

Abstract
Pulmonary arterial hypertension (PAH) is a rare disease characterised by pulmonary vascular remodelling and elevated pulmonary pressure, which eventually leads to right heart failure and death. Registries worldwide have noted a female predominance of the disease, spurring particular interest in hormonal involvement in the disease pathobiology. Several experimental models have shown both protective and deleterious effects of oestrogens, suggesting that complex mechanisms participate in PAH pathogenesis. In fact, oestrogen metabolites as well as receptors and enzymes implicated in oestrogen signalling pathways and associated conditions such as BMPR2 mutation contribute to PAH penetrance more specifically in women. Conversely, females have better right ventricular function, translating to a better prognosis. Along with right ventricular adaptation, women tend to respond to PAH treatment differently from men. As some young women suffer from PAH, contraception is of particular importance, considering that pregnancy in patients with PAH is strongly discouraged due to high risk of death. When contraception measures fail, pregnant women need a multidisciplinary team-based approach. This article aims to review epidemiology, mechanisms underlying the higher female predominance, but better prognosis and the intricacies in management of women affected by PAH.
Short abstract
Pulmonary arterial hypertension affects women more than men, although women have a better prognosis. Oestrogens might be the key to explain these gender differences. https://bit.ly/36KK1yX
Introduction
Pulmonary arterial hypertension (PAH) is a type of pre-capillary pulmonary hypertension (PH). The 6th World Symposium on Pulmonary Hypertension has recently redefined PAH as an elevated mean pulmonary arterial pressure >20 mmHg, a pulmonary artery wedge pressure (PAWP) ≤15 mmHg and pulmonary vascular resistance (PVR) ≥3 Wood units, assessed by right heart catheterisation (RHC) at rest [1]. PAH is group 1 PH among five groups classified mainly based on the underlying aetiology and mechanism of pulmonary pressure elevation. Group 1 PAH is a disease of the small pulmonary arterioles characterised by vascular injury, endothelial dysfunction, proliferation and remodelling [2]. Specific treatment has been developed for group 1 PAH targeting three pathways (endothelin, nitric oxide and prostacyclin) involved in PAH pathophysiology [2]. There are several subtypes of PAH (table 1) [1]. Idiopathic (i)PAH is the most common aetiology, accounting for 30–50% of cases. Heritable (h)PAH is diagnosed after extensive family history-taking and genetic testing, of which bone morphogenetic protein receptor-2 (BMPR2) mutations are the most frequent. Other subtypes include drug- and toxin-induced PAH, connective tissue disease (CTD)-PAH, HIV-PAH, portopulmonary hypertension, congenital heart disease (CHD)-PAH or schistosomiasis (which is rarely diagnosed in developed countries) [1]. In addition, group 1 PAH includes PAH with vasoreactivity, PAH with overt signs of venous/capillary involvement and persistent PH of the newborn syndrome [1].
TABLE 1.
Updated clinical classification of group 1 pulmonary arterial hypertension (PAH) adapted from the 6th World Symposium on Pulmonary Hypertension
|
1.1 Idiopathic PAH 1.2 Heritable PAH 1.3 Drug- and toxin-induced PAH 1.4 PAH associated with: 1.4.1 connective tissue disease 1.4.2 HIV infection 1.4.3 portal hypertension 1.4.4 congenital heart disease 1.4.5 schistosomiasis 1.5 PAH long-term responders to calcium channel blockers 1.6 PAH with overt features of venous/capillaries (PVOD/PCH) involvement 1.7 Persistent pulmonary hypertension of the newborn syndrome |
PVOD: pulmonary veno-occlusive disease; PCH: pulmonary capillary haemangiomatosis.
As PAH is a rare disease, precise estimates of prevalence and incidence are difficult to obtain accurately. Therefore, data collected from registries are of great interest. One notable feature of PAH is the female predominance; conversely, fewer males have PAH, but they have a worse prognosis [3–7]. Various mechanisms have been proposed to explain the differences in the incidence, physiology and prognosis between men and women with PAH including sex hormones and receptors, metabolites, immune-mediated mechanisms, genetic factors, comorbidities and sociocultural factors, which probably all play a complex role in disease expression [8]. Indeed, several animal and human models have shown the importance of sex hormones in PAH penetrance and pathogenesis. Synthesis and metabolism of sex hormones within the lung have been demonstrated to have both harmful and protective effects [9]. These findings give hope for potential new therapies for PAH that target hormonal signalling.
While pathophysiology differs between sexes partly because of oestrogen levels, other parameters such as haemodynamic profiles, right ventricle (RV) adaptation and survival are different in men and women (table 2) [8]. Additionally, observational studies have demonstrated a different response to PAH treatments between sexes in terms of quality of life and improvement in 6-min walk distance (6MWD) [10–12].
TABLE 2.
General findings from studies investigating gender differences in pulmonary arterial hypertension (PAH)
| Females with PAH compared to males |
| ↑ susceptibility to PAH development ↑ pulmonary vascular remodelling ↑ right ventricle contractility and ejection fraction ↓ haemodynamic alterations ↑ response to treatment with endothelin receptor antagonists ↑ improvement in right ventricle ejection fraction after PAH treatment ↑ survival |
Lastly, because group 1 PAH may also affect women of childbearing age [13], we discuss pregnancy outcomes, management and the importance of adequate contraception.
Epidemiology
PAH is a rare and severe disease with a prevalence estimated ∼10–50 cases per million inhabitants and an incidence of 5–10 cases per million per year [14]. Data from the Registry to Evaluate Early and Long-term Disease Management in PAH (REVEAL) between 2006 and 2009 in the United States suggests that, since the advent of PAH therapies, median survival has improved from 2.8 to 7 years with a 3-year survival rate of 68% in REVEAL compared to 48% in iPAH patients of the National Institutes of Health (NIH) Patient Registry for the Characterization of Primary Pulmonary Hypertension (1981–1985) [15]. More recent data from the French registry (2007–2013) show a 3-year survival rate of 83% in PAH patients when treated with upfront dual combination therapy (consisting of an endothelin receptor antagonist (ERA) and a phosphodiesterase type 5 inhibitor (PDE-5i)) [16].
Over the years and around the world, the age at diagnosis has increased, with a reported age of 36±15 years among iPAH patients in the first American NIH registry compared to mean ages of 50±17 to 65±15 years more recently [17]. The European Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) registry (2007–2011) showed that 64% of iPAH patients were aged >65 years [18]. This growing iPAH population could be explained by a greater awareness of PAH and improvement in noninvasive diagnostic tools and PAH treatment. Furthermore, because of the burden of cardiovascular and pulmonary comorbidities, PAH in the elderly could have been misdiagnosed previously. Nevertheless, a Chinese registry reported a mean age of 33±15 years in iPAH patients, which mirrors the early NIH registry data [19]. The question remains whether there is an actual drift in PAH demographics or if independent factors are implicated [17].
A greater proportion of female PAH patients has been noted in several registries, ranging from 56% to 86%, implicating female sex as a risk factor for PAH [18, 20–26]. As seen in the COMPERA registry, this higher female to male ratio 2.3:1 in younger patients (median 54 years) seems to decrease as age at diagnosis rises (1.2:1 in 75-year-old patients) along with the onset of menopause [18]. Yet sex differences in iPAH, hPAH and drug- and toxin-induced PAH have shown a better survival in women compared to men [25].
Like autoimmune diseases in general, CTD-PAH has a 9:1 female to male ratio [5]. PAH affects 3–13% of patients with CTD and is a major cause of death [5]. Moreover, CTD-PAH patients tend to be older than iPAH patients, probably because the onset of PAH varies over time after the first CTD symptoms, as described by Hachulla et al. [27] and Sundaram and Chung [28] in patients with systemic sclerosis (SSc). In the particular case of SSc-associated PAH, a lower survival rate is noted in men and in comparison to other CTDs (systemic lupus erythematous, rheumatoid arthritis or mixed-CTD), probably owing to mixed PH phenotypes (PAH, pulmonary venous involvement and concurrent interstitial lung disease) [5].
While cirrhosis has a known male predominance, females with liver diseases have a higher risk of developing portopulmonary hypertension [29]. Al-Naamani et al. [30] demonstrated that sex hormone signalling abnormalities, especially through genetic variation in aromatase, have been implicated in the development of PAH in chronic liver disease.
Regarding CHD-PAH, HIV-PAH and drug or toxin-associated PAH, gender and hormonal factors do not seem to influence the pathophysiological mechanisms [8].
Role of sex hormones in the pathogenesis of PAH
Oestrogen synthesis and signalling pathway
Oestrogens have several key roles in male and female physiology, including differentiation of several tissues and organs, modulation of inflammation and cardiovascular functions [31].
The three main oestrogens, 17β-oestradiol (E2), oestrone (E1) and oestriol (E3 or 16α-OH-E2) fluctuate in females depending on age, menstrual status and pregnancy. E2, secreted by the ovaries, is predominant in pre-menopausal women, while E1 is synthesised more in post-menopausal women. Sex hormones are synthesised in gonads and extragonadal tissues (brain, adipose tissue and vascular endothelium) with influence on one another depending on age. In men and post-menopausal women, extragonadal tissues are the main source of oestrogens [31]. In extragonadal tissues, such as the pulmonary arteries, aromatase is responsible for synthesis of local oestrogens, with autocrine and paracrine effects that may have more potent effects compared to circulating levels [32]. In the extragonadal tissues, oestrogens production is dependent on the availability of C19 steroid precursors: testosterone, androstenedione, dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS). Depending on the tissue, testosterone may be converted by aromatase to 5α-dihydrotestosterone or to E2 [31]. DHEAS and DHEA can also be converted to E2. The metabolism of DHEAS and DHEA is tissue-specific and depends on the distribution of the required enzymes. These observations make it impossible to dissociate oestrogen signalling from androgen signalling or to attribute sex-specific roles to each signalling cascade [31].
E2 acts on different cell types through interaction with oestrogen receptors (ERs) which then activate two distinct signalling pathways: the genomic and nongenomic pathways. Two nuclear ERs, ERα and ERβ, exhibit distinct transcriptional properties, forming homodimers and heterodimers that bind to oestrogen response elements located in the promoters of target genes corresponding to the genomic pathway. In addition, ERs can indirectly interact with other transcription factors. Regarding the nongenomic pathway, plasma membrane-associated ER activation can lead to cytoplasmic alterations and to regulation of gene expression [33]. In addition to ERα and ERβ, E2 binds to the G-protein coupled receptor GPR30, which is localised in the endoplasmic reticulum and plasma membrane. After E2 binds to GPR30, GPR30 exerts rapid nongenomic signalling, triggering intracellular signalling cascades that alter gene expression downstream [34, 35]. The contribution of each ER seems to be dependent on the stimulus; ERα modulates pulmonary artery (PA) vasoconstriction induced by phenylephrine and ERβ inhibits hypoxia-induced pulmonary vasoconstriction [36].
Oestrogen is metabolised by an oxidation reaction resulting in biologically active E2 metabolites. Among other pathways, it occurs via hydroxylation at the C-16, C-2 and C-4 position by P450 enzymes such as cytochrome P450 1B1 (CYP1B1) [37].
