

Abstract
Infectious agents can trigger autoimmune responses in a number of chronic inflammatory diseases. Lyme arthritis, which is caused by the tick-transmitted spirochaete Borrelia burgdorferi, is effectively treated in most patients with antibiotic therapy; however, in a subset of patients, arthritis can persist and worsen after the spirochaete has been killed (known as post-infectious Lyme arthritis). This Review details the current understanding of the pathogenetic events in Lyme arthritis, from initial infection in the skin, through infection of the joints, to post-infectious chronic inflammatory arthritis. The central feature of post-infectious Lyme arthritis is an excessive, dysregulated pro-inflammatory immune response during the infection phase that persists into the post-infectious period. This response is characterized by high amounts of IFNγ and inadequate amounts of the anti-inflammatory cytokine IL-10. The consequences of this dysregulated pro-inflammatory response in the synovium include impaired tissue repair, vascular damage, autoimmune and cytotoxic processes, and fibroblast proliferation and fibrosis. These synovial characteristics are similar to those in other chronic inflammatory arthritides, including rheumatoid arthritis. Thus, post-infectious Lyme arthritis provides a model for other chronic autoimmune or autoinflammatory arthritides in which complex immune responses can be triggered and shaped by an infectious agent in concert with host genetic factors.
Microbial infections have long been hypothesized to have a role in triggering autoimmunity in chronic inflammatory diseases1. However, the clinical onset of autoimmune disorders often develops over years or decades, making it difficult to establish a causal link between exposure to an infectious trigger and subsequent disease. Uniquely in Lyme arthritis, a late manifestation of Lyme disease, the triggering event, Borrelia burgdorferi infection, is known with certainty.
Lyme disease (also known as Lyme borreliosis) occurs in temperate regions of North America, Europe and Asia (FIG. 1), and causes ~300,000 cases annually in the USA2. Lyme disease is caused by the tick-transmitted spirochaete B. burgdorferi sensu lato (B. burgdorferi in the general sense), which consists of 20 different species2. However, the human infection is caused primarily by three species, B. burgdorferi sensu stricto (B. burgdorferi in the strict sense, hereafter called B. burgdorferi) in the USA, and Borrelia afzelii and Borrelia garinii in Europe and Asia. Less common species that can infect humans include Borrelia mayonii in upper midwestern USA, Borrelia bavariensis (which is closely related to B. garinii) in Europe and Asia, and B. burgdorferi in Europe. Each species or subtype is associated with distinct clinical features; for example, the common subtypes of B. burgdorferi that are found in north-eastern USA are particularly arthritogenic, whereas B. garinii and B. afzelii rarely cause Lyme arthritis2.
Fig. 1 |. Geographic distribution of Borrelia burgdorferi species relevant to human disease.
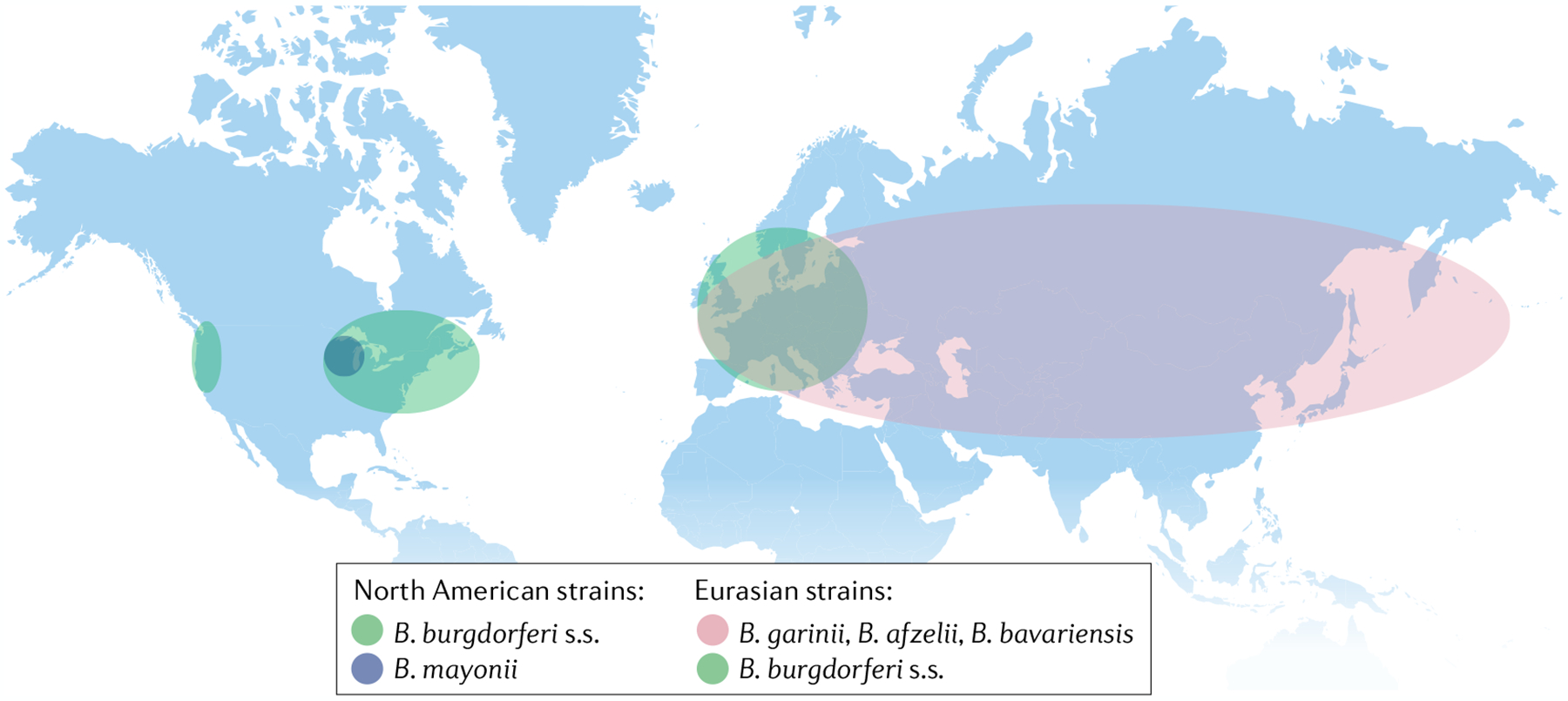
Borrelia burgdorferi sensu stricto (B. burgdorferi s.s.) is the major species in North America and is primarily found in the USA in the north-eastern and mid-Atlantic states, the upper Midwest, in northern California and, to a lesser degree, in Oregon and Washington. B. burgdorferi s.s. also extends into Canada at each of the bordering USA locations. Borrelia mayonii is much less common than B. burgdorferi s.s. and is restricted to the upper midwestern states in the USA. European strains include Borrelia garinii and Borrelia afzelii and, to a lesser extent, B. burgdorferi s.s. and Borrelia bavariensis, which is closely related to B. garinii. In Asia, B. garinii is the predominant species, but B. bavariensis and B. afzelii are also found there.
Infection with B. burgdorferi usually begins with an expanding erythema migrans skin lesion, which develops at the site of the tick bite2 (FIG. 2a). Within weeks, spirochaetal strains from north-eastern USA can disseminate to a number of sites3, a process that is often accompanied by flu-like symptoms and can be shortly followed by organ-specific involvement, particularly neurological or cardiac abnormalities4. Months later, many patients develop Lyme arthritis, which is characterized by intermittent or persistent joint swelling and pain, primarily in large joints (especially the knees) for a period of several years5,6. In some patients, early infection is asymptomatic and Lyme arthritis is the presenting manifestation of Lyme disease.
Fig. 2 |. Lyme arthritis stages and characteristics.
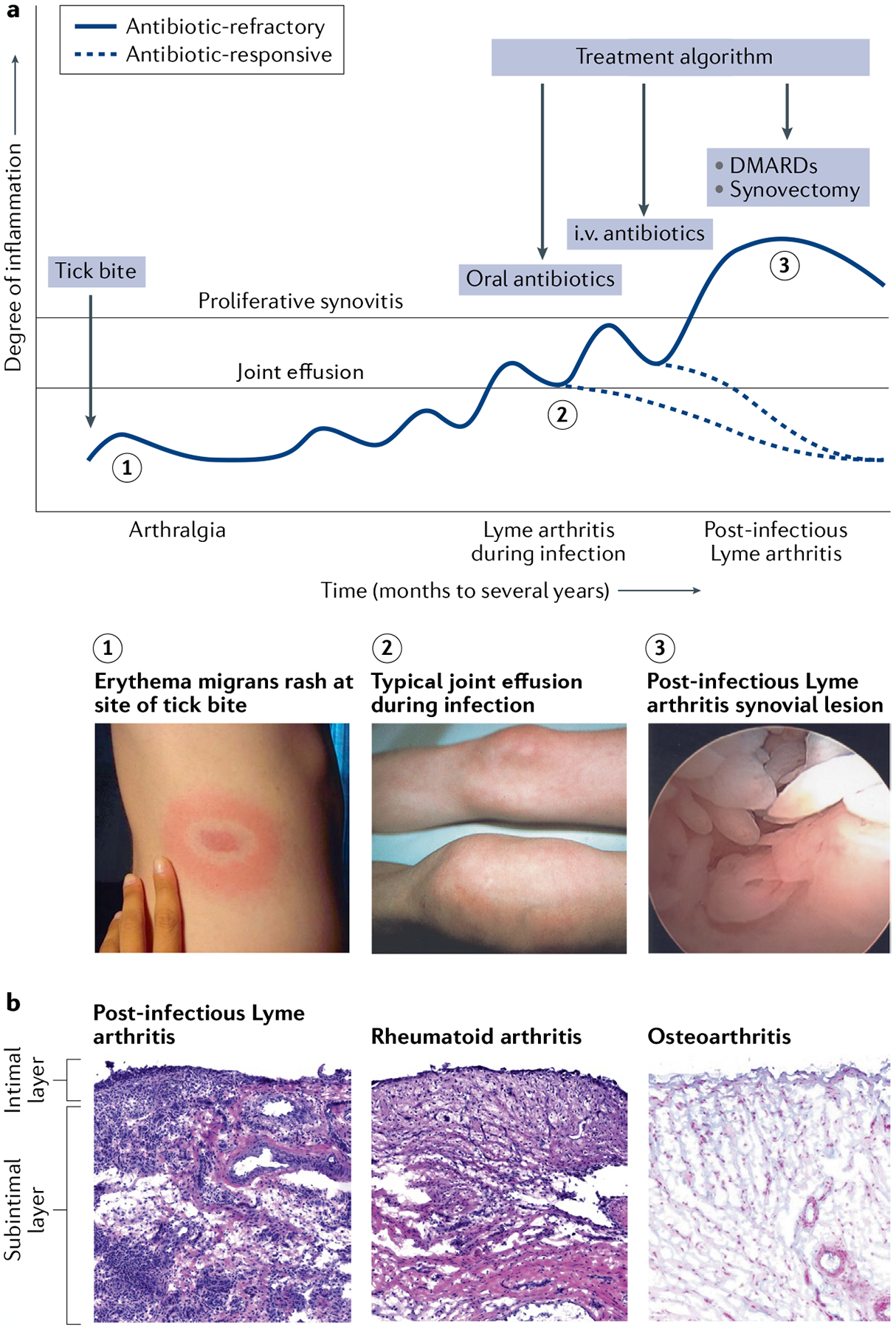
a | In untreated patients, Lyme disease occurs in stages, with different manifestations present at each stage. A slowly expanding erythema migrans rash commonly appears 3 to 32 days after a bite by a Borrelia burgdorferi-infected Ixodes tick (1), which can be accompanied by flu-like symptoms such as fever, headache, myalgias, arthralgias, malaise and fatigue. In the north-eastern states of the USA, Lyme arthritis typically causes large joint effusions, particularly affecting the knees (2), which develop a median of 6 months after the initial skin lesion. Arthritis usually resolves after 1–3 months of oral and, if necessary, intravenous (i.v.) antibiotic therapy (antibiotic-responsive Lyme arthritis). In a small subset of patients, arthritis persists or worsens despite 2–3 months of antibiotic therapy and apparent spirochaetal killing (post-infectious Lyme arthritis). These patients typically develop a highly proliferative synovial lesion (3) that does not respond to further courses of antibiotic therapy (antibiotic-refractory Lyme arthritis). Treatments such as DMARDs or arthroscopic synovectomy help to resolve their arthritis. b | The synovial lesion in post-infectious Lyme arthritis is similar to the lesion in rheumatoid arthritis and other inflammatory arthritides. By contrast, osteoarthritis synovium typically has minimal cellular infiltrate, the intimal layer is not inflamed or thickened, and the subintimal layer is composed of healthy, intact microvasculature and highly organized collagen fibres. In this figure, the synovial lesions from Lyme arthritis and rheumatoid arthritis are stained with haematoxylin and eosin (H&E), and osteoarthritis synovium is stained with H&E and Alcian blue to show acidic glycosaminoglycans on the outer surface of collagen fibre bundles and along the synovial lining. Image of the knee in part a reprinted with permission from REF.137, Elsevier.
