

Abstract
A variety of phenotypic categorisations have been developed for sarcoidosis. Phenotyping has been used for genetics studies and to guide treatment selection. The authors participated in a Delphi expert consensus panel to develop a proposed phenotype categorisation and treatment recommendations for pulmonary sarcoidosis patients. Panellists reached consensus that asymptomatic patients with normal pulmonary function and adenopathy alone or normal chest imaging do not require therapy, while symptomatic patients with impaired pulmonary function or infiltrates should be treated. The panel did not reach consensus on asymptomatic patients with abnormal chest imaging or reduced pulmonary function, or symptomatic patients with normal chest imaging and pulmonary function. The proposed phenotype categories and associated treatment recommendations are asymptomatic (no therapy), acute (disease duration <1–2 years, apparently self-limited, corticosteroids), chronic (antimetabolites and other second-line therapies) and advanced (biologics). Some clinical settings, such as dyspnoea/hypoxaemia at rest, severely impaired or rapidly decreasing pulmonary function tests, and severe cardiac, neurologic, ocular or renal involvement warrant immediate therapy.
Short abstract
Expert Delphi consensus recommendations on clinical phenotyping to guide therapy for pulmonary sarcoidosis – asymptomatic: no therapy; acute: corticosteroids; chronic: cytotoxics plus other second line; advanced: biologics plus other third line. http://bit.ly/38Lio7U
Introduction
Sarcoidosis is a multi-organ disease with a variable clinical outcome [1]. Because of this, researchers and healthcare providers have developed phenotypic categories to help identify distinct populations of sarcoidosis patients [2]. These phenotypic categories have been employed by both researchers and clinicians. For example, Löfgren syndrome was originally described because it was a clinical phenotype associated with an excellent prognosis. However, genetic studies demonstrated a high frequency of certain human leukocyte antigen (HLA) genotypes associated with a good clinical prognosis in patients with Löfgren syndrome [3]. Phenotyping is a dynamic process that evolves as further disease characteristics, biomarkers and genetic information are discovered. As our understanding of phenotypes improves, the use of phenotyping for clinical and/or research purposes may prove increasingly useful for many aspects of sarcoidosis.
History of clinical phenotyping in sarcoidosis
One of the most widely used clinical phenotype categorisations was developed based on chest radiograph patterns of patients with pulmonary sarcoidosis. In 1960 and 1961, Dr Wurm in Germany [4] and Dr Scadding in Scotland [5] reported independently that different combinations of the presence or absence of hilar adenopathy and parenchymal infiltrates were associated with different outcomes in chest radiograph over time. Scadding [5] added a fourth pattern and the Scadding stages of the chest radiograph have been used in practice for years.
Around the same time, Dr Löfgren noted that disease often resolved within 3–6 months in patients who presented with erythema nodosum and hilar adenopathy. A modification of Löfgren syndrome was suggested by Grunewald and Ecklund [6]. They found that periarticular arthralgia had the same prognosis as the presence of erythema nodosum [6]. They also noted that the use of HLA genotyping to predict prognosis was the same for those who presented with erythema nodosum as with periarticular arthralgias [3].
Another early proposed clinical phenotype was based on duration and clinical outcome of disease. Neville et al. [7] studied the clinical course of over 800 sarcoidosis patients followed at their clinic for >2 years. They noted that some patients had resolution of their disease within 2 years (acute disease) versus others who had persistent disease beyond 2 years (chronic disease) [7]. This simple division of patients into acute and chronic phenotypes has been used for many years by clinicians as they decide which patients may require long-term therapy.
In the past decade, several other phenotypes have been proposed [2]. Many of these were developed for genetic studies, but in other cases phenotypic categorisation has been used to define populations for specific treatments. A comparison of phenotype scales for sarcoidosis reported in literature is presented in table 1. From table 1, we note three papers which examined genotype/phenotype relations. Two studies by Grunewald and co-workers [3, 8] investigated HLA-DR markers in sarcoidosis patients with Löfgren syndrome and were able to differentiate patients with an excellent prognosis versus those with a 50% chance of having chronic disease. The study by Schupp et al. [15] identified clustering of symptoms so that further studies of genetic markers could be done on those patients with different clusters of organ disease.
TABLE 1.
