

Abstract
Introduction
The acceleration of biological aging is a risk factor for Alzheimer's disease (AD). Here, we performed weighted gene co‐expression network analysis (WGCNA) to identify modules and dysregulated genesinvolved in biological aging in AD.
Methods
We performed WGCNA to identify modules associated with biological clocks and hub genes of the module with the highest module significance. In addition, we performed differential expression analysis and association analysis with AD biomarkers.
Results
WGCNA identified five modules associated with biological clocks, with the module designated as “purple” showing the strongest association. Functional enrichment analysis revealed that the purple module was related to cell migration and death. Ten genes were identified as hub genes in purple modules, of which CX3CR1 was downregulated in AD and low levels of CX3CR1 expression were associated with AD biomarkers.
Conclusion
Network analysis identified genes associated with biological clocks, which suggests the genetic architecture underlying biological aging in AD.
Highlights
Examine links between Alzheimer's disease (AD) peripheral transcriptome and biological aging changes.
Weighted gene co‐expression network analysis (WGCNA) found five modules related to biological aging.
Among the hub genes of the module, CX3CR1 was downregulated in AD.
The CX3CR1 expression level was associated with cognitive performance and brain atrophy.
Keywords: Alzheimer's disease, AD biomarker, biological aging, CX3CR1, epigenetic clocks, telomere length, weighted gene co‐expression network analysis (WGCNA)
1. INTRODUCTION
Alzheimer's disease (AD) is the most common neurodegenerative disease, characterized by accumulation of amyloid beta (Aβ) plaques and neurofibrillary tangles in the brain. 1 , 2 AD develops from multiple factors, such as genetics, lifestyle factors, and aging. The actual cause of AD is still an open question, but the greatest known risk factor for AD is aging, although chronological age is not a sufficient marker of individual health outcomes and does not reflect sensitivity to aging‐related diseases. 3 Over the past decade there have been many studies to identify aging biomarkers that measure biological aging processes, also called biological age or biological clocks. 4 , 5 , 6 , 7 , 8 The acceleration of biological age has been reported to associate with biomarkers of AD including the presence of neuritic plaques, amyloid load, and the decline of global cognitive functioning, episodic, and working memory. 9 , 10 , 11 , 12 , 13 , 14
Telomere length (TL) and epigenetic clocks are the most promising biological clocks and have been reported to be associated with the risk of AD. Telomeres are repeated sequences at the end of chromosomes that are shortened with each cell cycle. 15 , 16 Telomere shortening was associated with aging, age‐related diseases, including AD, and cognitive performance in AD. 13 , 14 , 17 , 18 , 19 Epigenetic clocks are based on the changes in DNA methylation levels of multiple cytosine‐guanine dinucleotide (CpG) sites. Age‐related changes in methylation levels have been studied 20 , 21 , 22 , 23 and may cause various age‐related diseases including AD. 24 , 25 , 26 , 27 Moreover, epigenetic clocks have been linked to AD pathology, 10 and the acceleration of epigenetic clocks has been associated with neuritic plaques and amyloid load in the prefrontal cortex 10 and the decline of cognitive function, episodic memory, and working memory. 9 , 10 , 11 , 28
With the rapid development of high‐throughput screening techniques, gene expression analysis has become increasingly popular in studying the molecular mechanisms of human diseases. Previous studies have identified genes that are significantly dysregulated in AD. 29 , 30 , 31 , 32 In addition, individual genes do not work alone but interact with other genes. 33 , 34 Network analysis of gene expression profiles may offer the potential for identifying dysregulated genes that play important roles in complex diseases. Weighted gene co‐expression network analysis (WGCNA) has been developed to identify co‐expression modules of highly correlated genes, evaluate associations between modules and clinical outcomes, and identify potential disease‐associated gene sets. 35 , 36 , 37 , 38 We used WGCNA to identify modules by constructing a gene network using the correlation patterns between gene expression levels and evaluated the associations between modules and biological clocks.
To date, several biological clocks have been proposed. 4 , 5 , 39 , 40 Although previous studies reported the association between biological clocks and AD, transcriptomic factors influencing biological aging in AD are unknown. The objective of this study is to identify pathways and genes associated with biological aging and AD biomarkers in the AD continuum by integrating RNA profiling, DNA methylation, telomere lengths, cognition, and magnetic resonance imaging (MRI) scans from 551 non‐Hispanic White participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI). First, we used DNA methylation data to estimate DNA methylation–based epigenetic clocks 5 , 6 and DNA methylation–based TL. 4 In addition, we used TL measured using quantitative polymerase chain reaction (qPCR) to compare to DNA methylation–based estimated TL. After estimating biological clocks, we performed WGCNA on microarray messenger RNA (mRNA) expression profiles to identify modules (gene sets) as significantly associated with biological clocks. Then we identified hub genes in a module and performed differential expression analysis and association analysis of hub genes with AD diagnosis and AD biomarkers.
2. METHODS
2.1. Study participants
The data used in the study were downloaded from the ADNI database (http://adni.loni.usc.edu/). ADNI is a longitudinal study with more than 50 sites across the United States and Canada launched as a public‐private partnership in 2004, and details have been described previously. 41 , 42 In this study, a total of 551 ADNI participants including 181 cognitively normal older adults (CN), 272 mild cognitive impairment (MCI), and 93 AD were used for analysis.
2.2. Gene expression profiling
Gene expression profiling from blood samples was performed on the Affymetrix Human Genome U219 Array (www.affymetrix.com, Santa Clara, CA, USA). Raw microarray expression data were processed according to the standard quality control (QC) procedures described previously. 30 In brief, all probe sets were annotated with the reference human genome (hg19), and raw expression values were normalized. A sex discrepancy check was performed using expression levels of sex‐specific genes. After quality control, the RNA expression profiles contained 21,150 probes. To adjust for the confounding effects, we used batch effects and RNA integrity number (RIN) values as covariates.
2.3. Telomere length measurement
Telomere length (or TL) measurement was performed on DNA from blood or a buffy coat sample of ADNI participants using a real‐time qPCR) assay. TL was measured at the same visits with DNA methylation profiling and quantified by comparing the amount of the telomere amplification product (T) to that of a single copy gene (S). The relative telomere to single copy gene (T/S) ratio was calculated to yield a value that correlates with the average TL, and the T/S ratio was adjusted for experimental variables to correct for batch effects. The TL acquisition and quality control have been described in detail previously. 43
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using a PubMed and Google Scholar search. There is increasing evidence that the acceleration of biological aging has been reported to be associated with Alzheimer's disease (AD). It is, therefore, possible that expression levels of biological clock–associated genes could be associated with cognition and AD biomarkers.
Interpretation: This is the first study to show that biological clocks were associated with gene co‐expression network modules, whose hub genes were also associated with cognition and brain atrophy, providing the genetic architecture underlying biological aging in AD.
Future directions: Further replication and functional studies in larger independent cohorts and animal models should be performed to investigate the mechanistic roles of CX3CR1 in biological clocks and AD pathology.
