

Abstract
Granulomatous inflammation of the lung can be a manifestation of different conditions and can be caused by endogenous inflammation or external triggers. A multitude of different genetic mutations can either predispose patients to infections with granuloma-forming pathogens or cause autoinflammatory disorders, both leading to the phenotype of pulmonary granulomatosis. Based on a detailed patient history, physical examination and a diagnostic approach including laboratory workup, pulmonary function tests (PFTs), computed tomography (CT) scans, bronchoscopy with bronchoalveolar lavage (BAL), lung biopsies and specialised microbiological and immunological diagnostics, a correct diagnosis of an underlying cause of pulmonary granulomatosis of genetic origin can be made and appropriate therapy can be initiated. Depending on the underlying disorder, treatment approaches can include antimicrobial therapy, immunosuppression and even haematopoietic stem cell transplantation (HSCT). Patients with immunodeficiencies and autoinflammatory conditions are at the highest risk of developing pulmonary granulomatosis of genetic origin. Here we provide a review on these disorders and discuss pathogenesis, clinical presentation, diagnostic approach and treatment.
Short abstract
Pulmonary granulomatosis of genetic origin mostly occurs in immunodeficiency disorders and autoinflammatory conditions. In addition to specific approaches in this regard, the diagnostic workup needs to cover environmental and occupational aspects. https://bit.ly/31SqdHW
Introduction
Granulomas are defined as focal, organised inflammatory infiltrates of epithelioid histiocytes (macrophages). They may contain multinucleated giant cells, lymphocytes and plasma cells, as well as necrotic areas [1, 2]. Granulomas are formed to encapsulate material or pathogens that cannot be eliminated otherwise, but in some cases the exact causes for granuloma formation are still unclear [1, 2]. Granulomas can occur in different disorders and therefore warrant a careful evaluation of the clinical context [1]. Histologically, necrotising and non-necrotising granulomas are differentiated. Necrotising granulomas develop more commonly in association with an infectious cause [1]. Morphology, localisation and proof of infectious agents can give additional clues to the underlying diagnosis.
Pulmonary granulomatous inflammatory conditions comprise a heterogeneous group of diseases with different pathologies, phenotypes and prognoses [1]. Infectious and noninfectious causes have to be differentiated. Mycobacterial infections (both Mycobacterium tuberculosis and nontuberculosis (non-TB) mycobacteria) and fungal infections (histoplasma, cryptococcus, pneumocystis and aspergillus) are the most common infectious triggers associated with pulmonary granuloma formation. Noninfectious pulmonary granulomatous diseases encompass autoinflammatory conditions (e.g. sarcoidosis), granuloma formation after environmental exposure (e.g. chronic beryllium disease), vasculitis (e.g. granulomatosis with polyangiitis), autoimmune diseases and primary immunodeficiencies (see table 1 for an overview of the conditions discussed in this review) [1, 2]. Non-necrotising granulomas more commonly develop in cases without apparent infectious triggers, with the exception of granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis (table 1) [1, 2].
TABLE 1.
Immunodeficiencies and autoinflammatory disorders associated with pulmonary granulomatosis
| Condition | Exemplary clinical features | Exemplary laboratory features | Radiologic presentation | Histology |
| CVID |
|
Hypogammaglobulinaemia |
|
|
| LRBA/CTLA-4 deficiency |
|
|
See CVID | See CVID |
| CGD |
|
Reduced “respiratory burst” |
|
|
| STAT3 loss of function (AD HIES) |
|
|
|
|
| STAT3 gain of function | Autoimmunopathies (cytopenias, lymphoproliferation, enteropathy, diabetes) |
|
|
|
| MSMD |
|
Reduced IFN-γ production |
|
|
| Blau syndrome/early onset sarcoidosis |
|
|
|
Non-necrotising granulomas |
| NOD2-associated autoinflammatory disease |
|
|
|
Non-necrotising granulomas |
| Chronic beryllium disease |
|
|
|
|
| SAVI |
|
Constantly ↑ acute phase/inflammation parameters | Nodules Cavities Fixed infiltrates |
|
| Granulomatosis with polyangiitis |
|
↑ Cytoplasmic ANCAs |
|
|
| Eosinophilic granulomatosis with polyangiitis |
|
|
|
|
| Hypersensitivity pneumonitis |
|
↑ Specific IgGs against organic compounds |
|
|
CVID: common variable immunodeficiency; GLILD: granulomatous-lymphocytic interstitial lung disease; GGO: ground-glass opacity; LRBA: lipopolysaccharide-responsive beige-like anchor protein; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; CNS: central nervous system; CGD: chronic granulomatous disease; STAT-3: signal transducer and activator of transcription 3; Ig: immunoglobulin; AD: autosomal-dominant; HIES: hyper IgE syndrome; IL: interleukin; MSMD: Mendelian-susceptibility to mycobacterial disease; IFN: interferon; ACE: angiotensin-converting enzyme; BAL: bronchoalveolar lavage; NOD2: nucleotide-binding oligomerisation domain-containing protein 2; STING: stimulator of interferon genes; SAVI: STING-associated vasculopathy with onset in infancy; ANCA: anti-neutrophil cytoplasmic antibody.
Key for diagnosis of the underlying condition is not only the presence of pulmonary granulomas but a combination of typical clinical features, laboratory parameters, pulmonary function tests (PFTs), histologic assessment and imaging studies. Evaluation of affected lung tissue is the gold standard in accessing pulmonary granulomatous disease and might even be essential in identifying infectious causes in some conditions [1, 3]. Specimens can be obtained by bronchoscopy with bronchoalveolar lavage (BAL), transbronchial biopsy, cryobiopsy, or video-assisted thoracoscopic surgical (VATS) biopsy. VATS biopsy seems superior to transbronchial biopsy in establishing the correct diagnosis [4–6]; however, VATS should only be performed if a histological result is paramount for treatment decisions, especially in paediatric patients [3, 7, 8]. If granulomatous infiltration is present in organs that are more easily accessible for biopsy (e.g. the skin) then these sites should be preferred for sampling. As lung biopsies are not always readily available in the clinical setting, primary evaluation with high-resolution computed tomography (HRCT) chest scans is used to appreciate suspected granulomatous inflammation of the lung [8, 9]. Imaging techniques such as positron emission tomography (PET)–computed tomography (CT) might allow identification of additional sites with active lymphoproliferation that may be more easily accessible for biopsy (e.g. peripheral lymph nodes) [8]. Additional laboratory workup may reveal immunological abnormalities or hint at autoinflammatory conditions. PFTs are warranted to assess the clinical course of pulmonary involvement; however, there is no consensus on the best diagnostic algorithm for all cases of granulomatous lung disease and decisions need to be made on an individual basis.
This review focuses on genetic causes of pulmonary granulomatosis and respective disorders are presented here with regard to genetic, clinical, diagnostic, histologic and therapeutic aspects.
Primary immunodeficiencies associated with pulmonary granulomatosis
Common variable immunodeficiency
Common variable immunodeficiency (CVID) comprises a heterogeneous group of primary immunodeficiencies with hypogammaglobinaemia and variable T-lymphocyte/B-lymphocyte dysfunction [10]. CVID typically manifests in young adulthood and is the most common primary immunodeficiency with a prevalence of 0.7 per 10 000 [11, 12]. Monogenetic causes of CVID have been identified in up to 50% of cases, with mutations in nuclear factor κ light-chain-enhancer of activated B-cells 1 (NF-κB1) and transmembrane activator and calcium-modulating ligand (CAML) interactor (TACI) being the most common [13–18].
Diagnostic criteria for CVID include hypogammaglobulinaemia less than two standard deviations (sd) below the mean for age. Additionally, a reduction of immunoglobulin (Ig)A/IgM, onset of symptomatic immunodeficiency at greater than 2 years of age, absent isohaemagglutinins and/or poor response to vaccination, as well as exclusion of other defined causes of hypogammaglobulinaemia, constitute the criteria for CVID [19]. Patients typically suffer from recurrent bronchopulmonary infections and additional autoimmune phenomena are present in up to 40% of cases [10, 20]. A complication of CVID is granulomatous-lymphocytic interstitial lung disease (GLILD), characterised by granulomatous and lymphoproliferative inflammation of the small airways and pulmonary interstitium (figure 1) [1]. GLILD is considered to be a pulmonary manifestation of a systemic granulomatous disease that also includes other organs. Dyspnoea and recurrent bronchopulmonary infections are apparent in affected patients [8] and a restrictive pattern and reduced diffusing capacity of the lung for carbon monoxide (DLCO) can be observed in PFTs [21]. Up to one third of patients with CVID can develop GLILD [8] and GLILD in patients with CVID is associated with a poorer prognosis and higher mortality [22].
FIGURE 1.