Phenotype scales reported in sarcoidosis
| First author [ref.] | Phenotype | Description of developing phenotype | Patients n | Validation | Single versus multi-centre | Established association to genotype | Comments |
| Scadding [5] | Pulmonary | Chest radiograph stage | 136 | No | Single | No | Did not use computed tomography |
| Neville [7] | Acute versus chronic | Rate of resolution | 818 | No | Single | No | Did not account for more than one factor in patient |
| Grunewald [3, 8] | Löfgren versus non-Löfgren | Clinical outcome of Löfgren patients | 754 | Yes | Single | Yes | Series of studies, may not apply to other racial groups |
| Wasfi [9] | Severe versus non-severe | Backward regression based on clinical parameters | 104 | Two different groups of clinicians assessed same patients | Single | No | Based on expert opinion, no follow-up analysis |
| Prasse [10] | Acute versus chronic and duration of treatment | Evaluated patients seen within 1 year of presentation | 225 | No | Single | No | More useful for acute disease |
| Baughman[11] | Acute versus chronic including therapy | Developed criteria for long-term outcome | 500 | No | Multi-centre | No | Retrospective look based on expert opinion |
| Rodrigues[12] | Acute, relapse, fibrosis | Factor analysis | 137 | No | Multi-centre | No | Phenotypes were not distinct |
| Walsh [13] | Severe versus non-severe | Regression analysis of multiple factors | 251 | Yes (additional 252) | Single | No | Only focused on advanced pulmonary disease |
| Moller [14] | Various groups of patients | Expert opinion of grouping of patients | Not given | No | Multi-centre | Yes | Established criteria to be studied |
| Schupp [15] | Organ clustering | Multi-factor analysis based on gene expression | 2163 | No | Multi-centre | Yes | Used genetic profile to identify associated organ manifestations |
Phenotyping for genetic studies
As noted, clinical phenotyping can help better understand the genetics of a disease. For example, cystic fibrosis is the result of the mutation of one gene, cystic fibrosis transmembrane conductance regulator (CFTR). Over the past few years, thousands of mutations of this gene have been identified, and different mutations of CFTR have been shown to lead to different clinical outcomes. In addition, other genes may modify expression of CFTR [16]. Sarcoidosis is a condition in which multiple genes may be responsible for disease and prognosis [17]. Nevertheless, the first step would seem to be to define homogenous populations among sarcoidosis patients; then, certain genes may be identified which are associated with specific clinical presentations or outcomes.
One phenotype of sarcoidosis is Löfgren syndrome. Several groups found that Löfgren syndrome associated with HLA-DQB1*0201 had a good prognosis, especially in European patients [18, 19]. Further studies have shown that Swedish patients with Löfgren syndrome and another HLA marker, DRB1*0301, have a highly favourable prognosis and rarely require any therapy [3, 6]. This has led to the use of HLA phenotyping for treatment decisions in some clinics.
Two recent phenotypes have been specifically described to help with genetic studies. In the study by Schupp et al. [15], a large population of sarcoidosis patients from various clinics in Europe were evaluated. The authors grouped the clinical features of these patients and identified some associations. For example, they found that patients with eye disease often have neurologic disease. They also found that patients with neurologic disease had a higher frequency of cardiac disease, as noted by others [20]. These clusters may prove useful as researchers are trying to determine genotypes associated with phenotypes.
The Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis study developed a phenotype proposal predicting prognosis for patients in whom multiple specimens had been obtained [14]. This study promises to provide detailed genetic information regarding these different phenotypes.
Phenotyping for treatment decisions
Phenotyping patients may also prove useful for decisions about therapy. The authors participated in a Delphi expert consensus panel to develop consensus recommendations for phenotyping and appropriate therapies for different phenotypes. The design and results of the modified Delphi consensus process are presented elsewhere in this issue of the European Respiratory Review [21]. The Delphi group included 26 panellists. This panel size is typical for Delphi consensus studies and reflects a balance between the need to sample a wide range of expert opinion and the difficulty in recruiting experts and coordinating the panel. The Delphi process developed the proposed phenotype categorisation and treatment recommendations described here and shown in supplementary appendix 1.
The proposed phenotyping is based on clinical outcomes and is derived from the World Association of Sarcoidosis and other Granulomatous Disorders Clinical Outcome Status [11]. The Clinical Outcome Status evaluates patients 2–5 years after diagnosis and places them into nine general categories based on therapy and current status. Patients can be classified as having either no or minimal disease (<25% of maximal disease), persistent disease or worsening disease. The classifications for therapy include never, none in the prior year, on therapy and stable, or on therapy and increased in prior year.
