

Abstract
In addition to facilitating healthcare delivery planning, reliable information about prognosis is essential for treatment decisions in patients with idiopathic pulmonary fibrosis (IPF). This review aimed to evaluate the prognosis of patients with IPF without anti-fibrotic therapy. We included all cohort studies and the placebo arms of randomised controlled trials (RCTs) in IPF and follow-up of ≥12 months. Two reviewers independently evaluated studies for inclusion, assessed risk of bias and extracted data. A total of 154 cohort studies and 16 RCTs were included. The pooled proportions of mortality were 0.12 (95% CI 0.09–0.14) at 1–2 years, 0.38 (95% CI 0.34–0.42) between 2–5 years, and 0.69 (95% CI 0.59–0.78) at ≥5 years. The pooled mean overall survival was 4 years (95% CI 3.7–4.6) for studies with a follow-up duration of 10 years. At <2 years, forced vital capacity and diffusing capacity of the lung for carbon monoxide declined by a mean of 6.76% predicted (95% CI −8.92 −4.61) and 3% predicted (95% CI −5.14 −1.52), respectively. Although heterogeneity was high, subgroup analyses revealed lower pooled proportions of mortality at 1 year in the RCT participants (0.07 (95% CI 0.05–0.09)) versus cohort study participants (0.14 (95% CI 0.12–0.17)). This review provides comprehensive information on the prognosis of IPF, which can inform treatment discussions with patients and comparisons for future studies with new therapies.
Short abstract
Without anti-fibrotic therapy, patients with IPF have a mortality rate of 31% at ≥5 years, and a mean overall survival of 4 years over 10 years of follow-up http://bit.ly/2SDiZSb
Introduction
Idiopathic pulmonary fibrosis (IPF) is one of the most common interstitial lung diseases encountered in medical practice [1–6]. A recent systematic review of population-based studies in IPF estimated an incidence of up to nine cases per 100 000 population per year [7]. The British Lung Foundation estimated the prevalence of IPF at 50 per 100 000 people in the UK [8]. In the USA, healthcare utilisation by patients with IPF was approximately twice as high as that by matched controls [9].
Over time, criteria for diagnosing IPF have changed considerably, from a predominantly histopathological assessment to a multidisciplinary team approach based on clinical, radiologic and histopathologic correlation. Over the last couple of decades, the consensus statement for the diagnosis of IPF has been published and updated [10–12]. Although IPF has previously been reported to have a median survival of 3 years from time of diagnosis [13], this information has been derived mostly from studies performed prior to the publication of the recent consensus statements. It is uncertain whether the prognosis for IPF may have changed over time with changes in the diagnostic criteria.
With the availability of effective therapies for IPF [14, 15], a clear understanding of prognosis is crucial for proper evaluation of the potential impacts of these and other new approaches in the IPF management paradigm. Importantly, patients with IPF wish to have more information about their prognosis [16]. Knowledge of prognosis is essential for thoughtful discussions with patients regarding expectations, in order to ensure the timely offer of supportive care and avoidance of costly, unnecessary therapies with potentially significant adverse effects.
This systematic review and meta-analysis aimed to evaluate the prognosis for survival and respiratory-related outcomes in patients with IPF and without anti-fibrotic therapy.
Methods
This systematic review and meta-analysis were undertaken in collaboration with the Cochrane Airways and Prognosis Methods Groups. The review protocol was published prospectively on the Cochrane Library [17].
Inclusion and exclusion criteria
Eligible studies included cohort studies and randomised controlled trials (RCTs) with a placebo or best supportive therapy arm that enrolled participants with IPF aged ≥18 years who were either untreated or not treated with effective therapies (nintedanib or pirfenidone) and who were followed for ≥12 months. The active therapy arms of RCTs were excluded.
Search methods
We searched for relevant studies in the following databases (up to 3 May 2017): MEDLINE, Embase, CINAHL, PubMed and CENTRAL (part of the Cochrane Library via the Cochrane Register of Studies Online). For the full search strategies refer to supplementary material S1. The search was supplemented by reviewing reference lists of included studies and related review papers, clinical trial registries and annual conference abstracts from major respiratory societies. Authors of the consensus statements on IPF [10, 11] were contacted for unpublished studies. There was no restriction on language of publication.
Outcomes
Primary outcomes were the proportion of mortality and mean survival duration. Secondary outcomes included progression-free survival, respiratory-specific mortality, change in forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DLCO), proportions of patients with a significant decline in FVC or DLCO, dyspnoea, health-related quality of life (HRQoL), and 6-min walk distance (6MWD).
Study selection, data extraction and risk of bias assessment
Two authors (Y.H. Khor and Y. Ng or H. Barnes) independently identified eligible citations on the basis of sequential review of the title, abstract and full text. The agreement of the decisions between the two review authors was calculated using simple κ statistics. Data extraction and risk of bias assessment (supplementary material S2) were performed independently by two authors (Y.H. Khor and H. Barnes) using standardised forms. For RCTs, only data from the placebo or best supportive therapy arm were extracted. For each study, the two sets of data extracted by two review authors were cross-checked. Disagreements were resolved by consensus or a third review author if required. Study authors were contacted for clarification or to provide details of missing data where possible.
Statistical analysis
Meta-analysis was performed using Stata (v15.1; StataCorp, College Station, TX, USA). The pooled proportions of overall and respiratory-specific mortality were calculated under the random-effects model using the Freeman–Tukey transformation [18]. To perform meta-analyses of median survival and changes in secondary outcomes, the estimates of mean and standard error were first calculated according to published methods [19, 20] and then pooled using the Dersimonian–Laird random-effects model [21]. The magnitude of heterogeneity was measured through the I2 statistic, as described in the Cochrane Handbook [20]. The thresholds for I2 values were as follows: low (25–49%), moderate (50–74%) and high (≥75%).
Pre-specified subgroup analyses were performed based on the types of study design (cohort studies versus RCT, retrospective cohort studies versus prospective cohort studies) and diagnostic criteria for IPF: group 1: 2000 international consensus statement [10]; group 2: 2011 evidence-based guidelines [11]; group 3: other definitions (including physicians' diagnosis). Post-hoc sensitivity analyses was performed using studies with at least 50 participants who were diagnosed using either the 2000 or 2011 international consensus statements, to explore heterogeneity observed in the primary analyses.
Results
Search results
The literature search flow diagram is shown in figure 1. A total of 12 557 citations was considered, with 1531 being retrieved for full-text assessment. We excluded 1226 studies (supplementary material S3). Of the 305 included in this review, 170 were primary studies and 135 were additional reports of primary studies (supplementary martial S4). Inter-observer agreement for selection of studies was good, with κ=0.77 (95% CI 0.76–0.78).
FIGURE 1.
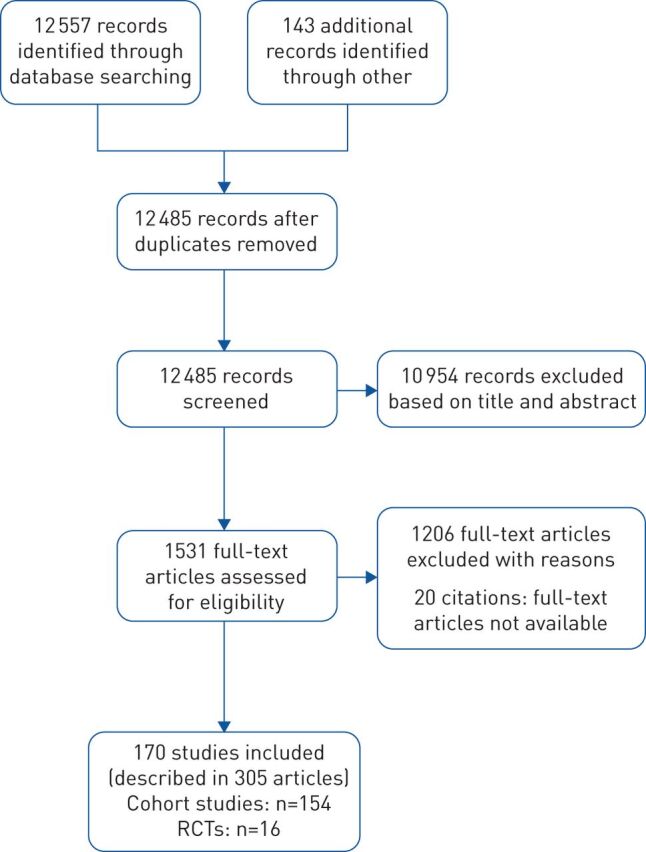
Study flow diagram. RCTs: randomised controlled trials.
Study characteristics
Of the 170 included studies, 154 were cohort studies [22–175] and 16 were RCTs [14, 15, 176–189]. Table 1 summarises the characteristics of included studies. Full details of included studies can be found in supplementary material S5. Data from studies with targeted selection of patients with acute exacerbation (n=7) [45, 50, 63, 108, 109, 135, 153] and lung transplants (n=2) [99, 131] were not included in pooled analyses for this review, given the established differences in the prognosis for these groups.
TABLE 1.
Characteristics of studies identified
| Characteristics | Details |
| Study type |
|
|
|
| Publication year |
|
|
|
|
|
|
|
| Location |
|
|
|
|
|
|
|
| IPF diagnostic criteria |
|
|
|
| Design | Cohort study# |
|
|
|
|
| RCT (n=16): all multicentre trials | |
| Participant number | Cohort study# |
|
|
|
|
| RCT (n=16): median (range) 94 (28–423)¶ | |
| Duration | Cohort study#,+ |
|
|
|
|
| RCT (n=16): all with a study duration of 1 year and <2 years |
IPF: idiopathic pulmonary fibrosis; ATS: American Thoracic Society; ERS: European Respiratory Society; JRS: Japanese Respiratory Society; ALAT: Latin American Thoracic Association; RCT: randomised controlled trial. #: not including Mura et al. [110]: a single-centre study with both prospective (n=70) and retrospective (n=68) cohorts, with a study duration ≥3 years; ¶: number of participants in the placebo arm only; +:: all studies had a study duration ≥1 year. Some studies provided mean or median follow-up duration which could not be incorporated into the table.