Most patients with Lyme arthritis respond to appropriate oral and, if needed, intravenous antibiotic therapy, and the arthritis resolves (termed antibiotic-responsive Lyme arthritis)7,8. However, in a small percentage of patients, joint swelling lessens but synovitis persists or worsens after spirochaetal killing with antibiotic therapy7,9. These patients develop massive synovial hyperplasia10, often accompanied by autoimmune T and B cell responses that can last for several years11–16, called post-infectious (or post-antibiotic or antibiotic-refractory) Lyme arthritis. After appropriate oral and intravenous antibiotic therapy, such patients are treated with DMARDs17, the standard of care for chronic autoimmune or autoinflammatory types of arthritis. As only one knee is usually affected in post-infectious Lyme arthritis, synovectomy is also an option18. The synovial lesion in post-infectious Lyme arthritis is similar to that seen in other forms of chronic inflammatory arthritis (FIG. 2b), including rheumatoid arthritis (RA)9,10,19.
In addition to relevant human studies, several in-bred, congenic and knockout strains of mice have provided critical insights into Lyme arthritis pathogenesis (TABLE 1). B. burgdorferi-infected C3H/HeN (C3H) mice develop severe arthritis of the tibiotarsal joint with thickening of the tibiotarsal tendon sheath, which peaks several weeks following infection and then spontaneously resolves20. By contrast, B. burgdorferi-infected C57BL/6 (B6) mice have only mild arthritis and quickly repair damaged tissue, leading to a reduction in all parameters of joint disease21. Comparison of how these two strains respond to B. burgdorferi infection has led to the identification of genetic and immune factors that are important for arthritis development22. However, important differences exist between mice and humans. Mice primarily rely on innate immune responses to control B. burgdorferi infection, whereas humans employ both innate and adaptive immune responses throughout infection. In humans, arthritis usually only develops after months of infection within the context of expanded innate and adaptive responses, which can become excessive and maladaptive. Immune responses to B. burgdorferi in mice and humans are discussed in more detail elsewhere23.
Table 1 |.
Characteristics of mouse models of lyme arthritis
| Mouse model | Immune defect | Effect on arthritis | Effect on host defence | Relevance to human disease | Refs |
|---|---|---|---|---|---|
| C57BL/6 (B6) | NA | Mild, self-resolving Lyme arthritis | NA | Probably mimics patients who develop only mild Lyme arthritis | 20 |
| C3H/HeN (C3H) | NA | Severe, acute, self-resolving Lyme arthritis | NA | Most similar to severe Lyme arthritis during active infection | 20 |
| Il10−/− (B6) | Dysregulated NF-κB and TH1 cell responses; impaired regulatory T cells | More severe (increased innate and adaptive inflammation) | Very few Borrelia burgdorferi in joints compared with B6 or C3H mice | Mimics dysregulated TH1 cell responses seen in patients who develop post-infectious Lyme arthritis9,10,43,70,71 | 101,109,110 |
| Tlr1−/− or Tlr2−/− (B6 or C3H) | Impaired response to Borrelia lipoproteins (such as OspA and OspC) | More severe (probably owing to impaired host defence) | ~100-foid more B. burgdorferi in joints compared with wild-type mice; OspA vaccine non-protective | Low TLR1 in vaccine low responders75; TLR1 hypomorph associated with severe Lyme arthritis41 | 74,75 |
| Mir146a−/− (B6) | Hyperactive NF-κB signalling | More severe (increased acute inflammation) | Slightly fewer B. burgdorferi in joints compared with wild-type mice | Probably reflects the central importance of NF-κB regulation in host defence and arthritis during infection9 | 100 |
| Ifnar−/− (C3H) | Defect in type I interferon signalling | Less severe (type I interferon is arthritogenic) | No effect | Unclear, might be important in early infection of skin | 59 |
| C3H Gusb allele (B6) | B6 mice with C3H Gusb allele are unable to clear ECM debris | More severe (accumulated glycosaminoglycans in joints) | No effect | Unclear, might be important in clearing B. burgdorferi peptidoglycan and host ECM debris | 77 |
ECM, extracellular matrix; NA, not applicable; Osp, outer-surface protein; TH1 cell, T helper 1 cell; TLR1, Toll-like receptor 1.
In this Review, we integrate human and mouse studies to detail the pathogenetic features of Lyme arthritis, from initial infection of the skin, to infection of joints, to post-infectious arthritis. We emphasize how, in genetically susceptible individuals, infection with certain B. burgdorferi strains can trigger an excessive, dysregulated immune response that results in post-infectious inflammatory synovitis similar to that seen in other forms of chronic autoimmune or inflammatory arthritis, including RA.
Skin infection and dissemination
After the injection of Borrelia spp. into the skin by an Ixodes tick, spirochaetes multiply in erythema migrans lesions24 (FIG. 3a). The immune response in the skin includes T cells, macrophages, dendritic cells and a small number of B cells25,26, and the main cytokines expressed are the pro-inflammatory cytokines IFNγ and IL-6, and the anti-inflammatory cytokine IL-10 (REFS25,27). In the USA, B. burgdorferi often disseminates in the blood during the first few weeks of infection in a process that requires the binding of Borrelia surface adhesins to host integrins on the vascular endothelium28–30 (FIG. 3b). As shown in mice, the spread of B. burgdorferi through the vasculature or lymphatics is dependent on the interactions of spirochaetal surface molecules and endothelial cell membrane proteins. Bacterial–endothelial cell interactions result in the loosening of tight junctions and migration of spirochaetes into the synovial extracellular matrix via small vascular lesions31,32 (FIG. 3c). In response, natural killer T (NKT) cells, tissue-resident macrophages, polymorphonuclear cells and stromal cells have an important role in maintaining endothelial cell barrier function, limiting spirochaetal invasion into extravascular tissues and suppressing tissue damage and arthritis development33. NKT cells secrete IFNγ in response to immunogenic B. burgdorferi glycolipids34,35 that are presented by CD1-expressing antigen-presenting cells36,37. Macrophages, polymorphonuclear cells, fibroblasts and endothelial cells respond to spirochaetal invasion by producing large amounts of innate immune response and tissue repair proteins. Notably, variations in these responses greatly affect arthritis severity and outcome.
Fig. 3 |. Spirochaete invasion into joint tissue.
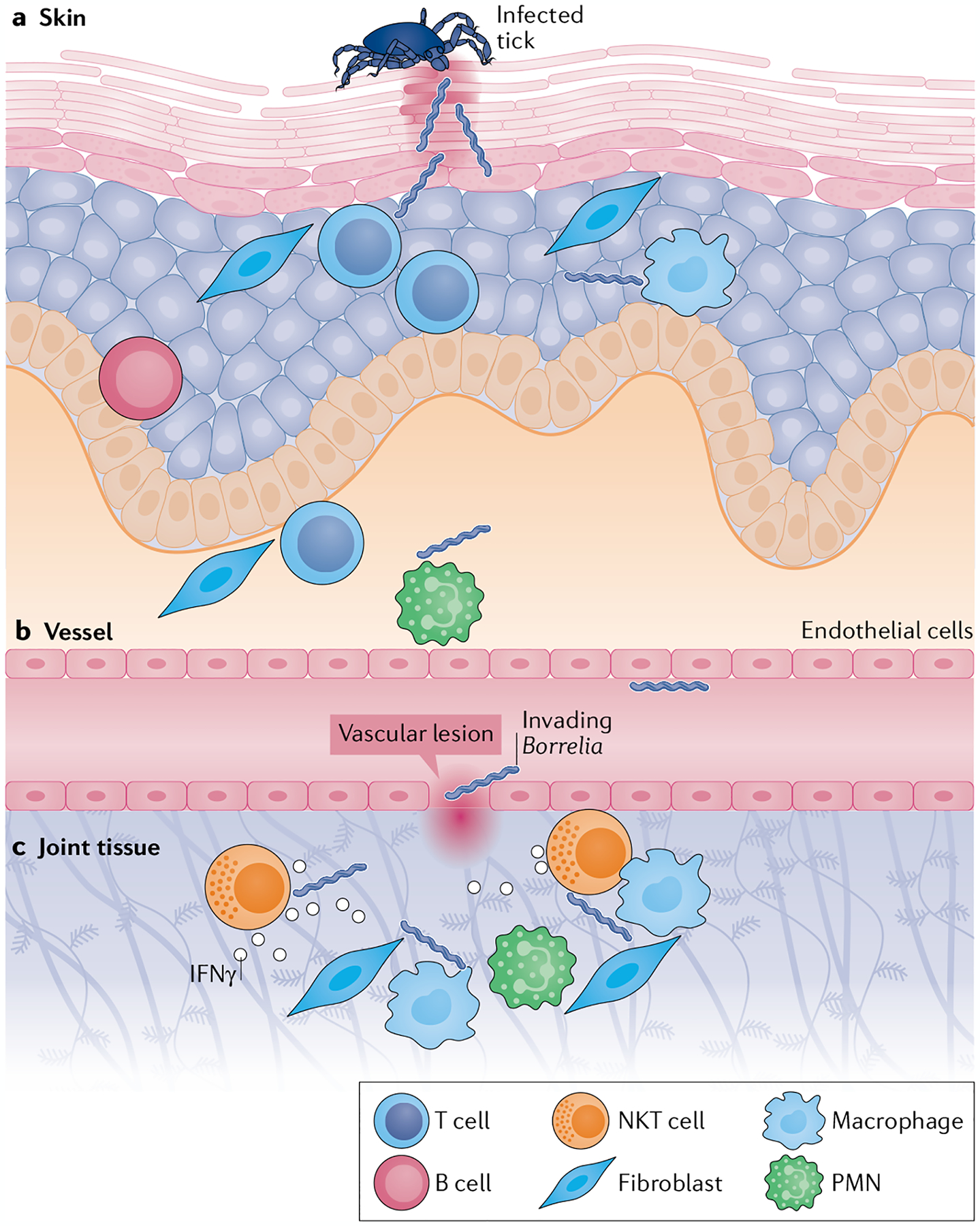
a | Spirochaetes invade the skin during the bloodmeal of an infected Ixodes tick. Upon entry, tissue-resident T cells, B cells, resident antigen-presenting cells (such as macrophages and dendritic cells), some polymorphonuclear cells (PMNs) and stromal cells (such as fibroblasts, keratinocytes, epithelial cells and endothelial cells) are responsible for early-acute immune responses to infection. b | A small number of spirochaetes escape the site of invasion and enter the vasculature, where Borrelia surface lipoproteins interact with vascular endothelial cells to induce the loosening of tight junctions. c | Spirochaetes enter the extracellular matrix of joint tissue through vascular lesions. Once in the joint tissue, they induce acute inflammatory responses by resident cells such as endothelial cells and synovial fibroblasts, which produce adhesion molecules, matrix metalloproteinases and innate immunity cytokines and chemokines. Natural killer T (NKT) cells produce IFNγ in response to CD1-presented immunogenic Borrelia glycolipids, thereby enhancing vascular barrier function and limiting spirochaetal invasion and chronic inflammation. The cytotoxic function of NKT cells might also directly contribute to spirochaetal killing. The nature and magnitude of these early immune responses in the skin and joint help to set the stage for subsequent arthritis development.