The phenotype categorisation in supplementary appendix 1 is derived from prior reported categories. One phenotype is the asymptomatic group. These patients have no symptoms and do not require any systemic therapy. The definition of symptomatic disease was left to the individual expert. Most patients who present with no symptoms rarely require therapy over time. This group is similar to the asymptomatic acute patients described by Prasse et al. [10]. It is also equivalent to groups 1, 3 and 5 from the World Association of Sarcoidosis and other Granulomatous Disorders Clinical Outcome Status [11].
The acute patients who present with symptoms are those within the first 1–2 years after diagnosis who appear to have self-limited disease. These patients are usually initially treated with prednisone. For these patients, the initial decision to start therapy is based on a combination of factors, including symptoms, chest imaging and pulmonary function testing.
The Delphi expert panel reached consensus regarding treatment for patients based on pulmonary function, chest imaging and symptoms (figure 1). Specifically, for symptomatic patients, there was consensus that a patient with a forced vital capacity (FVC) or diffusing capacity of the lung for carbon monoxide (DLCO) ≤80% should be considered for therapy. For the asymptomatic patient, there was consensus that those with normal DLCO or FVC did not need treatment. The panel did not reach consensus on asymptomatic patients with reduced pulmonary function or symptomatic patients with normal pulmonary function. Sarcoidosis patients should be evaluated initially and over time, and serial testing may indicate worsening disease. This was addressed in the Delphi and the experts felt that changes in pulmonary function testing were more important than the absolute value of the individual pulmonary function test.
FIGURE 1.

Delphi consensus on whether patients should be treated based on the presence of pulmonary symptoms, pulmonary function testing and imaging. Bold indicates consensus. HRCT: high-resolution computed tomography; PFT: pulmonary function test; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide.
With regard to imaging studies, there are no generally accepted criteria for an abnormal high-resolution computed tomography. Therefore, an abnormal high-resolution computed tomography was defined by the individual expert. The Delphi panel reached consensus that symptomatic patients with parenchymal infiltrates (Scadding stage 2 or 3) or fibrosis (Scadding stage 4) should be treated. Asymptomatic patients with adenopathy alone (Scadding stage 1) or normal chest radiograph do not require therapy. In evaluating the results of high-resolution computed tomography scans, the panel felt symptomatic patients with abnormal findings or fibrosis should be treated and asymptomatic patients with adenopathy alone do not require therapy. The panel did not reach consensus on asymptomatic patients with abnormal chest imaging or symptomatic patients with normal chest imaging. While we did explore criteria for moderate and severe individual findings in FVC, DLCO and Scadding stage, we did not further explore what criteria would be considered severe disease. We left this definition to the individual expert. Future studies aimed at defining specific thresholds for these criteria would be useful.
For sarcoidosis patients, the clinician should consider several factors, including extrapulmonary disease. The Delphi panel reached consensus on several statements dealing with extrapulmonary disease, summarised in figure 2. In general, the panel agreed that treatment should be considered for patients with symptomatic extrapulmonary disease, and that extrapulmonary disease can be a useful marker of sarcoidosis activity. The panellists also agreed that not all extrapulmonary manifestations were created equal. Cardiac, neurologic, calcium, eye and renal involvement should be treated regardless of whether the patient is symptomatic. However, skin or liver involvement does not require treatment unless the patient is symptomatic. Hypercalcaemia, a common cause of renal dysfunction in sarcoidosis, should also be treated.
FIGURE 2.
