

Abstract
Primary Sjogren syndrome (pSS) is a systemic autoimmune disease characterised by lymphocytic infiltration of exocrine glands and by a number of systemic manifestations, including those regarding the lung. Pulmonary involvement in pSS includes interstitial lung disease (ILD) and airway disease, together with lymphoproliferative disorders.
Patients with pSS-ILD report impaired health-related quality of life and a higher risk of death, suggesting the importance of early diagnosis and treatment of this type of pulmonary involvement. In contrast, airway disease usually has little effect on respiratory function and is rarely the cause of death in these patients.
More rare disorders can be also identified, such as pleural effusion, cysts or bullae.
Up to date, available data do not allow us to establish an evidence-based treatment strategy in pSS-ILD. No data are available regarding which patients should be treated, the timing to start therapy and better therapeutic options. The lack of knowledge about the natural history and prognosis of pSS-ILD is the main limitation to the development of clinical trials or shared recommendations on this topic. However, a recent trial showed the efficacy of the antifibrotic drug nintedanib in slowing progression of various ILDs, including those in pSS patients.
Short abstract
Primary Sjogren syndrome is a systemic autoimmune disease with a possible lung involvement, that it appears as polymorphic, including interstitial lung disease, airway disease and lymphoproliferative disorders with different degree of severity https://bit.ly/3akjk4b
Definition, epidemiology and classification criteria
Sjogren syndrome (SS) is a systemic autoimmune disease characterised by lymphocytic infiltration of the exocrine glands and by a wide number of systemic manifestations [1]. SS can occur as a primary disease (primary SS (pSS)) or be associated with another autoimmune disease, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), dermatomyositis, or systemic sclerosis (SSc) [1].
pSS shows a female-to-male predominance of 9:1 and a peak incidence at approximately 50 years of age [2]. The prevalence in the general population is estimated between 0.02% and 2.7% [3], according to the diagnostic criteria used and the ethnic background of the population.
The prevalence of pulmonary involvement in pSS patients varies widely [4]. Studies that define pulmonary involvement by respiratory symptoms associated with abnormal pulmonary function tests (PFTs) and/or abnormal imaging findings on high-resolution computed tomography (HRCT) [5] suggest a prevalence rate in the range 9–22% [6, 7], with a female predominance [8]. When pulmonary involvement is investigated systematically, prevalence ranges between 43% and 75% [9] and on HRCT of the chest abnormalities are found in 34–50% of the patients [10].
Generally, oral and ocular dryness is accompanied by involvement of other types of mucosa such as nose, pharynx, larynx and vagina, that can be the earliest clinical manifestations [1, 11]. Sometimes, the disease course can be also complicated by systemic manifestations that can represent the first signs of the disease, also preceding SS by many years [11].
Since 1965, 11 sets of classification/diagnostic criteria have been proposed for pSS.
In 2017, the “International Sjogren's Syndrome Criteria Working Group” published the American College of Rheumatology/European League Against Rheumatism classification criteria for pSS (table 1) [12]. Although these criteria were created for the purposes of classification, in the adequate clinical context they may also be useful for diagnosing pSS in clinical practice.
TABLE 1.
American College of Rheumatology/European League against Rheumatism classification criteria for primary Sjogren syndrome
| Item | Weight/score |
| Labial salivary gland with focal lymphocytic sialadenitis and focus score ≥1 foci·4 mm−2 | 3 |
| Anti-SSA/Ro-positive | 3 |
| Ocular staining score ≥5 (or van Bijsterveld score ≥4) in at least one eye | 1 |
| Schirmer's test ≤5 mm in 5 min in at least one eye | 1 |
| Unstimulated whole saliva flow rate ≤0.1 mL·min−1 | 1 |
A patient can be classified as having primary Sjogren syndrome with a score of ≥4 and without any of the conditions listed as exclusion criteria. Exclusion criteria include prior diagnosis of any of the following conditions: history of head and neck radiation treatment; active hepatitis C infection; AIDS; sarcoidosis; amyloidosis; graft-versus-host disease; immunoglobulin G4-related disease [12].
Pathogenesis and systemic manifestations
The pathogenesis of pSS is considered multifactorial, although not completely understood. Many of the SS risk genes identified to date are involved in interferon, lymphocyte migration, cytokine and cytokine receptor function and various other intracellular signalling pathways that are important in multiple immune cell subsets [13, 14]. Both T-cells and B-cells are involved in pSS pathogenesis, with a possible dominant role for the latter.
Type 17 helper T (Th17) cells and regulatory T-cells are also increased in the salivary glands of pSS patients compared to healthy controls, with a possible main pathogenetic role for Th17, as the co-expression of interleukin (IL)-17 and IL-18 seems to be correlated to pSS severity [14, 15].
In pSS, patients can also be observed with an increased level of serum immunoglobulin (Ig)A and/or IgG; hypergammaglobulinaemia may contribute to formation of immune complexes, while the autoantibodies (anti-Ro52, anti-Ro60 and anti-La) are associated with lymphocytic glandular infiltration [15, 16].
A wide range of systemic manifestations can occur in the course of pSS [1, 11, 13]. Fatigue is the most common symptom in pSS, after dry eye and dry mouth, and it occurs in around 70–80% of patients. The pathogenesis is unknown, and the cause may be multifactorial. Other systemic manifestations occur in approximately 30–40% of the patients with pSS [11]. Arthralgia and arthritis are common in pSS, occurring in around 40% of patients. Arthritis is reported in about 15% of pSS patients, mainly involving the hand joints, and it is more often symmetrical and generally, nonerosive [1, 11, 13].
Cryoglobulins have been detected in up to 10–20% of pSS patients, and cryoglobulinaemic vasculitis may be observed in 5–10% [11, 13]. It can manifest as purpura, urticaria and cutaneous ulcers, often involving the lower extremities [1, 11, 13]. Leukocytoclastic vasculitis is the most common pathological finding. Cutaneous vasculitis in pSS is associated with more severe disease with more systemic manifestations, lymphoma and poor prognosis. Annular erythema can also occur in about 10% of pSS patients, it is more frequent in females and it is associated with anti-SSA positivity [1, 11, 13]. Face, neck and upper arms are the most frequently involved sites.
The most common feature of renal involvement is tubulointerstitial nephritis, which is underdiagnosed because this condition progresses insidiously with minimal manifestations [1, 11, 13]. Tubulointerstitial nephritis is characterised by inflammation of the interstitium, which causes fibrosis and atrophy determining chronic kidney disease and, in many cases, renal tubular acidosis. Distal renal tubular acidosis occurs in around 10–40% of pSS patients, and it may be the only renal abnormality. Mild hypokalaemia, hyperchloraemia, nonanion-gap metabolic acidosis or a history of nephrolithiasis in a patient with pSS are the most common presenting manifestations. Glomerulonephritis is uncommon in pSS, its prevalence was found to be around 2–4%. The most common lesion is membranoproliferative glomerulonephritis, which is associated with cryoglobulinaemia and low complement levels [1, 11, 13].
The prevalence of neurologic involvement in pSS is around 20% and the peripheral nervous system is more frequently involved than the central nervous system. The two most common peripheral nervous systemic manifestations are distal axonal sensory polyneuropathy and small fibre neuropathy. Mononeuritis multiplex is a less common serious vasculitic manifestation, usually associated with serum cryoglobulinaemia. Cranial neuropathies, particularly sensory trigeminal neuralgia, may also occur. Central nervous system involvement varies, ranging from mild cognitive dysfunction, to transverse myelitis or demyelinating lesions mimicking multiple sclerosis [1, 11, 13, 17].
Other systemic manifestations are gastrointestinal manifestations, constitutional symptoms (fever and/or weight loss), haematologic abnormalities such as anaemia and leukopenia, and myositis.
Pregnancy
Pregnancy involving women with pSS has a higher risk of resulting in preterm delivery and low-birth-weight infants. Moreover, congenital heart block (CHB) is the most challenging complication for fetus and neonates delivered by mothers with pSS [18]. In fact, maternal anti-Ro/SSA and/or La/SSB autoantibodies may damage the conduction tissues during fetal development, leading to blockage of signal conduction at the atrioventricular node in an otherwise structurally normal heart [18]. These antibodies cross the placenta beginning at approximately 12 weeks of gestation and react with a fetal heart. When severe, this may be lethal, otherwise it can result in degeneration and endocardial fibroelastosis, disrupting conduction and leading to CHB [19], which is considered a major issue of fetal outcome. In fact, if left untreated, fetal and neonatal mortality due to CHB may develop [20].
Clinical manifestations and prognosis of pulmonary involvement
Pulmonary manifestations are polymorphic, representing a leading cause of morbidity and mortality in these patients, particularly when an interstitial lung disease (ILD) is diagnosed (pSS-ILD) [21].
Various studies suggest that the main risk factors for pulmonary involvement are male sex, being active smokers, late onset of the disease and having a long-lasting disease [6, 22, 23].
All compartments can be affected, including lung parenchyma, mainly with an ILD, airways, vasculature and pleura. Pulmonary manifestations also include lymphoproliferative disorders (and in general a higher incidence of malignancies), pulmonary infections and thromboembolic disorders. More rare disorders can be also identified, such as pleural effusion, cysts or bullae. Finally, many drugs normally used in pSS have been associated with lung toxicity.
Patients with pSS-ILD report impaired health-related quality of life (HRQoL), impaired physical functioning and higher risk of death [6], suggesting the importance of an early diagnosis and treatment.
In a recent systematic review, HRQoL appeared markedly reduced in pSS in multiple studies and across many countries when compared with HRQoL in healthy controls. The reduction in HRQoL was similar to that observed in other chronic diseases such as RA and SLE [24]. Using the Short Form-36 health survey, patients with lung involvement reported impaired HRQoL compared with the other patients with pSS [6].
Clinical evaluation of pulmonary involvement
The clinical evaluation of pulmonary involvement includes the detection of lung disease, the evaluation of disease severity, and the disease progression, with the aim to identify which patients should be treated.
Detection of lung disease
The multiorgan involvement of pSS makes it particularly difficult to detect pulmonary involvement on the basis of the respiratory symptoms: patients with mild ILD and/or airway disease may be asymptomatic at early stages of the disease, and often develop fatigue, dyspnoea on exertion and cough, that can be the consequence of nonpulmonary causes, such as anaemia, chest wall involvement, joint disease or muscle weakness. Nevertheless, respiratory symptoms may occur in cases of progression of ILD, airway disease, vascular disease or pleural involvement [25].
Physical examination is often unremarkable but, when an ILD occurs, may reveal fine bibasilar, end-inspiratory, “velcro-like” crackles at auscultation, which can precede the development of clinically overt ILD [26–28] and should prompt further investigations. In contrast, when a pSS patient is affected by airway disease, wheezing and/or rhonchi can be detected.
Nevertheless, chest radiography is usually insensitive in identifying both ILDs and airway disease in pSS patients.
