most or all of these genes. P. aerophilum shows clustering in two separate groups containing genes for steps 1, 2 and 6 and genes for steps 4 and 7 (data not shown). Therefore, the missing archaeal shikimate kinase gene seemed likely to be found among the unassigned ORFs (hypothetical proteins) clustered with genes encoding other enzymes of this pathway.
FIG. 3.

Alignment of selected chromosomal contigs containing chorismate biosynthetic genes, modified from data produced by the WIT tool Pinned Regions to visualize gene clustering on the chromosome. The display is created by aligning one specific gene from a number of organisms and depicting other orthologous genes that are conserved in the neighborhood at least in two different genomes. Contigs are aligned by 3-dehydroquinate synthase (the second gene of the pathway). ORFs with sequence similarity are outlined using the same pattern, and those with assigned functions in chorismate biosynthesis are marked with a number corresponding to the step in the pathway as in Fig. 1. Patterns are retained within gene fusions to show regions of homology with corresponding genes. The two genes marked with question marks are those predicted to encode an archaeal shikimate kinase.
The strongest candidate gene selected according to the criteria listed above is located immediately downstream of the shikimate dehydrogenase in A. pernix and P. abyssi (Fig. 3). Homologs of this gene are (i) embedded in large operon-like clusters in some archaeal genomes; (ii) conserved in all archaeal genomes, including that of M. jannaschii, where no clustering is observed (Fig. 1), but excluding that of P. horikoshii, where the entire pathway is missing; (iii) absent in nonarchaea; and (iv) characterized by sequence similarity with GHMP-kinases. No homology can be detected between this candidate gene (typified by RMJ07785 [gi|1500322]) and bacterial and eukaryotic shikimate kinases. Highest Psi-BLAST scores (http://www.ncbi.nlm.nih.gov/BLAST/) are observed between RMJ07785 and homoserine kinases, with the most pronounced conservation within one motif common for all GHMP-kinases (Fig. 4). Shikimate kinase activity was never detected with any representative of the GHMP-kinase superfamily or, more generally, with any other protein fold except that of the NMP-kinase family (19). All of these considerations taken together generated conflicting evidence regarding a functional assignment for RMJ07785; a homoserine kinase was suggested by the sequence similarity analysis, whereas a shikimate kinase was suggested by metabolic reconstruction and chromosomal clustering.
FIG. 4.

Amino acid sequence alignment of archaeal shikimate kinases. Conserved residues are highlighted. The segments bracketed with numbers 1 and 2 correspond to sites that are similarly conserved in homoserine kinase and involved in forming an ATP-binding site (42).
Two additional members of the GHMP-kinase superfamily in M. jannaschii, RMJ01903 (gi|1591748) and RMJ10221 (gi|1591731), were assigned as homoserine kinase (EC 2.7.1.39) and mevalonate kinase (EC 2.6.1.36), respectively. The predicted mevalonate kinase, RMJ10221, was recently cloned and expressed, and its activity was verified (13). Homoserine kinase is the fourth enzyme of the five-step threonine biosynthesis pathway (33). Genes for all enzymes of this pathway, including the putative homoserine kinase RMJ01903, are present in recognizable forms but not clustered in M. jannaschii. Orthologs of RMJ01903 in P. abyssi, P. furiosus, and P. aerophilum form chromosomal clusters with the other four enzymes of this pathway (not shown). Therefore, the assignment of RMJ01903, although never confirmed experimentally, was in complete agreement with metabolic reconstruction and the analysis of chromosomal arrangement. On the other hand, the unexpected sequence similarity between the predicted archaeal shikimate kinase and previously characterized homoserine kinases raised the possibility of overlapping activity. To address this question, we overexpressed both M. jannaschii proteins (RMJ07785 and RMJ01903) and characterized their substrate preferences.
Experimental verification.
RMJ07785 and RMJ01903 proteins were expressed in E. coli with a His6 tag and purified to homogeneity using a combination of chelating chromatography and gel filtration with a yield of pure proteins of ∼7 and 25 mg/liter, respectively. RMJ07785 had a tendency to precipitate at concentrations higher than 1 mg/ml. This precipitation was reversible, and solubility could be increased at least 10 times by the addition of 0.5 M NaCl. Both enzymes were stable in solution at high concentrations but rapidly lost activity at concentrations below 0.01 mg/ml. The latter effect may be a consequence of dissociation of the dimer, which is the predominant native form of both proteins, as revealed by gel filtration.