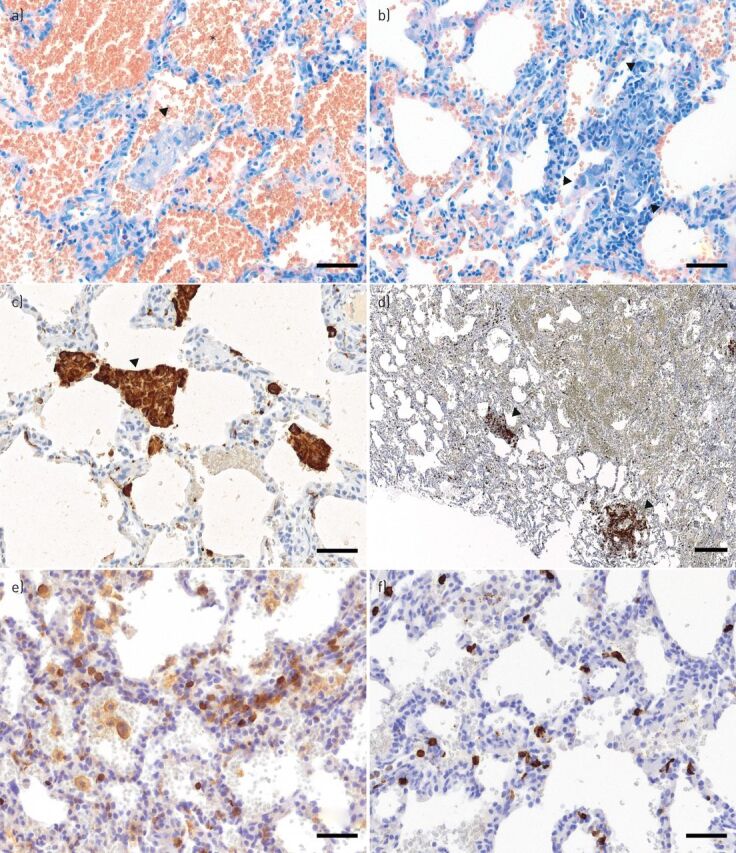
Alveolar haemorrhage: partly epithelioid cellular interstitial inflammation in the affected lung, suggestive of a mild granulomatous-lymphocytic interstitial lung disease (GLILD). Airspaces are filled with erythrocytes (a), as indicated by an exemplar asterisk), some cluster forming alveolar macrophages (a) and c), as indicated by arrowheads, the latter using CD68+ stain) and small non-necrotising interstitial granulomas (b), as indicated by arrowheads) consisting of a few epithelioid cells and lymphocytes. Interstitial lymphocytic infiltrate mainly consists of CD4+ T-cells, partly clustered d) and partly scattered e). CD8+ T-cells are scattered f) and form the minority of the lymphocytic infiltrations. Scale bars: a–c, e, f) 50 μm; d) 200 μm.
On histopathological evaluation, GLILD presents with lymphocytic interstitial pneumonitis, follicular bronchiolitis and non-necrotising granulomas [8]. These changes are typically found in the lower lobes of the lungs. A mix of both peribronchiolar and interstitial lymphoid infiltration is seen, predominantly by CD4+ T-cells but also by B-cells [23]. GLILD is also associated with cryptogenic organising pneumonia and interstitial fibrosis [23] and the latter may show a progressive course [22, 24].
Small and large nodules, consolidations and ground-glass abnormalities can be found on HRCT; additionally, a lymphadenopathy is often present [21]. Lung biopsies are recommended to establish a definitive diagnosis of GLILD and to rule out differential diagnoses including infectious causes, interstitial pneumonia, sarcoidosis, cryptogenic organising pneumonia, lymphoma and others [8].
In CVID, Ig replacement therapy is indicated to reduce susceptibility to bronchopulmonary infections. In GLILD, corticosteroids alone do not lead to durable improvement [8]; however, combined targeting of T-cells and B-cells with azathioprine and rituximab improves both radiographic pathology and PFTs in patients with CVID and GLILD [25]. Long-term therapy is needed to establish stable remission and several trials are currently exploring alternative immunosuppressive treatment strategies, including the use T-cell activation blocking agents like Abatacept.
Combined immunodeficiencies
Combined immunodeficiencies (CIDs) are a heterogeneous group of disorders with reduced but not absent T-cell immunity. CID-associated mutations have been found in Caspase10, PI3KCD, ITK, Dock8 and others [26]. There is considerable overlap with both severe combined immunodeficiencies (SCIDs) on one end of the spectrum and CVID on the other [19, 26]. Patients with CID may therefore present with SCID-like phenotypes, which require urgent haematopoietic stem cell transplantation (HSCT), but milder phenotypes can also occur, making treatment decisions difficult. If susceptibility to infection or autoimmunity are present, patients can be classified as having profound combined immunodeficiencies (PCIDs). Some 7% of patients with PCID have been found to suffer from chronic lung disease, including from GLILD [26]. Three disorders are discussed here in more detail, as follows: 1) null mutations in recombinase activating gene 1 or gene 2 (RAG 1 or RAG 2). These mutations cause SCID; however, hypomorphic mutations with residual RAG activity can lead to a PCID classified as atypical SCID [26–28]. Systemic granulomatous inflammation is a hallmark of the disorder. Interstitial pneumonia with noncaseating epithelioid-cell granulomas has also been reported in RAG deficiency [28]; 2) cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) deficiency; and 3) lipopolysaccharide-responsive beige-like anchor protein (LRBA) deficiency [20]. These latter two disorders are clinically very similar conditions that have been classified as CVID but may show some aspects of CID. LRBA regulates intracellular trafficking of CTLA-4 [29]. Both LRBA and CTLA-4 deficiency lead to impaired downregulation of immune processes [20, 29]. The clinical phenotype of both disorders is similar: autoimmune cytopenias as well as autoimmune inflammation in the intestines, the central nervous system (CNS) and other organs has been reported [30–32]. GLILD has been found in up to two thirds of patients with CTLA-4 deficiency and also in patients with LRBA deficiency (figures 2a–2c) [30, 31]. Diagnostic workup should follow the standards outlined above.
FIGURE 2.

Granulomatous-lymphocytic interstitial lung disease (GLILD). a) A 9-year old girl with atypical and confluent, partly spot-shaped infiltrates on chest computed tomography (CT) scan. b) A 20-year old woman showing nodules with adjacent ground-glass infiltrates. c) A chest CT scan of a 6-year old boy with cytotoxic T-lymphocyte-associated protein 4 deficiency. Multiple round infiltrates resembling “reversed halo” (or “atoll”) signs are evidence of GLILD. d) The same patient as in c) but 6 months after allogenic haematopoietic stem cell transplantation. Marked regression of GLILD is shown with mild residual, streaky interstitial consolidations. Scale bars: a, c, d) 15 mm; b) 20 mm.
CTLA-4 fusion proteins (e.g. abatacept) and mammalian target of rapamycin (mTOR) inhibitors have been used successfully in both LRBA and CTLA-4 deficiency and these treatments have had positive effects on GLILD as well [29, 30, 33]. Severe infectious or autoimmune complications in CIDs that cannot be controlled by Ig replacement or immunosuppressive therapy can be an indication for HSCT [26]. Improvement of GLILD after stem cell transplantation (SCT) has been demonstrated in CTLA-4 deficiency (figure 2d) [30]. Symptoms of mild RAG deficiency can be controlled by immunosuppression but long-term remission cannot be achieved in all patients. Some cases of partial RAG deficiency, especially atypical SCID, can only be cured with SCT [27, 28]. The diverse phenotype of (P)CIDs and the individual clinical course has therefore to be taken into account to decide whether SCT might be a viable option for affected patients [26, 27].
Chronic granulomatous disease
Chronic granulomatous disease (CGD) is caused by defects in superoxide-generating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase of phagocytes and lymphocytes [34, 35]. Currently, five different affected genes are known, while the most common findings are mutations in gp91phox (CYBB, cross-linked). Autosomal recessive forms involve mutations in p22phox (CYBA), p47phox (NCF1), p67phox (NCF2) and p40phox (NCF4) [34–36]. Both impaired killing of bacteria and fungi, as well as prolonged autoinflammatory reactions with granuloma formation, can occur [34, 35]. About one to three in 200 000 newborns are affected by CGD [36].
Pulmonary manifestations represent the most common organ involvement in CGD. Infectious complications are found in 80% of CGD patients, mostly in the form of pneumonia [37, 38]. Formation of abscesses, bronchiectasis and pulmonary granulomas has also been reported (figure 3) [3, 39].
FIGURE 3.

Pulmonary granulomas in a 4-year old boy with chronic granulomatous disease. A subpleural granuloma with a surrounding fuzzy rim is indicated (arrow). Scale bar: 10 mm.
Increased secretion of pro-inflammatory mediators or decreased production of anti-inflammatory cytokines causes inflammatory or auto-inflammatory manifestations in CGD [40]. This can lead to development of granulomas in multiple organs, such as the brain, liver, gastrointestinal tract, spleen, or lung. Granulomas are noncaseating and contain multinucleated giant cells [37]. Whether or not an infection is essential to trigger this inflammatory cascade is still debated [34]. Pulmonary symptoms in patients with CGD include dyspnoea, cough and reduced exercise tolerance [34, 35].
CT scans are important for visualisation of pulmonary pathologies in CGD (table 1); however, lung biopsies might be essential to correctly identify infectious triggers of disease exacerbation (as has been shown for Burkholderia species [3]). PFTs may show obstructive or restrictive patterns depending on the extent of lung involvement [34, 35].
