

Abstract
Pulmonary surfactant is a crucial and dynamic lung structure whose primary functions are to reduce alveolar surface tension and facilitate breathing. Though disruptions in surfactant homeostasis are typically thought of in the context of respiratory distress and premature infants, many lung diseases have been noted to have significant surfactant abnormalities. Nevertheless, preclinical and clinical studies of pulmonary disease too often overlook the potential contribution of surfactant alterations – whether in quantity, quality or composition – to disease pathogenesis and symptoms. In inflammatory lung diseases, whether these changes are cause or consequence remains a subject of debate. This review will outline 1) the importance of pulmonary surfactant in the maintenance of respiratory health, 2) the diseases associated with primary surfactant dysregulation, 3) the surfactant abnormalities observed in inflammatory pulmonary diseases and, finally, 4) the available research on the interplay between surfactant homeostasis and smoking-associated lung disease. From these published studies, we posit that changes in surfactant integrity and composition contribute more considerably to chronic inflammatory pulmonary diseases and that more work is required to determine the mechanisms underlying these alterations and their potential treatability.
Short abstract
Pulmonary surfactant is a crucial structure for lung function and gas exchange. Recent studies have documented pulmonary surfactant abnormalities in most chronic lung diseases, participating in disease pathology and symptoms. https://bit.ly/3CSaXeK
Introduction
Pulmonary surfactant has long been known to be an important component of the normal lung. It is mainly known for playing a role in facilitating inflation/deflation during breathing. In fact, breathing and proper gas exchange would simply not be possible without pulmonary surfactant. Furthermore, preclinical and clinical studies have demonstrated that surfactant is involved in many non-mechanical biological processes, such as host defence and lipid homeostasis, and that most chronic lung diseases are associated with significant alterations in pulmonary surfactant homeostasis. Although a lot is known about its function and how it is altered in pulmonary disease, the actual contribution of surfactant changes to the development of respiratory symptoms and to what extent surfactant can be targeted in chronic lung disease remains greatly underexplored. We strongly believe that surfactant alterations play a substantial role in the progression and/or resolution of several respiratory pathologies and that a better understanding of this could lead to novel therapeutic and patient management strategies. This review will outline the many facets of surfactant structure and function, the extent to which surfactant homeostasis is disrupted in inflammatory lung diseases, and the degree to which these changes may impact lung disease progression and symptoms, particularly in the context of environmental and tobacco smoke exposure.
Why is it so important to maintain pulmonary surfactant homeostasis?
Surfactant lines the pulmonary alveolar surface and is predominantly composed of lipids (90%; phospholipids and some neutral lipids such as cholesterol) and surfactant proteins (10%; SP-A, SP-B, SP-C and SP-D) (figure 1). Alveolar type II epithelial (ATII) cells are responsible for the secretion of surfactant complexes, in the form of lamellar bodies, as well as the bulk of its reuptake and recycling (figure 2a). Once lamellar bodies are secreted and mixed with surfactant proteins in the aqueous hypophase layer, tubular myelin structures are formed and act as a reservoir of phospholipids that are trafficked to a surfactant monolayer at the air–liquid interface of the alveolus. Over time or following direct damage – i.e. exposure to inhaled oxidants/toxins – surfactant phospholipids are taken up by ATII cells or alveolar macrophages for recycling or degradation, respectively. Although studies have shown that surfactant is involved in numerous homeostatic processes in the lung, from gas exchange to host defence, it is mostly known for its role in alveolar surface tension lowering. The reduction in surface tension – mediated by the major phospholipid component, phosphatidylcholine (PC), especially in its disaturated form dipalmitoyl-PC (DPPC) – facilitates lung inflation and deflation while preventing small airway collapse, making surfactant crucial for normal lung function. In addition, since oxygen and carbon dioxide exchange between the airspace and the lung parenchyma must pass through the surfactant layer at the air–liquid interface, the efficacy of this gas exchange can be greatly affected by changes in surfactant quantity, composition and structure [1–5].
FIGURE 1.
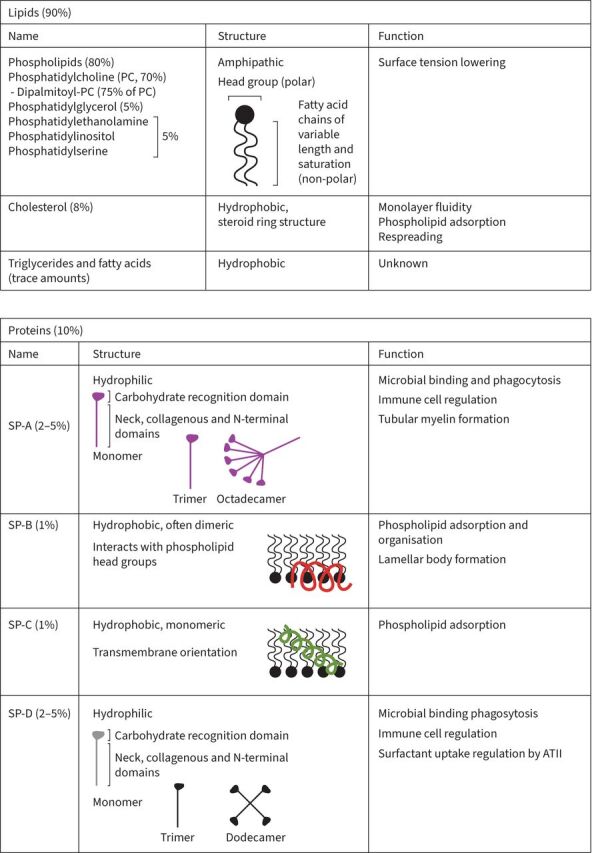
Components of pulmonary surfactant. ATII: alveolar type II epithelial cells; SP: surfactant protein.
FIGURE 2.

Schematic of the alveolus and pulmonary surfactant homeostasis in primary surfactant diseases. At the air–liquid interface, a surfactant monolayer helps reduce pulmonary surface tension and separates the alveolar airspace from the aqueous hypophase. Surfactant is produced by alveolar type II epithelial cells (ATII) and secreted as lamellar bodies which organise into tubular myelin structures. Surfactant is recycled and degraded by ATII cells and alveolar macrophages, respectively, and replaced using surfactant reserves stored in the tubular myelin. a) In patients suffering from pulmonary alveolar proteinosis (PAP), failure to catabolise and export surfactant, typically due to impaired granulocyte-monocyte colony stimulating factor (GM-CSF) signalling, leads to lipid-laden alveolar macrophages and surfactant accumulation. b) On the other hand, premature birth is associated with impaired ATII cell development leading to diminished surfactant production, thus reducing surfactant phospholipid and protein secretion and increasing alveolar surface tension. Despite their inverse impacts on surfactant pool size, patients suffering from both PAP and respiratory distress syndrome experience significant dyspnoea due to impaired gas exchange and increased difficulty breathing. ABCA3: ATP binding cassette subfamily A member 3; GM-CSFR: granulocyte-monocyte colony-stimulating factor receptor; SP: surfactant protein.
While the hydrophobic surfactant proteins SP-B and SP-C are mainly involved in surfactant organisation and its surface-active properties, hydrophilic SP-A and -D, also known as collectins, play a role in the innate immune response (figure 1). These collectins act as both positive and negative immunoregulators, depending on the context (reviewed in [6, 7]). In the absence of pathogen or apoptotic cells, SP-A and SP-D are thought to suppress inflammatory processes through the binding of the carbohydrate-recognition domain to the signal-regulatory protein α on alveolar macrophages, dendritic cells and ATII cells [8, 9]. Conversely, these proteins bind and opsonise infectious agents as well as apoptotic cells leading to enhanced bacterial aggregation, phagocytosis and pro-inflammatory mediator release [8–12]. The context-dependent roles of SP-A and SP-D become apparent in knockout animals, as they display not only impaired response to pathogens [13–17] but also increased basal inflammation [18].
Aside from ATII cells, alveolar macrophages also play a crucial role in regulating surfactant homeostasis. These phagocytes participate in the clearance and degradation of pulmonary surfactant and this specific activity is tightly controlled, granulocyte-monocyte colony stimulating factor (GM-CSF) being a prime regulator. It has also been suggested that other phagocytes recruited to the lungs following injury or insult, such as neutrophils, may contribute to surfactant homeostasis in the context of pulmonary inflammation [19, 20]. It is unsurprising that the processes of surfactant production, secretion and reuptake are dynamic and tightly regulated, as alterations in its quantity and/or quality can lead to important changes in its physical properties and surface activity, where both increased and decreased surfactant levels can have serious effects on pulmonary function (reviewed in [21, 22]). The important role of pulmonary surfactant homeostasis in the maintenance of normal lung function becomes patently clear when we look at pathologies caused by primary surfactant dysregulation.
Pulmonary diseases caused by primary surfactant dysregulation
Too little: respiratory distress syndrome (RDS) in pre-term infants
Typically affecting newborns and pre-term infants, RDS is characterised by incomplete ATII development and insufficient surfactant production (figure 2b), resulting in reduced lung compliance, increased risk of alveolar collapse, difficulty breathing and impaired gas exchange (reviewed in [23]). Not only do newborns with RDS exhibit significantly decreased surfactant pool size, but they also exhibit decreased bronchoalveolar lavage fluid (BALF) SP-A levels [24–26]. These pathological features can be significantly mitigated through exogenous surfactant administration, either synthetic or animal-derived [27], thus dramatically reducing the mortality rate of RDS [28]. Another therapeutic approach to prevent neonatal RDS during pre-term delivery is through maternal antenatal corticosteroid administration, which stimulates the development of fetal lung epithelial cells and increases endogenous surfactant production prior to birth (reviewed in [29]).
Too much: pulmonary alveolar proteinosis (PAP)
Diseases associated with impaired clearance or overproduction of surfactant can similarly lead to impaired lung function. Patients suffering from PAP typically present with difficulty breathing and exhibit surfactant accumulation in the alveoli, which impedes lung inflation and disrupts normal gas exchange [30, 31]. As shown in figure 2a, surfactant isolated from the BALF of PAP patients is highly abnormal: increased total phospholipid content, altered surfactant composition (increased % phosphatidylinositol (%PI), decreased %PC and % phosphatidylglycerol (%PG)) as well as increased SP-A and SP-D levels [32–36]. Primary PAP is typically caused by disruption of GM-CSF signalling such as mutations in the GM-CSF receptor or an autoimmune response generating antibodies against GM-CSF. Secondary or acquired PAP can be associated with several etiological causes – i.e. haematologic cancer, drugs, inhaled particulate matter, some infections, etc. (reviewed in [37–39]). Disruptions of GM-CSF signalling affect the development, maturation and function of alveolar macrophages (reviewed in [40]), further highlighting their critical role in the maintenance of surfactant homeostasis. The lack of GM-CSF signalling leads to impaired lipid catabolism and export from alveolar macrophages, leading to the development of an enlarged, foam cell-like phenotype and a reduced capacity for phagocytosis and surfactant processing [41, 42]. Although treatments rely heavily on whole lung lavage to remove excess surfactant, GM-CSF supplementation therapy has shown some mild benefit in certain patients [43–45]. As observed in surfactant-deficient pathologies, elevated surfactant levels are likewise associated with dyspnoea and reduced lung function, pointing to the importance of tightly regulated surfactant in the maintenance of pulmonary health.
