

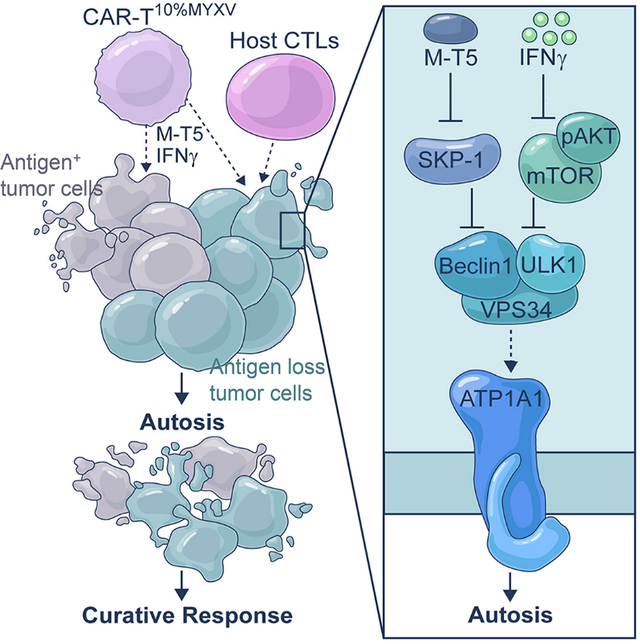
Summary
Cytotoxicity of tumor-specific T cells requires tumor cell-to-T cell contact-dependent induction of classic tumor cell apoptosis and pyroptosis. However, this may not trigger sufficient primary responses of solid tumors to adoptive cell therapy or prevent tumor antigen escape-mediated acquired resistance. Here, we test myxoma virus (MYXV)-infected tumor-specific T (TMYXV) cells, expressing chimeric-antigen-receptor (CAR) or T cell receptor (TCR), which systemically deliver MYXV into solid tumors to overcome primary resistance. In addition to T cell-induced apoptosis and pyroptosis, tumor eradication by CAR/TCR-TMYXV cells is also attributed to tumor cell autosis induction, a special type of cell death. Mechanistically, T cell-derived IFNγ-AKT signaling synergizes with MYXV-induced M-T5-SKP-1-VPS34 signaling to trigger robust tumor cell autosis. Moreover, CAR/TCR-TMYXV-elicited autosis functions as a potent bystander killing to restrain antigen escape. Collectively, we uncover an unexpected synergy between T cells and MYXV to bolster solid tumor cell autosis that reinforces tumor clearance.
eTOC Blurb:
Classic tumor cell apoptosis and pyroptosis induced by tumor-specific T cells may not trigger sufficient primary responses and prevent tumor antigen escape-mediated acquired resistance. Zheng et al. uncover that a special tumor cell death induced by CAR-TMYXV, autosis, which has not been attributed to any T cell killing mechanism before but can mediate a potent bystander killing for antigen-negative tumor cells.
Graphical Abstract
Introduction
Adoptively transferred T cells engineered to express chimeric antigen receptors (CAR) have shown some success in eliminating hematopoietic cancers, but have so far been limited in their efficacy against solid tumors, which account for most cancer deaths (June et al., 2018). Lack of primary response (initial tumor regression) is most likely multifactorial and includes limited homing to and penetration of tumors (Jin et al., 2016), T cell exhaustion, limited persistence, and an immunosuppressive tumor microenvironment (Jindal et al., 2018). Among those, interpatient and intratumoral heterogeneity of the tumor antigen may be one of the most serious impediments to efficacy against solid tumors and often lead to acquired resistance (recurrence after initial regression) (Hung T. Khong and Nicholas P. Restifo, 2002). Hence, it is urgent to develop novel strategies to overcome both primary and acquired resistance in solid tumors.
Oncolytic virotherapy is an emerging therapeutic modality for the treatment of cancer, but unfortunately, systemic delivery of oncolytic virus using standard intravenous infusion has thus far not achieved sufficient enrichment of virus in metastatic tumor beds (Power et al., 2007). Preclinical studies have also demonstrated the synergistic potential of combination oncolytic virotherapy with CAR-T adoptive cell therapy (ACT) for solid tumor treatment, in which local intratumor oncolytic virus injection may augment CAR-T cell trafficking into the virus-injected tumors, reshape local immunosuppression, and enhance CAR-T cell effector function (Ajina and Maher, 2017; Park et al., 2020). Although impressive responses can be obtained in the oncolytic virus-injected tumors, these approaches only induce limited or moderate responses to virus non-injectable metastatic tumors, which represents one major limitation for local virus administration (Hong and Yun, 2019; Park et al., 2020; Zheng et al., 2019).
One of the candidate oncolytic viruses is myxoma virus (MYXV), a DNA virus, which has a highly restricted host range and is only pathogenic to European rabbits (Stanford et al., 2007). MYXV is a promising, safe oncolytic virus (Rahman and McFadden, 2020) as it selectively infects and kills a wide variety of tumor cells while sparing normal cells and tissues (Sypula et al., 2004). Importantly, MYXV causes no overt disease in other host species tested, even in highly immunodeficient NSG mice (Kellish et al., 2019; Lun et al., 2010). Like other oncolytic viruses, intratumoral injection of MYXV tends to only eliminate the tumor mass in which the virus is administered (Lun et al., 2005).
Despite tremendous efforts, clinical trials of ACT have not produced desired therapeutic responses in solid tumors. This may be partly because classic tumor cell apoptosis and pyroptosis induced by tumor-specific T cells with the formation of an immunological synapse (Cazaux et al., 2019; Liu et al., 2020) are insufficient to trigger robust therapeutic responses in solid tumors. In this study, we investigated the potential to exploit CAR-T and TCR-T cells as MYXV-delivery carrier cells by pre-infecting the T cells with MYXV ex vivo by a spin-infection protocol (CAR-TMYXV and TCR-TMYXV). Tumor-specific CAR-TMYXV and TCR-TMYXV cells efficiently delivered MYXV into the cognate tumor cells, but not normal cells, in an antigen-specific manner. Unexpectedly, we observed a special tumor cell death induced by CAR-TMYXV, which has not been attributed to any T cell killing mechanism before but may contribute to the exciting observed antitumor potency. Differing from classic apoptosis and pyroptosis induced by T cells (Cazaux et al., 2019; Liu et al., 2020), this special type of cell death, called autosis, can also mediate a potent bystander killing for antigen-negative tumor cells. Therefore, we performed a series of studies to investigate the functions of these mechanisms.
Results
Efficient delivery of MYXV by tumor-specific T cells
Although MYXV is a promising and safe oncolytic virus that can selectively infect and kill various tumor cells (Sypula et al., 2004), it is unclear whether non-tumor cells, such as tumor-specific CAR-T cells, can be infected with MYXV. We prepared human mesothelin (MSLN) or CD19 CAR-T cells containing ~90% CAR+ cells (Figure 1A, S1A), and expanded them for 8 days. As seen in Figure 1B (top panel, Vehicle/No spin), directly adding tdTomato-expressing reporter MYXV at a multiplicity of infection (MOI) of 1:1, 3:1, 10:1 to CAR-T cells 8 days after CAR-T cell expansion, without spin infection, did not cause significant MYXV infection of T cells, even though T cells were exposed to MYXV for 48 hrs. To infect T cells with MYXV, we next adopted a related virus transduction protocol that is similar to the transduction of CAR-encoding γ-retrovirus to T cells in CAR-T cell preparation, which includes adding protamine and centrifuging for 2 hrs. Interestingly, the MYXV infection rate of CAR-T cells was bolstered by this alternative method to ~80% (Figure 1B, lower panel, Protamine/Spin). Although adding MYXV to CAR-T cells without spin infection had a limited effect on CAR-T cell survival (Figure 1C), MYXV-infected CAR-T (CAR-TMYXV, spin-infected) cells had relatively lower proliferation (Figure 1C) and gradually reduced viability (Figure 1D) about 7 days after MYXV infection, at which the majority of T cells were still MYXV positive (Figure S1B). These results indicate that a potential 7-day time frame exists for CAR-TMYXV cells to deliver MYXV into tumor cells.
Figure 1. Delivery of MYXV by CAR-T cells.
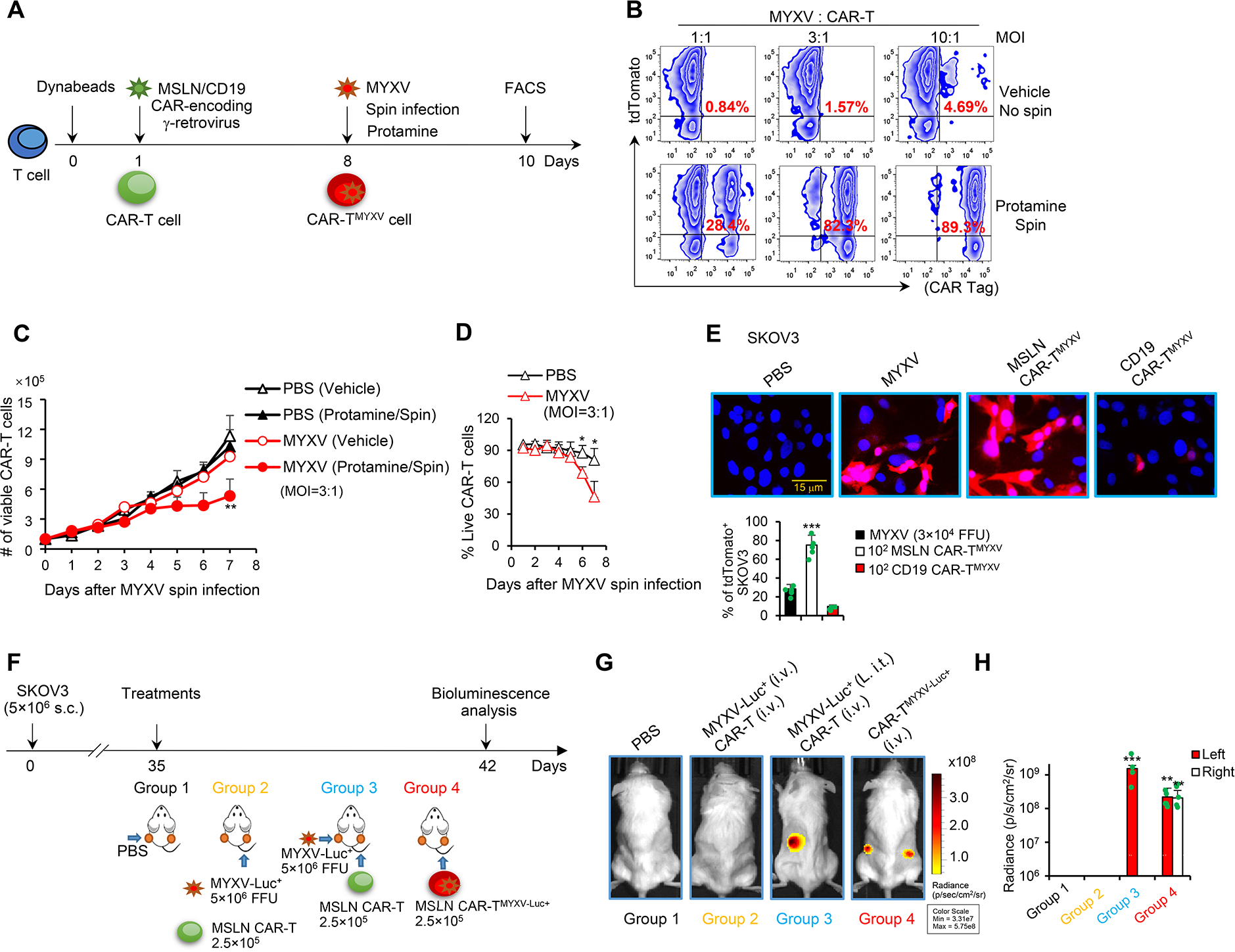
(A) Protocol to prepare CAR-TMYXV cells. On day 8, MYXV was added to CAR-T cells in the presence of 10 μg/ml protamine, and the culture was centrifuged at 1,800 rpm for 2 hrs for MYXV spin infection. (B) The infection rate was detected 48 hrs after adding MYXV by FACS. (C) The yield of viable uninfected (PBS) and CAR-TMYXV cells (n=3 donors/group). Data are mean ± SD. **P<0.01, MYXV (Protamine/Spin) compared with others, two-way ANOVA with posthoc Holm-Sidak test. (D) % of live CAR-TMYXV cells were measured by trypan blue exclusion test (n=3). Data are mean ± SD. *P<0.05, two-way ANOVA with posthoc Holm-Sidak test. (E) MYXV-tdTomato (3×104 FFUs), MSLN CAR-TMYXV-tdTomato (1×102), or CD19 CAR-TMYXV-tdTomato (1×102) was added to SKOV3 cells (1×104). Representative images and % of tdTomato+ SKOV3 cells are shown (24 hrs; n=3–5/group). Data are mean ± SD. ***P<0.001, MSLN CAR-TMYXV compared with any other groups, one-way ANOVA with Tukey test. (F-H) Treatment schema is shown (F). Indicated treatments were given when tumors reached ~7×6 mm on day 35. In groups 1 and 3, PBS and MYXV-Luc+, respectively, were i.t. injected only into tumors on the left flanks (L. i.t.), and tumors on the right flanks did not receive i.t. injection. In group 2, MYXV-Luc+ and MSLN CAR-T cells were i.v. injected. In groups 3 and 4, MSLN CAR-T and MSLN CAR-TMYXV-Luc+ cells were i.v. injected, respectively. Bioluminescence was measured on day 42. Representative images (G) and summarized data are shown (H) (n=5/group). Data are mean ± SD. ***P<0.001, group 3 (Left) compared with groups 1 & 2 (Left); **P<0.01, group 4 (Left) compared with groups 1 & 2 (Left); **P<0.01, group 4 (Right) compared with any other groups (Right), two-way ANOVA with posthoc Holm-Sidak test. Pooled results represent 2 independent experiments (E, H). See also Figure S1.