CGD is curable by HSCT and appropriate evaluations should be initiated as soon as the diagnosis is made [41, 42]. Until HSCT can be performed, control and prevention of infection is paramount in CGD. Removal of severely damaged pulmonary tissue is sometimes indicated and some experience exists in the use of steroids in combination with anti-infective drugs [43]. Some authors have suggested the use of tumour necrosis factor (TNF) inhibitors to control inflammation and granuloma formation [9].
Mendelian susceptibility to mycobacterial disease
Defects in the interferon-γ (IFNγ)/interleukin-12 (IL-12) pathway lead to Mendelian susceptibility to mycobacterial disease (MSMD). In MSMD, effective killing of intracellular pathogens is impaired [44, 45]. Currently, mutations in 15 different genes (IFNGR1, IFNGR2, STAT1, JAK1, IRF8, SPPL2A, IL12B, IL12RB1, IL12RB2, IL23R, ISG15, TYK2, RORC, CYBB and NEMO) have been reported in MSMD, although these account for only around 50% of cases [44]. Prevalence is estimated to be around one in 50 000 [46]. Patients present with mycobacterial infections, classically from Bacillus Calmette–Guérin (BCG) vaccine or non-TB mycobacteria. Infections by M. tuberculosis and other intracellular pathogens are common, as is chronic mucocutaneous candidiasis (for specific genetic defects) [44, 46]. Clinical presentation varies from localised to systemic manifestations and via an acute or chronic course. Pulmonary granuloma formation develops as frequently as in pulmonary mycobacterial infection and both necrotising and non-necrotising granulomas are found [44, 46]. Impaired cytokine secretion after leukocyte stimulation and reduced cell-surface/intracellular expression of receptors/proteins involved in the IFNγ pathway, can help in identifying specific targets for genetic analysis [45]. Prolonged treatment with antimycobacterial antibiotics is essential to control infections and additional IFNγ therapy can be helpful. In some cases, surgical removal of pulmonary lesions is indicated and in severe cases HSCT has been successful [46, 47].
STAT3 loss of function
Loss of function mutations in the signal transducer and activator of transcription 3 (STAT3) cause STAT3 autosomal-dominant (AD) hyper IgE syndrome (HIES) [48–51]. In STAT3 HIES, impaired neutrophil chemotaxis, reduced production of inflammatory cytokines and defective repair mechanisms of bronchiolar and alveolar epithelial cells are pathogenic factors [52–55]. STAT3 HIES is a rare immunodeficiency with an incidence of less than one in 1 000 000 [56].
STAT3 HIES patients present with typical coarse facial features, eczematous dermatitis, retention of primary teeth, scoliosis and joint hyperextensibility, as well as with immunodeficiency and recurrent bronchopulmonary infections [55]. Pulmonary granuloma formation as well as bronchiectasis, pneumatoceles and cavernas in these patients have been attributed to recurrent infections and to impaired lung remodelling mechanisms [55].
Imaging studies, especially chest CT scans, are essential in establishing the extent of pulmonary involvement [55]. Therapeutic options include antimicrobial prophylaxis with trimethoprimsulfamethoxazole, as well as early aggressive treatment of infections with antibiotics [52]. Ig replacement therapy has been shown to reduce pulmonary complications if IgG levels are low or serological responses to vaccination are missing [57]. Surgical excision of large pneumatoceles might be indicated [52].
STAT3 gain of function
Gain of function mutations in STAT3 cause early onset immunodeficiency with additional features such as autoimmune enteropathy, diabetes mellitus, autoimmune cytopenias, lymphadenopathy, splenomegaly, short stature and interstitial lung disease (ILD) [58–61]. Currently, less than 50 cases have been described [58]. Granulomatous lung disease has been reported in at least two affected individuals, but other forms of ILD seem to be a more common feature in patients with gain of function STAT3 mutations [58, 62, 63]. Therapeutic options include immunosuppression with anti-IL-6 agents and Janus kinase (JAK) inhibitors, while SCT has not yielded convincing results [58, 64]. STAT3 inhibitors are currently being tested clinically and might become a therapeutic option for patients with STAT3 gain of function in the future [58].
Other immunodeficiencies
Patients with XIAP and GATA2 deficiencies have been shown to develop GLILD, as have patients with Kabuki syndrome and those with IgA/IgG2 deficiencies [65–67]. GLILD was successfully treated with rituximab and azathioprine in at least one patient with XIAP deficiency [67].
Patients with caspase 8 deficiency, Rhoh deficiency, TAP1/TAP2 deficiency and Good's syndrome have been shown to develop pulmonary granulomas [66, 68–71]. Granulomatous lung disease has also been reported in some cases of haemophagocytic lymphohistiocytosis (HLH) [72–74]. Treatment decisions have to be individualised based on the specific defect and the clinical situation of the patient.
Autoimmune and autoinflammatory diseases associated with pulmonary granulomatosis
Sarcoidosis
Sarcoidosis is an inflammatory disorder of unknown cause that is characterised by granuloma formation in the affected organs, most often in the lungs [75] although any organ can be affected. The incidence and prevalence of sarcoidosis, as well as its clinical presentation, vary greatly across geographical regions and between the sexes, aw well as between different ethnicities and age groups. Its prevalence varies between two and 1160 per 100 000 and is highest in Scandinavia and in African-American populations [76–78].
The disease develops in genetically predisposed individuals with exposure to an as-yet unknown antigen. Genome-wide association studies have identified human leukocyte antigen (HLA) class II alleles and several non-HLA genes as susceptibility factors [79–82]. Most interestingly, a polymorphism in the TNF gene confers resistance to anti-TNF therapy [83]. Recently, defects in autophagy, JAK STAT signalling and mTOR pathways have been identified as playing a crucial role in ineffective clearance of infectious agents or nonorganic particles, triggering granuloma formation due to macrophage and T-cell dysfunction [84, 85]. Familial aggregation is known and having a family member with the disease is associated with a two-to-four-fold increased risk of developing sarcoidosis [86]. Although these findings are significant, there is no application of this genetic knowledge in everyday clinics.
The pathological hallmark of sarcoidosis is the presence of compact, epithelioid, non-necrotising granulomas with varying degrees of lymphocytic inflammation. This is used, in combination with a compatible clinical disease manifestation and typical radiological presentation, as a diagnostic parameter. Nevertheless, other causes of granuloma need to be excluded [87]. These inflammatory processes attract mononuclear and polymorphic nuclear cells to the lower respiratory tract, which can be probed by BAL. An increase in lymphocytes with an elevated CD4/CD8 ratio heralds a spontaneous resolution or a desirable course under therapy, but an increase in neutrophils is associated with progressive disease requiring therapy [88]. PFTs may reveal reduced diffusion capacity and a restrictive pattern with loss of vital capacity, but also obstructive changes [89].
Corticosteroids still constitute the first line treatment in cases with progressive organ damage [75, 87]. Corticosteroid-sparing agents, such as azathioprine or methotrexate, are frequently used when prolonged therapy is necessary [90, 91]. Studies and case series demonstrate the successful use of newer agents which manipulate the cytokine network, such as infliximab, rituximab, or JAK-inhibitors such as tofacitinib [92–97]. None of these are approved but off-label therapy is often initiated [75].
Blau syndrome/early-onset sarcoidosis
Both Blau syndrome and early-onset sarcoidosis are rare disorders caused by gain of function mutations in the nucleotide-binding oligomerisation domain-containing protein 2 (NOD2) pattern recognition receptor (also known as the caspase-recruitment domain-containing protein 15 (CARD-15)) [98–100]. NOD2 is involved in innate immune responses and the inflammative cascade after viral or bacterial infections (via NF-κB and TNF receptor-associated factor 3 (TRAF3)) [101, 102]. Gain of function mutations in NOD2 are associated with granulomatous inflammation of affected tissues, though a triggering infection is possibly essential [100, 103, 104]. Histologic evaluation reveals epithelioid cell-rich, noncaseating granulomas [105].
Blau syndrome is the inherited form of the disease and early-onset sarcoidosis is caused by de novo mutations in NOD2. The clinical course of the two entities is phenotypically indistinguishable and patients show a triad of granulomatous polyarthritis, dermatitis and uveitis [99, 103, 106–108] (figure 4). Most patients present under the age of 2 years [109] and about one third to one half of patients have additional manifestations. These include fever, lymphadenopathy, vasculitis, arterial hypertension, transient neuropathies, granulomatous kidney disease and granulomatous liver disease, as well as pulmonary embolisms [107, 110]. At least four patients with Blau syndrome and interstitial/granulomatous lung disease have been described [100, 107, 111]. Clinical signs were mild or not present and the pulmonary changes were found by chance on CT scan [111]. Pulmonary involvement in early-onset sarcoidosis is less frequent than in later-onset sarcoidosis. One patient has been reported with bronchial granulomas [112], one with bronchial granuloma and subsequent pulmonary haemorrhage [113] and one with pulmonary micronodules [114].
FIGURE 4.

Proposed algorithm to differentiate between early onset sarcoidosis and Blau syndrome. HRCT: high-resolution computed tomography; BAL: bronchoalveolar lavage; NOD2: nucleotide-binding oligomerisation domain-containing protein 2. Data from [103].