2.4. DNA methylation profiling
DNA methylation was profiled in blood or buffy coat samples using Illumina EPIC chips (Illumina, Inc., San Diego, CA, USA) according to the Illumina protocols. A detailed protocol describing DNA measurement has been described previously. 44 In brief, genomic DNA samples were obtained from NCRAD (National Centralized Repository for Alzheimer's Disease and Related Dementias), and normalization and quality control were conducted. 45 One sample was excluded because it had no CpG calls, and four samples were excluded because ≥1% of the CpG sites had a detection P‐value > .05. 46 The remaining 1915 samples were normalized using wateRmelon. 47 After quality control and normalization, the data set of comprised beta values was used for further analyses.
2.5. Prediction of DNA methylation–based epigenetic clocks and telomere length
DNA methylation–based epigenetic clocks (DNAmGrimAge and DNAmAgeSkinBlood) 5 , 6 and TL (DNAmTL) 4 were predicted using the online biological clock calculator (http://dnamage.genetics.ucla.edu/) (see Figure S1 for the correlation of biological clocks). DNAmGrimAge and DNAmAgeSkinBlood are biological clocks (in units of years), and DNAmTL predicts leukocyte TL (LTL in units of kilobase [kb]) based on DNA methylation at multiple CpG sites. It is well known that the abundance of different cell types in blood changes with age, 48 , 49 and DNA methylation–based epigenetic clocks and DNAmTL are also associated with age‐related changes in blood cell composition. 4 , 5 , 6 To adjust for the effect of blood cell composition, we estimated blood cell counts using DNA methylation profiles based on the Horvath method 40 and the Houseman method, 50 and we included the following seven blood cell counts in the multivariable regression to avoid multi‐collinearity between blood cell counts: B cells, CD8+ T cells, monocytes, plasma blasts, exhausted cytotoxic CD8+ T cells, naïve CD8+ T, and naïve CD4+ T.
2.6. Cognitive performance
Cognitive composite scores were downloaded from the ADNI database. Comprehensive neuropsychological evaluations were used to calculate composite measures for memory (MEM) and executive functioning (EF). 51 , 52 Briefly, composite scores for memory included the Rey Auditory Verbal Learning Test, Mini‐Mental State Examination, Logical Memory I‐II, and AD Assessment Scale‐Cognitive Test. Composite scores for EF included Digit Span Backwards, Category Fluency, WAIS‐R (Wechsler Adult Intelligence Scale‐Revised) Digit Symbol Substitution, Trail Making Tests A and B, and the Clock Drawing Test.
2.7. Magnetic resonance imaging processing
Three‐dimensional T1‐weighted MR images were downloaded from ADNI, and all MR images were processed with the FreeSurfer software (http://surfer.nmr.mgh.harvard.edu/). The detailed procedure has been described previously 42 , 53 Mean cortical thicknesses in regions of interest (ROIs) were estimated using Desikan‐Killiany‐Tourville ROIs. 54 , 55
2.8. Statistical analysis
2.8.1. Construction of gene co‐expression network and module detection
The gene co‐expression network was constructed using the WGCNA package 56 in R. WGCNA was used to identify the modules of highly correlated genes that are related to external traits. First, similarity matrices of gene expression were calculated using Pearson's correlation Sij = |cor(i,j)|. Next, according to the theory of the network construction algorithm described in 57 , 58 the adjacency matrix (aij) was constructed using the power function β to construct a scale‐free network aij = power (Sij, β) = |Sij|β. To satisfy scale‐free topology, scale independence and mean connectivity were tested using a gradient method ranging from 1 to 20, and an appropriate power value was used. In this study, the power of β = 8 was selected to construct a scale‐free network. Then the adjacency matrix (aij) was transformed into a topological overlap matrix (TOM). Finally, module detection was conducted by performing average linkage hierarchical clustering of 1‐TOM dissimilarity matrix; detected modules are designated by color. 35 , 36 , 56 , 59
2.8.2. Identification of modules associated with biological clocks
To identify functional modules in the gene co‐expression network, the module eigengene (ME) was summarized by the first principal component of a given module, and module‐trait relationships were estimated using the correlation between MEs and biological clocks. The associations between MEs and biological clocks were estimated after adjusting for age and sex. To adjust for multiple testing, we used false discovery rate (FDR) corrected P‐values. To evaluate correlation strength, we calculate module significance (MS), which is defined by the average of the absolute gene significance for all genes in a given module, where the gene significance is defined by the correlation between the trait and gene expression levels of the gene. The relationships between modules were estimated by correlations between MEs, and hierarchical clustering of MEs was performed. In the WGCNA, modules with the highest MS are usually defined as the key module and selected for further analysis. 36 , 56 , 59
2.8.3. Functional enrichment analysis of a module
To understand the biological relationships of genes within a module, gene‐set enrichment analysis (GSEA) 60 was performed to identify enriched biological pathways. We selected genes in a module of WGCNA and performed GSEA with pathways from the Gene Ontology (GO) database (http://geneontology.org/). Pathways with FDR‐corrected P‐value < .05 were considered as significant.
2.8.4. Construction of a protein‐protein interaction (PPI) network and identification of hub genes in a module
We constructed a protein‐protein interaction (PPI) network of genes in a module. To identify experimentally validated interactions in the module, PPIs from the STRING database (https://string‐db.org/) were used. Interactions with a confidence score >0.7 were used for network construction. To identify hub genes of the module, we used the maximal clique centrality (MCC) algorithm. 61 The MCC of each protein node was calculated using cytoHubba 61 in the Cytoscape platform (https://cytoscape.org/). In this study, genes with the top 10 MCC scores were considered hub genes of the module.
2.8.5. Associations of expression levels of hub genes with AD and AD endophenotypes
First, we performed differential expression analysis of probes belonging to hub genes between diagnostic groups using analysis of covariance (ANCOVA) with age and sex as covariates. Then we performed a post hoc analysis of probes significantly associated with the diagnostic groups using AD endophenotypes. We used composite scores for memory and EF and entorhinal cortical thickness measured from MRI scans as AD endophenotypes. We performed linear regression analysis to investigate the association between expression levels of probes and AD endophenotypes. Age, sex, education, and intracranial volume (ICV) were used as covariates, if appropriate.
3. RESULTS
3.1. Study participants
A total of 551 ADNI non‐Hispanic White participants were used for gene co‐expression network analysis (181 CN, 272 MCI, and 93 AD) and demographic information is presented in Table 1. Age and sex were significantly different between diagnostic groups, whereas education and RIN values did not show any significant differences between diagnostic groups. As expected, the number of apolipoprotein E (APOE) ε4 alleles was greater in MCI and AD.
TABLE 1.