We used an enzymatic assay similar to one previously described (14) to determine substrate preferences of both enzymes. The use of a continuous assay coupling the production of ADP to the oxidation of NADH allowed us to test various substrates in the same conditions without modifying the assay. The most important result was our verification that RMJ07785 is a novel shikimate kinase. Almost no pH dependence of shikimate kinase activity was observed in a range of pH 6.5 to 8.5. The specific activity of the pure RMJ07785 enzyme (150 U/mg at saturating shikimate) was comparable to that reported for the predominant E. coli shikimate kinase isozyme SK2 (100 U/mg) (5). No activity was detected with homoserine, galactose, or mevalonate, implying a >1,000-fold preference of this enzyme for shikimate over other known nonphosphorylated substrates of GHMP-kinases.
The same level of stringency in substrate specificity was observed for the predicted homoserine kinase RMJ01903. Its specific activity (60 U/mg) with homoserine was similar to that seen for the E. coli enzyme (74 U/mg) (14), and the preference for homoserine was at least 1,000-fold over that of shikimate, galactose, and mevalonate.
Steady-state kinetic data were generated for both enzymes in optimal conditions with their specific substrates (Fig. 5) and analyzed using the most general form of the rate equation for bireactant mechanisms (36). Kinetic parameters for both archaeal enzymes obtained with shikimate and homoserine are very similar to those of their bacterial counterparts. E. coli shikimate kinase SK2 (gene aroL) has an apparent Km,shikimate of 200 μM, but it is inhibited about sevenfold when the shikimate concentration is increased from 1 to 10 mM. The other isozyme, SK1 (gene aroK), is characterized by an apparent Km,shikimate higher than 5 mM (5). The archaeal shikimate kinase RMJ07785 has a Km,shikimate only about two times higher (414 ± 33 μM) than that of SK2. It is not inhibited by shikimate concentrations up to 10 mM, and its Km,ATP (48 ± 4 μM) is comparable to the E. coli SK2 apparent Km,ATP (160 μM). Homoserine kinase of E. coli (gene thrB), with a Km,homoserine of 140 μM, loses up to 70% of its activity when the substrate concentration is increased from 1 to 10 mM (15). The archaeal homoserine kinase RMJ01903 reveals a similar Km,homoserine (188 ± 37 μM) but no substrate inhibition up to 10 mM homoserine. In this respect, the archaeal enzyme is more similar to a recently characterized homoserine kinase from Arabidopsis thaliana (22). The values of Km,ATP are very similar between the archaeal homoserine kinase RMJ01903 and the E. coli homoserine kinase (101 ± 7 μM and 130 μM, respectively) (15).
FIG. 5.

Initial rate plots obtained for the shikimate kinase RMJ07785 versus shikimate concentration (A) and for the homoserine kinase RMJ01903 versus homoserine concentration (B). Symbols represent experimental data at various concentrations of ATP: 14 μM (▵), 35 μM (◊), 70 μM (□), 140 μM (▿), and 350 μM (○). Curves show global fits of the data using equation 2 for the shikimate kinase (parameters of the fit were KA = 48 ± 4 μM, KB = 414 ± 33 μM, and kcat = 57 ± 2 s−1) and equation 1 for the homoserine kinase (parameters of the fit were KA = 101 ± 7 μM, KB = 188 ± 37 μM, K1A = 475 ± 112 μM, and kcat = 28 ± 1 s−1).
DISCUSSION
After decades of molecular cloning, genes for many key metabolic enzymes remain unidentified, even in microorganisms with completely sequenced genomes. By various estimates, there are at least 100 such missing genes in the core metabolism of E. coli (1, 4). In addition to those genes that have not been identified, groups of organisms appear to encode enzymatic functions by genes structurally unrelated to their previously described counterparts from other sources. With a growing number of sequenced genomes, we see more and more examples of missing genes as a result of a nonorthologous displacement of the corresponding genes. Methods that rely solely on sequence comparison are of limited applicability for finding missing genes. Additional methods of comparative genomics that help to predict protein functionality beyond sequence comparison were recently reviewed (10).
