

Abstract
Pulmonary Langerhans cell histiocytosis (PLCH) is a rare sporadic cystic lung disease of unknown aetiology that is characterised by the infiltration and destruction of the wall of distal bronchioles by CD1a+ Langerhans-like cells. In adults, PLCH is frequently isolated and affects young smokers of both sexes. Recent multicentre studies have led to the more standardised management of patients in clinical practice. Smoking cessation is essential and is occasionally the only suitable intervention. Serial lung function testing is important because a significant proportion of patients may experience an early decline in forced expiratory volume in 1 s and develop airflow obstruction. Cladribine was reported to dramatically improve progressive PLCH in some patients. Its efficacy and tolerance are currently being evaluated. Patients who complain of unexplained dyspnoea with decreased diffusing capacity of the lung for carbon monoxide should be screened for pulmonary hypertension by Doppler echocardiography, which must be confirmed by right heart catheterisation. Lung transplantation is a therapeutic option for patients with advanced PLCH.
The identification of the BRAFV600E mutation in approximately half of Langerhans cell histiocytosis lesions, including PLCH, and other mutations of the mitogen-activated protein kinase (MAPK) pathway in a subset of lesions has led to targeted treatments (BRAF and MEK (MAPK kinase) inhibitors). These treatments need to be rigorously evaluated because of their potentially severe side-effects.
Short abstract
Recent advances have led to the more standardised management of patients with PLCH in clinical practice http://ow.ly/pyk930dWVxN
Introduction
Langerhans cell histiocytosis (LCH) is a rare disorder of unknown aetiology that is characterised by the infiltration of involved tissues by dendritic cells sharing phenotypic similarities with Langerhans cells, which are often organised into granulomas [1]. LCH may affect patients of all ages, ranging from neonates to the elderly [2, 3]. Although a few familial cases have been reported, LCH occurs sporadically [4]. The incidence in children under 15 years of age is approximately five cases per million [5] and it occurs rarely in adults.
The clinical presentation of LCH is widely heterogeneous. The disease may resolve spontaneously or lead to life-threatening situations [6, 7]. The clinical forms of LCH are classified according to the number and type of organs involved [6]. Multisystem LCH concerns two or more organs, including primarily bone, skin and the pituitary stalk, but also the lungs, lymph nodes and, more rarely, central nervous system. The liver, spleen and haematopoietic system, which are referred to as “risk organs”, are associated with a worse prognosis [6]. Single-system LCH affects only one tissue/organ, which is mainly bone (eosinophilic granuloma), but it also affects the lungs in adults and usually has a better prognosis [6, 7].
In adults, pulmonary LCH (PLCH) is often isolated or is a predominant feature [8, 9]. PLCH belongs to the spectrum of diffuse cystic interstitial pneumonias [10]. Its prevalence is unknown, but it is estimated to account for 3–5% of adult diffuse lung diseases [11, 12]. PLCH is mainly observed in young patients of both sexes, with a peak incidence at age 20–40 years, and females possibly develop PLCH at slightly older ages. It occurs almost exclusively in smokers (90–100% patients are current or ex-smokers in the largest series) [13, 14].
Over the last decade, significant progress has been made in expanding our knowledge of the natural history of PLCH, which has allowed more standardised management of patients in clinical practice [13, 15, 16]. Here, we first describe several clinical situations in which pulmonary physicians may be faced with decisions related to PLCH. We then review the key features of PLCH, including an important breakthrough in the understanding of the pathogenesis of this rare disease [17].
