

Abstract
The finding of collections of macrophages/histiocytes in lung biopsy and bronchoalveolar lavage is relatively common in routine practice. This morphological feature in itself is pathological, but the exact clinical significance and underlying disease should be evaluated together with clinical data, functional respiratory and laboratory tests and imaging studies.
Morphological characteristics of macrophages and their distribution along the different pulmonary structures should be examined carefully by pathologists. Indeed, haemosiderin-laden macrophages are associated with smoking-related diseases when pigment is fine and distribution is bronchiolocentric, while alveolar haemorrhage or pneumoconiosis are the main concerns when pigment is chunky or coarse and the macrophages show an intra-alveolar or perilymphatic location, respectively. In the same way, pulmonary accumulation of macrophages with foamy cytoplasm is generally associated with pathologies leading to broncho-bronchiolar obstruction (e.g. diffuse panbronchiolitis, hypersensitivity pneumonia or cryptogenic organising pneumonia) or alternatively to exogenous lipoid pneumonia, some drug toxicity (e.g. amiodarone exposure or toxicity) and metabolic disorders (e.g. type B Niemann–Pick disease).
This pathology-based perspectives article is aimed at concisely describing the diagnostic possibilities when faced with collection of macrophages in lung biopsy and cytology.
Short abstract
Morphology and localisation of macrophages in the lungs is helpful in the diagnosis of interstitial lung disease http://ow.ly/7bci30bBwmQ
Introduction
In the lung, macrophages and histiocytes play a key role in inflammatory and phagocytic processes through complex production of cytokines and cellular interaction [1, 2].
Recent studies have demonstrated dynamic changes of circulating monocytes, lung alveolar macrophages and interstitial macrophages with M1 (pro-inflammatory) and M2 (anti-inflammatory) phenotypes in preclinical models of interstitial lung diseases (ILDs) [3].
Nevertheless, the presence of collections of macrophages is considered a pathologic feature, although the exact clinical significance may range in a single case. Indeed, the occurrence of several macrophages in the lung often represent an incidental finding, while in other cases macrophages are indeed the problem.
Among ILDs, the careful evaluation of the morphological characteristics and the exact localisation of the macrophages disclosed in a lung biopsy may be fundamental diagnostic clues for pathologists. Pathologists have different diagnostic methodologies to identify macrophage-based lung lesions with various diagnostic yields, including bronchoalveolar lavage (BAL), transbronchial biopsy, transbronchial cryobiopsy and surgical lung biopsy (table 1) [4, 5].
TABLE 1.
Diagnostic yield of the different sampling techniques in macrophage-rich pulmonary diseases
| Bronchoalveolar lavage | Transbronchial biopsy | Transbronchial cryobiopsy | Surgical lung biopsy | |
| Respiratory bronchiolitis interstitial lung disease | + | ++ | +++ | +++ |
| Desquamative interstitial pneumonia | + | ++ | +++ | +++ |
| Langerhans cell histiocytosis | + | ++ | +++ | +++ |
| Extrinsic allergic alveolitis/hypersensitivity pneumonitis | + | ++ | +++ | +++ |
| Diffuse pan-bronchiolitis | + | ++ | +++ | +++ |
| Exogenous lipoid pneumonitis | ++ | ++ | +++ | +++ |
| Pneumoconiosis | + | ++ | +++ | +++ |
| Pulmonary alveolar proteinosis | +++ | ++ | +++ | +++ |
| Erdheim–Chester disease | − | + | ++ | +++ |
| Rosai–Dorfman disease | − | + | ++ | +++ |
| Gaucher disease | +# | + | ++ | +++ |
| Niemann–Pick disease | +# | + | ++ | +++ |
–: almost never diagnostic; +: weakly diagnostic; ++: moderately diagnostic; +++: almost always diagnostic. #: bronchoalveolar lavage is helpful in supporting the diagnosis once clinically suspected, but rarely diagnostic per se.
Several histochemical stains and immunohistochemical markers are used to recognise histiocytes in cytology and biopsy. Among histochemical stains oil red O or Sudan black are useful in recognising lipid-rich histiocytes on fresh material (alcohol-based fixation used for conventional haematoxylin–eosin stain digest the lipids that are not visible in formalin-fixed paraffin-embedded samples), while Perls’ Prussian blue stains the iron-laden macrophages. Periodic acid–Schiff (PAS) and May–Giemsa stains are helpful in differentiating some metabolic diseases. Among immunohistochemical markers, CD68, S-100 protein, CD1a, langerin, CD56, CD163, CD4, lysozyme, α1-antitrypsin and factor XIIIa are the most common primary antibodies used to recognise different histiocytic disorders [6]. Of note, there is evidence that human alveolar macrophages from donors are maintained in the lung parenchyma for several years after lung transplantation, possibly resulting in an important mechanism for the development of chronic graft rejection [7].
This article summarises the most important features of lung pathologies characterised by the accumulation of histiocytes (with the exclusion of granulomatous processes) and provides a practical approach to the diagnosis of ILDs from the “viewpoint” of the macrophages.
Pathologies consisting of accumulation of macrophages containing fine haemosiderin pigment
The accumulation of yellow-brown, haemosiderin-laden, pigmented macrophages (“smokers’ macrophages”) within the lumens of respiratory bronchioles and into the peribronchiolar alveolar spaces (figure 1) is seen very frequently and in the great majority of cases secondary to a cigarette smoking habit [8–11]. Macrophages initially fill the lumen of the respiratory bronchioles and peribronchiolar alveoli leading to a specific type of bronchiolitis, namely respiratory bronchiolitis. This histological finding is associated with a patchy submucosal and peribronchiolar chronic inflammation with or without mucostasis and occurs in virtually all cigarette smokers; patients are generally asymptomatic or paucisymptomatic and histological stigmata of respiratory bronchiolitis may persist for decades after smoking cessation [9].
FIGURE 1.

a) Smokers’ macrophages show a fine, golden haemosiderin cytoplasmic pigment; b) in respiratory bronchiolitis (RB), smokers’ macrophages are located in the bronchiolar lumen and peribronchiolar alveoli; in c) RB-associated interstitial lung disease and d) desquamative interstitial pneumonia, smokers’ macrophages progressively expand to the peripheral alveoli.
In some cases, particularly in heavy smokers, alveolar accumulation of pigmented macrophages extends from peribronchiolar zones to the periphery of the lobules, determining an ILD affecting the upper lung lobes with centrilobular nodules and ground-glass opacities at high-resolution computed tomography (HRCT) [12–14].
This phenomenon is defined as RB-ILD when the subpleural alveoli are not involved in the process, while the misnomer “desquamative interstitial pneumonia” (DIP) [15] is used when the entire lobule is involved by accumulation of smokers’ macrophages (although the differential diagnosis between RB-ILD and DIP is sometimes arbitrary and has no clinical significance). Respiratory bronchiolitis, RB-ILD and DIP do represent a continuous spectrum of histological lesions secondary to their accumulation of macrophages into the peripheral zone of the lungs and secondary to cigarette smoking in the great majority of cases (also collected under the umbrella term “smoking-related ILDs”).
A subset of patients with RB-ILD and DIP have an insidious presentation with exertional dyspnoea, symptomatic wheezing, persistent cough and concomitant functional impairment and imaging abnormalities at chest radiography and HRCT [13–15]. In most cases, RB-ILD and DIP show a relatively stable clinical course with long-term outcomes and good prognosis.
From the pathologist's viewpoint, it is important to underline that respiratory bronchiolitis, RB/ILD and DIP represent diagnoses of exclusion in all cases [13–15]. In other words, since the accumulation of smokers’ macrophages in a lung biopsy or BAL are nonspecific morphological features, it is mandatory to ascertain that they represent the real and unique problem of the patient. Indeed, respiratory bronchiolitis, RB-ILD and DIP may be observed concurrently in other more aggressive diseases, namely lung cancer and idiopathic pulmonary fibrosis. Keeping this in mind, a diagnosis of RB-ILD or DIP on small biopsy or BAL may be considered only in an adequate clinicoradiologic context.