Taken together, surfactant insufficiency or excess can significantly affect lung health and function, primarily due to changes in surface tension. Altered phospholipid content, whether increased or decreased, can substantially change the surfactant's surface activity and biophysical properties, leading to increased surface tension and impaired gas exchange [32, 46]. Furthermore, changes to surfactant protein content can disrupt normal surfactant function and may alter the response to pathogens. This is supported by research that found SP-D supplementation during surfactant replacement in pre-term lambs mitigated inflammation and improved alveolar surface tension [47, 48]. Given the severity of diseases caused by disrupted surfactant homeostasis, actively investigating how pulmonary surfactant is affected in inflammatory lung diseases and how it could cause and exacerbate symptoms appears crucial.
Pulmonary surfactant in inflammatory lung diseases
Besides the pulmonary pathologies directly associated with surfactant insufficiency or excess, the role of surfactant in the progression and development of other pulmonary diseases is often overlooked. Although many studies have found significant changes in surfactant homeostasis in inflammatory diseases of the lung (reviewed in [49–51]), few have explored the potential contribution of these changes to symptom presentation and disease progression/resolution, and even fewer have considered targeting these changes through therapeutic intervention. This section will discuss the relevant literature on the presence and extent of surfactant abnormalities in several inflammatory lung diseases (table 1 [52–113]) as well as outline the impact of these surfactant changes on disease progression and symptoms.
TABLE 1.
Summary of surfactant changes chronic pulmonary diseases
| Pulmonary disease | Model | Phospholipids | Surfactant proteins | Surface activity | References |
| Respiratory distress syndrome | Human (infants) | ↓↓↓ | ↓ SP-A | ↓↓↓ | [24–26, 52, 53] |
| Mice (SP-B−/−) | No data | ↓ | ↓↓↓ | [54, 55] | |
| Pulmonary alveolar proteinosis | Human | ↑↑↑ (↑ %PI, ↓ %PG and %PC) | ↑ all | ↓ in ABCA3 mutations | [31–35, 37, 39] |
| Mice (GM-CSF−/−) | ↑↑↑ | = or ↑ SP-A | = | [40, 56–59] | |
| Pulmonary infection | |||||
| Various | Human (children) | ↑ total (↓ PC) | ↓ SP-A | ↓↓↓ | [60, 61] |
| Respiratory syncytial virus | Human (infants) | ↑ total | ↓ SP-A, SP-B and SP-D | No data | [14, 62, 63] |
| Mice | = | No data | ↓↓↓ | [64] | |
| Pneumocystis jirovecii pneumonia | Mice | = | No data | ↓↓↓ | [65] |
| Asthma | |||||
| Baseline | Human (children and adults) | = total (↓ or = %DPPC) | No data | = | [60, 66–70] |
| Guinea pigs (OA) | = total, PC, PI, PE and PG | No data | ↓↓ | [71, 72, 73] | |
| Acute asthma attack and allergen challenge | Human (children and adults) | = total (↓ %DPPC) | No data | ↓↓↓ | [67, 70, 74] |
| Mice (Aspergillus fumigatus) | No data | ↓ SP-B | ↓↓↓ | [75] | |
| Pulmonary fibrosis | |||||
| Idiopathic pulmonary fibrosis | Human | ↓ total (↑ PI, ↓ PG and ↓ DPPC) | = SP-D, ↓ SP-A | No data | [36, 76–81] |
| Rats (bleomycin) | ↑ total | = SP-A | ↓↓ | [82, 83] | |
| Mice (bleomycin) | ↑ total (↑ PC and PG, ↓ PE) | No data | ↓↓ | [84–86, 87, 88] | |
| Mice (TGF-β1) | No data | ↓ SP-B and SP-C | ↓↓ | [89] | |
| Silicosis | Human | ↓ total | No data | No data | [76] |
| Mice (silica) | ↑ total | ↑ SP-A | No data | [90] | |
| Cigarette smoking | Mice | = or ↑ total (↓ %PC, ↑ oxPC) | = SP-B | No data | [91–93, 94, 95] |
| Rats | = total (↓ %DPPC) | ↓ SP-A, SP-B | ↓↓ | [96, 97, 98] | |
| Human (healthy smokers) | = or ↓ total (↑ %PE, ↑ % PG, ↓ %PC) | ↓ SP-A, SP-D | ↓↓ | [99, 100, 101–106, 107] | |
| COPD | Human | = or ↓ total (= or ↓ %PG, %PC) | ↓ SP-A, = SP-B, SP-C, SP-D | No data | [91, 102, 108, 109, 110, 111, 112] |
| E-cigarette vaping | Mice | ↑ total (↑ DSPC, MPPC, = DPPC) | ↓ SP-A, SP-D | No data | [113] |
| Human | No data | No data | No data | ||
ABCA3: ATP binding cassette subfamily A member 3; DPPC: dipalmitoyl-phosphatidylcholine; DSPC: distearoylphosphatidylcholine; GM-CSF: granulocyte-monocyte colony-stimulating factor; MPPC: monopalmitoylphosphatidylcholine; oxPC: oxidised phosphatidylcholine; PC: phosphatidylcholine; PE: phosphorylethanolamine; PG: phosphatidylglycerol; PI: phosphatidylinositol; SP: surfactant protein; TGF-β1: transforming growth factor-β1.
Pulmonary infections
As mentioned above, surfactant plays an important role in host-defence processes and there is some evidence that respiratory pathogens can affect surfactant homeostasis. A study looking at various pulmonary infections in children (i.e. Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa, etc.) found a significant increase in surfactant phospholipid content in BALF [60]. However, this was associated with decreased surface-active DPPC and subsequently reduced surface tension-lowering ability, where %DPPC positively correlated with forced expiratory volume in 1 s (FEV1) [60]. In infants with respiratory syncytial virus (RSV) infection, BALF total phospholipid levels were increased while surfactant proteins were decreased (SP-A, SP-B and SP-D) [62]. Another study found that children with bacterial or viral pneumonitis had decreased relative surfactant PC and SP-A levels upon admission to hospital, both of which were positively correlated to pulmonary compliance [61]. In a mouse model of Pneumocystis jirovecii pneumonia, there was no effect on total phospholipid levels although the surface tension-lowering activity of the BALF was reduced [65]. One study also found that RSV-infected mice had significant alterations in surfactant function and that in vitro co-incubation of calf surfactant with BALF from the infected mice significantly inhibited the exogenous surfactant's surface activity [64], suggesting that pulmonary infection may lead to the presence of inhibitory proteins in the alveoli. Several studies have since proposed that proteins of plasma origin, leaking into the lungs during inflammation due to increased vascular permeability and parenchymal damage can directly inhibit surfactant function: e.g. fibrinogen, haemoglobin and albumin [114, 115]. However, it remains unclear which components of surfactant are being interfered with and through what direct or indirect mechanisms these effects are mediated.
SP-A and SP-D are particularly important in host defence, as they have been shown in vitro to bind and opsonise a wide variety of infectious agents (reviewed in [116, 117]) to enhance their clearance while increasing inflammatory cytokine secretion [8–12]. The importance of SP-A and SP-D in inflammatory regulation has been explored using murine knockout models, which display ineffective responses to pathogens and increased susceptibility to various lung infections: i.e. Pneumocystis jirovecii [118, 119], Pseudomonas aeruginosa [15], group B Streptococcus [13, 120], influenza A virus [17, 121, 122] and non-typeable Haemophilus influenzae (NTHi) [16] and RSV [14]. Recently, it has also been shown that SP-D can limit neutrophil extracellular trap (NET) formation induced by bacterial endotoxin (lipopolysaccharide, LPS) and that SP-D-deficient mice exhibit excessive NET formation in the lungs following intratracheal LPS administration, an important finding since NETs can significantly modulate the surface-active properties of surfactant [123].
As a potential therapeutic approach, some studies have found that exogenous synthetic or natural surfactant treatment can mitigate pulmonary infection symptoms and improve bacterial clearance. Exogenous surfactant administration in children with severe RSV was found to ameliorate lung function outcomes – such as reduced resistance, increased compliance and improved gas exchange – where compliance and resistance improvements correlated with DPPC content in the BALF [124]. In addition, studies looking at the effect of surfactant lipids on NTHi bacteria found that they were able to bind NTHi in vitro, prevent their aggregation and thus inhibit their invasion into lung pneumocytes, as well as accelerate NTHi clearance in a mouse model of NTHi lung infection [125]. In vitro treatment with Survanta, a natural surfactant extract containing phospholipids and hydrophobic proteins (SP-B and SP-C), has also been shown to have an anti-inflammatory effect, leading to reduced NF-κB signalling and cytokine release (macrophage inflammatory protein-1α) by human alveolar macrophages following LPS stimulation [126]. Similarly, mice administered an SP-A-related peptide, SPA4, displayed a reduced inflammatory response following intratracheal LPS administration [127] as well as improved symptom and histological scores, increased bacterial clearance and reduced cytokine levels in mice infected with Pseudomonas aeruginosa [128]. Despite the growing body of research suggesting that surfactant plays an instrumental role in the pulmonary response to infection, it is often overlooked in the study of infectious lung diseases.
Asthma
Asthma is a heterogeneous and complex chronic lung disease, marked by chronic inflammation, reversible airflow obstruction and airway remodelling. Atopic asthma is tightly linked to allergies while the aetiology of non-atopic asthma remains poorly understood. With regard to pulmonary surfactant biology, it has been shown that eosinophil lysophospholipases (phospholipase A2) can hydrolyse and inactivate surfactant phospholipids in vitro [129]. However, the contribution of these phospholipid modifications on small airway closure in asthma remains speculative. In general, studies looking at surfactant levels in asthma patients have also found little to no difference in total phospholipid pool size [52, 66–69]. However, some changes in surfactant composition have been documented. For instance, sputum levels of DPPC in mild to severe asthmatics were significantly decreased compared to healthy volunteers and correlated with FEV1 [66]. On the other hand, asthmatic children showed no change in BALF DPPC content [60]. In another study of mild atopic asthma, researchers found that baseline BALF phospholipid levels were similar to healthy volunteers but, after an allergen challenge, asthmatics exhibited increased BALF small/large aggregate ratios and altered PC species composition, effects which were associated with reduced surface tension-lowering activity and impaired lung function [67, 70]. Similarly, sputum from asthmatics in a stable state displayed normal surfactant composition and surface activity, while during an acute asthma attack, they observed significantly blunted surface tension-lowering ability [69]. Therefore, despite normal baseline surfactant properties, it is clear from these studies that acute inflammatory exacerbations in asthmatics can significantly alter surfactant homeostasis, likely contributing to disease symptoms.