Sex differences in BMPR2 mutation carriers
In its heritable form, PAH is most frequently due to BMPR2 mutations. The BMPR2 gene encodes for a BMPR-II protein which is a serine/threonine receptor kinase involved in several processes including development, embryogenesis, proliferation and apoptosis [38]. A BMPR2 mutation is identified in ∼20% of patients presenting with a sporadic form of PAH and 75–90% of PAH patients with a family history of PAH [39]. The analysis of pedigrees of families with mutations in BMPR2 gives an estimated penetrance of hPAH in BMPR2 mutation carriers at 14% in males and 42% in females [40]. The recent DELPHI-2 study proposed annual screening in asymptomatic BMPR2 mutation carriers included clinical assessment, electrocardiogram, pulmonary function tests, 6MWD, cardiopulmonary exercise test, echocardiography and N-terminal pro-brain natriuretic peptide (NT-proBNP) level [41]. RHC was performed to confirm PAH based on pre-defined criteria. 55 subjects (median age 37 years) were included, and five patients developed PAH during long-term follow-up (>5 years). This study revealed a PAH incidence in BMPR2 mutation of carriers of 2.3% per year (0.99% per year in men and 3.5% per year in women) [41]. Despite the higher incidence in female mutation carriers, survival rate is similar in women and men affected with PAH due to a BMPR2 mutation [42].
The female predominance observed in hPAH patients could be explained by a sex-specific difference in expression and activity of BMPR-II. Indeed, pulmonary artery smooth muscle cells (PASMCs) isolated from females without PAH have reduced expression of BMPR-II (at mRNA and protein levels) when compared to PASMCs isolated from non-PAH males, with similar differences in the downstream BMPR-II signalling pathways [43]. BMPR-II expression is also reduced in lymphocytes from female patients compared to male patients [44]. Moreover, lower expression of CYP1B1 resulting in higher 16α-OH-E1 rates in the female PAH-affected BMPR2 carriers has been identified on isolated B-cells that are Epstein–Barr virus (EBV)-immortalised and cultured, compared to normal control patients and nonaffected carriers [45]. All these observations lead to the hypothesis that females may be predisposed to developing PAH through a synergistic interaction between oestrogens and their metabolites and the BMPR-II signalling pathway [46].
In addition, Al-Naamani et al. [30] showed that not only iPAH and hPAH, but also portopulmonary hypertension is associated with changes in genetic variation of aromatase and oestrogen metabolites.
Pre-clinical animal models and sex-specific differences in PAH
In women with PAH, outcomes are worse in those aged >60 years who are presumably post-menopausal [47]; however, there are no studies demonstrating an association between oestrogen levels, RV function and outcomes in patients with PAH. Experimental data from animal studies show that oestrogen can directly promote PAH development, while several other studies demonstrate a beneficial effect of oestrogen on pulmonary vascular remodelling and RV function, as recently reviewed by Hester et al. [8].
Despite the higher incidence of iPAH and hPAH in women, the common pre-clinical models of PAH show an increased male susceptibility [48]. While female sex and oestrogens show protective effects in rats in the experimental chronic hypoxia (CH)- and monocrotaline (MCT)- induced PAH models, recent findings suggest that classical models may not be appropriate to understand every effect of oestrogens in human PAH [49] (table 3).
TABLE 3.
Pulmonary hypertension (PH) experimental models used to assess sex hormone involvement in PH development
| Findings | First author, year [reference] | |
| Chronic hypoxia induced PH | Development of PH in male>female PH reduced on exogenous E2 |
Rabinovitch, 1981 [50] McMurty, 1973 [51] Lahm, 2012 [52] |
| Monocrotaline induced PH | Vascular lesion and RV hypertrophy reduced under ovariectomy PH reduced on exogenous E2 |
Yuan, 2013 [19] |
| Tofovic, 2006 [53] | ||
| Ahn, 2003 [54] | ||
| Sugen/hypoxia induced PH | Survival in female with PH>male PH severity in female>male PH severity in female under ovariectomy>no ovariectomy Vasoconstriction and remodelling reduced with exogenous E2 Exogenous E2 protects RV function |
Rafikova, 2015 [55] Tofovic, 2012 [56] Frump, 2015 [57] Philip, 2020 [58] Liu, 2017 [59] |
| Mitomycin/cyclophosphamide induced PH | PH severity in female>male | Perros, 2015 [60] Ranchoux, 2015 [61] |
| Dexfenfluramine/SERT+ induced PH | Development of PH in female>male | White, 2011 [62] Dempsie, 2011 [63] Dempsie, 2013 [64] |
| Overexpressed SERT+ | Development of PH in female with attenuation of PH under ovariectomy | White, 2011 [65] |
| Obesity | Development of PH in female and male | Mair, 2019 [66] |
| Nfu1g206c mutation | Development of PH in female>male | Niihori, 2020 [67] |
| SU5416-treatment and chronic hypoxia | PH severity in female>male | Tamosiuniene, 2018 [68] |
SERT+: serotonin transporter; E2: 17β-oestradiol; RV: right ventricle.
Classical models of experimental PH
Male rats are more susceptible to develop PH than females after CH exposure [50, 51]. Moreover, exogenous oestrogen administration reduces the development of PH in rats exposed to MCT and CH [19, 52, 53], as well as attenuating pulmonary vascular lesions and RV hypertrophy in ovariectomised rats injected with MCT [54].
Using the Sugen/hypoxia (Su/Hx)-PH rat model, Rafikova et al. [55] demonstrated that females survive longer than males because PH in female Su/Hx rats is associated with angioproliferative changes in the pulmonary arteries without inflammatory or fibrinogenic processes in lung or heart. These data suggest that the female population could be treated with antiproliferative drugs, whereas in males, therapies may also need to include anti-inflammatory and antifibrinogenic strategies. In addition, an unpublished study demonstrated that after Su/Hx-exposure, female rats develop more severe PH than males with more numerous plexiform lesions [56].
Frump et al. [57] found no significant differences in PH phenotype between male and female rats after Su/Hx-exposure. However, they demonstrated that the removal of endogenous oestrogens by ovariectomy worsened Su/Hx-induced PH without worsening of pulmonary vascular remodelling. Importantly, in these experiments E2-repletion in ovariectomised female PH rats strongly reduced the development of PH, suggesting that E2 has protective effects on PH development in females. Similarly, Philip et al. [58] confirmed that exogenous oestrogen treatment in Su/Hx-PH rats reduced PA vasoconstriction and remodelling after Su/Hx exposure. In addition, in Su/Hx rats, oestrogen treatment was found to protect RV function by preserving mitochondrial biogenesis, function and oxidative capacity, at least partially explaining the cardioprotective mechanism of oestrogens in female PH patients [59].
Additionally, female rats harbour a more severe PH phenotype than males after exposure to the chemotherapy agents mitomycin and cyclophosphamide [60, 61]. In mitomycin-exposed rats, serotonin production has also been shown to be increased [69]. Dexfenfluramine (an anorectic drug) was demonstrated to mediate PAH via a serotonergic mechanism [70]. Transgenic mice with overexpression of the serotonin transporter S100A4/Mts1, or that had exposure to dexfenfluramine, developed PH specifically in females [62–64]. Moreover, White et al. [65] demonstrated that the overexpression of the serotonin transporter (SERT+) in female mice induced spontaneous PH which was reduced by ovariectomy, while male mice overexpressing SERT+ did not develop spontaneous PH. Altogether, these results confirm that the serotonin pathway is a major actor and major determinant of the unequal sex ratio in PAH [48, 71]. More recently, Hood et al. [72] suggested that 5-hydroxytryptamine receptor 1B contributes to vascular remodelling in PAH by regulating Src-kinase and NADPH oxidase pathways.
Nonclassical models of experimental PH
Using an obese mouse model, Mair et al. [66] suggested that obesity can induce PH, partly due to the changes in oestrogen metabolism.
Recently, Niihori et al. [67] found that human mutations in NFU1 can predispose to the development of PAH. Patients with an autosomal-recessive NFU1 mutation G208C have reduced activity of the mitochondrial respiratory chain complex II and decreased levels of lipoic acid dependent enzymes, which facilitates the development of PAH. Using CRISPR/Cas9 genome editing, Niihori et al. [67] introduced a point mutation in rat Nfu1g206c (human G208C). They showed that Nfu1g206c-mutated rats developed spontaneous PH with a higher penetrance in females than in males, reproducing the established sex difference among patients with PAH. However, the expression and function of NFU1 have not been differentially evaluated in male and female PAH patients.
Exposure to the vascular endothelial growth factor-2 inhibitor SU5416 or CH induced the development of more severe PH in female than male athymic rats lacking regulatory T-cells (Tregs). In both models (Su5416 and CH), the authors found that Treg immune reconstitution protected against the development of PH [68], indicating that Tregs could contribute to the sex difference in PAH.
Studies on oestrogen signalling and metabolites involved in experimental PH
Lahm et al. [52] demonstrated that E2 has protective effects on PH induced by CH exposure, but these effects are reduced by a nonselective E2 receptor antagonist suggesting an ER-mediated mechanism of E2 action in CH-PH. In serotonin-dependent PH mice, ERα stimulates PASMC proliferation and decreases BMPR-II signalling, and ERα antagonists reverse experimental PH and increase BMPR-II expression [73]. This suggests a close relationship between ERα and BMPR-II expression [73]. Austin et al. [74] demonstrated the presence of an oestrogen response element in the promoter of BMPR2 gene. They observed that BMPR-II expression was higher in the lungs of male mice compared to ovariectomised female mice [74]. Wright et al. [73] demonstrated that ERα expression is higher in PASMCs isolated from female PAH patients compared to male patients. Since ERs were described to regulate BMPR-II expression via an oestrogen response element [74], these differences could be explained by an increased expression of ERα among females [35, 74]. Moreover, White et al. [65] found that female mice overexpressing the serotonin transporter developed severe PH, which is abolished by ovariectomy. The chronic in vivo administration of E2 re-establishes PH in ovariectomised mice overexpressing serotonin transporter, while in vitro serotonin exposure increases the expression of ERα. Additionally, Wright et al. [73] showed that ERα inhibition with the ERα antagonist MPP (1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole-dihydrochloride) reduced the development of PH induced by CH exposure, suggesting that inhibiting endogenous E2 is therapeutic, in contrast to the protective effects of exogenously stimulating the E2 system.
Regarding ERβ, Frump et al. [75] found that it is significantly increased in hypoxic conditions through the stabilisation of hypoxia inducible factor (HIF)-1α, and demonstrated that ERβ-deficiency in mice led to a reduced response to E2 during hypoxia exposure.
Conversely, Frump et al. [57] demonstrated that treatment with ERα-specific agonist reduces the development of PH, and Umar et al. [76] reported that ERβ agonist diarylpropionitrile treatment reduces the development of PH in male rats exposed to MCT. Two additional studies have suggested that ERα and ERβ cooperate to protect against the development of PH [36, 52]. In rats with PH induced by hypobaric CH exposure, the co-treatment with a nonselective ER inhibitor or ERα-specific antagonist prevents the protective effect of E2 treatment, whereas ERα- and ERβ-specific antagonists reduce the effects of E2 [52]. These observations support a potential protective role of E2 and ERα against PH and demonstrate the complexity of oestrogen involvement.
Finally, after treating rats with a GPR30-selective agonist G1, Alencar et al. [77] demonstrated that GPR30 activation reverses the development of PH in MCT-exposed male rats as well as in ovariectomised female rats exposed to MCT [78]. In addition, they found that GPR30 pharmacological activation had cardioprotective effects [77].