According to one subtyping system38, out of 23 B. burgdorferi outer-surface protein C (OspC) subtypes, types A, B, I and K are the most likely to disseminate in humans39. In patients with disseminated early infection, many interferon-associated genes are upregulated in peripheral blood mononuclear cells27,40. Serum samples often show high concentrations of the macrophage-recruiting chemokine CCL2 and of the innate immune mediators IL-6 and TNF, although the anti-inflammatory cytokine IL-10 is also prominent41. Patients with disseminated infection also have high serum concentrations of T helper 1 (TH1) cell-associated immune mediators, including IFNγ and the IFNγ-inducible T cell chemokines CXCL9 and CXCL10 (REFS41,42). Many patients’ sera contain numerous TH17 cell-associated mediators, particularly IL-23 (REF.43). Infection with the OspC type A (RST1) strain is particularly inflammatory, leading to more severe symptoms in patients with erythema migrans41,42. Similarly, strain-specific OspC also has an important role in spirochaetal joint invasion and colonization in mice44.
Lyme disease spirochaetes are only transiently present in the blood45 and rapidly migrate to extravascular tissues via transendothelial migration46. With their unique planar wave motion, these bacteria are highly adapted to move through dense connective tissue, which requires the binding of plasminogen or its activators to the surface of the organism47. The spirochaetal adhesins decorin binding protein A (DbpA) and DbpB bind to host decorin48, a proteoglycan that is bound to collagen, and spirochaetes can also bind directly to, invade and colonize native type I collagen lattices32. The binding of DbpA and DbpB to host decorin probably explains the alignment of spirochaetes with collagen fibrils in connective tissue in joints, heart or nerves49. Genetic variability in Borrelia outer-surface adhesins at least partially explains differences in tissue tropism between strains41,50,51. For example, in a mouse study, B. burgdorferi OspC subtypes that bound dermatan sulfate were associated with joint invasion44, which the authors suggest could explain the exceptional arthritogenicity of certain spirochaetal strains found in north-eastern USA.
The antibody response to B. burgdorferi develops slowly, and during the first few weeks of infection an IgM response is seen in only a minority of patients52. Total IgM concentrations can also be increased during early infection, suggestive of polyclonal activation of B cells53. As B. burgdorferi disseminates and infects host tissues, an increasingly higher percentage of patients develop IgM and IgG responses to the spirochaete52,54. To evade the host antibody response, spirochaetes seek protected niches and change the expression profile of their outer-surface proteins55. In particular, the lipoprotein VlsE undergoes extensive antigenic variation56. In addition, B. burgdorferi evade innate immune responses by binding host complement regulator proteins to their surface, which inactivate complement and induce innate immune tolerance57.
Dysregulation of innate immune responses during early disseminated infection might promote subsequent arthritis development. In C3H mice, type I interferons (IFNα and IFNβ) have a particularly important role during the first week of infection and set the stage for the subsequent development of arthritis58. Importantly, the type I interferon response (typically associated with anti-viral immunity) is maladaptive and has no effect on host defence59. This type I interferon response is accompanied by downregulation of numerous genes involved in tissue repair and wound healing, such as extracellular matrix proteins and transforming growth factor-β-inducible genes60. By contrast, B6 mice, which develop only mild Lyme arthritis, lack the robust interferon signature seen in arthritogenic C3H mice and exhibit marked upregulation of tissue repair and wound healing genes in joints at 1 week post-infection60.
As in C3H mice, early type I interferon responses are likely to be arthritogenic in humans with Lyme arthritis. Human peripheral blood mononuclear cells stimulated with the highly inflammatory OspC type A (RST1) strain of B. burgdorferi secrete type I interferons as well as type II interferon (IFNγ)42. In addition, type I and II interferons are predominant in erythema migrans skin lesions27. Moreover, type I interferons are known to be important in the development of a number of rheumatic and autoimmune diseases61. Given that patients with erythema migrans who are treated with antibiotics do not develop subsequent arthritis, it is difficult to directly test the importance of early type I interferon responses in the subsequent development of arthritis in humans. However, a role for type I interferons can be inferred from responses in C3H mice. On the basis of studies in this mouse model, we hypothesize that during early disease, dysregulated type I interferon responses to B. burgdorferi in the skin or joint set the stage for severe Lyme arthritis and autoimmunity later in the disease.
Lyme arthritis during active infection
Months after the initial infection, along with an expansion of the immune response to B. burgdorferi, untreated patients often develop marked joint swelling, frequently in one or both knees. B. burgdorferi has rarely been cultured from the synovial fluid of patients with Lyme arthritis, but prior to antibiotic treatment, B. burgdorferi DNA (but not mRNA) can be found in the synovial fluid of ~70% of these patients24,62. This finding suggests that live spirochaetes might survive only in protected tissue niches within joints and are killed if they escape into synovial fluid. During joint infection, immune responses are focused on spirochaetal killing, primarily through acute inflammatory responses to pathogen-associated molecular patterns (PAMPs), antibody production and the infiltration of polymorphonuclear cells into synovial fluid9, which might be the principal barrier preventing spirochaetal escape. In addition, large amounts of NF-κB-induced acute pro-inflammatory cytokines and chemokines are found in synovial tissue and synovial fluid from patients with Lyme arthritis41,42,63, typical of innate immune responses to bacterial infections.
Robust anti-B. burgdorferi antibody responses develop towards a large array of spirochaetal proteins52,64. Patients with Lyme arthritis can have antibody reactivity to as many as 89 spirochaetal proteins65, primarily outer-surface proteins, many of which are lipidated and might serve as immune adjuvants66. Two spirochaetal glycolipids, acylated cholesteryl galactoside (BbGL1) and monogalactosyl diacylglycerol (BbGL2), are also highly immunogenic34. Moreover, patients with Lyme arthritis can have antibody responses to spirochaetal antigens that are ordinarily expressed only in the tick, such as OspA, OspD and Borrelia iron and copper-binding protein A (BicA), a phenomenon found almost exclusively in the highly inflammatory milieu of joints in North American patients with Lyme arthritis67. Similarly, B. burgdorferi can be induced to express tick-specific proteins in mice in a highly inflammatory environment68.
Marked TH1 cell responses to B. burgdorferi antigens also occur in patients with Lyme arthritis, particularly among synovial fluid mononuclear cells, which produce large amounts of IFNγ69–71. The role of these cells might be primarily to help B cells to produce neutralizing antibodies against the spirochaete. Anti-borrelial antibodies are predominantly T cell-dependent IgG1 and IgG3 isotypes, which are capable of inducing opsonization and activating complement72. Most synovial fluid mononuclear cells also express memory markers71, which helps to explain why B. burgdorferi T cell and B cell responses typically persist for many years after the resolution of Lyme arthritis, and why reinfection occurs only rarely, if at all, after Lyme arthritis.
Animal model studies have provided insights into important innate immune effectors in Lyme arthritis (TABLE 1). Mice deficient in certain innate immune response pathways, particularly those involved in recognition of B. burgdorferi surface lipoproteins, including Toll-like receptor 2 (TLR2) and myeloid differentiation primary response protein MyD88, have impaired host defence and develop severe Lyme arthritis73–76. In addition, C3H mice have a hypomorphic allele (Bbaa2 locus on Chromosome 5) encoding the lysosomal enzyme β-glucuronidase, which allows the accumulation of arthritogenic glycosaminoglycans in infected joints77,78. Similarly, the Bbaa1 locus on Chromosome 4 in C3H mice, which contains the type I interferon locus, is involved in dysregulated type I interferon responses and severe Lyme arthritis79–81.
Untreated patients with Lyme arthritis often have intermittent flares of arthritis or persistent arthritis over a period of several years5. One theory is that spirochaetes might survive in relatively avascular sites, such as the tendons in and around joints, and then escape from these sites occasionally to repopulate the synovial tissue82,83. Consistent with this hypothesis, joint swelling might be more severe and prolonged in recurrent flares, and the very high antibody responses that occur in patients with Lyme arthritis are consistent with repeated waves of antigenic exposure to spirochaetes5. As affected joints are no longer swollen after treatment in antibiotic-responsive patients, post-infection immune responses cannot be assessed in these patients’ joints. However, we predict that antibacterial responses are downregulated after spirochaetal killing and wound repair genes are upregulated, leading to tissue repair, a return to joint homeostasis and arthritis resolution (FIG. 4), similar to the strong tissue repair signature that occurs in the joints of infected B6 mice60.
Fig. 4 |. Stages of arthritis and proposed tissue repair in antibiotic-responsive lyme arthritis.