As a potential therapeutic option for the mitigation of disease symptoms, a few preclinical and clinical studies have investigated the administration in asthma models and patients. In an ovalbumin-sensitised guinea pig model of asthma, prophylactic administration of calf surfactant extract (composed of surfactant lipids, SP-B and SP-C) prior to or immediately after antigen challenge led to decreased resistance and PCO2 as well as increased tidal volume, minute volume, flow rate, dynamic compliance and PO2 [71, 130]. In patients, one study found that during an acute asthma attack, asthmatics administered exogenous surfactant showed improved lung function (increased forced vital capacity (FVC) by approximately 12%, FEV1 by 27% and PaO2 by 13%) compared to untreated patients [74]. In addition, surfactant administration in asthmatic adults prior to allergen challenge was shown to mitigate early phase response and improve FEV1 without any effect on the late phase response [131]. Another study found that exogenous surfactant administration had no effect on bronchial responsiveness to histamine or lung function in nebulised asthmatic children [132]. In all, these studies suggest that in acute allergic asthma attacks, marked by increased pulmonary inflammation and surfactant dysfunction, administration of exogenous surfactant may improve some lung function outcomes but cannot alter primary disease presentations such as bronchial hyperresponsiveness.
Idiopathic pulmonary fibrosis (IPF)
Whether due to an unknown cause (idiopathic) or due to exposure to a specific agent (e.g. silica), pulmonary fibrosis is marked by inflammation, fibroblast overgrowth, irreversible matrix deposition and pulmonary remodelling, resulting in poor patient prognosis. Thus far, clinical studies have consistently found significant surfactant abnormalities in these patients. Although the surface-active properties of surfactant isolated from IPF patients have not been assessed, many researchers have observed decreased total phospholipid content in the BALF [76–80]. Similarly, a pattern of changes to the phospholipid composition in the BALF emerges from these studies: increased sphingomyelin and PI with reduced PG [77–81]. While some studies found that the levels of various PC subspecies were altered in IPF patient BALF [77, 81], others found that PC remained unchanged in these patients [76, 78, 79]. On the other hand, bleomycin- or silica-treated mice, the most commonly used animal models of pulmonary fibrosis, displayed a marked increase in lipid and phospholipid levels in the BALF, with increased PC, PS, cholesterol and triglycerides concentrations [82, 84–88, 90]. However, despite the observed increased in surfactant pool size, BALF isolated from bleomycin-treated rats displayed impaired surface tension-lowering activity [82, 83].
Animal models of pulmonary fibrosis strongly suggest that pulmonary lipid dysregulation directly contributes to disease pathology. One study found that genes associated with lipid transport and metabolism were downregulated in bleomycin- and silica-treated mice leading to oxidised PC accumulation in the BALF, which directly induced pulmonary fibrosis [86, 133]. Furthermore, treatment with GM-CSF, which stimulates surfactant reuptake and clearance, significantly mitigated pulmonary lipid alterations and reduced lung pathology scores [86]. Conversely, bleomycin administration in ATP binding cassette subfamily G member 1 (ABCG1)-knockout mice [86], displaying impaired lipid export, or mice lacking Elovl6 [134], leading to altered fatty acid composition, were both associated with increased oxidised phospholipid content in the BALF and worsened pulmonary fibrosis. In an effort to restore pulmonary lipid homeostasis, rats were administered exogenous natural surfactant repeatedly following bleomycin treatment, which resulted in decreased neutrophil infiltration, improved surfactant surface activity, increased airway opening and ameliorated lung function [135]. In all, it seems that changes to surfactant play an important role in the progression of pulmonary fibrosis and support further investigation into the clinical implications for IPF patients.
Smoking and COPD
Since as early as the 1960s and 1970s, pulmonary surfactant abnormalities have been noted in cigarette smokers and COPD patients [99, 100]. However, changes to surfactant quality, quantity and composition in the context of cigarette smoke-associated lung diseases, such as COPD, have often been overlooked as contributing factors to disease pathogenesis. In addition, the rising popularity of electronic (e)-cigarette vaping may also have implications on surfactant homeostasis and pulmonary health. This section outlines the available preclinical, clinical and in vitro data regarding the effects of cigarette smoking on pulmonary surfactant (figure 3), the interplay between the resultant surfactant abnormalities and development of COPD as well as the little that is known on the effects of e-cigarette use on surfactant homeostasis.
FIGURE 3.

Schematic of the alveolus and pulmonary surfactant homeostasis in the context of cigarette smoke exposure and chronic obstructive pulmonary disease (COPD). Chronic cigarette smoke exposure has directly cytotoxic and oxidative effects in the lungs, leading to increased oxidative damage and stress. a) Surfactant phospholipid damage and increased cellular debris stimulate surfactant uptake and recycling by alveolar macrophages and type I epithelial cells, respectively. As long-term exposure overwhelms these systems, alveolar macrophages from smokers and COPD patients become lipid-laden, exhibit impaired phagocytic ability and secrete a plethora of pro-inflammatory cytokines, which rapidly lead to the rapid recruitment of neutrophils from the circulation. It has recently been shown that recruited neutrophils also internalise pulmonary surfactant. b) Another hallmark of COPD is susceptibility to recurrent viral and bacterial infection, further exacerbating the pulmonary inflammatory response and adding an additional phagocytic burden on alveolar macrophages and neutrophils. Furthermore, increased lung tissue damage and inflammation leads to pulmonary vascular leak, making circulating surfactant protein (SP)-A and D levels a good indicator of disease severity in these patients.
Cigarette smoking
Despite the well-characterised harm to the respiratory system caused by tobacco smoking, it remains prevalent in the developed world and is in fact increasing in popularity in developing countries [136]. Smoking rapidly triggers an inflammatory response in the lungs, a major focus of the biomedical research pursued on the subject. However, it has been known for some time that cigarette smoke exposure also leads to significant dysregulation in surfactant homeostasis. Studies from as early as 1969 showed that active smokers have reduced surfactant levels in the BALF, even in the absence of pulmonary disease [99, 100, 101]. More recent clinical studies, mostly with small groups (10–23 smokers), have found conflicting results: either unchanged, reduced or increased total phospholipid levels in smokers with or without COPD compared to non-smokers, suggesting a significant heterogeneity in individual susceptibility to surfactant homeostasis disruption by smoking [102–104]. In terms of surfactant composition, however, many observed increased PE and PG as well as decreased PC and cholesterol in the BALF [103–106]. These alterations in pulmonary surfactant composition were also associated with significant reductions in surface activity in the BALF isolated from smokers compared to non-smokers [105, 137]. Interestingly, the reduction in BALF PC levels was found to be inversely correlated with alveolar macrophage number as well as the cumulative cigarette smoke exposure in active smokers (represented in pack years) [106], suggesting that alterations in surfactant composition may be due to increased damage and elevated rates of clearance by infiltrating inflammatory cells.
Of the few animal studies looking at the effects of cigarette smoke exposure on pulmonary surfactant, there seem to be significant differences in surfactant pool size and composition, likely due to the different animal models used and the exposure duration chosen. In mice exposed to cigarette smoke for 6 months, one study found that total phospholipid, cholesterol and PC levels in the BALF were decreased [91]. Conversely, after mice were acutely exposed to mainstream cigarette smoke for 4 days, no significant change in BALF PC was observed [92, 93]. In another study, while rats exposed to cigarette smoke over 60 weeks showed no change in surfactant composition or total phospholipid concentration, they found that surfactant isolated from these animals exhibited impaired respreadability and reduced surface tension-lowering activity compared to control animals [96]. It was proposed that cigarette smoke exposure increases phospholipid oxidation, which hinders its surface tension-lowering ability [138]. Indeed, BALF isolated from mice exposed to cigarette smoke for 2 weeks was found to contain high levels of oxidised PC and in vitro treatment of alveolar macrophages with oxidised lipids and phospholipids led to reduced cell viability, decreased respiratory burst upon LPS stimulation and impaired bacterial killing [94, 139]. Similarly, several studies have observed that direct in vitro treatment of surfactant with cigarette smoke led to dose-dependent alterations in surfactant interfacial properties and microstructure leading to reduced surface activity [140–143]. Furthermore, epithelial cells incubated in vitro with cigarette smoke extract showed high levels of lipid oxidation as well as significant changes to their lipidomic profile [139]. Interestingly, pulmonary administration of oxidised phospholipids reproduced several features of tobacco smoke-induced lung inflammation, suggesting that damaged surfactant plays a major role in driving the early immune response to smoking [93].
As discussed in the previous sections, surfactant proteins play a crucial role not only in pulmonary surfactant organisation and activity but also in host defence. In the BALF and sputum of active smokers, levels of SP-A and SP-D are reduced [102, 107, 144]. Likewise, rats chronically exposed to cigarette smoke exhibited a significant decrease in BALF SP-A and SP-B levels [97] and reduced SP-A expression in the lung [98]. One potential mechanism for the decreased SP-A and SP-D levels following chronic cigarette smoke exposure is that neutrophils recruited to the lungs secrete protease enzymes capable of cleaving and inactivating these proteins [145, 146]. Another study, however, found that SP-A and SP-D levels were increased in the BALF of mice acutely exposed to cigarette smoke exposure, an effect which was heightened when neutrophils were depleted [20]. In the lungs of mice exposed to cigarette smoke for just 5 days, chemically modified SP-A can be detected in the lungs [147]. In vitro experiments showed that both cigarette smoke extract and acrolein, a component of cigarette smoke, can modify SP-A leading to aldehyde adducts, impairing its ability to stimulate phagocytosis and inhibit bacterial growth [147]. These data suggest that cigarette smoke exposure not only reduces SP-A and SP-D levels but also modifies their host-defence effectiveness. How all these effects on surfactant quantity, quality and composition contribute to the development of cigarette smoke-associated lung diseases, such as COPD, remains to be fully elucidated.