CYP1B1 is highly expressed in the pulmonary arterial lesions of patients with PAH, while it is minimally expressed in pulmonary arteries from non-PAH patients [79]. Moreover, West et al. [45] found that mRNA expression of CYP1B1 is strongly reduced in PAH female lymphocytes isolated from whole blood and exposed to EBV to induce cell immortalisation, but this was not observed in lymphocytes from male PAH patients. These results implicated CYP1B1 as a candidate in the female predisposition to PAH. Austin et al. [80] linked the CYP1B1 N453S polymorphism to the development of PAH. This mutation changes the amino acid residue 453 (asparagine to serine), resulting in increased proteosomal-mediated degradation of CYP1B1. This polymorphism is well described in several oestrogen-regulated cancers. Patients with the homozygote CYP1B1 N453S polymorphism had a four-fold lower PAH penetrance compared with the wild-type CYP genotype. Moreover, a high 16α-OH-E1:2-OH-E2 ratio correlates with the development of PAH in human BMPR2 mutation carriers [80].
A CYP1B1 inhibitor, 2,3′,4′5′-tetramethoxystilbene (TMS) reversed PH in Su/Hx and CH-PH models [79]. In addition, in vivo TMS treatment improved the survival in the MCT-PH rat model [81], and TMS abolished oestrogen-induced proliferation in human (h)PASMCs from controls and patients [79]. The pharmacological inhibition of CYP1B1 (TMS) attenuates PH in obese mice [66].
CYP1B1 and CYP1A1 are described to metabolise E2 and oestrone (E1), generating active metabolites, which are increased in both animal models and human PAH. Indeed, in Su/Hx-PH rats, CYP1A1, which is activated by the aryl hydrocarbon receptor (AhR), is overexpressed in the lung (CYP1A1 and AhR), and the pharmacological inhibition of AhR reduced PH induced by Su/Hx exposure [82]. However, in nonpulmonary vascular cells, hypoxia induced a decrease in mRNA, protein and CYP1A1 activity [83].
Napoli et al. [84] highlighted the importance of the AhR/AhR nuclear translocator/CYP system in the regulation of the PAH epigenome, suggesting the potential role of the AhR/CYP1A1 pathway in PAH. Dempsie et al. [64] demonstrated that in mice exposed to dexfenfluramine for 28 days (5 mg·kg−1 per day), PAH developed only in females. Female mice developing PAH after dexfenfluramine exposure showed an increase in CYP1B1 lung expression, and cyp1b1-deficient mice are completely protected against the development of PAH after dexfenfluramine exposure [64]. These results highlighted that CYP1B1 and CYP1A1 could be potential therapeutic targets in PAH.
Using bmpr2-mutant mice, Fessel et al. [85] found that elevated 16α-OH-E1 increased the penetrance of PAH, while 2-methoxyoestradiol (ME2) administration did not confer any protection. In addition to an increased penetrance of PAH, it appears that 16α-OH-E1 also strongly reduces BMPR-II protein expression and phosphorylation of SMAD1/5/8 in bmpr2-mutant mice compared to wild-type mice. Furthermore, bmpr2-mutant cells become insensitive to oestrogen canonical signalling pathways. 4 weeks of daily injections of 16α-OH-E1 mediated a slight elevation in right ventricle systolic pressure in wild-type mice [79]. Indeed, 16α-OH-E1 (1.25 μg·h−1 for 4 weeks) promotes the development of PAH in bmpr2-mutant mice [86].
In vitro 2-ME2 treatment reduces the proliferation of hPASMCs and human lung fibroblasts in a dose-dependent manner [87], suggesting that 2-ME2 treatment could be beneficial in preventing pulmonary arterial remodelling. In an experimental PH model induced by MCT and bleomycin exposure, in vivo administration of 2-ME2 reversed PH [88–91]. The exact underlying antiproliferative mechanism of 2-ME2 is unclear. There is only evidence that 2-ME2 reduces the production of endothelin-1 in endothelial cells [92]. Docherty et al. [93] demonstrated in vivo and in vitro that 2-ME2 is an inhibitor of HIF-1α. In addition, they found that HIF-1α protein expression was higher in female hPASMCs compared with male hPASMCs. The higher HIF-1α expression in female hPASMCs could contribute to the increased susceptibility of females to develop PAH.
Human models and sex-specific differences in PAH
In a small cohort of iPAH patients, Denver et al. [94] found that altered oestrogen metabolism in males and post-menopausal females is correlated with an increased risk and severity of PAH, suggesting that oestradiol is not ovary-derived. They found that E1 levels are decreased while 16α-OH-E2 (oestriol) is increased in the serum of female iPAH patients. E1, E2 and 16α-OH-E1 are elevated in male iPAH patients. In female controls, the E1 level was higher compared with male controls, while in iPAH, E1 was higher in males. In female iPAH patients, increasing E1 and E2 levels were associated with higher PAWP, while increasing 16α-OH-E1 levels correlated with a reduction in cardiac output. In male iPAH patients, the increased E2 level correlated with an increased PAWP [94]. Ventetuolo and co-workers [95, 96] have reported elevated levels of E2 and lower levels of DHEA-S in male and post-menopausal female patients with PAH. Baird et al. [97] recently demonstrated that women with PAH have variations in cardiopulmonary function during menstruation driven by E2 and DHEA-S. In turn, these hormones influence transcription of extracellular vesicle miRNAs implicated in the pathobiology of pulmonary vascular disease and cancer.
These observations show that oestradiol metabolism is altered in iPAH and that 16α-OH-E1 and 16α-OH-E2 accumulate in iPAH patients, the magnitude of which is related to disease severity.
Future directions in targeting oestrogens pathway to treat PAH
Given the role of oestrogens in the development of PAH and pulmonary vascular remodelling, several studies have investigated oestrogen deprivation to treat PAH patients. Aromatase and CYP1B1 are key enzymes in the synthesis and metabolism of oestrogen (figure 1). Aromatase is known to be expressed in lung tissue, suggesting that the pulmonary vasculature could locally produce an oestrogenic microenvironment. A genetic polymorphism in the AROMATASE gene has been associated with an increased risk of portopulmonary hypertension, which leads to increased plasma oestrogen levels [98]. Anastrozole, an aromatase inhibitor that blocks conversion of androgens into oestrogen, attenuated experimental PAH in female animals but not males [99]. Anastrozole and fulvestrant (another aromatase inhibitor) both inhibit endogenous oestrogens that are produced in the pulmonary circulation, while all rat studies demonstrating protective effects of E2 do so by the artificial administration of exogenous E2. Of note, ovariectomy does not eliminate pulmonary-derived E2; only the circulating endogenous gonadal E2. This distinction is important to explain the beneficial effects of drugs that inhibit endogenous E2. Millions of women receive anastrozole for E2-sensitive cancers with no reports of adverse effects on the RV. This was explored in a small proof-of-concept randomised study of 18 men and post-menopausal women with PAH who received anastrozole or placebo [100]. Anastrozole was effective in reducing circulating oestrogen levels, but there were no significant effects on RV function on echocardiogram or on NT-proBNP levels. In vivo oestrogen inhibition by fulvestrant and anastrozole prevented and reduced the development of PH in bmpr2-mutant mice in association with a reduction in metabolism abnormalities, suggesting that fulvestrant and anastrozole could be useful in PAH [101]. This idea has been described more recently by clinical studies led by Kawut and co-workers [100, 102] and Mair et al. [32]. In Su/Hx-PH rats and CH-PH rats, in vivo inhibition of aromatase by anastrozole reversed the PH phenotype in female rats, in parallel with reduced plasma oestrogen levels, and a normalisation of BMPR-II signalling [103].
FIGURE 1.

Oestrogen biosynthesis and metabolites involved in pulmonary arterial hypertension (PAH) female predominance and possible targets for PAH treatment. Oestrogen and its metabolites (17β-oestradiol, 16α-hydroxyoestrone (OH-E1) and 2-methoxyoestradiol (ME2)) as well as enzymes (aromatase and CYP1B1) seem to play a major role in PAH female predominance, as many experimental models have tried to demonstrate. Potential targets for PAH treatment are shown in bold. DHEA: dehydroepiandrosterone; HSD: hydroxysteroidodehydrogenase; OH-E2: hydroxyoestradiol; 16α-OH-E2: 16α-hydroxyoestradiol (oestriol); ER: oestrogen receptor; BMPR: bone morphogenetic protein receptor; PASMC: pulmonary artery smooth muscle cell; TMS: 2,3′,4′5′-tetramethoxystilbene.
The therapeutic potential of anastrozole in PAH is promising, as it has been shown to be safe, well tolerated and effective in improving 6MWD and reducing oestrogen levels [100]. However, these results need to be confirmed in larger and longer-duration phase II trials.
Another trial of tamoxifen, a selective oestrogen receptor modulator, in PAH patients is ongoing (clinicaltrials.gov identifier NCT03528902).
Sex differences in right ventricular function and prognosis
Right ventricle and pulmonary artery coupling
RV function and adaptation to its haemodynamic load are critical determinants of survival in PAH [104–106]. The development of RV failure results in impaired delivery of blood to the pulmonary circulation and increased systemic venous pressures, which is a major cause of death in PAH [107]. RV–PA coupling reflects the efficiency of energy transfer between the RV and the pulmonary circulation, which is described using pressure–volume loops in the RV. Along with cardiac magnetic resonance imaging, this method is considered a gold standard for evaluating RV function in PH [105]. RV pressure–volume loops permit measurement of the end-systolic elastance (Ees) as an indicator of ventricular contractility, ventricular afterload is estimated from arterial elastance (Ea) and diastolic stiffness is deduced from the end-diastolic elastance [105]. In PAH, there is an initial compensation in RV function via hypertrophic changes and increased contractile function in response to the increased afterload (estimated from Ea). This early adaptation is beneficial as it facilitates maintenance of RV–PA coupling. With disease progression, these adaptations cannot maintain a normal pressure–volume relationship: RV–PA uncoupling occurs and RV failure ultimately ensues. The RV–PA uncoupling, as measured by a reduced Ees/Ea (<0.8; normal ratio ranging between 1.5 and 2), has been shown to be a predictor of clinical worsening and deterioration, and correlated with a significant reduction in RV ejection fraction (RVEF) [108, 109]. Labazi et al. [110] have recently hypothesised that pressure overload obtained by pulmonary trunk binding (PTB) leads to gene expression changes in the RV. This study shows that RV hypertrophy indices are significantly higher in male PTB rats than females with elevated natriuretic peptide A and B as well as collagen III, increasing the susceptibility to poorer outcome in males.
There are important sex differences in haemodynamic responses to therapy and RV adaptation to load in PAH. In healthy individuals, normal pulmonary arterial pressure at rest is 14±3 mmHg, which does not differ between men and women [111]. In contrast, there are sex differences in RV volumes and function in normal individuals. Healthy women have lower RV mass, smaller RV volumes and higher RVEF compared to men [112], which may be partially related to sex hormones and metabolites, as supported by Ventetuolo et al.’s [113] study on CYP1B1 single nucleotide polymorphism and RV function. In healthy post-menopausal women receiving hormone therapy, higher E2 levels were associated with higher RVEF and lower RV volumes, whereas in men, higher levels of androgens, such as testosterone and DHEA, were related to higher RV mass and RV volumes [114]. In patients with PAH, Tello et al. [115] showed that contractility (Ees) and RV–PA coupling were better in females compared to males, despite comparable afterload (Ea). In addition, early diastolic relaxation and RV mass index were lower in female patients, indicative of better diastolic function [115]. Females with PAH have better RV function and less-dilated RV volumes at baseline compared to males of the same age [116–118].