In addition, both primary and metastatic lung tumours are rarely associated with a peculiar discohesive growth pattern resulting in airspace filling by macrophage-like tumour cells, closely simulating RB-ILD and DIP. The malignant cytology of the tumour nuclei and immunohistochemical stains are the clues in differential diagnosis [16].
However, a subset of DIP occurs in nonsmokers and is related to occupational inhaled toxins, drug toxicity and connective tissue diseases [17]. In addition, the DIP pattern has been associated with genetic diseases, such as ABCA3 mutations [18].
Pathologies consisting of accumulation of macrophages containing coarse haemosiderin pigment
The finding of accumulations of macrophages with intracytoplasmic deposits of coarse or chunky haemosiderin pigment is entirely characteristic of various diseases, possibly leading to chronic haemorrhage [19] (figure 2).
FIGURE 2.

In chronic haemorrhage a) several pigmented macrophages fill the alveoli with dense fibrosis of the interstitium; b) the haemosiderin pigment in macrophages is coarser than that observed in smokers’ macrophages.
In chronic haemorrhage, apart from the presence of chunky haemosiderin pigment, there is some interstitial fibrosis and other features of alveolar bleeding, such as intra-alveolar fibrin intermingled with blood red cells, foci of organising pneumonia and capillaritis.
In some cases, the differential diagnosis between smoking-related ILD and chronic haemorrhage is very difficult and requires a striking integration with clinical, laboratory and imaging data.
Accumulation of macrophages with coarse haemosiderin pigment in the lung parenchyma may be observed in other pulmonary diseases, such as some pneumoconiosis (asbestosis and siderosis) and pulmonary vascular diseases (e.g. veno-occlusive disease) [20, 21].
The haemosiderin content of alveolar macrophages may be evaluated according to the Golde scoring system, considering the number of cells and the type of haemosiderin content using a special iron stain (e.g. Perls’ Prussian blue) [22].
Pathologies consisting of accumulation of macrophages containing exogenous material and crystals
Accumulation of macrophages is always present in pneumoconiosis (figure 3) such as silicosis [23, 24]. Indeed, the early phase of silicosis is characterised by the aggregation of “dirty macrophages” along the lymphatic routes. These macrophages show carbon particles and birefringent material into the cytoplasm, while the typical fibrotic nodules with “onion-skin” pattern appear only in the subsequent phases of the disease (figure 4). The increased risk of alveolar proteinosis and infection (e.g. tuberculosis) occurring in silicosis seems to be related to the lack of function of macrophages [24].
FIGURE 3.
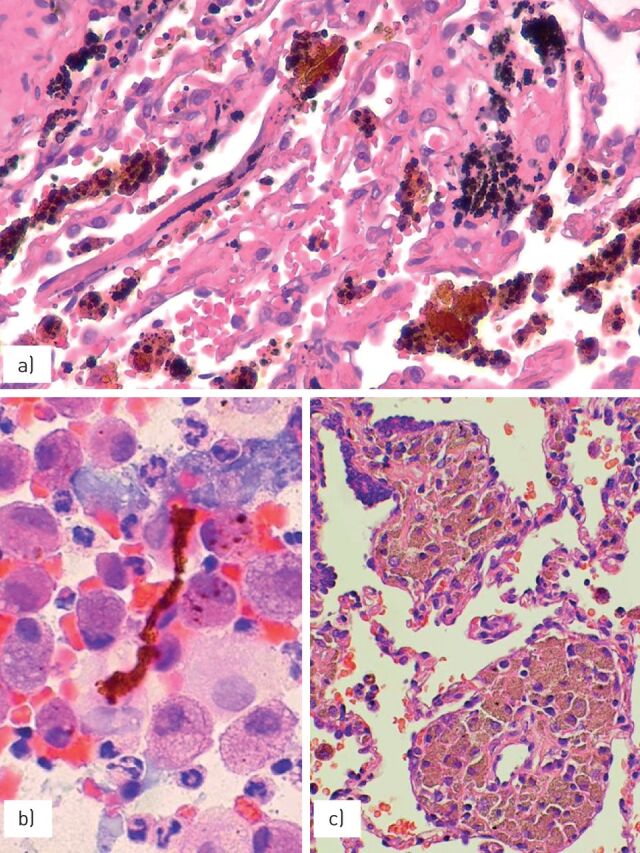
a) The accumulation of haemosiderin pigment may be observed in some cases of pneumoconiosis, such as b) asbestosis with the presence of asbestos bodies showing a linear non-birifringent “core” attracting ferrous material or c) pneumosiderosis (welder's lung) with bronchiolitis and sheets of macrophages having cytoplasmic iron pigment.
FIGURE 4.

Silicosis is characterised by a) irregular, stellate-shaped nodules along the lymphatic routes, with b) “dirty” macrophages and comparison of dense fibrosis in chronic phases.
In different forms of pneumoconiosis, the exogenous material is covered by iron leading to ferruginous bodies. The asbestos body is a peculiar type of ferruginous body in which the exogenous material is represented by the asbestos corpuscles. This is unique and consists of a transparent, not birefringent and linear corpuscle [8].
Intravenous talcosis is another form of pneumoconiosis caused by the injection of material containing silicates and characterised by the intravascular accumulation of exogenous material [25].
Chronic aspiration pneumonitis is a relatively common, but often underrated pathology characterised by the finding of mono-/multinucleated macrophages with intracytoplasmic alimentary digested debris or other exogenous material in the background of chronic bronchiolocentric damage with bronchiolar metaplasia and inflammatory reaction. Exogenous lipoid pneumonitis is a peculiar variant of aspiration pneumonitis due to prolonged aspiration of mineral oils leading to a pulmonary histological lesion identical to the subcutaneous paraffinoma with areas of dense fibrosis including clear vacuoles of different size, often surrounded by foamy macrophages (figure 5). Examination of BAL fluid may be used to diagnose exogenous lipoid pneumonitis when numerous lipid-laden macrophages are disclosed in the correct clinicoradiological setting [26].
FIGURE 5.

a) Lipoid exogenous pneumonia on bronchoalveolar lavage is characterised by macrophages with several intracytoplasmic vacuoles of various sizes; b) in chronic phases, the lesion is partially substituted by fibrosis and chronic inflammation, leading to a nodular mass known as paraffinoma.
The finding of multinucleated macrophages with intracytoplasmic cholesterol crystals/clefts is a frequent and nonspecific histological finding in lung biopsies. Nevertheless, their presence in the background of a cellular (lymphocyte and plasma cell-rich infiltrate) interstitial pneumonitis is an important morphological circumstantial proof favouring hypersensitivity pneumonitis (allergic extrinsic alveolitis), particularly when the inflammatory process is accentuated in the centrolobular regions [27, 28].
The finding of numerous macrophages with intracytoplasmic cholesterol clefts under the pleura or in the peripheral regions of the lung should raise the possibility of a pulmonary alveolar proteinosis in the healing phase. In this case, a meticulous search for focal areas including alveoli containing the characteristic eosinophilic, PAS-positive material is mandatory to confirm the diagnosis [29].
Rarely, primary pulmonary lymphomas (e.g. marginal zone or mucosa-associated lymphoid tissue lymphoma and lymphoplasmacytic lymphoma) or lung involvement by plasma cell proliferations (multiple myeloma or plasma cell disorders) and lymphomas are associated with aggregates of macrophages containing intracytoplasmic eosinophilic, refractile, rod-shaped crystals due to intralysosomal accumulation of immunoglobulin light chain crystals (figure 6). This condition is called crystal-storing histiocytosis [30] and has been described in some reactive processes secondary to non-neoplastic immune-mediated diseases (i.e. rheumatoid arthritis) or the use of clofazimine for the treatment of leprosy [31].
FIGURE 6.

a) Crystal-storing histiocytosis with sheets of macrophages containing intracytoplasmic eosinophilic crystal of immunoglobulin in a plasmacytoma and b) mucosa-associated lymphoid tissue-type lymphoma.
Chronic eosinophilic pneumonitis [32] is characterised by the accumulation of intra-alveolar eosinophilic granulocytes, macrophages and fibrin. Macrophages may contain eosinophilic granules leading to a brilliant pink staining of the cytoplasm. The finding of these macrophages, particularly when associated with fibrin deposits and organising pneumonia, is an important clue in suspecting a chronic eosinophilic pneumonia, even when eosinophilic granulocytes are very few or absent (possibly due to steroid treatment).