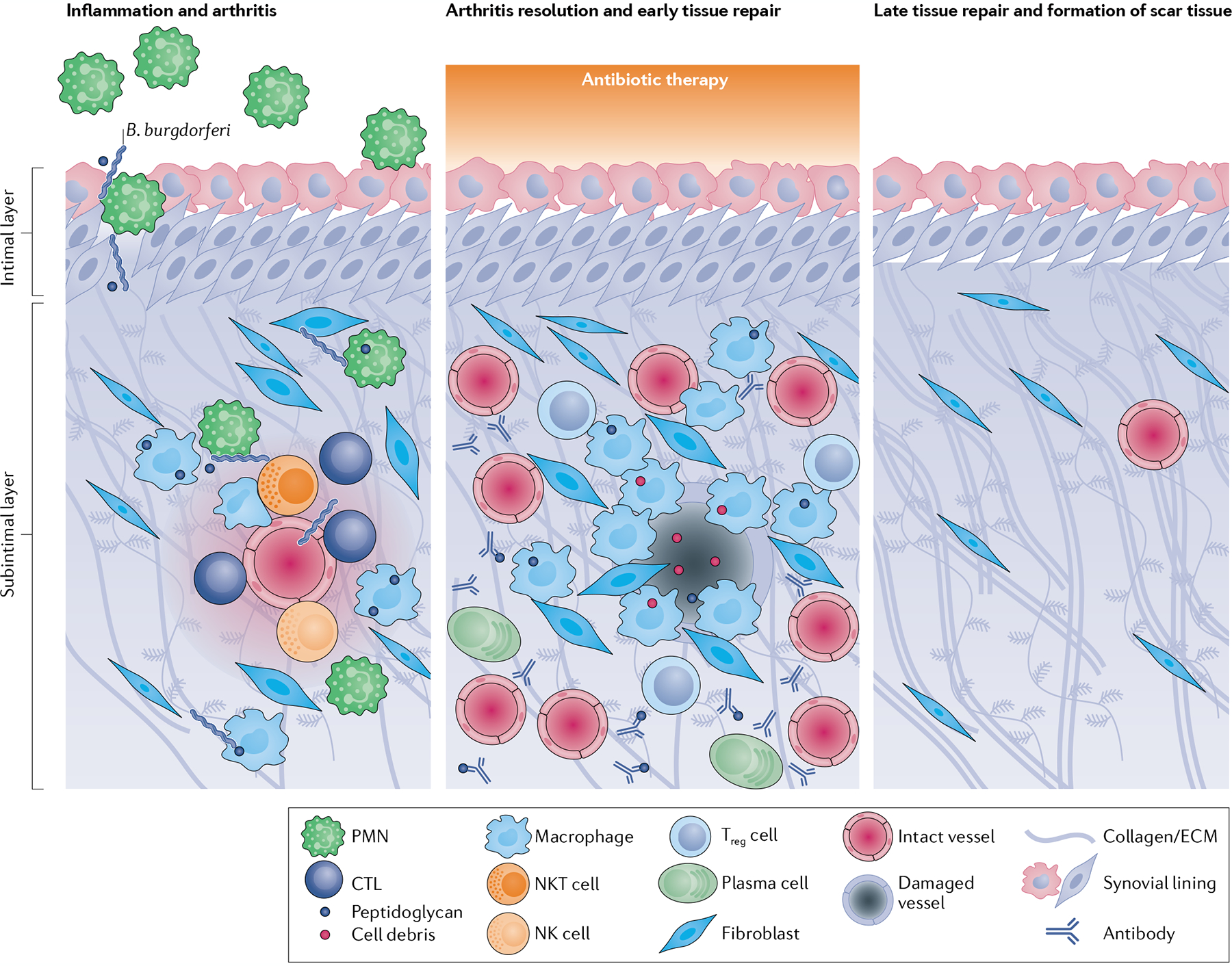
Immune responses to Borrelia burgdorferi and B. burgdorferi peptidoglycan by endothelial cells, fibroblasts, lymphocytes (such as natural killer (NK cells), natural killer T (NKT) cells and cytotoxic T lymphocytes (CTLs)) and myeloid cells (such as macrophages and polymorphonuclear cells (PMNs)) in the synovium trigger localized inflammation, tissue damage and arthritis. Antibiotic therapy is given during acute joint infection and inflammation, facilitating arthritis resolution and the initiation of early tissue repair responses. These responses are dominated by pro-angiogenic factors and the activation of tissue-repairing macrophages and fibroblasts, which remove bacterial debris and damaged cells from the damaged microvasculature, extracellular matrix (ECM) and fibrotic tissue. Other immune cells such as regulatory T (Treg) cells and plasma cells might also be present during arthritis resolution. Over several months, synovial fibroblasts differentiate into myofibroblasts and lay down collagen and form scar tissue, leading to full recovery.
Post-infectious Lyme arthritis
Rather than resolution of arthritis after antibiotic therapy, a small percentage of patients with Lyme arthritis have persistent synovitis that can worsen in the post-antibiotic period7. In these patients, the synovial lesion — the target of the immune response — shows massive synovial fibroblast proliferation and fibrosis, infiltration of mononuclear cells, large amounts of antigen presentation, marked vascular proliferation and, in some patients, obliterative microvascular lesions and massive fibrin deposition suggestive of microscopic bleeding84,85 (FIG. 5). These histological findings are similar to those that occur in other autoimmune or autoinflammatory forms of arthritis, including RA, albeit with a greater emphasis on microvascular damage in post-infectious Lyme arthritis10,12,84–86. Damage to the microvasculature, including obliterative microvascular lesions, seems to be a common feature of Lyme disease and can be found in other affected tissues, including the heart49,87,88, skeletal muscle49,89 and dura mater90. The inflammatory process in the joints can be accompanied by tendon sheath thickening (tenosynovitis) and tendon calcification (tendonitis) and, occasionally, by mild-to-moderate cartilage damage91. Although synovial fluid contains a very high percentage of neutrophils during active infection, in the post-infectious stage it contains relatively fewer neutrophils and proportionally more monocytes, macrophages and lymphocytes, suggestive of an expanded inflammatory response in the post-infectious phase9. In contrast with RA, post-infectious Lyme arthritis eventually resolves in all patients — often with the aid of DMARD therapy — usually within 1–2 years, but within a maximum of 4–5 years7,17. Presumably, without the immune stimuli provided by live spirochaetes, the immune response eventually regains homeostasis and the arthritis resolves.
Fig. 5 |. Microvascular involvement in the synovial lesion of post-infectious lyme arthritis.
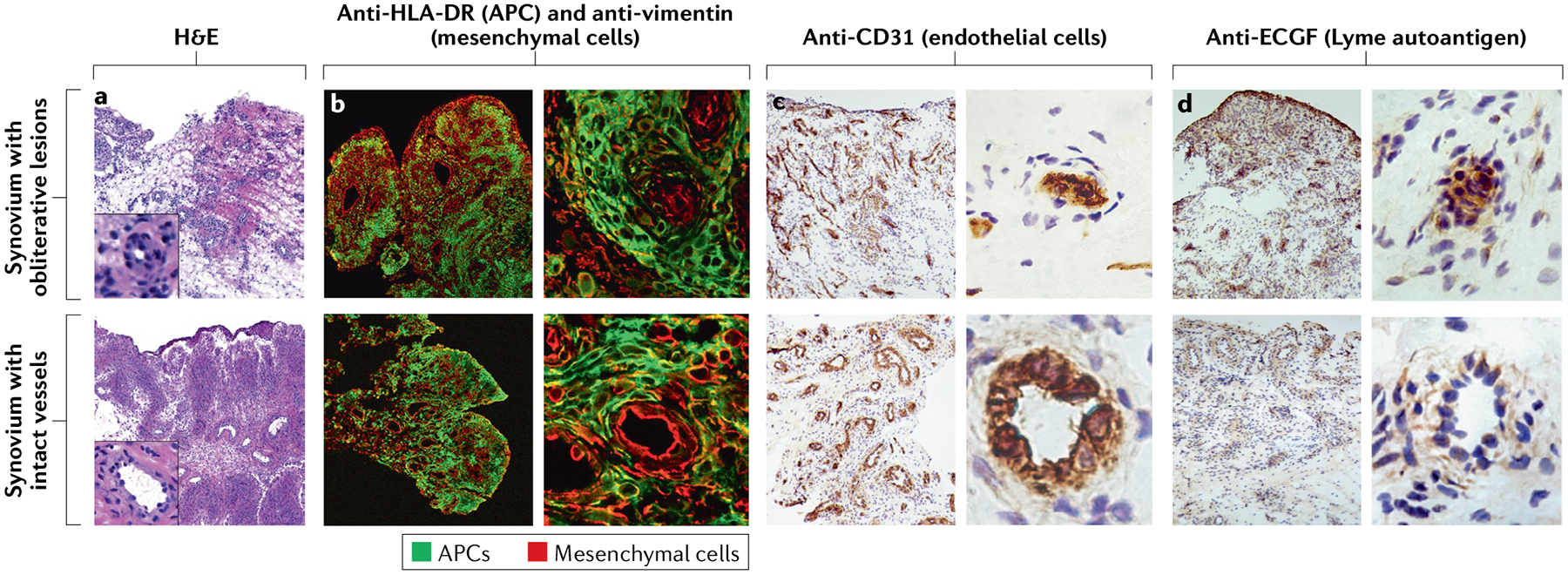
a | Haematoxylin and eosin (H&E)-stained sections of synovial tissue from representative patients with post-infectious Lyme arthritis with varying degrees of inflammation, fibrosis and vascular damage (insets show enlarged images of vessels). b | Synovial tissue sections stained with fluorescently labelled anti-HLA-DR (green) and anti-vimentin (red) antibodies, showing localization of antigen-presenting cells (APCs) and mesenchymal cells (such as fibroblasts and endothelial cells), respectively. c | Around half of patients with post-infectious Lyme arthritis have evidence of vascular damage, including obliterative microvascular lesions, as shown by staining with the endothelial cell marker CD31. d | Obliterative microvascular lesions are also enriched with the Lyme disease autoantigen endothelial cell growth factor (ECGF). In panels b–d, the panels on the left show the general architecture of the synovial tissue at low magnification, and the panels on the right show single blood vessels from the same sections at high magnification. Parts c and d adapted with permission from REF.12, Wiley. © 2014 by the American College of Rheumatology.
These changes in the cellular infiltrate in synovial fluid result from an inadequately restrained, excessive pro-inflammatory immune response that begins during infection and continues into the post-infectious period92. Infection with the highly inflammatory OspC Type A (RST1) strain of B. burgdorferi more commonly results in this outcome42,51. In studies of B. burgdorferi isolates from erythema migrans skin lesions, OspC type A strains were found in the USA in 21 of 58 isolates (36%) from New England states93, 46 of 291 isolates (16%) from New York state39 and only 2 of 65 isolates (3%) from Wisconsin, an upper midwestern state94, compared with 0 of 29 isolates from Slovenia95. These results might explain why post-infectious Lyme arthritis is most often found in New England. Nevertheless, a 2019 French study described patients with post-infectious Lyme arthritis that were similar to those found in the USA96, suggesting that highly inflammatory strains of B. burgdorferi might occur in certain regions in Europe.
Although the strain of B. burgdorferi is an important factor in stimulating excessive immune responses during infection, culture and PCR results have been uniformly negative in synovial tissue obtained from patients with Lyme arthritis months to years after antibiotic therapy24, hence the use of the term post-infectious Lyme arthritis10. Moreover, after oral and intravenous antibiotic therapy, re-emergence of infection has not been noted while patients are being treated with DMARDs7. However, spirochaetal remnants can persist during the post-infectious period82. A 2019 study found that B. burgdorferi peptidoglycan, a predominant cell wall component, is detectible in post-infectious Lyme arthritis synovial fluid up to several years after antibiotic treatment97. B. burgdorferi peptidoglycan is shed during cell replication and is uniquely difficult to clear97. Thus, uncleared peptidoglycan might be an important factor in promoting innate immune responses in the post-infectious period in genetically predisposed individuals.
Host factors associated with excessive immune responses.
Transcriptomic analysis of synovia from patients with post-infectious Lyme arthritis shows prominent gene signatures associated with innate immune responses, antigen presentation and cell-mediated immune activation10. As in erythema migrans skin lesions, a large number of interferon-response genes are highly or moderately enriched in synovial tissue from all patients with post-infectious Lyme arthritis10. Importantly, this high interferon signature correlates inversely with tissue repair response gene signatures10, indicating that high concentrations of interferons impair wound healing. Supporting the transcriptomic data, large percentages of T cells and natural killer (NK) cells isolated from synovial tissue or synovial fluid test positive for IFNγ by intracellular cytokine staining10,70,71. Thus, in patients with post-infectious Lyme arthritis, high numbers of IFNγ-producing lymphocytes present in synovial tissue might prevent appropriate repair of tissue damaged by B. burgdorferi infection, blocking the return to tissue homeostasis even after the bacteria themselves are cleared9.