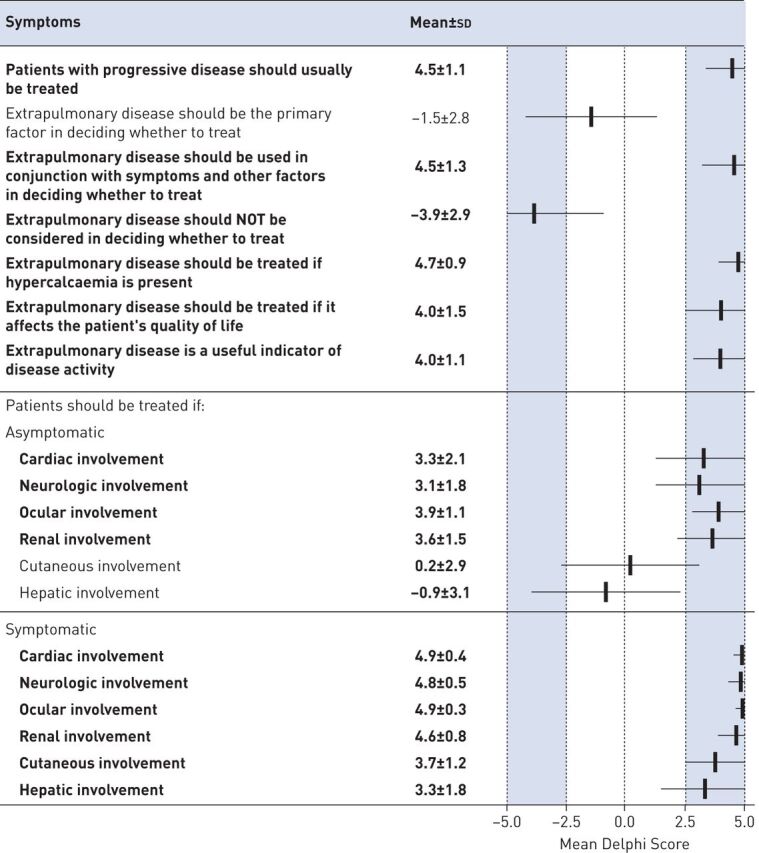
Delphi consensus on the role of extrapulmonary disease in treatment decisions. Bold indicates consensus.
Appendix 2 summaries features which the panel felt were important factors that should influence the decision to initiate therapy. These include patient preference, symptom severity and quality of life impact, Imaging studies, pulmonary function tests, extrapulmonary disease, pulmonary hypertension, need for oxygen, progressive disease, stability of disease, cardiac magnetic resonance imaging and ophthalmologic examination. The panel also reached consensus on several factors that should be considered in decision to treat. These are summarised in supplementary appendix 2. These factors included pulmonary function testing, chest imaging, cardiac magnetic resonance imaging and results of the eye examination. Factors for which there was no consensus regarding decision to treat include the following: ACE levels, CBC/lymphocyte panel, comprehensive metabolic panel, ECG, Holter monitoring, echocardiography, erythrocyte sedimentation rate and C-reactive protein, liver function tests, long-duration disease, lysozyme, positron emission tomography scan, soluble interleukin-2, urinalysis/urinary calcium, and vitamin D and metabolites. Figure 3 shows clinical situations for which the panel agreed that immediate therapy is required.
FIGURE 3.

Delphi consensus on clinical situations in which immediate therapy is warranted. PFT: pulmonary function test; DLCO: diffusing capacity of the lung for carbon monoxide; FVC: forced vital capacity; FEV1: forced expiratory volume in 1 s.
Prolonged use of corticosteroids is associated with significant toxicity, including impaired quality of life [22]. Therefore, the decision to begin steroid-sparing agents is usually made when it becomes clear that a patient will have chronic disease. Several studies have attempted to characterise patients with chronic disease, including those by Neville et al. [7], Wasfi et al. [9] and Prasse et al. [10]. For example, patients who have been started on corticosteroids have a >50% chance of requiring long-term therapy [23, 24]. Clinical Outcome Status categories 2, 4 and 6 include both acute and chronic phenotypes. Patients in these categories who were treated for >2 years would be considered chronic. It has been shown that patients who present with certain features, such as neurosarcoidosis or FVC <80%, are more likely to have chronic disease [25].
The Delphi panel reached consensus on when the patient should be considered to have chronic disease and be considered for second-line therapy, typically non-biologic cytotoxic therapies such as methotrexate, azathioprine, leflunomide or mycophenolate (figure 4). The dose and duration of glucocorticoid use were major determinants of when to switch to non-biologics. While methotrexate and azathioprine were the most commonly cited non-biologics, specific details regarding individual non-biologics are discussed elsewhere in this issue of the European Respiratory Review [26].
FIGURE 4.

Delphi consensus on clinical situations in which second-line therapies (usually non-biologic cytotoxic agents) and third-line therapies (usually biologic agents) should be considered. Bold indicates consensus.