Pathologies consisting of accumulation of foamy macrophages
In routine practice, the presence of foamy histiocytes in BAL fluid and lung biopsies is a very frequent finding (figure 7). Foamy macrophages represent an incidental and poorly significant sign in the great majority of cases. Indeed, it is very common to observe foamy macrophages akin to necrotic processes (e.g. resected lung tumours after neoadjuvant chemotherapy) peripherally to an obstructive pathology of the bronchial tree, along various levels and due to different causes, or in the setting of areas of parenchymal consolidation (“golden pneumonia”) at the periphery of a lung tumour occluding the bronchial lumen.
FIGURE 7.

Endoalveolar foamy histiocytes in a) hypersensitivity pneumonia and b) amiodarone-related toxicity. Images courtesy of T.V. Colby, from the Charles B. Carrington Collection, sited at the Dept of Laboratory Medicine and Pathology, Mayo Clinic, Scottsdale, AZ, USA.
However, in some circumstances foamy macrophages are an important “spy” of an underlying subtle obstructive respiratory pathology, possibly not considered at a first glance by the pathologist.
For example, endoalveolar foamy macrophages are frequently noted in bronchiolitis and small airway diseases, a complex group of diseases characterised by a clinically and functional obstructive picture coupled to subtle-to-evident inflammation and fibrosis of the small airways at histology [33, 34]. In addition, in the context of a diffuse infiltrative pneumonia, the finding of intra-alveolar foamy macrophages should lead to the consideration of hypersensitivity pneumonia [27, 28] and organising pneumonia (bronchiolitis obliterans organising pneumonia or cryptogenic organising pneumonia) [33–35], the ILDs most commonly associated with a prominent accentuation of peribronchiolar inflammation (bronchiolitis).
A peculiar variant of small airway disease is “diffuse panbronchiolitis”, a clinicopathological entity frequently observed in Japan and very rare in Western countries [36]. Patients present with productive cough, exertional dyspnoea, chronic sinusitis and elevated titres of cold hemagglutinins. At HRCT, diffuse panbronchiolitis shows bilateral diffuse centrolobular nodules with bronchiectasis and bronchiolectasis. Lung biopsy reveals accumulation of foamy macrophages in the lumen of the alveoli and peribronchiolar interstitium.
These histological features are incorrectly considered to be pathognomonic of diffuse panbronchiolitis, but rarely, interstitial aggregates of foamy histiocytes may be observed in other diseases, as in the peripheral lung portion of patients with cystic fibrosis, in lung involvement by idiopathic chronic inflammatory bowel diseases, some vasculitides and connective tissue diseases [37, 38]. As in many non-neoplastic lung pathologies, the diagnosis of diffuse panbronchiolitis requires a careful correlation between histology, clinical, laboratory and imaging data.
Apart from small-airway diseases, another group of pathologies characterised by the presence of foamy macrophages is represented by drug toxicities [8]. This is another poorly recognised and underrated rubric of lung diseases by clinicians and pathologists.
In the great majority of cases, drug toxicity in the lung consists of nonspecific histological features, ranging from acute (diffuse alveolar damage or haemorrhage) to chronic interstitial pneumonitis morphologically identical to nonspecific interstitial pneumonia or even idiopathic pulmonary fibrosis (www.pneumotox.com).
The definitive recognition of a pulmonary drug toxicity requires a close correlation of histological clues with clinical data and exclusion of other diseases justifying the lung injury. Despite this, it is quite true that drug toxicity should always be taken into account when pathologists face with an intra-alveolar accumulation of foamy macrophages with scattered, sparse eosinophils and foci of organising pneumonia.
Virtually all drugs may cause accumulation of foamy macrophages in the lung, but amiodarone toxicity is by far the most commonly encountered [39]. Amiodarone is an anti-arrhythmic agent leading to lung toxicity in ∼5–10% of cases. Amiodarone is often associated with foamy changes either in the macrophages and type II pneumocytes as a sign of drug exposure, not necessarily indicating drug toxicity. The real significance of these histological features should be evaluated in the light of clinical data.
Finally, the accumulation of foamy histiocytes is observed in exogenous lipoid pneumonia and in some histiocytic disorders, some infections and metabolic diseases.
Infections characterised by the accumulation of macrophages
Accumulation of pulmonary macrophages may be the morphological clue to some infections, particularly when associated with granulomas, necrosis and aggregates of neutrophils or in immunocompromised patients.
In all these situations, it is mandatory to rigorously exclude an infection using of specific stains (e.g. methenamine silver stain, Gram stain or Ziehl–Neelsen) and possibly microbiological cultures.
In this article, we focus on two unusual infections that occasionally involve the lung, namely Whipple's disease and malakoplakia.
Whipple's disease [40] is a rare infection involving several organs which is sustained by the Gram-positive bacterium Tropheryma whippelii. The disease mainly affects adult males and presents with systemic symptoms (arthralgia and weight loss) together with gastrointestinal and neurological manifestations. The pulmonary involvement is exceedingly rare and characterised by bronchiolocentric accumulation of intensely PAS-positive macrophages.
Malakoplakia is an uncommon inflammatory condition characterised by the accumulation of macrophages due to defective lysosomal activity and microtubular assembly leading to impairment of the normal phagocytic process with accumulation of benign macrophagesis. This rare condition occurs most commonly in the urinary tract and is due to the accumulation of macrophages unable to destroy phagocytosed bacteria. In the lung, malakoplakia [41, 42] is often secondary to Rhodococcus equi infection, Gram-positive coccobacilli common in the environment, which may cause fatal pneumonia in horses. Occasionally, other micro-organisms are responsible for pulmonary malakoplakia.
Malakoplakia may occur in immunocompromised and immunocompetent individuals concurrently to extrapulmonary involvement.
Chest radiography shows an opacity of the upper lobes tending to cavitate and the disease is often resistant to common antibiotic therapy. The histological features (figure 8) are characterised by abscesses containing the Gram-positive bacilli associated with dense accumulation of macrophages. Disease progression is characterised by a decrease in neutrophils and the number of bacilli coupled with an increase in macrophages. These peculiar macrophages tend to create confluent nodular tumour-like masses and are characterised by an ample eosinophilic cytoplasm (von Hansemann histiocytes) containing intracytoplasmic coccobacilli and scattered rounded von Kossa-positive inclusions consistent with Michaelis–Gutmann bodies.
FIGURE 8.
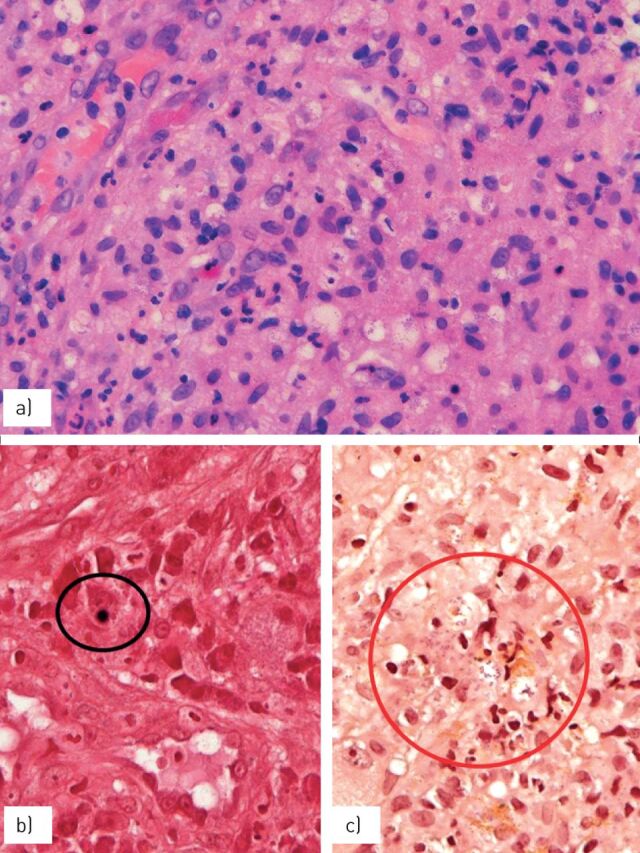
Dense clusters of a) eosinophilic macrophages (von Hansemann cells) containing b) von Kossa-positive, rounded bodies (Michaelis–Gutmann bodies) (circled) and c) Gram-positive coccobacilli (circled).