Both host and spirochaetal genetic factors can contribute to this exceptionally high IFNγ response. In individuals with a TLR1 single nucleotide polymorphism (1805GG) that affects the recognition of PAMPs by innate immune cells, infection with OspC type A (RST1) strains of B. burgdorferi leads to exceptionally high levels of IFNγ and signal transducer and activator of transcription 1 (STAT1)-dependent cytokines in joints41. In an initial study, this TLR1 polymorphism was present in 24 of 47 European Americans (51%), but in only 2 of 24 African Americans (8%) and 0 of 390 Vietnamese individuals98.
Among patients with Lyme arthritis in New England, the 18055GG polymorphism was present in 35 of 76 patients with antibiotic-responsive arthritis (47%) compared with 62 of 101 patients with post-infectious arthritis (62%)41. This polymorphism is within the portion of the gene that encodes the transmembrane region of TLR1 and might impair cell surface localization and downstream NF-κB signalling in response to the TLR1 and TLR2 ligand Pam3CSK4 (REF.99). Paradoxically, patients with Lyme arthritis who have this polymorphism have exceptionally high amounts of IFNγ, as well as STAT1-regulated and NF-κB-regulated pro-inflammatory immune mediators in their joints, but unremarkable amounts of IL-10 (REF.41). This polymorphism is hypothesized to be associated with excessive inflammatory responses to B. burgdorferi because it results in a deficiency in Janus kinase–STAT, NF-κB and mitogen-activated protein kinase feedback loop inhibitors, such as the regulatory microRNA miR-146a100 and the anti-inflammatory cytokine IL-10 (REF.101).
Antigen presentation of certain B. burgdorferi peptides by specific HLA-DR molecules might also lead to high IFNγ concentrations. In one study, 7 of 14 HLA-DRB molecules — HLA-DRB1*04:01 in particular — bound to a peptide from B. burgdorferi OspA (OspA163–175), whereas the other seven HLA-DRB molecules (including HLA-DRB1*11:01) did not102. Among patients with post-infectious Lyme arthritis, 56 of 71 (79%) had at least one HLA-DRB molecule that bound B. burgdorferi OspA163–175, compared with 23 of 50 patients with antibiotic-responsive Lyme arthritis (46%)102. As mentioned previously, immune responses to OspA are found primarily in the highly inflammatory joint milieu of patients with Lyme arthritis in North America67. In transgenic mice, those expressing the human HLA-DR4 allele had higher IFNγ responses and lower titres of anti-Borrelia antibodies than mice expressing the human HLA-DR11 allele, which had higher anti-Borrelia antibody titres but lower IFNγ responses103. Thus, presentation of OspA163–175 by certain HLA-DR molecules is associated with high IFNγ concentrations and with post-infectious Lyme arthritis.
Two NF-κB-regulated microRNAs, miR-146a and miR-155, which have been associated with a number of inflammatory joint diseases104, including RA, are also prominent in Lyme arthritis9,104. Experiments in mice have shown that these two microRNAs fine-tune the amplitude of inflammatory responses to B. burgdorferi to balance host defence and tissue damage in Lyme arthritis100,105. miRNA-146a functions as a feedback inhibitor of NF-κB signalling, and mice lacking miR-146a develop more severe Lyme arthritis than wild-type mice, despite having fewer bacteria in their joints100. By contrast, miR-155 enhances acute inflammation by potentiating NF-κB and STAT1 signal transduction105. In humans with post-infectious Lyme arthritis, miR-155 is particularly enriched in synovial fluid and correlates positively with arthritis duration, but is below or near the limit of detection in patients with antibiotic-responsive Lyme arthritis9. Concentrations of both miR-146a and miR-155 remain persistently elevated in synovial tissue and fluid from patients with post-infectious Lyme arthritis, providing further evidence of chronic NF-κB activation in the inflamed synovium9.
Several other types of immune regulation imbalance can result in high concentrations of IFNγ. In patients with post-infectious Lyme arthritis, a high percentage of CD4+CD25+ T cells, which are ordinarily regulatory T (Treg) cells, become effector cells that secrete large amounts of IFNγ, thereby skewing the TH1 cell–Treg cell balance71,106. By contrast, in patients with antibiotic-responsive Lyme arthritis, Treg cells secrete large amounts of anti-inflammatory IL-10 and negligible amounts of IFNγ71. IL-10 produced by Treg cells and other immune cells is critical to limiting NF-κB and STAT1 signalling, which is necessary for the development of innate and adaptive immune responses to B. burgdorferi. In mice, Treg cells are an important source of IL-10, and Treg cell-depleted mice develop more severe Lyme arthritis than immunocompetent mice107. Similarly, HLA-DR4 transgenic mice that lack the co-stimulatory molecule CD28, which greatly reduces the number of Treg cells, also develop persistent arthritis after spirochaetes have been killed108.
The critical role of the balance between IFNγ and IL-10 in post-infectious Lyme arthritis is underscored by studies in IL-10 knockout (Il10−/−) mice. Similar to patients with post-infectious Lyme arthritis, these mice have greatly increased innate and adaptive immune responses to infection with B. burgdorferi, resulting in severe arthritis despite having low to undetectable amounts of bacteria in inflamed joint tissues105,109,110. Longitudinal transcriptomic analysis of joints from infected B6 Il10−/− mice show marked upregulation in the transcription of IFNγ-stimulated genes and the pro-inflammatory microRNA miR-155, with a corresponding downregulation of mRNA transcripts and microRNAs involved in tissue repair and response to wounding, similar to human post-infectious Lyme arthritis60,105. This transcriptomic profile is probably the result of the impaired ability of IL-10 to regulate STAT1 activation in these mice105. The dysregulated IFNγ response in these mice is caused by TLR2-mediated bystander activation of both CD4+ and CD8+ T cells110. Importantly, spirochaetes are no longer detectable in synovial tissue at 16 weeks post-infection, and depletion of CD4+ and CD8+ T cells in B6 Il10−/− mice results in less severe Lyme arthritis110, demonstrating that a dysregulated TH1 cell response is arthritogenic. In the human disease, bystander activation of T cells could also cause a break in immune tolerance, providing a critical intermediate step on the path towards autoimmunity.
Consequences of excessive, dysregulated pro-inflammatory responses.
A model of the cellular architecture in the synovial lesion of post-infectious Lyme arthritis and the proposed roles of the main immune cells, peptidoglycan, autoantigens and IFNγ responses are summarized in FIG. 6. Immune dysregulation during microbial infection, particularly pathogenic TH17 cell responses, can trigger autoimmunity1,111. In a study in which HLA-DR-presented peptides were eluted from synovia of patients with post-infectious Lyme arthritis, four immunogenic peptides were identified that were derived from self-proteins112, including endothelial cell growth factor11, annexin A2 (REF.13), apolipoprotein B100 (REF.14) and matrix metalloproteinase 10 (REF.15). Similarly, HLA-DR molecules expressed on B. burgdorferi-stimulated dendritic cells obtained from healthy individuals presented peptides derived from all of these self-proteins, with the exception of matrix metalloproteinase 10 (REF.113). HLA-DR presentation of these self-proteins might reflect previous damage to endothelial cells and/or to the extracellular matrix by spirochaete invasion12. Moreover, autoantibodies to these self-proteins can sometimes be found in patients with early-stage B. burgdorferi infection, Lyme carditis, neuroborreliosis or antibiotic-responsive Lyme arthritis11–16, albeit usually without T cell responses. Thus, initial autoimmune responses might be triggered by increased TH17 cell responses during early infection, but T cell responses to autoantigens are not usually apparent at that time. By contrast, both T cell and B cell responses to these autoantigens are often found in patients with post-infectious Lyme arthritis, suggestive of further maturation of the immune response11,13–15. Amounts of TH17 cell-associated cytokines, particularly IL-23, that correlate with anti-B. burgdorferi antibody titres in early disease, correlate strongly with autoantibody titres in post-infectious Lyme arthritis43, suggesting a shift from protective anti-Borrelia responses to autoreactive immunity.
Fig. 6 |. Cellular architecture of the post-infectious lyme arthritis synovial lesion.
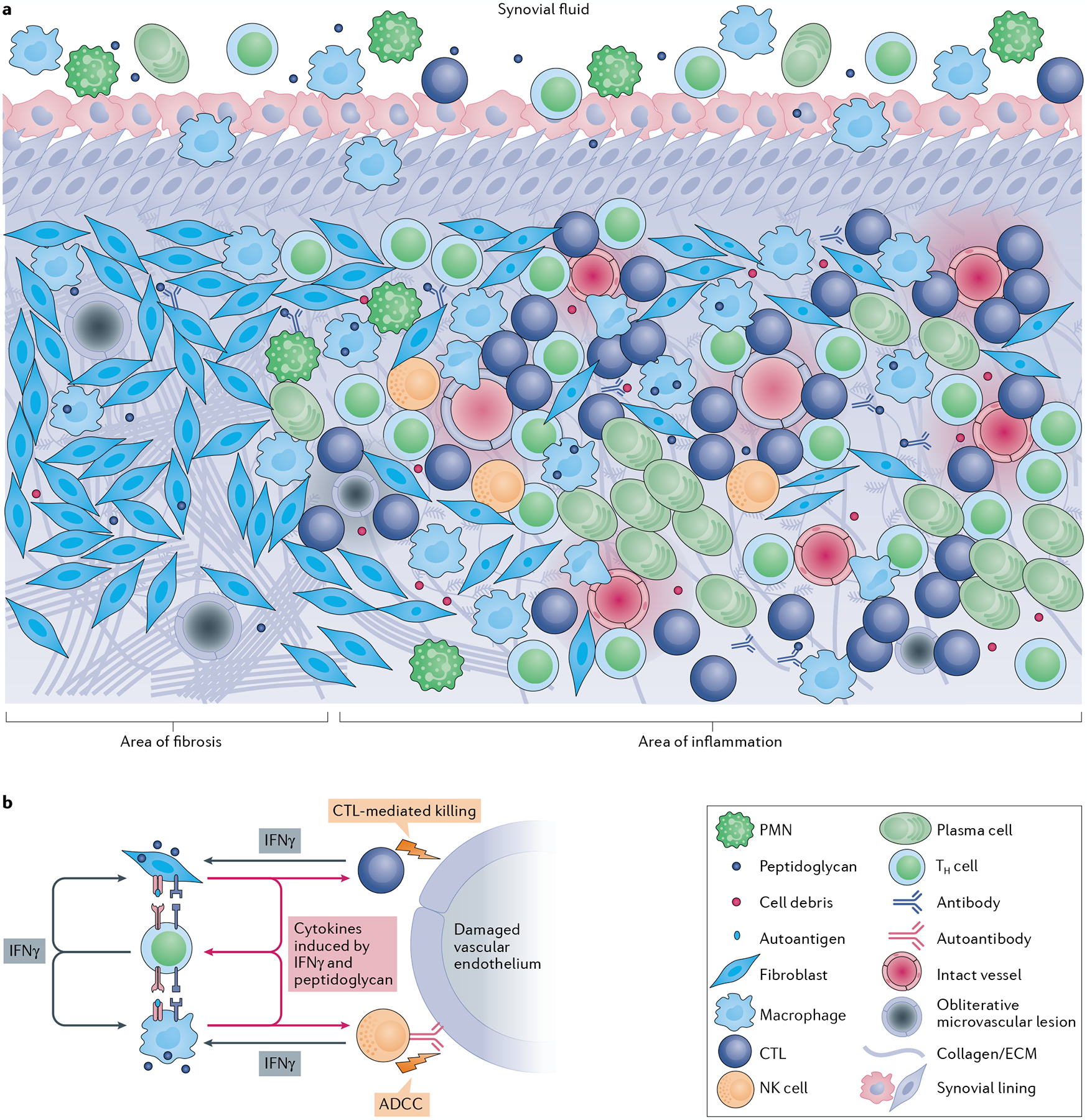
a | The post-infectious Lyme arthritis synovial lesion is characterized by widespread fibrosis and areas of marked inflammation. Fibrotic areas contain large numbers of synovial fibroblasts, obliterative microvascular lesions, disordered collagen and other extracellular matrix (ECM) proteins. Areas of inflammation are found primarily in highly vascularized synovial intimal and subintimal layers that can contain obliterative microvascular lesions and/or intact vessels. Immune cells, such as macrophages, CD4+ T helper (TH) cells, cytotoxic T lymphocytes (CTLs; mostly CD8+ T cells with a few γδ T cells), natural killer (NK) cells, and large numbers of antibody-producing plasma cells, are found primarily in vascularized areas, but can be found throughout the tissue. Vascularized areas also contain many HLA-DR-expressing synovial fibroblasts; however, they tend to have less fibrotic tissue. Bacterial peptidoglycan is present in synovial fluid and might additionally be present in inflamed tissue, along with degraded cellular and ECM debris. Only a few polymorphonuclear cells (PMNs) are present in post-infectious Lyme arthritis synovial tissue, but more are present in synovial fluid. b | In this panel, a hypothesis is developed regarding the roles of important cells and immune mediators in autoimmune-mediated damage to the endothelium in inflamed synovia. Large amounts of IFNγ produced by TH cells, CTLs and NK cells induce potent responses by HLA-DR-expressing synovial fibroblasts and macrophages, which upregulate proteins associated with antigen presentation, T cell activation and inflammation. Borrelia burgdorferi peptidoglycan and cell debris might amplify these responses. Synovial fibroblasts and macrophages present MHC class II-restricted peptides derived from Lyme autoantigens, which are abundant in synovial tissue, to autoreactive TH cells, perpetuating IFNγ responses in the tissue. Endothelial cells, which were damaged during infection with B. burgdorferi, can be targeted for killing by CTLs, either through direct CTL-mediated killing or through autoantibody-dependent cell-mediated cytotoxicity (ADCC), or both. Further damage to the microvasculature releases more autoantigens and debris, leading to a feedback loop of chronic inflammation and tissue damage.