Parameters that have been helpful in diagnosing sarcoidosis are serum levels of angiotensin-converting enzyme (ACE), soluble IL-2 receptor and serum amyloid A [103, 115]. For patients with high suspicion of Blau syndrome/early-onset sarcoidosis, genetic analysis should be performed (figure 4) [107]. A HRCT chest scan is paramount in identifying pulmonary involvement. If patients undergo bronchoscopy, the cellular composition of bronchoalveolar fluid should be analysed, as a lymphocytosis of >15% (as well as a CD4/CD8 ratio of >3.5: 1) suggests pulmonary sarcoidosis or pulmonary involvement in Blau syndrome [116].
Therapeutic approaches with corticosteroids, anti-TNF agents and anti-IL-1 therapy have yielded positive results in halting inflammation; however, there is no consensus regarding optimal therapy so far [109, 110, 117].
NOD2-associated autoinflammatory disease
A similar clinical picture to that in Blau syndrome can be found in NOD2-associated autoinflammatory disease (NAID), which is caused by the IVS8+ NOD2 variant or the heterozygous p.T189M and p.R703C NOD2 variants [118]. Less than 100 cases, mainly adult Caucasian patients, have been reported, who suffer from recurrent fever, weight loss, nonerosive arthritis, granulomatous dermatitis and granulomatous colitis [119, 120]. Pleuritis in several and non-necrotising pulmonary granulomas in at least one patient were also described [118, 120, 121]. Immunosuppression with corticosteroids or sulfasalazine has been effective, as has therapy with biologicals such as infliximab, tocilizumab and canakinumab [120].
Chronic beryllium disease
Inhaled beryllium can induce a cell-mediated or delayed hypersensitivity reaction in individuals with specific HLA-DPB1 polymorphisms and probably TNF-α polymorphisms. For the HLA-DPGlu69 variant, a direct interaction of beryllium has been shown via binding to the HLA molecule and eliciting of an IFNγ response [122–125]. Additional genetic risk factors might be present in individuals who develop beryllium sensitisation or chronic beryllium disease (CBD), as the above mentioned genotypes are also common in the general population [124, 126–129]. CBD is an occupational pulmonary granulomatous disease. Relevant exposure to beryllium can occur in manufacturing industries such as defence, aerospace, nuclear, automotive and electronics [130, 131]. Higher exposure puts patients at higher risk for CBD [129], but only 1–5% of exposed persons develop the disease, with a higher frequency in persons with the above mentioned polymorphisms [128, 132, 133]. CBD only rarely occurs in the general population, most frequently in persons living close to a beryllium processing plant or with family members that have been exposed to the contaminated clothes of beryllium workers [128, 134]. Patients are typically adults with a reported mean age at diagnosis of 44 years [131].
CBD is phenotypically indistinguishable from sarcoidosis and more than 6% of patients diagnosed with sarcoidosis might in fact suffer from CBD [131, 135]. Patients initially present with nonspecific symptoms, including dry cough and shortness of breath, that are similar to asthmatic symptoms, as well as less frequently with fever, fatigue, night sweats and weight loss [130, 136]. Imaging studies and PFTs reveal similar pathologies as in sarcoidosis. CBD should be considered if there is a history of beryllium exposure, beryllium sensitisation can be demonstrated (by a positive beryllium lymphocyte proliferation test) and there is evidence of noncaseating, poorly-formed granulomas and mononuclear cell infiltrates in lung biopsies [130].
Preventive measures have to be taken to reduce occupational beryllium exposure in persons with beryllium sensitisation [129]. The standard therapy for CBD is systemic glucocorticoids [130]. Patients with refractory disease or side-effects of glucocorticoid therapy might benefit from (additional) therapy with other immunosuppressive agents, such as methotrexate, azathioprine, or TNF antagonists, although evidence for this is limited [130, 137].
STING-associated vasculopathy of infancy
Gain of function mutations in the stimulator of IFN genes (STING) cause STING-associated vasculopathy with onset in infancy (SAVI) [138]. SAVI is considered rare with less than 20 cases reported so far [138–141].
Activation of STING by viral or bacterial triggers causes upregulation of IFNβ transcription, as well as upregulation of expression of other IFN-regulated genes leading to STAT1 phosphorylation. Patients with SAVI show uncontrolled STING activation, which causes early-onset constant fever, capillary vasculitis, lymphadenopathy, chronic anaemia, failure to thrive, interstitial and granulomatous lung disease, or pulmonary fibrosis [139, 140, 142]. Pulmonary involvement especially differentiates patients with SAVI from other interferonopathies [141].
Chronic cough and tachypnoea manifest in the first weeks of life. Cutaneous manifestations typically show within the first 6 months. They include teleangiectatic, pustular or blistering exanthemas, mostly on acral sites like the fingers, nose and ears, but also on the cheeks [138]. ILD is found on CT scan (sometimes with nodular infiltrates) and restrictive patterns in PFTs have also been demonstrated [143]. Histologically, a mixed lymphocytic infiltrate, interstitial fibrosis and emphysema, as well as vasculitis, have been found in lung biopsies [138]. Pulmonary involvement is life-limiting in a significant number of patients [138, 139].
Different immunosuppressive therapies, including corticosteroids, cyclophosphamide, azathioprine, methotrexate, rituximab and infliximab, have shown only moderate beneficial effects [139]. As JAK inhibitors target the phosphorylation of STAT1/STAT2 [144], this pharmacological approach might be helpful in the future for patients with SAVI [138, 145].
Granulomatosis with polyangiitis
Granulomatosis with polyangiitis is an autoinflammatory systemic vasculitic disorder linked to polymorphisms in HLA-DPB1, HLA-DPA1, PRTN3 and SERPINA1 [146]. Patients show elevated anti-neutrophil cytoplasmic antibodies (ANCAs) and, in 70–90% of cases, ANCAs that target proteinase-3 are detected [147]. The pathogenesis of granulomatosis with polyangiitis is not fully understood; however, a combination of genetic susceptibility factors and environmental triggers may lead to a dysregulation of innate and adaptive immune responses [148].
Cytoplasmic ANCA associated vasculitis has an incidence of about 13–20·(1 000 000)−1·year−1 in adults and 0.45–6.39·(1 000 000)−1·year−1 in children [149, 150]. The diagnosis of granulomatosis with polyangiitis can be established following the established classification criteria with a combination of histological, serological and clinical findings [151, 152]. Pulmonary involvement is common in granulomatosis with polyangiitis [152, 153]. Typical symptoms include rhinitis, persistent otitis media, cough, stridor, obstruction, dyspnoea and haemoptysis. Subglottic or bronchial stenosis might develop secondary to inflammation.
Pulmonary nodules, cavities or fixed infiltrates are found in imaging studies [149, 154] and fluorodeoxyglucose (FDG)-PET/CT scan can help to appreciate the extent of the disease and to identify occult sites of inflammation [155]. Bronchoscopy is indicated if tracheal or bronchial stenosis is suspected and to obtain biopsies [105]. On histologic examination of affected tissues, necrotising granulomas with necrotising vasculitis are found [1]. Infectious causes contribute to a high morbidity and mortality in granulomatosis with polyangiitis and need to be ruled out and treated aggressively [105].
Control of autoinflammation can be achieved with a combination of high-dose corticosteroids and other immunosuppressive agents, such as cyclophosphamide, azathioprine, methotrexate, mycophenolate mofetil or rituximab [152].
Eosinophilic granulomatosis with polyangiitis
Eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome) is associated with polymorphisms of the Fcγ receptor 3B that is expressed on neutrophils and contributes to the clearance of immune complexes [156]. ANCA antibodies, which usually target myeloperoxidase (perinuclear (p-)ANCAs), can be detected in 30–75% of adult patients and around 30% of affected children [157, 158].
The incidence in adults is around 1–3·(100 000)−1·year−1 and less than 100 paediatric cases have been reported [157]. Asthma and eosinophilia are present in almost all patients with eosinophilic granulomatosis with polyangiitis and the most common misdiagnosis is therapy-refractive asthma. Vasculitis and granuloma formation in the lungs, skin, digestive tract and heart are common in eosinophilic granulomatosis with polyangiitis [157, 159]. The condition has different phases of disease activity: a prodromal phase can be asymptomatic and is followed by an eosinophilic phase (where most paediatric patients are diagnosed); whereas adults are mainly diagnosed in the final vasculitic phase [157]. HRCT chest scan typically shows ground-glass opacities (GGOs), bronchial wall thickening, (micro)nodules and consolidations [157, 159]. Cytologic evaluation of BAL can demonstrate a mean eosinophilia of up to 33%, although patients may also present without eosinophilia [157, 159]. The histologic hallmark of eosinophilic granulomatosis with polyangiitis is necrotising granulomatous inflammation with eosinophilic infiltration, mainly of the small vessels of the upper and lower airways as well as the surrounding tissue, with formation of extravascular granulomas [159, 160].
Systemic corticosteroids, possibly in combination with azathioprine or cyclophosphamide, are recommended to control eosinophilic granulomatosis with polyangiitis [157, 161]. The IL-5 antibody mepolizumab has also yielded positive results in ANCA-positive patients with an eosinophilia of >150 cells·µL−1 [162].