Next, we determined whether CAR-TMYXV cells can deliver MYXV into tumor cells and whether such a delivery requires CAR-T cell tumor specificity. For CD19−MSLN+ human SKOV3 ovarian cancer (OvCa) cells (Figure S1C), adding MSLN CAR-TMYXV but not CD19 CAR-TMYXV cells efficiently delivered MYXV-tdTomato into SKOV3 cells (Figure 1E). Similarly, CD19 CAR-TMYXV but not MSLN CAR-TMYXV cells delivered MYXV into CD19+MSLN− Raji lymphoma cells (Figure S1D). Interestingly, we detected MYXV replication in TMYXV cells (Figure S1E). We also observed that optimal MYXV release requires CAR activation by antigen-positive tumor cells/beads or anti-CD3/28 bead-mediated T cell activation, and using CD3Z-truncated CAR (CAR-TΔZ) did not result in MYXV release (Figure S1F and S1G). Notably, adding MYXV-tdTomato to tumor cells (MOI=3) turned fewer tumor cells into tdTomato+ cells as compared with adding CAR-TMYXV cells (CAR-TMYXV to tumor cell=1:100) (Figure 1E), suggesting an efficient virus delivery by CAR-TMYXV cells. In addition, we used a Transwell assay to exclude trogocytosis (Hamieh et al., 2019), which may result in the transfer of tdTomato reporter gene. We observed tdTomato+ SKOV3 cells in the lower chamber (Figure S1H), which may be because MSLN CAR-TMYXV but not CD19 CAR-TMYXV cells were reactivated by SKOV3 cells in the upper chamber and triggered the subsequent release and spread of MYXV to the lower chamber tumor cells. However, adding MYXV or CAR-TMYXV to MSLN CAR-T cells did not cause the infection of MSLN CAR-T cells, indicating that CAR-TMYXV cannot propagate MYXV to other T cells (Figure S1I). These data highlight an efficient delivery of CAR-TMYXV cells to target cancer cells in an antigen-dependent manner.
To determine whether CAR-TMYXV cell transfer can systemically deliver MYXV in vivo, we intravenously (i.v.) injected MSLN CAR-TMYXV-luciferase (Luc)+ cells into MSLN+ SKOV3-bearing NSG mice (Figure 1F). As expected, intratumorally (i.t.) but not i.v. injected MYXV-Luc+ is capable of infecting SKOV3 tumors (Figure 1G and 1H). Moreover, after injection of MYXV-Luc+ into tumors inoculated on one flank of the mice, MYXV did not spread to tumors on the other, non-injected flank (Figure 1G and 1H). Remarkably, i.v. transfer of MSLN CAR-TMYXV-Luc+ cells efficiently delivered MYXV into SKOV3 tumors inoculated on both flanks of mice, as determined by markedly increased bilateral bioluminescence and the positive bioluminescent signal of isolated tumor cells (Figure 1G and 1H, S1J). This delivery required tumor antigen-mediated CAR signaling because truncation of CD3Z in CAR structure or KO of MSLN in SKOV3 abrogated MYXV delivery (Figure S1K and S1L). Overall, our results demonstrate that CAR-TMYXV may be used as efficient carrier cells to systemically deliver MYXV into cognate antigen-expressing tumors.
MYXV-infected T cells overcome primary therapeutic resistance
Next, we investigated the antitumor potential of MYXV-infected MSLN CAR-T cells in in vitro cytotoxicity assays. We used a high ratio of tumor cells to CAR-T cells (10:1), at which CAR-T cells can only lyse a small proportion of tumor cells. Surprisingly, MSLN CAR-TMYXV cells did not display higher antitumor killing ability than uninfected MSLN CAR-T cells (Figure S2A). Then we tested whether MSLN CAR-TMYXV cells could enhance the baseline antitumor ability of MSLN CAR-T cells. MSLN CAR-T cells were mixed with MSLN CAR-TMYXV cells at various ratios and tested for their function in in vitro cytotoxicity assays. Intriguingly, the optimal tumor-killing activity of CAR-T cells occurred with the formulation of MSLN CAR-T10%MYXV (90% MSLN CAR-T cells+10% MSLN CAR-TMYXV cells) even with a high ratio of tumor cells to CAR-T cells (10:1) (Figure S2B). Thereafter, we selected T10%MYXV cells to comprehensively evaluate their antitumor function (Figure 2A and 2B). Extraordinary tumor-killing activities were also obtained when using CD19 CAR-T10%MYXV cells to target CD19+ Raji cells or using MART-1 T10%MYXV cells to target human MART-1+ Mel-264 melanoma (Figure 2B). This synergistic antitumor function seems to require CAR-TMYXV tumor specificity because CD19 CAR-TMYXV cells largely failed to bolster antitumor function of MSLN CAR-T cells in targeting SKOV3 cells (Figure S2C). However, MSLN CAR-TMYXV cells had a limited capacity to infect non-tumor human monocytes, human umbilical vein endothelial cells (HUVECs), epithelial (RWPE-1) cells, or stromal (HS-5) cells with MYXV, although these cells have been overexpressed MSLN (Figure S2D). Furthermore, compared with MSLN CAR-T cells, MSLN CAR-T10%MYXV cells did not display increased killing activity against these non-tumor cells (Figure S2E).
Figure 2. Tumor-specific T10%MYXV display strong antitumor efficacy.
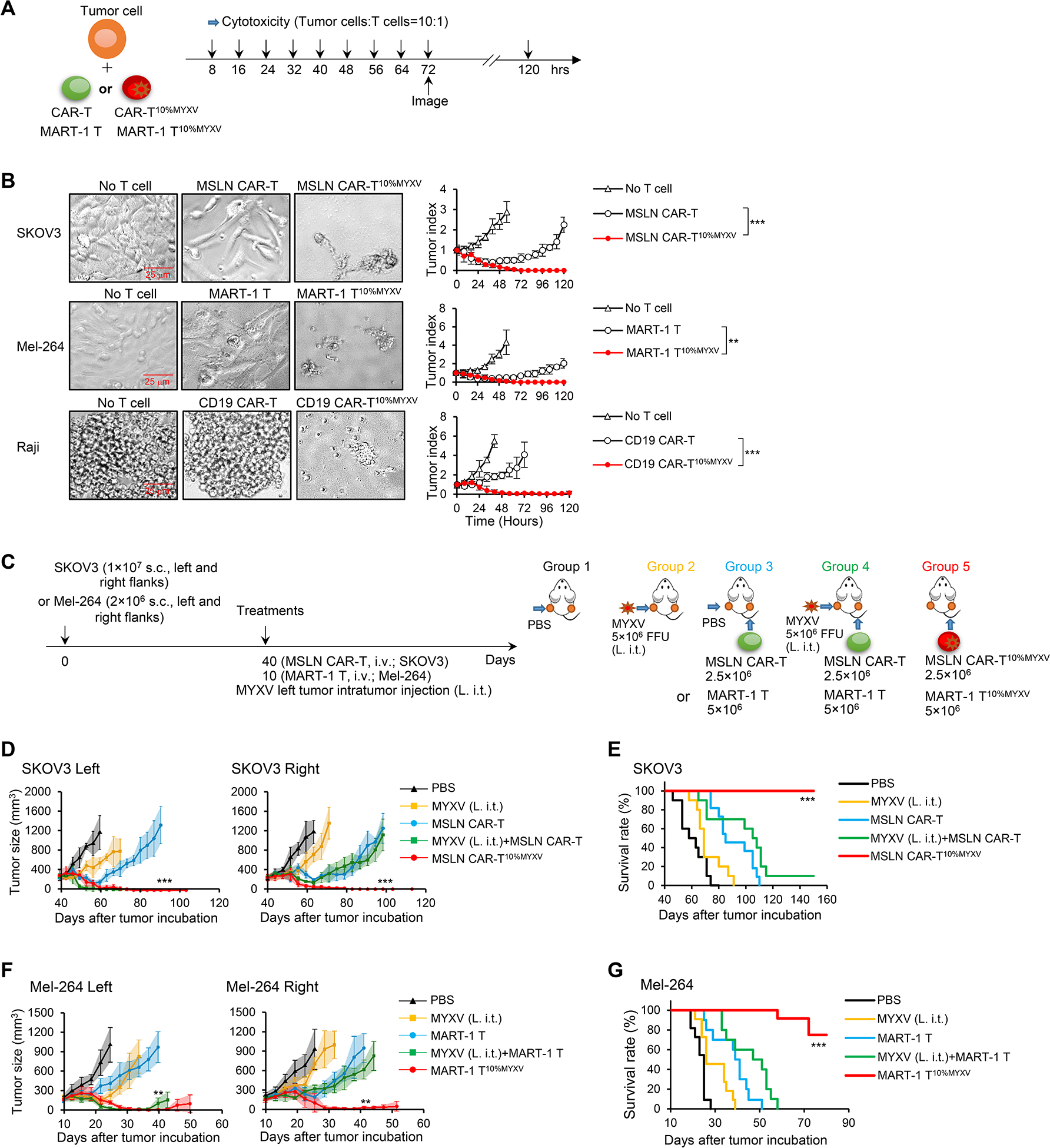
(A-B) Schema of in vitro cytolytic assays. (A) Indicated T cells (2×103) were cocultured with tumor cells (2×104). Cytotoxic assays were performed every 8 hrs. (B) Representative images (72 hrs) and summarized tumor-killing are shown (n=3/time point). Data are mean ± SD. ***P<0.001, SKOV3; **P<0.01, Mel-264; ***P<0.001, Raji, two-way ANOVA with posthoc Holm-Sidak test. (C-G) Treatment schema is shown (C). NSG mice bearing 40-day established SKOV3 tumors were treated when tumors reached ~9×8 mm on day 40 (C-E); NSG mice bearing 10-day established Mel-264 tumors were treated when tumors reached ~7×6 mm on day 10 (C, F-G). In groups 1, 2, 3 and 4, PBS or MYXV was i.t. injected only into tumors on the left flanks (L. i.t.), and tumors on the right flanks did not receive i.t. injection. In groups 3 and 4, MSLN CAR-T or MART-1 T cells were i.v. injected. In group 5, MART-1 T10%MYXV or MSLN CAR-T10%MYXV cells were i.v. injected. Tumor responses to ACT with MSLN CAR-T10%MYXV (D) and MART-1 T10%MYXV cells (F) are shown (n=5/group). Data are mean ± SD. ***P<0.001, MSLN CAR-T10%MYXV compared with any other groups (D); **P<0.01, MART-1 T10%MYXV compared with any other groups (F), two-way ANOVA with posthoc Holm-Sidak test. (E, G) Survival curves from 2 independent studies are summarized (n=10–12/group). ***P<0.001, compared with any other groups, survival analysis was conducted by log-rank test. See also Figure S2.
To test whether tumor-specific T10%MYXV cells can also eliminate established tumors in vivo, we injected MSLN CAR-T10%MYXV cells i.v. into MSLN+ SKOV3-bearing NSG mice (Figure 2C–2E). As a control, we i.t injected MYXV into tumors inoculated on the left flank of mice in combination with i.v. MSLN CAR-T ACT. I.t. injection of MYXV moderately inhibited tumor growth compared with PBS group, followed by aggressive recurrence (Figure 2D). Combination therapy with i.t injection of MYXV and i.v. MSLN CAR-T ACT largely improved responsiveness of MYXV-injected left tumors, but had no improved effect for right flank tumors without MYXV injection (Figure 2D), which is in line with the MYXV distribution (Figure 1H). Similarly, combination therapy with i.v. injection of MYXV and MSLN CAR-T ACT had no improved effect compared with MSLN CAR-T ACT (Figure S2F). Notably, optimal response was achieved with i.v. MSLN CAR-T10%MYXV cells and resulted in long-term survival (Figure 2D and 2E, S2G), which was associated with significantly increased CAR-T cell infiltration (Figure S2H), presence of CAR-TMYXV (Figure S2I), and MYXV replication (Figure S2J) in the tumor. However, ACT with the formulation of 90% MSLN CAR-T+10% CD19 CAR-TMYXV cells, or 90% CD19 CAR-T+10% CD19 CAR-TMYXV cells, had limited antitumor efficacy compared with MSLN CAR-T10%MYXV cells (Figure S2K). Robust antitumor efficacy was also observed with MART-1 T10%MYXV ACT for Mel-264 melanoma-bearing NSG mice (Figure 2F and 2G), which was accompanied by increased MART-1 T cell infiltration (Figure S2L) and MYXV replication (Figure S2M). Finally, CD19 CAR-T10%MYXV ACT displayed enhanced antitumor efficacy in Raji-bearing NSG mice (Figure S2N). Our results thus highlight that antigen specificities of both T and TMYXV cells are crucial for the optimal antitumor efficacy of tumor specific T10%MYXV cells.
CAR-T10%MYXV-induced tumor cell autosis contributes to tumor elimination
Elimination of tumor cells by T cells is reported to depend predominantly on the induction of tumor cell apoptosis and pyroptosis (Cazaux et al., 2019; Liu et al., 2020). In an in vitro killing assay, SKOV3 cells mainly underwent apoptosis and pyroptosis when cocultured with MSLN CAR-T cells (Figure 3A). However, an additional and different type of tumor cell death was observed during the robust killing activity induced by MSLN CAR-T10%MYXV cells, which stands in sharp contrast to what is typically observed with classic apoptosis and pyroptosis (Figure 3A). This type of tumor cell death has not been attributed to any known T cell-killing pathway before, and appears to be associated with a different morphologic feature that shows firm attachment to culture dishes in vitro (Figure 3A). Further, this cell death response can also function as bystander killing, since tumor cells separated from the main coculture system by Transwell were not exempt (Figure 3B, S3A). We also observed this type of tumor cell death when treated with MYXV alone, although it seemed to occur at a lower incidence (Figure S3B and S3C). Therefore, inclusion of MYXV-infected T cells in tumor-specific T cells promotes robust tumor clearance, which is associated with an additional type of cell death different from canonical apoptosis or pyroptosis.
Figure 3. CAR-T10%MYXV cells induce tumor cell autosis.