Right ventricle and response to PAH treatment
Haemodynamic markers of disease severity tend to be similar between sexes at the time of PAH diagnosis in most studies [24, 47, 117, 119]; however, several studies have demonstrated that RV function at the time of diagnosis and haemodynamic responses to PAH treatment are better in women [7, 120, 121]. The degree of improvement in RV function and haemodynamic variables with initial therapy, such as PVR and stroke volume index, are closely linked to long-term survival in newly diagnosed PAH patients [106, 122, 123]. Jacobs et al. [7] demonstrated that after adjusting for age, comorbidities and other baseline confounders, female and male patients with PAH demonstrated similar haemodynamic improvements, but RVEF improved in females by 3.6% and worsened by 1% in males. These findings suggest that females with PAH have a more adaptive RV phenotype which responds more favourably to therapies, and this key difference probably mediates observed sex differences in survival. It has also been shown that pulmonary arterial capacitance (stroke volume/pulmonary arterial pulse pressure) improved in both male and female PAH patients with treatment, but only females had decreases in RV end-diastolic pressure and right atrial pressure [47].
The right ventricle and sex hormones
Androgens and oestrogens have been implicated in both maladaptive and adaptive effects on the pulmonary circulation and the RV in animal and human studies of PAH [8]. There has been considerable research into targeting RV function by modulating sex hormone pathways in PAH. In a mouse model, testosterone had negative effects on the RV with increased myocyte size, fibrosis and RV hypertrophy in response to a haemodynamic load induced by pulmonary artery banding [124]. Deprivation of testosterone improved survival in this mouse model of high RV afterload [124]. In contrast, animal studies have demonstrated beneficial effects of DHEA in the prevention of PAH by directly improving RV function [125, 126]. In men, higher oestrogen levels were associated with the development of PAH, whereas higher DHEA levels were protective [95]. In a case–control study of post-menopausal female patients with PAH, lower DHEA levels and higher oestrogen levels were associated with higher risk of PAH (versus age-matched women without cardiovascular disease), worse clinical status and more severe haemodynamics among PAH patients [127]. Higher oestrogen levels were also associated with worse exercise capacity and symptoms, but were not associated with survival, whereas lower DHEA levels were associated with worse RV function and worse survival [127]. There is currently an ongoing clinical trial evaluating the effects of DHEA on RV function in PAH (clinicaltrials.gov identifier NCT03648385).
The cardioprotective effects of oestrogen on the RV have been well established in animal models of PAH. Liu et al. [128] demonstrated beneficial effects of oestradiol administration on RV function and contractility and RV–PA coupling in the rat Su/Hx model. Interestingly, there were no beneficial effects of oestradiol on RV diastolic function. In the study by Frump et al. [57], discussed earlier, oestrogen supplementation attenuated the development of pulmonary vascular remodelling in the Su/Hx rat model and prevented RV remodelling in ovariectomised female rats, whereas oestrogen supplementation in male rats was RV-protective, but did not prevent pulmonary vascular remodelling. Oestradiol supplementation could limit several maladaptive processes in the RV associated with PAH, including pro-apoptotic signalling, oxidative stress, cytoplasmic glycolysis and activation of pro-inflammatory cytokines, while upregulating procontractile signalling [57].
To date, there have been no studies investigating oestrogen as a therapeutic strategy to improve RV function in PAH patients, possibly because of safety concerns and balancing potential benefits on the RV with the potential detrimental effects on the pulmonary vasculature.
Pharmacology specificities in PAH
Female responses to PAH treatment
PAH therapies, such as PDE-5i, ERAs and prostanoids generally work by reducing PVR and improving cardiac output and RV stroke volume. Gender-specific differences in therapeutic response have been noted for some PAH treatments.
Endothelin-1 production and the type of endothelin receptor inhibition differ between men and women. With a higher production of endothelin-1 in men [129], vasodilation (as measured on forearm blood flow in middle-aged and older adults) is more pronounced in men with endothelin-A inhibition only. However, this difference is no longer observed for dual endothelin-A and -B receptor inhibition [130]. In clinical studies investigating ERAs, women appear to have a better response than men, with greater increases in 6MWD (+29.7 m, p=0.03) [10].
Regarding the nitric oxide (NO) pathway, men and post-menopausal women seem to benefit more from the effects of PDE-5i with a comparatively larger increase of 33 m (p=0.04) on tadalafil and 45 m on sildenafil (mean of the three studies’ dosage) [11, 12, 131]. Biosynthesis of NO in healthy women is higher than in healthy men [132]. Moreover, it has been shown that, in murine mesenteric arteries, female mice have an additional vasodilation mechanism independent from the NO-soluble guanylate cyclase pathway [133]. These observations could explain why men may respond better to PDE-5i. Studies on riociguat do not show any sex differences with regards to 6MWD [134].
Lastly, in the prostacyclin pathway, epoprostenol studies did not show any difference between men and women [135]; however, there was potentially a lack of statistical power.
Birth control
Pregnancy is associated with a substantial mortality rate in PAH [136]. Therefore, effective contraception is strongly recommended for women of childbearing age with PAH. Many birth control options are available, which include 1) barrier methods (condom, diaphragm or cervical cap); 2) pharmacological therapies (combination of oestrogen and progesterone such as combined pill or progesterone-only medications including implant, injection, vaginal ring, intrauterine device or copper intrauterine device (IUD)); and 3) permanent measures such as tubal ligation or vasectomies for male partners.
There is no consensus regarding the most effective method of birth control in PAH, but a combination of both barrier methods and pharmacological therapies is usually advised [137]. For long-term contraception, a combined hormonal contraceptive pill is usually not preferred because of the increased risk of venous thromboembolic disease [13]. Because of its lower risk of venous thromboembolism [138], a progesterone-only pill is potentially a better option. However, as seen with the combined hormonal contraceptive pill, attention to drug–drug interactions with PAH treatments is mandatory and additional barrier methods are required. This is especially the case with bosentan, which significantly decreases the efficacy of the oral contraception pill by inducing CYP3A4 and CYP2C9 activity [139]. Subcutaneous implants with progesterone are safe and effective, lasting up to 5 years. They are placed under local anaesthesia, but may sometimes cause drug–drug interactions as well [13]. An IUD (copper or progesterone-releasing) appears to be the more effective and appropriate option. However, given the higher risk of bleeding with copper, a progesterone-releasing IUD seems to be the best option for long-term contraception in patients treated for PAH. Close monitoring is important with IUD insertion, as this may induce vasovagal reactions, which can result in cardiovascular collapse in PAH patients.
Anticoagulants in women
Anticoagulation with vitamin K antagonists (VKA) is sometimes used as an adjunctive treatment in PAH, with observational data suggesting better survival, particularly in patients with iPAH [140, 141]. Currently, there are no strong recommendations on anticoagulant therapy in group 1 PAH and no data available for direct oral anticoagulants (DOACs) [142].
A direct pharmacological side-effect of anticoagulant therapy is the increased risk of bleeding, which is more frequent in women [143]. It may also occur with heavy menstrual bleeding or vaginal spotting at any point of the cycle. Menorrhagia seems to be more frequent and important with all DOACs in the general population, although published data mainly investigated rivaroxaban [144]. This potential adverse event should be discussed with the patient at the beginning of the treatment and regularly reassessed, because it may contribute to the cessation of the drug, or to anaemia that could worsen PAH.
Teratogenic agents
If a patient with PAH expresses a desire to become pregnant, she must be referred to a PAH specialist centre in order to optimise treatment. Some therapies must be avoided prior to pregnancy. The ERAs (bosentan, ambrisentan and macitentan) are all teratogenic and they, and riociguat, must not be used during pregnancy [145–148]. Moreover, VKAs are contraindicated during the first trimester because of a high risk of embryopathy, spontaneous abortion and fatal haemorrhage. VKAs can be used during the second and third trimesters, but must be stopped 2–4 weeks (depending on half-life) before delivery [149].
Pregnancy in PAH
Unfortunately, iPAH, hPAH, CHD-PAH and CTD-PAH often affect women of childbearing age [150]. Throughout pregnancy, but especially during the second and third trimesters, haemodynamic changes are poorly tolerated by PAH patients, resulting in a high pregnancy-related mortality rate. Indeed, the mortality of pregnant women with PAH has been shockingly high, though it seems to have improved over time from 30–56% in older series [136] to 5–33% more recently. The advances in PAH treatments as well as multidisciplinary approaches in the management of pregnant patients are probably responsible for the improvement in mortality [13]. Despite better PAH treatments and improved outcomes in pregnant patients, current recommendations still strongly discourage pregnancy [142].
Hormonal changes during pregnancy
During the first 8–10 weeks of gestation, oestrogens and progesterone are produced in the ovary (in the corpus luteum). Following this, the fetoplacental unit becomes competent as an endocrine organ as well [151]. However, the placenta lacks enzymes (CYP17A1, for example) to convert cholesterol into the immediate precursors of oestrogens. Therefore, it uses precursors from either the fetal adrenal or maternal sources to obtain oestradiol (E2), as shown in figure 2 [152]. E2 is the more potent oestrogen, but can be converted into oestrone (E1) through 17β- hydroxysteroidodehydrogenase (HSD)2 and reversed from E1 to E2 through 17β-HSD1. E1 and E2 can be catabolised to oestriol (E3) and oestetrol (E4), which are synthesised in larger amounts during pregnancy and are indicators of fetal liver function [152]. Throughout pregnancy, both oestrogens and progesterone exert cardiovascular effects in order to adequately respond to placental and fetal demands [153]. Progesterone, which is predominantly synthesised in the placenta before entering the maternal circulation, also has immunological effects to suppress the maternal response to fetal antigens, stimulates the endometrial glands to nourish the fetus, and later on acts to inhibit uterine contractions [151, 154].
FIGURE 2.
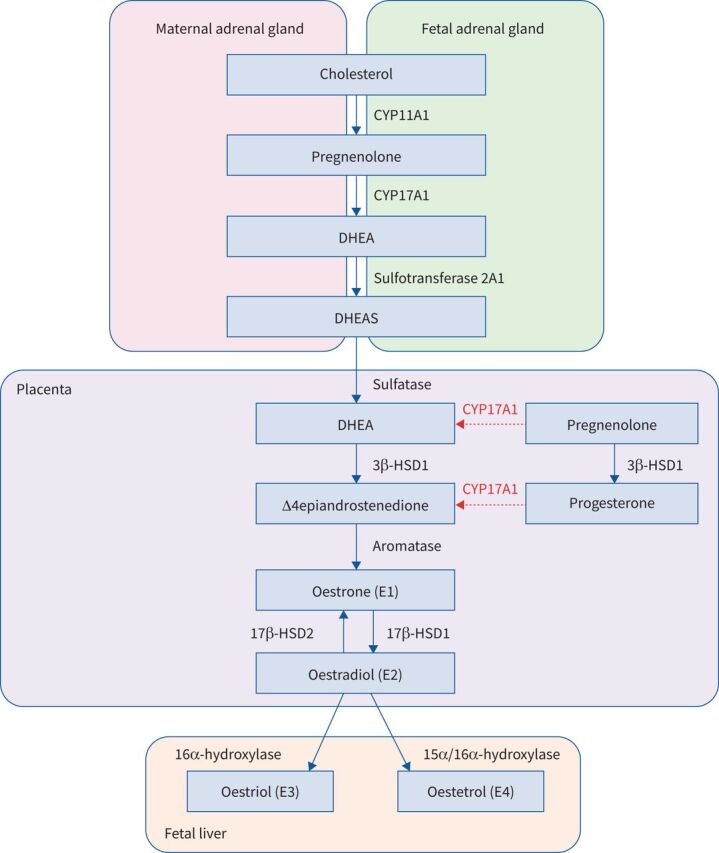
Oestrogen biosynthesis during pregnancy. After 8–10 weeks of gestation, the fetoplacental unit becomes an endocrine organ and is responsible for the growing amount of oestrogens and progesterone during pregnancy. Oestriol and oestetrol are synthesised predominantly during pregnancy and can be metabolised through several pathways, in particular through oestradiol. DHEA: dehydroepiandrosterone; DHEAS: dehydroepiandrosterone sulfate; 3β-HSD1: type 1 3β-hydroxysteroidodehydrogenase; 17β-HSD 1 and 2: types 1 and 2 17β-hydroxysteroidodehydrogenase.