Among the cohort studies, 98 were of retrospective design and the other 55 were prospective, while one study included both prospective and retrospective cohorts of participants [110]. The median (range) number of participants was 67 (7–13 615). Follow-up periods ranged from 1 to 10 years. Most studies were single-centre based (n=118), with the remaining using data from multiple centres (n=23) or national registry (n=11) and two being unclear. The diagnosis of IPF was based on the 2000 [10] and 2011 [11] consensus statements in 46 and 36 studies, respectively. Other nonstandardised diagnostic criteria were used for 72 studies. Different therapies used for management of IPF were documented, with the absence of details in 64 studies (supplementary material S6).
For the RCTs, the median (range) number of participants in the placebo arm was 94 (28–423). The study duration ranged from 52 to 96 weeks, with participants being diagnosed based on either the 2000 (n=11) [10] or 2011 (n=5) [11] consensus statements. Prohibited concomitant treatments for each RCT varied (supplementary material S7).
Risk of bias assessment
Assessments were performed based on the information available from study articles and clarification provided by study authors. Overall, the risk of bias in the 170 included studies was moderate. A summary of the risk of bias assessment is presented in supplementary material S8. Most cohort studies (97%) used a clinical source for selecting the study population. While the diagnosis of IPF for study participants was established at baseline for almost all studies (99.5%), only 35% of cohort studies enrolled study participants prospectively. The reasons for loss to follow-up were inadequately described in 79% of cohort studies. For RCTs, all study participants were selected from a clinical source. The percentage of dropouts (along with reasons for dropouts) were described in 81% and 88% of RCTs, respectively. The methods of blinding and use of intention-to-treat principle for data analysis were inadequately described in 19% of RCTs.
Overall survival outcomes
Proportion of mortality
Data on proportions of mortality at various timeframes were available from 100 cohort studies and 16 RCTs. The pooled proportions of mortality were 0.12 (95% CI 0.09–0.14) at 1 year to <2 years (figure 2), 0.38 (95% CI 0.34–0.42) between 2 and 5 years (figure 3), and 0.69 (95% CI 0.59–0.78) for ≥5 years (figure 4). Tests of heterogeneity were high for all analyses of pooled proportions of mortality.
FIGURE 2.
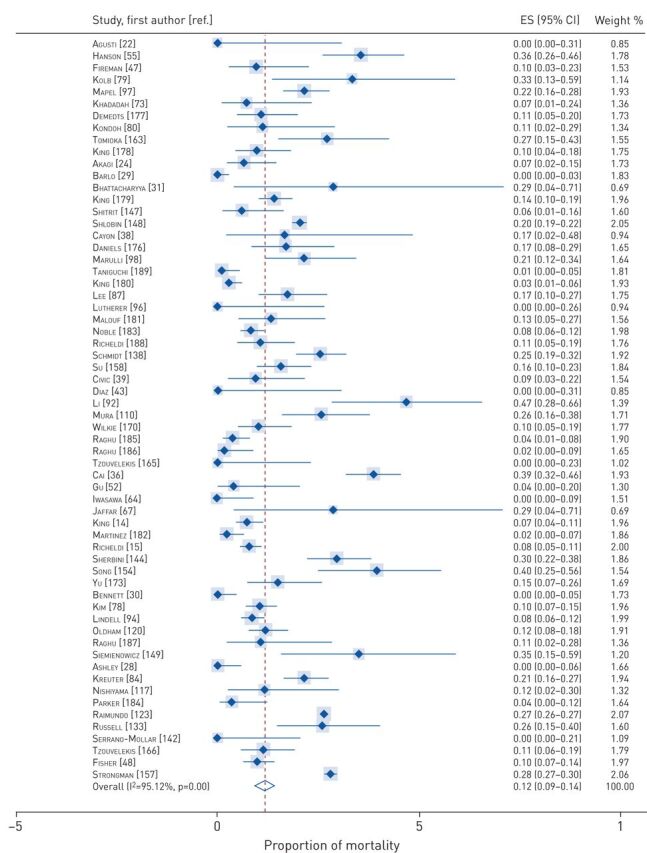
Pooled proportions of mortality at 1 year to <2 years. ES: effect size.
FIGURE 3.
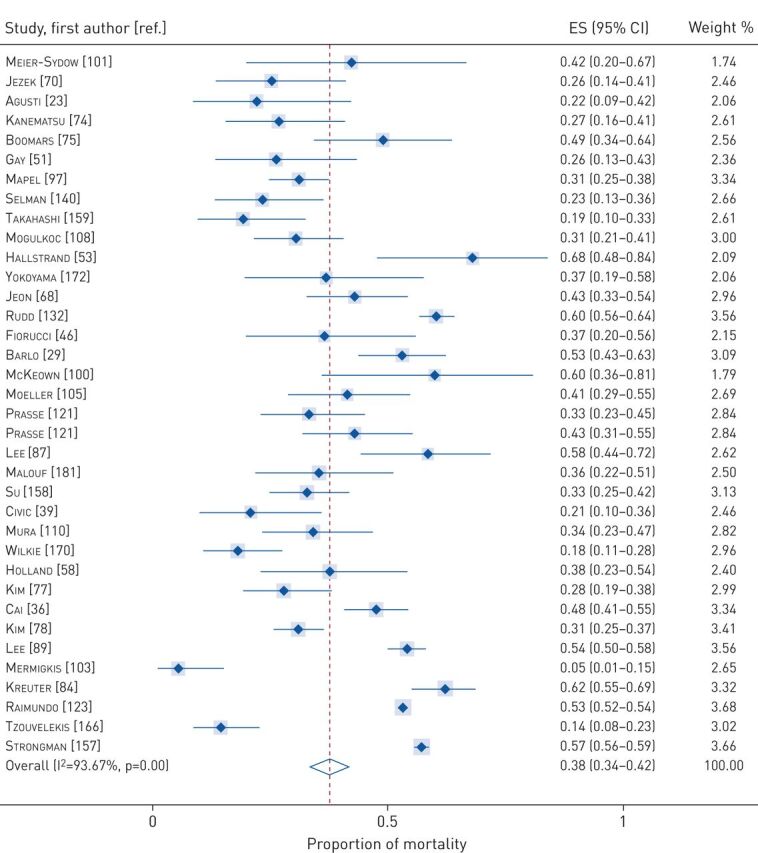
Pooled proportions of mortality between 2–5 years. ES: effect size.
FIGURE 4.

Pooled proportions of mortality at ≥5 years. ES: effect size.
Duration of survival
Data on the duration of survival were available from 72 cohort studies. Due to variable in follow-up duration, pooled analyse for durations of overall and transplant-free survival were not able to be performed using available data from 18 studies [34, 44, 53–55, 57, 76, 78, 79, 87, 88, 102, 104, 105, 107, 110, 121, 155] and three studies [33, 72, 73], respectively. For studies with a follow-up duration of 2 years, the pooled mean overall survival was 0.9 years (95% CI 0.4–1.4; high heterogeneity I2=94%). The pooled mean overall survival was 4 years (95% CI 3.7–4.6; low heterogeneity I2=0%) for studies with a follow-up duration of 10 years.
Respiratory-related outcomes
Changes in FVC were measured in 15 studies. At <2 years, pooled changes in FVC were −6.76% predicted (95% CI −8.92– −4.61; n=9 studies, 1000 participants; high heterogeneity I2=89%) (figure 5) [40, 42, 64, 179, 182–185, 188] and −0.18 L (95% CI −0.22– −0.13; n=9 studies, 629 participants; high heterogeneity I2=63%) (figure E2) [40, 73, 176, 180, 182, 184, 185, 187, 188]. In a study of 13 participants, Agusti et al. [23] reported a mean± sd decline of 0.46±0.09 L in 13 participants after 3 years of follow-up. Changes in DLCO were measured in seven studies. At 1 year to <2 years, pooled changes in DLCO were −3.33% predicted (95% CI −5.14– −1.52; n=5 studies, 277 participants; high heterogeneity I2=76%) (figure 6) [40, 176, 182, 184, 185] and −0.81 mL·mmHg−1·min−1 (95% CI −1.17– −0.46; n=4 studies, 428 participants; low heterogeneity I2=30%) (figure E3) [40, 180, 182, 186].
FIGURE 5.

Pooled mean changes in forced vital capacity (FVC) at 1 year to <2 years.
FIGURE 6.

Pooled mean changes in diffusing capacity of the lung for carbon monoxide (DLCO) at 1 year to <2 years.
Seven studies reported changes in 6MWD as an outcome measure. At 1 year to <2 years, the pooled mean change in 6MWD was −37 m (95% CI −88–15; n=4 studies, 444 participants; high heterogeneity I2=96%) (figure E4). Data pooling was not possible for three studies due to insufficient data [90, 182, 183]. Le Rouzic et al. [90] reported median changes of 6MWD of −35 m to 15 m in three subgroups of patients with IPF (total n=34) at 12 months. A study from the Idiopathic Pulmonary Fibrosis Clinical Research Network [182] reported a mean decline of 47.5 m in 6MWD in a 12-month study of 131 participants. In a study of 275 participants, Noble et al. [183] found a mean decline of 76.8 m in 6MWD at 72 weeks.
Due to insufficient data and use of different questionnaires, it was not possible to perform pooled analysis of change in dyspnoea. Using the University of California San Diego Shortness of Breath Questionnaire (UCSD SOBQ), King et al. [179] reported worsening dyspnoea with a mean± sd increment of 20.2±30.4 in 275 participants over a median follow-up duration of 77 weeks. the Idiopathic Pulmonary Fibrosis Clinical Research Network study reported [182] deterioration in dyspnoea with a mean increment of 5.8 in the UCSD SOBQ at 60 weeks in 131 patients. At 72 weeks, Noble et al. [183] and Parker et al. [184] reported worsening dyspnoea with a mean increment of 14.5 and 14.4 in the UCSD SOBQ in 347 and 57 patients, respectively.