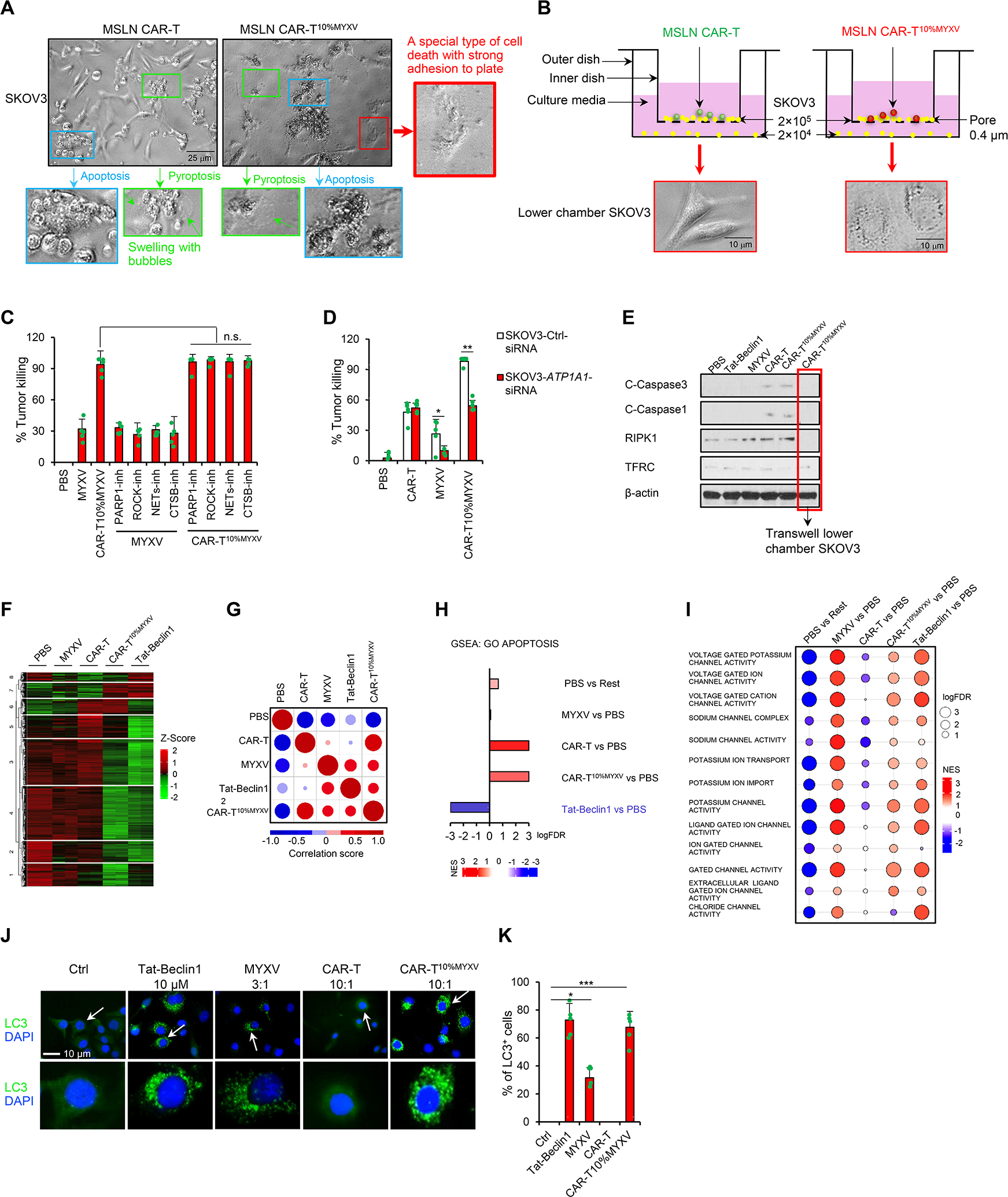
(A) MSLN CAR-T or MSLN CAR-T10%MYXV cells (2×103) were cocultured with SKOV3 cells (2×104). Various cell death types after 72 hrs coculture are shown (indicated with arrows), including apoptosis (blue), pyroptosis (green, swelling with bubbles) and special cell death (autosis) with a strong attachment to culture plate (red). (B) SKOV3 cells were seeded into the upper (2×105) and lower (2×104) Transwell chambers. MSLN CAR-T or MSLN CAR-T10%MYXV cells (2×104) were only added into upper chambers. Representative images for morphology of SKOV3 cells in the lower chamber are shown (72 hrs). (C) SKOV3 cells were pretreated with BYK204165 (parthanatos; PARP-1 inhibitor, 5 μM), Y-27632 (entotic cell death; ROCK inhibitor, 1 μM), Lactoferrin (netotic cell death; NETs inhibitor, 2.8 μM) or CA-074Me (lysosome-dependent cell death; CTSB inhibitor, 5 nM) for 24 hrs. MYXV (6×104 FFUs) or MSLN CAR-T10%MYXV cells (2×103) were then added to pretreated SKOV3 cells. % of killing was determined by an in vitro cytotoxicity assay (72 hrs, n=5). n.s. (not significant), one-way ANOVA with Tukey test. (D) Control (Ctrl) or ATP1A1 siRNA was transfected into SKOV3 cells 72 hrs before further treatments. MYXV (6×104 FFUs), MSLN CAR-T, or MSLN CAR-T10%MYXV cells (2×103) were seeded to SKOV3 cells. % of killing was determined by an in vitro cytotoxicity assay (72 hrs, n=5). Data are mean ± SD. *P<0.05, **P<0.01, two-way ANOVA with posthoc Holm-Sidak test. (E-I) MYXV (3×106 FFUs), MSLN CAR-T, or MSLN CAR-T10%MYXV cells (1×105) were cocultured with SKOV3 cells (1×106) for 24 hrs. Tat-Beclin1 was resuspended (10 μM) and coincubated with SKOV3 cells for 1.5 hrs. (E) Lower chamber SKOV3 cells were treated as shown in Figure 3B for 24 hrs. SKOV3 cell lysates (after T cell removal) or lower chamber SKOV3 cells were analyzed for protein expression levels by Western blot. (F-I) Total RNA was extracted from the SKOV3 cells for RNA-seq (n=2). (F) Hierarchical clustering of gene expression in SKOV3 cells. (G) Correlation matrix of Pearson correlation values (PCV) calculated pairwise between all groups (top ~250 most changed GO term pathways). Correlation plot, the size of circle shows the absolute value of corresponding correlation coefficients, and the color represents the correlation coefficients (ranging from −1 to 1). (H) GSEA of GO term apoptosis. (I) Dotplot heatmap of enriched ion channel-related signaling analyzed by GSEA. Balloon plot where the points size reflects the log-FDR and the color represent the NES value. (J-K) Representative images of LC3 staining in SKOV3 cells treated with Tat-Beclin1 (10 μM), MYXV (6×104 FFUs), MSLN CAR-T, or MSLN CAR-T10%MYXV cells (2×103) for 24 hrs. Arrows indicate representative LC3+ cells (J) and summarized data are shown (K, n=5). Data are mean ± SD. *P<0.05, ***P<0.001, one-way ANOVA with Tukey test. Pooled results represent 2 independent experiments. See also Figure S3.
To identify the potential death pathway(s) responsible for this unexpected type of cell death, we did a comprehensive screening of most known types of cell death (Tang et al., 2019). We found that PARP-1 inhibitor (parthanatos) (Andrabi et al., 2008), ROCK inhibitor (entotic cell death) (Sun et al., 2014), NETs inhibitor (netotic cell death) (Okubo et al., 2016), or CTSB inhibitor (lysosome-dependent cell death) (Aits and Jäättelä, 2013) did not inhibit MSLN CAR-T10%MYXV cell-killing activity (Figure 3C), whereas knockdown (KD) of the Na+, K+-ATPase α1 subunit (ATP1A1) required for autosis (Liu et al., 2013), blocked CAR-T10%MYXV but not CAR-T cell-killing activity (Figure 3D, S3D and S3E). Western blot analysis showed that expression levels of cleaved-caspase3 (apoptosis), cleaved-caspase1 (pyroptosis) (Miao et al., 2011), RIPK1 (necroptosis) (N. et al., 2000), and TFRC (ferroptosis) (Wu et al., 2019) in tumor cells did not increase after CAR-T10%MYXV cell treatment compared with CAR-T cells, further demonstrating that this special cell death does not belong to apoptosis, pyroptosis, necroptosis, or ferroptosis (Figure 3E). In line with other reports (Bartee et al., 2016), an exception for this effect is the induction of activated caspase 8-dependent apoptosis in human myeloma cells (Figure S3F and S3G). The above analyses strongly suggest that CAR-T10%MYXV cells trigger autosis for tumor clearance.
To further verify that this therapy induced autosis, we included Tat-Beclin1 peptide, an established method for autosis induction (Liu et al., 2013) that can also be blocked by KD of ATP1A1 in tumor cells (Figure S3H and S3I). We performed RNA-seq and cluster analyses which indicated that CAR-T10%MYXV cells and Tat-Beclin1-treated SKOV3 cells had relatively similar gene profiles (Figure 3F). In addition, CAR-T10%MYXV-treated tumor cells showed largely overlapping gene signatures with CAR-T cell treatment, including an apoptosis signature (Figure 3G and 3H). Interestingly, tumor cells treated with MYXV, CAR-T10%MYXV, and Tat-Beclin1 displayed ~30% shared gene signatures within the top ~200 enriched gene sets, which were distinct from those of cells treated with PBS and CAR-T cells (Figure 3G). Two prominent cellular changes associated with autosis, ‘ion channel activity’ and ‘ion transport’ signatures (Liu and Levine, 2015), were also only enriched in MYXV, CAR-T10%MYXV, and Tat-Beclin1 treatments (Figure 3I). Finally, consistent with the fact that LC3 puncta formation is essential for autosis (Liu et al., 2013), we detected largely increased LC3 puncta formation in tumor cells treated with MYXV, CAR-T10%MYXV, and Tat-Beclin1 (Figure 3J and 3K, S3J). Importantly, CAR-TMYXV had limited effects on baseline CAR-T killing activity (Figure S3K), survival (Figure S3L and S3M), T cell features (Figure S3N), and vice versa (Figure S3K–S3N). Thus, we have assembled multiple lines of evidence to support a potential mechanism underlying CAR-T10%MYXV cell-mediated robust killing activity, representing a mix of classic CAR-T cell-induced apoptosis and pyroptosis with an additional form cell-killing, autosis, to further reinforce antitumor immune responses and tumor clearance.
T cell-derived IFNγ synergizes with MYXV-derived M-T5-SKP-1-VPS34 signaling to induce tumor cell autosis
Autosis is a non-apoptotic and non-necrotic form of cell death initiated by excessive accumulation of autophagosomes and due to activation of the Na+/K+-ATPase pump, changes in membrane osmolarity, and ion transport (Liu and Levine, 2015). Also different from classic autophagic cell death, cell death in autosis has a distinct morphology and does not require autophagy flux or autolysosomal degradation (Liu and Levine, 2015). To investigate the potential pathway responsible for CAR-T10%MYXV cell promoted-autosis, we first focused on the MYXV-derived factor(s) that may contribute to autophagosome formation. Our published results demonstrated that MYXV-encoded M-T5 repeat protein interacts directly with SKP-1 in MYXV-infected cells (Werden et al., 2009). Since SKP-1 reportedly forms SCF (SKP-1-CUL1-F-BOX) to ubiquitinate and degrade vacuolar protein-sorting 34 (VPS34) (Xiao et al., 2015), a protein that is crucial for formation of the autophagosome (Kihara et al., 2001), we performed a series of analyses to uncover the potential role of MYXV-derived M-T5-SKP-1-VPS34 signaling. Indeed, the expression levels of VPS34 and ATP1A1 were substantially increased in SKOV3 cells treated with MYXV or CAR-T10%MYXV (Figure 4A). Selective inhibition of VPS34 by PIK-III (Figure S4A) or KD of ATP1A1 (Figure S4B) reduced the antitumor function of CAR-T10%MYXV but not CAR-T cells. Moreover, overexpression of SKP-1 in SKOV3 cells significantly reduced expression levels of VPS34 and ATP1A1 in SKOV3 cells treated with MYXV or CAR-T10%MYXV (Figure 4B), and decreased in vitro cytolytic activity of MYXV and CAR-T10%MYXV but not CAR-T cells (Figure 4C). As a confirmation, tumors established with SKP-1-overexpressing SKOV3 cells were more resistant to CAR-T10%MYXV ACT than SKOV3 cells, but they displayed similar sensitivity to CAR-T ACT (Figure 4D). These results indicate that tumor cell autosis induced by CAR-T10%MYXV may be triggered, at least in part, by MYXV-M-T5-mediated disruption of SKP-1 to upregulate VSP34 expression/functions.
Figure 4. T cell-derived IFNγ synergizes with MYXV-derived SKP-1-VPS34 signaling to induce tumor cell autosis.