Langerhans cell histiocytosis and other histiocytic disorders
Langerhans cell histiocytosis
Langerhans cell histiocytosis (LCH) may involve the lung as localised disease or, more rarely, as part of a systemic process [43]. Systemic disease is generally considered a clonal pathology [44, 45], while the pulmonary isolated form is generally considered a polyclonal reactive process secondary to active or passive cigarette smoking in virtually all cases [46, 47].
Pulmonary LCH is relatively rare and generally occurs in young smoking adults with a male prevalence. ∼25% of patients with LCH are asymptomatic, while cough, dyspnoea, chest pain, fever and weight loss are the most common and nonspecific symptoms. Pneumothorax is present in a quarter of cases. Respiratory function tests may return entirely normal results or show mild-to-moderate restrictive/obstructive abnormalities.
Imaging studies (chest radiographs and computed tomography scans) generally show pulmonary alterations characterised by multiple, tiny, peribronchiolar nodules (early phase) and cysts (chronic, advanced phase) of varying and irregular shapes. These lesions are preferentially located at the upper lobes, sparing the costophrenic angles [48, 49]. Ground-glass opacities and mediastinal lymphadenopathy may be observed [40], while solitary parenchymal or endobronchial nodules are very rare [50, 51]. Despite entirely characteristic imaging findings at computed tomography, the diagnosis usually requires a transbronchial or even surgical biopsy, particularly in atypical cases. BAL fluid analysis is very helpful when ≥5% of CD1a-positive Langerhans cells are found [52, 53] (figure 9).
FIGURE 9.

In early phases, Langerhans cell histiocytosis is characterised by a) bronchiocentric collection of peculiar macrophages with prominent grooves accompanied by smokers’ macrophages and eosinophils, leading to b) cellular bronchiolar nodules with irregular, stellate-shaped margins; c) Langerhans cell histiocytes express CD1a at immunohistochemistry; d) during disease progression, the nodules tend to form central cavitation surrounded by fibrosis and decrease of Langerhans cells; e) the disease generally has a good prognosis, leaving hypocellular bronchiolocentric scars of dense fibrosis without Langerhans cells. Images courtesy of T.V. Colby, from the Charles B. Carrington Collection, sited at the Dept of Laboratory Medicine and Pathology, Mayo Clinic, Scottsdale, AZ, USA.
At histology, the early phases of LCH are characterised by collections of Langerhans histiocytes in the peribronchiolar interstitial tissue; they tend to aggregate in small nests. In contrast, the presence of scattered and isolated Langerhans cells is a very common and nonspecific finding in smokers [51, 54]. Langerhans histiocytes show a peculiar morphology (discrete amounts of pale to slightly eosinophilic cytoplasm devoid of any pigment, irregular nuclei with prominent grooves and a delicate nuclear membrane) and a characteristic phenotype at immunohistochemistry (expression of S100 protein, CD1a, CD68 and Langerin and factor XIIIa-negative).
Langerhans cells proliferate along the bronchiolar wall and the alveolar septa leading to irregular, stellate-shaped, multiple nodules accompanied by a discrete quote of eosinophils and pigmented smokers' macrophages into the alveolar lumen and septa. With time, the central zone of the nodule develops a dense fibrosis and central cavitations due to progressive destruction of the bronchiolar wall [55].
In the chronic phase, the Langerhans cells are often visible only at the periphery of the lesions and absent in the central fibrotic areas. In this mature phase of LCH, the disease shows multiple stellate nodules, some of which are cavitated and characteristically separated by normal lung parenchyma. These histological alterations are easily recognisable at low magnification, and it is often possible to observe several lesions at difference stages of evolution (active CD1a-rich cellular nodules akin to “burnt-out” fibrotic and cavitated nodules) in the same histological slide. Occasionally, the nodules grow into the vascular walls, provoking pulmonary hypertension, which may be severe [56], or extend to the pleura, leading to pneumothorax and simulating an eosinophilic pleuritis [57].
Finally, the lesions of LCH tend to regress as small stellate, peribronchiolar scars that may be completely reabsorbed. Clinical history is quite variable, since the disease may regress spontaneously after the active phase or, more rarely, progresses to a fibrosing interstitial disease.
Treatment consists of smoking cessation with or without steroid therapy in the early phase of the disease [43, 47]. Patients with progressive fibrotic disease have been treated with chemotherapy or transplantation. Recurrence after lung transplant has been described rarely [58]. The clinical history of LCH is variable and sometimes complicated by the occurrence of malignancy, such as lung cancer or lymphomas [47, 59]. Nevertheless, the great majority of patients with LCH have a good prognosis, and only 10% died of respiratory failure [60]. Recently, the BRAF V600E mutation has been identified in ∼30% of Langerhans histiocytes with isolated pulmonary LCH and in ∼60% of multisystemic LCH cases [61]. These molecular features are promisingly open to targeted therapies with specific BRAF inhibitors in patients with refractory BRAF-mutated LCH [47, 61].
Alternatively, NRAS and MAP2K1 mutations have been reported in pulmonary LCH [62, 63].
The BRAF V600E mutation may be robustly detected using immunohistochemistry using the clone VE1 on formalin-fixed paraffin-embedded tissue [64], while future diagnosis of LCH could achieved by detecting mutations in circulating DNA using liquid biopsy.
An international classification of histiocytoses has been proposed [65]. The differential diagnosis between histiocytic disorders involving the lungs is summarised in table 2.
TABLE 2.
Main characteristics of Langerhans cell histiocytosis (LCH), Erdheim–Chester disease (ECD) and Rosai–Dorfman disease (RDD)
| Smoking habit | Histology | Chest CT | Special techniques | Molecular abnormalities | |
| LCH | Yes | Bronchiolocentric nodules with Langerhans cells, eosinophils and other inflammatory cells; multiple stellate-shaped nodules or fibrotic cysts separated by normal lung parenchyma |
Bronchiolocentric nodules and/or fibrotic cysts with upper lobes involvement and sparing of costophrenic angles | S100+, CD1a+, Langerin+ factor XIIIa- Birbeck granules at electron microscopy | BRAF (V600E) mutations in ∼30% |
| ECD | No | Expansion of pleura, interlobular septa and peribronchiolar tissue with xanthomatous/foamy histiocytes and interspersed inflammatory cells, multinucleate Touton-like giant cells in a fibrotic background | Thickening of pleura, interlobular septa and centrilobular nodules with/without pleural effusion and ground-glass opacities; mediastinal involvement | CD68+, CD163+, factor XIIIa+ CD1a-, S100-, Langerin- | BRAF (V600E) mutations in ≤60% |
| RDD | No | Expansion of pleura, interlobular septa and peribronchiolar tissue by histiocytic cells with mixed inflammatory infiltrates and emperipolesis phenomenon | Diffuse lymphadenopathies; expansion of pleura, interlobular septa and bronchiolocentric tissue | CD68+, S100+, CD14+ and CD11c1 CD1a-, Langerin-, factor XIIIa- | No BRAF V600E mutation |
CT: computed tomography.
Erdheim–Chester disease
Erdheim–Chester disease (ECD) is a rare, multisystemic, non-Langerhans form of histiocytosis of unknown origin, affecting adult patients (median age 53 years) with no sex prevalence [66, 67]. The disease is characterised by the accumulation of xanthomatous/foamy lipid-laden macrophages, with/without multinucleated macrophages (Touton-like giant cells) surrounded by fibrotic tissue.
These histiocytes show immunohistochemical expression of CD68, factor XIIIa, CD163 and occasionally S100 protein, whereas CD1a and Langerin are entirely negative. It is still unclear whether ECD is a clonal process [68, 69].