A pooled analysis of St Georges Respiratory Questionnaire (SGRQ) total scores in three studies (389 participants) showed a decline in HRQoL at 1 year to <2 years, with a mean increase of 3.65 (95% CI −1.46–8.76; high heterogeneity I2=96%) [179, 187, 188]. Two studies provided insufficient data for inclusion in the meta-analysis, although the reported mean changes in the SGRQ total score were similar [15, 184]. Richeldi et al. [15] reported a mean increase of 4.39 to 5.48 for SGRQ total scores in 414 patients at 1 year. The mean increase of SGRQ total score at 60 weeks was 4.06 in a study of 57 patients by Parker et al. [184]. Two studies were pooled for analysis of mean changes in different SGRQ domain scores at 1 year: symptoms domain: 4.14 (95% CI 2.10–6.18); activity domain: 6.67 (95% CI 5.28–8.05); and impacts domain: 4.88 (95% CI 3.49–6.27) [15, 188].
Results on progression-free survival, respiratory-specific mortality, proportions of patients with a significant decline in lung function, dyspnoea measured using the Transition Dyspnoea Index and HRQoL measured using the EuroQol-5 Dinemsion are available in supplementary material S10.
Subgroup analyses
Analysis by study design
The RCT subgroup had a significantly lower mortality rate than the cohort study subgroup (0.07% versus 0.14%, p<0.0001) (figure E5). However, there was substantial heterogeneity between the studies within each subgroup. Subgroup analyses for pooled estimates of the proportion of mortality at other timeframes and survival duration were not performed owing to the lack of RCTs for these outcome measures. There was no statistically significant difference in the proportion of mortality at different timeframes between retrospective and prospective cohort studies (table 2).
TABLE 2.
Subgroup analyses for pooled analyses of the proportion of mortality according to study design
| Studies | Participants | Proportion | 95% CI | Heterogeneity | |
| 1 year to <2 years | |||||
| Overall# | 62 | 25 770 | 0.12 | 0.09–0.14 | I2=95% |
| RCT | 16 | 2420 | 0.07 | 0.05–0.09 | I2=75% |
| Prospective study | 18 | 769 | 0.11 | 0.05–0.18 | I2=81% |
| Retrospective study | 28 | 22 581 | 0.16 | 0.13–0.19 | I2=95% |
| Test for heterogeneity between subgroups 0.36 | |||||
| 2 to <5 years | |||||
| Overall | 35 | 20 787 | 0.38 | 0.34–0.42 | I2=94% |
| Prospective study | 18 | 1650 | 0.33 | 0.25–0.43 | I2=92% |
| Retrospective study | 17 | 19 092 | 0.43 | 0.38–0.47 | I2=93% |
| Test for heterogeneity between subgroups=0.08 | |||||
| ≥5 years | |||||
| Overall | 32 | 6369 | 0.69 | 0.59–0.78 | I2=98% |
| Prospective study | 9 | 1060 | 0.58 | 0.38–0.78 | I2=97% |
| Retrospective study | 23 | 5309 | 0.73 | 0.61–0.83 | I2=98% |
| Test for heterogeneity between subgroups=0.21 |
Data are presented as n, unless otherwise stated. RCT: randomised controlled trial. #: included data from RCTs.
Analysis by diagnostic criteria
At <2 years, the pooled proportions of mortality were 0.07 (95% CI 0.05–0.09), 0.10 (95% CI 0.06–0.15) and 0.18 (95% CI 0.15–0.22) for studies which diagnosed IPF based on the 2011 and 2000 consensus statements and other criteria, respectively. The test of overall interaction among these subgroups was statistically significant (p<0.0001). There was no significant difference in the proportion of mortality at other timeframes according to the diagnostic criteria (table 3).
TABLE 3.
Subgroup analyses for pooled analyses of the proportion of mortality
| Studies | Participants | Proportion | 95% CI | Heterogeneity | |
| 1 year to <2 years | |||||
| Overall | 62 | 25 770 | 0.12 | 0.09–0.14 | I2=95% |
| 2011 ATS/ERS/JRS/ALAT guideline | 16 | 2283 | 0.07 | 0.05–0.09 | I2=72% |
| 2000 ATS/ERS consensus statement | 25 | 2382 | 0.10 | 0.06–0.15 | I2=92% |
| Other criteria | 21 | 21 105 | 0.18 | 0.15–0.22 | I2=92% |
| Test for difference between subgroups <0.0001 | |||||
| 2 to <5 years | |||||
| Overall | 35 | 20 787 | 0.38 | 0.34–0.42 | I2=94% |
| 2011 ATS/ERS/JRS/ALAT guideline | 4 | 1026 | 0.25 | 0.08–0.47 | I2=98% |
| 2000 ATS/ERS consensus statement | 13 | 995 | 0.42 | 0.37–0.47 | I2=54% |
| Other criteria | 18 | 18 766 | 0.38 | 0.33–0.43 | I2=94% |
| Test for difference between subgroups=0.17 | |||||
| ≥5 years | |||||
| Overall | 32 | 6369 | 0.69 | 0.59–0.78 | I2=98% |
| 2011 ATS/ERS/JRS/ALAT guideline | 4 | 326 | 0.62 | 0.38–0.83 | I2=93% |
| 2000 ATS/ERS consensus statement | 9 | 815 | 0.62 | 0.47–0.75 | I2=94% |
| Other criteria | 19 | 5228 | 0.74 | 0.62–0.84 | I2=98% |
| Test for difference between subgroups=0.37 |
Data are presented as n, unless otherwise stated. ATS: American Thoracic Society; ERS: European Respiratory Society; JRS: Japanese Respiratory Society; ALAT: Latin American Thoracic Association.
Sensitivity analysis
Sensitivity analyses using studies with at least 50 participants diagnosed based on the consensus statement criteria revealed pooled proportions of mortality of 0.10 (95% CI 0.07–0.13) at 1 year to <2 years, 0.36 (95% CI 0.27–0.44) between 2 and 5 years, and 0.58 (95% CI 0.43–0.71) for ≥5 years (figure E6). Tests of heterogeneity remained high in sensitivity analyses (range 92–95%). Sensitivity analyses for survival duration were not performed owing to the lack of studies meeting the specified criteria.
Discussion
This is the first systematic review with a meta-analysis of studies examining the prognosis of IPF. Collectively, overall survival rates for this population were 88% at 1 year to <2 years and 31% at ≥5 years. The pooled estimate of mean survival, derived from a meta-analysis of two studies with follow-up durations of 10 years was 4 years. Respiratory-related causes and IPF accounted for 50% and 30% of deaths at 1 year to <2 years, respectively. At <2 years, FVC and DLCO had fallen by an average of 7% predicted and 3% predicted, respectively. Dyspnoea worsened significantly over a period of <2 years, reaching the minimal important differences (MID) of 5 points using the UCSD SOBQ [190]. For HRQoL measured using the SGRQ, the mean activity domain score of 6.67 exceeded the MID of 5 at <2 years [191]. Although mean changes in total and other domain scores for SGRQ did not reach the MID, their CIs exceeded the MID. The mean decline in 6MWD was significant at 37 m, given a MID of 30 m [192].
This review confirmed the previously described poor prognosis of patients with IPF who have not received effective therapies. A recent editorial which incorporated a review of 24 study cohorts published between 2000 and 2017, each containing at least 50 consecutively enrolled participants with IPF, found an overall median survival of 3.2 years [193]. A meta-analysis of five clinical trials of pirfenidone versus placebo revealed 1-year overall mortality rate of 6.5% for the placebo group [194]. These results are similar to the findings from this review. The strength of this review lies in the large study populations from more than 28 countries on four continents, ranging from 6369 to 25 799 participants. Given that the majority of studies are longitudinal cohort studies with participants selected from clinical settings, the survival outcomes in this study likely reflect real world data.
At <2 years, the pooled survival rate was significantly higher in the RCT subgroup than in the cohort study subgroup. This difference is likely due to differences in disease severity and patient characteristics of the study populations in RCTs and cohort studies. Patients included in RCTs predominantly had mild-to-moderate disease severity and fewer comorbidities, while cohort studies included real-world populations, including patients with end-stage IPF and multimorbidities. Interventions for IPF also differs between these two study populations. Some of these interventions, such as the combined use of prednisolone, azathioprine and N-acetylcysteine [195], may affect survival outcomes in IPF. Similarly, such variations likely explain differences in the pooled survival rates according to the diagnostic criteria. Approximately one-third of the studies for subgroups using the consensus statement diagnostic criteria were RCTs, while all studies for the subgroup using other diagnostic criteria were cohort studies.
Limitations
There are limitations with this review. There was statistically significant heterogeneity in pooled results, which was not explained by the subgroup analyses. This observed heterogeneity may be attributed to variations of clinical characteristics across study populations for pooled analyses, including duration of diagnosis, disease severity, comorbidities and patient characteristics. The overall quality of included studies was moderate. It is important to note that almost all studies had used clinical sampling rather than population sampling, which may limit the generalisability of the findings to a wider population. The risk of bias was increased by differences in assessment methods and reporting timing of outcome measures, limiting the ability to perform pooled analyses. The included studies, particularly cohort studies, had variable follow-up periods, which may have affected reported outcomes. Due to the limited data reported, subgroup analyses of survival duration based on follow-up periods were not possible. The quality of evidence for mortality and survival outcomes was moderate due to significant heterogeneity with inconsistency between studies. Sensitivity analyses, including only studies of at least 50 participants diagnosed according to well-established criteria, did not show any notable changes in the overall survival outcomes and degrees of heterogeneity. This suggests that there are other, as yet undocumented, sources of heterogeneity contributing to these outcomes. The quality of evidence for all other secondary outcomes was low to moderate due to substantial heterogeneity or fewer studies for pooled analyses, or both. This review did not include any studies which diagnosed IPF based on the latest 2018 consensus statement [12]. However, given the similarities in the diagnostic criteria between the 2011 and 2018 consensus statements, findings from this review are relevant as references for IPF diagnosed using the 2018 consensus statement.
In conclusion, this review provides a comprehensive evaluation of all relevant studies regarding the prognosis of patients with IPF who either were untreated or had not received effective treatments. Without anti-fibrotic therapy, the overall survival rates for this condition are poor. Although limited by significant heterogeneity in study design and population, findings from this review can inform management discussions with patients and can provide a solid basis for comparisons in future studies with new therapies.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary_material ERR-0158-2019_Supplementary_material (3.7MB, pdf)
Acknowledgements
We thank Emma Jackson and Elizabeth Stovold from the Cochrane Airways Group, who were supported by the National Institute for Health Research, for their assistance with study conduct. We thank all study authors for provision of study clarification and/or additional data, and Cochrane reviewers for translation support.