COPD
Cigarette smoking is a major risk factor for the development of COPD, an insidious pulmonary disease affecting large and small airways as well as the lung parenchyma and generally characterised by fixed airflow obstruction, chronic lung inflammation, mucus overproduction, small airway loss and lung tissue destruction as well as frequent acute exacerbations (reviewed in [148]). Currently available treatments are not curative and mainly focus on optimising airflow and pulmonary inflammation management. Interestingly, COPD patients also seem to exhibit significant surfactant changes, although their contribution to pathogenesis or disease progression is unclear. Compared to healthy subjects, several studies have shown that COPD-patient BALF contains reduced total phospholipid levels [91, 108, 109]. However, no significant difference in total phospholipid pool size was observed when comparing smokers with or without COPD, with all smokers displaying lower levels of total phospholipid compared to non-smokers [102]. The composition of surfactant was found to be unchanged in many clinical studies compared to healthy non-smokers [108, 109], while one study found reduced cholesterol, PG and PC levels in the BALF, all of which negatively correlated with lung function outcomes [91]. As a potential therapeutic avenue, exogenous surfactant administration was tested in smokers with chronic bronchitis and led to ameliorated lung function (increased pre- and post-bronchodilator FEV1 and FVC) and decreased gas trapping [149]. Sadly, the surface activity and surface tension-lowering abilities of surfactant isolated from COPD patients was not measured in any of the aforementioned studies.
Although SP-A and SP-D have also been explored in the context of COPD, studies have mostly focussed on their potential as serum biomarkers, since advanced disease leads to increased vascular leak of pulmonary proteins from the alveoli into the circulation [144]. In line with this pulmonary leak, SP-A levels in exhalate [150], lung tissue and ATII pneumocytes [110] have been shown to be reduced in COPD patients while the percentage of SP-A positively alveolar macrophages increased [110], which inversely correlated with lung function. Similarly, while one study found BALF SP-D levels to be unchanged in COPD patients [102], Winkler et al. [111] found that SP-D was reduced in COPD patients compared to healthy smokers and non-smokers and that BALF SP-D levels were inversely correlated with lung function. Since we know that COPD patients exhibit pulmonary function decline as well as increased susceptibility to infection and we also understand the role of surfactant in the maintenance of lung function and the response to pathogen, it seems likely that the changes in surfactant homeostasis present in smokers and COPD patients contribute to the progression and development of lung disease.
Vaping and e-cigarette use
Recent case reports published across the United States have indicated that e-cigarette vaping can lead to vaping-associated lung injury (EVALI), marked by pulmonary inflammation, dramatic lung damage and even death. Patients presenting with EVALI were shown to have large, lipid-laden macrophages in the lavage [151, 152], suggesting that lipid components of e-cigarette liquid, such as vitamin E acetate and cannabis oil, are at the root of the lung injury [151–155]. However, even in animal models of e-cigarette vapour exposure without added lipids or lung injury, there seem to be significant pulmonary lipid abnormalities. Although mice exposed to e-cigarette vapours for 4 months did not develop any noticeable pulmonary inflammation or lung damage, it was shown that e-cigarette vapour-exposed mice exhibited enlarged alveolar macrophages with intracellular alterations in their lipidomic profile [113]. In addition, BALF levels of total and disaturated phospholipids (though not DPPC) were increased, accompanied by decreased SP-D in mice exposed to e-cigarette vapours compared to control animals [113].
Unfortunately, the effect of prolonged e-cigarette vapour exposure on surfactant surface activity was not assessed in any of these studies. In vitro experiments have noted that direct treatment of surfactant with e-cigarette vapours led to altered surfactant lateral structure without affecting its interfacial properties [140], in contrast to cigarette smoke exposure which significantly altered both aspects of surfactant structure/function. Similarly, it was found that only excessively elevated doses of e-cigarette liquid, equivalent to approximately 200 times a normal puffing session, altered the surface tension-lowering ability of bovine surfactant in vitro [156]. As many EVALI patients were using e-cigarette liquids containing vitamin E acetate, surfactant was treated in vitro with this additive to assess its effects on surface activity. Researchers found that vitamin E acetate altered surfactant arrangement and function, leading to impaired surface tension-lowering ability [157]. In all, it seems that further study into the potentially detrimental effects of e-cigarette vapour exposure on surfactant homeostasis and thus lung health requires thorough investigation, especially with e-cigarette liquids containing lipid additives.
Targeting surfactant homeostasis in lung diseases
In all, there are many pulmonary diseases that seem to be affected, either directly or indirectly, by alterations in surfactant function and we believe that significant changes in surfactant quantity, quality and composition should be considered in the study of any pulmonary pathology. As a dynamic yet tightly regulated structure, there are several pathways involved in the maintenance of surfactant homeostasis and therapeutic targeting of these pathways, though they may be of clinical benefit, could prove challenging. A prominent example of therapeutic surfactant modulation is corticosteroid administration, which hastens lung maturation thus improving surfactant production in pre-term infants. Unfortunately, this treatment does not affect surfactant production postnatally, even when administered only days after birth [158, 159]. Therefore, treatments that help restore surfactant function, through pharmacological modulation of production or recycling/degradation, may be worth exploring.
Supplementing the pulmonary surfactant pool
As outlined in the previous sections, several animal and clinical studies have explored exogenous surfactant administration as a potential therapeutic option in lung disease. Either synthetic or naturally derived surfactant preparations can effectively increase surfactant pool size; however, the effect is temporary, as surfactant turnover is rapid. Furthermore, the composition of the exogenously administered surfactant can have an important impact on the response to supplementation, as formulations differ in phospholipid profile/concentration, cholesterol levels as well as SP-B and -C content. It would be important to tailor the dose and formulation based on the specific disease presentations. Nevertheless, surfactant administration in the context pulmonary infection was found to ameliorate lung function outcomes in children with severe RSV [124] and in smokers with chronic bronchitis [149]. Similarly, in both animal models [71, 130] and in patients suffering acute asthma attacks [74, 131], exogenous surfactant was associated with short-term improvements in lung function. There is also the potential of supplementing with collectins, as one research group has shown in several mouse models that administration of SPA4, an SP-A-related protein, was able to significantly improve the response to infection in a mouse model [127, 128]. This highlights the importance of characterising the type, extent and cause of the surfactant dysfunction observed in a particular lung disease to best target the pathology.
Targeting surfactant recapture and degradation
Another way of modifying surfactant production is by targeting enzymes and transporters involved in surfactant production and clearance. For instance, ABC transporter proteins ATP binding cassette subfamily A member 1 and ABCG1, which are primarily involved in lipid export from ATII cells and alveolar macrophages, respectively, are crucial to surfactant homeostasis and mutations which limit their function led to impaired lipid processing and lung function (reviewed in [160]). Treatments which modulate the expression of these transporters and promote lipid export, such reverse cholesterol transport agonists, may be particularly useful in pulmonary pathologies marked by surfactant accumulation or increased surfactant damage necessitating increased clearance/degradation. For instance, treatment with the liver X receptor α agonist TO901317 led to a significant improvement in a murine model of lung fibrosis [133] and reduced pulmonary inflammation in response to LPS administration and Klebsiella pneumoniae infection [161]. On the other hand, in mice exposed to cigarette smoke, treatment with the same lipid export activator led to worsened lung inflammation and significantly depleted PC levels in the BALF [92]. This further suggests that upregulation of lipid export and surfactant turnover may not be beneficial in all lung pathologies and that a better understanding of the primary surfactant dysfunction is required to effectively target the specific surfactant changes in quantity, quantity or composition.
Targeting surfactant production
Many other endogenous or exogenous stimuli have been shown to enhance surfactant production, such as hyperventilation and pharmacological agonists such as isoproterenol, diacylglycerol, calcium ionophore, terbutaline, tetradecanoyl phorbol acetate and ATP (reviewed in [162, 163]). Some of these agents act by stimulating ATII cell exocytosis of lamellar bodies through upregulation of intracellular calcium signalling or through protein kinase C activation. There are also molecules which have been shown to inhibit surfactant secretion, SP-A and DPPC, which act through negative feedback pathways, as well as exocytosis inhibitors like cytochalasin D and vinblastine (reviewed in [164, 165]). Although the latter molecules are only research tools, it still suggests that the discovery of molecules that modulate surfactant production/secretion may be useful tools in mitigating the disruptions in surfactant homeostasis observed in pulmonary disease.
Closing remarks
In all, it is clear that maintenance of pulmonary surfactant homeostasis is significantly disrupted in a variety of chronic lung diseases as well as in the context of environmental and tobacco smoke exposure. Whether these effects on surfactant composition and integrity are a cause or a consequence of lung disease remains to be fully elucidated. Regardless, the evidence presented herein strongly suggests that the impact of surfactant alterations in the context of lung disease should not be disregarded as a contributor to disease progression and symptoms but rather should become a fixture of pulmonary research, as routine as measurements of inflammatory markers. There is also some evidence to suggest that surfactant modulation, either through direct exogenous administration or through pharmacological targeting, may mitigate the effects of surfactant dysregulation in certain pulmonary diseases.
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: N. Milad has nothing to disclose.
Conflict of interest: M.C. Morissette has nothing to disclose.