Clinical situations
Case 1
A 24-year-old man was referred for a nodulocystic pattern on lung high-resolution computed tomography (HRCT) that suggested PLCH (figure 1a). He had been smoking 10 cigarettes·day−1 since he was 17 years of age and had started complaining of a disabling dry cough 3 months before being referred. The physical examination was normal. Plethysmography showed normal total lung capacity (TLC) (89% predicted), a mild decrease in vital capacity (VC) (74% predicted), air trapping (residual volume (RV)/TLC 152% predicted) and no airflow obstruction. Diffusing capacity of the lung for carbon monoxide (DLCO) was decreased to 65% predicted. Bronchoalveolar lavage (BAL) recovered 240 000 cells·mL−1 with 90% macrophages, 6% lymphocytes, 2% neutrophils, 2% eosinophils and no pathogens. A presumptive diagnosis of isolated PLCH was made and the patient was strongly encouraged to quit smoking, which he managed to do. He was regularly seen at 3, 6 and 12 months during follow-up while he weaned himself off tobacco. He rapidly improved and became asymptomatic at 3 months of follow-up, and his chest radiograph and lung function improved. At his 1-year evaluation, plethysmography showed normal lung volume (TLC 91% predicted, VC 97% predicted and RV/TLC 88% predicted) and significant improvement of DLCO (76% predicted). Lung HRCT confirmed that there was virtually a complete resolution of pulmonary lesions (figure 1b).
FIGURE 1.

a) High-resolution computed tomography (HRCT) scan of the chest at the lung window level from patient 1 showing a characteristic nodulocystic pattern at the time of pulmonary Langerhans cell histiocytosis diagnosis. b) Serial HRCT 2 years later, while the patient was weaned off smoking, shows virtually complete resolution of pulmonary lesions.
Case 2
A 21-year-old man was referred for recurrent pneumothorax in the context of PLCH. The patient was a university student and had started smoking 3–5 cigarettes·day−1 1 year before the occurrence of a complete right-sided pneumothorax. He had previously presented with skin lesions that were diagnosed as psoriasis. Despite drainage, partial pneumothorax persisted and video-assisted surgical talc pleurodesis was performed. A lung biopsy at the same time confirmed the diagnosis of PLCH. At that time, lung HRCT showed diffuse thin-walled cystic lesions (figure 2a and b). The patient immediately stopped smoking, but right-sided pneumothorax recurred 10 days later, which was treated by thoracoscopic resection of apical bullae and a new talc pleurodesis. Three months later, a third right-sided partial basal pneumothorax occurred that resolved after drainage. Meanwhile, skin lesions appeared in the inguinal folds and scalp that corresponded to LCH lesions on biopsy and subsequently improved under dermal corticosteroids. A fourth pneumothorax recurred 3 months later on the same side with complex bullous lesions of the lung base on HRCT (figure 2c and d). The patient underwent a new thoracic surgical procedure with parietal pleurectomy. Subsequently, he has been free from pneumothorax during the 2 years of follow-up. At his last evaluation, he was asymptomatic, and his chest imaging (figure 2e and f) and lung function were considered satisfactory (TLC 101% predicted, VC 88% predicted, forced expiratory volume in 1 s (FEV1) 80% predicted and DLCO 83% predicted), except for air trapping (RV/TLC 139% predicted).
FIGURE 2.

Serial high-resolution computed tomography (HRCT) scans at the lung window from patient 2, who developed several episodes of right pneumothorax that required iterative surgical interventions. a, b) Thin-walled cystic lung pattern (note the presence of a drain for the first episode). c, d) Complex right pneumothorax with bullae in the right lung base. e, f) 18 months after the last thoracic surgery, there were residual thin-walled cystic lesions.
Case 3
A 55-year-old woman was referred for severe airway obstruction in the context of known PLCH and she was diagnosed by surgical lung biopsy when she was 26 years old. The patient had smoked 20 cigarettes·day−1 since she was 18 years old. During follow-up, her dyspnoea gradually worsened to New York Heart Association (NYHA) stage 3, which was not improved by inhaled combined corticosteroids and long-acting β2-agonists. She was examined several times for spastic exacerbation episodes that were treated with oral steroids and short-acting β2-agonist nebulisation. Between these episodes, lung auscultation was normal, as was her peripheral oxygen saturation. She walked 85% of the predicted distance during the 6-min walk test (6MWT) without oxygen desaturation. Her lung function showed severe irreversible airway obstruction (FEV1/forced vital capacity (FVC) 35% predicted, FEV1 38% predicted, VC 100% predicted, TLC 119% predicted and RV 116% predicted). DLCO was greatly reduced to 26% predicted without pulmonary hypertension on Doppler echocardiography. Lung HRCT showed a few thin-walled cysts and diffuse emphysema. The extent of the cystic lesions could not explain the importance of impairment of lung function. This patient actually suffered from chronic obstructive pulmonary disease (COPD) rather than progressive PLCH. She was put on a dedicated smoking cessation and rehabilitation programme.