The case of the missing archaeal shikimate kinase gene described here is an illustration of how this problem can be approached on the basis of reconstruction of metabolic pathways and the analysis of chromosomal arrangement of genes. Metabolic reconstruction from genomic data is a key step in defining a subset of organisms with a common requirement for a particular missing gene. Seven out of eight available archaeal genomes contain at least four genes of the chorismate pathway involved with the steps before and after shikimate kinase (Fig. 1). In WIT, this was treated as sufficient evidence for existence of the chorismate pathway (in its full or truncated version), even though a gene for shikimate kinase was not found in any archaea. Therefore, the subset of seven archaeal genomes was a source of candidates for a missing shikimate kinase gene. P. horikoshii, in which the chorismate pathway is absent, was not included in this subset.
At the next step, candidate genes revealed by neighborhood analysis are selected among hypothetical proteins, which are conserved in most genomes of the subset and not conserved in most of the other genomes. A display produced by the Pinned Regions tool in WIT, aligning chromosomal contigs by one gene of the pathway (Fig. 3), illustrates the efficacy of clustering analysis. As mentioned above, this tool searches for ORFs with sequence similarity, which occur in the neighborhood of the pinned gene, in any genome of the database. Remarkably, by selecting only one enzyme of the pathway (in this case, 3-dehydroquinate synthase), all of the other participating enzymes are revealed. Not a single false positive or false negative is produced, a situation which occurs frequently. Not surprisingly, the only strong candidate gene revealed by this tool in archaeal genomes (Fig. 1 and 3) was experimentally proven to encode a missing shikimate kinase.
All types of evidence taken together—metabolic reconstruction of the pathway, chromosomal clustering, and determined kinetic parameters similar to those of bacterial enzymes—suggest strongly that the RMJ07785 protein of M. jannaschii and its orthologs in other archaea perform a functional role of shikimate kinase in vivo (this possibility was also discussed by Graham et al. [12]). As mentioned above, the RMJ07785 protein belongs to the GHMP-kinase superfamily, while bacterial and eukaryotic shikimate kinases belong to a structurally unrelated NMP-kinase family (2, 19). Among various members of the GHMP-kinase superfamily, homoserine kinase displays the closest sequence similarity with RMJ07785. In M. jannaschii, a homoserine kinase function is assigned to another protein, RMJ01903. We verified this assignment experimentally and demonstrated that the two structurally related GHMP-kinases, RMJ07785 and RMJ01903, have no overlapping activity but rather display a very stringent preference for their specific substrates, shikimate and homoserine, correspondingly.
The identification of a new substrate specificity for the GHMP-kinase superfamily provides another illustration of the remarkable ability of this fold to accommodate different types of activity, including mevalonate pyrophosphate decarboxylase (40) and isopentenyl monophosphate kinase (21), which were recently added to the list. This suggests that members of this superfamily arose from a common ancestor by gene duplication, followed by development of divergent substrate preferences. In many cases such specialized genes are found within their functional clusters (operons). This plasticity with respect to the structure of a variable phosphoryl acceptor is reflected in very divergent sequences of GHMP-kinases. Only a few structural elements are conserved between archaeal shikimate kinases and homoserine kinases. Two major segments (segment 1, PX3GLGSSAA; segment 2, [S/T]GSGPS) conserved in homoserine kinases and involved with formation of ATP-binding site (42) are also well conserved in archaeal shikimate kinases, as seen in Fig. 4. Therefore, a sequence comparison with databases can effectively identify novel uncharacterized members of the GHMP-kinase superfamily, but it will most likely not indicate a specificity for a phosphoryl acceptor.
One approach to infer protein functionality based on detection of fused proteins, the so-called Rosetta Stone method, was recently described (26). This method is particularly useful for eukaryotic genomes, which appear to have a tendency to use fusion proteins instead of gene clusters. For example, five of the seven proteins of chorismate biosynthesis in Saccharomyces cerevisiae are fused into one pentafunctional protein (Fig. 1) (6). In prokaryotes, however, fusion of functionally related genes may be viewed simply as an extreme case of clustering on the chromosome. As illustrated in the Fig. 3, bifunctional fusion proteins occur in Chlamydia trachomatis and T. maritima, while in other microbial genomes the same genes are clustered on the chromosome. An attempt to apply the Rosetta Stone method would fail to produce a candidate for a shikimate kinase within the available data for archaeal genomes.
Proximity on the chromosome was traditionally used to predict gene functionality mostly within a concept of operons. Our approach of using clustering on the chromosome to infer functional coupling is related but not equivalent to the traditional paradigm. The operon evidence is usually considered in a context of transcriptional coregulation and only within a particular organism of interest. However, clustering of functionally related genes in any given genome is a possibility, not a certainty. Therefore, we use an overall tendency of genes to cluster on the chromosome as statistically accumulated evidence across the whole phylogenetic space, without necessarily implying any coregulation (31).