References
- 1.Olmeda B, Villen L, Cruz A, et al. Pulmonary surfactant layers accelerate O2 diffusion through the air-water interface. Biochim Biophys Acta 2010; 1798: 1281–1284. doi: 10.1016/j.bbamem.2010.03.008 [DOI] [PubMed] [Google Scholar]
- 2.Schermuly R, Schmehl T, Gunther A, et al. Ultrasonic nebulization for efficient delivery of surfactant in a model of acute lung injury – impact on gas exchange. Am J Respir Crit Care Med 1997; 156: 445–453. doi: 10.1164/ajrccm.156.2.9609092 [DOI] [PubMed] [Google Scholar]
- 3.van Zyl JM, Smith J, Hawtrey A. The effect of a peptide-containing synthetic lung surfactant on gas exchange and lung mechanics in a rabbit model of surfactant depletion. Drug Des Devel Ther 2013; 7: 139–148. doi: 10.2147/DDDT.S40622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walther FJ, Hernandez-Juviel J, Bruni R, et al. Protein composition of synthetic surfactant affects gas exchange in surfactant-deficient rats. Pediatr Res 1998; 43: 666–673. doi: 10.1203/00006450-199805000-00016 [DOI] [PubMed] [Google Scholar]
- 5.Krause MF, Schulte-Monting J, Hoehn T. Rate of surfactant administration influences lung function and gas exchange in a surfactant-deficient rabbit model. Pediatr Pulmonol 1998; 25: 196–204. doi: [DOI] [PubMed] [Google Scholar]
- 6.Glasser JR, Mallampalli RK. Surfactant and its role in the pathobiology of pulmonary infection. Microbes Infect 2012; 14: 17–25. doi: 10.1016/j.micinf.2011.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korfhagen TR. Surfactant protein A (SP-A)-mediated bacterial clearance: SP-A and cystic fibrosis. Am J Respir Cell Mol Biol 2001; 25: 668–672. doi: 10.1165/ajrcmb.25.6.f221 [DOI] [PubMed] [Google Scholar]
- 8.Gardai SJ, Xiao Y-Q, Dickinson M, et al. By binding SIRPα or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003; 115: 13–23. doi: 10.1016/S0092-8674(03)00758-X [DOI] [PubMed] [Google Scholar]
- 9.Janssen WJ, McPhillips KA, Dickinson MG, et al. Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am J Respir Crit Care Med 2008; 178: 158–167. doi: 10.1164/rccm.200711-1661OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schagat TL, Wofford JA, Wright JR. Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. J Immunol 2001; 166: 2727–2733. doi: 10.4049/jimmunol.166.4.2727 [DOI] [PubMed] [Google Scholar]
- 11.Vandivier RW, Ogden CA, Fadok VA, et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol 2002; 169: 3978–3986. doi: 10.4049/jimmunol.169.7.3978 [DOI] [PubMed] [Google Scholar]
- 12.Hartshorn KL, White MR, Crouch EC. Contributions of the N- and C-terminal domains of surfactant protein d to the binding, aggregation, and phagocytic uptake of bacteria. Infect Immun 2002; 70: 6129–6139. doi: 10.1128/IAI.70.11.6129-6139.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeVine AM, Bruno MD, Huelsman KM, et al. Surfactant protein A-deficient mice are susceptible to group B streptococcal infection. J Immunol 1997; 158: 4336–4340. [PubMed] [Google Scholar]
- 14.LeVine AM, Gwozdz J, Stark J, et al. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J Clin Invest 1999; 103: 1015–1021. doi: 10.1172/JCI5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LeVine AM, Kurak KE, Bruno MD, et al. Surfactant protein-A-deficient mice are susceptible to Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol 1998; 19: 700–708. doi: 10.1165/ajrcmb.19.4.3254 [DOI] [PubMed] [Google Scholar]
- 16.LeVine AM, Whitsett JA, Gwozdz JA, et al. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol 2000; 165: 3934–3940. doi: 10.4049/jimmunol.165.7.3934 [DOI] [PubMed] [Google Scholar]
- 17.LeVine AM, Whitsett JA, Hartshorn KL, et al. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol 2001; 167: 5868–5873. doi: 10.4049/jimmunol.167.10.5868 [DOI] [PubMed] [Google Scholar]
- 18.Hawgood S, Ochs M, Jung A, et al. Sequential targeted deficiency of SP-A and -D leads to progressive alveolar lipoproteinosis and emphysema. Am J Physiol Lung Cell Mol Physiol 2002; 283: L1002–L1010. doi: 10.1152/ajplung.00118.2002 [DOI] [PubMed] [Google Scholar]
- 19.Quintero OA, Wright JR. Clearance of surfactant lipids by neutrophils and macrophages isolated from the acutely inflamed lung. Am J Physiol Lung Cell Mol Physiol 2002; 282: 330–339. doi: 10.1152/ajplung.00190.2001 [DOI] [PubMed] [Google Scholar]
- 20.Milad N, Pineault M, Lechasseur A, et al. Neutrophils and IL-1α regulate surfactant homeostasis during cigarette smoking. J Immunol 2021; 207: ji2001182. doi: 10.4049/jimmunol.2001182 [DOI] [PubMed] [Google Scholar]
- 21.Goss V, Hunt AN, Postle AD. Regulation of lung surfactant phospholipid synthesis and metabolism. Biochim Biophys Acta 2013; 1831: 448–458. doi: 10.1016/j.bbalip.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 22.Perez-Gil J. Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim Biophys Acta 2008; 1778: 1676–1695. doi: 10.1016/j.bbamem.2008.05.003 [DOI] [PubMed] [Google Scholar]
- 23.Ma CC-H, Ma S. The role of surfactant in respiratory distress syndrome. Open Respir Med J 2012; 6: 44–53. doi: 10.2174/1874306401206010044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevens PA, Scadow B, Bartholain S, et al. Surfactant protein A in the course of respiratory distress syndrome. Eur J Pediatr 1992; 151: 596–600. doi: 10.1007/BF01957730 [DOI] [PubMed] [Google Scholar]
- 25.Griese M, Dietrich P, Reinhardt D. Pharmacokinetics of bovine surfactant in neonatal respiratory distress syndrome. Am J Respir Crit Care Med 1995; 152: 1050–1054. doi: 10.1164/ajrccm.152.3.7663782 [DOI] [PubMed] [Google Scholar]
- 26.Taïeb J, Francoual J, Magny J-F, et al. Surfactant associated protein A determination using a chemiluminescence system - application to tracheal aspirates from newborns. Clin Chim Acta 1995; 235: 229–234. doi: 10.1016/0009-8981(95)06019-0 [DOI] [PubMed] [Google Scholar]
- 27.Hallman M, Merritt TA, Pohjavuori M, et al. Effect of surfactant substitution on lung effluent phospholipids in respiratory distress syndrome: evaluation of surfactant phospholipid turnover, pool size, and the relationship to severity of respiratory failure. Pediatric Res 1986; 20: 1228–1235. doi: 10.1203/00006450-198612000-00008 [DOI] [PubMed] [Google Scholar]
- 28.Malloy MH, Freeman DH. Respiratory distress syndrome mortality in the United States, 1987 to 1995. J Perinatol 2000; 20: 414–420. doi: 10.1038/sj.jp.7200420 [DOI] [PubMed] [Google Scholar]
- 29.Robertson B. Corticosteroids and surfactant for prevention of neonatal RDS. Ann Med 1993; 25: 285–288. doi: 10.3109/07853899309147876 [DOI] [PubMed] [Google Scholar]
- 30.Vymazal T, Krecmerova M. Respiratory strategies and airway management in patients with pulmonary alveolar proteinosis: a review. Biomed Res Int 2015; 2015: 639543. doi: 10.1155/2015/639543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griese M. Pulmonary alveolar proteinosis: a comprehensive clinical perspective. Pediatrics 2017; 140: e20170610. doi: 10.1542/peds.2017-0610 [DOI] [PubMed] [Google Scholar]
- 32.Ramirez J, Harlan WR, Jr. Pulmonary alveolar proteinosis. Nature and origin of alveolar lipid. Am J Med 1968; 45: 502–512. doi: 10.1016/0002-9343(68)90166-6 [DOI] [PubMed] [Google Scholar]
- 33.Brasch F, Birzele J, Ochs M, et al. Surfactant proteins in pulmonary alveolar proteinosis in adults. Eur Respir J 2004; 24: 426–435. doi: 10.1183/09031936.04.00076403 [DOI] [PubMed] [Google Scholar]
- 34.Doyle IR, Davidson KG, Barr HA, et al. Quantity and structure of surfactant proteins vary among patients with alveolar proteinosis. Am J Respir Crit Care Med 1998; 157: 658–664. doi: 10.1164/ajrccm.157.2.9701090 [DOI] [PubMed] [Google Scholar]
- 35.Honda Y, Kataoka K, Hayashi H, et al. Alterations of acidic phospholipids in bronchoalveolar lavage fluids of patients with pulmonary alveolar proteinosis. Clin Chim Acta 1989; 181: 11–18. doi: 10.1016/0009-8981(89)90312-4 [DOI] [PubMed] [Google Scholar]
- 36.Honda Y, Kuroki Y, Matsuura E, et al. Pulmonary surfactant protein D in sera and bronchoalveolar lavage fluids. Am J Respir Crit Care Med 1995; 152: 6 Pt 1, 1860–1866. doi: 10.1164/ajrccm.152.6.8520747 [DOI] [PubMed] [Google Scholar]
- 37.Trapnell BC, Carey BC, Uchida K, et al. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol 2009; 21: 514–521. doi: 10.1016/j.coi.2009.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salvaterra E, Campo I. Pulmonary alveolar proteinosis: from classification to therapy. Breathe 2020; 16: 200018. doi: 10.1183/20734735.0018-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell BC, Nakata K, Bonella F, et al. Pulmonary alveolar proteinosis. Nat Rev Dis Primers 2019; 5: 16. doi: 10.1038/s41572-019-0066-3 [DOI] [PubMed] [Google Scholar]
- 40.Trapnell BC, Whitsett JA. GM-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol 2002; 64: 775–802. doi: 10.1146/annurev.physiol.64.090601.113847 [DOI] [PubMed] [Google Scholar]
- 41.Uchida K, Nakata K, Suzuki T, et al. Granulocyte/macrophage-colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood 2009; 113: 2547–2556. doi: 10.1182/blood-2008-05-155689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenhill SR, Kotton DN. Pulmonary alveolar proteinosis: a bench-to-bedside story of granulocyte-macrophage colony-stimulating factor dysfunction. Chest 2009; 136: 571–577. doi: 10.1378/chest.08-2943 [DOI] [PubMed] [Google Scholar]
- 43.Sheng G, Chen P, Wei Y, et al. Better approach for autoimmune pulmonary alveolar proteinosis treatment: inhaled or subcutaneous granulocyte-macrophage colony-stimulating factor: a meta-analyses. Respir Res 2018; 19: 163. doi: 10.1186/s12931-018-0862-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papiris SA, Tsirigotis P, Kolilekas L, et al. Long-term inhaled granulocyte macrophage-colony-stimulating factor in autoimmune pulmonary alveolar proteinosis: effectiveness, safety, and lowest effective dose. Clin Drug Investig 2014; 34: 553–564. doi: 10.1007/s40261-014-0208-z [DOI] [PubMed] [Google Scholar]