The pregnancy-induced changes in serum concentration of oestrogens and progesterone induce major haemodynamic changes: expansion of total plasma volume, enlargement of the heart ventricles, increase in cardiac output and decrease in pulmonary and vascular resistances, among others [155]. Pregnant patients with PAH cannot cope well with an increase in plasma volume and cardiac output. If cardiac output cannot increase, the PVR may further increase, eventually leading to higher RV afterload, precipitating acute right heart failure [156]. It is unclear whether the impact of sex hormones on vascular remodelling are responsible for PAH worsening during pregnancy or if this is merely a volume-dependent issue. Studies are required to evaluate the haemodynamic role of oestrogens in this specific setting [13].
Management of PAH during pregnancy
Women with an intermediate- or high-risk PAH profile should be counselled carefully and advised on early termination during the first trimester and up to 22 weeks’ gestation. Uterine dilation and surgical evacuation is the safest procedure, but a medical approach using prostaglandins E1 and E2 can also be considered. In patients with a low risk profile or near-normal pulmonary pressure at rest (frequently seen in inhaled NO responders), individual counselling and shared decision-making on the course of pregnancy are recommended [157–159].
After 22 weeks of gestation, complications related to termination are similar to those during childbirth [160]. Thus, management must involve PAH specialists, gynaecologists, anaesthesiologists and intensive care doctors in tertiary hospitals. Appropriate surveillance must be offered with a tailored PAH treatment strategy, regular echocardiographic and/or haemodynamic assessment, routine visits and a well-defined plan for the timing and mode of delivery.
Increased mortality is linked to both severe or poorly controlled PAH and late diagnosis, hence PAH treatment needs to be implemented without delay [136, 157, 161]. While ERAs and soluble guanylate cyclase stimulators (e.g. riociguat) are prohibited during pregnancy due to harmful effects on fetal development, PDE-5i drugs (e.g. sildenafil, tadalafil) have shown beneficial effects and success in pregnant women [162–164]. Intravenous prostacyclin can be used safely and remains a first-line therapy, alone or combined with PDE-5i, for severe PAH with New York Heart Association functional class III or IV dyspnoea [163, 165].
If there are signs of high central venous pressure, the use of diuretics (furosemide) is advised to relieve the RV pre-load; however, spironolactone is not suggested because of its anti-androgenic effect.
Furthermore, if used in iPAH and hPAH patients, VKAs must be withdrawn and changed to low molecular weight heparin to avoid teratogenic effects. Prophylactic heparin is considered in the peripartum period due to the increased risk of venous thromboembolism [13].
Management of labour and delivery
In addition to abortion, the peripartum and post-partum periods bear the maximum risk of complications due to dramatic shifts in blood volume and abrupt decreases in sex hormones [150, 157]. Labour and delivery management requires a plan of action with a qualified multidisciplinary team, anticipation and access to both advanced cardiopulmonary support such as veno-arterial extracorporeal membrane oxygenation (ECMO) and urgent lung transplant capability. Emergency deliveries must be avoided at all cost. When delivery is scheduled between 32 and 34 weeks of gestation, sufficient fetal maturity is present and maternal physiological deterioration is less likely [13, 166].
Adequate control of pain is important during labour and delivery. General anaesthesia is not recommended on a routine basis because of a four times higher mortality risk due to the haemodynamic effects of increasing intrathoracic pressure with mechanical ventilation [150, 157]. A single-shot spinal anaesthetic dose should not be administered, in order to avoid hypotension and its effect on the RV pre-load. Therefore, incremental regional anaesthesia is preferred which consists of a gradual dose of epidural anaesthesia with or without a small single spinal dose [167, 168].
Vaginal delivery is reported to have good outcomes with fewer infections, lower thromboembolic risks and reduced blood loss if labour progresses easily [169]. Nonetheless, unexpected, difficult and painful labour can exacerbate haemodynamic changes, which is why caesarean section is possibly safer [13]. Uterotonic medications such as oxytocin and prostaglandin F2, infused to reduce bleeding after delivery, are contraindicated owing to vasoconstriction and resulting haemodynamic disturbances [170].
Post-partum monitoring is critical, because most maternal deaths occur 2–30 days following delivery. Leading causes of death are acute decompensated PH and thromboembolic events. Recommendations include monitoring patients in an intensive care unit during the 72 h following delivery, monitoring fluid balance and avoiding any physiological stress that could lead to an increase in PVR. Intensifying PAH treatment, use of inotropic agents and/or NO inhalation may be needed for patients who are unstable after delivery. In some dramatic cases, ECMO must be considered, either while waiting for lung transplantation or as bridge to recovery [13] (figure 3).
FIGURE 3.

Proposed algorithm for the management of pregnant women with pulmonary arterial hypertension (PAH). CCB: calcium channel blocker; PH: pulmonary hypertension; i.v.: intravenous; PDE-5i: phosphodiesterase type-5 inhibitor.
In conclusion, it appears necessary that pregnancy remains contraindicated in intermediate- and high-risk PAH patients due to high risk of haemodynamic collapse. Birth control must be implemented and termination is advised before 22 weeks of gestation. Pregnancy management must involve a multidisciplinary team in expert centres and delivery must be well planned with an individualised approach.
Conclusion
Women are more susceptible to developing PAH than men, although paradoxically they have better RV function, and consequently better survival. Dysregulation of oestrogen synthesis and metabolism seems to play a major role in these sex-related differences. Many studies and experimental models have explored the oestrogen pathways in order to establish new targeted treatments. Several clinical trials evaluating therapies that target hormonal pathways are ongoing. The female preponderance in PAH raises the issue of appropriate contraception to avoid pregnancy because of the high mortality rate in PAH patients who become pregnant. Better understanding of the penetrance of PAH in women may translate to a better prognosis and a better quality of life.
Footnotes
Number 1 in the Series “Sex and gender in lung disease” Edited by Jason Weatherald, Marc Humbert and Renata Riha
This article has an editorial commentary: https://doi.org/10.1183/16000617.0217-2021
Provenance: Commissioned article, peer reviewed.
Authors contribution: All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of interest: C. Cheron has nothing to disclose.
Conflict of interest: S.A. McBride has nothing to disclose.
Conflict of interest: F Antigny has nothing to disclose.
Conflict of interest: B. Girerd has nothing to disclose.
Conflict of interest: M. Chouchana has nothing to disclose.
Conflict of interest: M-C. Chaumais has nothing to disclose.
Conflict of interest: X. Jaïs reports grants, personal fees and non-financial support from Actelion/Janssen, personal fees and non-financial support from MSD, and grants from Bayer, outside the submitted work.
Conflict of interest: L. Bertoletti reports grants, personal fees and non-financial support from Actelion, MSD, Bayer, BMS/Pfizer and Léo-Pharma, outside the submitted work.
Conflict of interest: O. Sitbon reports grants, personal fees and non-financial support from Actelion; personal fees from Acceleron, Ferrer and Gossamer Bio; grants and personal fees from Bayer and MSD; and grants from GlaxoSmithKline, outside the submitted work.
Conflict of interest: J. Weatherald reports grants, personal fees and non-financial support from Janssen Inc. and Actelion, personal fees and non-financial support from Bayer, and personal fees from Novartis, outside the submitted work.
Conflict of interest: M. Humbert reports grants, personal fees and non-financial support from GlaxoSmithKline; personal fees from AstraZeneca, Novartis, Roche, Sanofi, Teva and Merck; grants and personal fees from Acceleron, Actelion and Bayer, outside the submitted work.
Conflict of interest: D. Montani reports grants and personal fees from Actelion and Bayer; personal fees from GSK, Pfizer, Chiesi and Boerhinger; grants, personal fees and non-financial support from MSD; and non-financial support from Acceleron, outside the submitted work.