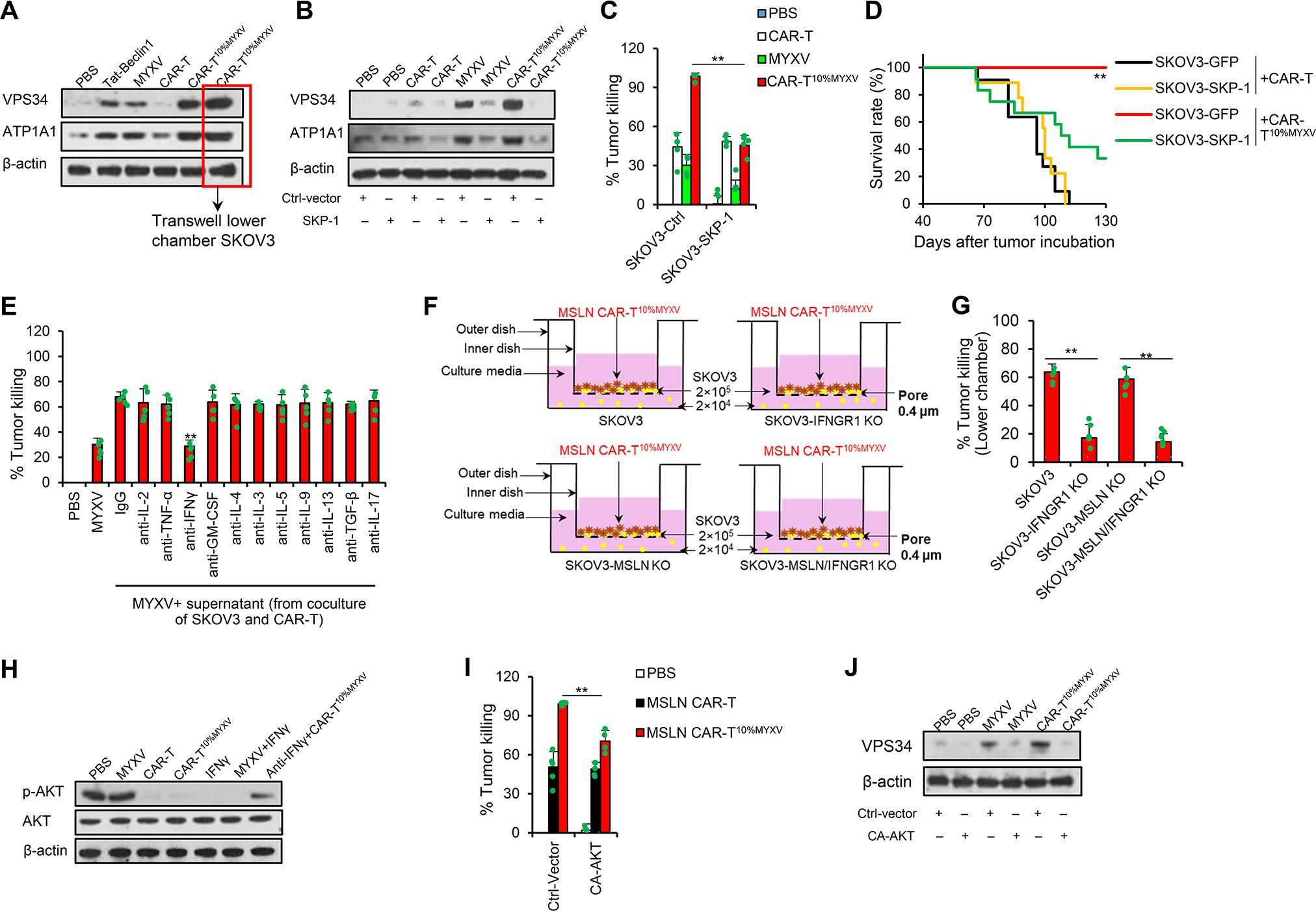
(A) SKOV3 cells were treated as shown in Figure 3E. SKOV3 cell lysates were analyzed by Western blots. (B-D) SKOV3 cells were transduced with Ctrl or human SKP-1 expression plasmids. (B) SKOV3 cell lysates (24 hrs) were analyzed by Western blots. (C) % of killing was determined by an in vitro cytotoxicity assay (72 hrs, n=5/group). Data are mean ± SD. **P<0.01, two-way ANOVA with posthoc Holm-Sidak test. (D) MSLN CAR-T or MSLN CAR-T10%MYXV cells (2.5×106) were transferred i.v. into NSG mice bearing SKOV3 tumors when tumors reached ~9×8 mm (1×107 SKOV3-GFP cells or SKOV3-SKP-1 cells challenged s.c. 40 days before ACT). Survival curves from two independent studies are summarized (n=9–12/group). **P<0.01 compared with any other groups, survival analysis was conducted by log-rank test. (E) In vitro cytotoxicity assay (72 hrs, n=5/group), SKOV3 cells (2×104) were treated with MYXV (6×104 FFUs) plus supernatant from SKOV3 and MSLN CAR-T cell coculture, and 10 μg/ml mAbs (n=5/group). Data are mean ± SD. **P<0.01, anti-IFNγ compared with IgG, one-way ANOVA with Tukey test. (F-G) SKOV3 cells (2×105) were seeded into the upper chambers. SKOV3 or gene-modified SKOV3 cells (2×104) were seeded into lower chambers. MSLN CAR-T10%MYXV cells (2×104) were only added to upper chambers. % of killing was determined by an in vitro cytotoxicity assay (72 hrs, n=5). Data are mean ± SD. **P<0.01, one-way ANOVA with Tukey test. (H) Western blot analysis of SKOV3 cells with indicated treatments after T cell removal. (24 hrs, 10 ng/ml hIFNγ, 10 μg/ml anti-hIFNγ mAb). (I-J) Ctrl vector or CA-AKT vector was transduced into SKOV3 cells. (I) Indicated treatment of MSLN CAR-T or MSLN CAR-T10%MYXV cells (2×103) was given for 24 hrs after transduction. % of killing was determined by an in vitro cytotoxicity assays (72 hrs; n=5/group). Data are mean ± SD. **P<0.01, two-way ANOVA with posthoc Holm-Sidak test. (J) MYXV (6×104 FFUs) or MSLN CAR-T10%MYXV (2×103) cells were given for 24 hrs after transduction. Western blot analysis of SKOV3 cells with indicated treatments after T cell removal. Representative results from one of two (E-G, I) repeated experiments are shown. See also Figure S4.
Because CAR-T10%MYXV cells seem to induce tumor cell autosis more potently than infection with MYXV (Figure S3B), we further hypothesized that T cell-derived soluble factor(s) may also contribute to autosis induction. We treated SKOV3 cells with MYXV and supernatant from SKOV3 and CAR-T cell coculture and observed significantly bolstered tumor cell death (Figure 4E). IFNγ in the supernatant seems crucial for this effect, as neutralization of IFNγ but not other cytokines nullified enhanced antitumor activity (Figure 4E, S4C and S4D), and reciprocally, MYXV antitumor function was enhanced by IFNγ (Figure S4E and S4F). However, inhibition of VPS34 by PIK-III (Figure S4E) or KD of ATP1A1 (Figure S4F) abrogated this effect. These results may also explain the insufficient antitumor activity of 100% MSLN CAR-TMYXV cells because they produced a low amount of IFNγ when cocultured with tumor cells (Figure S4G).
Furthermore, IFNγ and MYXV are indispensable for CAR-T10%MYXV cell-mediated bystander killing, since KO of IFNGR1 in SKOV3 cells or removal of MYXV by 0.22-μm filtration largely abrogated tumor eradication in an in vitro killing assay and in tumor-bearing mice (Figure 4F and 4G, S4H and S4I). Because the mTOR-AKT pathway suppresses autophagosome formation, but can be inhibited by IFNγ (Su et al., 2015; Wang et al., 2012), we tested whether AKT can be inhibited by CAR-T10%MYXV cells. Indeed, treatment with IFNγ or IFNγ-producing CAR-T or CAR-T10%MYXV cells markedly reduced p-AKT levels in tumor cells (Figure 4H). Overexpression of constitutively activate AKT (CA-AKT) largely negated the antitumor activity of CAR-T10%MYXV but not CAR-T cells (Figure 4I). Moreover, the expression levels of VPS34 were substantially reduced in CA-AKT-overexpressing SKOV3 cells treated with CAR-T10%MYXV (Figure 4J). Taken together, our results highlight a synergy between MYXV and CAR-T cells to induce tumor autosis death by inhibiting the role of SKP-1 and AKT signaling, respectively, while the elimination of tumor cells remains dependent on T cell tumor specificity.
Murine tumor-specific T10%MYXV ACT overcomes acquired resistance in solid tumors
Antigen loss variant (ALV) escape is likely to emerge as a major barrier to durability of CAR-T cell therapy in solid tumors (Majzner and Mackall, 2018). Tumor-specific T10%MYXV ACT may promote bystander killing of tumor cells with ALV to overcome this obstacle by inducing host antitumor immune activation and autosis via MYXV delivery into tumor beds. To test this hypothesis, we employed syngeneic tumor models in immunocompetent C57BL/6 (B6) mice. We used a mixture of 80% antigen-positive and 20% antigen-negative tumor cells to establish solid tumors with increased tumor antigen heterogeneity. We used murine MSLN CAR-T cells to target human MSLN-overexpressed murine ID-8 (ID-8hMSLN) OvCa, and created ID-880%hMSLN ‘chimeric tumor’ cells containing 20% of MSLN− ID-8 cells serving as ALV cells. ACT with murine MSLN CAR-T10%MYXV cells also eliminated tumors inoculated on both flanks of mice and resulted in long-term survival, whereas relapse due to ALV cell outgrowth seemed inevitable after MSLN CAR-T ACT (Figure 5A–5C, S5A and S5B). However, such antitumor efficacy was slightly reduced when targeting tumors made of 50% ALV cells (Figure S5C) and no antitumor function was detected if tumors were made of 100% ALV cells (Figure S5D).
Figure 5. Murine tumor-specific T10%MYXV ACT eradicates solid tumors with antigen heterogeneity.
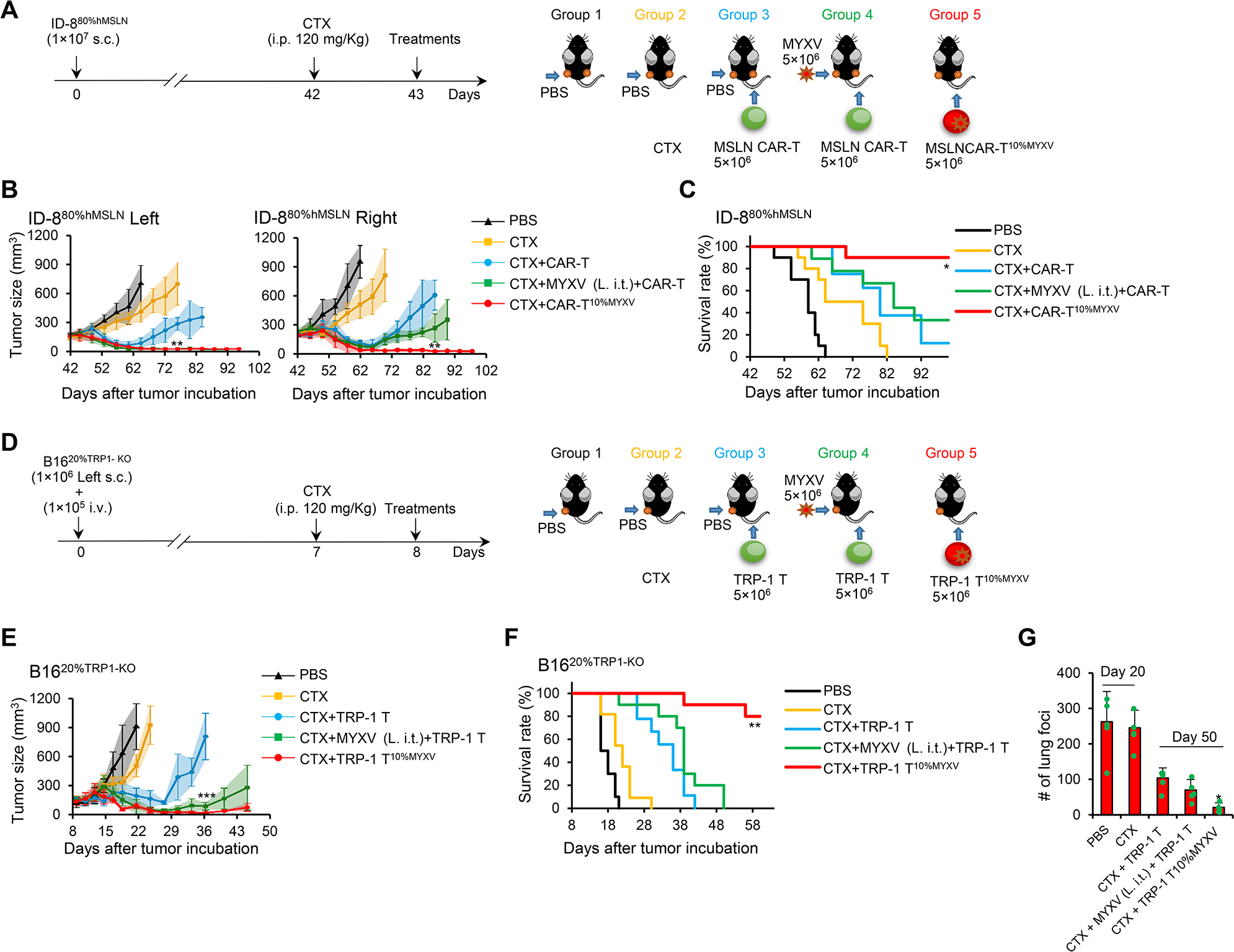
(A-C) Indicated treatments were given to B6 mice bearing s.c. established ID-880%hMSLN tumors on day 43 when tumors reached ~9×7 mm. (A) Treatment schema is shown. In groups 1, 2 and 3, PBS was i.t. injected only into tumors on the left flank (L. i.t.). In group 4, MYXV was i.t. injected only into tumors on the left flank (L. i.t.). In groups 3 and 4, MSLN CAR-T cells were i.v. injected. In group 5, MSLN CAR-T10%MYXV cells were i.v. injected. (B) Tumor responses are shown (n=5/group). Data are mean ± SD. **P<0.01, MSLN CAR-T10%MYXV compared with any other groups, two-way ANOVA with posthoc Holm-Sidak test. (C) Survival curves from two independent studies are summarized (n=8–10/group). *P<0.05, compared with any other groups, survival analysis was conducted by log-rank test. (D-G) B6 mice were inoculated s.c. with 1×106 B1620%TRP−1-KO cells only on the left flank and injected i.v. with 1×105 B1620%TRP−1-KO cells to induce lung metastatic tumors. Treatments were administrated on day 8 when s.c. tumors reached ~7×6 mm. (D) Treatment schema is shown. In groups 1, 2, and 3, PBS was i.t. injected only into tumors on the left flank (L. i.t.). In group 4, MYXV was i.t. injected only into tumors on the left flank (L. i.t.). In groups 3 and 4, TRP-1 T cells were i.v. injected. In group 5, TRP-1 T10%MYXV cells were i.v. injected. (E) Tumor responses are shown (n=5/group). Data are mean ± SD. ***P<0.001, TRP-1 T10%MYXV compared with TRP-1 T group, two-way ANOVA with posthoc Holm-Sidak test. (F) Survival curves from two independent studies are summarized (n=9–11/group). **P<0.01, compared with any other groups, survival analysis was conducted by log-rank test. (G) The counts of lung foci (n=5/group). Data are mean ± SD. *P<0.05, TRP-1 T10%MYXV compared with any other groups (Day 50), one-way ANOVA with Tukey test. See also Figure S5.
To strengthen our findings, we also used a tumor model of TRP-1 T cells targeting murine B16 melanoma established with subcutaneous (s.c.) and i.v. injection of B1620%TRP−1-KO tumor cells (80% of B16+20% TRP-1 KO B16 tumor cells) (Figure 5D). TRP-1 T10%MYXV treatment was highly effective against both s.c. (Figure 5E and 5F) and lung metastatic (Figure 5G) tumors in mice compared with TRP-1 T cell ACT. Remarkably, TRP-1 T10%MYXV ACT also extended protection to mice upon tumor-specific rechallenge (Figure S5E). Relapsed tumors following TRP-1 T cell ACT appeared to experience ALV cell outgrowth although tumors composed of exclusively B16 or TRP-1-KO B16 cells had similar growth rates in vivo (Figure S5F and S5G). Collectively, our results pinpoint a pivotal role of CAR/TCR-T10%MYXV ACT that allows the clearance of solid tumors containing ALV cells.