Demographic characteristics of the study participants
| CN | MCI | AD | P | |
|---|---|---|---|---|
| N (N = 551) | 186 | 272 | 93 | |
| Age | 76.2 (6.44) | 72.6 (7.72) | 77.1 (7.91) | 4.66 × 10–9 |
| Sex (female/male) | 92/94 | 120/152 | 35/58 | 1.64 × 10–1 |
| Education | 16.4 (2.71) | 16.2 (2.71) | 15.9 (3.03) | 4.43 × 10–1 |
| Number of APOE ε4 alleles (0/1/2) | 137/45/4 | 157/95/20 | 30/46/17 | 1.52 × 10–10 |
| RIN | 6.90 (0.52) | 7.01 (0.52) | 6.98 (0.64) | 1.35 × 10–1 |
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E gene; CN, cognitive normal; MCI, mild cognitive impairment; RIN, RNA integrity number.
Note: Boldface indicates P‐values less than .05. The mean and standard deviation of age, education, RIN values are shown in the table.
3.2. Construction of gene co‐expression networks and identification of modules
To identify gene modules, the gene co‐expression networks were constructed using WGCNA. To ensure a scale‐free network, scale independence and mean connectivity were tested using the gradient method, with the power value ranging from 1 to 20, and the power of β = 8 was selected. After the construction of the scale‐free network, the modules were detected using a hierarchical clustering dendrogram (Figure S2). As the result, 13,302 probes were grouped into 21 modules using the average linkage hierarchical clustering, and 7848 un‐clustered probes (included in the gray module) were removed for subsequent analysis. Twenty‐one modules were labeled by color, and the number of genes in each module is shown in Table S1.
3.3. Identification of gene co‐expression modules associated with biological clocks
The module‐trait relationships of 21 modules were tested by calculating the correlation between the MEs and biological clocks. Among the 21 modules, five modules (purple, blue, brown, tan, and magenta) were associated significantly with TL, DNAmTL, and DNAmAgeSkinBlood after adjusting for multiple testing using FDR correction (Figure 1A). In particular, the purple module was significantly associated with TL (r = −0.19, FDR corrected P = 1.59 × 10–4), DNAmTL (r = −0.22, FDR corrected P = 6.25 × 10–6), and DNAmAgeSkinBlood (r = 0.2, FDR corrected P = 6.23 × 10–5) (see Tables S2, S3, and S4 for module‐trait relationships in each diagnostic group). MS is defined by the average of the absolute gene significance for all genes in a given module, where the gene significance is defined by the correlation between the trait and expression levels of the gene. WGCNA usually defined the module with the highest MS as the key module, and it was selected for further analysis. 36 , 56 , 59 In this study, the purple module has the highest mean MS across the biological clocks (Figure 1B). In addition, the relationships between modules were tested. As shown in Figure S3, modules were divided primarily into two clusters according to the correlations between MEs. The results showed that, of the five modules that are significantly associated with biological clocks, the tan and magenta modules were clustered with the purple module, whereas the brown and blue modules were not. Therefore, based on the relationships between modules, further analysis was performed on the purple module, a key module, as well as two modules, blue and brown, which did not cluster with the purple module.
FIGURE 1.
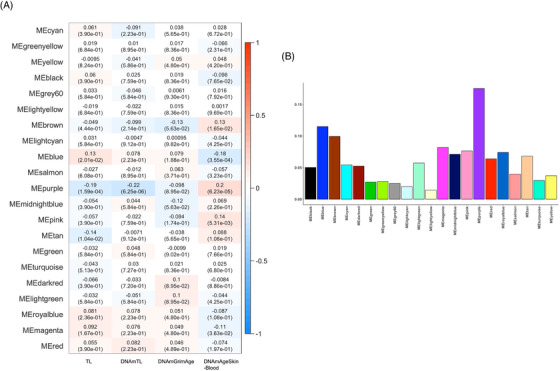
Identification of modules associated with biological clocks. (A) Relationship between modules and biological clocks. Heatmap displays the correlation coefficient (r) between module eigengenes (MEs) and biological clocks and false discovery rate (FDR)–corrected P‐values. (B) Average of the module significance (MS) of biological clocks.
3.4. Gene set enrichment analysis of genes within the modules
To understand the biological relationship of genes in the modules, GSEA was performed with probes belonging to each module. The GSEA analysis with pathways from GO showed 50 enriched pathways for the purple module (Table 2). For the measured TL, the purple module was significantly enriched in a nuclear protein‐containing complex (FDR corrected P = 1.90 × 10–2). For the DNAmTL, the purple module was enriched in 11 pathways including leukocyte migration. Finally, for the DNAmAgeSkinBlood, the purple module was significantly enriched in 39 pathways including cell and intracellular transport. The GSEA results for the blue and brown modules are shown in Tables S6 and S7. In the blue module, 25 pathways were enriched for TL and DNAmAgeSkinBlood, including transferase complex, leukocyte mediate immunity, and cell activation (Table S6). In the brown module, five pathways were enriched for DNAmGrimAge and DNAmAgeSkinBlood (Table S7).
TABLE 2.