The disease generally presents with bilateral and symmetric cortical osteosclerosis of the diaphyseal and metaphyseal regions in the long bones, but it may involve central nervous system, orbit, pituitary gland and hypothalamic areas, kidney, retroperitoneum, skin (often misdiagnosed as xanthoma) and lung (14% of cases) [66, 70].
Interestingly, patients concurrently or metachronously affected by ECD and LCH are reported in literature, thus suggesting a relationship between these diseases [65]. Pulmonary involvement was not found to be a major prognostic factor in ECD and an overall mortality rate of 59% has been reported [66]. Pulmonary involvement is characterised by dyspnoea [70–75] and thickness of the interlobular septa and pleura with centrolobular nodular and ground-glass opacities.
At histology, lung biopsy shows a diffuse infiltration and expansion of the lymphatic bundles (pleura, interlobular septa and peribronchiolar connective tissue) by xanthomatous/foamy histiocytes surrounded by dense fibrosis and a lymphoplasmacytic inflammatory infiltrate (figure 10).
FIGURE 10.

Erdheim–Chester disease is characterised by a) thickening of the pleura and interlobular septa with b) dense fibrosis and a proliferation of lymphocytes and macrophages.
At low magnification, the marked fibroinflammatory dilatation of the lymphatic bundles in absence of “dirty macrophages” (pathognomonic of silicosis) and granulomas (suggestive of sarcoidosis) should alert the pathologist to the possibility of pulmonary involvement of ECD.
The BRAF V600E mutation is reported in ≤60% of patients with ECD. Significant, rapid clinical and biological improvements with the BRAF inhibitor vemurafenib have been reported in patients with refractory and severe forms of ECD [65, 70].
Rosai–Dorfman disease
Sinus histiocytosis with massive lymphadenopathy (commonly known as Rosai–Dorfman disease (RDD)) is a histiocytic disorder of unknown cause, primarily characterised by lymphadenopathy (particularly of the cervical region) and, less frequently, involvement of skin, nasal sinuses, soft tissue, orbit, bones, salivary glands and central nervous system. Among 423 patients included in an international registry [76], only nine (2%) cases (six females and three males; median age 14 years) had tracheal, bronchial or pulmonary involvement, while the lung parenchyma was affected in only three cases. Concurrent lymphadenopathy and nasal involvement was observed in all nine cases.
Given the rarity of lung involvement, the morphological characteristics of pulmonary RDD are poorly recognized. In our single case, there was a marked expansion of the pleural, peribronchiolar and connective tissue of interlobular septa. In these locations, the characteristic histiocytes (macrophages with large and pale cytoplasm with emperipolesis phenomenon, expression of S100 protein and CD68 with negativity for CD1a, factor XIIIa and CD163) were intermingled with a mixed inflammatory infiltrate and dense fibrosis [76]. The differential diagnosis between ECD, RDD and other histiocytic disorders not otherwise specified is often problematic, requiring a close correlation with clinical data. No BRAF V600E mutation has been reported in RDD [65, 70].
Metabolic diseases
Gaucher disease
Gaucher disease is an autosomal recessive lysosomal storage disease caused by mutations in GBA1 with resultant defective function of the expressed enzyme, acid β-glucosidase (glucocerebrosidase or GCase) leading to excess accumulation of glucosylceramide and glucosylsphingosine in macrophages and other lineage cells with multiorgan involvement [77, 78]. Three types of Gaucher disease are described, based upon the presence and severity of neurological involvement. Type 1, lacking central nervous system involvement, accounts for ∼90% of cases, with features usually limited to the visceral organs (e.g. hepatosplenomegaly). Types 2 and 3 are neuronopathic variants distinguished by the age of onset and biological progression of neurological disease: type 2 is the acute variant with early onset at age 6 months and severe, progressive neurological and visceral involvement leading to death in the first 1–2 years of life; and type 3 is the subacute neuronopathic variant associated with later-onset neurological and visceral disease of varying severity [79].
Lung involvement is frequent, particularly in infantile forms and patients with L444P homozygosis [80], with accumulations of Gaucher cells into the alveoli, interstitial space and pleura, around arteries and in the alveolar capillaries. Collection of Gaucher macrophages in pulmonary capillaries and arteries may cause pulmonary hypertension [81]. Gaucher cells may show various patterns, including intracapillary involvement, lymphangitic pattern with patchy aggregated into the alveolar interstitium, diffuse thickening or alveolar septa and intra-alveolar accumulation. Gaucher cells are CD68-positive and have an eccentric nucleus and abundant granular or fibrillary blue-grey cytoplasm with a wrinkled tissue paper-like appearance with abundant lightly PAS-positive and iron-stained fibrillary material in the cytoplasm [82]. Diagnosis is confirmed by the levels of serum glucocerebrosidase.
Niemann–Pick disease
Niemann–Pick disease is a rare autosomal recessive lysosomal storage disease with three subtypes and accumulation of sphingomyelin-rich macrophages. Types A and B result from a deficiency of acid sphingomyelinase activity, while type C is a complex lipid storage disorder. Type A is a fatal neurodegenerative disorder of infancy; type B is a less severe form. Pulmonary involvement occurs in all three types [83, 84], but most frequently in type B.
Imaging findings consist of ground-glass opacities and smooth interlobular septal thickening, and intralobular lines involving mainly the lower lobes [84]. Histology is quite characteristic [85, 86] with finely vacuolated foamy macrophages (Niemann–Pick cells) filling the alveolar spaces, alveolar septa and pleura. The ciliated bronchiolar epithelium presents a cytoplasm with fine vacuolation or clear change (figure 11). May–Giemsa stain and the Schmorl reaction evidences intensely blue-stained Niemann–Pick cells (“sea-blue histiocytes”) [82]. BAL is helpful in the diagnosis of Niemann–Pick's disease, showing the presence of the characteristic clear macrophages [87]. The diagnosis is confirmed by the analysis of sphingomyelinase in the serum and in vitro fibroblasts.
FIGURE 11.

Niemann–Pick disease, type B, with a) diffuse accumulation of intra-alveolar foamy histiocytes and b) clarification of the bronchiolar epithelium.
Fabry disease
Fabry disease is an X-linked lysosomal storage recessive disorder caused by deficiency of α-galactosidase A, which leads to storage of neutral glycosphingolipids in virtually all tissues and cells [88]. Cutaneous and mucosal angiokeratomas, hypohydrosis and paraesthaesia of the extremities are usually present. The progressive involvement of vascular system may lead to renal and cardiac failure. The obstructive lung disease is the main pulmonary abnormality in these patients (36% of cases) [89], secondary to accumulation of neutral glycosphingolipids (leading to dense granular inclusion bodies) along the bronchial tree with progressive narrowing of the airways by accumulated glycosphingolipids in the bronchial cells and smooth muscle cells.
Hermansky–Pudlak disease
Hermansky–Pudlak disease is an autosomal recessive disorder characterised by oculocutaneous albinism, defects of platelet aggregation with bleeding diathesis, granulomatous colitis and systemic accumulation of macrophages containing ceroid pigment (amorphous lipid–protein complexes) [90, 91]. Pulmonary involvement (the most important complication of this disease) at chest computed tomography shows increased reticular opacities, thickened interlobular septa, and ground-glass infiltrates in addition to fibrotic changes, including traction bronchiectasis and honeycombing [92].
Histological lesions in the lung reveal an interstitial pneumonia with a nonspecific interstitial pneumonia-like or usual interstitial pneumonia-like pattern. Collection of macrophages with intracytoplasmic ceroid pigment are present in lung biopsy and upon BAL [93], as well as in other organs (e.g. intestine) [94]. The presence of surfactant engulfment at electron microscopy with secondary marked vacuolisation of pneumocytes has been noted [95].
Miscellanea
Several other exceedingly rare disorders characterised by the accumulation of macrophages may involve the lungs. Patients with mucopolysaccharidoses often show a pulmonary involvement with obstruction of the airways and recurrent infections. Other rare metabolic diseases may show pulmonary alterations, such as infantile gangliosidosis GM1 [96], leukodystrophies (Krabbe disease) [97], glicogenosis (Pompe disease) [98], Farber disease [99], cystinosis, Marfan syndrome and Ehlers–Danlos syndrome [100, 101].