PFTs are helpful for diagnosing and tracking pulmonary involvement in pSS patients. Typically, in the presence of an ILD, PFTs will detect a restrictive ventilatory failure, characterised by reduced total lung capacity (TLC) and forced vital capacity (FVC), with a normal forced expiratory volume in 1 s (FEV1)/FVC ratio [29, 30]. In contrast, when pulmonary involvement in pSS results in airway disease, an obstructive ventilatory failure may be detected, resulting in an increased TLC, with a reduced FEV1/FVC ratio [9, 31]. Diffusing capacity of the lung for carbon monoxide (DLCO) may be reduced in both these conditions, because it can be lowered by the occurrence of ILD, pulmonary arterial hypertension (PAH) and emphysema. ILDs and airway disease may coexist in pSS patients; in these cases, spirometry can be normal [32], but a disproportionate reduction in DLCO may occur [33].
However, the most sensitive tool to identify ILD is HRCT, which is both sensitive and specific in the identification of ILD [34]. Although subclinical abnormalities are often detectable, they do not necessarily evolve to clinically significant ILD. In contrast, HRCT should be used by protocol to confirm or exclude a connective tissue disease (CTD)-ILD only in higher risk groups or when there is reason to suspect the presence of pSS-ILD, according to symptoms, clinical examination, chest radiography or pulmonary function abnormalities [35].
Evaluation of disease severity
The detection of pulmonary involvement in pSS, should be followed by an evaluation of disease severity, that can include symptoms, pulmonary function impairment and the extent of disease on HRCT [34].
Amongst these measures, PFTs provide the most precise measurement of disease severity, both in the cases of airway or lung parenchyma involvement. However, pulmonary function variables should not be interpreted in isolation due to the confounding effect of the normal range, which is particularly misleading when there is mild pulmonary function impairment [34].
Evaluation of disease progression
The most accurate tool for evaluating CTD-ILD progression is focused on serial PFT. As FVC is highly reproducible, in the absence of major extrapulmonary restriction due to pleural disease or muscle weakness, changes in FVC are specific to ILD [36]. As for systemic sclerosis, disease progression can be detected in pSS patients by changes over time that include a decline in FVC of ≥10% or a decrease in the DLCO of ≥15% over 6–12 months [37].
Several studies have shown that 6-min walk distance (6MWD) and/or decline in 6MWD are strong independent predictors of mortality in patients with idiopathic pulmonary fibrosis (IPF) [38, 39] and in CTD-ILD [40], possibly including pSS-ILD patients. Desaturation (oxygen saturation measured by pulse oximetry ≤88%) during or at the end of a 6MWD and change in oxygen saturation measured by pulse oximetry during a 6MWD have been found to be significant predictors of mortality [41]. A baseline 6MWD <250 m was associated with a two-fold increase in mortality, and a decline in 6MWD over 50 m between baseline and 24 weeks was associated with an almost three-fold increase in mortality [42]. However, exercise limitation in pSS can be considered multifactorial, with contributions including impairment of gas exchange and pulmonary hypertension, ventilatory dysfunction and muscle dysfunction [43]. However, disease progression measurement requires the integration of symptoms, pulmonary function variables and, in selected individuals, serial HRCT, worsening in 6MWD and echocardiographic data.
In the case of airway involvement, the useful tools for evaluation of disease progression can be more elusive: in various cases, more frequently when large airway involvement occurs, neither radiological nor lung function tests/6MWD can be detected in patients with uncomplicated clinical features. Similarly, HRCT and symptoms usually appear insensitive in evaluating disease progression in the case of airway involvement.
Pulmonary manifestations
Interstitial lung disease
Histological and clinical features
pSS-ILD is the most common form of pulmonary involvement, ranging between 6–70% in different studies [44]. Lymphocytic interstitial pneumonia (LIP) has a classic association with pSS [45], although it does not appear as the most common pathological subtype. Nonspecific interstitial pneumonia (NSIP) is recognised as the most common ILD disorder [5, 23] followed by organising pneumonia (OP) and usual interstitial pneumonia (UIP), the latter considered particularly frequent in some case series with early onset of ILD [46]. Rare cases of acute fibrinous and organising pneumonia and pleuroparenchymal fibroelastosis in SS are also reported [47–49]. Some manifestations in pSS patients can appear as cystic lesions of the lung, representing a broad range of diseases, including amyloidosis [50], malignant lymphoma [29] and LIP [51]. Some examples of lung biopsies in SS are illustrated in figure 1.
FIGURE 1.
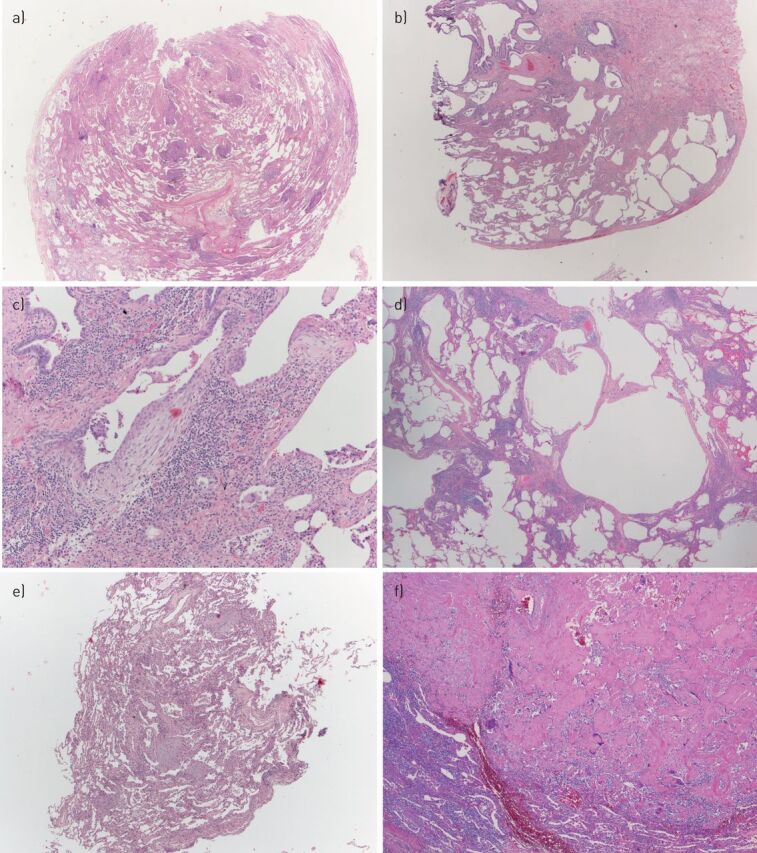
Some examples of surgical lung biopsies showing different interstitial lung disease in patients with Sjogren syndrome. a) Mixed cellular/fibrosing nonspecific interstitial pneumonia (NSIP), consisting of a combination of lymphoplasmacytic infiltrate and uniform interstitial fibrosis. b,c) A fibrosing process with usual interstitial pneumonia pattern, consisting of patchy scars (b) and fibroblastic foci (c). The lymphoplasmacytic infiltrate present in (c) is a clue, suggesting an underlying autoimmune disease. d) A lymphoplasmacytic interstitial infiltrate consistent with lymphocytic interstitial pneumonia, associated with small cysts of probable bronchiolar/alveolar duct origin. In this case, the limit with cellular NSIP is blurred; when the infiltrate is denser, the differential diagnosis with lymphoma (particularly mucosa-associated lymphoid tissue (MALT) lymphoma) can be difficult and requires immunohistochemical and sometimes molecular evaluation. e) Transbronchial biopsy showing organising pneumonia, consisting of intra-alveolar plugs of loose connective tissue. Some fibrin is frequently present in organising pneumonia; when fibrin is prominent, the term acute fibrinous and organising pneumonia can be applied. f) Nodular amyloidosis, consisting of deposition of acellular jaline material. The peripheral giant cell reaction is a clue pointing to the correct diagnosis. In nodular amyloidosis, any associated lymphoid infiltrate should be regarded with suspicion and should induce the pathologist to order the appropriate immunohistochemical stains to evaluate the possibility of a concomitant indolent B-cell lymphoma, particularly of the MALT type: in this case, the lymphoid infiltrate was polyclonal.
The clinical manifestations of pSS-ILD resemble those of IPF, with exertional dyspnoea and nonproductive cough as the most frequent respiratory symptoms [8]. The time of onset of pulmonary involvement in these patients is variable, being the first manifestation of pSS before diagnosis of the autoimmune disease, but it can sometimes begin at the same time as other systemic manifestations (10% of cases) [52] or, in other cases, late in the disease course [53].
Data on the natural history of pSS-ILD are limited, although a few studies suggest that ILD is associated with a premature mortality [54].
Acute exacerbation of ILD is reported to be developed not only in patients with IPF, but also in those with CTD-ILD, including pSS-ILD [8, 55]. Acute exacerbation is defined as an acute, clinically significant respiratory deterioration characterised by evidence of new widespread alveolar abnormalities [56]. Histologically, it generally consists of diffuse alveolar damage (but sometimes in OP or in particularly exuberant fibroblastic foci) superimposed on a chronic ILD. A few studies compared the prevalence of acute exacerbation across the spectrum of CTD-ILD [57], showing that a UIP pattern seems to be a higher risk for this complication irrespective of the underlying disease [58]. The incidence and the clinical characteristics of acute exacerbation in CTD-ILD appear to be similar to those of IPF patients, specifically in regard to a high mortality rate (about 80%) [55, 57].
Imaging
Because of the complexity of pulmonary abnormalities in pSS, it may not be diagnosed on the basis of imaging features alone. Even when the main disease pattern is a type of interstitial pneumonia, areas of other patterns of interstitial pneumonia or airway abnormalities often coexist (figure 2) [59].
FIGURE 2.

A 63-year-old woman with Sjogren syndrome. Coronal computed tomography image shows numerous cysts admixed with fine reticular abnormalities in the lower lobes. In the upper lobes, a few centrilobular branching opacities (circle) consistent with bronchiolitis (likely follicular) can be appreciated. This patient also suffered from pulmonary hypertension, which is responsible for pulmonary artery enlargement (arrow).
pSS-related ILDs may show the same radiological patterns (e.g. NSIP, UIP or LIP) as those observed in idiopathic interstitial pneumonias on HRCT. In fact, the most common HRCT findings are ground-glass opacities, reticular abnormalities, consolidation, honeycombing, cysts, nodules and also bronchiectasis (figure 3) [29, 60, 61]. The nodules are consistent with follicular bronchiolitis, pulmonary nodular hyperplasia or cancer, though is difficult to predict histologic correlates [59, 62]. The cysts are consistent with LIP [63]. The association of LIP and amyloidosis (manifesting on HRCT as multiple irregular nodules) in patients with pSS is also recognised, but as these patients are also at increased risk of pulmonary lymphoma, the finding of LIP on HRCT in conjunction with multiple nodules in a patient with pSS should at least prompt the consideration of a neoplastic process [64, 65]. A recent study showed that labial gland biopsy scores as well as dry mouth may reflect lymphoproliferative activity in the lungs in patients with pSS [66].
FIGURE 3.

A 51-year-old woman with Sjogren syndrome. Axial computed tomography shows peribronchovascular reticular and ground-glass opacities in the lower lobes. This pattern is consistent with nonspecific interstitial pneumonia.
Signs of airways disease can be frequently seen on HRCT. Indeed, either small or large airways disease affected more than half of a study cohort of 35 patients [67]. Bronchiectasis has been reported in 4–38% of patients with pSS [61, 68, 69]. Other HRCT signs of chronic inflammation include bronchial wall thickening and centrilobular branching opacities.