Although the pathogenic nature of Lyme autoantibodies has not yet been delineated, IgG4 Lyme disease autoantibody titres correlate with the magnitude of obliterative microvascular lesions and fibrosis in synovial tissue72. Curiously, a 2020 study using humanized mice indicated that loss of the inhibitory Fc receptor Fcγ-receptor IIb, which binds to IgG4 immune complexes, might contribute to infection-induced autoantibody responses in Lyme arthritis114. Although IgG4 responses are typically considered to be anti-inflammatory, clinical data imply a pathogenic role for these autoantibodies.
The timeline for the development of putative pathogenic autoantibodies in Lyme arthritis could have parallels with RA. In RA, anti-citrullinated protein antibodies (ACPAs) typically develop years before inflammatory arthritis manifests115. Prior to arthritis development, ACPAs can undergo epitope spreading, which, together with the appearance of innate immune mediators including IL-1, IL-6 and TNF, can lead to the development of clinical arthritis116. Moreover, in a study from the Netherlands, IgG4 ACPAs were noted in 104 of 373 patients with RA (28%), and anti-carbamylated protein antibodies were found in 209 of the 373 patients (56%)117. Similarly, in a study from France, 35 of 141 patients with RA (25%) had IgG4 antibodies that recognized citrullinated fibrinogen, a common target of ACPAs118. In a Chinese study, patients with IgG4 antibodies were more difficult to treat successfully with DMARDs119, suggesting that serum IgG4 autoantibodies might define a specific clinical phenotype associated with more severe disease.
Cytotoxic immune responses had not previously been thought to be involved in synovial pathology, yet a 2019 transcriptomic analysis of synovial tissue from patients with post-infectious Lyme arthritis or RA showed marked upregulation of genes associated with cell-mediated cytotoxicity10. Cells associated with cytotoxic potential include CD8+ T cells, NK cells and, less commonly, γδ T cells120. However, each of these cell types can also secrete cytokines, which has been thought to be their primary function in chronic inflammatory arthritides. During spirochaete dissemination and in the infectious phase of Lyme arthritis, these cells probably function as part of a complex web of inflammatory responses that are important for spirochaetal killing. For example, innate-like cytotoxic lymphocytes, such as NK cells and NKT cells, could initially have a role in trapping spirochaetes in obliterative microvascular lesions35,84,121. However, the role of cells with cytotoxic potential and the cellular targets of such responses are yet to be clarified in post-infectious Lyme arthritis and in other chronic inflammatory arthritides.
Synovial fibroblasts, the most common cells in the synovial lesion, function as important immune effector cells in inflamed synovial tissue70. When stimulated with IFNγ and B. burgdorferi in vitro, primary synovial fibroblasts derived from patients with post-infectious Lyme arthritis secrete large amounts of NF-κB-regulated and STAT1-regulated cytokines, chemokines and pattern recognition receptors70. These cells also secrete TH1 cell-promoting immune mediators and proteins involved in antigen presentation to T cells, including HLA-DR molecules and co-stimulatory molecules. These data70 suggest that synovial fibroblasts function as non-professional antigen-presenting cells and might contribute to T cell reactivity to HLA-DR-presented Lyme autoantigens. Furthermore, when synovial fibroblasts obtained from patients with post-infectious Lyme arthritis were grown in culture, the gene signature of IFNγ-stimulated cells in vitro was quite similar to that found in vivo in the synovia from such patients10, confirming a central role for IFNγ and synovial fibroblasts in post-infectious Lyme arthritis. Thus, immune dysregulation in post-infectious Lyme arthritis leads to pro-inflammatory and tumour-like proliferative responses by synovial fibroblasts, rather than the wound healing and appropriate tissue repair responses that are probably orchestrated by these cells in antibiotic-responsive Lyme arthritis following antibiotic therapy. In RA, genetic, epigenetic and phenotypic changes in synovial fibroblasts are likely to contribute to inflammatory synovitis and the development of autoimmunity122–124. Taken together, these studies indicate a central role for synovial fibroblasts in the pathogenesis of a number of forms of chronic inflammatory arthritis, including Lyme arthritis and RA125.
Linking infection and autoimmunity
The main message from post-infectious Lyme arthritis for other forms of chronic autoimmune or autoinflammatory arthritis is that this complex immune response can begin with an antimicrobial immune response, and is shaped by complex interactions between pathogen and host. Such an immune response could be triggered by an invading pathogen, as is the case in Lyme arthritis, or by commensals in the host microbiome. In RA, evidence is emerging that bacteria-induced inflammation at mucosal sites in the periodontium, lung or bowel might trigger or enhance autoimmunity and joint disease in predisposed individuals126. For example, the periodontal pathogen Porphyromonas gingivalis is associated with RA127,128, as is the gut commensal Prevotella copri129,130. In ankylosing spondylitis and Crohn’s disease-associated spondyloarthritis, strains of Escherichia coli or Prevotella spp. that adhere to the bowel mucosa have been implicated in the pathogenesis of joint disease131,132. Similarly, in psoriatic arthritis, skin flora might have a role in pathogenesis133,134. As with other arthritides, changes in host microflora could also affect the pathogenesis of Lyme arthritis. B. burgdorferi modulate the host microbiomes of their tick vectors to facilitate colonization135 and this process could also occur during tick-to-mammal transmission, disrupting the normal skin flora and altering the local immune environment.
Interestingly, patients have been reported to develop systemic autoimmune diseases, including RA and spondyloarthritis, within months of having Lyme disease136. Although these occurrences could be coincidental, we speculate that latent autoimmunity might be induced non-specifically by the adjuvant effects of infection, or alternatively, that autoimmune-promoting conditions that develop during Lyme disease might trigger other systemic autoimmune diseases. The Lyme arthritis story underscores the importance of research into the potential role of specific infectious agents in various forms of chronic inflammatory arthritis, research that is hoped to provide breakthroughs in approaches to diagnosis and treatment.
Conclusions
After B. burgdorferi infection of the skin, early dissemination of spirochaetes to joints accompanied by dysregulation of innate immune responses (particularly type I interferon responses), might promote subsequent arthritis development. Months later, clinical arthritis develops within the context of an expanded adaptive immune response to the spirochaete. Rather than the arthritis resolving following antibiotic therapy, a small percentage of patients have persistent synovitis that can worsen in the post-antibiotic period. In these patients, the central pathogenetic feature is an excessive, dysregulated pro-inflammatory immune response characterized by exceptionally high amounts of IFNγ coupled with inadequate amounts of the anti-inflammatory cytokine IL-10. The consequences of this dysregulated response in synovia include chronic vascular damage and impaired tissue repair, autoimmune T cell and B cell responses, and tumour-like fibroblast proliferation and fibrosis. These histological characteristics are similar to those seen in other chronic inflammatory arthritides, including RA. Thus, post-infectious Lyme arthritis might serve as a model to aid our understanding of other forms of arthritis in which an infectious agent triggers or shapes the complex interactions between pathogen and host immune responses, leading to joint inflammation. However, important gaps remain in understanding the link between infection and autoimmunity in Lyme arthritis. Future research should focus on determining the initial steps in the break in immune tolerance during infection, on elucidating the role of B. burgdorferi peptidoglycan or other spirochaetal remnants in the pathogenesis of Lyme arthritis, on studying antibody specificities and function, and on identifying how autoimmune responses seem to evolve over time to become increasingly T cell dependent and more pathogenic.
Key points.
A combination of spirochaetal and host genetic factors shape the outcome of Lyme arthritis, which ranges from mild, antibiotic-responsive joint inflammation to persistent, antibiotic-refractory autoinflammatory or autoimmune synovitis.
Certain highly inflammatory strains of Borrelia burgdorferi most commonly found in north-eastern USA are present at an increased frequency among patients who subsequently develop post-infectious Lyme arthritis.
The histology of post-infectious Lyme arthritis synovia is similar to that in other chronic inflammatory arthritides, such as rheumatoid arthritis, but there is greater microvascular damage in Lyme arthritis.
B. burgdorferi is no longer present in synovia after treatment with antibiotics, but B. burgdorferi peptidoglycan might persist and could be an important promoter of innate immune responses.
Dysregulated, excessive IFNγ responses and inadequate amounts of the anti-inflammatory cytokine IL-10 are a central feature of post-infectious Lyme arthritis, and contribute to persistent inflammation and the development of autoimmunity.