Other conditions
Apart from the immunodeficiencies and autoinflammatory diseases presented here, there is growing evidence that other conditions that have some genetic background might predispose to pulmonary granulomatous inflammation. One example is histiocytosis X/Langerhan's cell histiocytosis (LCH), a neoplastic process with inflammatory characteristics. Somatic mutations in BRAF and NRAS have been described in most patients with pulmonary LCH [163, 164]. Incidence of pulmonary LCH is around 0.27 per 100 000 and mostly affects young adults who smoke [165]. Clinical presentation is unspecific, involving dry cough, dyspnoea and fatigue [165]. Chest CT scans typically reveal stellate nodules, nodular opacities, cysts, or honeycombing [166] and these changes mainly occur in the middle and upper lobes of the lungs. BAL may yield CD1a and CD207 positive cells supporting the diagnosis [167]. Cessation of smoking is essential. Glucocorticoid therapy is successful only in some patients and others might need more aggressive chemotherapy (e.g. cytarabine or vinblastine) [167].
Conclusions and further directions
In this review we discuss the broad clinical and genetic spectrum of pulmonary granulomatosis of genetic origin. The diversity of conditions that can manifest in infants, children, adolescents and adults, based on specific genetic defects, is evident in the varying amount and type of granulomatous inflammation as well as the accompanying symptoms. These symptoms may give clues to the underlying disorder but can be nonspecific in many cases. Pathophysiology of granuloma formation is well understood in some of the diseases presented here and may be based on endogenous inflammation, impaired control of environmental pathogens, or pathological immune responses to harmless antigens. In other conditions, the pathways leading to pulmonary granuloma formation are less clear. Nevertheless, in all cases of suspected pulmonary granulomatosis it is paramount to exclude infectious causes before considering the rarer, noninfectious reasons for their development, while simultaneously keeping in mind that some of the genetic defects discussed here specifically predispose to infections that lead to the pulmonary granuloma. Certain immunodeficiencies and autoimmune disorders have been classically associated with pulmonary granulomatosis. As the understanding of the genetic basis of many disorders is expanded, so is the possible list of differential diagnoses of pulmonary granulomatosis of genetic origin. Decisive indicators of the underlying disorder can be elicited by pulmonologists, immunologists, rheumatologists, radiologists and pathologists; therefore, a multidisciplinary approach is paramount in correctly diagnosing affected patients.
Early identification of individuals at risk for pulmonary granulomatosis of genetic origin can help to avoid environmental or infectious triggers, support aggressive antimicrobial or anti-inflammatory therapy in patient subgroups and might identify individuals eligible for SCT. A combined diagnostic approach taking clinical presentation, laboratory workup, imaging techniques, histologic findings and genetics results into consideration can therefore help to shape the way to personalised diagnosis and treatment. With the advance of new sequencing techniques, the genetic background of other causes of pulmonary granulomatous inflammation might be discovered in the future.
Footnotes
Previous articles in the Series: No. 1: Daccord C, Good J-M, Morren M-A, et al. Brit–Hogg–Dubé syndrome. Eur Respir Rev 2020; 29: 200042. No. 2: Hadchouel A, Drummond D, Abou Taam R, et al. Alveolar proteinosis of genetic origins. Eur Respir Rev 2020; 29: 200187. No. 3: Cazzato S, Omenetti A, Ravaglia C, et al. Lung involvement in monogenetic interferonopathies. Eur Respir Rev 2021; 30: 200001. No. 4: Yokoyama T, Gochuico BR. Hermansky–Pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease. Eur Respir Rev 2021; 30: 200193. No. 5: van Moorsel CHM, Van der Vis JJ, Grutters JC. Genetic disorders of the surfactant system: focus on adult disease. Eur Respir Rev 2021; 30: 200085.
Provenance: Commissioned article, peer reviewed.
Author contributions: S.F.N. Bode conceptualised the review and prepared the first draft of the manuscript. M. Seidl provided the histological figures and descriptions. All authors critically revised and edited the manuscript and agreed to the submitted version.
Number 6 in the Series “Rare genetic interstitial lung diseases” Edited by Bruno Crestani and Raphaël Borie
Conflict of interest: S.F.N. Bode has nothing to disclose.
Conflict of interest: J. Rohr has nothing to disclose.
Conflict of interest: J.M. Müller-Quernheim reports grants and non-financial support from Bristol Myers Squibb; personal fees from Roche and Novartis; personal fees and non-financial support from Boehringer Ingelheim; and non-financial support from CLS Behring, outside the submitted work. In addition, J.M. Müller-Quernheim has a patent “Use of inhaled VIP for treatment of immune checkpoint inhibitor-induced pneumonitis” pending and is co-founder of AdVita Lifescience GmbH, a spin-off of the University of Freiburg. Advita reports a pending intellectual property concerning the use of Aviptadil for the treatment of immune checkpoint inhibitor (ICI) pneumonitis.
Conflict of interest: M. Seidl has nothing to disclose.
Conflict of interest: C. Speckmann has nothing to disclose.
Conflict of interest: A. Heinzmann has nothing to disclose.
References
- 1.Ohshimo S, Guzman J, Costabel U, et al. . Differential diagnosis of granulomatous lung disease: clues and pitfalls. Eur Respir Rev 2017; 26: 170012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mukhopadhyay S, Gal AA. Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med 2010; 134: 667–690. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg DE, Goldberg JB, Stock F, et al. . Recurrent Burkholderia infection in patients with chronic granulomatous disease: 11-year experience at a large referral center. Clin Infect Dis 2009; 48: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lieberman S, Gleason JB, Ilyas MIM, et al. . Assessing the safety and clinical impact of thoracoscopic lung biopsy in patients with interstitial lung disease. J Clin Diagn Res 2017; 11: OC57–OC59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blackhall V, Asif M, Renieri A, et al. . The role of surgical lung biopsy in the management of interstitial lung disease: experience from a single institution in the UK. Interact Cardiovasc Thorac Surg 2013; 17: 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YC, Wu CT, Hsu HH, et al. . Surgical lung biopsy for diffuse pulmonary disease: experience of 196 patients. J Thorac Cardiovasc Surg 2005; 129: 984–990. [DOI] [PubMed] [Google Scholar]
- 7.Bush A, Cunningham S, de Blic J, et al. . European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax 2015; 70: 1078–1084. [DOI] [PubMed] [Google Scholar]
- 8.Baumann U, Routes JM, Soler-Palacin P, et al. . The lung in primary immunodeficiencies: new concepts in infection and inflammation. Front Immunol 2018; 9: 1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahdaviani SA, Rezaei N. Pulmonary manifestations of primary immunodeficiency diseases. Basel, Springer, 2019. [Google Scholar]
- 10.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999; 92: 34–48. [DOI] [PubMed] [Google Scholar]
- 11.Selenius JS, Martelius T, Pikkarainen S, et al. . Unexpectedly high prevalence of common variable immunodeficiency in Finland. Front Immunol 2017; 8: 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Mousa H, Al-Saud B. Primary immunodeficiency diseases in highly consanguineous populations from Middle East and North Africa: epidemiology, diagnosis, and care. Front Immunol 2017; 8: 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aggarwal V, Banday AZ, Jindal AK, et al. . Recent advances in elucidating the genetics of common variable immunodeficiency. Genes Dis 2020; 7: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimbacher B, Hutloff A, Schlesier M, et al. . Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol 2003; 4: 261–268. [DOI] [PubMed] [Google Scholar]
- 15.Castigli E, Wilson SA, Garibyan L, et al. . TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet 2005; 37: 829–834. [DOI] [PubMed] [Google Scholar]
- 16.van Zelm MC, Reisli I, van der Burg M, et al. . An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med 2006; 354: 1901–1912. [DOI] [PubMed] [Google Scholar]
- 17.Abolhassani H, Hammarstrom L, Cunningham-Rundles C. Current genetic landscape in common variable immune deficiency. Blood 2020; 135: 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuijnenburg P, Lango Allen H, Burns SO, et al. . Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol 2018; 142: 1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.European Society for Immunodeficiencies (ESID) . Diagnostic criteria for primary immunodeficiencies. Geneva, ESID, 2020. https://esid.org/Working-Parties/Clinical-Working-Party/Resources/Diagnostic-criteria-for-PID2 [Google Scholar]
- 20.Sun D, Heimall J. Disorders of CTLA-4 expression, how they lead to CVID and dysregulated immune responses. Curr Opin Allergy Clin Immunol 2019; 19: 578–585. [DOI] [PubMed] [Google Scholar]