There are also a few descriptions of air-trapping in pSS. Air-trapping can be also elicited on an expiratory HRCT scan, more frequently than in healthy controls [70, 71]. It probably reflects constrictive obliterative bronchiolitis, which frequently represents an accompanying feature of bronchiectasis. There is scarce pathological–radiological correlation data on follicular bronchiolitis in pSS. The cardinal HRCT features of follicular bronchiolitis are centrilobular nodules of 1–12 mm in diameter, variably associated with peribronchial nodules and patchy areas of ground-glass opacity [72].
Pulmonary function tests
In patients with pSS-ILD, PFTs are frequently abnormal, showing a restrictive ventilatory failure with decreased DLCO, even in the absence of symptoms [8].
DLCO is highly sensitive for predicting the presence of ILD, whereas lung volumes may be more useful for assessing disease extent [73].
Bronchoalveolar lavage
Findings in bronchoalveolar lavage (BAL) fluid obtained from patients with pSS-ILD are frequently abnormal, showing, in the majority of the cases, a lymphocytosis, characterised mainly by T-cells, even among patients who are asymptomatic [74, 75].
However, abnormalities in the cellular constituents of BAL fluid are not useful for predicting outcome or response to treatment. As a result, BAL is not routinely performed in the diagnostic work-up of patients with pSS-ILD. In patients with an acute onset or worsening of respiratory symptoms and radiographical abnormalities, BAL is useful for excluding ILDs other than pSS- ILD, such as malignancy or infection [25].
Airway disease
Clinical features
Airways seem to be involved in about 20% of pSS patients [76]. Airways involvement can be caused either as a consequence of destruction of exocrine glands, leading to sicca syndrome or to cell infiltration, involving the trachea, bronchi or bronchioles [30]. Histopathological studies showed, even in asymptomatic patients and without radiographic abnormalities, an infiltration characterised by CD4+ T-lymphocytes was detected both in bronchial and in bronchiolar submucosa [77, 78]. Consistently, BAL reveals CD4+ lymphocytic alveolitis in 55% of patients with pSS [74, 78].
Involvement of the trachea and large airways manifests as dryness of the mucosa causing xerotrachea and xerobronchitis, respectively, the latter occurring when large airways are involved [31]. In these cases, clinical presentation is generally characterised as a nonproductive cough that may precede CTD diagnosis by several years, and often affecting quality of life [79].
Chronic cough may indeed be not only secondary to airway dryness, but also the consequence of abnormal mucociliary clearance [80], bronchial or bronchiolar inflammation, gastro-oesophageal reflux and bronchial hyperresponsiveness, frequently reported in pSS [81–83].
Bronchiolitis (i.e. small airways involvement) is a frequent finding in nonsmoking patients with pSS, but is rarely responsible for a severe clinical picture [84]; indeed, these patients are usually asymptomatic [68]. When symptoms occur, they present with cough, dyspnoea and wheezing and may be isolated or associated with interstitial pneumonia [29]. Although lung biopsy does not have to be performed routinely in pSS patients with pulmonary involvement, particularly in patients with a suspected bronchiolitis, different types of bronchiolitis exist: the main type is cellular/follicular bronchiolitis [85], whereas constrictive bronchiolitis is rare (figure 4). Patients with follicular bronchiolitis usually respond to corticosteroids [85]. In steroid-resistant cases, rituximab (RTX) [87, 88] or macrolides [84] may be utilised, depending on the severity of the disease.
FIGURE 4.

Histological findings of airway pathology in Sjogren syndrome. a) Bronchial biopsy showing a dense periglandular lymphoid infiltrate, similar to that seen in minor salivary gland biopsies in Sjogren syndrome. b) Surgical lung biopsy showing cellular bronchiolitis, consisting of a centrilobular cellular lymphoplasmacytic infiltrate, associated with bronchioloectasia. In the upper part of the picture a small granuloma is present (shown at higher magnification in c). Small non-necrotising granulomas are more frequent in Sjogren syndrome than in other connective tissue disease, and the histologic distinction with other granulomatous diseases (particularly infections and sarcoidosis) can be difficult. Occasionally Sjogren syndrome and sarcoidosis coexist in the same patient [86]. d) Surgical lung biopsy showing follicular bronchiolitis, a variant of cellular bronchiolitis in which the lymphoid infiltrate is organised in follicles with germinal centres. In Sjogren syndrome, cellular/follicular bronchiolitis can be the only lesion present in the biopsy, or can coexist with other lesions (cellular nonspecific interstitial pneumonia in this case). e) Surgical lung biopsy showing constrictive bronchiolitis, consisting of granulation tissue obliterating the bronchiolar lumen. Constrictive bronchiolitis is quite rare in Sjogren syndrome, cellular/follicular bronchiolitis can be the only lesion present in the biopsy, or can coexist, and clinically corresponds to the most severe forms of airway disease.
Pulmonary function tests
Rarely, airway disease shows lung function test impairment that, in contrast, can be assessed in the case of a distal airway disease (bronchiolitis) [68]. The occurrence of an obstructive ventilatory failure seems to be related to severity of the disease [84]. When a concomitant airway and ILD occurs a mixed, restrictive and obstructive respiratory failure may be observed [30].
Cysts or bullae
Cysts or bullae have been described in patients with pSS and are often associated with lymphoproliferative disorders, including LIP.
Diffuse cystic lung disease is an uncommon clinical and radiographic presentation with a broad differential diagnosis, including lymphangioleiomyomatosis and pulmonary Langerhans cell histiocytosis [89]. However, a cystic-predominant LIP-follicular bronchiolitis can also be considered in pSS patients [90].
Pleural involvement
In pSS, the incidence of pleural effusion is rare, occurring in <1% of patients and mostly observed in Japan, with a few cases described in Europe [91–94]. The occurrence of pleural effusion in pSS should always raise the possibility of an overlap syndrome with SLE or RA (secondary SS), considering that in the latter CTDs, pleural effusion is more frequent than in pSS [44]. The main differential diagnosis is with pleural infection or lymphoma.
Pleural effusion is more frequently bilateral [92, 94, 95]. It is usually exudative, with a predominant lymphocytic cell count. The pleural fluid analysis may also show an increased pleural and/or serum levels of rheumatoid factor, anti-SSA/Ro, anti-SSB/La and immune complexes. Decreased pleural and/or serum levels of complement may be also detected [44, 91]; in these cases, overlapping with SLE or RA (secondary SS) needs to be excluded.
In a few case reports, pleural effusions have regressed with corticosteroid therapy [96, 97] or improved spontaneously.
The detection of pleural thickening in patients with pSS has been more frequently associated with recurrent pneumonias and atelectasis.
Pulmonary infections
Patients with pSS are at increased risk of serious pulmonary infections, mainly in relation to a combination of immunosuppressive treatment and abnormalities of the immune system, clinically manifesting with pneumonia in 10–35% of pSS patients [30]. Mechanisms promoting the occurrence of lung infections in patients with pSS include impaired mucociliary clearance, abnormal sputum, deficit of local immunity, gastro-oesophageal reflux, bronchiectasis and parodontopathy [98].
Pulmonary embolism
It has been shown that there is an increased risk for venous thromboembolism or pulmonary embolism in patients with pSS [99].
In about one-third of patients, this prothrombotic state may be related to an increased prevalence of antiphospholipid antibodies, such as anticardiolipin, anti-β2GP1 or lupus anticoagulant [100]. However, this may also occur in response to chronic inflammation, typically observed in pSS.
Drug toxicity
Disease-modifying antirheumatic drugs are not frequently used to treat pSS. Hydroxychloroquine and methotrexate (MTX) alone or in combination therapy are particularly used to treat patients with inflammatory musculoskeletal pain [101]. Acute pneumonitis is the main lung complication related to MTX, it is a hypersensitivity reaction and usually occurs early after starting MTX, it is also dose-independent. Recent observations in RA suggest that MTX may not be associated with an increased risk of ILD, but on the contrary MTX may delay the onset of ILD [102, 103]. However, the clinician should be cautious when commencing MTX in patients with pre-existing lung disease. RTX is the only biological therapy recommended for pSS [101], particularly for patients with severe organ manifestations. In patients with RA and concomitant ILD, RTX seems to be relatively safe, does not lead to progression of ILD and demonstrates promising therapeutic results for ILD [104]. No data on possible RTX-induced lung toxicity in pSS are available. No cases of hydroxychloroquine-related pulmonary toxicity were described in pSS and in other autoimmune diseases. Therefore, hydroxychloroquine and RTX can be safely administered in patients with ILD related to pSS. As B-cells are also involved in the pathogenesis of pulmonary involvement in pSS, RTX could have a positive impact on this organ complication. Treatment of drug-induced pulmonary toxicity consists of drug cessation and sometimes corticosteroid therapy. The general prognosis of acute and subacute lung toxicity is usually favourable.
Malignancy
Among systemic autoimmune diseases, pSS has the higher risk to develop B-cell lymphoproliferative disorders [105], mainly non-Hodgkin lymphomas. Patients with pSS have a 14–20-fold increased risk of developing lymphomas than general population [105, 106].
The risk of lymphoma increases with disease duration, with a cumulative risk of 3.4% and 9.8% at 5 and 15 years from diagnosis, respectively [107, 108]. Multiple histologic types of non-Hodgkin lymphomas have been described, such as follicular, lymphoplasmacytoid and diffuse large B-cell lymphoma, with extranodal marginal zone B-cell lymphoma (mucosa-associated lymphoid tissue (MALT) lymphoma) as the most common subtype [109]. The pathogenetic link between pSS and lymphoma is unclear, but chronic antigenic activation of B-cells is believed to play a role [110].
Primary lymphoma of the lung is a rare entity representing about 4% of all extranodal lymphomas and 0.4% of non-Hodgkin lymphomas [111]. The prevalence of primary pulmonary lymphoma seems to be higher in pSS patients and it is estimated to be 1–2% [112]; it is usually a low-grade extranodal marginal B-cell lymphoma of the MALT type and rarely associated with an infectious agent, such as human herpesvirus-6 or Epstein–Barr virus [112]. However, to date, only few monocentric case series have been described [113, 114].
Recurrent swelling of parotid glands, purpura, low serum C4 level, cryoglobulinaemia, lymphopenia, monoclonal gammopathy and hypergammaglobulinaemia are all associated with lymphoma appearance [109, 115, 116].
Lymphoma slightly increases the mortality rate of pSS patients compared to the general population; usually pSS-related lymphomas show a favourable prognosis with an average 5-year survival rate of about 65–90% [115, 117–119], although, in a low proportion of patients, low-grade lymphomas can evolve to high-grade disease, mainly a diffuse large B-cell lymphoma [112].
When the lung is primarily involved, lymphoma clinically manifests with cough, dyspnoea, weight loss, sweats and fatigue [120]. Radiographically, it can present as solitary or multifocal nodules, bilateral alveolar infiltrates or interstitial markings randomly distributed with a mild predilection for the lower lobes. Mediastinal lymphadenopathy and pleural effusions may accompany the parenchymal abnormalities [113, 121]. Treatment of lung lymphoma should be personalised; watchful waiting, without specific drug treatment, can be reserved for asymptomatic nonevolutive cases, while progressive and symptomatic lymphomas require specific haematological treatment [122, 123].