Footnotes
Contributors: Y.H. Khor, Y. Ng, N.S.L. Goh, C.F. McDonald and A.E. Holland were responsible for review conception and development of the review protocol. Y.H. Khor, Y. Ng and H. Barnes were responsible for study selection, quality assessment and data extraction. Y.H. Khor was responsible for data analysis. Y.H. Khor, N.S.L. Goh, C.F. McDonald and A.E. Holland interpreted the data. Y.H. Khor drafted the manuscript. All authors revised and approved the manuscript for submission.
This article has supplementary material available from err.ersjournals.com
Provenance: Submitted article, peer reviewed.
Conflict of interest: Y.H. Khor reports grants from National Health and Medical Research Council Postgraduate Scholarship, during the conduct of the study; other from Air Liquide Healthcare, grants and personal fees from Boehringer Ingelheim, personal fees from Roche, outside the submitted work.
Conflict of interest: Y. Ng has nothing to disclose.
Conflict of interest: H. Barnes has nothing to disclose.
Conflict of interest: N.S.L. Goh reports grants and personal fees from Boehringer Ingelheim, non-financial support from Air liquide, personal fees from Roche, outside the submitted work.
Conflict of interest: C.F. McDonald reports non-financial support from Air Liquide Healthcare, other from AstraZeneca and Menarini, grants from Boehringer Ingelheim, outside the submitted work.
Conflict of interest: A.E. Holland reports grants from Roche, personal fees from Boehringer Ingelheim, non-financial support from BOC Healthcare and Air Liquide, outside the submitted work.
Support statement: Y.H. Khor has received research fellowship support from the National Health and Medical Research Council (APP1093506). Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Coultas DB, Hughes MP. Accuracy of mortality data for interstitial lung diseases in New Mexico, USA. Thorax 1996; 5: 717–720. doi: 10.1136/thx.51.7.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomeer MJ, Costabel U, Rizzato G, et al. . Comparison of registries of interstitial lung diseases in three European Countries. Eur Respir J 2001; 18: Suppl. 32, 114s–118s. [PubMed] [Google Scholar]
- 3.Ohno S, Nakaya T, Bando M, et al. . Idiopathic pulmonary fibrosis – results from a Japanese nationwide epidemiological survey using individual clinical records. Respirology 2008; 13: 926–928. doi: 10.1111/j.1440-1843.2008.01349.x [DOI] [PubMed] [Google Scholar]
- 4.Karakatsani A, Papakosta D, Rapti A, et al. . Epidemiology of interstitial lung diseases in Greece. Respir Med 2009; 103: 1122–1129. doi: 10.1016/j.rmed.2009.03.001 [DOI] [PubMed] [Google Scholar]
- 5.Musellim B, Okumus G, Uzaslan E, et al. . Epidemiology and distribution of interstitial lung diseases in Turkey. Clin Respir J 2014; 8: 55–62. doi: 10.1111/crj.12035 [DOI] [PubMed] [Google Scholar]
- 6.Bando M, Sugiyama Y, Azuma A, et al. . A prospective survey of idiopathic interstitial pneumonias in a web registry in Japan. Respir Investig 2015; 53: 51–59. doi: 10.1016/j.resinv.2014.11.001 [DOI] [PubMed] [Google Scholar]
- 7.Hutchinson J, Fogarty A, Hubbard R, et al. . Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46: 795–806. doi: 10.1183/09031936.00185114 [DOI] [PubMed] [Google Scholar]
- 8.British Lung Foundation . The Battle for Breath – The impact of lung disease in the UK. 2016. www.blf.org.uk/policy/the-battle-for-breath-2016. Date last accessed: 14 September 2019.
- 9.Collard HR, Ward AJ, Lanes S, et al. . Burden of illness in idiopathic pulmonary fibrosis. J Med Econ 2012; 15: 829–835. doi: 10.3111/13696998.2012.680553 [DOI] [PubMed] [Google Scholar]
- 10.American Thoracic Society, European Respiratory Society . Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. Am J Respir Crit Care Med 2000; 161: 646–664. doi: 10.1164/ajrccm.161.2.ats3-00 [DOI] [PubMed] [Google Scholar]
- 11.Raghu G, Collard HR, Egan JJ, et al. . An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. doi: 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raghu G, Remy-Jardin M, Myers JL, et al. . Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2018; 198: e44–e68. doi: 10.1164/rccm.201807-1255ST [DOI] [PubMed] [Google Scholar]
- 13.Vancheri C, Failla M, Crimi N, et al. . Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J 2010; 35: 496–504. doi: 10.1183/09031936.00077309 [DOI] [PubMed] [Google Scholar]
- 14.King TE Jr, Bradford WZ, Castro-Bernardini S, et al. . A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083–2092. doi: 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 15.Richeldi L, Du Bois RM, Raghu G, et al. . Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. doi: 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 16.Holland AE, Fiore JF Jr, Goh N, et al. . Be honest and help me prepare for the future: what people with interstitial lung disease want from education in pulmonary rehabilitation. Chron Respir Dis 2015; 12: 93–101. doi: 10.1177/1479972315571925 [DOI] [PubMed] [Google Scholar]
- 17.Khor YH, Ng Y, Goh NSL, et al. . Prognosis of adults with idiopathic pulmonary fibrosis without treatment or without effective therapies (Protocol). Cochrane Database Syst Rev 2017; 2017: CD012647. [Google Scholar]
- 18.Nyaga VN, Arbyn M, Aerts M. Metaprop: a Stata command to perform meta-analysis of binomial data. Arch Public Health 2014; 72: 39. doi: 10.1186/2049-3258-72-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hozo SP, Djulbegovic B, Hozo I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med Res Meth 2005; 5: 13. doi: 10.1186/1471-2288-5-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0. Chichester, Cochrane Collaboration, 2011. [Google Scholar]
- 21.Kontopantelis E, Reeves D. Metaan: random-effects meta-analysis. Stata J 2010; 10: 395–407. doi: 10.1177/1536867X1001000307 [DOI] [Google Scholar]
- 22.Agusti C, Xaubet A, Ballester E, et al. . Aerosolised ribavirin in patients with advanced cryptogenic fibrosing alveolitis: a pilot study. Thorax 1993; 48: 68–69. doi: 10.1136/thx.48.1.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agusti C, Xaubet A, Agusti AG, et al. . Clinical and functional assessment of patients with idiopathic pulmonary fibrosis: results of a 3-year follow-up. Eur Respir J 1994; 7: 643–650. doi: 10.1183/09031936.94.07040643 [DOI] [PubMed] [Google Scholar]
- 24.Akagi T, Matsumoto T, Harada T, et al. . Coexistent emphysema delays the decrease of vital capacity in idiopathic pulmonary fibrosis. Respir Med 2009; 103: 1209–1215. doi: 10.1016/j.rmed.2009.02.001 [DOI] [PubMed] [Google Scholar]
- 25.Alhamad EH, Masood M, Shaik SA, et al. . Clinical and functional outcomes in Middle Eastern patients with idiopathic pulmonary fibrosis. Clin Respir J 2008; 2: 220–226. doi: 10.1111/j.1752-699X.2008.00070.x [DOI] [PubMed] [Google Scholar]
- 26.Antoniou KM, Hansell DM, Rubens MB, et al. . Idiopathic pulmonary fibrosis: outcome in relation to smoking status. Am J Respir Crit Care Med 2008; 177: 190–194. doi: 10.1164/rccm.200612-1759OC [DOI] [PubMed] [Google Scholar]
- 27.Araki T, Katsura H, Sawabe M, et al. . A clinical study of idiopathic pulmonary fibrosis based on autopsy studies in elderly patients. Intern Med 2003; 42: 483–489. doi: 10.2169/internalmedicine.42.483 [DOI] [PubMed] [Google Scholar]
- 28.Ashley SL, Xia M, Murray S, et al. . Six-SOMAmer index relating to immune, protease and angiogenic functions predicts progression in IPF. PLoS One 2016; 11: e0159878. doi: 10.1371/journal.pone.0159878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barlo NP, van Moorsel CH, van den Bosch JM, et al. . Idiopathic pulmonary fibrosis; description of a Dutch cohort. Ned Tijdschr Geneeskd 2009; 153: B425. [PubMed] [Google Scholar]
- 30.Bennett D, Fossi A, Bargagli E, et al. . Mortality on the waiting list for lung transplantation in patients with idiopathic pulmonary fibrosis: a single-centre experience. Lung 2015; 193: 677–681. doi: 10.1007/s00408-015-9767-x [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharyya P, Nag S, Bardhan S, et al. . The role of long-term doxycycline in patients of idiopathic pulmonary fibrosis: the results of an open prospective trial. Lung India 2009; 26: 81–85. doi: 10.4103/0970-2113.53231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bjoraker JA, Ryu JH, Edwin MK, et al. . Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998; 157: 199–203. doi: 10.1164/ajrccm.157.1.9704130 [DOI] [PubMed] [Google Scholar]
- 33.Bjurstrom L, Andersen KH, Kjærgaard J, et al. . Pulmonary hypertension in end-stage pulmonary fibrosis: prevalence, predictors and survival before and after lung transplantation. J Heart Lung Transplant 2013; 1: S18–S19. doi: 10.1016/j.healun.2013.01.028 [DOI] [Google Scholar]
- 34.Boomars KA, Wagenaar SS, Mulder PG, et al. . Relationship between cells obtained by bronchoalveolar lavage and survival in idiopathic pulmonary fibrosis. Thorax 1995; 50: 1087–1092. doi: 10.1136/thx.50.10.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bournazos S, Bournazou I, Murchison JT, et al. . Copy number variation of FCGR3B is associated with susceptibility to idiopathic pulmonary fibrosis. Respiration 2011; 81: 142–149. doi: 10.1159/000321997 [DOI] [PubMed] [Google Scholar]