- 45.Tazawa R, Ueda T, Abe M, et al. Inhaled GM-CSF for pulmonary alveolar proteinosis. N Engl J Med 2019; 381: 923–932. doi: 10.1056/NEJMoa1816216 [DOI] [PubMed] [Google Scholar]
- 46.Ikegami M, Jacobs H, Jobe A. Surfactant function in respiratory distress syndrome. J Pediatr 1983; 102: 443–447. doi: 10.1016/S0022-3476(83)80673-8 [DOI] [PubMed] [Google Scholar]
- 47.Ikegami M, Carter K, Bishop K, et al. Intratracheal recombinant surfactant protein d prevents endotoxin shock in the newborn preterm lamb. Am J Respir Crit Care Med 2006; 173: 1342–1347. doi: 10.1164/rccm.200509-1485OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato A, Whitsett JA, Scheule RK, et al. Surfactant protein-d inhibits lung inflammation caused by ventilation in premature newborn lambs. Am J Respir Crit Care Med 2010; 181: 1098–1105. doi: 10.1164/rccm.200912-1818OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopez-Rodriguez E, Gay-Jordi G, Mucci A, et al. Lung surfactant metabolism: early in life, early in disease and target in cell therapy. Cell Tissue Res 2017; 367: 721–735. doi: 10.1007/s00441-016-2520-9 [DOI] [PubMed] [Google Scholar]
- 50.Han S, Mallampalli RK. The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc 2015; 12: 765–774. doi: 10.1513/AnnalsATS.201411-507FR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griese M. Pulmonary surfactant in health and human lung diseases: state of the art. Eur Respir J 1999; 13: 1455–1476. doi: 10.1183/09031936.99.13614779 [DOI] [PubMed] [Google Scholar]
- 52.Garmany TH, Moxley MA, White FV, et al. Surfactant composition and function in patients with ABCA3 mutations. Pediatr Res 2006; 59: 801–805. doi: 10.1203/01.pdr.0000219311.14291.df [DOI] [PubMed] [Google Scholar]
- 53.Nogee LM. Alterations in SP-B and SP-C expression in neonatal lung disease. Annu Rev Physiol 2004; 66: 601–623. doi: 10.1146/annurev.physiol.66.032102.134711 [DOI] [PubMed] [Google Scholar]
- 54.Clark JC, Wert SE, Bachurski CJ, et al. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc Natl Acad Sci USA 1995; 92: 7794–7798. doi: 10.1073/pnas.92.17.7794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akinbi HT, Breslin JS, Ikegami M, et al. Rescue of SP-B knockout mice with a truncated SP-B proprotein. Function of the C-terminal propeptide. J Biol Chem 1997; 272: 9640–9647. doi: 10.1074/jbc.272.15.9640 [DOI] [PubMed] [Google Scholar]
- 56.Dranoff G, Crawford AD, Sadelain M, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994; 264: 713–716. doi: 10.1126/science.8171324 [DOI] [PubMed] [Google Scholar]
- 57.Ikegami M, Jobe AH, Huffman Reed JA, et al. Surfactant metabolic consequences of overexpression of GM-CSF in the epithelium of GM-CSF-deficient mice. Am J Physiol 1997; 273: L709–L714. [DOI] [PubMed] [Google Scholar]
- 58.Ikegami M, Ueda T, Hull W, et al. Surfactant metabolism in transgenic mice after granulocyte macrophage-colony stimulating factor ablation. Am J Physiol 1996; 270: L650–L658. [DOI] [PubMed] [Google Scholar]
- 59.Ikegami M, Hull WM, Yoshida M, et al. SP-D and GM-CSF regulate surfactant homeostasis via distinct mechanisms. Am J Physiol Lung Cell Mol Physiol 2001; 281: L697–L703. doi: 10.1152/ajplung.2001.281.3.L697 [DOI] [PubMed] [Google Scholar]
- 60.Mander A, Langton-Hewer S, Bernhard W, et al. Altered phospholipid composition and aggregate structure of lung surfactant is associated with impaired lung function in young children with respiratory infections. Am J Respir Cell Mol Biol 2002; 27: 714–721. doi: 10.1165/rcmb.4746 [DOI] [PubMed] [Google Scholar]
- 61.LeVine AM, Lotze A, Stanley S, et al. Surfactant content in children with inflammatory lung disease. Crit Care Med 1996; 24: 1062–1067. doi: 10.1097/00003246-199606000-00029 [DOI] [PubMed] [Google Scholar]
- 62.Kerr MH, Paton JY. Surfactant protein levels in severe respiratory syncytial virus infection. Am J Respir Crit Care Med 1999; 159: 4 Pt 1, 1115–1118. doi: 10.1164/ajrccm.159.4.9709065 [DOI] [PubMed] [Google Scholar]
- 63.Griese M. Respiratory syncytial virus and pulmonary surfactant. Viral Immunol 2002; 15: 357–363. doi: 10.1089/08828240260066279 [DOI] [PubMed] [Google Scholar]
- 64.Van Schaik SM, Vargas I, Welliver RC, et al. Surfactant dysfunction develops in BALB/c mice infected with respiratory syncytial virus. Pediatr Res 1997; 42: 169–173. doi: 10.1203/00006450-199708000-00007 [DOI] [PubMed] [Google Scholar]
- 65.Wright TW, Notter RH, Wang Z, et al. Pulmonary inflammation disrupts surfactant function during Pneumocystis carinii pneumonia. Infect Immun 2001; 69: 758–764. doi: 10.1128/IAI.69.2.758-764.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wright SM, Hockey PM, Enhorning G, et al. Altered airway surfactant phospholipid composition and reduced lung function in asthma. J Appl Physiol 2000; 89: 1283–1292. doi: 10.1152/jappl.2000.89.4.1283 [DOI] [PubMed] [Google Scholar]
- 67.Hohlfeld JM, Ahlf K, Enhorning G, et al. Dysfunction of pulmonary surfactant in asthmatics after segmental allergen challenge. Am J Respir Crit Care Med 1999; 159: 1803–1809. doi: 10.1164/ajrccm.159.6.9806145 [DOI] [PubMed] [Google Scholar]
- 68.Jarjour NN, Enhorning G. Antigen-induced airway inflammation in atopic subjects generates dysfunction of pulmonary surfactant. Am J Respir Crit Care Med 1999; 160: 336–341. doi: 10.1164/ajrccm.160.1.9806155 [DOI] [PubMed] [Google Scholar]
- 69.Kurashima K, Fujimura M, Matsuda T, et al. Surface activity of sputum from acute asthmatic patients. Am J Respir Crit Care Med 1997; 155: 1254–1259. doi: 10.1164/ajrccm.155.4.9105063 [DOI] [PubMed] [Google Scholar]
- 70.Heeley EL, Hohlfeld JM, Krug N, et al. Phospholipid molecular species of bronchoalveolar lavage fluid after local allergen challenge in asthma. Am J Physiol Lung Cell Mol Physiol 2000; 278: L305–L311. doi: 10.1152/ajplung.2000.278.2.L305 [DOI] [PubMed] [Google Scholar]
- 71.Kurashima K, Fujimura M, Tsujiura M, et al. Effect of surfactant inhalation on allergic bronchoconstriction in guinea pigs. Clin Exp Allergy 1997; 27: 337–342. doi: 10.1111/j.1365-2222.1997.tb00713.x [DOI] [PubMed] [Google Scholar]
- 72.Liu M, Wang L, Enhorning G. Surfactant dysfunction develops when the immunized guinea-pig is challenged with ovalbumin aerosol. Clin Exp Allergy 1995; 25: 1053–1060. doi: 10.1111/j.1365-2222.1995.tb03251.x [DOI] [PubMed] [Google Scholar]
- 73.Liu M, Wang L, Holm BA, et al. Dysfunction of guinea-pig pulmonary surfactant and type II pneumocytes after repetitive challenge with aerosolized ovalbumin. Clin Exp Allergy 1997; 27: 802–807. doi: 10.1046/j.1365-2222.1997.420885.x [DOI] [PubMed] [Google Scholar]
- 74.Kurashima K, Ogawa H, Ohka T, et al. A pilot study of surfactant inhalation in the treatment of asthmatic attack. Arerugi 1991; 40: 160–163. [PubMed] [Google Scholar]
- 75.Haczku A, Atochina EN, Tomer Y, et al. The late asthmatic response is linked with increased surface tension and reduced surfactant protein B in mice. Am J Physiol Lung Cell Mol Physiol 2002; 283: L755–L765. doi: 10.1152/ajplung.00062.2002 [DOI] [PubMed] [Google Scholar]
- 76.Bégin R, Lesur O, Bouhadiba T, et al. Phospholipid content of bronchoalveolar lavage fluid in granite workers with silicosis in Quebec. Thorax 1993; 48: 840–844. doi: 10.1136/thx.48.8.840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Honda Y, Tsunematsu K, Suzuki A, et al. Changes in phospholipids in bronchoalveolar lavage fluid of patients with interstitial lung diseases. Lung 1988; 166: 293–301. doi: 10.1007/BF02714060 [DOI] [PubMed] [Google Scholar]
- 78.Hughes DA, Haslam PL. Changes in phosphatidylglycerol in bronchoalveolar lavage fluids from patients with cryptogenic fibrosing alveolitis. Chest 1989; 95: 82–89. doi: 10.1378/chest.95.1.82 [DOI] [PubMed] [Google Scholar]
- 79.McCormack FX, King TE, Jr, Voelker DR, et al. Idiopathic pulmonary fibrosis. Abnormalities in the bronchoalveolar lavage content of surfactant protein A. Am Rev Respir Dis 1991; 144: 160–166. doi: 10.1164/ajrccm/144.1.160 [DOI] [PubMed] [Google Scholar]
- 80.Robinson PC, Watters LC, King TE, et al. Idiopathic pulmonary fibrosis. Abnormalities in bronchoalveolar lavage fluid phospholipids. Am Rev Respir Dis 1988; 137: 585–591. doi: 10.1164/ajrccm/137.3.585 [DOI] [PubMed] [Google Scholar]
- 81.Lesur O, Mancini NM, Janot C, et al. Loss of lymphocyte modulatory control by surfactant lipid extracts from acute hypersensitivity pneumonitis: comparison with sarcoidosis and idiopathic pulmonary fibrosis. Eur Respir J 1994; 7: 1944–1949. [PubMed] [Google Scholar]
- 82.Horiuchi T, Mason RJ, Kuroki Y, et al. Surface and tissue forces, surfactant protein A, and the phospholipid components of pulmonary surfactant in bleomycin-induced pulmonary fibrosis in the rat. Am Rev Respir Dis 1990; 141: 1006–1013. doi: 10.1164/ajrccm/141.4_Pt_1.1006 [DOI] [PubMed] [Google Scholar]
- 83.Horiuchi T, Ikegami M, Cherniack RM, et al. Increased surface tension of the lung and surfactant in bleomycin-induced pulmonary fibrosis in rats. Am J Respir Crit Care Med 1996; 154: 1002–1005. doi: 10.1164/ajrccm.154.4.8887598 [DOI] [PubMed] [Google Scholar]
- 84.Low RB, Adler KB, Woodcock-Mitchell J, et al. Bronchoalveolar lavage lipids during development of bleomycin-induced fibrosis in rats. Relationship to altered epithelial cell morphology. Am Rev Respir Dis 1988; 138: 709–713. doi: 10.1164/ajrccm/138.3.709 [DOI] [PubMed] [Google Scholar]
- 85.Thrall RS, Swendsen CL, Shannon TH, et al. Correlation of changes in pulmonary surfactant phospholipids with compliance in bleomycin-induced pulmonary fibrosis in the rat. Am Rev Respir Dis 1987; 136: 113–118. doi: 10.1164/ajrccm/136.1.113 [DOI] [PubMed] [Google Scholar]