Case 4
A 23-year-old man was referred because of progressive multisystem LCH despite treatment with vinblastine. He had presented multisystem LCH at 4 years of age, which involved the skin, peripheral lymph nodes and pituitary stalk with diabetes insipidus. At that time, he received two lines of treatment with vinblastine, became disease-free at 6 years of age and was maintained on desmopressin for diabetes insipidus. At 22 years of age, cutaneous and peripheral lymph node LCH lesions recurred and were histologically confirmed. Lung involvement was also present and responsible for NYHA stage 3 dyspnoea and important lung function impairment (FEV1/FVC 68% predicted, FEV1 37% predicted and DLCO 37% predicted). Lung HRCT showed bullocystic lesions that were predominant in lung bases (figure 3a and b). Notably, the patient had started smoking occasionally when he was 17 years old and stopped when LCH recurred. As skin and lymph node lesions did not significantly improve after two induction courses of vinblastine, he was switched to subcutaneous cladribine as a second-line treatment for multisystem LCH, which was associated with infection prophylaxis with trimethoprim/sulfamethoxazole and valaciclovir. The treatment was well tolerated, and the skin and lymph node LCH lesions were resolved. The lung involvement also improved clinically and in lung imaging (figure 3c and d); lung function at 6 months after initiation of cladribine also improved (FEV1 57% predicted) [18]. Subsequently, the patient continued to improve, and at the last follow-up 4 years later he was still asymptomatic and received only desmopressin for diabetes insipidus and had satisfactory lung function (FEV1/FVC 84% predicted, FEV1 74% predicted and DLCO 61% predicted). A concomitant lung HRCT showed impressive regression of bullocystic pulmonary lesions (figure 3e and f).
FIGURE 3.

Serial high-resolution computed tomography (HRCT) scans at the lung window from patient 4, who was treated with cladribine for progressive multisystem Langerhans cell histiocytosis. a, b) A predominant bullocystic pattern is observed in the lung bases. c, d) Six months after treatment began, there was a substantial reduction in the extent of cystic lung lesions. e, f) Four years after treatment, lung abnormalities regressed substantially, with only residual fibrocystic lesions in the left lung base.
Case 5
A 40-year-old woman was seen at the reference centre for the management of PLCH. The diagnosis was obtained 6 years previously following a surgical lung biopsy. She had been a heavy cigarette smoker (average 30 cigarettes·day−1) since she was 16 years old and continued to smoke 10 cigarettes·day−1 when she was referred for treatment. At that time, she complained of mild NYHA stage 2 dyspnoea, and HRCT showed a nodulocystic pattern and some linear densities (figure 4a). Her lung function parameters were within the normal ranges, except for DLCO (54% predicted). Her arterial blood gases while breathing room air and the 6MWT were normal. Skeletal survey radiography showed a unifocal asymptomatic small lytic lesion of the left femur. The patient was strongly encouraged to stop smoking, which she managed to do transiently several times during follow-up. Unfortunately, she resumed smoking each time and finally persisted in smoking 8 cigarettes·day−1. She became progressively more dyspnoeic. After 3 years of follow-up, her lung function deteriorated (TLC 98% predicted, VC 82% predicted, FEV1 66% predicted, FEV1/FVC 69% predicted and DLCO 40% predicted). HRCT showed diffuse cystic lesions (figure 4b). Cladribine was proposed, but the patient declined this treatment.
FIGURE 4.

Serial high-resolution computed tomography (HRCT) scans at the lung window from patient 5. a) At initial evaluation, there was a nodulocystic pattern with scattered linear densities. b) Three years later, there were diffuse confluent lung cystic lesions.