T10%MYXV ACT induces autosis and adaptive immunity that restrains ALVs
To determine the underlying mechanism within tumor-specific T10%MYXV cells that eliminates solid tumors with antigen heterogeneity (Figure S6A), we analyzed global RNA expression of treated B1620%TRP−1-KO tumors using RNA-seq (Figure 6A). Clustering analysis indicated that TRP-1 T10%MYXV ACT resulted in a relatively similar gene profile as MYXV (i.t.) combined with TRP-1 T cell ACT [‘MYXV (L. i.t.)’], whereas these two groups were completely different from the ‘remote tumor (no MYXV)’ and control (CTX) treatment (Figure 6B). Specifically, GSEA revealed that tumors treated with TRP-1 T10%MYXV or ‘MYXV (L. i.t.)’ were highly enriched for ‘inflammatory response’ and ‘adaptive immune response’ signatures (Figure 6C and 6D). Mean differential expression analyses further revealed many highly and significantly expressed cytokine and chemokine-related genes and genes suggesting T cell activation in TRP-1 T10%MYXV-treated tumors versus CTX control (Figure 6E, S6B).
Figure 6. ACT with T10%MYXV induces a robust adaptive immunity to restrain ALVs.
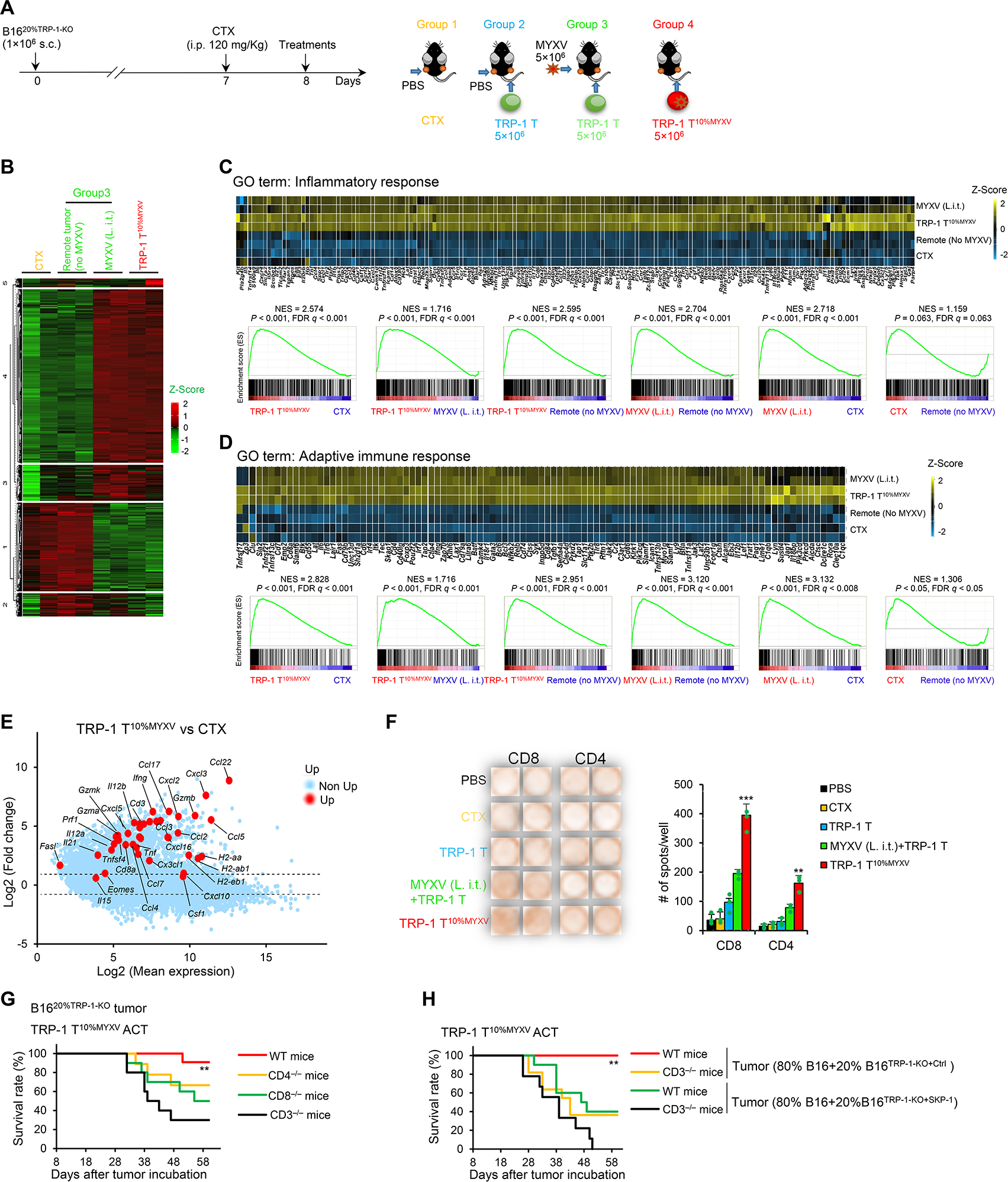
(A-F) B1620%TRP−1-KO cells were s.c. injected into both flanks of the mice. Treatments were administrated on day 8 when tumors reached ~7×6 mm. (A) Treatment schema is shown. In groups 1 and 2, PBS was i.t. injected only into tumors on the left flank (L. i.t.). In group 3, MYXV was i.t. injected only into tumors on the left flank (L. i.t.). In groups 2 and 3, TRP-1 T cells were i.v. injected. In group 4, TRP-1 T10%MYXV cells were i.v. injected. (B-D) Tumor tissues (~100 mg/mice, 12 days after ACT) were harvested. Total RNA was extracted from the tumor tissues for RNA-seq (n=2). (B) Hierarchical clustering of expression levels of 25,240 genes. Heatmap of enriched genes of GO terms ‘inflammatory response’ (C) and ‘adaptive immune response’ (D) and GSEA between indicated treatments are shown. (E) Log fold-change ratio versus mean average expression plot of differentially expressed genes suggesting antitumor immunity in tumors after TRP-110%MYXV ACT or CTX treatment. (F) IFNγ ELISpot analysis of tumor-infiltrating CD4+ and CD8+ host T cells restimulated with irradiated B16TRP−1-KO ALVs. Representative images and statistical results are shown (n=5/group). Data are mean ± SD. ***P<0.001, TRP-1 T10%MYXV compared with any other groups (CD8+ T cells); **P<0.01, TRP-1 T10%MYXV compared with any other groups (CD4+ T cells), one-way ANOVA with Tukey test. Representative results from two repeated experiments are shown. (G) TRP-1 T10%MYXV cells (5×106) were transferred i.v. into WT, CD4−/−, CD8−/− and CD3−/− B6 mice when B1620%TRP−1-KO tumors reached ~7×6 mm (similar to Figure 6A). Survival curves from 2 independent studies are summarized (n=9–11/group). **P<0.01, compared with any other groups, survival analysis was conducted by log-rank test. (H) WT and CD3−/− B6 mice were s.c. inoculated with 1×106 (80% B16+20% B16TRP−1-KO+Ctrl) tumor cells. Some groups of mice were s.c. inoculated with 1×106 (80% B16+20% B16TRP−1-KO+SKP−1) tumor cells. TRP-1 T10%MYXV cells (5×106) were transferred i.v. into mice when tumors reached ~7×6 mm (similar to Figure 6A). Survival curves from two independent studies are summarized (n=9–11/group). **P<0.01, compared with any other groups, survival analysis was conducted by log-rank test. See also Figure S6.
Given that MYXV can be delivered into tumors, we defined host antitumor immunity by IFNγ ELISpot analysis of tumor-infiltrating CD4+ or CD8+ host T cells (CD45.1+) isolated and restimulated with irradiated B16TRP−1-KO tumor cells ex vivo. Our results demonstrated that TRP-1 T10%MYXV ACT induced strong host CD4+ and CD8+ T cell activation (Figure 6F). Although host T cells do not seem to contribute significantly to the efficacy of TRP-1 T cell ACT targeting B1620%TRP−1-KO tumors (Figure S6D) or TRP-1 T10%MYXV ACT targeting B16 tumors (Figure S6E), deficiency of host CD4+, CD8+, or CD3+ T cells significantly reduced the antitumor effect of TRP-1 T10%MYXV ACT targeting B1620%TRP−1-KO tumors (Figure 6G). Finally, autosis induction in the tumor bed may also contribute to anti-ALV activity of TRP-1 T10%MYXV ACT, because survival was significantly reduced in mice bearing chimeric tumors containing SKP-1-overexpressing autosis-resistant TRP-1-KO B16 tumor cells (Figure 6H). Taken together, our results demonstrate that both TRP-1 T10%MYXV ACT-promoted host T cell activation and tumor cell autosis may be required to control the clonal expansion of ALV cells.
Discussion
In this study, we developed a special strategy for systemically delivering oncolytic MYXV into solid tumors by transferring tumor-specific T10%MYXV cells. Remarkably, eradication of solid tumors by tumor specific T10%MYXV cells was associated with induction of tumor cell autosis, which is unlike the classic cytolytic machinery of T cell cytotoxicity and causes a potent bystander killing of tumor cells. Therefore, our study uncovers an unexpected, T cell-killing mechanism of tumor cells by T10%MYXV, which could catalyze a paradigm shift in ACT to overcome therapeutic resistance in solid tumors.
MYXV can selectively infect and kill a broad variety of non-rabbit cancerous cells while sparing the normal cell and tissue counterparts (Sypula et al., 2004). We found that directly adding MYXV did not efficiently infect 8-day cultured CAR-T cells in vitro. However, successful transduction of MYXV to CAR-T cells ex vivo was achieved by using a modified protamine spin infection protocol similar to CAR-encoding γ-retrovirus transfection to T cells. Thus, we demonstrate that CAR-T cells can be efficiently infected with MYXV under such non-physiological conditions, which turns them into MYXV carriers. Interestingly, CAR-TMYXV cells had only moderate antitumor function, similar to non-infected CAR-T cells. Infection with MYXV may have somehow reduced the effector functions of CAR-TMYXV cells, because we observed decreased IFNγ production when cocultured with tumor cells. We hypothesized that CAR-TMYXV plus non-infected CAR-T cells might achieve dual-functional effects, maintaining full cytotoxicity of CAR-T cells and augmenting with MYXV delivery into the tumor beds. In fact, a formulation of CAR-T cells containing 10% CAR-TMYXV displayed remarkably potent antitumor functions.
The cytolytic function of T cells mainly induces apoptosis and pyroptosis of cancer cells by recruiting cytolytic machinery to the T cell/cancer cell synapse and the subsequent release of lytic granules containing perforin and granzymes (Jenkins and Griffiths, 2010; Liu et al., 2020). In our study, induction of tumor cell apoptosis and pyroptosis by a suboptimal dose of CAR-T cell coculture (i.e., tumor cell-to-T cell ratio of 10:1) was insufficient to kill the majority of tumor cells. An intriguing finding is that, even at such a low tumor cell-to-T cell ratio, CAR-T10%MYXV cells appeared to be endowed with unprecedented cytolytic function to eliminate tumor cells. In sharp contrast to apoptosis and pyroptosis, following which solid tumor cells usually detach from the culture dish, a different type of tumor cell death was observed, featuring a unique morphological feature that includes continued firm attachment to culture dishes. We further identified this tumor cell death as autosis, and in this scenario, the formation of an immunological cell-cell synapse between tumor and T cells seemed unnecessary for induced tumor cell autosis. Instead, CAR-TMYXV-released MYXV infection of tumor cells initiates autosis, while optimal levels of tumor cell autosis induction require synergy from CAR-T cell-derived IFNγ. Hence, it is possible that engineering IFNγ-producing MYXV can also enhance MYXV antitumor activity.
Collectively, our results highlight a pivotal role of CAR-T10%MYXV cells in improving the efficacy of ACT by: (1) delivering MYXV into tumor beds; (2) recognizing and inducing classic tumor cell apoptosis and pyroptosis to antigen-positive tumor cells accompanied by IFNγ secretion; (3) inducing autosis in antigen-positive and -negative cancer cells; and (4) potentially eliminating ALV cells by autosis and adaptive antitumor immunity. Thus far, our data suggest the existence of a tumor cell autosis-triggering strategy dependent on both MYXV and antigen-programmed CAR-T cells, which strategically incorporates MYXV and tumor-specific T cells to overcome therapeutic resistance in solid tumors.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yong Lu (ylu2@houstonmethodist.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The data reported in this paper is deposited in the Gene Expression Omnibus (GEO) database under accession number GSE199777 and GSE200305.
Experimental models and subject details
Mice
C57BL/6 (B6), DKO-NSG (MHC-I/II-double knockout; NOD.Cg-B2mtm1Unc Prkdcscid H2dlAb1-Ea Il2rgtm1Wjl/SzJ), Cd4−/− (B6.129S2-Cd4tm1Mak/J), Cd8−/− (B6.129S2-Cd8atm1Mak/J), Cd3−/− (B6;129-Cd3etm1Lov/J), CD4+ TRP-1 (B6.Cg-Rag1tm1Mom Tyrp1B-w Tg(Tcra,Tcrb)9Rest/J), CD8+ TRP-1 (B6.Cg-Rag2tm1.1Cgn TcrbLn4Sdou TcraLn2Sdou/J) mice were purchased from Jackson Laboratory. Mice were housed in individually ventilated cages within barrier facilities (≤5 mice/cage). Male and female 6- to 8-week-old mice were used for each animal experiment. Animals bearing engrafted tumors were randomized assigned into cohorts, to ensure a similar mean tumor volume/group based on caliper measurements at study enrollment. The studies fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All animal handling, surveillance, and experimentation was performed in accordance with and approval from the the Institutional Animal Care and Use Committee and Institutional Review Board of the Wake Forest School of Medicine.