Gene‐set enrichment analysis of the purple module for biological clocks
| Biological age | Gene Ontology | N genes | NES | FDR P |
|---|---|---|---|---|
| TL | Nuclear protein–containing complex | 6 | −1.130 | 1.90 × 10–2 |
| DNAmTL | Regulation of response to biotic stimulus | 13 | 1.210 | 4.10 × 10–2 |
| Response to bacterium | 8 | 1.200 | 3.60 × 10–2 | |
| Granulocyte migration | 6 | −1.138 | 4.66 × 10–2 | |
| Leukocyte migration | 14 | −1.141 | 4.67 × 10–2 | |
| Positive regulation of intracellular signal transduction | 16 | −1.127 | 4.68 × 10–2 | |
| Positive regulation of MAPK cascade | 7 | −1.132 | 4.75 × 10–2 | |
| Myeloid cell differentiation | 6 | −1.128 | 4.80 × 10–2 | |
| Neutrophil migration | 5 | −1.142 | 4.82 × 10–2 | |
| Regulation of leukocyte migration | 7 | −1.132 | 4.83 × 10–2 | |
| Protein dimerization activity | 6 | −1.119 | 4.58 × 10–2 | |
| Identical protein binding | 21 | −1.101 | 4.72 × 10–2 | |
| DNAmAgeSkinBlood | Positive regulation of intracellular transport | 6 | 2.405 | 2.95 × 10–4 |
| Regulation of intracellular transport | 6 | 2.405 | 3.69 × 10–4 | |
| Positive regulation of locomotion | 10 | 2.422 | 4.92 × 10–4 | |
| Positive regulation of phosphorus metabolic process | 13 | 2.441 | 7.38 × 10–4 | |
| Taxis | 14 | 2.326 | 9.98 × 10–4 | |
| Regulation of cellular component movement | 17 | 2.456 | 1.48 × 10–3 | |
| Locomotion | 31 | 2.249 | 1.72 × 10–3 | |
| Positive regulation of intracellular protein transport | 5 | 2.258 | 1.94 × 10–3 | |
| Regulation of intracellular protein transport | 5 | 2.258 | 2.22 × 10–3 | |
| Positive regulation of protein localization to membrane | 5 | 2.135 | 3.99 × 10–3 | |
| Regulation of protein localization to membrane | 5 | 2.135 | 4.35 × 10–3 | |
| Regulation of anatomical structure morphogenesis | 7 | 2.104 | 4.36 × 10–3 | |
| Regulation of vasculature development | 5 | 2.090 | 4.47 × 10–3 | |
| Positive regulation of protein phosphorylation | 12 | 2.144 | 4.52 × 10–3 | |
| Second messenger mediated signaling | 9 | 2.047 | 5.50 × 10–3 | |
| Calcium mediated signaling | 9 | 2.047 | 5.87 × 10–3 | |
| Cell migration | 27 | 1.992 | 6.83 × 10–3 | |
| Protein kinase b signaling | 8 | 2.003 | 6.87 × 10–3 | |
| Gliogenesis | 6 | 1.981 | 7.41 × 10–3 | |
| Regulation of intracellular signal transduction | 23 | 1.928 | 1.12 × 10–2 | |
| Positive regulation of protein kinase b signaling | 5 | 1.902 | 1.33 × 10–2 | |
| Intracellular transport | 13 | 1.878 | 1.64 × 10–2 | |
| Positive regulation of catalytic activity | 14 | 1.869 | 1.72 × 10–2 | |
| Organophosphate metabolic process | 6 | 1.864 | 1.72 × 10–2 | |
| Blood vessel morphogenesis | 10 | 1.857 | 1.79 × 10–2 | |
| Cell chemotaxis | 8 | 1.841 | 1.90 × 10–2 | |
| Regulation of cell death | 13 | 1.798 | 2.45 × 10–2 | |
| G protein–coupled receptor signaling pathway | 14 | 1.798 | 2.52 × 10–2 | |
| Negative regulation of cell death | 8 | 1.800 | 2.57 × 10–2 | |
| Positive regulation of chemotaxis | 5 | 1.773 | 2.70 × 10–2 | |
| Regulation of chemotaxis | 5 | 1.773 | 2.79 × 10–2 | |
| Growth | 8 | 1.775 | 2.86 × 10–2 | |
| Anatomical structure formation involved in morphogenesis | 13 | 1.743 | 3.10 × 10–2 | |
| Positive regulation of protein modification process | 15 | 1.736 | 3.14 × 10–2 | |
| Cellular component morphogenesis | 8 | 1.745 | 3.16 × 10–2 | |
| Biological process involved in symbiotic interaction | 17 | 1.701 | 4.22 × 10–2 | |
| Positive regulation of intracellular signal transduction | 16 | 1.685 | 4.63 × 10–2 | |
| Vasculature development | 11 | 1.669 | 4.99 × 10–2 | |
| Cell morphogenesis | 8 | 1.671 | 5.00 × 10–2 |
Abbreviations: FDR P, false discovery rate (FDR)–corrected P‐value; N genes, the number of genes in gene sets; NES, normalized enrichment score.
3.5. Identification of hub genes in the modules
The interaction networks in each module were constructed using the PPI network. The hub genes within a network were determined by the MCC algorithm. The PPI network with the genes in the purple module consisted of 49 nodes and 114 edges (Figure 2A). Genes with the top 10 higher MCC scores were selected as hub genes of the purple module (Figure 2B and Table S5): CCR5 (C‐C motif chemokine receptor 5), CCL4 (C‐C motif chemokine ligand 4), CXCR6 (C‐X‐C motif chemokine receptor 6), CX3CR1 (C‐X3‐C motif chemokine receptor 1), NMUR1 (neuromedin U receptor 1), S1PR5 (sphingosine‐1‐phosphate receptor 5), CCL5 (C‐C motif chemokine ligand 5), PRF1 (perforin 1), IL2RB (interleukin 2 receptor subunit beta), and GZMB (granzyme B). The PPI network with the genes in the blue module consisted of 1439 nodes and 5752 edges, and the network with the genes in the brown module consisted of 1398 nodes and 7874 edges. The hub genes in the blue and brown modules and their first connected genes are shown in Figures S4 and S5.
FIGURE 2.
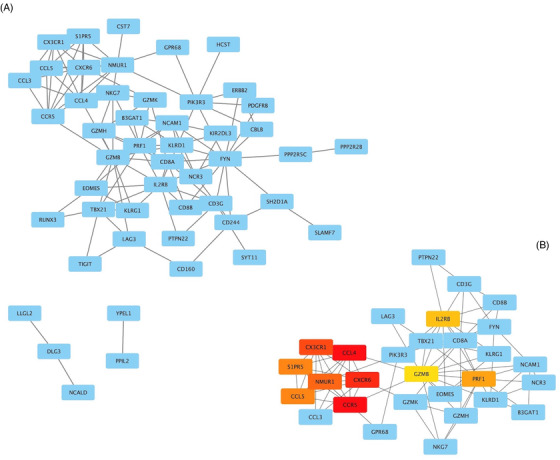
Visualization of the protein‐protein interaction (PPI) network and hub genes. (A) PPI network with genes in the purple module. The blue nodes represent the genes and edges present the associations between genes. (B) 10 Hub genes with highest maximal clique centrality (MCC) scores and their first connected genes. The MCC scores are displayed in colors from red to yellow on the node.
3.6. Association of blood expression levels of hub genes with AD diagnosis and AD biomarkers
We performed differential expression analysis of probes belonging to the 10 hub genes in each module. In total, 14 probes in the purple module, 17 probes in blue, and 21 probes in the brown module were used for the differential expression analysis. Among the 14 probes in the purple module, one probe annotated on CX3CR1 gene showed a significant difference of expression levels between diagnostic groups (Table 3 and Figure S6). CX3CR1 was significantly downregulated in AD compared to MCI and CN (P = 3.27 × 10–2, AD‐MCI P = 1.62 × 10–3, AD‐CN P = 1.40 × 10–3, MCI‐CN P = 4.81 × 10–1). There were no differentially expressed genes between diagnostic groups in the blue and brown modules (Tables S8 and S9). We evaluated the associations with AD biomarkers for the differentially expressed gene in the purple module. The expression levels of CX3CR1 were significantly associated with composite scores for MEM (β = 0.301, P = 2.27 × 10–2) and for EF (β = 0.407, P = 5.22 × 10–3) (Figure 3A,B). Higher expression levels of CX3CR1 were associated with higher composite scores for both MEM and EF. In addition, higher expression levels of CX3CR1 were also significantly associated with thicker entorhinal cortical thickness (β = 0.153, P = 2.40 × 10–2) (Figure 3C).
TABLE 3.