Synovial fibroblasts, the most common cell in the synovial lesion, become immune effector cells capable of altering the innate and adaptive immune microenvironment in Lyme arthritis.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.von Herrath MG, Fujinami RS & Whitton JL Microorganisms and autoimmunity: making the barren field fertile? Nat. Rev. Microbiol 1, 151–157 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Steere AC et al. Lyme borreliosis. Nat. Rev. Dis. Prim 2, 16090 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steere AC Lyme disease. N. Engl. J. Med 321, 586–596 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Radolf JD, Strle K, Lemieux JE & Strle F Lyme disease in humans. Curr. Issues Mol. Biol 42, 333–384 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steere AC, Schoen RT & Taylor E The clinical evolution of Lyme arthritis. Ann. Intern. Med 107, 725–731 (1987). [DOI] [PubMed] [Google Scholar]
- 6.Miller JB & Aucott JN Stages of Lyme arthritis. J. Clin. Rheumatol 10.1097/RHU.0000000000001513 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Arvikar SL & Steere AC Diagnosis and treatment of Lyme arthritis. Infect. Dis. Clin. North. Am 29, 269–280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steere AC & Angelis SM Therapy for Lyme arthritis: strategies for the treatment of antibiotic-refractory arthritis. Arthritis Rheum. 54, 3079–3086 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Lochhead RB et al. MicroRNA expression shows inflammatory dysregulation and tumor-like proliferative responses in joints of patients with post-infectious Lyme arthritis. Arthritis Rheumatol. 69, 1100–1110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lochhead RB et al. Robust interferon signature and suppressed tissue repair gene expression in synovial tissue from patients with postinfectious, Borrelia burgdorferi-induced Lyme arthritis. Cell Microbiol. 21, e12954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drouin EE et al. A novel human autoantigen, endothelial cell growth factor, is a target of T and B cell responses in patients with Lyme disease. Arthritis Rheum. 65, 186–196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Londono D et al. Antibodies to endothelial cell growth factor and obliterative microvascular lesions in the synovium of patients with antibiotic-refractory Lyme arthritis. Arthritis Rheumatol. 66, 2124–2133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pianta A et al. Annexin A2 is a target of autoimmune T and B cell responses associated with synovial fibroblast proliferation in patients with antibiotic-refractory Lyme arthritis. Clin. Immunol 160, 336–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crowley JT et al. A highly expressed human protein, apolipoprotein B-100, serves as an autoantigen in a subgroup of patients with Lyme disease. J. Infect. Dis 212, 1841–1850 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crowley JT et al. Matrix metalloproteinase-10 is a target of T and B cell responses that correlate with synovial pathology in patients with antibiotic-refractory Lyme arthritis. J. Autoimmun 69, 24–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang KS, Klempner MS, Wormser GP, Marques AR & Alaedini A Association of immune response to endothelial cell growth factor with early disseminated and late manifestations of Lyme disease but not posttreatment Lyme disease syndrome. Clin. Infect. Dis 61, 1703–1706 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lantos PM et al. Clinical practice guidelines by the Infectious Diseases Society of America (IDSA), American Academy of Neurology (AAN), and American College of Rheumatology (ACR): 2020 guidelines for the prevention, diagnosis and treatment of Lyme disease. Clin. Infect. Dis 72, e1–e48 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Schoen RT, Aversa JM, Rahn DW & Steere AC Treatment of refractory chronic Lyme arthritis with arthroscopic synovectomy. Arthritis Rheum. 34, 1056–1060 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Steere AC & Glickstein L Elucidation of Lyme arthritis. Nat. Rev. Immunol 4, 143–152 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Barthold SW, Beck DS, Hansen GM, Terwilliger GA & Moody KD Lyme borreliosis in selected strains and ages of laboratory mice. J. Infect. Dis 162, 133–138 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Ma Y et al. Distinct characteristics of resistance to Borrelia burgdorferi-induced arthritis in C57BL/6 N mice. Infect. Immun 66, 161–168 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y et al. Interval-specific congenic lines reveal quantitative trait loci with penetrant lyme arthritis phenotypes on chromosomes 5, 11, and 12. Infect. Immun 77, 3302–3311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bockenstedt LK, Wooten RM & Baumgarth N Immune response to Borrelia: lessons from Lyme disease spirochetes. Curr. Issues Mol. Biol 42, 145–190 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X et al. Burden and viability of Borrelia burgdorferi in skin and joints of patients with erythema migrans or Lyme arthritis. Arthritis Rheum. 63, 2238–2247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mullegger RR et al. Differential expression of cytokine mRNA in skin specimens from patients with erythema migrans or acrodermatitis chronica atrophicans. J. Invest. Dermatol 115, 1115–1123 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Salazar JC et al. Coevolution of markers of innate and adaptive immunity in skin and peripheral blood of patients with erythema migrans. J. Immunol 171, 2660–2670 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Marques A et al. Transcriptome assessment of erythema migrans skin lesions in patients with early Lyme disease reveals predominant interferon signaling. J. Infect. Dis 217, 158–167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coburn J, Magoun L, Bodary SC & Leong JM Integrins αvβ3 and α5β1 mediate attachment of Lyme disease spirochetes to human cells. Infect. Immun 66, 1946–1952 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caine JA & Coburn J Multifunctional and redundant roles of Borrelia burgdorferi outer surface proteins in tissue adhesion, colonization, and complement evasion. Front. Immunol 7, 442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ristow LC et al. Integrin binding by Borrelia burgdorferi P66 facilitates dissemination but is not required for infectivity. Cell Microbiol. 17, 1021–1036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comstock LE & Thomas DD Characterization of Borrelia burgdorferi invasion of cultured endothelial cells. Microb. Pathog 10, 137–148 (1991). [DOI] [PubMed] [Google Scholar]
- 32.Zambrano MC, Beklemisheva AA, Bryksin AV, Newman SA & Cabello FC Borrelia burgdorferi binds to, invades, and colonizes native type I collagen lattices. Infect. Immun 72, 3138–3146 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tupin E et al. NKT cells prevent chronic joint inflammation after infection with Borrelia burgdorferi. Proc. Natl Acad. Sci. USA 105, 19863–19868 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones KL et al. Strong IgG antibody responses to Borrelia burgdorferi glycolipids in patients with Lyme arthritis, a late manifestation of the infection. Clin. Immunol 132, 93–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee WY et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat. Immunol 11, 295–302 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kinjo Y et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat. Immunol 7, 978–986 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Reinink P et al. CD1b presents self and Borrelia burgdorferi diacylglycerols to human T cells. Eur. J. Immunol 49, 737–746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seinost G et al. Four clones of Borrelia burgdorferi sensu stricto cause invasive infection in humans. Infect. Immun 67, 3518–3524 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wormser GP et al. Borrelia burgdorferi genotype predicts the capacity for hematogenous dissemination during early Lyme disease. J. Infect. Dis 198, 1358–1364 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petzke MM et al. Global transcriptome analysis identifies a diagnostic signature for early disseminated Lyme disease and its resolution. mBio 11, e00047–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strle K, Shin JJ, Glickstein LJ & Steere AC Association of a Toll-like receptor 1 polymorphism with heightened Th1 inflammatory responses and antibiotic-refractory Lyme arthritis. Arthritis Rheum. 64, 1497–1507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strle K, Jones KL, Drouin EE, Li X & Steere AC Borrelia burgdorferi RST1 (OspC type A) genotype is associated with greater inflammation and more severe Lyme disease. Am. J. Pathol 178, 2726–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strle K et al. T-helper 17 cell cytokine responses in Lyme disease correlate with Borrelia burgdorferi antibodies during early infection and with autoantibodies late in the illness in patients with antibiotic-refractory Lyme arthritis. Clin. Infect. Dis 64, 930–938 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin YP et al. Strain-specific joint invasion and colonization by Lyme disease spirochetes is promoted by outer surface protein C. PLoS Pathog. 16, e1008516 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang L et al. Rapid clearance of Borrelia burgdorferi from the blood circulation. Parasit. Vectors 13, 191 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moriarty TJ et al. Real-time high resolution 3D imaging of the Lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog. 4, e1000090 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hyde JA Borrelia burgdorferi keeps moving and carries on: a review of borrelial dissemination and invasion. Front. Immunol 8, 114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo BP, Brown EL, Dorward DW, Rosenberg LC & Hook M Decorin-binding adhesins from Borrelia burgdorferi. Mol. Microbiol 30, 711–723 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Duray PH & Steere AC Clinical pathologic correlations of Lyme disease by stage. Ann. N. Y. Acad. Sci 539, 65–79 (1988). [DOI] [PubMed] [Google Scholar]
- 50.Lin YP et al. Strain-specific variation of the decorin-binding adhesin DbpA influences the tissue tropism of the Lyme disease spirochete. PLoS Pathog. 10, e1004238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones KL, McHugh GA, Glickstein LJ & Steere AC Analysis of Borrelia burgdorferi genotypes in patients with Lyme arthritis: high frequency of ribosomal RNA intergenic spacer type 1 strains in antibiotic-refractory arthritis. Arthritis Rheum. 60, 2174–2182 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steere AC, McHugh G, Damle N & Sikand VK Prospective study of serologic tests for Lyme disease. Clin. Infect. Dis 47, 188–195 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steere AC, Hardin JA, Ruddy S, Mummaw JG & Malawista SE Lyme arthritis: correlation of serum and cryoglobulin IgM with activity, and serum IgG with remission. Arthritis Rheum. 22, 471–483 (1979). [DOI] [PubMed] [Google Scholar]
- 54.Craft JE, Fischer DK, Shimamoto GT & Steere AC Antigens of Borrelia burgdorferi recognized during Lyme disease. Appearance of a new immunoglobulin M response and expansion of the immunoglobulin G response late in the illness. J. Clin. Invest 78, 934–939 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alverson J, Bundle SF, Sohaskey CD, Lybecker MC & Samuels DS Transcriptional regulation of the ospAB and ospC promoters from Borrelia burgdorferi. Mol. Microbiol 48, 1665–1677 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Zhang JR, Hardham JM, Barbour AG & Norris SJ Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89, 275–285 (1997). [DOI] [PubMed] [Google Scholar]
- 57.Skare JT & Garcia BL Complement evasion by Lyme disease spirochetes. Trends Microbiol. 28, 889–899 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JC, Maylor-Hagen H, Ma Y, Weis JH & Weis JJ The Lyme disease spirochete Borrelia burgdorferi utilizes multiple ligands, including RNA, for interferon regulatory factor 3-dependent induction of type I interferon-responsive genes. Infect. Immun 78, 3144–3153 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lochhead RB et al. Endothelial cells and fibroblasts amplify the arthritogenic type I IFN response in murine Lyme disease and are major sources of chemokines in Borrelia burgdorferi-infected joint tissue. J. Immunol 189, 2488–2501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crandall H et al. Gene expression profiling reveals unique pathways associated with differential severity of Lyme arthritis. J. Immunol 177, 7930–7942 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Crow MK & Ronnblom L Type I interferons in host defence and inflammatory diseases. Lupus Sci. Med 6, e000336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nocton JJ et al. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in synovial fluid from patients with Lyme arthritis. N. Engl. J. Med 330, 229–234 (1994). [DOI] [PubMed] [Google Scholar]
- 63.Shin JJ, Glickstein LJ & Steere AC High levels of inflammatory chemokines and cytokines in joint fluid and synovial tissue throughout the course of antibiotic-refractory Lyme arthritis. Arthritis Rheum. 56, 1325–1335 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Kannian P et al. Antibody responses to Borrelia burgdorferi in patients with antibiotic-refractory, antibiotic-responsive, or non-antibiotic-treated Lyme arthritis. Arthritis Rheum. 56, 4216–4225 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Barbour AG et al. A genome-wide proteome array reveals a limited set of immunogens in natural infections of humans and white-footed mice with Borrelia burgdorferi. Infect. Immun 76, 3374–3389 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Y, Bruno JF & Luft BJ Profiling the humoral immune response to Borrelia burgdorferi infection with protein microarrays. Microb. Pathog 45, 403–407 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Li X et al. Tick-specific borrelial antigens appear to be upregulated in American but not European patients with Lyme arthritis, a late manifestation of Lyme borreliosis. J. Infect. Dis 208, 934–941 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crowley H & Huber BT Host-adapted Borrelia burgdorferi in mice expresses OspA during inflammation. Infect. Immun 71, 4003–4010 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gross DM, Steere AC & Huber BT T helper 1 response is dominant and localized to the synovial fluid in patients with Lyme arthritis. J. Immunol 160, 1022–1028 (1998). [PubMed] [Google Scholar]