Final remarks
A careful morphological evaluation of the features and localisation of macrophages in the lung is often helpful in reducing the diagnostic possibilities and differential diagnosis. Obviously, these findings need a striking correlation with clinical, laboratory and imaging data.
Faced with accumulation of haemosiderin-laden macrophages, the first concern should be for smoking-related lesions (when pigment is fine) or alveolar haemorrhage and some pneumoconiosis (when pigment is chunky and coarse).
Faced with collection of foamy macrophages, pathologists first should take into account all the pathologies associated with broncho-bronchiolar obstruction, or alternatively exogenous lipoid pneumonia, drug toxicity and metabolic disorders. When foamy macrophages are sited in the alveoli, encrusted in the interstitium and coupled to bronchiolitis, a “diffuse panbronchiolitis” should be considered. If the disease presents clinically as an interstitial pneumonia, organising pneumonia (cryptogenic organising pneumonia or secondary organising pneumonia), hypersensitivity pneumonia, drug toxicity and some metabolic diseases are at the top of the diagnostic list. Collections of PAS-positive macrophages in the pleura and interlobular septa associated with cytoplasmic clear changes of bronchiolar epithelium (particularly when alveolar accumulation of foamy macrophages is diffuse without inflammation and fibrosis) are quite consistent with the possibility of type B Niemann–Pick disease.
Multiple and stellate nodules separated by normal lung is quite suggestive of LCH. Expansion of the pleural and peri-broncho-vascular connective tissue associated with fibrosis and mixed inflammatory cells with histiocytes in the interlobular septa is strongly suspicious of ECD or RDD. Finally, any unclear accumulation of macrophages in the lung raises the possibility of a systemic metabolic disorder, even in adulthood.
Footnotes
Previous articles in this series: No. 1: Ghigna MR, Mooi WJ, Grünberg K. Pulmonary hypertensive vasculopathy in parenchymal lung diseases and/or hypoxia. Eur Respir Rev 2017; 26: 170003. No. 2: Bubendorf L, Lantuejoul S, de Langen AJ, et al. Nonsmall cell lung carcinoma: diagnostic difficulties in small biopsies and cytological specimens. Eur Respir Rev 2017; 26: 170007.
Conflict of interest: None declared.
Provenance: Commissioned article, peer reviewed.
References
- 1.Coalson JJ. The adult lung: structure and function. In: Saldana MJ, ed. Pathology of Pulmonary Disease. Philadelphia, Lippincott, 1994; pp. 3–14. [Google Scholar]
- 2.Marten K, Hansell DM. Imaging of macrophage-related lung diseases. Eur Radiol 2005; 15: 727–741. [DOI] [PubMed] [Google Scholar]
- 3.Xiang G, Zhang Y, Su C, et al. Dynamic changes of mononuclear phagocytes in circulating, pulmonary alveolar and interstitial compartments in a mouse model of experimental silicosis. Inhal Toxicol 2016; 28: 393–402. [DOI] [PubMed] [Google Scholar]
- 4.Poletti V, Chilosi M, Olivieri D. Diagnostic invasive procedures in diffuse infiltrative lung diseases. Respiration 2004; 71: 107–119. [DOI] [PubMed] [Google Scholar]
- 5.Poletti V, Ravaglia C, Gurioli C, et al. Invasive diagnostic techniques in idiopathic interstitial pneumonias. Respirology 2016; 21: 44–50. [DOI] [PubMed] [Google Scholar]
- 6.Wang CW, Colby TV. Histiocytic lesions and proliferations in the lung. Semin Diagn Pathol 2007; 24: 162–182. [DOI] [PubMed] [Google Scholar]
- 7.Eguíluz-Gracia I, Schultz HH, Sikkeland LI, et al. Long-term persistence of human donor alveolar macrophages in lung transplant recipients. Thorax 2016; 71: 1006–1011. [DOI] [PubMed] [Google Scholar]
- 8.Hasleton P, Flieder DB. Spencer's Pathology of the Lung. 6th Edn. Cambridge, Cambridge University Press, 2013. [Google Scholar]
- 9.Fraig M, Shreesha U, Savici D, et al. Respiratory bronchiolitis: a clinicopathologic study in current smokers, ex-smokers, and never-smokers. Am J Surg Pathol 2002; 26: 647–653. [DOI] [PubMed] [Google Scholar]
- 10.Ryu JH, Myers JL, Capizzi S, et al. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest 2005; 127: 178–184. [DOI] [PubMed] [Google Scholar]
- 11.Travis WD, King TE, Bateman ED, et al. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304. [DOI] [PubMed] [Google Scholar]
- 12.Sieminska A, Kuziemski K. Respiratory bronchiolitis-interstitial lung disease. Orphanet J Rare Dis 2014; 9: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katzenstein AA. Smoking-related interstitial fibrosis (SRIF), pathogenesis and treatment of usual interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Mod Pathol 2012; 25: Suppl. 1, S68–S78. [DOI] [PubMed] [Google Scholar]
- 14.Pipavath SJ, Lynch DA, Cool C, et al. Radiologic and pathologic features of bronchiolitis. AJR Am J Roentgenol 2005; 185: 354–363. [DOI] [PubMed] [Google Scholar]
- 15.Tazelaar HD, Wright JL, Churg A. Desquamative interstitial pneumonia. Histopathology 2011; 58: 509–516. [DOI] [PubMed] [Google Scholar]
- 16.Raparia K, Ketterer J, Dalurzo ML, et al. Lung tumors masquerading as desquamative interstitial pneumonia (DIP): report of 7 cases and review of the literature. Am J Surg Pathol 2014; 38: 921–924. [DOI] [PubMed] [Google Scholar]
- 17.Godbert B, Wissler MP, Vignaud JM. Desquamative interstitial pneumonia: an analytic review with an emphasis on aetiology. Eur Respir J 2013; 22: 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doan ML, Guillerman RP, Dishop MK, et al. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax 2008; 63: 366–373. [DOI] [PubMed] [Google Scholar]
- 19.Cavazza A, Rossi G, Barbareschi M, et al. Emorragia polmonare. Malattie associate, alterazioni morfologiche e criteri diagnostici. [Pulmonary haemorrhage. A review of the literature from the pathologist's perspective.] Pathologica 2003; 95: 422–435. [PubMed] [Google Scholar]
- 20.Churg A, Green FH. Pathology of Occupational Lung Disease. Baltimore, Williams and Wilkins, 1998. [Google Scholar]
- 21.Lederer H, Muggli B, Speich R, et al. Haemosiderin-laden sputum macrophages for diagnosis in pulmonary veno-occlusive disease. PLoS One 2014; 9: e115219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golde DW, Drew WL, Klein HZ, et al. Occult pulmonary haemorrhage in leukaemia. Br Med J 1975; 2: 166–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crouch E, Churg A. Ferruginous bodies and the histologic evaluation of dust exposure. Am J Surg Pathol 1984; 8: 109–116. [DOI] [PubMed] [Google Scholar]
- 24.Travis WD, Colby TV, Koss MN, et al. Non-neoplastic Disorders of the Lower Respiratory Tract. Atlas of Nontumor Pathology. Washington, AFIP/ARP, 2002. [Google Scholar]
- 25.Crouch E, Churg A. Progressive massive fibrosis of the lung secondary to intravenous injection of talc. A pathologic and mineralogic analysis. Am J Clin Pathol 1983; 80: 520–526. [DOI] [PubMed] [Google Scholar]