- 21.Hurst JR, Verma N, Lowe D, et al. . British Lung Foundation/United Kingdom Primary Immunodeficiency Network consensus statement on the definition, diagnosis, and management of granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency disorders. J Allergy Clin Immunol Pract 2017; 5: 938–945. [DOI] [PubMed] [Google Scholar]
- 22.Bates CA, Ellison MC, Lynch DA, et al. . Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol 2004; 114: 415–421. [DOI] [PubMed] [Google Scholar]
- 23.Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency–histologic and immunohistochemical analyses of 16 cases. Hum Pathol 2015; 46: 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maglione PJ, Overbey JR, Cunningham-Rundles C. Progression of common variable immunodeficiency interstitial lung disease accompanies distinct pulmonary and laboratory findings. J Allergy Clin Immunol Pract 2015; 3: 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chase NM, Verbsky JW, Hintermeyer MK, et al. . Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol 2013; 33: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Speckmann C, Doerken S, Aiuti A, et al. . A prospective study on the natural history of patients with profound combined immunodeficiency: an interim analysis. J Allergy Clin Immunol 2017; 139: 1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulkhi AA, Dasso JF, Schuetz C, et al. . Approaches to patients with variants in RAG genes: from diagnosis to timely treatment. Expert Rev Clin Immunol 2019; 15: 1033–1046. [DOI] [PubMed] [Google Scholar]
- 28.Schuetz C, Huck K, Gudowius S, et al. . An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med 2008; 358: 2030–2038. [DOI] [PubMed] [Google Scholar]
- 29.Lo B, Zhang K, Lu W, et al. . Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015; 349: 436–440. [DOI] [PubMed] [Google Scholar]
- 30.Schwab C, Gabrysch A, Olbrich P, et al. . Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol 2018; 142: 1932–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schubert D, Bode C, Kenefeck R, et al. . Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 2014; 20: 1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, et al. . Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 2012; 90: 986–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S, Moon JS, Lee CR, et al. . Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol 2016; 137: 327–330. [DOI] [PubMed] [Google Scholar]
- 34.Segal BH, Leto TL, Gallin JI, et al. . Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000; 79: 170–200. [DOI] [PubMed] [Google Scholar]
- 35.Winkelstein JA, Marino MC, Johnston RB,Jr, et al. . Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000; 79: 155–169. [DOI] [PubMed] [Google Scholar]
- 36.Rider NL, Jameson MB, Creech CB. Chronic granulomatous disease: epidemiology, pathophysiology, and genetic basis of disease. J Pediatric Infect Dis Soc 2018; 7: Suppl. 1, S2–S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahdaviani SA, Mohajerani SA, Rezaei N, et al. . Pulmonary manifestations of chronic granulomatous disease. Expert Rev Clin Immunol 2013; 9: 153–160. [DOI] [PubMed] [Google Scholar]
- 38.Mansouri D, Adimi P, Mirsaedi M, et al. . Primary immune deficiencies presenting in adults: seven years of experience from Iran. J Clin Immunol 2005; 25: 385–391. [DOI] [PubMed] [Google Scholar]
- 39.van den Berg JM, van Koppen E, Ahlin A, et al. . Chronic granulomatous disease: the European experience. PLoS One 2009; 4: e5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Ravin SS, Naumann N, Cowen EW, et al. . Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol 2008; 122: 1097–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gungor T, Teira P, Slatter M, et al. . Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet 2014; 383: 436–448. [DOI] [PubMed] [Google Scholar]
- 42.Morillo-Gutierrez B, Beier R, Rao K, et al. . Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood 2016; 128: 440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamazaki-Nakashimada MA, Stiehm ER, Pietropaolo-Cienfuegos D, et al. . Corticosteroid therapy for refractory infections in chronic granulomatous disease: case reports and review of the literature. Ann Allergy Asthma Immunol 2006; 97: 257–261. [DOI] [PubMed] [Google Scholar]
- 44.Bustamante J. Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet 2020; 139: 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Esteve-Sole A, Sologuren I, Martinez-Saavedra MT, et al. . Laboratory evaluation of the IFN-γ circuit for the molecular diagnosis of Mendelian susceptibility to mycobacterial disease. Crit Rev Clin Lab Sci 2018; 55: 184–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosain J, Kong XF, Martinez-Barricarte R, et al. . Mendelian susceptibility to mycobacterial disease: 2014–2018 update. Immunol Cell Biol 2019; 97: 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reed B, Dolen WK. The child with recurrent mycobacterial disease. Curr Allergy Asthma Rep 2018; 18: 44. [DOI] [PubMed] [Google Scholar]
- 48.Hagl B, Heinz V, Schlesinger A, et al. . Key findings to expedite the diagnosis of hyper-IgE syndromes in infants and young children. Pediatr Allergy Immunol 2016; 27: 177–184. [DOI] [PubMed] [Google Scholar]
- 49.Holland SM, DeLeo FR, Elloumi HZ, et al. . STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 2007; 357: 1608–1619. [DOI] [PubMed] [Google Scholar]
- 50.Minegishi Y, Saito M, Tsuchiya S, et al. . Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007; 448: 1058–1062. [DOI] [PubMed] [Google Scholar]
- 51.Renner ED, Torgerson TR, Rylaarsdam S, et al. . STAT3 mutation in the original patient with Job's syndrome. N Engl J Med 2007; 357: 1667–1668. [DOI] [PubMed] [Google Scholar]
- 52.Mogensen TH. STAT3 and the hyper-IgE syndrome: clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. JAKSTAT 2013; 2: e23435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tadokoro T, Wang Y, Barak LS, et al. . IL-6/STAT3 promotes regeneration of airway ciliated cells from basal stem cells. Proc Natl Acad Sci USA 2014; 111: E3641–E3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan C, Naltner A, Martin M, et al. . Transcriptional stimulation of the surfactant protein B gene by STAT3 in respiratory epithelial cells. J Biol Chem 2002; 277: 10967–10972. [DOI] [PubMed] [Google Scholar]
- 55.Kroner C, Neumann J, Ley-Zaporozhan J, et al. . Lung disease in STAT3 hyper-IgE syndrome requires intense therapy. Allergy 2019; 74: 1691–1702. [DOI] [PubMed] [Google Scholar]
- 56.Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am 2008; 28: 277–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chandesris MO, Melki I, Natividad A, et al. . Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) 2012; 91: e1–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fabre A, Marchal S, Barlogis V, et al. . Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J Allergy Clin Immunol Pract 2019; 7: 1958–1969. [DOI] [PubMed] [Google Scholar]
- 59.Flanagan SE, Haapaniemi E, Russell MA, et al. . Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet 2014; 46: 812–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haapaniemi EM, Kaustio M, Rajala HL, et al. . Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood 2015; 125: 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hillmer EJ, Zhang H, Li HS, et al. . STAT3 signaling in immunity. Cytokine Growth Factor Rev 2016; 31: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maffucci P, Filion CA, Boisson B, et al. . Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol 2016; 7: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gutierrez M, Scaglia P, Keselman A, et al. . Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol Cell Endocrinol 2018; 473: 166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Milner JD, Vogel TP, Forbes L, et al. . Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015; 125: 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schussler E, Beasley MB, Maglione PJ. Lung disease in primary antibody deficiencies. J Allergy Clin Immunol Pract 2016; 4: 1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seppänen M, Rezaei N. General considerations of pulmonary manifestations of primary immunodeficiency diseases. In:Mahdaviani SA, Rezaei N, eds. Pulmonary manifestations of primary immunodeficiency diseases. 1st Edn. Basel, Springer, 2019; pp. 1–36. [Google Scholar]
- 67.Steele CL, Dore M, Ammann S, et al. . X-linked inhibitor of apoptosis complicated by granulomatous lymphocytic interstitial lung disease (GLILD) and granulomatous hepatitis. J Clin Immunol 2016; 36: 733–738. [DOI] [PubMed] [Google Scholar]