- 36.Cai M, Zhu M, Ban C, et al. . Clinical features and outcomes of 210 patients with idiopathic pulmonary fibrosis. Chin Med J 2014; 127: 1868–1873. [PubMed] [Google Scholar]
- 37.Castria D, Refini RM, Bargagli E, et al. . Pulmonary hypertension in idiopathic pulmonary fibrosis: prevalence and clinical progress. Int J Immunopathol Pharmacol 2012; 25: 681–689. doi: 10.1177/039463201202500314 [DOI] [PubMed] [Google Scholar]
- 38.Cayon I, Gonzalez L, Garcia I, et al. . Gamma interferon and prednisone decreasing-dose therapy in patients with idiopathic pulmonary fibrosis. Biotecnologia Aplicada 2010; 27: 29–35. [Google Scholar]
- 39.Civic B, Swift I, Panetta N, et al. . Upper lobe emphysema is associated with lower survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012; 185: A4387. [Google Scholar]
- 40.Collins BF, Shaw M, Ho LA, et al. . Clinical patterns and pulmonary physiology among patients with autoimmune interstitial lung disease (AI-ILD), connective tissue disease associated interstitial lung disease (CTD-ILD) and idiopathic pulmonary fibrosis (IPF) over 12 months. Am J Respir Crit Care Med 2015; 191: A1589. [Google Scholar]
- 41.Cottin V, Schmidt A, Catella L, et al. . Burden of idiopathic pulmonary fibrosis progression: a 5-year longitudinal follow-up study. PLoS One 2017; 12: e0166462. doi: 10.1371/journal.pone.0166462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Couluris M, Kinder BW, Xu P, et al. . Treatment of idiopathic pulmonary fibrosis with losartan: a pilot project. Lung 2012; 190: 523–527. doi: 10.1007/s00408-012-9410-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diaz KT, Skaria S, Harris K, et al. . Delivery and safety of inhaled interferon-gamma in idiopathic pulmonary fibrosis. J Aerosol Med Pulm Drug Deliv 2012; 25: 79–87. doi: 10.1089/jamp.2011.0919 [DOI] [PubMed] [Google Scholar]
- 44.Erbes R, Schaberg T, Loddenkemper R. Lung function tests in patients with idiopathic pulmonary fibrosis. Are they helpful for predicting outcome? Chest 1997; 111: 51–57. doi: 10.1378/chest.111.1.51 [DOI] [PubMed] [Google Scholar]
- 45.Faverio P, Ruggiero R, Paolini V, et al. . Intravenous pulse doses of high-dose corticosteroid and cyclophosphamide in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J 2015; 46: Suppl. 59, PA3045. [Google Scholar]
- 46.Fiorucci E, Lucantoni G, Paone G, et al. . Colchicine, cyclophosphamide and prednisone in the treatment of mild-moderate idiopathic pulmonary fibrosis: comparison of three currently available therapeutic regimens. Eur Rev Med Pharmacol Sci 2008; 12: 105–111. [PubMed] [Google Scholar]
- 47.Fireman E, Vardinon N, Burke M, et al. . Predictive value of response to treatment of T-lymphocyte subpopulations in idiopathic pulmonary fibrosis. Eur Respir J 1998; 11: 706–711. [PubMed] [Google Scholar]
- 48.Fisher JH, Al-Hejaili F, Kandel S, et al. . Multi-dimensional scores to predict mortality in patients with idiopathic pulmonary fibrosis undergoing lung transplantation assessment. Respir Med 2017; 125: 65–71. doi: 10.1016/j.rmed.2017.03.006 [DOI] [PubMed] [Google Scholar]
- 49.Fisher M, Nathan SD, Hill C, et al. . Predicting life expectancy for pirfenidone in idiopathic pulmonary fibrosis. J Manag Care Spec Pharm 2017; 23: S17–S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujimoto K, Taniguchi H, Johkoh T, et al. . Acute exacerbation of idiopathic pulmonary fibrosis: high-resolution CT scores predict mortality. Eur Radiol 2012; 22: 83–92. doi: 10.1007/s00330-011-2211-6 [DOI] [PubMed] [Google Scholar]
- 51.Gay SE, Kazerooni EA, Toews GB, et al. . Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Criti Care Med 1998; 157: 1063–1072. doi: 10.1164/ajrccm.157.4.9703022 [DOI] [PubMed] [Google Scholar]
- 52.Gu P, Luo B, Yi X, et al. . The expressions and meanings of BMP-7 and TGF-beta in idiopathic pulmonary fibrosis and idiopathic nonspecific interstitial pneumonia. Zhonghua Jie He He Hu Xi Za Zhi 2014; 37: 664–670. [PubMed] [Google Scholar]
- 53.Hallstrand TS, Boitano LJ, Johnson WC, et al. . The timed walk test as a measure of severity and survival in idiopathic pulmonary fibrosis. Eur Respir J 2005; 25: 96–103. doi: 10.1183/09031936.04.00137203 [DOI] [PubMed] [Google Scholar]
- 54.Hamada K, Nagai S, Tanaka S, et al. . Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest 2007; 131: 650–656. doi: 10.1378/chest.06-1466 [DOI] [PubMed] [Google Scholar]
- 55.Hanson D, Winterbauer RH, Kirtland SH, et al. . Changes in pulmonary function test results after 1 year of therapy as predictors of survival in patients with idiopathic pulmonary fibrosis. Chest 1995; 108: 305–310. doi: 10.1378/chest.108.2.305 [DOI] [PubMed] [Google Scholar]
- 56.Harris JM, Johnston ID, Rudd R, et al. . Cryptogenic fibrosing alveolitis and lung cancer: the BTS study. Thorax 2010; 65: 70–76. doi: 10.1136/thx.2009.121962 [DOI] [PubMed] [Google Scholar]
- 57.Hiwatari N, Shimura S, Yamauchi K, et al. . Significance of elevated procollagen-III-peptide and transforming growth factor-beta levels of bronchoalveolar lavage fluids from idiopathic pulmonary fibrosis patients. Tohoku J Exp Med 1997; 181: 285–295. doi: 10.1620/tjem.181.285 [DOI] [PubMed] [Google Scholar]
- 58.Holland AE, Hill CJ, Glaspole I, et al. . Impaired chronotropic response to 6-min walk test and reduced survival in interstitial lung disease. Respir Med 2013; 107: 1066–1072. doi: 10.1016/j.rmed.2013.04.002 [DOI] [PubMed] [Google Scholar]
- 59.Hopkins RB, Burke N, Fell C, et al. . Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur Respir J 2016; 48: 187–195. doi: 10.1183/13993003.01504-2015 [DOI] [PubMed] [Google Scholar]
- 60.Hosein K, Le J, Mura M. Assessing the therapeutic response to pirfenidone in idiopathic pulmonary fibrosis: can we do better than with forced vital capacity alone? Lung 2017; 195: 101–105. doi: 10.1007/s00408-016-9963-3 [DOI] [PubMed] [Google Scholar]
- 61.Hubbard R, Johnston I, Britton J. Survival in patients with cryptogenic fibrosing alveolitis: a population-based cohort study. Chest 1998; 113: 396–400. doi: 10.1378/chest.113.2.396 [DOI] [PubMed] [Google Scholar]
- 62.Huynh RH, Shino MY, Derhovanessian A, et al. . Th2 products IL-4 and IL-13 are associated with rapidly progressive idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2015; 191: A2166. [Google Scholar]
- 63.Inase N, Sawada M, Ohtani Y, et al. . Cyclosporin A followed by the treatment of acute exacerbation of idiopathic pulmonary fibrosis with corticosteroid. Intern Med 2003; 42: 565–570. doi: 10.2169/internalmedicine.42.565 [DOI] [PubMed] [Google Scholar]
- 64.Iwasawa T, Ogura T, Sakai F, et al. . CT analysis of the effect of pirfenidone in patients with idiopathic pulmonary fibrosis. Eur J Radiol 2014; 83: 32–38. doi: 10.1016/j.ejrad.2012.02.014 [DOI] [PubMed] [Google Scholar]
- 65.Izumi T, Nagai S, Kondo Y, et al. . Ten-year follow up of 222 patients with idiopathic pulmonary fibrosis (IPF)-a nation-wide survey report in Japan. Am Rev Respir Dis 1992; 145: A218. [Google Scholar]
- 66.Jacob J, Bartholmai BJ, Rajagopalan S, et al. . Mortality prediction in idiopathic pulmonary fibrosis: evaluation of computer-based CT analysis with conventional severity measures. Eur Respir J 2016; 49: 1601011. doi: 10.1183/13993003.01011-2016 [DOI] [PubMed] [Google Scholar]
- 67.Jaffar J, Unger S, Corte TJ, et al. . Fibulin-1 predicts disease progression in patients with idiopathic pulmonary fibrosis. Chest 2014; 146: 1055–1063. doi: 10.1378/chest.13-2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jeon K, Chung MP, Lee KS, et al. . Prognostic factors and causes of death in Korean patients with idiopathic pulmonary fibrosis. Respir Med 2006; 100: 451–457. doi: 10.1016/j.rmed.2005.06.013 [DOI] [PubMed] [Google Scholar]
- 69.Jezek V, Fucik J. [Prognosis of pulmonary hypertension in cryptogenic fibrosing alveolitis of the lung]. Vnitr Lek 1979; 25: 635–640. [PubMed] [Google Scholar]
- 70.Jezek V, Fucik J, Michaljanic A, et al. . The prognostic significance of functional tests in kryptogenic fibrosing alveolitis. Bull Eur Physiopathol Respir 1980; 16: 711–720. [PubMed] [Google Scholar]
- 71.Jezkova L, Fucik J, Michaljanic A, et al. . ECG and its significance for pulmonary hypertension diagnosis and for the estimation of prognosis in cryptogenic fibrosing pulmonary alveolitis. Cas Lek Cesk 1981; 120: 1050–1054. [PubMed] [Google Scholar]
- 72.Jo HE, Glaspole I, Grainge C, et al. . Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J 2017; 49: 1601592. doi: 10.1183/13993003.01592-2016 [DOI] [PubMed] [Google Scholar]
- 73.Justet A, Laurent-Bellue A, Thabut G, et al. . [18F]FDG PET/CT predicts progression-free survival in patients with idiopathic pulmonary fibrosis. Respir Res 2017; 18: 74. doi: 10.1186/s12931-017-0556-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kanematsu T, Kitaichi M, Nishimura K, et al. . Clubbing of the fingers and smooth-muscle proliferation in fibrotic changes in the lung in patients with idiopathic pulmonary fibrosis. Chest 1994; 105: 339–342. doi: 10.1378/chest.105.2.339 [DOI] [PubMed] [Google Scholar]