References
- 1.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913. doi: 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351: 1425–1436. doi: 10.1056/NEJMra040291 [DOI] [PubMed] [Google Scholar]
- 3.Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987; 107: 216–223. doi: 10.7326/0003-4819-107-2-216 [DOI] [PubMed] [Google Scholar]
- 4.Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006; 173: 1023–1030. doi: 10.1164/rccm.200510-1668OC [DOI] [PubMed] [Google Scholar]
- 5.Chung L, Liu J, Parsons L, et al. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest 2010; 138: 1383–1394. doi: 10.1378/chest.10-0260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weatherald J, Boucly A, Chemla D, et al. Prognostic value of follow-up hemodynamic variables after initial management in pulmonary arterial hypertension. Circulation 2018; 137: 693–704. doi: 10.1161/CIRCULATIONAHA.117.029254 [DOI] [PubMed] [Google Scholar]
- 7.Jacobs W, van de Veerdonk MC, Trip P, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014; 145: 1230–1236. doi: 10.1378/chest.13-1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hester J, Ventetuolo C, Lahm T. Sex, gender, and sex hormones in pulmonary hypertension and right ventricular failure. Compr Physiol 2019; 10: 125–170. doi: 10.1002/cphy.c190011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin YN, Pabelick CM. Sex differences in the pulmonary circulation: implications for pulmonary hypertension. Am J Physiol Heart Circ Physiol 2014; 306: H1253–H1264. doi: 10.1152/ajpheart.00857.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabler NB, French B, Strom BL, et al. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest 2012; 141: 20–26. doi: 10.1378/chest.11-0404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathai SC, Hassoun PM, Puhan MA, et al. Sex differences in response to tadalafil in pulmonary arterial hypertension. Chest 2015; 147: 188–197. doi: 10.1378/chest.14-0263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rusiecki J, Rao Y, Cleveland J, et al. Sex and menopause differences in response to tadalafil: 6-minute walk distance and time to clinical worsening. Pulm Circ 2015; 5: 701–706. doi: 10.1086/683829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemnes AR, Kiely DG, Cockrill BA, et al. Statement on pregnancy in pulmonary hypertension from the Pulmonary Vascular Research Institute. Pulm Circ 2015; 5: 435–465. doi: 10.1086/682230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lau EMT, Giannoulatou E, Celermajer DS, et al. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol 2017; 14: 603–614. doi: 10.1038/nrcardio.2017.84 [DOI] [PubMed] [Google Scholar]
- 15.McGoon MD, Benza RL, Escribano-Subias P, et al. Pulmonary arterial hypertension: epidemiology and registries. J Am Coll Cardiol 2013; 62: D51–D59. doi: 10.1016/j.jacc.2013.10.023 [DOI] [PubMed] [Google Scholar]
- 16.Sitbon O, Sattler C, Bertoletti L, et al. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur Respir J 2016; 47: 1727–1736. doi: 10.1183/13993003.02043-2015 [DOI] [PubMed] [Google Scholar]
- 17.Hoeper MM, Simon R, Gibbs J. The changing landscape of pulmonary arterial hypertension and implications for patient care. Eur Respir Rev 2014; 23: 450–457. doi: 10.1183/09059180.00007814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoeper MM, Huscher D, Ghofrani HA, et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol 2013; 168: 871–880. doi: 10.1016/j.ijcard.2012.10.026 [DOI] [PubMed] [Google Scholar]
- 19.Yuan P, Wu W-H, Gao L, et al. Oestradiol ameliorates monocrotaline pulmonary hypertension via NO, prostacyclin and endothelin-1 pathways. Eur Respir J 2013; 41: 1116–1125. doi: 10.1183/09031936.00044112 [DOI] [PubMed] [Google Scholar]
- 20.Valverde AB, Soares JM, Viana KP, et al. Pulmonary arterial hypertension in Latin America: epidemiological data from local studies. BMC Pulm Med 2018; 18: 106. doi: 10.1186/s12890-018-0667-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Wang K. The changing landscape of pulmonary arterial hypertension in 21st century. Acta Cardiol Sin 2017; 33: 510–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaid HM, Camacho X, Granton JT, et al. The characteristics of treated pulmonary arterial hypertension patients in Ontario. Can Respir J 2016; 2016: 6279250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kjellström B, Nisell M, Kylhammar D, et al. Sex-specific differences and survival in patients with idiopathic pulmonary arterial hypertension 2008–2016. ERJ Open Res 2019; 5: 00075-2019. doi: 10.1183/23120541.00075-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012; 186: 790–796. doi: 10.1164/rccm.201203-0383OC [DOI] [PubMed] [Google Scholar]
- 25.Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010; 122: 156–163. doi: 10.1161/CIRCULATIONAHA.109.911818 [DOI] [PubMed] [Google Scholar]
- 26.Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010; 122: 164–172. doi: 10.1161/CIRCULATIONAHA.109.898122 [DOI] [PubMed] [Google Scholar]
- 27.Hachulla E, Launay D, Mouthon L, et al. Is pulmonary arterial hypertension really a late complication of systemic sclerosis? Chest 2009; 136: 1211–1219. doi: 10.1378/chest.08-3042 [DOI] [PubMed] [Google Scholar]
- 28.Sundaram SM, Chung L. An update on systemic sclerosis-associated pulmonary arterial hypertension: a review of the current literature. Curr Rheumatol Rep 2018; 20: 10. doi: 10.1007/s11926-018-0709-5 [DOI] [PubMed] [Google Scholar]
- 29.Kawut SM, Krowka MJ, Trotter JF, et al. Clinical risk factors for portopulmonary hypertension. Hepatology 2008; 48: 196–203. doi: 10.1002/hep.22275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Naamani N, Krowka MJ, Forde KA, et al. Estrogen signaling and portopulmonary hypertension: the Pulmonary Vascular Complications of Liver Disease Study (PVCLD2). Hepatology 2021; 73: 726–737. doi: 10.1002/hep.31314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vrtačnik P, Ostanek B, Mencej-Bedrač S, et al. The many faces of estrogen signaling. Biochem Med 2014; 24: 329–342. doi: 10.11613/BM.2014.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mair KM, Wright AF, Duggan N, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med 2014; 190: 456–467. doi: 10.1164/rccm.201403-0483OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics 2006; 7: 497–508. doi: 10.2174/138920206779315737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res 2016; 118: 994–1007. doi: 10.1161/CIRCRESAHA.115.305376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aryan L, Younessi D, Zargari M, et al. The role of estrogen receptors in cardiovascular disease. Int J Mol Sci 2020; 21: 4314. doi: 10.3390/ijms21124314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lahm T, Crisostomo PR, Markel TA, et al. Selective estrogen receptor-α and estrogen receptor-β agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. Am J Physiol Regul Integr Comp Physiol 2008; 295: R1486–R1493. doi: 10.1152/ajpregu.90667.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med 2006; 354: 270–282. doi: 10.1056/NEJMra050776 [DOI] [PubMed] [Google Scholar]
- 38.Guignabert C, Bailly S, Humbert M. Restoring BMPRII functions in pulmonary arterial hypertension: opportunities, challenges and limitations. Expert Opin Ther Targets 2017; 21: 181–190. doi: 10.1080/14728222.2017.1275567 [DOI] [PubMed] [Google Scholar]
- 39.Evans JDW, Girerd B, Montani D, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med 2016; 4: 129–137. doi: 10.1016/S2213-2600(15)00544-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larkin EK, Newman JH, Austin ED, et al. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2012; 186: 892–896. doi: 10.1164/rccm.201205-0886OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montani D, Girerd B, Jaïs X, et al. Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation. Eur Respir J 2020; 58: 2004229. doi: 10.1183/13993003.04229-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans JDW, Girerd B, Montani D, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med 2016; 4: 129–137. doi: 10.1016/S2213-2600(15)00544-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mair KM, Yang XD, Long L, et al. Sex affects bone morphogenetic protein type II receptor signaling in pulmonary artery smooth muscle cells. Am J Respir Crit Care Med 2015; 191: 693–703. doi: 10.1164/rccm.201410-1802OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Austin ED, Hamid R, Hemnes AR, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ 2012; 3: 6. doi: 10.1186/2042-6410-3-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.West J, Cogan J, Geraci M, et al. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med Genomics 2008; 1: 45. doi: 10.1186/1755-8794-1-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cirulis MM, Dodson MW, Brown LM, et al. At the X-roads of sex and genetics in pulmonary arterial hypertension. Genes 2020; 11: 1371. doi: 10.3390/genes11111371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kozu K, Sugimura K, Aoki T, et al. Sex differences in hemodynamic responses and long-term survival to optimal medical therapy in patients with pulmonary arterial hypertension. Heart Vessels 2018; 33: 939–947. doi: 10.1007/s00380-018-1140-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dempsie Y, MacLean MR. The influence of gender on the development of pulmonary arterial hypertension. Exp Physiol 2013; 98: 1257–1261. doi: 10.1113/expphysiol.2012.069120 [DOI] [PubMed] [Google Scholar]
- 49.Tofovic SP. Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol 2010; 56: 696–708. doi: 10.1097/FJC.0b013e3181f9ea8d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rabinovitch M, Gamble WJ, Miettinen OS, et al. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol 1981; 240: H62–H72. [DOI] [PubMed] [Google Scholar]
- 51.McMurtry IF, Frith CH, Will DH. Cardiopulmonary responses of male and female swine to simulated high altitude. J Appl Physiol 1973; 35: 459–462. doi: 10.1152/jappl.1973.35.4.459 [DOI] [PubMed] [Google Scholar]
- 52.Lahm T, Albrecht M, Fisher AJ, et al. 17β-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med 2012; 185: 965–980. doi: 10.1164/rccm.201107-1293OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tofovic SP, Zhang X, Jackson EK, et al. 2-Methoxyestradiol mediates the protective effects of estradiol in monocrotaline-induced pulmonary hypertension. Vascul Pharmacol 2006; 45: 358–367. doi: 10.1016/j.vph.2006.05.007 [DOI] [PubMed] [Google Scholar]
- 54.Ahn BH, Park HK, Cho HG, et al. Estrogen and enalapril attenuate the development of right ventricular hypertrophy induced by monocrotaline in ovariectomized rats. J Korean Med Sci 2003; 18: 641–648. doi: 10.3346/jkms.2003.18.5.641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rafikova O, Rafikov R, Meadows ML, et al. The sexual dimorphism associated with pulmonary hypertension corresponds to a fibrotic phenotype. Pulm Circ 2015; 5: 184–197. doi: 10.1086/679724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tofovic SP, Rafikova O, Champion H, et al. Estrogens exacerbate development of occlusive pulmonary arterial hypertension and formation of plexiform lesions. Am J Respir Crit Care Med 2012; 185: A6803. [Google Scholar]
- 57.Frump AL, Goss KN, Vayl A, et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol 2015; 308: L873–L890. doi: 10.1152/ajplung.00006.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Philip JL, Tabima DM, Wolf GD, et al. Exogenous estrogen preserves distal pulmonary arterial mechanics and prevents pulmonary hypertension in rats. Am J Respir Crit Care Med 2020; 201: 371–374. doi: 10.1164/rccm.201906-1217LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu A, Philip J, Vinnakota KC, et al. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol Rep 2017; 5: e13157. doi: 10.14814/phy2.13157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perros F, Günther S, Ranchoux B, et al. Mitomycin-induced pulmonary veno-occlusive disease: evidence from human disease and animal models. Circulation 2015; 132: 834–847. doi: 10.1161/CIRCULATIONAHA.115.014207 [DOI] [PubMed] [Google Scholar]
- 61.Ranchoux B, Günther S, Quarck R, et al. Chemotherapy-induced pulmonary hypertension: role of alkylating agents. Am J Pathol 2015; 185: 356–371. doi: 10.1016/j.ajpath.2014.10.021 [DOI] [PubMed] [Google Scholar]