The number and phylogenetic diversity of analyzed genomes are the key factor behind the efficiency of our approach. The strong overall tendency of chorismate biosynthetic genes to cluster as discussed above is almost undetectable in E. coli or Bacillus subtilis (Fig. 1). Similarly, no candidate gene for shikimate kinase could be found in analyses of the M. jannaschii and a few other available archaeal genomes, in which chorismate biosynthetic genes were scattered randomly along the chromosome. Later addition of such genomes as those of P. abyssi and A. pernix produced sufficient evidence that could be extrapolated to the whole subset of archaeal genomes.
Finally, extensive operon-like clustering, as observed in this case, is very helpful, but it is not a strict requirement for a successful implementation of the technique. For example, with very limited clustering evidence, we were able to predict and experimentally verify a candidate gene for a missing bacterial nicotinate mononucleotide adenylyltransferase (O. Kurnasov and A. Osterman, unpublished results). Thus, in general, analysis of gene clustering on the chromosome provides an efficient technique to address chemical and biological functions of hypothetical proteins.
ACKNOWLEDGMENTS
We greatly appreciate the productive discussion and structural alignments provided by Nick Grishin and Hong Zhang. We also thank Iain Anderson for critical reading of the manuscript and discussion. We are grateful to Alice Park and Scott Mackey for technical assistance with DNA sequencing and for use of robotic equipment for kinetic analysis.
REFERENCES
- 1.Blattner F R, Plunkett III G, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, Gregor J, Davis N W, Kirkpatrick H A, Goeden M A, Rose D J, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 2.Bork P, Sander C, Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2:31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, Sutton G G, Blake J A, FitzGerald L M, Clayton R A, Gocayne J D, Kerlavage A R, Dougherty B A, Tomb J F, Adams M D, Reich C I, Overbeek R, Kirkness E F, Weinstock K G, Merrick J M, Glodek A, Scott J L, Geoghagen N S M, Venter J C. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 4.Cordwell S J. Microbial genomes and “missing” enzymes: redefining biochemical pathways. Arch Microbiol. 1999;172:269–279. doi: 10.1007/s002030050780. [DOI] [PubMed] [Google Scholar]
- 5.De Feyter R. Shikimate kinases from Escherichia coli K12. Methods Enzymol. 1987;142:355–361. doi: 10.1016/s0076-6879(87)42047-8. [DOI] [PubMed] [Google Scholar]
- 6.Duncan K, Edwards R M, Coggins J R. The pentafunctional arom enzyme of Saccharomyces cerevisiae is a mosaic of monofunctional domains. Biochem J. 1987;246:375–386. doi: 10.1042/bj2460375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ely B, Pittard J. Aromatic amino acid biosynthesis: regulation of shikimate kinase in Escherichia coli K-12. J Bacteriol. 1979;138:933–943. doi: 10.1128/jb.138.3.933-943.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraser C M, Norris S J, Weinstock G M, White O, Sutton G G, Dodson R, Gwinn M, Hickey E K, Clayton R, Ketchum K A, Sodergren E, Hardham J M, McLeod M P, Salzberg S, Peterson J, Khalak H, Richardson D, Howell J K, Chidambaram M, Utterback T, McDonald L, Artiach P, Bowman C, Cotton M D, Venter J C, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 9.Galperin M Y, Koonin E V. Functional genomics and enzyme evolution. Homologous and analogous enzymes encoded in microbial genomes. Genetica. 1999;106:159–170. doi: 10.1023/a:1003705601428. [DOI] [PubMed] [Google Scholar]
- 10.Galperin M Y, Koonin E V. Who's your neighbor? New computational approaches for functional genomics. Nat Biotechnol. 2000;18:609–613. doi: 10.1038/76443. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez J M, Masuchi Y, Robb F T, Ammerman J W, Maeder D L, Yanagibayashi M, Tamaoka J, Kato C. Pyrococcus horikoshii sp. nov., a hyperthermophilic archaeon isolated from a hydrothermal vent at the Okinawa Trough. Extremophiles. 1998;2:123–130. doi: 10.1007/s007920050051. [DOI] [PubMed] [Google Scholar]