The detection of enlarged mediastinal nodes in pSS patients may be of uncertain clinical significance: systemic lymphoplasmacytic inflammation and mediastinal lymphoma may cause it; however, other diseases, such as sarcoidosis [124], Lyme's disease [125] or more common infections may be part of the differential diagnosis.
Pulmonary amyloidosis
Pulmonary amyloidosis associated with pSS usually affects women presenting with cough, dyspnoea, weakness, haemoptysis and/or pleuritic chest pain [30]. As stated in a recent systematic review by Sambataro et al. [21], this condition can be found in about 10% of ILD-pSS patients, alone or as a secondary pattern and the AA subtype, associated with chronic inflammatory disease, is the usual amyloidosis subtype observed in pSS. Surgical lung biopsy is usually required to establish the diagnosis and to rule out lymphoma [30]. The prognosis of pSS-related pulmonary amyloidosis is unknown, and no specific therapy has been reported [4].
Pulmonary hypertension
PAH is associated with various autoimmune disorders, particularly scleroderma [126]. Pulmonary hypertension is rare in pSS, although it appears as one of the most severe complications in these patients, with a survival rate of about 70% at 1 year and about 65% at 3 years [127].
PAH in pSS can be the result of various pathophysiological mechanisms, including arteriopathy, pulmonary veno-occlusive disease, valvular heart disease or ILDs [44].
Launay et al. [126] found that pSS patients who developed PAH are more frequently women in their third and fourth decades of life and that they were more likely to have Raynaud's phenomenon, cutaneous vasculitis and ILDs. Furthermore, no correlations have been observed between the severity of sicca syndrome and the degree of PAH. Patients with pSS-PAH also more frequently had antinuclear, anti-Ro/SSA and anti-RNP autoantibodies, as well as positive rheumatoid factor and hypergammaglobulinaemia [127]. Hypocomplementaemia and cryoglobulinaemia have also been shown to be strong predictors of pulmonary artery systolic pressure [128].
Histologic lesions are nonspecific, and consist of small arteries and arterioles with concentric intimal proliferation, medial hypertrophy and sometimes plexiform lesions.
The best treatment strategy is unknown. Current PAH therapy has been shown to be effective in some patients; however, failures with these drugs have also been observed [127].
Some case reports showed the efficacy of pSS-PAH treatment with immunosuppressants as monotherapy, with initial improvement [44], but second-line standard PAH therapy was subsequently added to all cases.
Finally, patients with pSS-PAH seem to be nonresponders to acute vasodilator testing, suggesting that calcium channel blockers are inappropriate to treat this specific form of PH [127].
Management
The lack of knowledge about the natural history and the prognosis of pSS-ILD and the heterogeneity of this condition are the main limitations to the development of an evidence-based treatment strategy.
The treatment of pSS-ILD depends on the severity of symptoms and effects on lung function. For patients with asymptomatic mild or nonprogressive ILD without significant abnormalities on PFTs, a “see and wait” approach could be acceptable, while in patients with progressive or severe disease, first-line therapy is usually based on glucocorticoids, alone or in combination with immunosuppressive drugs. The radiological pattern of ILD could influence the response to therapy: NSIP, OP and LIP patterns seem to better respond to steroid or immunosuppressive therapies than the UIP pattern [8, 129].
Glucocorticoids are commonly used at the initial dose of 0.5–1 mg·kg−1 of prednisone daily, according to the severity of ILD [129, 130]. As for other connective tissue diseases, glucocorticoids can be associated with other immunosuppressive drugs such as cyclophosphamide (CYC), mycophenolate mofetil (MMF) or azathioprine (AZA). The association with these drugs should reduce the cumulative dose of steroids and improve the effectiveness of the treatment [8, 131–133]. However, due to the absence of randomised controlled trials, the treatment of pSS-ILD remains mainly empirical, because data are extrapolated from small case series, usually including not only patients with pSS but also other CTDs. Table 2 includes ongoing and completed clinical trials in pSS and ongoing trials on CTD-ILD.
TABLE 2.
Possible future therapeutic options in patients with primary Sjogren syndrome and connective tissue disease-related interstitial lung disease (ILD)
| Molecule | Mechanism of action | Type of trial | Development phase study | Primary outcome |
| Ongoing trial in primary Sjogren's syndrome | ||||
| Belimumab/rituximab [134] | Anti-BAFF/anti-CD20 | Phase 2 | Ongoing | Safety and efficacy of sequential therapy with belimumab followed by rituximab or their co-administration for primary Sjogren's syndrome-related B-cell lymphoproliferation |
| Prezalumab [134] | Anti-B7RP-1 (ICOSL) | Phase 2 | Ongoing | Change from baseline in ESSDAI score at day 99 |
| Tocilizumab [135] | Anti-IL-6R | Phase 2/3 | Ongoing | Improvement of the ESSDAI score ≥3 points compared to enrolment |
| Low-dose recombinant human IL-2 [136] | Low dose of IL-2 | Phase 2 | Ongoing | Improvement in ESSDAI at 24 weeks |
| Filgotinib, lanraplenib, tirabrutinib [137] | Selective JAK-inhibitors | Phase 2 | Ongoing | Improvement in ESSDAI at 24 weeks |
| Iguratimod [138] | Inhibitor of nuclear factor κB activation | Phase 1/2 | Ongoing | Open-label study, improvement in ESSDAI at 24 weeks |
| Tibulizumab [139] | Anti-BAFF and anti-IL-17A | Phase 1 | Ongoing | Evaluation of safety, tolerability, pharmacokinetics and pharmacodynamics |
| Completed trial in primary Sjogren's syndrome | ||||
| RSLV-132 [140] | RNase-Fc fusion protein | Phase 2 | Completed | Significant reduction in fatigue |
| Ianalumab [141] | Anti-BAFF-R | Phase 2 | Completed | Trend in reduction of ESSDAI |
| CFZ533 [142] | Anti-CD40 | Phase 2 | Completed | Trend in reduction of ESSDAI |
| Ongoing trials on ILD secondary to connective tissue diseases | ||||
| Pirfenidone [143] | RELIEF | Phase 2 | Completed | Efficacy and safety of pirfenidone as add-on to existing treatment for progressive, non-IPF lung fibrosis |
| NCT02808871 [144] | TRAIL1 | Phase 2 | Ongoing | Efficacy and safety of pirfenidone as add-on to existing treatment in patients with rheumatoid arthritis related ILD |
| NCT03843892 [145] | Not available | Phase 2 | Ongoing | Expanded access programme to provide nintedanib to patients with non-IPF ILD who have no alternative treatment possibilities |
| Rituximab versus cyclophosphamide [146] | RECITAL | Phase 3 | Ongoing | Change in FVC at 24 weeks in patients with ILD due to systemic sclerosis, inflammatory idiopathic myopathies, mixed connective tissue diseases |
| Pirfenidone versus mycophenolate mofetil [147] | SLS III | Phase 2 | Ongoing | Treatment of patients with active and symptomatic systemic sclerosis related ILD |
| Abatacept [148] | ATtackMy-ILD | Phase 2 | Ongoing | Efficacy and safety of s.c. abatacept in treating myositis related ILD in comparison to placebo |
IL: interleukin; BAFF: B-cell activating factor; ICOSL: inducible T-cell co-stimulator; ESSDAI: EULAR Sjogren syndrome disease activity index; IPF: idiopathic pulmonary fibrosis; FVC: forced vital capacity.
Among immunosuppressants, the use of AZA has been described in some case series: in seven of 11 pSS patients with ILD, FVC values improved after a 6-month follow-up, remained unchanged in three patients and worsened in one [132]. Roca et al. [129] reported an improvement of pulmonary disease in three out of four patients with steroid-refractory ILD.
Treatment with MMF was associated with either stable or improved pulmonary findings over a median 2.5 years of follow-up and with a low rate of discontinuation in 125 patients with different CTD-ILDs, including pSS, but specifically no considerations could be made regarding the effectiveness in pSS patients [133]. Finally, the use of CYC has been described in isolated cases and small case series with good results. In fact, Roca et al. [129] reported an improvement of pulmonary status in one patient treated with CYC (0.7 g·m−2 every 4 weeks for six cycles), while Shi et al. [149] reported improvement or stability during the follow-up period (median 38 months) in 14 patients with pSS and biopsy-proven pulmonary involvement treated with prednisone and CYC.
Some evidence suggest that RTX may be effective and safe in the treatment of systemic manifestations of pSS, in particular vasculitis and arthritis [101], but data regarding the treatment of ILD are more limited [15, 130, 132]. A registry-based study from France reported improvement in lung function in six out of eight patients with pSS-ILD treated with RTX, already evident after the first cycle of the therapy [150].
IPF is invariably progressive and is associated with a median survival of only 3–4 years [151]; pirfenidone and nintedanib, two oral antifibrotic therapies, reduce lung function decline in IPF patients [152–154]. A progressive fibrosing phenotype has also been observed to develop in a proportion of patients with other ILDs, including those associated with CTD and pSS. The open-label LOTUSS study suggested that pirfenidone associated with MMF also had an acceptable safety and tolerability profile in subjects with SSc-ILD, but no conclusions could be drawn on its effects on lung function because of the lack of a comparator group [155]. The efficacy and safety of pirfenidone in subjects with SSc-ILD (in combination with MMF versus MMF alone), is currently being investigated in clinical trials.
The efficacy and safety of nintedanib in subjects with SSc-ILD was explored in the SENSCIS trial, in which a reduction in the rate of FVC decline was observed in patients treated with nintedanib compared to patients enrolled in the placebo arm. The effect of nintedanib was lower in patients with SSc-ILD than in patients with IPF (INPULSIS trials), but the relative reduction in the rate of FVC decline observed with nintedanib versus placebo was similar (44% and 49%, respectively) [156]. In this study, approximately half of the trial population received MMF. The decline in FVC in the placebo group and the magnitude of the effect of nintedanib differed depending on MMF use, suggesting a potential benefit of MMF on lung function.
In the INBUILD trial (ClinicalTrials.gov identifier: NCT02999178), the efficacy and safety of nintedanib versus placebo was assessed in patients with a physician-diagnosed ILD other than IPF, who have features of diffuse progressive, fibrosing lung disease [157], including those diagnosed in patients with CTD [158].
The patients who received nintedanib had a slower progression of ILD than those receiving placebo, as shown by a lower annual rate of decline in the FVC over the 52-week period, both in the overall trial population and in patients with a UIP-like fibrotic pattern on HRCT. An interesting result of this trial was that the decline in the FVC of placebo-treated patients with non-UIP-like fibrotic patterns was only slightly lower than that in patients with a UIP-like fibrotic pattern, suggesting that progressive fibrosing ILDs, regardless of clinical diagnosis, have a similar pathobiological mechanism. Therefore, antifibrotic therapies, may have beneficial effect also in a heterogenous group of patients with progressive fibrosing ILD, including those associated to pSS. A therapeutic association between an antifibrotic drug and a traditional immunosuppressive agent as MMF could retard the progression of ILD related to CTD.