Differentially expressed probes and related hub genes in the purple module
| probe ID | Gene | CN | MCI | AD | P | FDR P |
|---|---|---|---|---|---|---|
| 11723048_at | CX3CR1 | 0.032 | 0.005 | −0.081 | 2.33 × 10–3 | 3.27 × 10–2 |
| 11752490_x_at | NMUR1 | 0.000 | 0.013 | −0.044 | 5.54 × 10–1 | 7.65 × 10–1 |
| 11740639_at | NMUR1 | −0.013 | 0.018 | −0.033 | 6.52 × 10–1 | 7.65 × 10–1 |
| 11729977_a_at | CXCR6 | 0.033 | −0.017 | −0.012 | 6.55 × 10–1 | 7.65 × 10–1 |
| 11747295_a_at | PRF1 | 0.006 | 0.010 | −0.046 | 6.02 × 10–1 | 7.65 × 10–1 |
| 11730909_s_at | CCR5 | 0.043 | −0.014 | −0.040 | 2.74 × 10–1 | 7.65 × 10–1 |
| 11718983_x_at | CCL4 | 0.027 | −0.005 | −0.038 | 3.49 × 10–1 | 7.65 × 10–1 |
| 11752664_a_at | S1PR5 | 0.004 | 0.007 | −0.031 | 8.35 × 10–1 | 8.74 × 10–1 |
| 11722635_at | IL2RB | 0.006 | 0.018 | −0.073 | 1.15 × 10–1 | 7.65 × 10–1 |
| 11732275_at | CCL5 | 0.018 | 0.004 | −0.050 | 4.79 × 10–1 | 7.65 × 10–1 |
| 11718982_s_at | CCL4 | 0.021 | −0.005 | −0.025 | 3.84 × 10–1 | 7.65 × 10–1 |
| 11753810_a_at | CCL5 | 0.021 | 0.004 | −0.057 | 3.13 × 10–1 | 7.65 × 10–1 |
| 11732276_x_at | CCL5 | 0.022 | 0.007 | −0.069 | 1.99 × 10–1 | 7.65 × 10–1 |
| 11724900_a_at | GZMB | 0.001 | 0.005 | −0.018 | 8.74 × 10–1 | 8.74 × 10–1 |
Abbreviations: AD, Alzheimer's disease; CN, cognitively normal old adults; FDR P, false discovery rate (FDR)−corrected P‐value; MCI, mild cognitive impairment; P, P‐value.
Note: Boldface indicates FDR P‐values less than 0.05.
FIGURE 3.
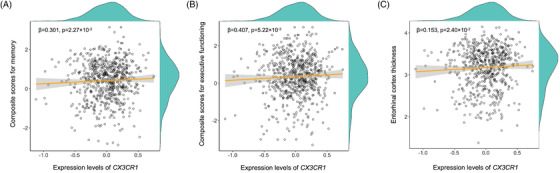
Associations between expression levels of CX3CR1 gene and Alzheimer's disease (AD) biomarkers. Expression levels of CX3CR1 versus composite scores for memory (A), executive functioning (B), and entorhinal cortical thickness (C).
4. DISCUSSION
Accumulating evidence suggests that acceleration of biological aging may contribute to increasing risk for AD. 10 , 11 , 13 , 14 , 19 Here, we conducted WGCNA of transcriptomics profiles to identify modules and hub genes of the modules associated with biological aging in the continuum of AD (MCI, AD) using four types of biological clocks including measured TL, two DNA methylation–based clocks (DNAmGrimAge, DNAmAgeSkinBlood), and DNA methylation–based estimated telomere length (DNAmTL).
Gene co‐expression network analysis identified five modules significantly associated with biological aging, and among them the purple module showed the strongest associations with four biological clocks. GSEA of the purple module identified 50 significantly enriched pathways related to the acceleration of biological clocks. Our analysis revealed the enriched pathways related to leukocyte migration and intracellular transport. Leukocyte migration plays an important role in inflammation, which is linked to AD development, 62 and leukocyte filtration in AD brain is stimulated by Aβ. 63 The disruption of the intracellular transport of enzymes involved in the production of Aβ has been associated with the onset of AD. 64 Furthermore, pathway analysis also revealed pathways related to cell migration, cell death, chemotaxis, and gliogenesis. Cell death and migration are related to Aβ, a key molecule in the pathogenesis of AD. 65 The migration of monocytes into the AD brain begins with Aβ deposition and contributes to neuronal cell death that causes brain atrophy. 66 Microglial chemotaxis is related to the concentration of soluble Aβ, 67 and migration of microglia induced by Aβ constitutes a chemotactic response of neuroimmune brain cells. 68 Gliogenesis in the adult brain regulates brain function and is involved in neurological conditions such as AD. 69 , 70
Therefore, genes in the purple module are likely linked to biological processes relevant to AD. Furthermore, according to the MCC algorithm, the top 10 genes with higher MCC scores were considered as hub genes of the purple module, and their expression was found to be downregulated in patients with AD compared with cognitively normal older adults (or CN). Among them, CX3CR1 expression was significantly downregulated in AD and was also significantly associated with cognitive performance and brain atrophy.
The CX3CR1 (CX3C chemokine receptor 1) gene is a protein‐coding gene that binds to CX3CL1 (CX3C chemokine fractalkine), also known as fractalkine. The chemokine is involved in the adhesion and migration of leukocytes 71 and chemokine, and chemokine receptors play a role in the development of AD. 72 Genetic mutations in the CX3CR1 gene are associated with the number of cells in lymphocytes, 73 , 74 leukocytes, 75 , 76 , 77 and myeloid white cells. 74 CX3CL1/CX3CR1, one type of chemokine and chemokine receptors, has been reported to play a role in AD. 78 , 79 In the central nervous system (CNS), CX3CL1 is highly expressed in neurons, 80 and CX3CR1 is mainly expressed in microglia. 81 In our study, the CX3CR1 gene was downregulated in AD, which is consistent with the previous study. 82 Previous studies suggest that the interruption of the CX3CL1/CX3CR1 signaling, which plays an important role in neuro‐microglia interaction, has beneficial as well as detrimental effects on AD pathology. 78 CX3CL1/CX3CR1 signaling has a neuroprotective effect in the CNS by reducing neurotoxicity and microglial activation 83 , 84 and has a beneficial effect on Aβ clearance. 82 , 85 , 86 , 87 , 88 Conversely, CX3CL1/CX3CR1 signaling has also been reported to have a negative effect on AD pathology. Disrupting the CX3CL1/CX3CR1 signaling pathway by ablation of the CX3CR1 resulted in enhancement of tau accumulation. 82 , 85 , 86 , 87 , 88 In addition, the disruption of CX3CL1/CX3CR1 signaling has been associated with the dysregulation of microglial responses, neuronal damage, 81 , 89 alleviation of Aβ neurotoxicity, and memory deficiency. 90 , 91
Among the hub genes in the purple module, five genes (CX3CR1, CCL4, CCR5, CCL5, CXCR6) are chemokine or chemokine receptors. Chemokines and their receptors have been reported to be related to neuroinflammation in AD. 72 CCR5 (C‐C chemokine receptor type 5) is involved in neuroinflammation processes associated with AD 72 , 92 , 93 and related to long‐term and spatial memory functions in mice. 94 In addition, the absence of CCR5 modulates the expression of CCR2, which is related to the activation of astrocytes, which may lead to Aβ deposition. 93 CCL4 (C‐C motif chemokine ligand 4), also known as macrophage inflammatory protein‐1β (MIP‐1β), binds to CCR5 and CCR8 (C‐C chemokine receptor 8). The expression of CCL4 is modulated by Aβ deposition. 95
This study is limited due to its modest sample size. Replication studies in independent cohorts with large sample sizes are needed to validate our findings. In addition, we used only non‐Hispanic Whites from the ADNI cohort to avoid confounding by population stratification. Further studies in racially diverse samples will be important for generalization of these findings to community‐based and ancestrally diverse populations.