- 62.White K, Loughlin L, Maqbool Z, et al. Serotonin transporter, sex, and hypoxia: microarray analysis in the pulmonary arteries of mice identifies genes with relevance to human PAH. Physiol Genomics 2011; 43: 417–437. doi: 10.1152/physiolgenomics.00249.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dempsie Y, Nilsen M, White K, et al. Development of pulmonary arterial hypertension in mice over-expressing S100A4/Mts1 is specific to females. Respir Res 2011; 12: 159. doi: 10.1186/1465-9921-12-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dempsie Y, MacRitchie NA, White K, et al. Dexfenfluramine and the oestrogen-metabolizing enzyme CYP1B1 in the development of pulmonary arterial hypertension. Cardiovasc Res 2013; 99: 24–34. doi: 10.1093/cvr/cvt064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White K, Dempsie Y, Nilsen M, et al. The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 2011; 90: 373–382. doi: 10.1093/cvr/cvq408 [DOI] [PubMed] [Google Scholar]
- 66.Mair KM, Harvey KY, Henry AD, et al. Obesity alters oestrogen metabolism and contributes to pulmonary arterial hypertension. Eur Respir J 2019; 53: 1801524. doi: 10.1183/13993003.01524-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Niihori M, Eccles CA, Kurdyukov S, et al. Rats with a human mutation of NFU1 develop pulmonary hypertension. Am J Respir Cell Mol Biol 2020; 62: 231–242. doi: 10.1165/rcmb.2019-0065OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tamosiuniene R, Manouvakhova O, Mesange P, et al. Dominant role for regulatory T cells in protecting females against pulmonary hypertension. Circ Res 2018; 122: 1689–1702. doi: 10.1161/CIRCRESAHA.117.312058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manaud G, Nossent EJ, Lambert M, et al. Comparison of human and experimental pulmonary veno-occlusive disease. Am J Respir Cell Mol Biol 2020; 63: 118–131. doi: 10.1165/rcmb.2019-0015OC [DOI] [PubMed] [Google Scholar]
- 70.Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 1996; 335: 609–616. doi: 10.1056/NEJM199608293350901 [DOI] [PubMed] [Google Scholar]
- 71.Eddahibi S, Guignabert C, Barlier-Mur A-M, et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation 2006; 113: 1857–1864. doi: 10.1161/CIRCULATIONAHA.105.591321 [DOI] [PubMed] [Google Scholar]
- 72.Hood KY, Mair KM, Harvey AP, et al. Serotonin signaling through the 5-HT1B receptor and NADPH oxidase 1 in pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 2017; 37: 1361–1370. doi: 10.1161/ATVBAHA.116.308929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wright AF, Ewart M-A, Mair K, et al. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc Res 2015; 106: 206–216. doi: 10.1093/cvr/cvv106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Austin ED, Hamid R, Hemnes AR, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ 2012; 3: 6. doi: 10.1186/2042-6410-3-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frump AL, Selej M, Wood JA, et al. Hypoxia upregulates estrogen receptor β in pulmonary artery endothelial cells in a HIF-1α-dependent manner. Am J Respir Cell Mol Biol 2018; 59: 114–126. doi: 10.1165/rcmb.2017-0167OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Umar S, Iorga A, Matori H, et al. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med 2011; 184: 715–723. doi: 10.1164/rccm.201101-0078OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alencar AK, Montes GC, Montagnoli T, et al. Activation of GPER ameliorates experimental pulmonary hypertension in male rats. Eur J Pharm Sci 2017; 97: 208–217. doi: 10.1016/j.ejps.2016.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alencar AKN, Montes GC, Costa DG, et al. Cardioprotection induced by activation of GPER in ovariectomized rats with pulmonary hypertension. J Gerontol A Biol Sci Med Sci 2018; 73: 1158–1166. doi: 10.1093/gerona/gly068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.White K, Johansen AK, Nilsen M, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation 2012; 126: 1087–1098. doi: 10.1161/CIRCULATIONAHA.111.062927 [DOI] [PubMed] [Google Scholar]
- 80.Austin ED, Cogan JD, West JD, et al. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J 2009; 34: 1093–1099. doi: 10.1183/09031936.00010409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johansen AKZ, Dean A, Morecroft I, et al. The serotonin transporter promotes a pathological estrogen metabolic pathway in pulmonary hypertension via cytochrome P450 1B1. Pulm Circ 2016; 6: 82–92. doi: 10.1086/685023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dean A, Gregorc T, Docherty CK, et al. Role of the aryl hydrocarbon receptor in Sugen 5416-induced experimental pulmonary hypertension. Am J Respir Cell Mol Biol 2018; 58: 320–330. doi: 10.1165/rcmb.2017-0260OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vorrink SU, Severson PL, Kulak MV, et al. Hypoxia perturbs aryl hydrocarbon receptor signaling and CYP1A1 expression induced by PCB 126 in human skin and liver-derived cell lines. Toxicol Appl Pharmacol 2014; 274: 408–416. doi: 10.1016/j.taap.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Napoli C, Benincasa G, Loscalzo J. Epigenetic inheritance underlying pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 2019; 39: 653–664. doi: 10.1161/ATVBAHA.118.312262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fessel JP, Chen X, Frump A, et al. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm Circ 2013; 3: 564–577. doi: 10.1086/674312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen X, Talati M, Fessel JP, et al. Estrogen metabolite 16α-hydroxyestrone exacerbates bone morphogenetic protein receptor type II-associated pulmonary arterial hypertension through microRNA-29-mediated modulation of cellular metabolism. Circulation 2016; 133: 82–97. doi: 10.1161/CIRCULATIONAHA.115.016133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tofovic SP, Zhang X, Jackson EK, et al. 2-Methoxyestradiol attenuates bleomycin-induced pulmonary hypertension and fibrosis in estrogen-deficient rats. Vascul Pharmacol 2009; 51: 190–197. doi: 10.1016/j.vph.2009.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tofovic SP, Jones T, Petrusevska G. Dose-dependent therapeutic effects of 2-methoxyestradiol on monocrotaline-induced pulmonary hypertension and vascular remodelling. Prilozi 2010; 31: 279–295. [PubMed] [Google Scholar]
- 89.Tofovic SP, Salah EM, Mady HH, et al. Estradiol metabolites attenuate monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol 2005; 46: 430–437. doi: 10.1097/01.fjc.0000175878.32920.17 [DOI] [PubMed] [Google Scholar]
- 90.Tofovic SP, Zhang X, Zhu H, et al. 2-Ethoxyestradiol is antimitogenic and attenuates monocrotaline-induced pulmonary hypertension and vascular remodeling. Vascul Pharmacol 2008; 48: 174–183. doi: 10.1016/j.vph.2008.02.001 [DOI] [PubMed] [Google Scholar]
- 91.Tofovic SP, Jones TJ, Bilan VP, et al. Synergistic therapeutic effects of 2-methoxyestradiol with either sildenafil or bosentan on amelioration of monocrotaline-induced pulmonary hypertension and vascular remodeling. J Cardiovasc Pharmacol 2010; 56: 475–483. doi: 10.1097/FJC.0b013e3181f215e7 [DOI] [PubMed] [Google Scholar]
- 92.Dubey RK, Jackson EK, Keller PJ, et al. Estradiol metabolites inhibit endothelin synthesis by an estrogen receptor-independent mechanism. Hypertension 2001; 37: 640–644. [DOI] [PubMed] [Google Scholar]
- 93.Docherty CK, Nilsen M, MacLean MR. Influence of 2-methoxyestradiol and sex on hypoxia-induced pulmonary hypertension and hypoxia-inducible factor-1-α. J Am Heart Assoc 2019; 8: e011628. doi: 10.1161/JAHA.118.011628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Denver N, Homer NZM, Andrew R, et al. Estrogen metabolites in a small cohort of patients with idiopathic pulmonary arterial hypertension. Pulm Circ 2020; 10: 2045894020908783. doi: 10.1177/2045894020908783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ventetuolo CE, Baird GL, Barr RG, et al. Higher estradiol and lower dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med 2016; 193: 1168–1175. doi: 10.1164/rccm.201509-1785OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baird GL, Archer-Chicko C, Barr RG, et al. Lower DHEA-S levels predict disease and worse outcomes in post-menopausal women with idiopathic, connective tissue disease- and congenital heart disease-associated pulmonary arterial hypertension. Eur Respir J 2018; 51: 1800467. doi: 10.1183/13993003.00467-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baird GL, Walsh T, Aliotta J, et al. Insights from the menstrual cycle in pulmonary arterial hypertension. Ann Am Thorac Soc 2021; 18: 218–228. doi: 10.1513/AnnalsATS.202006-671OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roberts KE, Fallon MB, Krowka MJ, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med 2009; 179: 835–842. doi: 10.1164/rccm.200809-1472OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mair KM, Wright AF, Duggan N, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med 2014; 190: 456–467. doi: 10.1164/rccm.201403-0483OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kawut SM, Archer-Chicko CL, DeMichele A, et al. Anastrozole in pulmonary arterial hypertension. A randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med 2017; 195: 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen X, Austin ED, Talati M, et al. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur Respir J 2017; 50: 1602337. doi: 10.1183/13993003.02337-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kawut SM, Pinder D, Al-Naamani N, et al. Fulvestrant for the treatment of pulmonary arterial hypertension. Ann Am Thorac Soc 2019; 16: 1456–1459. doi: 10.1513/AnnalsATS.201904-328RL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tofovic SP, Jackson EK. Complexities of oestradiol pharmacology in pulmonary arterial hypertension. Eur Respir J 2013; 41: 1465–1466. doi: 10.1183/09031936.00164912 [DOI] [PubMed] [Google Scholar]
- 104.Vonk Noordegraaf A, Chin KM, Haddad F, et al. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J 2019; 53: 1801900. doi: 10.1183/13993003.01900-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vonk Noordegraaf A, Westerhof BE, Westerhof N. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol 2017; 69: 236–243. doi: 10.1016/j.jacc.2016.10.047 [DOI] [PubMed] [Google Scholar]
- 106.van de Veerdonk MC, Marcus JT, Westerhof N, et al. Signs of right ventricular deterioration in clinically stable patients with pulmonary arterial hypertension. Chest 2015; 147: 1063–1071. doi: 10.1378/chest.14-0701 [DOI] [PubMed] [Google Scholar]
- 107.Tonelli AR, Arelli V, Minai OA, et al. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013; 188: 365–369. doi: 10.1164/rccm.201209-1640OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tello K, Dalmer A, Axmann J, et al. Reserve of right ventricular-arterial coupling in the setting of chronic overload. Circ Heart Fail 2019; 12: e005512. doi: 10.1161/CIRCHEARTFAILURE.118.005512 [DOI] [PubMed] [Google Scholar]
- 109.Richter MJ, Peters D, Ghofrani HA, et al. Evaluation and prognostic relevance of right ventricular-arterial coupling in pulmonary hypertension. Am J Respir Crit Care Med 2020; 201: 116–119. doi: 10.1164/rccm.201906-1195LE [DOI] [PubMed] [Google Scholar]
- 110.Labazi H, Axelsen JB, Hillyard D, et al. Sex-dependent changes in right ventricular gene expression in response to pressure overload in a rat model of pulmonary trunk banding. Biomedicines 2020; 8: 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kovacs G, Berghold A, Scheidl S, et al. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009; 34: 888–894. doi: 10.1183/09031936.00145608 [DOI] [PubMed] [Google Scholar]