Moreover, best supportive care (or palliative care) aimed to improve HRQoL, has also an important role in the management of chronic diseases, such as pSS-ILD, mainly for the control of symptoms, such as cough and dyspnoea, side-effects caused by treatment of lung involvement, and psychological, social, and spiritual problems related to the disease or its treatment [159].
Palliation of dyspnoea and anxiety is generally managed with opioids and benzodiazepines [58]. Also, management of cough is important, using various types of drugs to palliate this symptom, such as oral steroids, opioids and gabapentin [159].
Oxygen therapy increases survival and improves HRQoL of hypoxaemic patients and is usually prescribed when oxygen saturation is <88% at rest [160]. A recent study suggests that oxygen seems to be associated with improved HRQoL in patients with ILD with isolated exertional hypoxia and can, therefore, be considered an effective intervention in this patient group [161].
Finally, data regarding survival after lung transplantation in CTD and specifically in patients with pSS are poor and conflicting [162]. However, recent studies seem to suggest that post-transplant outcomes in patients with CTD-ILD, including pSS, do not differ significantly from those in patients with non-CTD-ILD [162]. Possible negative prognostic factors include renal impairment, possible onset of skin ulcerations and consequent complicated surgical wound repair, arrhythmias and older age.
Conclusions
Improved understanding of the pathogenesis of pSS will permit the development of new therapeutic agents, particularly for pulmonary involvement.
pSS can potentially affect any organ; therefore, comprehensive treatment is related to the evaluation of the whole spectrum of this condition, including pulmonary involvement. However, there are no data on the clinical utility of using HRCT in all patients with pSS to confirm or exclude CTD-ILD. Some patients with pSS may have a subclinical nonprogressive lung disease that does not require specific therapy. An important unmet clinical need is the identification at diagnosis of predictors able to identify the patients who will develop progressive lung disease and therefore will need more accurate pulmonary screening and more aggressive therapies.
Antifibrotic drugs are effective in reducing lung function decline in patients with ILD, including also those with CTD (particularly in patients with SSc). Although there are no direct therapeutic experiences, traditional immunosuppressive agents in association with antifibrotic drugs could also become the preferential treatment in patients with pSS and progressive fibrotic ILD.
Footnotes
Provenance: Submitted article, peer reviewed
Conflict of interest: F. Luppi reports personal fees from Roche and Boheringer-Ingelheim, outside the submitted work.
Conflict of interest: M. Sebastiani has nothing to disclose.
Conflict of interest: N. Sverzellati has nothing to disclose.
Conflict of interest: A. Cavazza has nothing to disclose.
Conflict of interest: C. Salvarani has nothing to disclose.
Conflict of interest: A. Manfredi has nothing to disclose.
Support statement: This research was partially supported by the Italian Ministry of University and Research (MIUR) Department of Excellence project PREMIA (PREcision MedIcine Approach: bringing biomarker research to clinic).
References
- 1.Fox RI. Sjögren's syndrome. Lancet 2005; 366: 321–331. doi: 10.1016/S0140-6736(05)66990-5 [DOI] [PubMed] [Google Scholar]
- 2.Qin B, Wang J, Yang Z, et al. Epidemiology of primary Sjogren's syndrome: a systematic review and meta-analysis. Ann Rheum Dis 2015; 74: 1983–1989. doi: 10.1136/annrheumdis-2014-205375 [DOI] [PubMed] [Google Scholar]
- 3.Patel R, Shahane A. The epidemiology of Sjögren's syndrome. Clin Epidemiol 2014; 6: 247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Natalini J, Johr C, Kreider M. Pulmonary involvement in Sjögren syndrome. Clin Chest Med 2019; 40: 531–544. doi: 10.1016/j.ccm.2019.05.002 [DOI] [PubMed] [Google Scholar]
- 5.Ramos-Casals M, Brito-Zerón P, Seror R, et al. Characterization of systemic disease in primary Sjögren's syndrome: EULAR-SS task force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology (Oxford) 2015; 54: 2230–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palm Ø, Garen T, Enger TB, et al. Clinical pulmonary involvement in primary Sjögren's syndrome: prevalence, quality of life and mortality - A retrospective study based on registry data. Rheumatology (Oxford) 2013; 52: 173–179. [DOI] [PubMed] [Google Scholar]
- 7.Yazisiz V, Arslan G, Ozbudak IH, et al. Lung involvement in patients with primary Sjögren's syndrome: what are the predictors? Rheumatol Int 2010; 30: 1317–1324. doi: 10.1007/s00296-009-1152-8 [DOI] [PubMed] [Google Scholar]
- 8.Parambil JG, Myers JL, Lindell RM, et al. Interstitial lung disease in primary Sjogren syndrome. Chest 2006; 130: 1489–1495. doi: 10.1378/chest.130.5.1489 [DOI] [PubMed] [Google Scholar]
- 9.Kelly C, Gardiner P, Pal B, et al. Lung function in primary Sjogren's syndrome: a cross sectional and longitudinal study. Thorax 1991; 46: 180–183. doi: 10.1136/thx.46.3.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuyama N, Ashizawa K, Okimoto T, et al. Pulmonary lesions associated with Sjögren's syndrome: radiographic and CT findings. Br J Radiol 2003; 76: 880–884. doi: 10.1259/bjr/18937619 [DOI] [PubMed] [Google Scholar]
- 11.Mariette X, Criswell LA. Primary Sjögren's syndrome. New Engl J Med 2018; 378: 931–939. doi: 10.1056/NEJMcp1702514 [DOI] [PubMed] [Google Scholar]
- 12.Shiboski CH, Shiboski SC, Seror R, et al. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for primary Sjögren's syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol 2017; 69: 35–45. doi: 10.1002/art.39859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vivino FB, Bunya VY, Massaro-Giordano G, et al. Sjogren's syndrome: an update on disease pathogenesis, clinical manifestations and treatment. Clin Immunol 2019; 203: 81–121. doi: 10.1016/j.clim.2019.04.009 [DOI] [PubMed] [Google Scholar]
- 14.Sandhya P, Kurien B, Danda D, et al. Update on pathogenesis of Sjogren's syndrome. Curr Rheumatol Rev 2017; 13: 5–22. doi: 10.2174/1573397112666160714164149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Both T, Dalm VASH, Martin van Hagen P, et al. Reviewing primary Sjögren's syndrome: beyond the dryness - from pathophysiology to diagnosis and treatment. Int J Med Sci 2017; 14: 191–200. doi: 10.7150/ijms.17718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bodewes ILA, Björk A, Versnel MA, et al. Innate immunity and interferons in the pathogenesis of Sjögren's syndrome. Rheumatology (Oxford) 2019; 10.1093/rheumatology/key360. [DOI] [PubMed] [Google Scholar]
- 17.Margaretten M. Neurologic manifestations of primary Sjögren syndrome. Rheum Dis Clin North Am 2017; 43: 519–529. doi: 10.1016/j.rdc.2017.06.002 [DOI] [PubMed] [Google Scholar]
- 18.Iijima S. Fetal and neonatal involvement in maternal rheumatologic disease. J Matern Neonatal Med 2018; 31: 2079–2085. doi: 10.1080/14767058.2017.1334048 [DOI] [PubMed] [Google Scholar]
- 19.Gupta S, Gupta N. Sjögren syndrome and pregnancy: a literature review. Perm J 2017; 21: 16-047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baruteau AE, Pass RH, Thambo JB, et al. Congenital and childhood atrioventricular blocks: pathophysiology and contemporary management. Eur J Pediatr 2016; 175: 1235–1248. doi: 10.1007/s00431-016-2748-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambataro G, Ferro F, Orlandi M, et al. Clinical, morphological features and prognostic factors associated with interstitial lung disease in primary Sjögren's syndrome: a systematic review from the Italian Society of Rheumatology. Autoimmun Rev 2020: 102447. doi: 10.1016/j.autrev.2019.102447 [DOI] [PubMed] [Google Scholar]
- 22.García-Carrasco M, Ramos-Casals M, Rosas J, et al. Primary Sjögren syndrome: clinical and immunologic disease patterns in a cohort of 400 patients. Medicine (Baltimore) 2002; 81: 270–280. doi: 10.1097/00005792-200207000-00003 [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Hou Z, Qiu M, et al. Risk factors for primary Sjögren syndrome-associated interstitial lung disease. J Thorac Dis 2018; 10: 2108–2117. doi: 10.21037/jtd.2018.03.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyamoto ST, Valim V, Fisher BA. Health-related quality of life and costs in Sjögren's syndrome. Rheumatology (Oxford) 2019; in press [ 10.1093/rheumatology/key370]. [DOI] [PubMed] [Google Scholar]
- 25.Spagnolo P, Cordier JF, Cottin V. Connective tissue diseases, multimorbidity and the ageing lung. Eur Respir J 2016; 47: 1535–1558. doi: 10.1183/13993003.00829-2015 [DOI] [PubMed] [Google Scholar]
- 26.Manfredi A, Cassone G, Cerri S, et al. Diagnostic accuracy of a Velcro sound detector (VECTOR) for interstitial lung disease in rheumatoid arthritis patients: the InSPIRAtE validation study (INterStitial pneumonia in rheumatoid ArThritis with an electronic device). BMC Pulm Med 2019; 19: 111. doi: 10.1186/s12890-019-0875-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sgalla G, Walsh SLF, Sverzellati N, et al. “Velcro-type” crackles predict specific radiologic features of fibrotic interstitial lung disease. BMC Pulm Med 2018; 18: 103. doi: 10.1186/s12890-018-0670-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pancaldi F, Sebastiani M, Cassone G, et al. Analysis of pulmonary sounds for the diagnosis of interstitial lung diseases secondary to rheumatoid arthritis. Comput Biol Med 2018; 96: 91–97. doi: 10.1016/j.compbiomed.2018.03.006 [DOI] [PubMed] [Google Scholar]
- 29.Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of primary Sjögren's syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med 2005; 171: 632–638. doi: 10.1164/rccm.200403-417OC [DOI] [PubMed] [Google Scholar]