- 12.Graham, D. E., N. Kyrpides, I. J. Anderson, R. Overbeek, and W. B. Whitman. Genome of Methanocaldococcus (Methanococcus) jannaschii. Methods Enzymol., in press. [DOI] [PubMed]
- 13.Huang K X, Scott A I, Bennett G N. Overexpression, purification, and characterization of the thermostable mevalonate kinase from Methanococcus jannaschii. Protein Expr Purif. 1999;17:33–40. doi: 10.1006/prep.1999.1106. [DOI] [PubMed] [Google Scholar]
- 14.Huo X, Viola R E. Functional group characterization of homoserine kinase from Escherichia coli. Arch Biochem Biophys. 1996;330:373–379. doi: 10.1006/abbi.1996.0264. [DOI] [PubMed] [Google Scholar]
- 15.Huo X, Viola R E. Substrate specificity and identification of functional groups of homoserine kinase from Escherichia coli. Biochemistry. 1996;35:16180–16185. doi: 10.1021/bi962203z. [DOI] [PubMed] [Google Scholar]
- 16.Kawarabayasi Y, Hino Y, Horikawa H, Yamazaki S, Haikawa Y, Jin-no K, Takahashi M, Sekine M, Baba S, Ankai A, Kosugi H, Hosoyama A, Fukui S, Nagai Y, Nishijima K, Nakazawa H, Takamiya M, Masuda S, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Kikuchi H, et al. Complete genome sequence of an aerobic hyper-thermophilic crenarchaeon, Aeropyrum pernix K1. DNA Res. 1999;6:83–101. doi: 10.1093/dnares/6.2.83. , 145–152. [DOI] [PubMed] [Google Scholar]
- 17.Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, Yamamoto S, Sekine M, Baba S, Kosugi H, Hosoyama A, Nagai Y, Sakai M, Ogura K, Otsuka R, Nakazawa H, Takamiya M, Ohfuku Y, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Aoki K, Kikuchi H. Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res. 1998;5:55–76. doi: 10.1093/dnares/5.2.55. [DOI] [PubMed] [Google Scholar]
- 18.Klenk H P, Clayton R A, Tomb J F, White O, Nelson K E, Ketchum K A, Dodson R J, Gwinn M, Hickey E K, Peterson J D, Richardson D L, Kerlavage A R, Graham D E, Kyrpides N C, Fleischmann R D, Quackenbush J, Lee N H, Sutton G G, Gill S, Kirkness E F, Dougherty B A, McKenney K, Adams M D, Loftus B, Venter J C, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 19.Krell T, Coggins J R, Lapthorn A J. The three-dimensional structure of shikimate kinase. J Mol Biol. 1998;278:983–997. doi: 10.1006/jmbi.1998.1755. [DOI] [PubMed] [Google Scholar]
- 20.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, Bertero M G, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell S C, Bron S, Brouillet S, Bruschi C V, Caldwell B, Capuano V, Carter N M, Choi S K, Codani J J, Connerton I F, Danchin A, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 21.Lange B M, Croteau R. Isopentenyl diphosphate biosynthesis via a mevalonate-independent pathway: isopentenyl monophosphate kinase catalyzes the terminal enzymatic step. Proc Natl Acad Sci USA. 1999;96:13714–13719. doi: 10.1073/pnas.96.24.13714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee M, Leustek T. Identification of the gene encoding homoserine kinase from Arabidopsis thaliana and characterization of the recombinant enzyme derived from the gene. Arch Biochem Biophys. 1999;372:135–142. doi: 10.1006/abbi.1999.1481. [DOI] [PubMed] [Google Scholar]
- 23.Lobner-Olesen A, Marinus M G. Identification of the gene (aroK) encoding shikimic acid kinase I of Escherichia coli. J Bacteriol. 1992;174:525–529. doi: 10.1128/jb.174.2.525-529.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maeder D L, Weiss R B, Dunn D M, Cherry J L, Gonzalez J M, DiRuggiero J, Robb F T. Divergence of the hyperthermophilic archaea Pyrococcus furiosus and P. horikoshii inferred from complete genomic sequences. Genetics. 1999;152:1299–1305. doi: 10.1093/genetics/152.4.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makarova K S, Aravind L, Galperin M Y, Grishin N V, Tatusov R L, Wolf Y I, Koonin E V. Comparative genomics of the Archaea (Euryarchaeota): evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 1999;9:608–628. [PubMed] [Google Scholar]