- 70.Lochhead RB et al. Interferon-gamma production in Lyme arthritis synovial tissue promotes differentiation of fibroblast-like synoviocytes into immune effector cells. Cell Microbiol. 21, e12992 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vudattu NK, Strle K, Steere AC & Drouin EE Dysregulation of CD4+CD25 high T cells in the synovial fluid of patients with antibiotic-refractory Lyme arthritis. Arthritis Rheum. 65, 1643–1653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sulka KB et al. Lyme disease-associated IgG4 autoantibodies correlate with synovial pathology in antibiotic-refractory Lyme arthritis. Arthritis Rheumatol. 70, 1835–1846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bolz DD et al. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J. Immunol 173, 2003–2010 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Wooten RM et al. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J. Immunol 168, 348–355 (2002). [DOI] [PubMed] [Google Scholar]
- 75.Alexopoulou L et al. Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat. Med 8, 878–884 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Hirschfeld M et al. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by Toll-like receptor 2. J. Immunol 163, 2382–2386 (1999). [PubMed] [Google Scholar]
- 77.Bramwell KK et al. Lysosomal β-glucuronidase regulates Lyme and rheumatoid arthritis severity. J. Clin. Invest 124, 311–320 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bramwell KK et al. β-Glucuronidase, a regulator of Lyme arthritis severity, modulates lysosomal trafficking and MMP-9 secretion in response to inflammatory stimuli. J. Immunol 195, 1647–1656 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma Y et al. Borrelia burgdorferi arthritis-associated locus Bbaa1 regulates Lyme arthritis and K/BxN serum transfer arthritis through intrinsic control of type I IFN production. J. Immunol 193, 6050–6060 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paquette JK et al. Genetic control of Lyme arthritis by Borrelia burgdorferi arthritis-associated locus 1 Is dependent on localized differential production of IFN-β and requires upregulation of myostatin. J. Immunol 199, 3525–3534 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller JC et al. A critical role for type I IFN in arthritis development following Borrelia burgdorferi infection of mice. J. Immunol 181, 8492–8503 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bockenstedt LK, Gonzalez DG, Haberman AM & Belperron AA Spirochete antigens persist near cartilage after murine Lyme borreliosis therapy. J. Clin. Invest 122, 2652–2660 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bockenstedt LK & Wormser GP Review: unraveling Lyme disease. Arthritis Rheumatol. 66, 2313–2323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnston YE et al. Lyme arthritis. Spirochetes found in synovial microangiopathic lesions. Am. J. Pathol 118, 26–34 (1985). [PMC free article] [PubMed] [Google Scholar]
- 85.Steere AC, Duray PH & Butcher EC Spirochetal antigens and lymphoid cell surface markers in Lyme synovitis. Comparison with rheumatoid synovium and tonsillar lymphoid tissue. Arthritis Rheum. 31, 487–495 (1988). [DOI] [PubMed] [Google Scholar]
- 86.Akin E, Aversa J & Steere AC Expression of adhesion molecules in synovia of patients with treatment-resistant Lyme arthritis. Infect. Immun 69, 1774–1780 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muehlenbachs A et al. Cardiac tropism of Borrelia burgdorferi: An autopsy study of sudden cardiac death associated with Lyme carditis. Am. J. Pathol 186, 1195–1205 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Cadavid D et al. Cardiac involvement in non-human primates infected with the Lyme disease spirochete Borrelia burgdorferi. Lab. Invest 84, 1439–1450 (2004). [DOI] [PubMed] [Google Scholar]
- 89.Cadavid D et al. Infection and inflammation in skeletal muscle from nonhuman primates infected with different genospecies of the Lyme disease spirochete Borrelia burgdorferi. Infect. Immun 71, 7087–7098 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Casselli T et al. A murine model of Lyme disease demonstrates that Borrelia burgdorferi colonizes the dura mater and induces inflammation in the central nervous system. PLoS Pathog. 17, e1009256 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lawson JP & Steere AC Lyme arthritis: radiologic findings. Radiology 154, 37–43 (1985). [DOI] [PubMed] [Google Scholar]
- 92.Steere AC Posttreatment Lyme disease syndromes: distinct pathogenesis caused by maladaptive host responses. J. Clin. Invest 130, 2148–2151 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jones KL et al. Borrelia burgdorferi genetic markers and disseminated disease in patients with early Lyme disease. J. Clin. Microbiol 44, 4407–4413 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hanincova K et al. Multilocus sequence typing of Borrelia burgdorferi suggests existence of lineages with differential pathogenic properties in humans. PLoS ONE 8, e73066 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cerar T et al. Differences in genotype, clinical features, and inflammatory potential of Borrelia burgdorferi sensu stricto strains from Europe and the United States. Emerg. Infect. Dis 22, 818–827 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grillon A et al. Characteristics and clinical outcomes after treatment of a national cohort of PCR-positive Lyme arthritis. Semin. Arthritis Rheum 48, 1105–1112 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Jutras BL et al. Borrelia burgdorferi peptidoglycan is a persistent antigen in patients with Lyme arthritis. Proc. Natl Acad. Sci. USA 116, 13498–13507 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hawn TR et al. A common human TLR1 polymorphism regulates the innate immune response to lipopeptides. Eur. J. Immunol 37, 2280–2289 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Johnson CM et al. Cutting edge: a common polymorphism impairs cell surface trafficking and functional responses of TLR1 but protects against leprosy. J. Immunol 178, 7520–7524 (2007). [DOI] [PubMed] [Google Scholar]
- 100.Lochhead RB et al. MicroRNA-146a provides feedback regulation of Lyme arthritis but not carditis during infection with Borrelia burgdorferi. PLoS Pathog. 10, e1004212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sahay B et al. Induction of interleukin 10 by Borrelia burgdorferi is regulated by the action of CD14-dependent p38 mitogen-activated protein kinase and cAMP-mediated chromatin remodeling. Infect. Immun 86, e00781–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Steere AC et al. Antibiotic-refractory Lyme arthritis is associated with HLA-DR molecules that bind a Borrelia burgdorferi peptide. J. Exp. Med 203, 961–971 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iliopoulou BP, Guerau-De-Arellano M & Huber BT HLA-DR alleles determine responsiveness to Borrelia burgdorferi antigens in a mouse model of self-perpetuating arthritis. Arthritis Rheum. 60, 3831–3840 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vicente R, Noel D, Pers YM, Apparailly F & Jorgensen C Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat. Rev. Rheumatol 12, 211–220 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Lochhead RB et al. Antagonistic interplay between microRNA-155 and IL-10 during Lyme carditis and arthritis. PLoS ONE 10, e0135142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shen S et al. Treg cell numbers and function in patients with antibiotic-refractory or antibiotic-responsive Lyme arthritis. Arthritis Rheum. 62, 2127–2137 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Siebers EM, Liedhegner ES, Lawlor MW, Schell RF & Nardelli DT Regulatory T cells contribute to resistance against Lyme arthritis. Infect. Immun 88, e00160–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iliopoulou BP, Alroy J & Huber BT Persistent arthritis in Borrelia burgdorferi-infected HLA-DR4-positive CD28-negative mice post-antibiotic treatment. Arthritis Rheum. 58, 3892–3901 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sonderegger FL et al. Localized production of IL-10 suppresses early inflammatory cell infiltration and subsequent development of IFN-γ-mediated Lyme arthritis. J. Immunol 188, 1381–1393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Whiteside SK et al. IL-10 deficiency reveals a role for TLR2-dependent bystander activation of T cells in Lyme arthritis. J. Immunol 200, 1457–1470 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Park H et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol 6, 1133–1141 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang Q et al. Immunogenic HLA-DR-presented self-peptides identified directly from clinical samples of synovial tissue, synovial fluid, or peripheral blood in patients with rheumatoid arthritis or Lyme arthritis. J. Proteome Res 16, 122–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gutierrez-Hoffmann MG et al. Borrelia burgdorferi-induced changes in the class II self-immunopeptidome displayed on HLA-DR molecules expressed by dendritic cells. Front. Med 7, 568 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Danzer H et al. Human Fcγ-receptor IIb modulates pathogen-specific versus self-reactive antibody responses in Lyme arthritis. eLife 9, e55319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deane KD & Holers VM Rheumatoid arthritis pathogenesis, prediction, and prevention: an emerging paradigm shift. Arthritis Rheumatol. 73, 181–193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.O’Neil LJ et al. Association of a serum protein signature with rheumatoid arthritis development. Arthritis Rheumatol. 73, 78–88 (2021). [DOI] [PubMed] [Google Scholar]
- 117.van Delft MAM et al. The isotype and IgG subclass distribution of anti-carbamylated protein antibodies in rheumatoid arthritis patients. Arthritis Res. Ther 19, 190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chapuy-Regaud S et al. IgG subclass distribution of the rheumatoid arthritis-specific autoantibodies to citrullinated fibrin. Clin. Exp. Immunol 139, 542–550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen LF et al. Elevated serum IgG4 defines specific clinical phenotype of rheumatoid arthritis. Mediators Inflamm. 2014, 635293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Divan A, Budd RC, Tobin RP & Newell-Rogers MK γδ T Cells and dendritic cells in refractory Lyme arthritis. J. Leukoc. Biol 97, 653–663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Katchar K, Drouin EE & Steere AC Natural killer cells and natural killer T cells in Lyme arthritis. Arthritis Res. Ther 15, R183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Viatte S, Plant D & Raychaudhuri S Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol 9, 141–153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carmona-Rivera C et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci. Immunol 2, eaag3358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ai R et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat. Commun 9, 1921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang F et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol 20, 928–942 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Klareskog L, Catrina AI & Paget S Rheumatoid arthritis. Lancet 373, 659–672 (2009). [DOI] [PubMed] [Google Scholar]
- 127.Potempa J, Mydel P & Koziel J The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol 13, 606–620 (2017). [DOI] [PubMed] [Google Scholar]
- 128.Arvikar SL et al. Periodontal inflammation and distinct inflammatory profiles in saliva and GCF compared with serum and joints in rheumatoid arthritis patients. J. Periodontol 10.1002/JPER.20-0051 (2021). [DOI] [PubMed] [Google Scholar]
- 129.Scher JU et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2, e01202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pianta A et al. Two rheumatoid arthritis-specific autoantigens correlate microbial immunity with autoimmune responses in joints. J. Clin. Invest 127, 2946–2956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gracey E et al. Revisiting the gut-joint axis: links between gut inflammation and spondyloarthritis. Nat. Rev. Rheumatol 16, 415–433 (2020). [DOI] [PubMed] [Google Scholar]
- 132.Viladomiu M et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci. Transl. Med 9, eaaf9655 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yan D et al. The role of the skin and gut microbiome in psoriatic disease. Curr. Dermatol. Rep 6, 94–103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gladman DD, Antoni C, Mease P, Clegg DO & Nash P Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann. Rheum. Dis 64, ii14–ii17 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kurokawa C et al. Interactions between Borrelia burgdorferi and ticks. Nat. Rev. Microbiol 18, 587–600 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Arvikar SL, Crowley JT, Sulka KB & Steere AC Autoimmune arthritides, rheumatoid arthritis, psoriatic arthritis, or peripheral spondyloarthritis following Lyme disease. Arthritis Rheumatol. 69, 194–202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Steere AC in Rheumatology 7th Edn (eds Hochberg M et al. ) Vol. 963 (Elsevier, 2019). [Google Scholar]