- 30.Flament T, Bigot A, Chaigne B, et al. Pulmonary manifestations of Sjögren's syndrome. Eur Respir Rev 2016; 25: 110–123. doi: 10.1183/16000617.0011-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papiris SA, Maniati M, Constantopoulos SH, et al. Lung involvement in primary Sjogren's syndrome is mainly related to the small airway disease. Ann Rheum Dis 1999; 58: 61–64. doi: 10.1136/ard.58.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cottin V, Nunes H, Mouthon L, et al. Combined pulmonary fibrosis and emphysema syndrome in connective tissue disease. Arthritis Rheum 2011; 63: 295–304. doi: 10.1002/art.30077 [DOI] [PubMed] [Google Scholar]
- 33.Mejía M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest 2009; 136: 10–15. doi: 10.1378/chest.08-2306 [DOI] [PubMed] [Google Scholar]
- 34.Wells AU, Denton CP. Interstitial lung disease in connective tissue disease - mechanisms and management. Nat Rev Rheumatol 2014; 10: 728–739. doi: 10.1038/nrrheum.2014.149 [DOI] [PubMed] [Google Scholar]
- 35.Fischer A, du Bois RM. Interstitial lung disease in connective tissue disorders. Lancet 2012; 380: 689–698. doi: 10.1016/S0140-6736(12)61079-4 [DOI] [PubMed] [Google Scholar]
- 36.Davidson BKS, Kelly CA, Griffiths ID. Ten year follow up of pulmonary function in patients with primary Sjogren's syndrome. Ann Rheum Dis 2000; 59: 709–712. doi: 10.1136/ard.59.9.709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goh NS, Hoyles RK, Denton CP, et al. Short-term pulmonary function trends are predictive of mortality in interstitial lung disease associated with systemic sclerosis. Arthritis Rheumatol 2017; 69: 1670–1678. doi: 10.1002/art.40130 [DOI] [PubMed] [Google Scholar]
- 38.Du Bois RM, Albera C, Bradford WZ, et al. 6-minute walk distance is an independent predictor of mortality in patients with idiopathic pulmonary fibrosis. Eur Respir J 2014; 43: 1421–1429. doi: 10.1183/09031936.00131813 [DOI] [PubMed] [Google Scholar]
- 39.Caminati A, Bianchi A, Cassandro R, et al. Walking distance on 6-MWT is a prognostic factor in idiopathic pulmonary fibrosis. Respir Med 2009; 103: 117–123. doi: 10.1016/j.rmed.2008.07.022 [DOI] [PubMed] [Google Scholar]
- 40.Vandecasteele E, De Pauw M, De Keyser F, et al. Six-minute walk test in systemic sclerosis: a systematic review and meta-analysis. Int J Cardiol 2016; 212: 265–273. doi: 10.1016/j.ijcard.2016.03.084 [DOI] [PubMed] [Google Scholar]
- 41.Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174: 803–809. doi: 10.1164/rccm.200604-488OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakazawa A, Cox NS, Holland AE. Current best practice in rehabilitation in interstitial lung disease. Ther Adv Respir Dis 2017; 11: 115–128. doi: 10.1177/1753465816676048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rizzi M, Radovanovic D, Santus P, et al. Usefulness of six-minute walk test in systemic sclerosis. Clin Exp Rheumatol 2018; 36: Suppl. 1, 161–167. [PubMed] [Google Scholar]
- 44.Kreider M, Highland K. Pulmonary involvement in Sjögren syndrome. Semin Respir Crit Care Med 2014; 35: 255–264. doi: 10.1055/s-0034-1371529 [DOI] [PubMed] [Google Scholar]
- 45.Liebow AA, Carrington CB. Diffuse pulmonary lymphoreticular infiltrations associated with dysproteinemia. Med Clin North Am 1973; 57: 809–843. doi: 10.1016/S0025-7125(16)32278-7 [DOI] [PubMed] [Google Scholar]
- 46.Manfredi A, Sebastiani M, Cerri S, et al. Prevalence and characterization of non-sicca onset primary Sjögren syndrome with interstitial lung involvement. Clin Rheumatol 2017; 36: 1261–1268. doi: 10.1007/s10067-017-3601-1 [DOI] [PubMed] [Google Scholar]
- 47.Enomoto Y, Nakamura Y, Colby TV, et al. Radiologic pleuroparenchymal fibroelastosis-like lesion in connective tissue disease-related interstitial lung disease. PLoS One 2017; 12: e0180283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Zhao S, Du G, et al. Acute fibrinous and organizing pneumonia as initial presentation of primary Sjögren's syndrome: a case report and literature review. Clin Rheumatol 2018; 37: 2001–2005. doi: 10.1007/s10067-018-4128-9 [DOI] [PubMed] [Google Scholar]
- 49.Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19: 453–459. doi: 10.1097/MCP.0b013e328363f48d [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi H, Matsuoka R, Kitamura S, et al. Sjogren's syndrome with multiple bullae and pulmonary nodular amyloidosis. Chest 1988; 94: 438–440. doi: 10.1378/chest.94.2.438 [DOI] [PubMed] [Google Scholar]
- 51.Hubscher O, Re R, Iotti R. Cystic lung disease in Sjögren's syndrome. J Rheumatol 2002; 29: 2235–2236. [PubMed] [Google Scholar]
- 52.Ramos-Casals M, Solans R, Rosas J, et al. Primary Sjögren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine (Baltimore) 2008; 87: 210–219. doi: 10.1097/MD.0b013e318181e6af [DOI] [PubMed] [Google Scholar]
- 53.Cain HC, Noble PW MR. Pulmonary manifestations of Sjögren's syndrome. Clin Chest Med 1998; 19: 687–699. doi: 10.1016/S0272-5231(05)70110-6 [DOI] [PubMed] [Google Scholar]
- 54.Gao H, Zhang XW, He J, et al. Prevalence, risk factors, and prognosis of interstitial lung disease in a large cohort of Chinese primary Sjögren syndrome patients: a case-control study. Med (United States) 2018; 97: e11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suda T, Kaida Y, Nakamura Y, et al. Acute exacerbation of interstitial pneumonia associated with collagen vascular diseases. Respir Med 2009; 103: 846–853. doi: 10.1016/j.rmed.2008.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis an international working group report. Am J Respir Crit Care Med 2016; 194: 265–275. doi: 10.1164/rccm.201604-0801CI [DOI] [PubMed] [Google Scholar]
- 57.Manfredi A, Sebastiani M, Cerri S, et al. Acute exacerbation of interstitial lung diseases secondary to systemic rheumatic diseases: a prospective study and review of the literature. J Thorac Dis 2019; 11: 1621–1628. doi: 10.21037/jtd.2019.03.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luppi F, Cerri S, Taddei S, et al. Acute exacerbation of idiopathic pulmonary fibrosis: a clinical review. Intern Emerg Med 2015; 10: 401–411. doi: 10.1007/s11739-015-1204-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Egashira R, Kondo T, Hirai T, et al. CT findings of thoracic manifestations of primary Sjögren syndrome: radiologic-pathologic correlation. Radiographics 2013; 33: 1933–1949. doi: 10.1148/rg.337125107 [DOI] [PubMed] [Google Scholar]
- 60.Lohrmann C, Uhl M, Warnatz K, et al. High-resolution CT imaging of the lung for patients with primary Sjögren's syndrome. Eur J Radiol 2004; 52: 137–143. doi: 10.1016/j.ejrad.2004.01.006 [DOI] [PubMed] [Google Scholar]
- 61.Franquet T, Giménez A, Monill JM, et al. Primary Sjogren's syndrome and associated lung disease: CT findings in 50 patients. Am J Roentgenol 1997; 169: 655–658. doi: 10.2214/ajr.169.3.9275871 [DOI] [PubMed] [Google Scholar]
- 62.Song MK, Seol YM, Park YE, et al. Pulmonary nodular lymphoid hyperplasia associated with Sjögren's syndrome. Korean J Intern Med 2007; 22: 192–196. doi: 10.3904/kjim.2007.22.3.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martínez-Balzano CD, Touray S, Kopec S. Cystic lung disease among patients with Sjögren syndrome: frequency, natural history, and associated risk factors. Chest 2016; 150: 631–639. doi: 10.1016/j.chest.2016.05.015 [DOI] [PubMed] [Google Scholar]
- 64.Jeong YJ, Lee KS, Chung MP, et al. Amyloidosis and Lymphoproliferative disease in Sjögren syndrome: thin-section computed tomography findings and histopathologic comparisons. J Comput Assist Tomogr 2004; 28: 776–781. doi: 10.1097/00004728-200411000-00008 [DOI] [PubMed] [Google Scholar]
- 65.Masaki Y, Sugai S. Lymphoproliferative disorders in Sjögren's syndrome. Autoimmun Rev 2004; 3: 175–182. doi: 10.1016/S1568-9972(03)00102-2 [DOI] [PubMed] [Google Scholar]
- 66.Kakugawa T, Sakamoto N, Ishimoto H, et al. Lymphocytic focus score is positively related to airway and interstitial lung diseases in primary Sjögren's syndrome. Respir Med 2018; 137: 95–102. doi: 10.1016/j.rmed.2018.02.023 [DOI] [PubMed] [Google Scholar]
- 67.Taouli B, Brauner MW, Mourey I, et al. Thin-section chest CT findings of primary Sjögren's syndrome: correlation with pulmonary function. Eur Radiol 2002; 12: 1504–1511. doi: 10.1007/s00330-001-1236-7 [DOI] [PubMed] [Google Scholar]
- 68.Uffmann M, Kiener HP, Bankier AA, et al. Lung manifestation in asymptomatic patients with primary Sjögren syndrome: assessment with high resolution CT and pulmonary function tests. J Thorac Imag 2001; 16: 282–289. doi: 10.1097/00005382-200110000-00009 [DOI] [PubMed] [Google Scholar]
- 69.Koyama M, Johkoh T, Honda O, et al. Pulmonary involvement in primary Sjögren's syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imag 2001; 16: 290–296. doi: 10.1097/00005382-200110000-00010 [DOI] [PubMed] [Google Scholar]
- 70.Franquet T, Díaz C, Domingo P, et al. Air trapping in primary Sjogren syndrome: correlation of expiratory CT with pulmonary function tests. J Comput Assist Tomogr 1999; 23: 169–173. doi: 10.1097/00004728-199903000-00002 [DOI] [PubMed] [Google Scholar]
- 71.Nakanishi M, Fukuoka J, Tanaka T, et al. Small airway disease associated with Sjögren's syndrome: clinico-pathological correlations. Respir Med 2011; 105: 1931–1938. doi: 10.1016/j.rmed.2011.08.009 [DOI] [PubMed] [Google Scholar]
- 72.Hare SS, Souza CA, Bain G, et al. The radiological spectrum of pulmonary lymphoproliferative disease. Br J Radiol 2012; 85: 848–864. doi: 10.1259/bjr/16420165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ciancio N, Pavone M, Torrisi SE, et al. Contribution of pulmonary function tests (PFTs) to the diagnosis and follow up of connective tissue diseases. Multidiscip Respir Med 2019; 14: 17. doi: 10.1186/s40248-019-0179-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hatron P-Y, Wallaert B, Gosset D, et al. Subclinical lung inflammation in primary Sjögren's syndrome: relationship between bronchoalveolar lavage cellular analysis findings and characteristics of the disease. Arthritis Rheum 1987; 30: 1226–1231. doi: 10.1002/art.1780301104 [DOI] [PubMed] [Google Scholar]