The final phenotypic category in supplementary appendix 1 is the patient with advanced disease. This group includes those who have stable disease but require ≥10 mg of prednisone, often with other, non-biologic treatments as well as those refractory patients who required increasing treatment in the prior year, corresponding to Clinical Outcome Status groups 7, 8 and 9 [11]. For these patients, third-line treatments are often used, including infliximab, adalimumab, rituximab and repository corticotrophin injection. For the purpose of the Delphi consensus, all four of these agents were classified as biologics. Figure 4 shows those factors for which the panellists reached consensus about adding or switching to a biologic agent. Failure or toxicity of steroids with non-biologics was the major factor driving panellists to consider biologics.
Conclusion
Clinical phenotyping of sarcoidosis patients has many potential benefits in studying and treating sarcoidosis patients. In our Delphi study, we developed a clinical phenotype schema which can be used to help direct therapy. The Delphi participants reached consensus on when patients should be considered for different levels of treatment. These levels of therapy, including specific agents to be considered, are summarised in supplementary appendix 3.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary_appendix ERR-0145-2019_Supplementary_appendix (120.6KB, pdf)
Footnotes
This article has supplementary material available from err.ersjournals.com
Provenance: Publication of this peer-reviewed article was sponsored by Mallinckrodt Pharmaceuticals, USA (principal sponsor, European Respiratory Review issue 155).
Conflict of interest: R.P. Baughman reports grants and personal fees from Malllinckrodt, Novartis and Celgene, grants from Gilead, Genentech and Bayer, and personal fees from West Pharmaceutical, during the conduct of the study.
Conflict of interest: M.B. Scholand reports other from Boehringer Ingelheim, Genentech, Fibrogen and Global Blood Therapeutics, outside the submitted work. In addition, M.B. Scholand has a patent Apparatus, Compositions and Methods for Assessment of Chronic Obstructive Pulmonary Disease Progression among Rapid and Slow Decline Conditions issued.
Conflict of interest: F.F. Rahaghi reports grants and consulting fees from Mallinckrodt, during the conduct of the study.
References
- 1.Hunninghake GW, Costabel U, Ando M, et al. . ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999; 16: 149–173. [PubMed] [Google Scholar]
- 2.Pereira CA, Dornfeld MC, Baughman R, et al. . Clinical phenotypes in sarcoidosis. Curr Opin Pulm Med 2014; 20: 496–502. doi: 10.1097/MCP.0000000000000077 [DOI] [PubMed] [Google Scholar]
- 3.Grunewald J, Brynedal B, Darlington P, et al. . Different HLA-DRB1 allele distributions in distinct clinical subgroups of sarcoidosis patients. Respir Res 2010; 11: 25. doi: 10.1186/1465-9921-11-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wurm K. The significance of stage classification of sarcoidosis (Boeck's disease). Dtsch Med Wochenschr 1960; 85: 1541–1548. doi: 10.1055/s-0028-1112616 [DOI] [PubMed] [Google Scholar]
- 5.Scadding JG. Prognosis of intrathoracic sarcoidosis in England. A review of 136 cases after five years’ observation. Br Med J 1961; 2: 1165–1172. doi: 10.1136/bmj.2.5261.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grunewald J, Eklund A. Sex-specific manifestations of Lofgren's syndrome. Am J Respir Crit Care Med 2007; 175: 40–44. doi: 10.1164/rccm.200608-1197OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med 1983; 52: 525–533. [PubMed] [Google Scholar]
- 8.Grunewald J, Eklund A. Lofgren's syndrome: human leukocyte antigen strongly influences the disease course. Am J Respir Crit Care Med 2009; 179: 307–312. doi: 10.1164/rccm.200807-1082OC [DOI] [PubMed] [Google Scholar]
- 9.Wasfi YS, Rose CS, Murphy JR, et al. . A new tool to assess sarcoidosis severity. Chest 2006; 129: 1234–1245. doi: 10.1378/chest.129.5.1234 [DOI] [PubMed] [Google Scholar]