- 68.Gadola SD, Moins-Teisserenc HT, Trowsdale J, et al. . TAP deficiency syndrome. Clin Exp Immunol 2000; 121: 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crequer A, Troeger A, Patin E, et al. . Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J Clin Invest 2012; 122: 3239–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee SH, Lee SM, Yang SC, et al. . A case of granulomatous lung disease in a patient with Good's syndrome. Korean J Intern Med 2008; 23: 219–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niemela J, Kuehn HS, Kelly C, et al. . Caspase-8 deficiency presenting as late-onset multi-organ lymphocytic infiltration with granulomas in two adult siblings. J Clin Immunol 2015; 35: 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fitzgerald NE, MacClain KL. Imaging characteristics of hemophagocytic lymphohistiocytosis. Pediatr Radiol 2003; 33: 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin YK, Xie ZD, Yang S, et al. . Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis: a retrospective study of 78 pediatric cases in mainland of China. Chin Med J 2010; 123: 1426–1430. [PubMed] [Google Scholar]
- 74.Rohr J, Beutel K, Maul-Pavicic A, et al. . Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica 2010; 95: 2080–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grunewald J, Grutters JC, Arkema EV, et al. . Sarcoidosis. Nat Rev Dis Primers 2019; 5: 45. [DOI] [PubMed] [Google Scholar]
- 76.Arkema EV, Grunewald J, Kullberg S, et al. . Sarcoidosis incidence and prevalence: a nationwide register-based assessment in Sweden. Eur Respir J 2016; 48: 1690–1699. [DOI] [PubMed] [Google Scholar]
- 77.Dumas O, Abramovitz L, Wiley AS, et al. . Epidemiology of sarcoidosis in a prospective cohort study of U.S. women. Ann Am Thorac Soc 2016; 13: 67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cozier YC, Berman JS, Palmer JR, et al. . Sarcoidosis in black women in the United States: data from the Black Women's Health Study. Chest 2011; 139: 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hofmann S, Franke A, Fischer A, et al. . Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat Genet 2008; 40: 1103–1106. [DOI] [PubMed] [Google Scholar]
- 80.Rivera NV, Ronninger M, Shchetynsky K, et al. . High-density genetic papping identifies new susceptibility variants in sarcoidosis phenotypes and shows genomic-driven phenotypic differences. Am J Respir Crit Care Med 2016; 193: 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Valentonyte R, Hampe J, Huse K, et al. . Sarcoidosis is associated with a truncating splice site mutation in BTNL2. Nat Genet 2005; 37: 357–364. [DOI] [PubMed] [Google Scholar]
- 82.Schurmann M, Lympany PA, Reichel P, et al. . Familial sarcoidosis is linked to the major histocompatibility complex region. Am J Respir Crit Care Med 2000; 162: 861–864. [DOI] [PubMed] [Google Scholar]
- 83.Wijnen PA, Cremers JP, Nelemans PJ, et al. . Association of the TNF-α G-308A polymorphism with TNF-inhibitor response in sarcoidosis. Eur Respir J 2014; 43: 1730–1739. [DOI] [PubMed] [Google Scholar]
- 84.Pacheco Y, Lim CX, Weichhart T, et al. . Sarcoidosis and the mTOR, Rac1, and autophagy triad. Trends Immunol 2020; 41: 286–299. [DOI] [PubMed] [Google Scholar]
- 85.Zhou T, Casanova N, Pouladi N, et al. . Identification of Jak-STAT signalling involvement in sarcoidosis severity via a novel microRNA-regulated peripheral blood mononuclear cell gene signature. Sci Rep 2017; 7: 4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grunewald J, Spagnolo P, Wahlstrom J, et al. . Immunogenetics of disease-causing inflammation in sarcoidosis. Clin Rev Allergy Immunol 2015; 49: 19–35. [DOI] [PubMed] [Google Scholar]
- 87.Valeyre D, Prasse A, Nunes H, et al. . Sarcoidosis. Lancet 2014; 383: 1155–1167. [DOI] [PubMed] [Google Scholar]
- 88.Ziegenhagen MW, Rothe ME, Schlaak M, et al. . Bronchoalveolar and serological parameters reflecting the severity of sarcoidosis. Eur Respir J 2003; 21: 407–413. [DOI] [PubMed] [Google Scholar]
- 89.Rasheed A, Viswanath V, Arjomand F. Patterns of pulmonary function test (PFT) abnormalities in sarcoidosis. Chest 2012; 142: 446A. https://journal.chestnet.org/article/S0012-3692(16)55316-1/fulltext [Google Scholar]
- 90.Müller-Quernheim J, Kienast K, Held M, et al. . Treatment of chronic sarcoidosis with an azathioprine/prednisolone regimen. Eur Respir J 1999; 14: 1117–1122. [DOI] [PubMed] [Google Scholar]
- 91.Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 2000; 17: 60–66. [PubMed] [Google Scholar]
- 92.Damsky W, Thakral D, Emeagwali N, et al. . Tofacitinib treatment and molecular analysis of cutaneous sarcoidosis. N Engl J Med 2018; 379: 2540–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Damsky W, Thakral D, McGeary MK, et al. . Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol 2020; 82: 612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Judson MA, Baughman RP, Costabel U, et al. . Efficacy of infliximab in extrapulmonary sarcoidosis: results from a randomised trial. Eur Respir J 2008; 31: 1189–1196. [DOI] [PubMed] [Google Scholar]
- 95.Sweiss NJ, Lower EE, Mirsaeidi M, et al. . Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J 2014; 43: 1525–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korsten P, Mirsaeidi M, Sweiss NJ. Nonsteroidal therapy of sarcoidosis. Curr Opin Pulm Med 2013; 19: 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sweiss NJ, Curran J, Baughman RP. Sarcoidosis, role of tumor necrosis factor inhibitors and other biologic agents, past, present, and future concepts. Clin Dermatol 2007; 25: 341–346. [DOI] [PubMed] [Google Scholar]
- 98.Miceli-Richard C, Lesage S, Rybojad M, et al. . CARD15 mutations in Blau syndrome. Nat Genet 2001; 29: 19–20. [DOI] [PubMed] [Google Scholar]
- 99.Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr 1985; 107: 689–693. [DOI] [PubMed] [Google Scholar]
- 100.Wouters CH, Maes A, Foley KP, et al. . Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J 2014; 12: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhong Y, Kinio A, Saleh M. Functions of NOD-like receptors in human diseases. Front Immunol 2013; 4: 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moreira LO, Zamboni DS. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol 2012; 3: 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chiu B, Chan J, Das S, et al. . Pediatric sarcoidosis: a review with emphasis on early onset and high-risk sarcoidosis and diagnostic challenges. Diagnostics (Basel) 2019; 9: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sidiqi A, Pegado V. Blau syndrome following a bacterial infection. J AAPOS 2020; 24: 118–120. [DOI] [PubMed] [Google Scholar]
- 105.Richardson AE, Warrier K, Vyas H. Respiratory complications of the rheumatological diseases in childhood. Arch Dis Child 2016; 101: 752–758. [DOI] [PubMed] [Google Scholar]
- 106.Gedalia A, Khan TA, Shetty AK, et al. . Childhood sarcoidosis: Louisiana experience. Clin Rheumatol 2016; 35: 1879–1884. [DOI] [PubMed] [Google Scholar]
- 107.Rose CD, Pans S, Casteels I, et al. . Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford) 2015; 54: 1008–1016. [DOI] [PubMed] [Google Scholar]
- 108.Rose CD, Wouters CH, Meiorin S, et al. . Pediatric granulomatous arthritis: an international registry. Arthritis Rheum 2006; 54: 3337–3344. [DOI] [PubMed] [Google Scholar]
- 109.Takeuchi Y, Shigemura T, Kobayashi N, et al. . Early diagnosis of early-onset sarcoidosis: a case report with functional analysis and review of the literature. Clin Rheumatol 2017; 36: 1189–1196. [DOI] [PubMed] [Google Scholar]
- 110.Rose CD, Arostegui JI, Martin TM, et al. . NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum 2009; 60: 1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Becker ML, Martin TM, Doyle TM, et al. . Interstitial pneumonitis in Blau syndrome with documented mutation in CARD15. Arthritis Rheum 2007; 56: 1292–1294. [DOI] [PubMed] [Google Scholar]
- 112.Kanazawa N, Matsushima S, Kambe N, et al. . Presence of a sporadic case of systemic granulomatosis syndrome with a CARD15 mutation. J Invest Dermatol 2004; 122: 851–852. [DOI] [PubMed] [Google Scholar]
- 113.Okafuji I, Nishikomori R, Kanazawa N, et al. . Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis. Arthritis Rheum 2009; 60: 242–250. [DOI] [PubMed] [Google Scholar]
- 114.Chauhan K, Michet C. A case of Blau syndrome. Case Rep Rheumatol 2014; 2014: 216056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Govender P, Berman JS. The diagnosis of sarcoidosis. Clin Chest Med 2015; 36: 585–602. [DOI] [PubMed] [Google Scholar]
- 116.Drent M, Mansour K, Linssen C. Bronchoalveolar lavage in sarcoidosis. Semin Respir Crit Care Med 2007; 28: 486–495. [DOI] [PubMed] [Google Scholar]
- 117.Milman N, Andersen CB, Hansen A, et al. . Favourable effect of TNF-α inhibitor (infliximab) on Blau syndrome in monozygotic twins with a de novo CARD15 mutation. APMIS 2006; 114: 912–919. [DOI] [PubMed] [Google Scholar]
- 118.Yao Q, Zhou L, Cusumano P, et al. . A new category of autoinflammatory disease associated with NOD2 gene mutations. Arthritis Res Ther 2011; 13: R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yao Q, Su LC, Tomecki KJ, et al. . Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J Am Acad Dermatol 2013; 68: 624–631. [DOI] [PubMed] [Google Scholar]
- 120.Yao Q, Shen M, McDonald C, et al. . NOD2-associated autoinflammatory disease: a large cohort study. Rheumatology (Oxford) 2015; 54: 1904–1912. [DOI] [PubMed] [Google Scholar]
- 121.Yao Q, Piliang M, Nicolacakis K, et al. . Granulomatous pneumonitis associated with adult-onset Blau-like syndrome. Am J Respir Crit Care Med 2012; 186: 465–466. [DOI] [PubMed] [Google Scholar]