Cell lines
The human OvCa cell line SKOV3; Burkitt’s lymphoma cell line Raji; human pancreatic ductal cell line PANC1; human breast cancer cell line SK-BR-3; human multiple myeloma cell line RPMI 8226; murine melanoma cell lines B16; and BSC40 cells, were purchased from ATCC. Human melanoma cell line, Mel-264 (HLA-A2+ MART-1+) was a gift from Dr. Steven Rosenberg (Hughes et al., 2005). Human glioblastoma cell line, U251, was a gift from Dr. Waldemar Debinski. Murine OvCa cell line, ID-8, was a gift from Dr. Neveen Said at Wake Forest School of Medicine. SKP-1-overexpressing SKOV3 (SKOV3-SKP-1) tumor cells were generated by transduction with lentivirus vectors encoding human SKP-1. SKOV3-IFNGR1-KO tumor cells were generated using CRISPR/Cas9 for IFNGR1 deletion. SKOV3-MSLN-KO tumor cells were generated using CRISPR/Cas9 for Msln deletion. B16TRP−1-KO cells were generated using CRISPR/Cas9 for Tyrp1 (TRP-1) deletion. ID-8 tumor cells were KO of Tp53 gene by CRISPR/Cas9 to recapitulate the human high-grade serous OvCa (Walton et al., 2016). hMSLN-expressing ID-8 tumor cells were generated by transduction with lentivirus vectors encoding hMSLN to ID-8p53-KO cells. Cells were cultured in RPMI 1640 Medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Thermo Scientific), and 100 U/ml penicillin-streptomycin and 2 mM L-glutamine (Invitrogen). Human umbilical vein endothelial cells (HUVECs), human epithelial cells (RWPE-1), and human stromal cells (HS-5) were purchased from ATCC. hMSLN-expressing HUVECs, hMSLN-expressing RWPE-1, and hMSLN-expressing HS-5 were generated by transduction with lentivirus vectors encoding hMSLN.
Myxoma virus
Myxoma virus (MYXV) stocks were grown using RK13 or Vero cells purchased from ATCC and purified by centrifugation on a sucrose cushion or sucrose gradient as described previously (Smallwood et al., 2010). Construction of Myxv-tdTomato (wild-type MYXV expressing tdTomato under a poxvirus synthetic early/late promoter) and Myxv-Fluc (wild-type MYXV expressing firefly luciferase under a poxvirus synthetic early/late promoter and tdTomato under a poxvirus p11 late promoter) were described previously (Yamada and Liu, 2009; Zemp et al., 2013).
Method details
Viral titer measurement
MYXV titer measurement was performed as described before (Wennier et al., 2012). The amount of infectious virus in each sample (culture supernatant or cell lysate) was quantified using foci formation assay on BSC40 cells. In some studies, MSLN CAR-TMYXV-tdTomato or MART-1 TMYXV-tdTomato cells were cocultured with SKOV3 cells, Mel-264 cells, or beads, and culture supernatants (virus containing media) were collected 72 hrs after reactivation. In some studies, treated TMYXV-tdTomato cells were lysed by 2 rounds of frozen and thawed cycles, and cell lysates containing virus were collected. About 48 hrs after adding diluted culture supernatants or cell lysates to BSC40 cells, the numbers of red fluorescent foci were counted from each dilution and calculated for the virus titer.
Human T cell preparation
Human T cells were activated by priming human CD3+ T cells isolated from PBMCs of the healthy donor with αCD3/CD28 Dynabeads and 200 U/ml hIL-2 for 24 hrs. During the activation, T cells were transduced with hMSLN-(SS1) (Ho et al., 2011)-hBBZ-CAR or CD19-(FMC63) (Sommermeyer et al., 2017)-hBBZ-CAR encoding γ-retrovirus to make T cells targeting human MSLN or CD19 in the presence of 10 μg/ml protamine sulfate (Sigma) by centrifugation for 2 hrs at 1,800 rpm, at room temperature. T cells may be transduced with CD3Z-truncated hMSLN-hBBΔZ-CAR as a control. In some studies, T cells were transduced with human HLA-A*02-restricted MART-127–35 specific TCR lentivector (DMF5) to make T cells targeting melanoma-associated antigen MART-1 (Hughes et al., 2005) in the presence of 10 μg/ml protamine sulfate (Sigma) by centrifugation for 2 hrs at 1,800 rpm, at room temperature. T cells were then expanded in the presence of 200 U/ml hIL-2 for an additional 7–8 days before use. Transduction rates of CAR or TCR vectors were >85%.
Murine T cells preparation
Murine T cells were activated by priming CD3+ T cells isolated from the spleens of C57BL/6 (B6) mice with αCD3/CD28 Dynabeads and 200 U/ml hIL-2 for 24 hrs. During the activation, T cells were transduced with hMSLN-(SS1) (Ho et al., 2011)-mBBZ-CAR encoding γ-retrovirus to make T cells targeting MSLN in the presence of 10 μg/ml protamine sulfate (Sigma) by centrifugation for 2 hrs at 1,800 rpm at room temperature. CAR+ (GFP+) T cells were sorted and then expanded in the presence of 200 U/ml hIL-2 for an additional 7–10 days before use. In some studies, CD4+ TRP-1-specific T cells were prepared by culturing splenocytes of CD4+ TRP-1 mice with TRP-1 peptide (SGHNCGTCRPGWRGAACNQKILTVR; 5 μg/ml) (Muranski et al., 2008) and hIL-2 (200 U/ml). CD8+ TRP-1-specific T cells were prepared by culturing splenocytes of CD8+ TRP-1 mice with TRP-1 peptide (TAPDNLGYA; 5 μg/ml) (Dougan et al., 2013) and hIL-2 (200 U/ml). After culturing for a total of 5 days, CD4+ TRP-1 T and CD8+ TRP-1 T cells were depleted of dead cells and mixed in a ratio of 1:1 for use in animal studies.
Infecting of MYXV to tumor-specific T cells
To infect MYXV to the tumor-specific T cells, MYXV was added at MOI of 3:1 to T cells 7–8 days after T cell expansion, in the presence of 10 μg/ml protamine sulfate (Sigma) by centrifugation for 2 hrs at 1,800 rpm, at room temperature. Tumor-specific T10%MYXVcells were formulated with tumor-specific T and tumor-specific TMYXV at a ratio of 9:1. % of live tumor-specific TMYXV cells were determined by immunofluorescence. The relative count of viable MSLN CAR-T cells was determined by trypan blue exclusion test.
To test the infectious activity of MYXV, MYXV-tdTomato (3×104 FFU), CD19 CAR-TMYXV-tdTomato (1×102), or MSLN CAR-TMYXV-Tdtomato (1×102) was added to Raji or SKOV3 cells (1×104). Relative tdTomato fluorescence intensities were determined with an Eclipse TE300 Inverted Microscope.
Tumor models and treatments
DKO-NSG mice received s.c. injections of SKOV3 cells (1×107), SKOV3-MSLN-KO cells (1×107), SKOV3-SKP-1 cells (1×107), or SKOV3-IFNGR1-KO cells (1×107). MSLN CAR-T cells (2.5×106) or MSLN CAR-T10%MYXV (2.5×106; contains 90% of MSLN CAR-T cells and 10% of MSLN CAR-TMYXV cells) cells were i.v. injected when tumors reached ~9×8 mm (or ~8×7 mm) on day 40. In some experiments, PBS or MYXV was i.t. injected only into tumors on the left flanks (L. i.t.); tumors on the right flanks did not receive i.t. injections. MART-1+ Mel-264 cells (2×106) were s.c. injected into DKO-NSG mice. MART-1 T cells (5×106) or MART-1 T10%MYXV cells (5×106; contains 90% of MART-1 T cells and 10% of MART-1 TMYXV cells) were i.v. injected when tumors reached ~7×6 mm on day 10. In some experiments, PBS or MYXV was i.t. injected only into the tumors on the left flanks (L. i.t.), and tumors on the right flanks did not receive i.t. injections. Raji tumor cells (1×105) were i.v. injected into DKO-NSG mice. CD19 CAR-T cells (2.5×106) or CD19 CAR-T10%MYXV cells (2.5×106; contains 90% of CD19 CAR-T cells and 10% of CD19 CAR-TMYXV cells) were i.v. injected on day 10. In some experiments, PBS or MYXV was i.v. injected. Mice were euthanized at indicated days or the endpoint.
B6 mice were received s.c. injection with ID-880%hMSLN tumors (containing 20% WT ID-8 cells as ALVs). At 43 days (tumors reached ~9×7 mm) after tumor injection, mice were treated with i.v. injection with MSLN CAR-T (5×106) or MSLN CAR-T10%MYXV (5×106) cells. In some studies, B6 mice were inoculated s.c. with 1×106 B1620%TRP−1-KO cells (left flank, containing 20% B16TRP−1-KO ALVs; no injection on the right flank) and injected i.v. with 1×105 B1620%TRP−1-KO cells to induce lung metastatic tumors. TRP-1 T (5×106) or TRP-1 T10%MYXV (5×106) cells were i.v. injected on day 10 when s.c. tumors reached ~7×6 mm. In some studies, PBS or MYXV was i.t. injected only into the tumors on the left flanks (L. i.t.), and tumors on the right flanks did not receive intratumorally injection. In some studies, WT, CD4−/−, CD8−/− and CD3−/− B6 mice were received s.c. B1620%TRP−1-KO tumors (1×106 B1620%TRP−1-KO challenged s.c. 8 days before ACT). TRP-1 T10%MYXV (5×106) cells were transferred i.v. into the mice when the tumor reached ~7×6 mm. In some studies, WT and CD3–/– B6 mice were s.c. inoculated with B1620%TRP−1-KO+Ctrl (1×106; 80%B16+20%B16TRP−1-KO+Ctrl), B1620%TRP−1-KO+SKP−1 (1×106; 80%B16+20%B16TRP−1-KO+SKP−1), or B16 (1×106) tumor cells. TRP-1 T10%MYXV (5×106) cells were i.v. transferred into mice when tumors reached ~7×6 mm (1×106 tumor cells challenged s.c. 8 days before ACT). Cyclophosphamide was given i.p. as a single dose at 120 mg/kg 1 day before T-cell transfer. Mice were euthanized at indicated days or the endpoint.
In vivo bioluminescence imaging
Before imaging, mice were anesthetized with isoflurane and i.p. injected with 100 μl of 20 mg/ml D-Luciferin (Xenogen Corp.). After 3 min, animals were imaged using an IVIS 200 system (Xenogen), according to the manufacturer’s instructions. Living Image software (Xenogen) was used to analyze data.
In vitro cytotoxic assays
The functionality of tumor-specific T10%MYXV cells was assessed by coculturing with target cells in 96-well plates, e.g., SKOV3-Luc+, Mel-264-Luc+, and Raji-Luc+ tumor cells. Unless otherwise stated, 2×103 sorted TCR/CAR-T cells or 2×103 sorted TCR/CAR-T10%MYXV cells were mixed with 2×104 target cells in a total volume of 200 μl complete medium. After coculture for 8 to 120 hrs, 1 μl of 20 mg/ml D-Luciferin (Xenogen Corp.) was added to each well, and luminescent signals were analyzed using POLARstar Omega Plate Reader (BMG LABTECH), according to the manufacturer’s instructions. Luminescent signals were used to calculate cytotoxicity.
In some studies, human monocytes were isolated from peripheral blood mononuclear cells of the healthy donor using EasySep™ Direct Human Monocyte Isolation Kit (STEMCELL; catalog# 19669). hMSLN-expressing monocytes were generated by transduction with expression plasmid encoding hMSLN.
In some studies, SKOV3-Luc+ tumor cells were seeded into the upper (2×105) and lower (2×104) Transwell chamber with 400 μl of the completed medium. MSLN CAR-T (2×104) or MSLN CAR-T10%MYXV (2×104) cells were added to the upper Transwell chamber. The pore size of Transwell inserts is 0.4 μM. After 72 hrs of coculture, luminescent signals of SKOV3-Luc+ cells in the lower chamber were detected using POLARstar Omega Plate Reader (BMG LABTECH), according to the manufacturer’s instructions.
RNA-sequencing
SKOV3 cells were treated with MYXV, MSLN CAR-T cells, MSLN CAR-T10%MYXV cells, or Tat-Beclin1 for 24 hrs in vitro. Total RNA was extracted with the RNeasy Mini kit (Qiagen). In some studies, B1620%TRP−1-KO cells (1×106) were s.c. injected in both flanks of the mice. TRP-1 T (5×106) or TRP-1 T10%MYXV (5×106) cells were administrated on day 7 when tumors reached ~7×6 mm. MYXV was i.t. injected only into tumors on the left flanks and tumors on the right flanks did not receive i.t. injections. Adjuvant cyclophosphamide (i.p.) was administered to mice one day before ACT. Tumor tissues (~100 mg/mice, 12 days after ACT) were harvested and total RNA was extracted with the RNeasy Mini kit (Qiagen). RNA was transferred to cDNA followed by ligation of adapters and the cDNA was sequenced. RNA-seq analyses were performed by BGI Genomics Co., Ltd.
Gene set enrichment analyses
Gene set enrichment analyses (GSEA) of the gene expression profiles were implemented using GSEA software (gsea-v3.0, http://software.broadinstitute.org/gsea/downinfects.jsp). P-values were calculated with the Kolmogorov-Smirnov test (threshold = 0.01). The false discovery rate (FDR), q value, is the estimated probability that a gene set with a given NES represents a false-positive finding. The threshold for q value in GSEA is 0.25. Gene sets for the mature effector were derived from a publicly available study of the genes differentially expressed by >2 fold in quaternary versus primary cells (Gu et al., 2016).
Flow cytometry and Western blot analyses
FITC- or PE-conjugated mAbs (1:100 dilution) were used for staining and analyzed using a FACS Fortessa flow cytometer.