- 26.Corwin RW, Irwin RS. The lipid-laden alveolar macrophage as a marker of aspiration in parenchymal lung disease. Am Rev Respir Dis 1985; 132: 576–581. [DOI] [PubMed] [Google Scholar]
- 27.Coleman A, Colby TV. Histologic diagnosis of extrinsic allergic alveolitis. Am J Surg Pathol 1988; 12: 514–518. [DOI] [PubMed] [Google Scholar]
- 28.Colby TV, Coleman A. The histologic diagnosis of extrinsic allergic alveolitis and its differential diagnosis. In: Fenoglio-Preiser M, Wolff M, Rilke F, eds. Progress in Surgical Pathology. Berlin, Springer, 1989; pp. 11–25. [Google Scholar]
- 29.Suzuki T, Trapnell BC. Pulmonary alveolar proteinosis syndrome. Clin Chest Med 2016; 37: 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossi G, De Rosa N, Cavazza A, et al. Localized pleuropulmonary crystal-storing histiocytosis: 5 cases of a rare histiocytic disorder with variable clinicoradiologic features. Am J Surg Pathol 2013; 37: 906–912. [DOI] [PubMed] [Google Scholar]
- 31.Ionescu DN, Pierson DM, Qing G, et al. Pulmonary crystal-storing histiocytoma. Arch Pathol Lab Med 2005; 129: 1159–1163. [DOI] [PubMed] [Google Scholar]
- 32.Marchand E, Reynaud-Gaubert M, Lauque D, et al. Idiopathic chronic eosinophilic pneumonia. A clinical and follow-up study of 62 patients. Medicine 1998; 77: 299–312. [DOI] [PubMed] [Google Scholar]
- 33.Colby TV. Bronchiolitis. Pathologic considerations. Am J Clin Pathol 1998; 109: 101–109. [DOI] [PubMed] [Google Scholar]
- 34.Ryu JH, Myers JL, Swensen SJ. Bronchiolar disorders. Am J Respir Crit Care Med 2003; 168: 1277–1292. [DOI] [PubMed] [Google Scholar]
- 35.Colby TV. Pathologic aspects of bronchiolitis obliterans organizing pneumonia. Chest 1992; 102: Suppl. 1, 38S–43S. [DOI] [PubMed] [Google Scholar]
- 36.Poletti V, Chilosi M, Casoni G, et al. Diffuse panbronchiolitis. Sarcoidosis Vasc Diffuse Lung Dis 2004; 21: 94–104. [PubMed] [Google Scholar]
- 37.Iwata M, Colby TV, Kitaichi M. Diffuse panbronchiolitis: diagnosis and distinction from various pulmonary diseases with centrilobular interstitial foam cell accumulations. Hum Pathol 1994; 25: 357–363. [DOI] [PubMed] [Google Scholar]
- 38.Camus P, Piard F, Ashcroft T, et al. The lung in inflammatory bowel disease. Medicine 1993; 72: 151–183. [PubMed] [Google Scholar]
- 39.Malhotra A, Muse VV, Mark EJ. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 12–2003. An 82-year-old man with dyspnea and pulmonary abnormalities. N Engl J Med 2003; 348: 1574–1585. [DOI] [PubMed] [Google Scholar]
- 40.Durand DV, Lecomte C, Cathebras P, et al. Whipple disease. Clinical review of 52 cases. Medicine 1997; 76: 170–184. [DOI] [PubMed] [Google Scholar]
- 41.Kwon KY, Colby TV. Rhodococcus equi pneumonia and pulmonary malakoplakia in acquired immunodeficiency syndrome. Pathologic features. Arch Pathol Lab Med 1994; 118: 744–748. [PubMed] [Google Scholar]
- 42.Colby TV, Kwon KY. Rhodococcus equi – an old bug with a new name; relevance for the anatomic pathologist? Adv Anat Pathol 1995; 4: 263–269. [Google Scholar]
- 43.Vassallo R, Ryu JH, Colby TV, et al. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med 2000; 342: 1969–1978. [DOI] [PubMed] [Google Scholar]
- 44.Willman CL, Busque L, Griffith BB, et al. Langerhans’-cell histiocytosis (histiocytosis X) – a clonal proliferative disease. N Engl J Med 1994; 331: 154–160. [DOI] [PubMed] [Google Scholar]
- 45.Yu RC, Chu C, Buluwela L, et al. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 1994; 343: 767–768. [DOI] [PubMed] [Google Scholar]
- 46.Yousem SA, Colby TV, Chen YY, et al. Pulmonary Langerhans’ cell histiocytosis: molecular analysis of clonality. Am J Surg Pathol 2001; 25: 630–636. [DOI] [PubMed] [Google Scholar]
- 47.DeMartino E, Go RS, Vassallo R. Langerhans cell histiocytosis and other histiocytic diseases of the lung. Clin Chest Med 2016; 37: 421–430. [DOI] [PubMed] [Google Scholar]
- 48.Moore DA, Godwin JD, Müller NL, et al. Pulmonary histiocytosis X: comparison of radiographic and CT findings. Radiology 1989; 172: 249–254. [DOI] [PubMed] [Google Scholar]
- 49.Brauner MW, Grenier P, Mouelhi MM, et al. Pulmonary histiocytosis X: evaluation with high-resolution CT. Radiology 1989; 172: 255–258. [DOI] [PubMed] [Google Scholar]
- 50.Khoor A, Myers JL, Tazelaar HD, et al. Pulmonary Langerhans cell histiocytosis presenting as a solitary nodule. Mayo Clin Proc 2001; 76: 209–211. [DOI] [PubMed] [Google Scholar]
- 51.O'Donnell AE, Tsou E, Awh C, et al. Endobronchial eosinophilic granuloma: a rare cause of total lung atelectasis. Am Rev Respir Dis 1987; 136: 1478–1480. [DOI] [PubMed] [Google Scholar]
- 52.Colby TV, Lombard C. Histiocytosis X in the lung. Hum Pathol 1983; 14: 847–856. [DOI] [PubMed] [Google Scholar]
- 53.Travis WD, Borok Z, Roum JH, et al. Pulmonary Langerhans cell granulomatosis (histiocytosis X). A clinicopathologic study of 48 cases. Am J Surg Pathol 1993; 17: 971–986. [DOI] [PubMed] [Google Scholar]
- 54.Casolaro MA, Bernaudin JF, Saltini C, et al. Accumulation of Langerhans’ cells on the epithelial surface of the lower respiratory tract in normal subjects in association with cigarette smoking. Am Rev Respir Dis 1988; 137: 406–411. [DOI] [PubMed] [Google Scholar]
- 55.Kambouchner M, Basset F, Marchal J, et al. Three-dimensional characterization of pathologic lesions in pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med 2002; 166: 1483–1490. [DOI] [PubMed] [Google Scholar]
- 56.Hamada K, Teramoto S, Narita N, et al. Pulmonary veno-occlusive disease in pulmonary Langerhans’ cell granulomatosis. Eur Respir J 2000; 15: 421–423. [DOI] [PubMed] [Google Scholar]
- 57.Askin FB, McCann BG, Kuhn C. Reactive eosinophilic pleuritis: a lesion to be distinguished from pulmonary eosinophilic granuloma. Arch Pathol Lab Med 1977; 101: 187–191. [PubMed] [Google Scholar]
- 58.Etienne B, Bertocchi M, Gamondes JP, et al. Relapsing pulmonary Langerhans cell histiocytosis after lung transplantation. Am J Respir Crit Care Med 1998; 157: 288–291. [DOI] [PubMed] [Google Scholar]
- 59.Lombard CM, Medeiros J, Colby TV. Pulmonary histiocytosis X and carcinoma. Arch Pathol Lab Med 1987; 111: 339–341. [PubMed] [Google Scholar]
- 60.Vassallo R, Ryu JH, Schroeder DR, et al. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med 2002; 346: 484–490. [DOI] [PubMed] [Google Scholar]
- 61.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012; 120: 2700–2703. [DOI] [PubMed] [Google Scholar]
- 62.Mourah S, How-Kit A, Meignin V, et al. Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J 2016; 47: 1785–1796. [DOI] [PubMed] [Google Scholar]
- 63.Zeng K, Ohshima K, Liu Y, et al. BRAFV600E and MAP2K1 mutations in Langerhans cell histiocytosis occur predominantly in children. Hematol Oncol 2016; in press [ 10.1002/hon.2344]. [DOI] [PubMed] [Google Scholar]