In conclusion, we performed a co‐expression network analysis of blood gene expression profiles to identify modules and dysregulated genes associated with biological clocks in AD. We identified five modules related to biological clocks, where the purple module showed the most significant association with biological clocks. Among hub genes of the purple module, CX3CR1 was downregulated in AD and associated with cognitive performance and brain atrophy. The biological role of CX3CR1 in biological clocks merits further investigation. This study may contribute to the understanding of the genetic architecture underlying biological aging and its role in relation to AD pathophysiology.
CONFLICT OF INTEREST
Qingqin S. Li is an employee of Janssen Research & Development, LLC, an operating company of Johnson & Johnson, and holds company equity. The authors declare that they have no non‐financial competing interests. Author disclosures are available in the supporting information.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
Data collection and sharing for this project were funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging and the National Institute of Biomedical Imaging and Bioengineering, and has received generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private sector contributions are made by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Additional support for data analysis was provided in part by grants: P30 AG010133, P30 AG072976, R01 AG019771, R01 AG057739, U01 AG024904, R01 LM013463, R01 AG068193, T32 AG071444, U01 AG068057, U01 AG072177, R01 LM012535, and R03 AG063250.
Kim B‐H, Vasanthakumar A, Li QS, et al. Integrative analysis of DNA methylation and gene expression identifies genes associated with biological aging in Alzheimer's disease. Alzheimer's Dement. 2022;14:e12354. 10.1002/dad2.12354
REFERENCES
- 1. Wang J, Gu BJ, Masters CL, Wang YJ. A systemic view of Alzheimer disease – Insights from amyloid‐β metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612‐623. doi: 10.1038/nrneurol.2017.111 [DOI] [PubMed] [Google Scholar]
- 2. Kumar A, Singh A, Ekavali. A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol Reports. 2015;67(2):195‐203. doi: 10.1016/j.pharep.2014.09.004 [DOI] [PubMed] [Google Scholar]
- 3. Sebastiani P, Thyagarajan B, Sun F, et al. Biomarker signatures of aging. Aging Cell. 2017;16(2):329‐338. doi: 10.1111/acel.12557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu AT, Seeboth A, Tsai PC, et al. DNA methylation‐based estimator of telomere length. Aging (Albany NY). 2019;11(16):5895‐5923. doi: 10.18632/aging.102173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu AT, Quach A, Wilson JG, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019;11(2):303‐327. doi: 10.18632/aging.101684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horvath S, Oshima J, Martin GM, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY). 2018;10(7):1758‐1775. doi: 10.18632/aging.101508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006;12(10):1133‐1138. doi: 10.1038/nm1006-1133 [DOI] [PubMed] [Google Scholar]
- 8. Holly AC, Melzer D, Pilling LC, et al. Towards a gene expression biomarker set for human biological age. Aging Cell. 2013;12(2):324‐326. doi: 10.1111/acel.12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marioni RE, Shah S, McRae AF, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 2015;44(4):1388‐1396. doi: 10.1093/ije/dyu277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre‐frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer's disease related cognitive functioning. Aging (Albany NY). 2015;7(12):1198‐211. doi: 10.18632/aging.100864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McCartney DL, Stevenson AJ, Walker RM, et al. Investigating the relationship between DNA methylation age acceleration and risk factors for Alzheimer's disease. Alzheimers Dement (Amst). 2018;21(10):429‐437. doi: 10.1016/j.dadm.2018.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forero DA, González‐Giraldo Y, López‐Quintero C, Castro‐Vega LJ, Barreto GE, Perry G. Meta‐analysis of telomere length in Alzheimer's disease. J Gerontol A Biol Sci Med Sci. 2016;71(8):1069‐1073. doi: 10.1093/gerona/glw053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mahoney ER, Dumitrescu L, Seto M, et al. Telomere length associations with cognition depend on Alzheimer's disease biomarkers. Alzheimers Dement Transl Res Clin Interv. 2019;5:883‐890. doi: 10.1016/j.trci.2019.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Honig LS, Kang MS, Schupf N, Lee JH, Mayeux R. Association of shorter leukocyte telomere repeat length with dementia and mortality. Arch Neurol. 2012;69(10):1332‐1339. doi: 10.1001/archneurol.2012.1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blackburn EH. Structure and function of telomeres. Nature. 1991;350(6319):569‐573. doi: 10.1038/350569a0 [DOI] [PubMed] [Google Scholar]
- 16. Blackburn EH. Telomere states and cell fates. Nature. 2000;408(6808):53‐56. doi: 10.1038/35040500 [DOI] [PubMed] [Google Scholar]
- 17. Kimura M, Hjelmborg JVB, Gardner JP, et al. Telomere length and mortality: a study of leukocytes in elderly danish twins. Am J Epidemiol. 2008;167(7):799‐806. doi: 10.1093/aje/kwm380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Panossian LA, Porter VR, Valenzuela HF, et al. Telomere shortening in T cells correlates with Alzheimer's disease status. Neurobiol Aging. 2003;24(1):77‐84. doi: 10.1016/S0197-4580(02)00043-X [DOI] [PubMed] [Google Scholar]
- 19. Martin‐Ruiz C, Dickinson HO, Keys B, Rowan E, Kenny RA, Von Zglinicki T. Telomere length predicts poststroke mortality, dementia, and cognitive decline. Ann Neurol. 2006;60(2):174‐180. doi: 10.1002/ana.20869 [DOI] [PubMed] [Google Scholar]
- 20. Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23(8):413‐418. doi: 10.1016/j.tig.2007.05.008 [DOI] [PubMed] [Google Scholar]
- 21. Rakyan VK, Down TA, Maslau S, et al. Human aging‐associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20(4):434‐439. doi: 10.1101/gr.103101.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Teschendorff AE, Menon U, Gentry‐Maharaj A, et al. Age‐dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20(4):440‐446. doi: 10.1101/gr.103606.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jung M, Pfeifer GP. Aging and DNA methylation. BMC Biol. 2015;13:7. doi: 10.1186/s12915-015-0118-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057‐1068. doi: 10.1038/nbt.1685 [DOI] [PubMed] [Google Scholar]