- 112.Kawut SM, Lima JAC, Barr RG, et al. Sex and race differences in right ventricular structure and function: the multi-ethnic study of atherosclerosis-right ventricle study. Circulation 2011; 123: 2542–2551. doi: 10.1161/CIRCULATIONAHA.110.985515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ventetuolo CE, Mitra N, Wan F, et al. Oestradiol metabolism and androgen receptor genotypes are associated with right ventricular function. Eur Respir J 2016; 47: 553–563. doi: 10.1183/13993003.01083-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ventetuolo CE, Ouyang P, Bluemke DA, et al. Sex hormones are associated with right ventricular structure and function: the MESA-right ventricle study. Am J Respir Crit Care Med 2011; 183: 659–667. doi: 10.1164/rccm.201007-1027OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tello K, Richter MJ, Yogeswaran A, et al. Sex differences in right ventricular-pulmonary arterial coupling in pulmonary arterial hypertension. Am J Respir Crit Care Med 2020; 202: 1042–1046. doi: 10.1164/rccm.202003-0807LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Swift AJ, Rajaram S, Campbell MJ, et al. Prognostic value of cardiovascular magnetic resonance imaging measurements corrected for age and sex in idiopathic pulmonary arterial hypertension. Circ Cardiovasc Imaging 2014; 7: 100–106. doi: 10.1161/CIRCIMAGING.113.000338 [DOI] [PubMed] [Google Scholar]
- 117.Jacobs W, van de Veerdonk MC, Trip P, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014; 145: 1230–1236. doi: 10.1378/chest.13-1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kawut SM, Al-Naamani N, Agerstrand C, et al. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest 2009; 135: 752–759. doi: 10.1378/chest.08-1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Swift AJ, Capener D, Hammerton C, et al. Right ventricular sex differences in patients with idiopathic pulmonary arterial hypertension characterised by magnetic resonance imaging: pair-matched case controlled study. PLoS One 2015; 10: e0127415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ventetuolo CE, Hess E, Austin ED, et al. Sex-based differences in veterans with pulmonary hypertension: results from the Veterans Affairs-Clinical Assessment Reporting and Tracking Database. PLoS One 2017; 12: e0187734. doi: 10.1371/journal.pone.0187734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ventetuolo CE, Praestgaard A, Palevsky HI, et al. Sex and haemodynamics in pulmonary arterial hypertension. Eur Respir J 2014; 43: 523–530. doi: 10.1183/09031936.00027613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weatherald J, Boucly A, Launay D, et al. Haemodynamics and serial risk assessment in systemic sclerosis associated pulmonary arterial hypertension. Eur Respir J 2018; 52: 1800678. doi: 10.1183/13993003.00678-2018 [DOI] [PubMed] [Google Scholar]
- 123.van Wolferen SA, Marcus JT, Boonstra A, et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007; 28: 1250–1257. doi: 10.1093/eurheartj/ehl477 [DOI] [PubMed] [Google Scholar]
- 124.Hemnes AR, Maynard KB, Champion HC, et al. Testosterone negatively regulates right ventricular load stress responses in mice. Pulm Circ 2012; 2: 352–358. doi: 10.4103/2045-8932.101647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alzoubi A, Toba M, Abe K, et al. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2013; 304: H1708–H1718. doi: 10.1152/ajpheart.00746.2012 [DOI] [PubMed] [Google Scholar]
- 126.Dumas de La Roque E, Bellance N, Rossignol R, et al. Dehydroepiandrosterone reverses chronic hypoxia/reoxygenation-induced right ventricular dysfunction in rats. Eur Respir J 2012; 40: 1420–1429. doi: 10.1183/09031936.00011511 [DOI] [PubMed] [Google Scholar]
- 127.Baird GL, Archer-Chicko C, Barr RG, et al. Lower DHEA-S levels predict disease and worse outcomes in post-menopausal women with idiopathic, connective tissue disease- and congenital heart disease-associated pulmonary arterial hypertension. Eur Respir J 2018; 51: 1800467. doi: 10.1183/13993003.00467-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu A, Schreier D, Tian L, et al. Direct and indirect protection of right ventricular function by estrogen in an experimental model of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2014; 307: H273–H283. doi: 10.1152/ajpheart.00758.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Miyauchi T, Yanagisawa M, Iida K, et al. Age- and sex-related variation of plasma endothelin-1 concentration in normal and hypertensive subjects. Am Heart J 1992; 123: 1092–1093. doi: 10.1016/0002-8703(92)90734-D [DOI] [PubMed] [Google Scholar]
- 130.Stauffer BL, Westby CM, Greiner JJ, et al. Sex differences in endothelin-1-mediated vasoconstrictor tone in middle-aged and older adults. Am J Physiol Regul Integr Comp Physiol 2010; 298: R261–R265. doi: 10.1152/ajpregu.00626.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005; 353: 2148–2157. doi: 10.1056/NEJMoa050010 [DOI] [PubMed] [Google Scholar]
- 132.Forte P, Kneale BJ, Milne E, et al. Evidence for a difference in nitric oxide biosynthesis between healthy women and men. Hypertension 1998; 32: 730–734. [DOI] [PubMed] [Google Scholar]
- 133.Chan MV, Bubb KJ, Noyce A, et al. Distinct endothelial pathways underlie sexual dimorphism in vascular auto-regulation. Br J Pharmacol 2012; 167: 805–817. doi: 10.1111/j.1476-5381.2012.02012.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ghofrani HA, D'Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013; 369: 319–329. 10.1056/NEJMoa1209657. [DOI] [PubMed] [Google Scholar]
- 135.Frantz RP, Schilz RJ, Chakinala MM, et al. Hospitalization and survival in patients using epoprostenol for injection in the PROSPECT observational study. Chest 2015; 147: 484–494. 10.1378/chest.14-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weiss BM, Zemp L, Seifert B, et al. Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol 1998; 31: 1650–1657. doi: 10.1016/S0735-1097(98)00162-4 [DOI] [PubMed] [Google Scholar]
- 137.Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30: 2493–2537. doi: 10.1093/eurheartj/ehp297 [DOI] [PubMed] [Google Scholar]
- 138.Lidegaard Ø, Nielsen LH, Skovlund CW, et al. Risk of venous thromboembolism from use of oral contraceptives containing different progestogens and oestrogen doses: Danish cohort study, 2001–2009. BMJ 2011; 343: d6423. doi: 10.1136/bmj.d6423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.van Giersbergen PLM, Halabi A, Dingemanse J. Pharmacokinetic interaction between bosentan and the oral contraceptives norethisterone and ethinyl estradiol. Int J Clin Pharmacol Ther 2006; 44: 113–118. doi: 10.5414/CPP44113 [DOI] [PubMed] [Google Scholar]
- 140.Khan MS, Usman MS, Siddiqi TJ, et al. Is anticoagulation beneficial in pulmonary arterial hypertension? Circ Cardiovasc Qual Outcomes 2018; 11: e004757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bertoletti L, Mismetti V, Giannakoulas G. Use of anticoagulants in patients with pulmonary hypertension. Hamostaseologie 2020; 40: 348–355. doi: 10.1055/a-1171-3995 [DOI] [PubMed] [Google Scholar]
- 142.Galiè N, Humbert M, Vachiery J-L, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37: 67–119. doi: 10.1093/eurheartj/ehv317 [DOI] [PubMed] [Google Scholar]
- 143.Blanco-Molina A, Enea I, Gadelha T, et al. Sex differences in patients receiving anticoagulant therapy for venous thromboembolism. Medicine 2014; 93: 309–317. doi: 10.1097/MD.0000000000000114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ferreira M, Barsam S, Patel JP, et al. Heavy menstrual bleeding on rivaroxaban. Br J Haematol 2016; 173: 314–315. doi: 10.1111/bjh.13583 [DOI] [PubMed] [Google Scholar]
- 145.European Medicines Agency . Adempas. https://www.ema.europa.eu/en/medicines/human/EPAR/adempas Date last updated: 29 April 2019.
- 146.European Medicines Agency . Volibris. https://www.ema.europa.eu/en/medicines/human/EPAR/volibris Date last updated: 16 July 2019.
- 147.European Medicines Agency . Tracleer. https://www.ema.europa.eu/en/medicines/human/EPAR/tracleer Date last updated: 16 October 2020.
- 148.European Medicines Agency . Opsumit. https://www.ema.europa.eu/en/medicines/human/EPAR/opsumit Date last updated: 9 April 2021.
- 149.Schaefer C, Hannemann D, Meister R, et al. Vitamin K antagonists and pregnancy outcome. A multi-centre prospective study. Thromb Haemost 2006; 95: 949–957. doi: 10.1160/TH06-02-0108 [DOI] [PubMed] [Google Scholar]
- 150.Bédard E, Dimopoulos K, Gatzoulis MA. Has there been any progress made on pregnancy outcomes among women with pulmonary arterial hypertension? Eur Heart J 2009; 30: 256–265. doi: 10.1093/eurheartj/ehn597 [DOI] [PubMed] [Google Scholar]
- 151.Kumar P, Magon N. Hormones in pregnancy. Niger Med J 2012; 53: 179–183. doi: 10.4103/0300-1652.107549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Berkane N, Liere P, Oudinet J-P, et al. From pregnancy to preeclampsia: a key role for estrogens. Endocr Rev 2017; 38: 123–144. doi: 10.1210/er.2016-1065 [DOI] [PubMed] [Google Scholar]
- 153.Kodogo V, Azibani F, Sliwa K. Role of pregnancy hormones and hormonal interaction on the maternal cardiovascular system: a literature review. Clin Res Cardiol 2019; 108: 831–846. doi: 10.1007/s00392-019-01441-x [DOI] [PubMed] [Google Scholar]
- 154.Tuckey RC. Progesterone synthesis by the human placenta. Placenta 2005; 26: 273–281. doi: 10.1016/j.placenta.2004.06.012 [DOI] [PubMed] [Google Scholar]
- 155.Hill CC, Pickinpaugh J. Physiologic changes in pregnancy. Surg Clin North Am 2008; 88: 391–401. doi: 10.1016/j.suc.2007.12.005 [DOI] [PubMed] [Google Scholar]
- 156.Banerjee D, Ventetuolo CE. Pulmonary hypertension in pregnancy. Semin Respir Crit Care Med 2017; 38: 148–159. doi: 10.1055/s-0037-1602242 [DOI] [PubMed] [Google Scholar]
- 157.Jaïs X, Olsson KM, Barbera JA, et al. Pregnancy outcomes in pulmonary arterial hypertension in the modern management era. Eur Respir J 2012; 40: 881–885. doi: 10.1183/09031936.00141211 [DOI] [PubMed] [Google Scholar]
- 158.Bonnin M, Mercier FJ, Sitbon O, et al. Severe pulmonary hypertension during pregnancy: mode of delivery and anaesthetic management of 15 consecutive cases. Anesthesiology 2005; 102: 1133–1137. doi: 10.1097/00000542-200506000-00012 [DOI] [PubMed] [Google Scholar]
- 159.Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111: 3105–3111. doi: 10.1161/CIRCULATIONAHA.104.488486 [DOI] [PubMed] [Google Scholar]
- 160.Olsson KM, Jais X. Birth control and pregnancy management in pulmonary hypertension. Semin Respir Crit Care Med 2013; 34: 681–688. doi: 10.1055/s-0033-1355438 [DOI] [PubMed] [Google Scholar]
- 161.Katsuragi S, Yamanaka K, Neki R, et al. Maternal outcome in pregnancy complicated with pulmonary arterial hypertension. Circ J 2012; 76: 2249–2254. doi: 10.1253/circj.CJ-12-0235 [DOI] [PubMed] [Google Scholar]
- 162.Kamp JC, von Kaisenberg C, Greve S, et al. Pregnancy in pulmonary arterial hypertension: mid-term outcomes of mothers and offspring. J Heart Lung Transplant 2020; 40: 229–233. doi: 10.1016/j.healun.2020.12.002 [DOI] [PubMed] [Google Scholar]
- 163.Huang S, DeSantis ERH. Treatment of pulmonary arterial hypertension in pregnancy. Am J Health Syst Pharm 2007; 64: 1922–1926. doi: 10.2146/ajhp060391 [DOI] [PubMed] [Google Scholar]
- 164.Goland S, Tsai F, Habib M, et al. Favorable outcome of pregnancy with an elective use of epoprostenol and sildenafil in women with severe pulmonary hypertension. Cardiology 2010; 115: 205–208. doi: 10.1159/000287638 [DOI] [PubMed] [Google Scholar]
- 165.Easterling TR, Ralph DD, Schmucker BC. Pulmonary hypertension in pregnancy: treatment with pulmonary vasodilators. Obstet Gynecol 1999; 93: 494–498. [DOI] [PubMed] [Google Scholar]
- 166.Olsson KM, Channick R. Pregnancy in pulmonary arterial hypertension. Eur Respir Rev 2016; 25: 431–437. doi: 10.1183/16000617.0079-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Martin SR, Edwards A. Pulmonary hypertension and pregnancy. Obstet Gynecol 2019; 134: 974–987. doi: 10.1097/AOG.0000000000003549 [DOI] [PubMed] [Google Scholar]
- 168.Han H, Francis JK, Spitzer Y, et al. Anesthetic management of parturients with pulmonary hypertension undergoing cesarean deliveries. J Clin Anesth 2019; 57: 79. doi: 10.1016/j.jclinane.2019.03.008 [DOI] [PubMed] [Google Scholar]
- 169.Keepanasseril A, Pillai AA, Yavanasuriya J, et al. Outcome of pregnancies in women with pulmonary hypertension: a single-centre experience from South India. BJOG 2019; 126: Suppl. 4, 43–49. doi: 10.1111/1471-0528.15681 [DOI] [PubMed] [Google Scholar]
- 170.Zhang J, Lu J, Zhou X, et al. Perioperative management of pregnant women with idiopathic pulmonary arterial hypertension: an observational case series study from China. J Cardiothorac Vasc Anesth 2018; 32: 2547–2559. doi: 10.1053/j.jvca.2018.01.043 [DOI] [PubMed] [Google Scholar]