- 10.Prasse A, Katic C, Germann M, et al. . Phenotyping sarcoidosis from a pulmonary perspective. Am J Respir Crit Care Med 2008; 177: 330–336. doi: 10.1164/rccm.200705-742OC [DOI] [PubMed] [Google Scholar]
- 11.Baughman RP, Nagai S, Balter M, et al. . Defining the clinical outcome status (COS) in sarcoidosis: results of WASOG Task Force. Sarcoidosis Vasc Diffuse Lung Dis 2011; 28: 56–64. [PubMed] [Google Scholar]
- 12.Rodrigues SC, Rocha NA, Lima MS, et al. . Factor analysis of sarcoidosis phenotypes at two referral centers in Brazil. Sarcoidosis Vasc Diffuse Lung Dis 2011; 28: 34–43. [PubMed] [Google Scholar]
- 13.Walsh SL, Wells AU, Sverzellati N, et al. . An integrated clinicoradiological staging system for pulmonary sarcoidosis: a case-cohort study. Lancet Respir Med 2014; 2: 123–130. doi: 10.1016/S2213-2600(13)70276-5 [DOI] [PubMed] [Google Scholar]
- 14.Moller DR, Koth LL, Maier LA, et al. . Rationale and design of the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) study. Sarcoidosis protocol. Ann Am Thorac Soc 2015; 12: 1561–1571. doi: 10.1513/AnnalsATS.201503-172OT [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schupp JC, Freitag-Wolf S, Bargagli E, et al. . Phenotypes of organ involvement in sarcoidosis. Eur Respir J 2018; 51: 1700991. doi: 10.1183/13993003.00991-2017 [DOI] [PubMed] [Google Scholar]
- 16.Bombieri C, Seia M, Castellani C. Genotypes and phenotypes in cystic fibrosis and cystic fibrosis transmembrane regulator-related disorders. Semin Respir Crit Care Med 2015; 36: 180–193. doi: 10.1055/s-0035-1547318 [DOI] [PubMed] [Google Scholar]
- 17.Iannuzzi MC, Rybicki BA, Maliarik M, et al. . Finding disease genes. From cystic fibrosis to sarcoidosis. Thomas A. Neff Lecture. Chest 1997; 111: Suppl. 6, 70S–73S. [DOI] [PubMed] [Google Scholar]
- 18.Sato H, Grutters JC, Pantelidis P, et al. . HLA-DQB1*0201: a marker for good prognosis in British and Dutch patients with sarcoidosis. Am J Respir Cell Mol Biol 2002; 27: 406–412. doi: 10.1165/rcmb.4782 [DOI] [PubMed] [Google Scholar]
- 19.Grubic Z, Zunec R, Peros-Golubicic T, et al. . HLA class I and class II frequencies in patients with sarcoidosis from Croatia: role of HLA-B8, -DRB1*0301, and -DQB1*0201 haplotype in clinical variations of the disease. Tissue Antigens 2007; 70: 301–306. doi: 10.1111/j.1399-0039.2007.00904.x [DOI] [PubMed] [Google Scholar]
- 20.Lower EE, Broderick JP, Brott TG, et al. . Diagnosis and management of neurological sarcoidosis. Arch Intern Med 1997; 157: 1864–1868. doi: 10.1001/archinte.1997.00440370104011 [DOI] [PubMed] [Google Scholar]
- 21.Rahaghi FF, Baughman RP, Saketkoo LA, et al. . Delphi consensus recommendations for a treatment algorithm in pulmonary sarcoidosis. Eur Respir Rev 2020; 29: 190146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Judson MA, Chaudhry H, Louis A, et al. . The effect of corticosteroids on quality of life in a sarcoidosis clinic: the results of a propensity analysis. Respir Med 2015; 109: 526–531. doi: 10.1016/j.rmed.2015.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baughman RP, Judson MA, Teirstein A, et al. . Presenting characteristics as predictors of duration of treatment in sarcoidosis. QJM 2006; 99: 307–315. doi: 10.1093/qjmed/hcl038 [DOI] [PubMed] [Google Scholar]
- 24.Gottlieb JE, Israel HL, Steiner RM, et al. . Outcome in sarcoidosis. The relationship of relapse to corticosteroid therapy. Chest 1997; 111: 623–631. doi: 10.1378/chest.111.3.623 [DOI] [PubMed] [Google Scholar]
- 25.Baughman RP, Lower EE. Features of sarcoidosis associated with chronic disease. Sarcoidosis Vasc Diffuse Lung Dis 2015; 31: 275–281. [PubMed] [Google Scholar]
- 26.Rahaghi FF, Sweiss NJ, Saketkoo LA, et al. . Management of repository corticotrophin injection therapy for pulmonary sarcoidosis: a Delphi study. Eur Respir Rev 2020; 29: 190147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary_appendix ERR-0145-2019_Supplementary_appendix (120.6KB, pdf)