- 86.Romero F, Shah D, Duong M, et al. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am J Respir Cell Mol Biol 2015; 53: 74–86. doi: 10.1165/rcmb.2014-0343OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Swendsen CL, Skita V, Thrall RS. Alterations in surfactant neutral lipid composition during the development of bleomycin-induced pulmonary fibrosis. Biochim Biophys Acta 1996; 1301: 90–96. doi: 10.1016/0005-2760(96)00023-9 [DOI] [PubMed] [Google Scholar]
- 88.Saito K, Tanaka N, Ikari J, et al. Comprehensive lipid profiling of bleomycin-induced lung injury. J Appl Toxicol 2019; 39: 658–671. doi: 10.1002/jat.3758 [DOI] [PubMed] [Google Scholar]
- 89.Lopez-Rodriguez E, Boden C, Echaide M, et al. Surfactant dysfunction during overexpression of TGF-β1 precedes profibrotic lung remodeling in vivo. Am J Physiol Lung Cell Mol Physiol 2016; 310: L1260–L1271. doi: 10.1152/ajplung.00065.2016 [DOI] [PubMed] [Google Scholar]
- 90.Viviano CJ, Bakewell WE, Dixon D, et al. Altered regulation of surfactant phospholipid and protein A during acute pulmonary inflammation. Biochim Biophys Acta 1995; 1259: 235–244. doi: 10.1016/0005-2760(95)00167-0 [DOI] [PubMed] [Google Scholar]
- 91.Agudelo CW, Kumley BK, Area-Gomez E, et al. Decreased surfactant lipids correlate with lung function in chronic obstructive pulmonary disease (COPD). PLoS ONE 2020; 15: e0228279. doi: 10.1371/journal.pone.0228279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jubinville É, Routhier J, Maranda-Robitaille M, et al. Pharmacological activation of liver X receptor during cigarette smoke exposure adversely affects alveolar macrophages and pulmonary surfactant homeostasis. Am J Physiol Lung Cell Mol Physiol 2019; 316: L669–L678. doi: 10.1152/ajplung.00482.2018 [DOI] [PubMed] [Google Scholar]
- 93.Morissette MC, Shen P, Thayaparan D, et al. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur Respir J 2015; 46: 1451–1460. doi: 10.1183/09031936.00216914 [DOI] [PubMed] [Google Scholar]
- 94.Kimura T, Shibata Y, Yamauchi K, et al. Oxidized phospholipid, 1-palmitoyl-2-(9'-oxo-nonanoyl)-glycerophosphocholine (PON-GPC), produced in the lung due to cigarette smoking, impairs immune function in macrophages. Lung 2012; 190: 169–182. doi: 10.1007/s00408-011-9331-2 [DOI] [PubMed] [Google Scholar]
- 95.Jubinville E, Talbot M, Berube JC, et al. Interplay between cigarette smoking and pulmonary reverse lipid transport. Eur Respir J 2017; 50: 1700681. doi: 10.1183/13993003.00681-2017 [DOI] [PubMed] [Google Scholar]
- 96.Subramaniam S, Bummer P, Gairola CG. Biochemical and biophysical characterization of pulmonary surfactant in rats exposed chronically to cigarette smoke. Fundam Appl Toxicol 1995; 27: 63–69. doi: 10.1006/faat.1995.1108 [DOI] [PubMed] [Google Scholar]
- 97.Subramaniam S, Whitsett JA, Hull W, et al. Alteration of pulmonary surfactant proteins in rats chronically exposed to cigarette smoke. Toxicol Appl Pharmacol 1996; 140: 274–280. doi: 10.1006/taap.1996.0222 [DOI] [PubMed] [Google Scholar]
- 98.Hu Q, Zhang H, Xiong S, et al. The alteration and significance of surfactant protein A in rats chronically exposed to cigarette smoke. J Huazhong Univ Sci Technolog Med Sci 2008; 28: 128–131. doi: 10.1007/s11596-008-0203-9 [DOI] [PubMed] [Google Scholar]
- 99.Finley TN, Ladman AJ. Low yield of pulmonary surfactant in cigarette smokers. N Engl J Med 1972; 286: 223–227. doi: 10.1056/NEJM197202032860501 [DOI] [PubMed] [Google Scholar]
- 100.Pratt SA, Finley TN, Smith MH, et al. A comparison of alveolar macrophages and pulmonary surfactant (?) obtained from the lungs of human smokers and nonsmokers by endobronchial lavage. Anat Rec 1969; 163: 497–507. doi: 10.1002/ar.1091630402 [DOI] [PubMed] [Google Scholar]
- 101.Low RB, Davis GS, Giancola MS. Biochemical analyses of bronchoalveolar lavage fluids of healthy human volunteer smokers and nonsmokers. Am Rev Respir Dis 1978; 118: 863–875. [DOI] [PubMed] [Google Scholar]
- 102.Moré JM, Voelker DR, Silveira LJ, et al. Smoking reduces surfactant protein D and phospholipids in patients with and without chronic obstructive pulmonary disease. BMC Pulm Med 2010; 10: 53. doi: 10.1186/1471-2466-10-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mancini NM, Béné MC, Gérard H, et al. Early effects of short-time cigarette smoking on the human lung: a study of bronchoalveolar lavage fluids. Lung 1993; 171: 277–291. doi: 10.1007/BF03215871 [DOI] [PubMed] [Google Scholar]
- 104.Hughes DA, Haslam PL. Effect of smoking on the lipid composition of lung lining fluid and relationship between immunostimulatory lipids, inflammatory cells and foamy macrophages in extrinsic allergic alveolitis. Eur Respir J 1990; 3: 1128–1139. [PubMed] [Google Scholar]
- 105.Schmekel B, Bos JA, Khan AR, et al. Integrity of the alveolar-capillary barrier and alveolar surfactant system in smokers. Thorax 1992; 47: 603–608. doi: 10.1136/thx.47.8.603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schmekel B, Khan AR, Linden M, et al. Recoveries of phosphatidylcholine and alveolar macrophages in lung lavage from healthy light smokers. Clin Physiol 1991; 11: 431–438. doi: 10.1111/j.1475-097X.1991.tb00815.x [DOI] [PubMed] [Google Scholar]
- 107.Honda Y, Takahashi H, Kuroki Y, et al. Decreased contents of surfactant proteins A and D in BAL fluids of healthy smokers. Chest 1996; 109: 1006–1009. doi: 10.1378/chest.109.4.1006 [DOI] [PubMed] [Google Scholar]
- 108.Hallman M, Spragg R, Harrell JH, et al. Evidence of lung surfactant abnormality in respiratory failure. Study of BAL phospholipids, surface activity, phospholipase activity and plasma myoinositol. J Clin Invest 1982; 70: 673–683. doi: 10.1172/JCI110662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lusuardi M, Capelli A, Carli S, et al. Role of surfactant in chronic obstructive pulmonary disease: therapeutic implications. Respiration 1992; 59: Suppl. 1, 28–32. doi: 10.1159/000196100 [DOI] [PubMed] [Google Scholar]
- 110.Vlachaki EM, Koutsopoulos AV, Tzanakis N, et al. Altered surfactant protein-A expression in type II pneumocytes in COPD. Chest 2010; 137: 37–45. doi: 10.1378/chest.09-1029 [DOI] [PubMed] [Google Scholar]
- 111.Winkler C, Atochina-Vasserman EN, Holz O, et al. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir Res 2011; 12: 29. doi: 10.1186/1465-9921-12-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tafel O, Latzin P, Paul K, et al. Surfactant proteins SP-B and SP-C and their precursors in bronchoalveolar lavages from children with acute and chronic inflammatory airway disease. BMC Pulm Med 2008; 8: 6. doi: 10.1186/1471-2466-8-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Madison MC, Landers CT, Gu BH, et al. Electronic cigarettes disrupt lung lipid homeostasis and innate immunity independent of nicotine. J Clin Invest 2019; 129: 4290–4304. doi: 10.1172/JCI128531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fuchimukai T, Fujiwara T, Takahashi A, et al. Artificial pulmonary surfactant inhibited by proteins. J Appl Physiol (1985) 1987; 62: 429–437. doi: 10.1152/jappl.1987.62.2.429 [DOI] [PubMed] [Google Scholar]
- 115.Seeger W, Grube C, Günther A, et al. Surfactant inhibition by plasma proteins: differential sensitivity of various surfactant preparations. Eur Respir J 1993; 6: 971–977. [PubMed] [Google Scholar]
- 116.Shepherd VL. Distinct roles for lung collectins in pulmonary host defense. Am J Respir Cell Mol Biol 2002; 26: 257–260. doi: 10.1165/ajrcmb.26.3.f227 [DOI] [PubMed] [Google Scholar]
- 117.LeVine AM, Whitsett JA. Pulmonary collectins and innate host defense of the lung. Microbes Infect 2001; 3: 161–166. doi: 10.1016/S1286-4579(00)01363-0 [DOI] [PubMed] [Google Scholar]
- 118.Atochina EN, Gow AJ, Beck JM, et al. Delayed clearance of pneumocystis carinii infection, increased inflammation, and altered nitric oxide metabolism in lungs of surfactant protein-D knockout mice. J Infect Dis 2004; 189: 1528–1539. doi: 10.1086/383130 [DOI] [PubMed] [Google Scholar]
- 119.Linke MJ, Harris CE, Korfhagen TR, et al. Immunosuppressed surfactant protein A-deficient mice have increased susceptibility to Pneumocystis carinii infection. J Infect Dis 2001; 183: 943–952. doi: 10.1086/319252 [DOI] [PubMed] [Google Scholar]
- 120.LeVine AM, Kurak KE, Wright JR, et al. Surfactant protein-A binds group B Streptococcus enhancing phagocytosis and clearance from lungs of surfactant protein-A-deficient mice. Am J Respir Cell Mol Biol 1999; 20: 279–286. doi: 10.1165/ajrcmb.20.2.3303 [DOI] [PubMed] [Google Scholar]
- 121.LeVine AM, Hartshorn K, Elliott J, et al. Absence of SP-A modulates innate and adaptive defense responses to pulmonary influenza infection. Am J Physiol Lung Cell Mol Physiol 2002; 282: L563–L572. doi: 10.1152/ajplung.00280.2001 [DOI] [PubMed] [Google Scholar]
- 122.Li G, Siddiqui J, Hendry M, et al. Surfactant protein-A-deficient mice display an exaggerated early inflammatory response to a beta-resistant strain of influenza A virus. Am J Respir Cell Mol Biol 2002; 26: 277–282. doi: 10.1165/ajrcmb.26.3.4584 [DOI] [PubMed] [Google Scholar]
- 123.Arroyo R, Khan MA, Echaide M, et al. SP-D attenuates LPS-induced formation of human neutrophil extracellular traps (NETs), protecting pulmonary surfactant inactivation by NETs. Commun Biol 2019; 2: 470. doi: 10.1038/s42003-019-0662-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tibby SM, Hatherill M, Wright SM, et al. Exogenous surfactant supplementation in infants with respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med 2000; 162: 1251–1256. doi: 10.1164/ajrccm.162.4.9909004 [DOI] [PubMed] [Google Scholar]