For Western blot analysis, β-Actin (8H10D10) (catalog# 3700T) mouse mAb and Cleaved Caspase-3 Ab (catalog# 4926), Cleaved Caspase-1 (catalog# 89332), RIP (E8S7U) XP® (RIPK1, catalog# 73271), CD71 (D7G9X) XP® (TFRC, catalog# 13113), Phospho-Akt (Ser473) (D9E) XP® (catalog# 4060), and Akt (pan) (11E7) (catalog# 4685) rabbit mAbs from Cell Signaling Technology were used at a 1:1000 dilution. Anti-PI 3-kinase p100 (F-11) (catalog# sc-365404) and anti-alpha 1 sodium potassium ATPase/ATP1A1 (F-2) (catalog# sc-514614) mouse mAbs from Santa Cruz Biotechnology were used at a 1:500 dilution. In some experiments, SKOV3 tumor cells were seeded into upper (2×105) and lower (2×104) Transwell chambers with 400 μl of the completed medium. MSLN CAR-T (2×104) or MSLN CAR-T10%MYXV (2×104) cells were added to the upper Transwell chamber. The pore size of Transwell inserts is 0.4 μM. After 24 hrs of coculture, the SKOV3 cells in the upper (after T cell removal) and lower chambers were lysed and used for Western blot analyses.
Real-time PCR
Total RNA was extracted from murine tumors and tumor cell lines using the RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions. Genes were expressed with specific primers and analyzed using SYBR green real-time PCR (Applied Biosystems). Expression was normalized to the expression of the housekeeping gene GAPDH; mGAPDH F: 5’-TTGATGGCAACAATCTCCAC-3’, mGAPDH R: 5’-CGTCCCGTAGACAAAATGGT-3’; mTyrp1 F: 5’-CCCCTAGCCTATATCTCCCTTTT-3’, mTyrp1 R: 5’-TACCATCGTGGGGATAATGGC-3’. mCd3e F: 5’-TCAGCCTCCTAGCTGTTGG-3’, mCd3e R: 5’-GTCAACTCTACACTGGTTCCTG-3’. mCd8a F: 5’-CCGTTGACCCGCTTTCTGT-3’, mCd8a R: 5’-TTCGGCGTCCATTTTCTTTGG-3’. mCd4 F: 5’-CTAGCTGTCACTCAAGGGAAGA-3’, mCd4 R: 5’-CGAAGGCGAACCTCCTCTAA-3’. mCxcl10 F: 5’-CCAAGTGCTGCCGTCATTTTC-3’, mCxcl10 R: 5’-GGCTCGCAGGGATGATTTCAA-3’. mCcr7 F: 5’-CAGGTGTGCTTCTGCCAAGAT-3’, mCcr7 R: 5’-GGTAGGTATCCGTCATGGTCT-3’. mIl12a F: 5’-CAATCACGCTACCTCCTCTTTT-3’, mIl12a R: 5’-CAGCAGTGCAGGAATAATGTTTC-3’. mIl12b F: 5’-GTCCTCAGAAGCTAACCATCTCC-3’, mIl12b R: 5’-CCAGAGCCTATGACTCCATGTC-3’. mGzmb F: 5’-TCTCGACCCTACATGGCCTTA-3’, mGzmb R: 5’-TCCTGTTCTTTGATGTTGTGGG-3’. mSlc11a1 F: 5’-GCAGGCCCAGTTATGGCTC-3’, mSlc11a1 R: 5’-CAGGCTGAATGTACCCTGGTC-3’. mIfng F: 5’-GCCACGGCACAGTCATTGA-3’, mIfng R: 5’-TGCTGATGGCCTGATTGTCTT-3’. mLef1 F: 5’-GCCACCGATGAGATGATCCC-3’, mLef1 R: 5’-TTGATGTCGGCTAAGTCGCC-3’. hMSLN F: 5’-CCAACCCACCTAACATTTCCAG-3’, hMSLN R: 5’-CAGCAGGTCCAATGGGAGG-3’. hAtp1a1 F: 5’-ACAGACTTGAGCCGGGGATTA-3’, hAtp1a1 R: 5’-TCCATTCAGGAGTAGTGGGAG-3’. hSell F: 5’-ACCCAGAGGGACTTATGGAAC-3’, hSell R: 5’-GCAGAATCTTCTAGCCCTTTGC-3’. hIl7r F: 5’-CCCTCGTGGAGGTAAAGTGC-3’, hIl7r R: 5’-CCTTCCCGATAGACGACACTC-3’. hTcf7 F: 5’CTGGCTTCTACTCCCTGACCT-3’, hTcf7 R: 5’-ACCAGAACCTAGCATCAAGGA-3’. hGzmb F: 5’-CCCTGGGAAAACACTCACACA-3’, hGzmb R: 5’-GCACAACTCAATGGTACTGTCG-3’. hPrf1 F: 5’-GGCTGGACGTGACTCCTAAG-3’, hPrf1 R: 5’-CTGGGTGGAGGCGTTGAAG-3’. hLag3 F: 5’-GCGGGGACTTCTCGCTATG-3’, hLag3 R: 5’-GGCTCTGAGAGATCCTGGGG-3’. hCtla4 F: 5’-GCCCTGCACTCTCCTGTTTTT-3’, hCtla4 R: 5’-GGTTGCCGCACAGACTTCA-3’. hPdcd1 F: 5’CCAGGATGGTTCTTAGACTCCC-3’, hPdcd1 R: 5’-TTTAGCACGAAGCTCTCCGAT-3’. hGAPDH F: 5′-AGTCAACGGATTTGGTCGTATTGGG-3′, hGAPDH R: 5′-ACGTACTCAGCGCCAGCATCG-3′.
Immunofluorescence
Cells were fixed with 4% PFA for 15 min at room temperature and blocked with blocking buffer (1X PBS/5% normal serum/0.3% Triton™ X-100) for 1 hr, followed by incubation with LC3A/B rabbit pAb with 1:200 (Cell Signaling Technology, catalog# 4108) overnight at 4°C. The cells were then incubated with goat anti-rabbit IgG Alexa Fluor® 488 Conjugate (Cell Signaling Technology, catalog# 4412). DAPI, which fluoresces blue (Invitrogen, catalog# D1306) was used for nuclear counterstaining. Fluorescence microscopy assessments were performed on an Eclipse TE300 Inverted Microscope (Nikon Microscopy).
Enzyme-linked immunosorbent spot (ELISpot) assays
B1620%TRP−1-KO cells (1×106) were s.c. injected into both flanks of B6 mice. TRP-1 T cells (5×106) or TRP-1 T10%MYXV cells (5×106) were i.v. injected on day 7 when tumors reached ~7×6 mm. In some mice, free MYXV was i.t. injected only into the tumors on the left flanks. Mice were sacrificed on ~20 days after tumor inoculation and tumor tissues were minced and digested using a tumor dissociation kit (Miltenyi Biotec). Each host immune cell subset in about 200 mg tumor tissues was isolated by a bead positive selection kit (CD8+ or CD4+). Isolated cells per 200 mg of tumor tissues were cocultured with irradiated B16TRP−1-KO tumor cells on IFNγ ELISpot Kit plates (Mouse IFN-gamma ELISpot Kit, R&D Systems) for 48 hrs following the manufacturer’s instructions. The plates were imaged and evaluated by a Cellular Technology Limited ELISPOT Analyzer.
Imaging mass cytometry (IMC) analysis
IMC was performed on the Hyperion™ Imaging System using Maxpar metal-tagged antibodies. In brief, mouse tissue samples were fixed in 4% formalin and 6 μm thick sections were made and placed on slides. Each antibody was used at manufacturer-recommended concentrations. The IMC data were bead-normalized and de-barcoded by mass cytometry, and visualized by MCD viewer. Imaging data were analyzed by customized software using Python 3.7 (www.python.org). An imaging processing package, opencv-python (https://pypi.org/project/opencv-python/) was used to quantify total areas in red and blue; these were then used to calculate relative antigen positivity.
Statistical analyses
For statistical analysis, Student’s t-test or ANOVA was used. A P value less than 0.05 was considered statistically significant. Results are presented as mean ± s.d. unless otherwise indicated. The log rank test was used to compare survival curves. The test compares the entire survival experience between groups and can be thought of as a test of whether the survival curves are identical (overlapping) or not. Pairwise comparisons between group levels were calculated with corrections for multiple testing. The analysis was conducted using survival package in software R (https://www.r-project.org/).
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human IgG1 isotype control | BioXcell | Cat#BE0297 |
| αTNFα | BioXcell | Cat#SIM0006 |
| αIL-4 | BioXcell | Cat#BE0240 |
| αIL-9 | BioXcell | Cat#BE0327 |
| αIL-5 | BioXcell | Cat#BE0198 |
| αIFN-γ | BioXcell | Cat#BE0235 |
| αTGF-β | BioXcell | Cat#BE0057 |
| αIL-3 | R&D Systems | Cat#MAB603 |
| αIL-13 | R&D Systems | Cat#MAB213 |
| αIL-17 | R&D Systems | Cat#AF-317-NA |
| αGM-CSF | R&D Systems | Cat#MAB215 |
| αIL-2 | R&D Systems | Cat#MAB202 |
| PE anti-DYKDDDDK Tag Antibody | BioLegend | Cat#637309 |
| Biotin anti-human Mesothelin | BioLegend | Cat#530203 |
| Bacterial and virus strains | ||
| Myxoma virus (MYXV) | MYXV production by Dr. Grant McFadden’s lab | N/A |
| vMyx-tdTomato (MYXV-tdTomato) | MYXV-tdTomato production by Dr. Grant McFadden’s lab | N/A |
| vMyx-Fluc (MYXV-Luc+) | MYXV- Luc+ production by Dr. Grant McFadden’s lab | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| BYK204165 (PARP-1 inhibitor) | MilliporeSigma | Cat#B3188 |
| Y-27632 (ROCK inhibitor) | MilliporeSigma | Cat#Y0503 |
| lactoferrin (NETs inhibitor) | MilliporeSigma | Cat#L9507 |
| CA-074Me (CTSB inhibitor) | MilliporeSigma | Cat#205530 |
| PIK-III (VPS34 inhibitor) | VWR | Cat#103546-876 |
| Tat-Beclin1 D11 | Novus Biologicals | Cat#NBP2-49888 |
| siRNAs targeting the human Na+, K+-ATPase α1 subunit | Invitrogen | Cat#4390824 |
| Nontargeting siRNA | Thermo Scientific | Cat#D-001210-02-20 |
| MHC class I–restricted TRP1 (TAPDNLGYA) | GenScript | N/A |
| MHC class II-restricted TRP1 (SGHNCGTCRPGWRGAACNQKILTVR) | GenScript | N/A |
| Human IFN-γ | R&D Systems | Cat#285-IF |
| Human IL-2 | R&D Systems | Cat#202-IL |
| Critical commercial assays | ||
| Mouse IFN-gamma ELISpot Kit | R&D Systems | EL485 |
| Caspase-8 Activity Assay Kit (Colorimetric) | Novus Biologicals | Cat#NBP2-54817 |
| Human CD3+ T Cell Isolation Kit | STEMCELL | Cat#17751 |
| Human CD4+ T Cell Isolation Kit | STEMCELL | Cat#17952 |
| Human CD8+ T Cell Isolation Kit | STEMCELL | Cat#17953 |
| Deposited data | ||
| RNA-Sequencing | NCBI Gene Expression Omnibus | GSE199777 |
| RNA-Sequencing | NCBI Gene Expression Omnibus | GSE200305 |
| Experimental models: Cell lines | ||
| SK-OV-3 | ATCC | Cat#HTB-77 |
| Raji | ATCC | Cat#CCL-86 |
| PANC-1 | ATCC | Cat#CRL-1469 |
| SK-BR-3 | ATCC | Cat#HTB-30 |
| RPMI 8226 | ATCC | Cat#CRM-CCL-155 |
| B16 | ATCC | Cat#CRL-6323 |
| ID-8 | A gift from Dr. Neveen Said at Wake Forest School of Medicine | N/A |
| Mel-264 | A gift from Dr. Steven Rosenberg | N/A |
| U251 | A gift from Dr. Waldemar Debinski. | N/A |
| HUVECs | ATCC | Cat#PCS-100-013 |
| RWPE-1 | ATCC | Cat#CRL-11609 |
| HS-5 | ATCC | Cat#CRL-11882 |
| BSC40 | ATCC | Cat#CRL-2761 |
| 293T | ATCC | Cat#CRL-3216 |
| Experimental models: Organisms/strains | ||
| C57BL/6 | The Jackson Laboratory | Cat#000664 |
| Ifnar1−/− (B6(Cg)-Ifnar1tm1.2Ees/J) | The Jackson Laboratory | Cat#028288 |
| DKO-NSG (MHC-I/II-double knockout; NOD.Cg-B2mtm1Unc Prkdcscid H2dlAb1-Ea Il2rgtm1Wjl/SzJ) | The Jackson Laboratory | Cat#030547 |
| Cd4−/− (B6.129S2-Cd4tm1Mak/J) | The Jackson Laboratory | Cat#002663 |
| Cd8−/− (B6.129S2-Cd8atm1Mak/J) | The Jackson Laboratory | Cat#002665 |
| Cd3−/− (B6;129-Cd3etm1Lov/J) | The Jackson Laboratory | Cat#004177 |
| CD4+ TRP-1 (B6.Cg-Rag1tm1Mom Tyrp1B-w Tg(Tcra,Tcrb)9Rest/J) | The Jackson Laboratory | Cat#008684 |
| CD8+ TRP-1 (B6.Cg-Rag2tm1.1Cgn TcrbLn4Sdou TcraLn2Sdou/J) | The Jackson Laboratory | Cat#030957 |
| Oligonucleotides | ||
| mGAPDH F: 5’-TTGATGGCAACAATCTCCAC-3’ | Sigma | N/A |
| mGAPDH R: 5’-CGTCCCGTAGACAAAATGGT-3’ | Sigma | N/A |
| mTyrp1 F: 5’-CCCCTAGCCTATATCTCCCTTTT-3’ | Sigma | N/A |
| mTyrp1 R: 5’-TACCATCGTGGGGATAATGGC-3’ | Sigma | N/A |
| mCd3e F: 5’-TCAGCCTCCTAGCTGTTGG-3’ | Sigma | N/A |
| mCd3e R: 5’-GTCAACTCTACACTGGTTCCTG-3’ | Sigma | N/A |
| mCd8a F: 5’-CCGTTGACCCGCTTTCTGT-3’ | Sigma | N/A |
| mCd8a R: 5’-TTCGGCGTCCATTTTCTTTGG-3’ | Sigma | N/A |
| mCd4 F: 5’-CTAGCTGTCACTCAAGGGAAGA-3’ | Sigma | N/A |
| mCd4 R: 5’-CGAAGGCGAACCTCCTCTAA-3’ | Sigma | N/A |
| mCxcl10 F: 5’-CCAAGTGCTGCCGTCATTTTC-3’ | Sigma | N/A |
| mCxcl10 R: 5’-GGCTCGCAGGGATGATTTCAA-3’ | Sigma | N/A |
| mCcr7 F: 5’-CAGGTGTGCTTCTGCCAAGAT-3’ | Sigma | N/A |
| mCcr7 R: 5’-GGTAGGTATCCGTCATGGTCT-3’ | Sigma | N/A |
| mIl12a F: 5’-CAATCACGCTACCTCCTCTTTT-3’ | Sigma | N/A |
| mIl12a R: 5’-CAGCAGTGCAGGAATAATGTTTC-3’ | Sigma | N/A |
| mIl12b F: 5’-GTCCTCAGAAGCTAACCATCTCC-3’ | Sigma | N/A |
| mIl12b R: 5’-CCAGAGCCTATGACTCCATGTC-3’ | Sigma | N/A |
| mGzmb F: 5’-TCTCGACCCTACATGGCCTTA-3’ | Sigma | N/A |
| mGzmb R: 5’-TCCTGTTCTTTGATGTTGTGGG-3’ | Sigma | N/A |
| mSlc11a1 F: 5’-GCAGGCCCAGTTATGGCTC-3’ | Sigma | N/A |
| mSlc11a1 R: 5’-CAGGCTGAATGTACCCTGGTC-3’ | Sigma | N/A |
| mIfng F: 5’-GCCACGGCACAGTCATTGA-3’ | Sigma | N/A |
| mIfng R: 5’-TGCTGATGGCCTGATTGTCTT-3’ | Sigma | N/A |
| mLef1 F: 5’-GCCACCGATGAGATGATCCC-3’ | Sigma | N/A |
| mLef1 R: 5’-TTGATGTCGGCTAAGTCGCC-3’ | Sigma | N/A |
| hMSLN F: 5’-CCAACCCACCTAACATTTCCAG-3’ | Sigma | N/A |
| hMSLN R: 5’-CAGCAGGTCCAATGGGAGG-3’ | Sigma | N/A |
| hAtp1a1 F: 5’-ACAGACTTGAGCCGGGGATTA-3’ | Sigma | N/A |
| hAtp1a1 R: 5’-TCCATTCAGGAGTAGTGGGAG-3’ | Sigma | N/A |
| hSell F: 5’-ACCCAGAGGGACTTATGGAAC-3’ | Sigma | N/A |
| hSell R: 5’-GCAGAATCTTCTAGCCCTTTGC-3’ | Sigma | N/A |
| hIl7r F: 5’-CCCTCGTGGAGGTAAAGTGC-3’ | Sigma | N/A |
| hIl7r R: 5’-CCTTCCCGATAGACGACACTC-3’ | Sigma | N/A |
| hTcf7 F: 5’-CTGGCTTCTACTCCCTGACCT-3’ | Sigma | N/A |
| hTcf7 R: 5’-ACCAGAACCTAGCATCAAGGA-3’ | Sigma | N/A |
| hGzmb F: 5’-CCCTGGGAAAACACTCACACA-3’ | Sigma | N/A |
| hGzmb R: 5’-GCACAACTCAATGGTACTGTCG-3’ | Sigma | N/A |
| hPrf1 F: 5’-GGCTGGACGTGACTCCTAAG-3’ | Sigma | N/A |