- 75.Khadadah ME, Onadeko BO, Abul AT, et al. . Clinicopathological and therapeutic patterns of idiopathic pulmonary fibrosis in Kuwait: a prospective study. Int J Clin Pract 2003; 57: 879–884. [PubMed] [Google Scholar]
- 76.Kim JH, Lee JH, Ryu YJ, et al. . Clinical predictors of survival in idiopathic pulmonary fibrosis. Tuberc Respir Dis 2012; 73: 162–168. doi: 10.4046/trd.2012.73.3.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim HJ, Gillen K, Tomic R. Idiopathic pulmonary fibrosis: typical versus atypical survival characteristics. Am J Respir Crit Care Med 2013; 187: A4333. [Google Scholar]
- 78.Kim ES, Choi SM, Lee J, et al. . Validation of the GAP score in Korean patients with idiopathic pulmonary fibrosis. Chest 2015; 147: 430–437. doi: 10.1378/chest.14-0453 [DOI] [PubMed] [Google Scholar]
- 79.Kolb M, Kirschner J, Riedel W, et al. . Cyclophosphamide pulse therapy in idiopathic pulmonary fibrosis. Eur Respir J 1998; 12: 1409–1414. doi: 10.1183/09031936.98.12061409 [DOI] [PubMed] [Google Scholar]
- 80.Kondoh Y, Taniguchi H, Yokoi T, et al. . Cyclophosphamide and low-dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. Eur Respir J 2005; 25: 528–533. doi: 10.1183/09031936.05.00071004 [DOI] [PubMed] [Google Scholar]
- 81.Kondoh Y, Taniguchi H, Katsuta T, et al. . Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27: 103–110. [PubMed] [Google Scholar]
- 82.Korthagen NM, van Moorsel CH, Barlo NP, et al. . Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir Med 2011; 105: 106–113. doi: 10.1016/j.rmed.2010.09.012 [DOI] [PubMed] [Google Scholar]
- 83.Kotecha J, Shulgina L, Sexton DW, et al. . Plasma vascular endothelial growth factor concentration and alveolar nitric oxide as potential predictors of disease progression and mortality in idiopathic pulmonary fibrosis. J Clin Med 2016; 5: E80. doi: 10.3390/jcm5090080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kreuter M, Ehlers-Tenenbaum S, Palmowski K, et al. . Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS One 2016; 11: e0151425. doi: 10.1371/journal.pone.0151425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kurashima K, Takayanagi N, Tsuchiya N, et al. . The effect of emphysema on lung function and survival in patients with idiopathic pulmonary fibrosis. Respirology 2010; 15: 843–848. doi: 10.1111/j.1440-1843.2010.01778.x [DOI] [PubMed] [Google Scholar]
- 86.Lederer DJ, Arcasoy SM, Barr RG, et al. . Racial and ethnic disparities in idiopathic pulmonary fibrosis: a UNOS/OPTN database analysis. Am J Transplant 2006; 6: 2436–2442. doi: 10.1111/j.1600-6143.2006.01480.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee SH, Shim HS, Cho SH, et al. . Prognostic factors for idiopathic pulmonary fibrosis: clinical, physiologic, pathologic, and molecular aspects. Sarcoidosis Vasc Diffuse Lung Dis 2011; 28: 102–112. [PubMed] [Google Scholar]
- 88.Lee HY, Lee KS, Jeong YJ, et al. . High-resolution CT findings in fibrotic idiopathic interstitial pneumonias with little honeycombing: serial changes and prognostic implications. AJR Am J Roentgenol 2012; 199: 982–989. doi: 10.2214/AJR.11.8192 [DOI] [PubMed] [Google Scholar]
- 89.Lee JW, Shehu E, Gjonbrataj J, et al. . Clinical findings and outcomes in patients with possible usual interstitial pneumonia. Respir Med 2015; 109: 510–516. doi: 10.1016/j.rmed.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 90.Le Rouzic O, Bendaoud S, Chenivesse C, et al. . Prognostic value of the initial chest high-resolution CT pattern in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2015; 32: 353–359. [PubMed] [Google Scholar]
- 91.Li ZH, Peng SC, Kang J, et al. . A retrospective cohort study of prognostic factors for death in patients with idiopathic pulmonary fibrosis. Zhonghua Jie He He Hu Xi Za Zhi 2010; 33: 887–891. [PubMed] [Google Scholar]
- 92.Li H, Liu S, Liu Z, et al. . The efficacy and safety of Chinese herb on the treatment of idiopathic pulmonary fibrosis. Chest 2012; 142: 4 Suppl. 1, 956A. doi: 10.1378/chest.1390834 [DOI] [Google Scholar]
- 93.Li X, Peng S, Wei L, et al. . Relevance analysis of clinical and lung function parameters changing and prognosis of idiopathic pulmonary fibrosis. Int J Clin Exp Med 2015; 7: 4759–4769. [PMC free article] [PubMed] [Google Scholar]
- 94.Lindell KO, Liang Z, Hoffman LA, et al. . Palliative care and location of death in decedents with idiopathic pulmonary fibrosis. Chest 2015; 147: 423–429. doi: 10.1378/chest.14-1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu B, Su F, Xu N, et al. . Chronic use of anti-reflux therapy improves survival of patients with pulmonary fibrosis. Int J Clin Exp Med 2017; 10: 5805–5810. [Google Scholar]
- 96.Lutherer LO, Nugent KM, Schoettle BW, et al. . Low-dose oral interferon alpha possibly retards the progression of idiopathic pulmonary fibrosis and alleviates associated cough in some patients. Thorax 2011; 66: 446–447. doi: 10.1136/thx.2010.135947 [DOI] [PubMed] [Google Scholar]
- 97.Mapel DW, Hunt WC, Utton R, et al. . Idiopathic pulmonary fibrosis: survival in population based and hospital based cohorts. Thorax 1998; 53: 469–476. doi: 10.1136/thx.53.6.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marulli G, Lay M, Di Chiara F, et al. . End stage idiopathic pulmonary fibrosis: which patients are at higher risk of mortality awaiting lung transplantation? J Heart Lung Transplant 2010; 1: S114. doi: 10.1016/j.healun.2009.11.351 [DOI] [Google Scholar]
- 99.Mason DP, Brizzio ME, Alster JM, et al. . Lung transplantation for idiopathic pulmonary fibrosis. Ann Thorac Surg 2007; 84: 1121–1128. doi: 10.1016/j.athoracsur.2007.04.096 [DOI] [PubMed] [Google Scholar]
- 100.McKeown S, Richter AG, O'Kane C, et al. . MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur Respir J 2009; 33: 77–84. doi: 10.1183/09031936.00060708 [DOI] [PubMed] [Google Scholar]
- 101.Meier-Sydow J, Connette B, Hubel E, et al. . Long term development of idiopathic pulmonary fibrosis. Combined therapy with immunodepressors, D-penicillamine and corticosteroids. Broncho-Pneumologie 1979; 29: 249–254. [PubMed] [Google Scholar]
- 102.Meier-Sydow J, Rust M, Kronenberger H, et al. . Survival of patients with idiopathic pulmonary fibrosis following treatment with azathioprine, d-penicillamine or prednisone. Chest 1990; 98: 18S. doi: 10.1378/chest.98.2_Supplement.18S [DOI] [Google Scholar]
- 103.Mermigkis C, Bouloukaki I, Antoniou K, et al. . Obstructive sleep apnea should be treated in patients with idiopathic pulmonary fibrosis. Sleep Breath 2015; 19: 385–391. doi: 10.1007/s11325-014-1033-6 [DOI] [PubMed] [Google Scholar]
- 104.Mirrani G, Wootton C, Foley NM. Median survival and cause of death in unselected patients with idiopathic pulmonary fibrosis in the UK. Am J Respir Crit Care Med 2012; 185: A4373. [Google Scholar]
- 105.Moeller A, Gilpin SE, Ask K, et al. . Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179: 588–594. doi: 10.1164/rccm.200810-1534OC [DOI] [PubMed] [Google Scholar]
- 106.Mogulkoc N, Brutsche MH, Bishop PW, et al. . Pulmonary function in idiopathic pulmonary fibrosis and referral for lung transplantation. Am J Respir Crit Care Med 2001; 164: 103–108. doi: 10.1164/ajrccm.164.1.2007077 [DOI] [PubMed] [Google Scholar]
- 107.Moore A, Dave K, Vergnaud S, et al. . Differing clinical courses of IPF at an acute district general hospital. Eur Respir J 2013; 42: P3379. [Google Scholar]
- 108.Morawiec E, Tillie-Leblond I, Pansini V, et al. . Exacerbations of idiopathic pulmonary fibrosis treated with corticosteroids and cyclophosphamide pulses. Eur Respir J 2011; 38: 1487–1489. doi: 10.1183/09031936.00127311 [DOI] [PubMed] [Google Scholar]
- 109.Moua T, Westerly BD, Dulohery MM, et al. . Patients with fibrotic interstitial lung disease hospitalized for acute respiratory worsening: a large cohort analysis. Chest 2016; 149: 1205–1214. doi: 10.1016/j.chest.2015.12.026 [DOI] [PubMed] [Google Scholar]
- 110.Mura M, Porretta MA, Bargagli E, et al. . Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J 2012; 40: 101–109. doi: 10.1183/09031936.00106011 [DOI] [PubMed] [Google Scholar]
- 111.Nadrous HF, Ryu JH, Douglas WW, et al. . Impact of angiotensin-converting enzyme inhibitors and statins on survival in idiopathic pulmonary fibrosis. Chest 2004; 126: 438–446. doi: 10.1016/S0012-3692(15)31155-7 [DOI] [PubMed] [Google Scholar]
- 112.Nagai S, Kitaichi M, Itoh H, et al. . Idiopathic nonspecific interstitial pneumonia/fibrosis: comparison with idiopathic pulmonary fibrosis and BOOP. Eur Respir J 1998; 12: 1010–1019. doi: 10.1183/09031936.98.12051010 [DOI] [PubMed] [Google Scholar]
- 113.Nambiar AM, Anzueto AR, Peters JI. Effectiveness and safety of mycophenolate mofetil in idiopathic pulmonary fibrosis. PLoS One 2017; 12: e0176312. doi: 10.1371/journal.pone.0176312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Natsuizaka M, Chiba H, Kuronuma K, et al. . Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med 2014; 190: 773–779. doi: 10.1164/rccm.201403-0566OC [DOI] [PubMed] [Google Scholar]
- 115.Nicholson AG, Colby TV, du Bois RM, et al. . The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162: 2213–2217. doi: 10.1164/ajrccm.162.6.2003049 [DOI] [PubMed] [Google Scholar]