- 125.García-Fojeda B, González-Carnicero Z, de Lorenzo A, et al. Lung surfactant lipids provide immune protection against haemophilus influenzae respiratory infection. Front Immunol 2019; 10: 458. doi: 10.3389/fimmu.2019.00458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Raychaudhuri B, Abraham S, Bonfield TL, et al. Surfactant blocks lipopolysaccharide signaling by inhibiting both mitogen-activated protein and IkappaB kinases in human alveolar macrophages. Am J Respir Cell Mol Biol 2004; 30: 228–232. doi: 10.1165/rcmb.2003-0263OC [DOI] [PubMed] [Google Scholar]
- 127.Awasthi S, Rahman N, Rui B, et al. Lung and general health effects of Toll-like receptor-4 (TLR4)-interacting SPA4 peptide. BMC Pulm Med 2020; 20: 179. doi: 10.1186/s12890-020-01187-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Awasthi S, Singh B, Ramani V, et al. TLR4-interacting SPA4 peptide improves host defense and alleviates tissue injury in a mouse model of Pseudomonas aeruginosa lung infection. PLoS ONE 2019; 14: e0210979. doi: 10.1371/journal.pone.0210979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ackerman SJ, Kwatia MA, Doyle CB, et al. Hydrolysis of surfactant phospholipids catalyzed by phospholipase A2 and eosinophil lysophospholipases causes surfactant dysfunction: a mechanism for small airway closure in asthma. Chest 2003; 123: 3 Suppl, 355s. doi: 10.1378/chest.123.3_suppl.355S [DOI] [PubMed] [Google Scholar]
- 130.Liu M, Wang L, Li E, et al. Pulmonary surfactant given prophylactically alleviates an asthma attack in guinea-pigs. Clin Exp Allergy 1996; 26: 270–275. doi: 10.1111/j.1365-2222.1996.tb00091.x [DOI] [PubMed] [Google Scholar]
- 131.Babu KS, Woodcock DA, Smith SE, et al. Inhaled synthetic surfactant abolishes the early allergen-induced response in asthma. Eur Respir J 2003; 21: 1046–1049. doi: 10.1183/09031936.03.00069202 [DOI] [PubMed] [Google Scholar]
- 132.Oetomo SB, Dorrepaal C, Bos H, et al. Surfactant nebulization does not alter airflow obstruction and bronchial responsiveness to histamine in asthmatic children. Am J Respir Crit Care Med 1996; 153: 1148–1152. doi: 10.1164/ajrccm.153.3.8630559 [DOI] [PubMed] [Google Scholar]
- 133.Romero F, Hong X, Shah D, et al. Lipid synthesis is required to resolve endoplasmic reticulum stress and limit fibrotic responses in the lung. Am J Respir Cell Mol Biol 2018; 59: 225–236. doi: 10.1165/rcmb.2017-0340OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sunaga H, Matsui H, Ueno M, et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat Commun 2013; 4: 2563. doi: 10.1038/ncomms3563 [DOI] [PubMed] [Google Scholar]
- 135.Steffen L, Ruppert C, Hoymann HG, et al. Surfactant replacement therapy reduces acute lung injury and collapse induration-related lung remodeling in the bleomycin model. Am J Physiol Lung Cell Mol Physiol 2017; 313: L313–L327. doi: 10.1152/ajplung.00033.2017 [DOI] [PubMed] [Google Scholar]
- 136.GBD 2015 Chronic Respiratory Disease Collaborators . Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med 2017; 5: 691–706. doi: 10.1016/S2213-2600(17)30293-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cook WA, Webb WR. Surfactant in chronic smokers. Ann Thorac Surg 1966; 2: 327–333. doi: 10.1016/S0003-4975(10)66584-8 [DOI] [PubMed] [Google Scholar]
- 138.Stachowicz-Kuśnierz A, Cwiklik L, Korchowiec J, et al. The impact of lipid oxidation on the functioning of a lung surfactant model. Phys Chem Chem Phys 2018; 20: 24968–24978. doi: 10.1039/C8CP04496A [DOI] [PubMed] [Google Scholar]
- 139.Ween MP, White JB, Tran HB, et al. The role of oxidised self-lipids and alveolar macrophage CD1b expression in COPD. Sci Rep 2021; 11: 4106. doi: 10.1038/s41598-021-82481-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Przybyla RJ, Wright J, Parthiban R, et al. Electronic cigarette vapor alters the lateral structure but not tensiometric properties of calf lung surfactant. Respir Res 2017; 18: 193. doi: 10.1186/s12931-017-0676-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bringezu F, Pinkerton KE, Zasadzinski JA. Environmental tobacco smoke effects on the primary lipids of lung surfactant. Langmuir 2003; 19: 2900–2907. doi: 10.1021/la026455e [DOI] [Google Scholar]
- 142.Stenger PC, Alonso C, Zasadzinski JA, et al. Environmental tobacco smoke effects on lung surfactant film organization. Biochim Biophys Acta 2009; 1788: 358–370. doi: 10.1016/j.bbamem.2008.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Davies MJ, Birkett JW, Kotwa M, et al. The impact of cigarette/e-cigarette vapour on simulated pulmonary surfactant monolayers under physiologically relevant conditions. Surface Interface Anal 2017; 49: 654–665. doi: 10.1002/sia.6205 [DOI] [Google Scholar]
- 144.Mazur W, Toljamo T, Ohlmeier S, et al. Elevation of surfactant protein A in plasma and sputum in cigarette smokers. Eur Respir J 2011; 38: 277–284. doi: 10.1183/09031936.00110510 [DOI] [PubMed] [Google Scholar]
- 145.Hirche TO, Crouch EC, Espinola M, et al. Neutrophil serine proteinases inactivate surfactant protein D by cleaving within a conserved subregion of the carbohydrate recognition domain. J Biol Chem 2004; 279: 27688–27698. doi: 10.1074/jbc.M402936200 [DOI] [PubMed] [Google Scholar]
- 146.Baker CS, Evans TW, Randle BJ, et al. Damage to surfactant-specific protein in acute respiratory distress syndrome. Lancet 1999; 353: 1232–1237. doi: 10.1016/S0140-6736(98)09449-5 [DOI] [PubMed] [Google Scholar]
- 147.Takamiya R, Uchida K, Shibata T, et al. Disruption of the structural and functional features of surfactant protein A by acrolein in cigarette smoke. Sci Rep 2017; 7: 8304. doi: 10.1038/s41598-017-08588-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet 2017; 389: 1931–1940. doi: 10.1016/S0140-6736(17)31222-9 [DOI] [PubMed] [Google Scholar]
- 149.Anzueto A, Jubran A, Ohar JA, et al. Effects of aerosolized surfactant in patients with stable chronic bronchitis: a prospective randomized controlled trial. JAMA 1997; 278: 1426–1431. doi: 10.1001/jama.1997.03550170056032 [DOI] [PubMed] [Google Scholar]
- 150.Lärstad M, Almstrand AC, Larsson P, et al. Surfactant protein A in exhaled endogenous particles is decreased in chronic obstructive pulmonary disease (COPD) patients: a pilot study. PLoS ONE 2015; 10: e0144463. doi: 10.1371/journal.pone.0144463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Blagev DP, Harris D, Dunn AC, et al. Clinical presentation, treatment, and short-term outcomes of lung injury associated with e-cigarettes or vaping: a prospective observational cohort study. Lancet 2019; 394: 2073–2083. doi: 10.1016/S0140-6736(19)32679-0 [DOI] [PubMed] [Google Scholar]
- 152.Butt YM, Smith ML, Tazelaar HD, et al. Pathology of vaping-associated lung injury. N Engl J Med 2019; 381: 1780–1781. doi: 10.1056/NEJMc1913069 [DOI] [PubMed] [Google Scholar]
- 153.Blount BC, Karwowski MP, Morel-Espinosa M, et al. Evaluation of bronchoalveolar lavage fluid from patients in an outbreak of e-cigarette, or vaping, product use-associated lung injury – 10 states, August–October 2019. MMWR Morb Mortal Wkly Rep 2019; 68: 1040–1041. doi: 10.15585/mmwr.mm6845e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Abeles M, Popofsky S, Wen A, et al. Vaping-associated lung injury caused by inhalation of cannabis oil. Pediatr Pulmonol 2020; 55: 226–228. doi: 10.1002/ppul.24579 [DOI] [PubMed] [Google Scholar]
- 155.Kalininskiy A, Bach CT, Nacca NE, et al. E-cigarette, or vaping, product use associated lung injury (EVALI): case series and diagnostic approach. Lancet Respir Med 2019; 7: 1017–1026. doi: 10.1016/S2213-2600(19)30415-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sosnowski TR, Jabłczyńska K, Odziomek M, et al. Physicochemical studies of direct interactions between lung surfactant and components of electronic cigarettes liquid mixtures. Inhal Toxicol 2018; 30: 159–168. doi: 10.1080/08958378.2018.1478916 [DOI] [PubMed] [Google Scholar]
- 157.DiPasquale M, Gbadamosi O, Nguyen MHL, et al. A mechanical mechanism for vitamin E acetate in e-cigarette/vaping-associated lung injury. Chem Res Toxicol 2020; 33: 2432–2440. doi: 10.1021/acs.chemrestox.0c00212 [DOI] [PubMed] [Google Scholar]
- 158.Tschanz SA, Damke BM, Burri PH. Influence of postnatally administered glucocorticoids on rat lung growth. Biol Neonate 1995; 68: 229–245. doi: 10.1159/000244241 [DOI] [PubMed] [Google Scholar]
- 159.Vyas J, Kotecha S. Effects of antenatal and postnatal corticosteroids on the preterm lung. Arch Dis Child Fetal Neonatal Ed 1997; 77: F147–F150. doi: 10.1136/fn.77.2.F147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chai AB, Ammit AJ, Gelissen IC. Examining the role of ABC lipid transporters in pulmonary lipid homeostasis and inflammation. Respir Res 2017; 18: 41. doi: 10.1186/s12931-017-0526-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Smoak K, Madenspacher J, Jeyaseelan S, et al. Effects of liver X receptor agonist treatment on pulmonary inflammation and host defense. J Immunol 2008; 180: 3305–3312. doi: 10.4049/jimmunol.180.5.3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mason RJ, Voelker DR. Regulatory mechanisms of surfactant secretion. Biochim Biophys Acta 1998; 1408: 226–240. doi: 10.1016/S0925-4439(98)00070-2 [DOI] [PubMed] [Google Scholar]
- 163.Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. Regulation of surfactant secretion in alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 2007; 293: L259–L271. doi: 10.1152/ajplung.00112.2007 [DOI] [PubMed] [Google Scholar]
- 164.Tsilibary EC, Williams MC. Actin and secretion of surfactant. J Histochem Cytochem 1983; 31: 1298–1304. doi: 10.1177/31.11.6688627 [DOI] [PubMed] [Google Scholar]
- 165.Brown LA, Pasquale SM, Longmore WJ. Role of microtubules in surfactant secretion. J Appl Physiol 1985; 58: 1866–1873. doi: 10.1152/jappl.1985.58.6.1866 [DOI] [PubMed] [Google Scholar]