| hPrf1 R: 5’-CTGGGTGGAGGCGTTGAAG-3’ | Sigma | N/A |
| hLag3 F: 5’-GCGGGGACTTCTCGCTATG-3’ | Sigma | N/A |
| hLag3 R: 5’-GGCTCTGAGAGATCCTGGGG-3’ | Sigma | N/A |
| hCtla4 F: 5’-GCCCTGCACTCTCCTGTTTTT-3’ | Sigma | N/A |
| hCtla4 R: 5’-GGTTGCCGCACAGACTTCA-3’ | Sigma | N/A |
| hPdcd1 F: 5’-CCAGGATGGTTCTTAGACTCCC-3’ | Sigma | N/A |
| hPdcd1 R: 5’-TTTAGCACGAAGCTCTCCGAT-3’ | Sigma | N/A |
| hGAPDH F: 5′ -AGTCAACGGATTTGGTCGTATTGGG-3′ | Sigma | N/A |
| hGAPDH R: 5′ -ACGTACTCAGCGCCAGCATCG-3′ | Sigma | N/A |
| Recombinant DNA | ||
| MART-127-35 specific TCR lentivector (DMF5) | a gift from Dr. Steven Rosenberg | N/A |
| PMKO.1 | Addgene | Cat#10676 |
| MigR1 | Addgene | Cat#27490 |
| MSCV-IRES | Addgene | Cat#20672 |
| pUltra | Addgene | Cat#24129 |
| lentiCRISPR v2 | Addgene | Cat#52961 |
| Software and algorithms | ||
| GSEA v2.2.2 | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| R | https://www.r-project.org/ | |
| Other | ||
Highlights:
CAR-TMYXV cells systemically deliver MYXV into cognate antigen-expressing tumors
T cell-derived IFNγ synergizes with MYXV-derived M-T5 to trigger tumor cell autosis
Autosis is a potent bystander killing to eradicate antigen-negative tumor cells
CAR/TCR-T10%MYXV induces autosis and adaptive immunity to restrain antigen escape
Acknowledgments
This work was supported by National Cancer Institute (NCI, 1R37CA251318-01, 1R01CA248111-01A1, R01CA258477-01, 1R01CA264102-01), and CPRIT Scholar (RR210067). We thank Weiskoff Amanda M. for editing assistance.
Footnotes
Declaration of Interests
Previously, GM was a co-founder and equity holder of OncoMyx Therapeutics, but it was operationally dissolved on 07/2022 and it’s assets no long hold any value. GM continues to hold patents related to the use of transgene-armed MYXV to treat cancer.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aits S, and Jäättelä M (2013). Lysosomal cell death at a glance. Journal of Cell Science 126, 1905–1912. [DOI] [PubMed] [Google Scholar]
- Ajina A, and Maher J (2017). Prospects for combined use of oncolytic viruses and CAR T-cells. Journal for ImmunoTherapy of Cancer 5, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Dawson TM, and Dawson VL (2008). Mitochondrial and nuclear cross talk in cell death: Parthanatos. Annals of the New York Academy of Sciences 1147, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee MY, Dunlap KM, and Bartee E (2016). Myxoma Virus Induces Ligand Independent Extrinsic Apoptosis in Human Myeloma Cells. Clinical Lymphoma, Myeloma and Leukemia 16, 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazaux M, Grandjean CL, Lemaître F, Garcia Z, Beck RJ, Milo I, Postat J, Beltman JB, Cheadle EJ, and Bousso P (2019). Single-cell imaging of CAR T cell activity in vivo reveals extensive functional and anatomical heterogeneity. Journal of Experimental Medicine 216, 1038–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan SK, Dougan M, Kim J, Turner JA, Ogata S, Cho H Il, Jaenisch R, Celis E, and Ploegh HL (2013). Transnuclear TRP1-specific CD8 T cells with high or low affinity TCRs show equivalent antitumor activity. Cancer Immunology Research 1, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, Eyquem J, Zhao Z, Whitlock BM, Miele MM, et al. (2019). CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M, Feng M, Fisher RJ, Rader C, and Pastan I (2011). A novel high-affinity human monoclonal antibody to mesothelin. International Journal of Cancer 128, 2020–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, and Yun C-O (2019). Overcoming the limitations of locally administered oncolytic virotherapy. BMC Biomedical Engineering 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MS, Yu YYL, Dudley ME, Zheng Z, Robbins PF, Li Y, Wunderlich J, Hawley RG, Moayeri M, Rosenberg SA, et al. (2005). Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Human Gene Therapy 16, 457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong Hung T. and Restifo Nicholas P. (2002). Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nature Immunology 3, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MR, and Griffiths GM (2010). The synapse and cytolytic machinery of cytotoxic T cells. Current Opinion in Immunology 22, 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Yu D, and Essand M (2016). Prospects to improve chimeric antigen receptor T-cell therapy for solid tumors. Immunotherapy 8, 1355–1361. [DOI] [PubMed] [Google Scholar]
- Jindal V, Arora E, and Gupta S (2018). Challenges and prospects of chimeric antigen receptor T cell therapy in solid tumors. Medical Oncology 35, 87. [DOI] [PubMed] [Google Scholar]
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S, and Milone MC (2018). CAR T cell immunotherapy for human cancer. Science 359, 1361–1365. [DOI] [PubMed] [Google Scholar]
- Kellish P, Shabashvili D, Rahman MM, Nawab A, Guijarro MV, Zhang M, Cao C, Moussatche N, Boyle T, Antonia S, et al. (2019). Oncolytic virotherapy for small-cell lung cancer induces immune infiltration and prolongs survival. Journal of Clinical Investigation 129, 2279–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Noda T, Ishihara N, and Ohsumi Y (2001). Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase y sorting in Saccharomyces cerevisiae. Journal of Cell Biology 152, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, and Levine B (2015). Autosis and autophagic cell death: The dark side of autophagy. Cell Death and Differentiation 22, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shoji-Kawata S, Sumpter RM, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al. (2013). Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proceedings of the National Academy of Sciences of the United States of America 110, 20364–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, Liu M, Zhou N, Lv J, Tang K, et al. (2020). Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Science Immunology 5, eaax7969. [DOI] [PubMed] [Google Scholar]
- Lun X, Yang W, Alain T, Shi ZQ, Muzik H, Barrett JW, McFadden G, Bell J, Hamilton MG, Senger DL, et al. (2005). Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Research 65, 9982–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun XQ, Alain T, Zemp FJ, Zhou H, Rahman MM, Hamilton MG, McFadden G, Bell J, Senger DL, and Forsyth PA (2010). Myxoma virus virotherapy for glioma in immunocompetent animal models: Optimizing administration routes and synergy with rapamycin. Cancer Research 70, 598–608. [DOI] [PubMed] [Google Scholar]
- Majzner RG, and Mackall CL (2018). Tumor antigen escape from car t-cell therapy. Cancer Discovery 8, 1219–1226. [DOI] [PubMed] [Google Scholar]
- Miao EA, Rajan JV, and Aderem A (2011). Caspase-1-induced pyroptotic cell death. Immunological Reviews 243, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, et al. (2008). Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 112, 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N. H, R. Z, O. M, M. T, A. A, S. V, J. B, P., S, B. S, and J. T (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature Immunology 1, 489–495. [DOI] [PubMed] [Google Scholar]
- Okubo K, Kamiya M, Urano Y, Nishi H, Herter JM, Mayadas T, Hirohama D, Suzuki K, Kawakami H, Tanaka M, et al. (2016). Lactoferrin Suppresses Neutrophil Extracellular Traps Release in Inflammation. EBioMedicine 10, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park AK, Fong Y, Kim SI, Yang J, Murad JP, Lu J, Jeang B, Chang WC, Chen NG, Thomas SH, et al. (2020). Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Science Translational Medicine 12, eaaz1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, Stojdl DF, Forsyth PAJ, Atkins H, and Bell JC (2007). Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Molecular Therapy 15, 123–130. [DOI] [PubMed] [Google Scholar]
- Rahman MM, and McFadden G (2020). Oncolytic Virotherapy with Myxoma Virus. Journal of Clinical Medicine 9, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SE, Rahman MM, Smith DW, and McFadden G (2010). Myxoma virus: Propagation, purification, quantification, and storage. Current Protocols in Microbiology 14, Unit–14A.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommermeyer D, Hill T, Shamah SM, Salter AI, Chen Y, Mohler KM, and Riddell SR (2017). Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 31, 2191–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford MM, Werden SJ, and McFadden G (2007). Myxoma virus in the European rabbit: Interactions between the virus and its susceptible host. Veterinary Research 38, 299–318. [DOI] [PubMed] [Google Scholar]
- Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, Cross JR, Rätsch G, Rice CM, and Ivashkiv LB (2015). Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nature Immunology 16, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Dotti G, Huye LE, Foster AE, Savoldo B, Gramatges MM, Spencer DM, and Rooney CM (2010). T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Molecular Therapy 18, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Cibas ES, Huang H, Hodgson L, and Overholtzer M (2014). Induction of entosis by epithelial cadherin expression. Cell Research 24, 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sypula J, Wang F, Ma Y, Bell J, and Mcfadden G (2004). Myxoma virus tropism in human tumor cells. Gene Therapy and Molecular Biology 8, 103–114. [Google Scholar]
- Tang D, Kang R, Berghe T. Vanden, Vandenabeele P, and Kroemer G (2019). The molecular machinery of regulated cell death. Cell Research 29, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton J, Blagih J, Ennis D, Leung E, Dowson S, Farquharson M, Tookman LA, Orange C, Athineos D, Mason S, et al. (2016). CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Research 76, 6118–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, and Levine B (2012). Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338, 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennier ST, Liu J, Li S, Rahman MM, Mona M, and McFadden G (2012). Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Molecular Therapy 20, 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werden SJ, Lanchbury J, Shattuck D, Neff C, Dufford M, and McFadden G (2009). The Myxoma Virus M-T5 Ankyrin Repeat Host Range Protein Is a Novel Adaptor That Coordinately Links the Cellular Signaling Pathways Mediated by Akt and Skp1 in Virus-Infected Cells. Journal of Virology 83, 12068–12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen ZN, and Jiang X (2019). Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature 572, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Zhang T, Xu D, Wang H, Cai Y, Jin T, Liu M, Jin M, Wu K, and Yuan J (2015). FBXL20-mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes and Development 29, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, and Liu DX (2009). Proteolytic Activation of the Spike Protein at a Novel RRRR/S Motif Is Implicated in Furin-Dependent Entry, Syncytium Formation, and Infectivity of Coronavirus Infectious Bronchitis Virus in Cultured Cells. Journal of Virology 83, 8744–8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemp FJ, Lun X, McKenzie BA, Zhou H, Maxwell L, Sun B, Kelly JJP, Stechishin O, Luchman A, Weiss S, et al. (2013). Treating brain tumor-initiating cells using a combination of myxoma virus and rapamycin. Neuro-Oncology 15, 904–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Huang J, Tong A, and Yang H (2019). Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Molecular Therapy - Oncolytics 15, 234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data reported in this paper is deposited in the Gene Expression Omnibus (GEO) database under accession number GSE199777 and GSE200305.