- 75.Dalavanga YA, Voulgari p V, Georgiadis AN, et al. Lymphocytic alveolitis: a surprising index of poor prognosis in patients with primary Sjogren's syndrome. Rheumatol Int 2006; 26: 799–804. doi: 10.1007/s00296-005-0092-1 [DOI] [PubMed] [Google Scholar]
- 76.Constantopoulos SH, Papadimitriou CS, Moutsopoulos HM. Respiratory manifestations in primary Sjogren's syndrome. A clinical, functional, and histologic study. Chest 1985; 88: 226–229. doi: 10.1378/chest.88.2.226 [DOI] [PubMed] [Google Scholar]
- 77.Gardiner P, Ward C, Allison A, et al. Pleuropulmonary abnormalities in primary Sjogren's syndrome. J Rheumatol 1993; 20: 831–837. [PubMed] [Google Scholar]
- 78.Papiris SA, Saetta M, Turato G, et al. CD4-positive T-lymphocytes infiltrate the bronchial mucosa of patients with Sjogren's syndrome. Am J Respir Crit Care Med 1997; 156: 637–641. doi: 10.1164/ajrccm.156.2.9610076 [DOI] [PubMed] [Google Scholar]
- 79.Mialon P, Barthélémy L, Sébert P, et al. A longitudinal study of lung impairment in patients with primary Sjogren's syndrome. Clin Exp Rheumatol 1997; 15: 349–354. [PubMed] [Google Scholar]
- 80.Mathieu A, Cauli A, Pala R, et al. Tracheo-bronchial mucociliary clearance in patients with primary and secondary Sjögren's syndrome. Scand J Rheumatol 1995; 24: 300–304. doi: 10.3109/03009749509095167 [DOI] [PubMed] [Google Scholar]
- 81.Potena A, La Corte R, Fabbri LM, et al. Increased bronchial responsiveness in primary and secondary Sjogren's syndrome. Eur Respir J 1990; 3: 548–553. [PubMed] [Google Scholar]
- 82.Gudbjornsson B, Hedenstrom H, Stalenheim G, et al. Bronchial hyperresponsiveness to methacholine in patients with primary Sjogren's syndrome. Ann Rheum Dis 1991; 50: 36–40. doi: 10.1136/ard.50.1.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.La Corte R, Potena A, Bajocchi G, et al. Increased bronchial responsiveness in primary Sjogren's syndrome. A sign of tracheobronchial involvement. Clin Exp Rheumatol 1991; 9: 125–130. [PubMed] [Google Scholar]
- 84.Borie R, Schneider S, Debray MP, et al. Severe chronic bronchiolitis as the presenting feature of primary Sjögren's syndrome. Respir Med 2011; 105: 130–136. doi: 10.1016/j.rmed.2010.07.017 [DOI] [PubMed] [Google Scholar]
- 85.Wells AU, Du Bois RM. Bronchiolitis in association with connective tissue disorders. Clin Chest Med 1993; 14: 655–666. [PubMed] [Google Scholar]
- 86.Ramos-Casals M, Brito-Zerón P, García-Carrasco M, et al. Sarcoidosis or Sjogren's syndrome? Clues to defining mimicry or coexistence in 59 cases. Medicine (Baltimore) 2004; 83: 85–95. [DOI] [PubMed] [Google Scholar]
- 87.Devauchelle-Pensec V, Pennec Y, Morvan J, et al. Improvement of Sjögren's syndrome after two infusions of rituximab (anti-CD20). Arthritis Rheum 2007; 57: 310–317. doi: 10.1002/art.22536. [DOI] [PubMed] [Google Scholar]
- 88.Seror R, Sordet C, Guillevin L, et al. Tolerance and efficacy of rituximab and changes in serum B-cell biomarkers in patients with systemic complications of primary Sjögren's syndrome. Ann Rheum Dis 2007; 66: 351–357. doi: 10.1136/ard.2006.057919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gupta N, Vassallo R, Wikenheiser-Brokamp KA, et al. Diffuse cystic lung disease: Part I. Am J Respir Crit Care Med 2015; 191: 1354–1366. doi: 10.1164/rccm.201411-2094CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gupta N, Wikenheiser-Brokamp KA, Fischer A, et al. Diffuse cystic lung disease as the presenting manifestation of Sjögren syndrome. Ann Am Thorac Soc 2016; 13: 371–375. doi: 10.1513/AnnalsATS.201511-759BC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kawamata K, Haraoka H, Hirohata S, et al. Pleurisy in primary Sjogren's syndrome: T-cell receptor β-chain variable region gene bias and local autoantibody production in the pleural effusion. Clin Exp Rheumatol 1997; 15: 193–196. [PubMed] [Google Scholar]
- 92.Ma D, Lu H, Qu Y, et al. Primary Sjögren's syndrome accompanied by pleural effusion: a case report and literature review. Int J Clin Exp Pathol 2015; 8: 15322–15327. [PMC free article] [PubMed] [Google Scholar]
- 93.Baenas DF, Retamozo S, Pirola JP, et al. Shrinking lung syndrome and pleural effusion as an initial manifestation of primary Sjögren's syndrome. Reumatol Clínica (English Ed) 2019; 16: 65–68. [DOI] [PubMed] [Google Scholar]
- 94.Hosoda C, Hosaka Y, Ryu K, et al. Pleuritis associated with primary Sjogren syndrome. Respirol Case Rep 2018; 6: e00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Teshigawara K, Kakizaki S, Horiya M, et al. Primary Sjogren's syndrome complicated by bilateral pleural effusion. Respirology 2008; 13: 155–158. [DOI] [PubMed] [Google Scholar]
- 96.Cruz-Pérez Fdel P, Doval-Cortés A, Jaume-Anselmi F, et al. Pleural effusion in a patient with primary Sjögren's syndrome successfully treated with corticosteroids. Bol Asoc Med P R 2015; 107: 13–16. [PubMed] [Google Scholar]
- 97.Mutsukura K, Nakamura H, Iwanaga N, et al. Successful treatment of a patient with primary Sjögren's syndrome complicated with pericarditis during pregnancy. Intern Med 2007; 46: 1143–1147. doi: 10.2169/internalmedicine.46.0062 [DOI] [PubMed] [Google Scholar]
- 98.Soto-Cardenas MJ, Perez-De-lis M, Bove A, et al. Bronchiectasis in primary Sjögren's syndrome: prevalence and clinical significance. Clin Exp Rheumatol 2010; 28: 647–653. [PubMed] [Google Scholar]
- 99.Ahlehoff O, Wu JJ, Raunsø J, et al. Cutaneous lupus erythematosus and the risk of deep venous thrombosis and pulmonary embolism: a Danish nationwide cohort study. Lupus 2017; 26: 1435–1439. doi: 10.1177/0961203317716306 [DOI] [PubMed] [Google Scholar]
- 100.Fauchais AL, Lambert M, Launay D, et al. Antiphospholipid antibodies in primary Sjögren's syndrome: prevalence and clinical significance in a series of 74 patients. Lupus 2004; 13: 245–248. doi: 10.1191/0961203304lu1006oa [DOI] [PubMed] [Google Scholar]
- 101.Carsons SE, Vivino FB, Parke A, et al. Treatment guidelines for rheumatologic manifestations of Sjögren's syndrome: use of biologic agents, management of fatigue, and inflammatory musculoskeletal pain. Arthritis Care Res 2017; 69: 517–527. doi: 10.1002/acr.22968 [DOI] [PubMed] [Google Scholar]
- 102.Fragoulis GE, Conway R, Nikiphorou E. Methotrexate and interstitial lung disease: controversies and questions. A narrative review of the literature. Rheumatol (United Kingdom) 2019; 58: 1900–1906. [DOI] [PubMed] [Google Scholar]
- 103.Kiely P, Busby AD, Nikiphorou E, et al. Is incident rheumatoid arthritis interstitial lung disease associated with methotrexate treatment? Results from a multivariate analysis in the ERAS and ERAN inception cohorts. BMJ Open 2019; 9: e028466. doi: 10.1136/bmjopen-2018-028466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duarte AC, Porter JC, Leandro MJ. The lung in a cohort of rheumatoid arthritis patients-an overview of different types of involvement and treatment. Rheumatol (United Kingdom) 2019; 58: 2031–2038. [DOI] [PubMed] [Google Scholar]
- 105.Zintzaras E, Voulgarelis M, Moutsopoulos HM. The risk of lymphoma development in autoimmune diseases: a meta-analysis. Arch Intern Med 2005; 165: 2337–2344. doi: 10.1001/archinte.165.20.2337 [DOI] [PubMed] [Google Scholar]
- 106.Theander E, Henriksson G, Ljungberg O, et al. Lymphoma and other malignancies in primary Sjögren's syndrome: a cohort study on cancer incidence and lymphoma predictors. Ann Rheum Dis 2006; 65: 796–803. doi: 10.1136/ard.2005.041186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Solans-Laqué R, López-Hernandez A, Angel Bosch-Gil J, et al. Risk, predictors, and clinical characteristics of lymphoma development in primary Sjögren's syndrome. Semin Arthritis Rheum 2011; 41: 415–423. doi: 10.1016/j.semarthrit.2011.04.006 [DOI] [PubMed] [Google Scholar]
- 108.Ioannidis JPA, Vassiliou VA, Moutsopoulos HM. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjögren's syndrome. Arthritis Rheum 2002; 46: 741–747. doi: 10.1002/art.10221 [DOI] [PubMed] [Google Scholar]
- 109.Liang Y, Yang Z, Qin B, et al. Primary Sjögren's syndrome and malignancy risk: a systematic review and meta-analysis. Ann Rheum Dis 2014; 73: 1151–1156. doi: 10.1136/annrheumdis-2013-203305 [DOI] [PubMed] [Google Scholar]
- 110.Bernatsky S, Ramsey-Goldman R, Clarke A. Malignancy and autoimmunity. Curr Opin Rheumatol 2006; 18: 129–134. doi: 10.1097/01.bor.0000209423.39033.94 [DOI] [PubMed] [Google Scholar]
- 111.Schreuder MI, van den Brand M, Hebeda KM, et al. Novel developments in the pathogenesis and diagnosis of extranodal marginal zone lymphoma. J Hematop 2017; 10: 91–107. doi: 10.1007/s12308-017-0302-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kokosi M, Riemer EC HK. Pulmonary involvement in Sjögren syndrome. Clin Chest Med 2010; 31: 489–500. doi: 10.1016/j.ccm.2010.05.007 [DOI] [PubMed] [Google Scholar]
- 113.Papiris SA, Kalomenidis I, Malagari K, et al. Extranodal marginal zone B-cell lymphoma of the lung in Sjögren's syndrome patients: reappraisal of clinical, radiological, and pathology findings. Respir Med 2007; 101: 84–92. doi: 10.1016/j.rmed.2006.04.005 [DOI] [PubMed] [Google Scholar]
- 114.Yachoui R, Leon C, Sitwala K, et al. Pulmonary MALT lymphoma in patients with Sjögren's syndrome. Clin Med Res 2017; 15: 6–12. doi: 10.3121/cmr.2017.1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Goules AV, Tzioufas AG. Lymphomagenesis in Sjögren's syndrome: predictive biomarkers towards precision medicine. Autoimmun Rev 2019; 18: 137–143. doi: 10.1016/j.autrev.2018.08.007 [DOI] [PubMed] [Google Scholar]
- 116.Goules AV, Tzioufas AG. Primary Sjögren's syndrome: clinical phenotypes, outcome and the development of biomarkers. Immunol Res 2017; 65: 331–344. doi: 10.1007/s12026-016-8844-4 [DOI] [PubMed] [Google Scholar]
- 117.Baimpa E, Dahabreh IJ, Voulgarelis M, et al. Hematologic manifestations and predictors of lymphoma development in primary Sjögren syndrome: clinical and pathophysiologic aspects. Medicine (Baltimore) 2009; 88: 284–293. doi: 10.1097/MD.0b013e3181b76ab5 [DOI] [PubMed] [Google Scholar]