- 64.Ballester LY, Cantu MD, Lim KPH, et al. The use of BRAF V600E mutation-specific immunohistochemistry in pediatric Langerhans cell histiocytosis. Hematol Oncol 2017; in press [ 10.1002/hon.2388]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016; 127: 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim–Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine 1996; 75: 157–169. [DOI] [PubMed] [Google Scholar]
- 67.Bisceglia M, Cammisa M, Suster S, et al. Erdheim–Chester disease: clinical and pathologic spectrum of four cases from the Arkadi M. Rywlin slide seminars. Adv Anat Pathol 2003; 10: 160–171. [DOI] [PubMed] [Google Scholar]
- 68.Chettritt J, Paradis V, Dargere D, et al. Chester–Erdheim disease: a neoplastic disorder. Hum Pathol 1999; 30: 1093–1096. [DOI] [PubMed] [Google Scholar]
- 69.Al-Quran S, Reith J, Bradley J, et al. Erdheim–Chester disease: case report, PCR-based analysis of clonality, and review of literature. Mod Pathol 2002; 15: 666–672. [DOI] [PubMed] [Google Scholar]
- 70.Haroche J, Arnaud L, Cohen-Aubart F, et al. Erdheim–Chester Disease. Rheum Dis Clin North Am 2013; 39: 299–311. [DOI] [PubMed] [Google Scholar]
- 71.Wittemberg KH, Swensen SJ, Myers JL. Pulmonary involvement with Erdheim–Chester disease: radiographic and CT findings. AJR Am J Roengtenol 2000; 174: 1327–1331. [DOI] [PubMed] [Google Scholar]
- 72.Devouassoux G, Lantuejoul S, Chatelain P, et al. Erdheim–Chester disease: a primary macrophage cell disorder. Am J Respir Crit Care Med 1998; 157: 650–653. [DOI] [PubMed] [Google Scholar]
- 73.Egan AJM, Boardman LA, Tazelaar HD, et al. Erdheim–Chester disease: clinical, radiologic, and histopathologic findings in five patients with interstitial lung disease. Am J Surg Pathol 1999; 23: 17–26. [DOI] [PubMed] [Google Scholar]
- 74.Rush WL, Andriko JAW, Galateau-Salle F, et al. Pulmonary pathology of Erdheim–Chester disease. Mod Pathol 2000; 13: 747–754. [DOI] [PubMed] [Google Scholar]
- 75.Allen CT, Chevez-Barrios P, Shetlar DJ, et al. Pulmonary and ophthalmic involvement with Erdheim–Chester disease: a case report and review of the literature. Arch Pathol Lab Med 2004; 128: 1428–1431. [DOI] [PubMed] [Google Scholar]
- 76.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): review of the entity. Semin Diagn Pathol 1990; 7: 19–73. [PubMed] [Google Scholar]
- 77.Adachi M. Pulmonary involvement in some inborn errors of metabolism. In: Saldana MJ, ed. Pathology of Pulmonary Disease. Philadelphia, Lippincott, 1994; pp. 105–113. [Google Scholar]
- 78.Amir G, Ron N. Pulmonary pathology in Gaucher's disease. Hum Pathol 1999; 30: 666–670. [DOI] [PubMed] [Google Scholar]
- 79.Burrow TA, Sun Y, Prada CE, et al. CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11 years of therapy: clinical, histopathologic, and biochemical findings. Mol Genet Metab 2015; 114: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Santamaria F, Parenti G, Guidi G, et al. Pulmonary manifestations of Gaucher disease: an increased risk for L444P homozygotes? Am J Respir Crit Care Med 1998; 157: 985–989. [DOI] [PubMed] [Google Scholar]
- 81.Harats D, Pauzner R, Elstein D, et al. Pulmonary hypertension in two patients with type I Gaucher disease while on alglucerase therapy. Acta Haematol 1997; 98: 47–50. [DOI] [PubMed] [Google Scholar]
- 82.Chen M, Wang J. Gaucher disease: review of the literature. Arch Pathol Lab Med 2008; 132: 851–853. [DOI] [PubMed] [Google Scholar]
- 83.Mendelson DS, Wasserstein MP, Desnick RJ, et al. Type B Niemann–Pick disease: findings at chest radiography, thin-section CT, and pulmonary function testing. Radiology 2006; 238: 339–345. [DOI] [PubMed] [Google Scholar]
- 84.von Ranke FM, Pereira Freitas HM, Mançano AD, et al. Pulmonary involvement in Niemann–Pick disease: a state-of-the-art review. Lung 2016; 194: 511–518. [DOI] [PubMed] [Google Scholar]
- 85.Falco F, Bembi B, Cavazza A, et al. Pulmonary involvement in an adult female affected by type B Niemann Pick disease. Sarcoidosis Vasc Diffuse Lung Dis 2005; 22: 229–233. [PubMed] [Google Scholar]
- 86.Nicholson AG, Florio R, Hansell DM, et al. Pulmonary involvement by Niemann–Pick disease. A report of six cases. Histopathology 2006; 48: 596–603. [DOI] [PubMed] [Google Scholar]
- 87.Nicholson AG, Wells AU, Hooper J, et al. Successful treatment of endogenous lipoid pneumonia due to Niemann–Pick type B disease with whole-lung lavage. Am J Respir Crit Care Med 2002; 165: 128–131. [DOI] [PubMed] [Google Scholar]
- 88.Franzen D, Krayenbuehl PA, Lidove O, et al. Pulmonary involvement in Fabry disease: overview and perspectives. Eur J Intern Med 2013; 24: 707–713. [DOI] [PubMed] [Google Scholar]
- 89.Brown LK, Miller A, Bhuptani A, et al. Pulmonary involvement in Fabry disease. Am J Respir Crit Care Med 1997; 155: 1004–1010. [DOI] [PubMed] [Google Scholar]
- 90.El-Chemaly S, Young LR. Hermansky–Pudlak syndrome. Clin Chest Med 2016; 37: 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schinella RA, Greco MA, Garay SM, et al. Hermansky–Pudlak syndrome: a clinicopathologic study. Hum Pathol 1985; 16: 366–376. [DOI] [PubMed] [Google Scholar]
- 92.Vicary GW, Vergne Y, Santiago-Cornier A, et al. Pulmonary fibrosis in Hermansky–Pudlak syndrome. Ann Am Thorac Soc 2016; 13: 1839–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.White DA, Walker Smith GJ, Cooper JA, et al. Hermansky–Pudlak syndrome and interstitial lung disease: report of a case with lavage findings. Am Rev Respir Dis 1984; 138–141. [DOI] [PubMed] [Google Scholar]
- 94.Schinella RA, Greco MA, Cobert BL, et al. Hermansky–Pudlak syndrome with granulomatous colitis. Ann Intern Med 1980; 92: 20–23. [DOI] [PubMed] [Google Scholar]
- 95.Nakatani Y, Nakamura N, Sano J, et al. Interstitial pneumonia in Hermansky–Pudlak syndrome: significance of florid foamy swelling/degeneration (giant lamellar body degeneration) of type-2 pneumocytes. Virchows Arch 2000; 437: 304–313. [DOI] [PubMed] [Google Scholar]
- 96.Matsumoto T, Matsumori H, Taki T, et al. Infantile GM1-gangliosidosis with marked manifestation of lungs. Acta Pathol Jpn 1979; 29: 269–276. [DOI] [PubMed] [Google Scholar]
- 97.Clarke JTR, Ozere RL, Krause VW. Early infantile variant of Krabbe globoid cell leucodystrophy with lung involvement. Arch Dis Child 1981; 56: 640–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lightman NI, Schooley RT. Adult-onset acid maltase deficiency: case report of an adult with severe respiratory difficulty. Chest 1977; 72: 250–252. [DOI] [PubMed] [Google Scholar]
- 99.Rutsaert J, Tondeur M, Vamos-Hurwitz E, et al. The cellular lesions of Farber's disease and their experimental reproduction in tissue culture. Lab Invest 1977; 36: 474–480. [PubMed] [Google Scholar]
- 100.Murray RA, Poulton TB, Saltarelli MG, et al. Rare pulmonary manifestation of Ehlers–Danlos syndrome. J Thorac Imaging 1995; 10: 138–141. [DOI] [PubMed] [Google Scholar]
- 101.Yost BA, Vogelsang JP, Lie JT. Fatal hemoptysis in Ehlers–Danlos syndrome. Old malady with a new curse. Chest 1995; 107: 1465–1467. [DOI] [PubMed] [Google Scholar]