- 116.Nicol LM, Muralidharan V, Mills R, et al. . The clinical value and feasibility of repeat bronchoalveolar lavage in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2015; 191: A2526. [Google Scholar]
- 117.Nishiyama O, Yamazaki R, Sano A, et al. . Prognostic value of forced expiratory volume in 1 second/forced vital capacity in idiopathic pulmonary fibrosis. Chron Respir Dis 2016; 13: 40–47. doi: 10.1177/1479972315603714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nishiyama O, Yamazaki R, Sano H, et al. . Fat-free mass index predicts survival in patients with idiopathic pulmonary fibrosis. Respirology 2017; 22: 480–485. doi: 10.1111/resp.12941 [DOI] [PubMed] [Google Scholar]
- 119.Oda K, Ishimoto H, Yatera K, et al. . High-resolution CT scoring system-based grading scale predicts the clinical outcomes in patients with idiopathic pulmonary fibrosis. Respir Res 2014; 15: 10. doi: 10.1186/1465-9921-15-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Oldham JM, Kumar D, Lee C, et al. . Thyroid disease is prevalent and predicts survival in patients with idiopathic pulmonary fibrosis. Chest 2015; 148: 692–700. doi: 10.1378/chest.14-2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Prasse A, Probst C, Bargagli E, et al. . Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179: 717–723. doi: 10.1164/rccm.200808-1201OC [DOI] [PubMed] [Google Scholar]
- 122.Raghu G, Chen SY, Yeh WS, et al. . Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2: 566–572. doi: 10.1016/S2213-2600(14)70101-8 [DOI] [PubMed] [Google Scholar]
- 123.Raimundo K, Broder M, Chang E, et al. . Mortality risk of patients with idiopathic pulmonary fibrosis. Chest 2016; 150: 484A. doi: 10.1016/j.chest.2016.08.498 [DOI] [Google Scholar]
- 124.Rajasekaran A, Shovlin D, Saravanan V, et al. . Interstitial lung disease in patients with rheumatoid arthritis: comparison with cryptogenic fibrosing alveolitis over 5 years. J Rheumatol 2006; 33: 1250–1253. [PubMed] [Google Scholar]
- 125.Rangappa P, Moran JL. Outcomes of patients admitted to the intensive care unit with idiopathic pulmonary fibrosis. Crit Care Resusc 2009; 11: 102–109. [PubMed] [Google Scholar]
- 126.Reid T, Vennelle M, McKinley M, et al. . Sleep-disordered breathing and idiopathic pulmonary fibrosis--is there an association? Sleep Breath 2015; 19: 719–721. doi: 10.1007/s11325-014-1117-3 [DOI] [PubMed] [Google Scholar]
- 127.Richards TJ, Kaminski N, Baribaud F, et al. . Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012; 185: 67–76. doi: 10.1164/rccm.201101-0058OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rogers K, Hadinnapola C, Sylvester K, et al. . Identification of clinical prognostic parameters in patients with idiopathic pulmonary fibrosis. Thorax 2016; 71: A14. doi: 10.1136/thoraxjnl-2016-209333.27 [DOI] [Google Scholar]
- 129.Roig V, Herrero A, Arroyo-Cozar M, et al. . Comparative study between oral azathioprine and intravenous cyclophosphamide pulses in the treatment of idiopathic pulmonary fibrosis. Arch Bronconeumol 2010; 46: 15–19. doi: 10.1016/j.arbres.2009.09.015 [DOI] [PubMed] [Google Scholar]
- 130.Roskell N, Saunders O, Lee D, et al. . Long-term survival analysis: pirfenidone compared to standard care for the treatment of patients with idiopathic pulmonary fibrosis. Eur Respir J 2014; 44: 1905. [Google Scholar]
- 131.Rubin AS, Nascimento DZ, Sanchez L, et al. . Functional improvement in patients with idiopathic pulmonary fibrosis undergoing single lung transplantation. J Bras Pneumol 2015; 41: 299–304. doi: 10.1590/S1806-37132015000000057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rudd RM, Prescott RJ, Chalmers JC, et al. . British Thoracic Society Study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62: 62–66. doi: 10.1136/thx.2005.045591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Russell AM, Adamali H, Molyneaux PL, et al. . Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016; 194: 989–997. doi: 10.1164/rccm.201511-2152OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ryerson CJ, Vittinghoff E, Ley B, et al. . Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest 2014; 145: 723–728. doi: 10.1378/chest.13-1474 [DOI] [PubMed] [Google Scholar]
- 135.Sakamoto S, Homma S, Miyamoto A, et al. . Cyclosporin A in the treatment of acute exacerbation of idiopathic pulmonary fibrosis. Intern Med 2010; 49: 109–115. doi: 10.2169/internalmedicine.49.2359 [DOI] [PubMed] [Google Scholar]
- 136.Sato S, Tanino Y, Misa K, et al. . Identification of clinical phenotypes in idiopathic interstitial pneumonia with pulmonary emphysema. Intern Med 2016; 55: 1529–1535. doi: 10.2169/internalmedicine.55.6009 [DOI] [PubMed] [Google Scholar]
- 137.Saydain G, Islam A, Afessa B, et al. . Outcome of patients with idiopathic pulmonary fibrosis admitted to the intensive care unit. Am J Respir Crit Care Med 2002; 166: 839–842. doi: 10.1164/rccm.2104038 [DOI] [PubMed] [Google Scholar]
- 138.Schmidt SL, Nambiar AM, Tayob N, et al. . Pulmonary function measures predict mortality differently in IPF versus combined pulmonary fibrosis and emphysema. Eur Respir J 2011; 38: 176–183. doi: 10.1183/09031936.00114010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schupp JC, Binder H, Jager B, et al. . Macrophage activation in acute exacerbation of idiopathic pulmonary fibrosis. PLoS One 2015; 10: e0116775. doi: 10.1371/journal.pone.0116775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Selman M, Carrillo G, Salas J, et al. . Colchicine, D-penicillamine, and prednisone in the treatment of idiopathic pulmonary fibrosis: a controlled clinical trial. Chest 1998; 114: 507–512. doi: 10.1378/chest.114.2.507 [DOI] [PubMed] [Google Scholar]
- 141.Selman M, Carrillo G, Estrada A, et al. . Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One 2007; 2: e482. doi: 10.1371/journal.pone.0000482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Serrano-Mollar A, Mapel;-Jordi G, Guillamat-Prats R, et al. . Safety and tolerability of alveolar type II cell transplantation in idiopathic pulmonary fibrosis. Chest 2016; 150: 533–543. doi: 10.1016/j.chest.2016.03.021 [DOI] [PubMed] [Google Scholar]
- 143.Shafiq M, Muzaffar B, Esper AM, et al. . Progression of disease in connective tissue-related interstitial lung disease vs idiopathic pulmonary fibrosis. Chest 2010; 138: 861A. doi: 10.1378/chest.10164 [DOI] [Google Scholar]
- 144.Sherbini N, Feteih MN, Wali SO, et al. . Idiopathic pulmonary fibrosis in Saudi Arabia: demographic, clinical, and survival data from two tertiary care hospitals. Ann Thorac Med 2014; 9: 168–172. doi: 10.4103/1817-1737.134073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shin KM, Lee KS, Chung MP, et al. . Prognostic determinants among clinical, thin-section CT, and histopathologic findings for fibrotic idiopathic interstitial pneumonias: Tertiary hospital study. Radiology 2008; 249: 328–337. doi: 10.1148/radiol.2483071378 [DOI] [PubMed] [Google Scholar]
- 146.Shinoda H, Tasaka S, Fujishima S, et al. . Elevated CC chemokine level in bronchoalveolar lavage fluid is predictive of a poor outcome of idiopathic pulmonary fibrosis. Respiration 2009; 78: 285–292. doi: 10.1159/000207617 [DOI] [PubMed] [Google Scholar]
- 147.Shitrit D, Rusanov V, Peled N, et al. . The 15-step oximetry test: a reliable tool to identify candidates for lung transplantation among patients with idiopathic pulmonary fibrosis. J Heart Lung Transplant 2009; 28: 328–333. doi: 10.1016/j.healun.2008.12.019 [DOI] [PubMed] [Google Scholar]
- 148.Shlobin OA, Edwards E, Nathan SD. Waiting times and mortality for IPF patients listed for bilateral or single lung transplantation. J Heart Lung Transplant 2009; 1: S168. doi: 10.1016/j.healun.2008.11.302 [DOI] [PubMed] [Google Scholar]
- 149.Siemienowicz ML, Kruger SJ, Goh NS, et al. . Agreement and mortality prediction in high-resolution CT of diffuse fibrotic lung disease. J Med Imaging Radiat Oncol 2015; 59: 555–563. doi: 10.1111/1754-9485.12314 [DOI] [PubMed] [Google Scholar]
- 150.Soares Pires F, Caetano Mota P, Melo N, et al. . Idiopathic pulmonary fibrosis – clinical presentation, outcome and baseline prognostic factors in a Portuguese cohort. Rev Port Pneumol 2013; 19: 19–27. doi: 10.1016/j.rppneu.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 151.Soares MR, Pereira C, Ferreira R, et al. . A score for estimating survival in idiopathic pulmonary fibrosis with rest SpO2>88. Sarcoidosis Vasc Diffuse Lung Dis 2015; 32: 121–128. [PubMed] [Google Scholar]
- 152.Sobiecka M, Onish K, Boros P, et al. . Clinical characteristics and survival in patients with idiopathic pulmonary fibrosis with and without associated emphysema. Eur Respir J 2013; 42: Suppl. 57, P3376. [Google Scholar]
- 153.Song JW, Hong SB, Lim CM, et al. . Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37: 356–363. doi: 10.1183/09031936.00159709 [DOI] [PubMed] [Google Scholar]