- 118.Hansen LA, Prakash UBCT. Pulmonary lymphoma in Sjögren's syndrome. Mayo Clin Proc 1989; 64: 920–931. doi: 10.1016/S0025-6196(12)61219-1 [DOI] [PubMed] [Google Scholar]
- 119.Graham BB, Mathisen DJ, Mark EJ TR. Primary pulmonary lymphoma. Ann Thorac Surg 2005; 80: 1248–1253. doi: 10.1016/j.athoracsur.2005.04.014 [DOI] [PubMed] [Google Scholar]
- 120.Zinzani PL, Tani M, Gabriele A, et al. Extranodal marginal zone B-cell lymphoma of MALT-type of the lung: single-center experience with 12 patients. Leuk Lymphoma 2003; 44: 821–824. doi: 10.1080/1042819031000067972 [DOI] [PubMed] [Google Scholar]
- 121.Sirajuddin A, Raparia K, Lewis VA, et al. Primary pulmonary lymphoid lesions: radiologic and pathologic findings. RadioGraphics 2016; 36: 53–70. doi: 10.1148/rg.2016140339 [DOI] [PubMed] [Google Scholar]
- 122.Gribben JG. How I treat indolent lymphoma. Blood 2007; 109: 4617–4626. doi: 10.1182/blood-2006-10-041863 [DOI] [PubMed] [Google Scholar]
- 123.Borie R, Wislez M, Thabut G, et al. Clinical characteristics and prognostic factors of pulmonary MALT lymphoma. Eur Respir J 2009; 34: 1408–1416. doi: 10.1183/09031936.00039309 [DOI] [PubMed] [Google Scholar]
- 124.Ahmad Y. Case review of sarcoidosis resembling Sjogren's syndrome. J Clin Med Res 2010; 2: 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Smiyan S, Galaychuk I, Zhulkevych I, et al. Sjögren's syndrome and lymphadenopathy unraveling the diagnosis of Lyme disease. Reumatologia 2019; 57: 59–62. doi: 10.5114/reum.2019.83242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Launay D, Sobanski V, Hachulla E, et al. Pulmonary hypertension in systemic sclerosis: different phenotypes. Eur Respir Rev 2017; 26: 170056. doi: 10.1183/16000617.0056-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Launay D, Hachulla E, Hatron PY, et al. Pulmonary arterial hypertension: a rare complication of primary Sjögren syndrome - report of 9 new cases and review of the literature. Medicine (Baltimore) 2007; 86: 299–315. doi: 10.1097/MD.0b013e3181579781 [DOI] [PubMed] [Google Scholar]
- 128.Vassiliou VA, Moyssakis I, Boki KA, et al. Is the heart affected in primary Sjögren's syndrome? An echocardiographic study. Clin Exp Rheumatol 2008; 26: 109–112. [PubMed] [Google Scholar]
- 129.Roca F, Dominique S, Schmidt J, et al. Interstitial lung disease in primary Sjögren's syndrome. Autoimmun Rev 2017; 16: 48–54. doi: 10.1016/j.autrev.2016.09.017 [DOI] [PubMed] [Google Scholar]
- 130.Ramos-Casals M, Brito-Zerón P, Sisó-Almirall A, et al. Topical and systemic medications for the treatment of primary Sjögren's syndrome. Nat Rev Rheumatol 2012; 8: 399–411. doi: 10.1038/nrrheum.2012.53 [DOI] [PubMed] [Google Scholar]
- 131.Deheinzelin D, Capelozzi VL, Kairalla RA, et al. Interstitial lung disease in primary Sjogren's syndrome: a clinical-pathological evaluation and response to treatment. Am J Respir Crit Care Med 1996; 154: 794–799. doi: 10.1164/ajrccm.154.3.8810621 [DOI] [PubMed] [Google Scholar]
- 132.Saraux A, Pers JO, Devauchelle-Pensec V. Treatment of primary Sjögren syndrome. Nat Rev Rheumatol 2016; 12: 456–471. doi: 10.1038/nrrheum.2016.100 [DOI] [PubMed] [Google Scholar]
- 133.Fischer A, Brown KK, Du Bois RM, et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J Rheumatol 2013; 40: 640–646. doi: 10.3899/jrheum.121043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Felten R, Dervovic E, Chasset F, et al. The 2018 pipeline of targeted therapies under clinical development for systemic lupus erythematosus: a systematic review of trials. Autoimmun Rev 2018; 18: 576–582. doi: 10.1016/j.autrev.2018.12.008 [DOI] [PubMed] [Google Scholar]
- 135.Mavragani CP, Moutsopoulos HM. Sjögren's syndrome: old and new therapeutic targets. J Autoimmun 2019: 102364. doi: 10.1016/j.jaut.2019.102364 [DOI] [PubMed] [Google Scholar]
- 136.Yu A, Zhu L, Altman NH, et al. A Low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity 2009; 30: 204–217. doi: 10.1016/j.immuni.2008.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Namour F, Diderichsen PM, Cox E, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIb dose selection. Clin Pharmacokinet 2015; 54: 859–874. doi: 10.1007/s40262-015-0240-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li G, Yamasaki R, Fang M, et al. Novel disease-modifying anti-rheumatic drug iguratimod suppresses chronic experimental autoimmune encephalomyelitis by down-regulating activation of macrophages/microglia through an NF-κB pathway. Sci Rep 2018; 8: 1933. doi: 10.1038/s41598-018-20390-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang LW, Zhou PR, Wei P, et al. Expression of interleukin-17 in primary Sjögren's syndrome and the correlation with disease severity: a systematic review and meta-analysis. Scand J Immunol 2018; 87: e12649. doi: 10.1111/sji.12649 [DOI] [PubMed] [Google Scholar]
- 140.Burge DJ, Eisenman J, Byrnes-Blake K, et al. Safety, pharmacokinetics, and pharmacodynamics of RSLV-132, an RNase-Fc fusion protein in systemic lupus erythematosus: a randomized, double-blind, placebo-controlled study. Lupus 2017; 26: 825–834. doi: 10.1177/0961203316678675 [DOI] [PubMed] [Google Scholar]
- 141.Dörner T, Posch MG, Li Y, et al. Treatment of primary Sjögren's syndrome with ianalumab (vay736) targeting b cells by baff receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann Rheum Dis 2019; 78: 641–647. doi: 10.1136/annrheumdis-2018-214720 [DOI] [PubMed] [Google Scholar]
- 142.Fischer B, Margit Z, Ng W-F, et al. The novel anti-CD40 monoclonal antibody CFZ533 shows beneficial effects in patients with primary Sjögren's syndrome: a phase IIa double-blind, placebo-controlled randomized trial. Arthritis Rheumatol 2017; 69; Suppl. 10, 1784.28544596 [Google Scholar]
- 143.Behr J, Neuser P, Prasse A, et al. Exploring efficacy and safety of oral pirfenidone for progressive, non-IPF lung fibrosis (RELIEF) - a randomized, double-blind, placebo-controlled, parallel group, multi-center, phase II trial. BMC Pulm Med 2017; 17: 122. doi: 10.1186/s12890-017-0462-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Solomon JJ, Danoff SK, Goldberg HJ, et al. The design and rationale of the Trail1 trial: a randomized double-blind phase 2 clinical trial of pirfenidone in rheumatoid arthritis-associated interstitial lung disease. Adv Ther 2019; 36: 3279–3287. doi: 10.1007/s12325-019-01086-2 [DOI] [PubMed] [Google Scholar]
- 145.ClinicalTrials.gov . An Expanded Access Program to Provide Nintedanib to Patients with Non-IPF ILD Who Have no Alternative Treatment PossibilitiesDate last updated: 8 April 2020; date last accessed: 8 April 2020. https://clinicaltrials.gov/ct2/show/study/NCT03843892.
- 146.Saunders P, Tsipouri V, Keir GJ, et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials 2017; 18: 275. doi: 10.1186/s13063-017-2016-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.ClinicalTrials.gov . Scleroderma Lung Study III - Combining Pirfenidone With Mycophenolate (SLSIII). https://clinicaltrials.gov/ct2/show/NCT03221257 Date last updated: 21 February 2020; date last accessed: 7 December 2019. [Google Scholar]
- 148.ClinicalTrials.gov . Abatacept for the Treatment of Myositis-associated Interstitial Lung Disease (ATtackMy-ILD). www.clinicaltrials.gov/ct2/show/NCT03215927 Date last updated: 14 July 2020; date last accessed: 15 January 2020. [Google Scholar]
- 149.Shi JH, Liu HR, Xu WB, et al. Pulmonary manifestations of Sjögren's syndrome. Respiration 2009; 78: 377–386. doi: 10.1159/000214841 [DOI] [PubMed] [Google Scholar]
- 150.Gottenberg JE, Cinquetti G, Larroche C, et al. Efficacy of rituximab in systemic manifestations of primary Sjögren's syndrome: results in 78 patients of the autoimmune and rituximab registry. Ann Rheum Dis 2013; 72: 1026–1031. doi: 10.1136/annrheumdis-2012-202293 [DOI] [PubMed] [Google Scholar]
- 151.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. doi: 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Luppi F, Spagnolo P, Cerri S, et al. The big clinical trials in idiopathic pulmonary fibrosis. Curr Opin Pulm Med 2012; 18: 428–432. doi: 10.1097/MCP.0b013e3283567ff9 [DOI] [PubMed] [Google Scholar]
- 153.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. doi: 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 154.King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083–2092. doi: 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 155.Khanna D, Albera C, Fischer A, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J Rheumatol 2016; 43: 1672–1679. doi: 10.3899/jrheum.151322 [DOI] [PubMed] [Google Scholar]
- 156.Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med 2019; 380: 2518–2528. doi: 10.1056/NEJMoa1903076 [DOI] [PubMed] [Google Scholar]
- 157.Flaherty KR, Brown KK, Wells AU, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res 2017; 4: e000212. doi: 10.1136/bmjresp-2017-000212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019; 381: 1718–1727. doi: 10.1056/NEJMoa1908681 [DOI] [PubMed] [Google Scholar]
- 159.Ferrara G, Luppi F, Birring SS, et al. Best supportive care for idiopathic pulmonary fibrosis: current gaps and future directions. Eur Respir Rev 2018; 27: 170076. doi: 10.1183/16000617.0076-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hardinge M, Annandale J, Bourne S, et al. British Thoracic Society guidelines for home oxygen use in adults. Thorax 2015; 70: Suppl. 1, i1–43. doi: 10.1136/thoraxjnl-2015-206865 [DOI] [PubMed] [Google Scholar]
- 161.Visca D, Mori L, Tsipouri V, et al. Effect of ambulatory oxygen on quality of life for patients with fibrotic lung disease (AmbOx): a prospective, open-label, mixed-method, crossover randomised controlled trial. Lancet Respir Med 2018; 6: 759–770. doi: 10.1016/S2213-2600(18)30289-3 [DOI] [PubMed] [Google Scholar]
- 162.Mathai SC, Danoff SK. Management of interstitial lung disease associated with connective tissue disease. BMJ 2016; 352: h6819. doi: 10.1136/bmj.h6819 [DOI] [PMC free article] [PubMed] [Google Scholar]