- 122.Amicosante M, Berretta F, Dweik R, et al. . Role of high-affinity HLA-DP specific CLIP-derived peptides in beryllium binding to the HLA-DPGlu69 berylliosis-associated molecules and presentation to beryllium-sensitized T cells. Immunology 2009; 128: Suppl. 1, e462–e470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Amicosante M, Sanarico N, Berretta F, et al. . Beryllium binding to HLA-DP molecule carrying the marker of susceptibility to berylliosis glutamate β69. Hum Immunol 2001; 62: 686–693. [DOI] [PubMed] [Google Scholar]
- 124.McCanlies EC, Ensey JS, Schuler CR, et al. . The association between HLA-DPB1Glu69 and chronic beryllium disease and beryllium sensitization. Am J Ind Med 2004; 46: 95–103. [DOI] [PubMed] [Google Scholar]
- 125.McCanlies EC, Kreiss K, Andrew M, et al. . HLA-DPB1 and chronic beryllium disease: a HuGE review. Am J Epidemiol 2003; 157: 388–398. [DOI] [PubMed] [Google Scholar]
- 126.Maier LA, Sawyer RT, Bauer RA, et al. . High beryllium-stimulated TNF-α is associated with the −308 TNF-α promoter polymorphism and with clinical severity in chronic beryllium disease. Am J Respir Crit Care Med 2001; 164: 1192–1199. [DOI] [PubMed] [Google Scholar]
- 127.Silveira LJ, McCanlies EC, Fingerlin TE, et al. . Chronic beryllium disease, HLA-DPB1, and the DP peptide binding groove. J Immunol 2012; 189: 4014–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mayer A, Hamzeh N. Beryllium and other metal-induced lung disease. Curr Opin Pulm Med 2015; 21: 178–184. [DOI] [PubMed] [Google Scholar]
- 129.Van Dyke MV, Martyny JW, Mroz MM, et al. . Risk of chronic beryllium disease by HLA-DPB1 E69 genotype and beryllium exposure in nuclear workers. Am J Respir Crit Care Med 2011; 183: 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Balmes JR, Abraham JL, Dweik RA, et al. . An official American Thoracic Society statement: diagnosis and management of beryllium sensitivity and chronic beryllium disease. Am J Respir Crit Care Med 2014; 190: 34–59. [DOI] [PubMed] [Google Scholar]
- 131.Muller-Quernheim J, Gaede KI, Fireman E, et al. . Diagnoses of chronic beryllium disease within cohorts of sarcoidosis patients. Eur Respir J 2006; 27: 1190–1195. [DOI] [PubMed] [Google Scholar]
- 132.Sackett HM, Maier LA, Silveira LJ, et al. . Beryllium medical surveillance at a former nuclear weapons facility during cleanup operations. J Occup Environ Med 2004; 46: 953–961. [DOI] [PubMed] [Google Scholar]
- 133.Henneberger PK, Cumro D, Deubner DD, et al. . Beryllium sensitization and disease among long-term and short-term workers in a beryllium ceramics plant. Int Arch Occup Environ Health 2001; 74: 167–176. [DOI] [PubMed] [Google Scholar]
- 134.Newman LS, Kreiss K. Nonoccupational beryllium disease masquerading as sarcoidosis: identification by blood lymphocyte proliferative response to beryllium. Am Rev Respir Dis 1992; 145: 1212–1214. [DOI] [PubMed] [Google Scholar]
- 135.Fireman E, Haimsky E, Noiderfer M, et al. . Misdiagnosis of sarcoidosis in patients with chronic beryllium disease. Sarcoidosis Vasc Diffuse Lung Dis 2003; 20: 144–148. [PubMed] [Google Scholar]
- 136.Marchand-Adam S, El Khatib A, Guillon F, et al. . Short- and long-term response to corticosteroid therapy in chronic beryllium disease. Eur Respir J 2008; 32: 687–693. [DOI] [PubMed] [Google Scholar]
- 137.Maier LA, Barkes BQ, Mroz M, et al. . Infliximab therapy modulates an antigen-specific immune response in chronic beryllium disease. Respir Med 2012; 106: 1810–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu Y, Jesus AA, Marrero B, et al. . Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014; 371: 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Omoyinmi E, Melo Gomes S, Nanthapisal S, et al. . Stimulator of interferon genes-associated vasculitis of infancy. Arthritis Rheumatol 2015; 67: 808. [DOI] [PubMed] [Google Scholar]
- 140.Xirotagaros G, Hernandez-Ostiz S, Arostegui JI, et al. . Newly described autoinflammatory diseases in pediatric dermatology. Pediatr Dermatol 2016; 33: 602–614. [DOI] [PubMed] [Google Scholar]
- 141.Balci S, Ekinci RMK, de Jesus AA, et al. . Baricitinib experience on STING-associated vasculopathy with onset in infancy: a representative case from Turkey. Clin Immunol 2020; 212: 108273. [DOI] [PubMed] [Google Scholar]
- 142.Clarke SL, Pellowe EJ, de Jesus AA, et al. . Interstitial lung disease caused by STING-associated vasculopathy with onset in infancy. Am J Respir Crit Care Med 2016; 194: 639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Malle L, Marrero B, Liu Y, et al. . Interstitial lung disease in STING-associated vasculopathy with onset in infancy (SAVI): preliminary genotype-phenotype correlation. Pediatr Rheumatol Online J 2015; 13: Suppl. 1, O32. [Google Scholar]
- 144.O'Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med 2013; 368: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sanchez GAM, Reinhardt A, Ramsey S, et al. . JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 2018; 128: 3041–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Merkel PA, Xie G, Monach PA, et al. . Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Rheumatol 2017; 69: 1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kallenberg CG, Mulder AH, Tervaert JW. Antineutrophil cytoplasmic antibodies: a still-growing class of autoantibodies in inflammatory disorders. Am J Med 1992; 93: 675–682. [DOI] [PubMed] [Google Scholar]
- 148.Jariwala MP, Laxer RM. Primary vasculitis in childhood: GPA and MPA in childhood. Front Pediatr 2018; 6: 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Watts RA, Mahr A, Mohammad AJ, et al. . Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrol Dial Transplant 2015; 30: Suppl. 1, i14–i22. [DOI] [PubMed] [Google Scholar]
- 150.Grisaru S, Yuen GW, Miettunen PM, et al. . Incidence of Wegener's granulomatosis in children. J Rheumatol 2010; 37: 440–442. [DOI] [PubMed] [Google Scholar]
- 151.Uribe AG, Huber AM, Kim S, et al. . Increased sensitivity of the European medicines agency algorithm for classification of childhood granulomatosis with polyangiitis. J Rheumatol 2012; 39: 1687–1697. [DOI] [PubMed] [Google Scholar]
- 152.Cabral DA, Uribe AG, Benseler S, et al. . Classification, presentation, and initial treatment of Wegener's granulomatosis in childhood. Arthritis Rheum 2009; 60: 3413–3424. [DOI] [PubMed] [Google Scholar]
- 153.Rottem M, Fauci AS, Hallahan CW, et al. . Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr 1993; 122: 26–31. [DOI] [PubMed] [Google Scholar]
- 154.Frankel SK, Schwarz MI. The pulmonary vasculitides. Am J Respir Crit Care Med 2012; 186: 216–224. [DOI] [PubMed] [Google Scholar]
- 155.Nelson DR, Johnson GB, Cartin-Ceba R, et al. . Characterization of F-18 fluorodeoxyglucose PET/CT in granulomatosis with polyangiitis. Sarcoidosis Vasc Diffuse Lung Dis 2016; 32: 342–352. [PubMed] [Google Scholar]
- 156.Martorana D, Bonatti F, Alberici F, et al. . Fcγ-receptor 3B (FCGR3B) copy number variations in patients with eosinophilic granulomatosis with polyangiitis. J Allergy Clin Immunol 2016; 137: 1597–1599. [DOI] [PubMed] [Google Scholar]
- 157.Fina A, Dubus JC, Tran A, et al. . Eosinophilic granulomatosis with polyangiitis in children: data from the French RespiRare® cohort. Pediatr Pulmonol 2018; 53: 1640–1650. [DOI] [PubMed] [Google Scholar]
- 158.Sinico RA, Di Toma L, Maggiore U, et al. . Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg–Strauss syndrome. Arthritis Rheum 2005; 52: 2926–2935. [DOI] [PubMed] [Google Scholar]
- 159.Cottin V, Bel E, Bottero P, et al. . Respiratory manifestations of eosinophilic granulomatosis with polyangiitis (Churg–Strauss). Eur Respir J 2016; 48: 1429–1441. [DOI] [PubMed] [Google Scholar]
- 160.Jennette JC, Falk RJ, Bacon PA, et al. . 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum 2013; 65: 1–11. [DOI] [PubMed] [Google Scholar]
- 161.Groh M, Pagnoux C, Baldini C, et al. . Eosinophilic granulomatosis with polyangiitis (Churg–Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med 2015; 26: 545–553. [DOI] [PubMed] [Google Scholar]
- 162.Ennis D, Lee JK, Pagnoux C. Mepolizumab for the treatment of eosinophilic granulomatosis with polyangiitis. Expert Opin Biol Ther 2019; 19: 617–630. [DOI] [PubMed] [Google Scholar]
- 163.Mourah S, How-Kit A, Meignin V, et al. . Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J 2016; 47: 1785–1796. [DOI] [PubMed] [Google Scholar]
- 164.Badalian-Very G, Vergilio JA, Degar BA, et al. . Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010; 116: 1919–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sundar KM, Gosselin MV, Chung HL, et al. . Pulmonary Langerhans cell histiocytosis: emerging concepts in pathobiology, radiology, and clinical evolution of disease. Chest 2003; 123: 1673–1683. [DOI] [PubMed] [Google Scholar]
- 166.Castoldi MC, Verrioli A, De Juli E, et al. . Pulmonary Langerhans cell histiocytosis: the many faces of presentation at initial CT scan. Insights Imaging 2014; 5: 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Roden AC, Yi ES. Pulmonary Langerhans cell histiocytosis: an update from the pathologists’ perspective. Arch Pathol Lab Med 2016; 140: 230–240. [DOI] [PubMed] [Google Scholar]