- 25. Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic mechanisms in Alzheimer's disease. Neurobiol Aging. 2011;32(7):1161‐1180. doi: 10.1016/j.neurobiolaging.2010.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yokoyama AS, Rutledge JC, Medici V. DNA methylation alterations in Alzheimer's disease. Environ Epigenetics. 2017;3(2):dvx008. doi: 10.1093/eep/dvx008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156‐1163. doi: 10.1038/nn.3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hillary RF, Stevenson AJ, Cox SR, et al. An epigenetic predictor of death captures multi‐modal measures of brain health. Mol Psychiatry. 2019;26(8):3806‐3816. doi: 10.1038/s41380-019-0616-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Puthiyedth N, Riveros C, Berretta R, Moscato P. Identification of differentially expressed genes through integrated study of Alzheimer's disease affected brain regions. PLoS One. 2016;11(4):e0152342. doi: 10.1371/journal.pone.0152342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nho K, Nudelman K, Allen M, et al. Genome‐wide transcriptome analysis identifies novel dysregulated genes implicated in Alzheimer's pathology. Alzheimers Dement. 2020;16(9):1213‐1223. doi: 10.1002/alz.12092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang WS, Reiman EM, Valla J, et al. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A. 2008;105(11):4441‐4446. doi: 10.1073/pnas.0709259105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang WS, Dunckley T, Beach TG, et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiol Genomics. 2008;33(2):240‐256. doi: 10.1152/physiolgenomics.00242.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arnone MI, Davidson EH. The hardwiring of development: organization and function of genomic regulatory systems. Development. 1997;124(10):1851‐1864. doi: 10.1242/dev.124.10.1851 [DOI] [PubMed] [Google Scholar]
- 34. Li J, Zhou D, Qiu W, et al. Application of weighted gene co‐expression network analysis for data from paired design. Sci Rep. 2018;8(1):622. doi: 10.1038/s41598-017-18705-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li CY, Cai JH, Tsai JJP, Wang CCN. Identification of hub genes associated with development of head and neck squamous cell carcinoma by integrated bioinformatics analysis. Front Oncol. 2020;10:681. doi: 10.3389/fonc.2020.00681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Niu X, Zhang J, Zhang L, et al. Weighted gene co‐expression network analysis identifies critical genes in the development of heart failure after acute myocardial infarction. Front Genet. 2019;10:1214. doi: 10.3389/fgene.2019.01214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liang JW, Fang ZY, Huang Y, et al. Application of weighted gene co‐expression network analysis to explore the key genes in Alzheimer's disease. J Alzheimers Dis. 2018;65(4):1353‐1364. doi: 10.3233/JAD-180400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jin X, Li J, Li W, et al. Weighted gene co‐expression network analysis reveals specific modules and biomarkers in Parkinson's disease. Neurosci Lett. 2020;728:134950. doi: 10.1016/j.neulet.2020.134950 [DOI] [PubMed] [Google Scholar]
- 39. Levine ME, Lu AT, Quach A, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10(4):573‐591. doi: 10.18632/aging.101414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mueller SG, Weiner MW, Thal LJ, et al. The Alzheimer's disease neuroimaging initiative. Neuroimaging Clin N Am. 2005;15:869‐877. doi: 10.1016/j.nic.2005.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jack CR, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685‐691. doi: 10.1002/jmri.21049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nudelman KNH, Lin J, Lane KA, et al. Telomere shortening in the Alzheimer's disease neuroimaging initiative cohort. J Alzheimers Dis. 2019;71(1):33‐43. doi: 10.3233/JAD-190010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vasanthakumar A, Davis JW, Idler K, et al. Harnessing peripheral DNA methylation differences in the Alzheimer's Disease Neuroimaging Initiative (ADNI) to reveal novel biomarkers of disease. Clin Epigenetics. 2020;12(1):84. doi: 10.1186/s13148-020-00864-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113):2673‐2734. doi: 10.1016/S0140-6736(17)31363-6 [DOI] [PubMed] [Google Scholar]
- 46. Jazwinski SM, Kim S. Examination of the dimensions of biological age. Front Genet. 2019;10:263. doi: 10.3389/fgene.2019.00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data‐driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gibson J, Russ TC, Clarke TK, et al. A meta‐analysis of genome‐wide association studies of epigenetic age acceleration. PLoS Genet. 2019;15(11):e1008104. doi: 10.1371/journal.pgen.1008104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fagnoni FF, Vescovini R, Passeri G, et al. Shortage of circulating naive CD8+ T cells provides new insights on immunodeficiency in aging. Blood. 2000;95(9):2860‐2868. doi: 10.1182/blood.v95.9.2860.009k35_2860_2868 [DOI] [PubMed] [Google Scholar]
- 50. Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crane PK, Carle A, Gibbons LE, et al. Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav. 2012;6(4):502‐516. doi: 10.1007/s11682-012-9186-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gibbons LE, Carle AC, Mackin RS, et al. A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav. 2012;6(4):517‐527. doi: 10.1007/s11682-012-9176-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Risacher SL, Shen L, West JD, et al. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging. 2010;31(8):1401‐1418. doi: 10.1016/j.neurobiolaging.2010.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Klein A, Tourville J. 101 labeled brain images and a consistent human cortical labeling protocol. Front Neurosci. 2012;6:171. doi: 10.3389/fnins.2012.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968‐980. doi: 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 56. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ghazalpour A, Doss S, Zhang B, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2(8):e130. doi: 10.1371/journal.pgen.0020130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang B, Horvath S. A general framework for weighted gene co‐expression network analysis. Stat Appl Genet Mol Biol. 2005;4(1):1‐45. doi: 10.2202/1544-6115.1128 [DOI] [PubMed] [Google Scholar]
- 59. Wang CCN, Li CY, Cai JH, et al. Identification of prognostic candidate genes in breast cancer by integrated bioinformatic analysis. J Clin Med. 2019;8(8):1160. doi: 10.3390/jcm8081160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545‐15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub‐networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi: 10.1186/1752-0509-8-S4-S11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21(3):38. doi: 10.1016/S0197-4580(00)00124-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rezai‐Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D‐Day for neurodegenerative disease? J Neuroimmune Pharmacol. 2009;4(4):462‐475. doi: 10.1007/s11481-009-9166-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Miyagawa T, Ebinuma I, Morohashi Y, et al. BIN1 regulates BACE1 intracellular trafficking and amyloid‐β production. Hum Mol Genet. 2016;25(14):2948‐2958. doi: 10.1093/hmg/ddw146 [DOI] [PubMed] [Google Scholar]