- 154.Song DH, Choi IH, Ha SY, et al. . Usual interstitial pneumonia with lung cancer: clinicopathological analysis of 43 cases. Korean J Pathol 2014; 48: 10–16. doi: 10.4132/KoreanJPathol.2014.48.1.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Stack BH, Choo-Kang YF, Heard BE. The prognosis of cryptogenic fibrosing alveolitis. Thorax 1972; 27: 535–542. doi: 10.1136/thx.27.5.535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Strand MJ, Sprunger D, Cosgrove GP, et al. . Pulmonary function and survival in idiopathic vs secondary usual interstitial pneumonia. Chest 2014; 146: 775–785. doi: 10.1378/chest.13-2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Strongman H, Kausar I, Maher TM. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv Ther 2018; 35: 724–736. doi: 10.1007/s12325-018-0693-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Su R, Bennett M, Jacobs S, et al. . An analysis of connective tissue disease-associated interstitial lung disease at a US Tertiary Care Center: better survival in patients with systemic sclerosis. J Rheumatol 2011; 38: 693–701. doi: 10.3899/jrheum.100675 [DOI] [PubMed] [Google Scholar]
- 159.Takahashi H, Fujishima T, Koba H, et al. . Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med 2000; 162: 1109–1114. doi: 10.1164/ajrccm.162.3.9910080 [DOI] [PubMed] [Google Scholar]
- 160.Ten Klooster L, van Moorsel CH, Kwakkel-van Erp JM, et al. . Immunoglobulin A in serum: an old acquaintance as a new prognostic biomarker in idiopathic pulmonary fibrosis. Clin Exp Immunol 2015; 181: 357–361. doi: 10.1111/cei.12636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tokgoz Akyil F, Sevim T, Akman C, et al. . The predictors of mortality in IPF. Does emphysema change the prognosis? Sarcoidosis Vasc Diffuse Lung Dis 2016; 33: 267–274. [PubMed] [Google Scholar]
- 162.Tolle LB, Tayob N, Nambiar A, et al. . The role of high resolution CT patterns in prognosticating IPF. Am J Respir Crit Care Med 2014; 189: 1–3.24381984 [Google Scholar]
- 163.Tomioka H, Imanaka K, Hashimoto K, et al. . Health-related quality of life in patients with idiopathic pulmonary fibrosis – cross-sectional and longitudinal study. Intern Med 2007; 46: 1533–1542. doi: 10.2169/internalmedicine.46.6218 [DOI] [PubMed] [Google Scholar]
- 164.Tryfon S, Saroglou M, Stanopoulos I, et al. . Survival prognostic validity of cardiac output and central venous saturation measured during ergometry in idiopathic pulmonary fibrosis patients. Chest 2009; 136: 54S. doi: 10.1378/chest.136.4_MeetingAbstracts.54S-f [DOI] [Google Scholar]
- 165.Tzouvelekis A, Paspaliaris V, Koliakos G, et al. . A prospective, non-randomized, no placebo-controlled, phase Ib clinical trial to study the safety of the adipose derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. J Transl Med 2013; 11: 171. doi: 10.1186/1479-5876-11-171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tzouvelekis A, Herazo-Maya JD, Slade M, et al. . Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology 2017; 22: 486–493. doi: 10.1111/resp.12920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Umeda Y, Demura Y, Morikawa M, et al. . Prognostic value of dual-time-point 18F-FDG PET for idiopathic pulmonary fibrosis. J Nucl Med 2015; 56: 1869–1875. doi: 10.2967/jnumed.115.163360 [DOI] [PubMed] [Google Scholar]
- 168.Vysehradsky R, Rozborilova E, Strakova J, et al. . Potential predictors of therapeutic response in patients with cryptogenic fibrosing alveolitis. Vnitr Lek 2002; 48: 185–191. [PubMed] [Google Scholar]
- 169.Wallaert B, Guetta A, Wemeau-Stervinou L, et al. . Prognostic value of clinical exercise testing in idiopathic pulmonary fibrosis. Rev Mal Respir 2011; 28: 290–296. doi: 10.1016/j.rmr.2010.08.016 [DOI] [PubMed] [Google Scholar]
- 170.Wilkie MEM, Chalmers JD, Smith RP, et al. . Comparison of two prognostic tools for identifying high risk patients with idiopathic pulmonary fibrosis. Thorax 2012; 67: A120. [Google Scholar]
- 171.Wright PH, Heard BE, Steel SJ, et al. . Cryptogenic fibrosing alveolitis: assessment by graded trephine lung biopsy histology compared with clinical, radiographic, and physiological features. Br J Dis Chest 1981; 75: 61–70. doi: 10.1016/S0007-0971(81)80008-3 [DOI] [PubMed] [Google Scholar]
- 172.Yokoyama A, Kondo K, Nakajima M, et al. . Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 2006; 11: 164–168. doi: 10.1111/j.1440-1843.2006.00834.x [DOI] [PubMed] [Google Scholar]
- 173.Yu Y, Goehring E, Nguyen-Khoa BA, et al. . Comorbidity burden and healthcare resource use in patients with idiopathic pulmonary fibrosis (IPF) in the United States (US) military health system. Chest 2014; 146: 373A. doi: 10.1378/chest.199163524626819 [DOI] [Google Scholar]
- 174.Zhang L, Zhang C, Dong F, et al. . Combined pulmonary fibrosis and emphysema: a retrospective analysis of clinical characteristics, treatment and prognosis. BMC Pulm Med 2016; 16: 137. doi: 10.1186/s12890-016-0300-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zurkova M, Kolek V, Lostakova V, et al. . The effect of pirfenidone on impaired lung function in patients with idiopathic pulmonary fibrosis (IPF) from Czech IPF registry. Eur Respir J 2016; 48: Suppl. 60, PA2087. [Google Scholar]
- 176.Daniels CE, Lasky JA, Limper AH, et al. . Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results. Am J Respir Crit Care Med 2010; 181: 604–610. doi: 10.1164/rccm.200906-0964OC [DOI] [PubMed] [Google Scholar]
- 177.Demedts M, Behr J, Buhl R, et al. . High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2005; 353: 2229–2242. doi: 10.1056/NEJMoa042976 [DOI] [PubMed] [Google Scholar]
- 178.King TE Jr., Behr J, Brown KK, et al. . BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008; 177: 75–81. doi: 10.1164/rccm.200705-732OC [DOI] [PubMed] [Google Scholar]
- 179.King TE Jr, Albera C, Bradford WZ, et al. . Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet 2009; 374: 222–228. doi: 10.1016/S0140-6736(09)60551-1 [DOI] [PubMed] [Google Scholar]
- 180.King TE Jr.Brown KK, Raghu G, et al. . BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 184: 92–99. doi: 10.1164/rccm.201011-1874OC [DOI] [PubMed] [Google Scholar]
- 181.Malouf MA, Hopkins P, Snell G, et al. . An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology 2011; 16: 776–783. doi: 10.1111/j.1440-1843.2011.01955.x [DOI] [PubMed] [Google Scholar]
- 182.Idiopathic Pulmonary Fibrosis Clinical Research Network , Martinez FJ, de Andrade JA, et al. . Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2093–2101. doi: 10.1056/NEJMoa1401739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Noble PW, Albera C, Bradford WZ, et al. . Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377: 1760–1769. doi: 10.1016/S0140-6736(11)60405-4 [DOI] [PubMed] [Google Scholar]
- 184.Parker J, Glaspole I, Lancaster L, et al. . A phase 2 randomised placebo-controlled study of tralokinumab in subjects with idiopathic pulmonary fibrosis (IPF). QJM 2016; 109: S46. [DOI] [PubMed] [Google Scholar]
- 185.Raghu G, Behr J, Brown KK, et al. . Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med 2013; 158: 641–649. doi: 10.7326/0003-4819-158-9-201305070-00003 [DOI] [PubMed] [Google Scholar]
- 186.Raghu G, Million-Rousseau R, Morganti A, et al. . Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J 2013; 42: 1622–1632. doi: 10.1183/09031936.00104612 [DOI] [PubMed] [Google Scholar]
- 187.Raghu G, Martinez FJ, Brown KK, et al. . CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur Respir J 2015; 46: 1740–1750. doi: 10.1183/13993003.01558-2014 [DOI] [PubMed] [Google Scholar]
- 188.Richeldi L, Costabel U, Selman M, et al. . Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 2011; 365: 1079–1087. doi: 10.1056/NEJMoa1103690 [DOI] [PubMed] [Google Scholar]
- 189.Taniguchi H, Ebina M, Kondoh Y, et al. . Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2010; 35: 821–829. doi: 10.1183/09031936.00005209 [DOI] [PubMed] [Google Scholar]
- 190.Kupferberg DH, Kaplan RM, Slymen DJ, et al. . Minimal clinically important difference for the UCSD Shortness of Breath Questionnaire. J Cardiopulm Rehabil 2005; 25: 370–377. doi: 10.1097/00008483-200511000-00011 [DOI] [PubMed] [Google Scholar]
- 191.Swigris JJ, Brown KK, Behr J, et al. . The SF-36 and SGRQ: Validity and first look at minimum important differences in IPF. Respir Med 2010; 104: 296–304. doi: 10.1016/j.rmed.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Holland AE, Spruit MA, Troosters T, et al. . An official European Respiratory Society/American Thoracic Society technical standard: field walking tests in chronic respiratory disease. Eur Respir J 2014; 44: 1428–1446. doi: 10.1183/09031936.00150314 [DOI] [PubMed] [Google Scholar]
- 193.Ryerson CJ, Kolb M. The increasing mortality of idiopathic pulmonary fibrosis: fact or fallacy? Eur Respir J 2018; 51: 1702420. doi: 10.1183/13993003.02420-2017 [DOI] [PubMed] [Google Scholar]
- 194.Nathan SD, Albera C, Bradford WZ, et al. . Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5: 33–41. doi: 10.1016/S2213-2600(16)30326-5 [DOI] [PubMed] [Google Scholar]
- 195.Idiopathic Pulmonary Fibrosis Clinical Research Network , Raghu G, Anstrom KJ, et al. . Prednisolone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366: 1968–1977. doi: 10.1056/NEJMoa1113354 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary_material ERR-0158-2019_Supplementary_material (3.7MB, pdf)