- 26.Marcotte E M, Pellegrini M, Ng H L, Rice D W, Yeates T O, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 27.McConkey G A. Targeting the shikimate pathway in the malaria parasite Plasmodium falciparum. Antimicrob Agents Chemother. 1999;43:175–177. doi: 10.1128/aac.43.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mewes H W, Albermann K, Bahr M, Frishman D, Gleissner A, Hani J, Heumann K, Kleine K, Maierl A, Oliver S G, Pfeiffer F, Zollner A. Overview of the yeast genome. Nature. 1997;387:7–65. doi: 10.1038/42755. [DOI] [PubMed] [Google Scholar]
- 29.Nelson K E, Clayton R A, Gill S R, Gwinn M L, Dodson R J, Haft D H, Hickey E K, Peterson J D, Nelson W C, Ketchum K A, McDonald L, Utterback T R, Malek J A, Linher K D, Garrett M M, Stewart A M, Cotton M D, Pratt M S, Phillips C A, Richardson D, Heidelberg J, Sutton G G, Fleischmann R D, Eisen J A, Fraser C M, et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 30.Osterman A, Grishin N V, Kinch L N, Phillips M A. Formation of functional cross-species heterodimers of ornithine decarboxylase. Biochemistry. 1994;33:13662–13667. doi: 10.1021/bi00250a016. [DOI] [PubMed] [Google Scholar]
- 31.Overbeek R, Fonstein M, D'Souza M, Pusch G D, Maltsev N. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Overbeek R, Larsen N, Pusch G D, D'Souza M, Selkov E, Jr, Kyrpides N, Fonstein M, Maltsev N, Selkov E. WIT: integrated system for high-throughput genome sequence analysis and metabolic reconstruction. Nucleic Acids Res. 2000;28:123–125. doi: 10.1093/nar/28.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patte J C. Biosynthesis of threonine and lysine. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C.: ASM Press; 1996. pp. 528–541. [Google Scholar]
- 34.Pittard A J. Biosynthesis of the aromatic amino acids. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C.: ASM Press; 1996. pp. 458–484. [Google Scholar]
- 35.Roberts F, Roberts C W, Johnson J J, Kyle D E, Krell T, Coggins J R, Coombs G H, Milhous W K, Tzipori S, Ferguson D J, Chakrabarti D, McLeod R. Evidence for the shikimate pathway in apicomplexan parasites. Nature. 1998;393:801–805. doi: 10.1038/31723. [DOI] [PubMed] [Google Scholar]
- 36.Rudolph F B, Fromm H J. Plotting methods for analyzing enzyme rate data. Methods Enzymol. 1979;63:138–159. doi: 10.1016/0076-6879(79)63009-4. [DOI] [PubMed] [Google Scholar]
- 37.Selkov E, Maltsev N, Olsen G J, Overbeek R, Whitman W B. A reconstruction of the metabolism of Methanococcus jannaschii from sequence data. Gene. 1997;197:GC11–GC26. doi: 10.1016/s0378-1119(97)00307-7. [DOI] [PubMed] [Google Scholar]
- 38.Smith D R, Doucette-Stamm L A, Deloughery C, Lee H, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbert K, Harrison D, Hoang L, Keagle P, Lumm W, Pothier B, Qiu D, Spadafora R, Vicaire R, Wang Y, Wierzbowski J, Gibson R, Jiwani N, Caruso A, Bush D, Reeve J N, et al. Complete genome sequence of Methanobacterium thermoautotrophicum ΔH: functional analysis and comparative genomics. J Bacteriol. 1997;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephens R S, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov R L, Zhao Q, Koonin E V, Davis R W. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 40.Toth M J, Huwyler L. Molecular cloning and expression of the cDNAs encoding human and yeast mevalonate pyrophosphate decarboxylase. J Biol Chem. 1996;271:7895–7898. doi: 10.1074/jbc.271.14.7895. [DOI] [PubMed] [Google Scholar]
- 41.Tumbula D L, Teng Q, Bartlett M G, Whitman W B. Ribose biosynthesis and evidence for an alternative first step in the common aromatic amino acid pathway in Methanococcus maripaludis. J Bacteriol. 1997;179:6010–6013. doi: 10.1128/jb.179.19.6010-6013.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou, T., M. Daugherty, N. V. Grishin, A. L. Osterman, and H. Zhang. Structure, in press.