- 65. Niikura T, Tajima H, Kita Y. Neuronal cell death in Alzheimers disease and a neuroprotective factor, humanin. Curr Neuropharmacol. 2006;4(2):139‐147. doi: 10.2174/157015906776359577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hohsfield LA, Humpel C. Migration of blood cells to β‐amyloid plaques in Alzheimer's disease. Exp Gerontol. 2015;65:8‐15. doi: 10.1016/j.exger.2015.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cho H, Hashimoto T, Wong E, et al. Microfluidic chemotaxis platform for differentiating the roles of soluble and bound amyloid‐β on microglial accumulation. Sci Rep 2013;3:1823. doi: 10.1038/srep01823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McLarnon JG. Microglial chemotactic signaling factors in Alzheimer's disease. Am J Neurodegener Dis 2012;1(3):199‐204. [PMC free article] [PubMed] [Google Scholar]
- 69. Choi SS, Lee S‐R, Lee HJ. Neurorestorative role of stem cells in Alzheimer's disease: astrocyte involvement. Curr Alzheimer Res. 2016;13(4):419‐27. doi: 10.2174/156720501304160314162812 [DOI] [PubMed] [Google Scholar]
- 70. Rusznák Z, Henskens W, Schofield E, Kim WS, Fu YH. Adult neurogenesis and gliogenesis: possible mechanisms for neurorestoration. Exp Neurobiol. 2016;25(3):103‐12. doi: 10.5607/en.2016.25.3.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moser B, Wolf M, Walz A, Loetscher P. Chemokines: Multiple levels of leukocyte migration control. Trends Immunol. 2004;25(2):75‐84. doi: 10.1016/j.it.2003.12.005 [DOI] [PubMed] [Google Scholar]
- 72. Liu C, Cui G, Zhu M, Kang X, Guo H. Neuroinflammation in Alzheimer's disease: chemokines produced by astrocytes and chemokine receptors. Int J Clin Exp Pathol. 2014;7(12):8342‐8355. [PMC free article] [PubMed] [Google Scholar]
- 73. Astle WJ, Elding H, Jiang T, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167(5):1415‐1429.e19. doi: 10.1016/j.cell.2016.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen MH, Raffield LM, Mousas A, et al. Trans‐ethnic and ancestry‐specific blood‐cell genetics in 746,667 individuals from 5 global populations. Cell. 2020;182(5):1198‐1213.e14. doi: 10.1016/j.cell.2020.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kichaev G, Bhatia G, Loh PR, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. 2019;104(1):65‐75. doi: 10.1016/j.ajhg.2018.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vuckovic D, Bao EL, Akbari P, et al. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182(5):1214‐1231.e11. doi: 10.1016/j.cell.2020.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sakaue S, Kanai M, Tanigawa Y, et al. A cross‐population atlas of genetic associations for 220 human phenotypes. Nat Genet 2021;53(10):1415‐1424. doi: 10.1038/s41588-021-00931-x [DOI] [PubMed] [Google Scholar]
- 78. Pawelec P, Ziemka‐Nalecz M, Sypecka J, Zalewska T. The impact of the CX3CL1/CX3CR1 axis in neurological disorders. Cells. 2020;9(10):2277. doi: 10.3390/cells9102277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chen P, Zhao W, Guo Y, Xu J, Yin M. CX3CL1/CX3CR1 in Alzheimer's disease: a target for neuroprotection. Biomed Res Int. 2016;2016:8090918 doi: 10.1155/2016/8090918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1‐expressing microglia. Proc Natl Acad Sci U S A. 1998;95(18):10896‐10901. doi: 10.1073/pnas.95.18.10896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9(7):917‐924. doi: 10.1038/nn1715 [DOI] [PubMed] [Google Scholar]
- 82. Cho SH, Sun B, Zhou Y, et al. CX3CR1 protein signaling modulates microglial activation and protects against plaque‐independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286(37):32713‐32722. doi: 10.1074/jbc.M111.254268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979(1‐2):65‐70. doi: 10.1016/S0006-8993(03)02867-1 [DOI] [PubMed] [Google Scholar]
- 84. Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson's disease. J Neuroinflammation. 2011;8:9. doi: 10.1186/1742-2094-8-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Joaquín Merino J, Muñetón‐Gómez V, Alvárez M‐I, Toledano‐Díaz A. Effects of CX3CR1 and Fractalkine chemokines in amyloid beta clearance and p‐Tau accumulation in Alzheimer's disease (AD) rodent models: is Fractalkine a systemic biomarker for AD? Curr Alzheimer Res. 2016;13(4):403‐412. doi: 10.2174/1567205013666151116125714 [DOI] [PubMed] [Google Scholar]
- 86. Bhaskar K, Konerth M, Kokiko‐Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19‐31. doi: 10.1016/j.neuron.2010.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lee S, Varvel NH, Konerth ME, et al. CX3CR1 deficiency alters microglial activation and reduces beta‐amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177(5):2549‐2562. doi: 10.2353/ajpath.2010.100265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid‐β phagocytosis. J Neurosci. 2010;30(50):17091‐17101. doi: 10.1523/JNEUROSCI.4403-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Febinger HY, Thomasy HE, Pavlova MN, et al. Time‐dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. J Neuroinflammation. 2015;12:154. doi: 10.1186/s12974-015-0386-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dworzak J, Renvoisé B, Habchi J, et al. Neuronal Cx3cr1 deficiency protects against amyloid β‐induced neurotoxicity. PLoS One. 2015;10(6):e0127730. doi: 10.1371/journal.pone.0127730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wu J, Bie B, Yang H, Xu JJ, Brown DL, Naguib M. Suppression of central chemokine fractalkine receptor signaling alleviates amyloid‐induced memory deficiency. Neurobiol Aging. 2013;34(12):2843‐2852. doi: 10.1016/j.neurobiolaging.2013.06.003 [DOI] [PubMed] [Google Scholar]
- 92. Man SM, Ma YR, Shang DS, et al. Peripheral T cells overexpress MIP‐1α to enhance its transendothelial migration in Alzheimer's disease. Neurobiol Aging. 2007;28(4):485‐496. doi: 10.1016/j.neurobiolaging.2006.02.013 [DOI] [PubMed] [Google Scholar]
- 93. Combarros O, Infante J, Llorca J, Peña N, Fernández‐Viadero C, Berciano J. The chemokine receptor CCR5‐Δ32 gene mutation is not protective against Alzheimer's disease. Neurosci Lett. 2004;366(3):312‐314. doi: 10.1016/j.neulet.2004.05.058 [DOI] [PubMed] [Google Scholar]
- 94. Lee YK, Kwak DH, Oh KW, et al. CCR5 deficiency induces astrocyte activation, Aβ deposit and impaired memory function. Neurobiol Learn Mem. 2009;92(3):356‐363. doi: 10.1016/j.nlm.2009.04.003 [DOI] [PubMed] [Google Scholar]
- 95. Ito S, Sawada M, Haneda M, Ishida Y, Isobe K ichi. Amyloid‐β peptides induce several chemokine mRNA expressions in the primary microglia and Ra2 cell line via the PI3K/Akt and/or ERK pathway. Neurosci Res. 2006;56(3):294‐299. doi: 10.1016/j.neures.2006.07.009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information