After 5 years of follow-up, her lung function parameters were: TLC 97% predicted, VC 75% predicted, FEV1 65% predicted and DLCO 30% predicted. Doppler echocardiography measured systolic pulmonary arterial pressure (PAP) at 48 mmHg with mild dilatation of the right ventricle. Pre-capillary pulmonary hypertension was confirmed through right heart catheterisation (mean PAP 33 mmHg and cardiac index 3.38 L·min−1·m−2) and was considered secondary to hypoxaemia (arterial oxygen tension 55 mmHg breathing room air), for which the patient received oxygen and furosemide. Two years later, the patient required higher oxygen flow. The lung volumes were virtually stable, but DLCO had decreased to 15% predicted. Lung transplantation was considered, but unfortunately smoking cessation was not achieved. A new right heart catheterisation showed severe pulmonary hypertension (mean PAP 62 mmHg and cardiac index 2.8 L·min−1·m−2). Her furosemide dosage was increased and bosentan was initiated, but not well tolerated and was associated with worsening hypoxaemia. Ultimately, as the patient was weaned from tobacco, she received a lung transplant with an uneventful surgery 9 years after her initial evaluation at the centre.
Key features of adult PLCH
Clinical presentation
At clinical presentation [13, 14], PLCH is diagnosed by three main symptoms. 1) Respiratory symptoms (cough and dyspnoea on exertion), which occur in approximately two-thirds of all such cases. Constitutional manifestations (asthenia, fever, night sweats and weight loss) may be associated with 10–20% of these patients. 2) Spontaneous pneumothorax, which is observed in 15–20% of cases. It may occur at any time during the course of the disease and may be bilateral and/or recurrent, and should be suspected in any patient complaining of increased dyspnoea or chest pain. 3) Lesions in routine chest radiography, which occur in 10–25% of asymptomatic patients.
Haemoptysis is uncommon and should not be attributed to PLCH until other aetiologies, such as acute bronchitis, bronchogenic carcinoma and the rare Aspergillus colonisation of a cystic lung cavity, and alternative diagnoses have been ruled out.
Adult PLCH is generally isolated. When present, LCH extrathoracic lesions usually involve the bones (lytic lesion), pituitary stalk (diabetes insipidus) and, more rarely, the skin. Physical examination is generally normal, except in cases of pneumothorax, advanced stages or when associated with extrathoracic involvement. Rales are rarely present and clubbing is extremely rare.
Imaging
Chest radiography
Standard chest radiography typically shows bilateral symmetric reticulo-micronodular infiltration, in which cysts may be identified, predominantly involving the upper and middle lung fields but sparing the costophrenic angles [8, 9]. Lung volumes are usually preserved or increased. A pneumothorax or, more rarely, a lytic lesion in a rib may be visible, which can provide diagnostic clues. Pleural effusion is not a feature and mediastinal adenopathy is unusual. Hilar enlargement suggests pulmonary hypertension. In later stages, chest radiography may show an emphysema-like appearance. Rarely, chest radiography may be normal.
High-resolution computed tomography
HRCT is mandatory for every suspected case of PLCH [8, 9]. It can visualise parenchymal lesions, such as nodules or cysts that are not readily visible on standard chest radiography. The typical HRCT pattern combines small, poorly limited nodules, cavitated nodules and thick- and thin-walled cysts predominantly in the upper and middle lung fields, with a relative sparing of the bases. The lesions are focal and are separated by apparently normal parenchyma, and they affect both the peripheral and the central parts of the lung fields.
The lung lesions vary with disease duration. As the disease evolves, cystic lesions become a predominant finding. Cysts vary in size and may coalesce to form irregular shapes (“bizarre cysts”). Other findings in PLCH may include linear opacities or ground-glass opacities and localised emphysema secondary to cigarette smoking [19]. Significantly enlarged mediastinal lymph nodes are rarely observed and should suggest another diagnosis. Pulmonary artery enlargement suggests that pulmonary hypertension is present.
Notably, in patients who developed PLCH as teenagers and are seen when they are adults, pulmonary lesions are common in the lung bases [20]. Finally, HRCT is essential for selecting an optimal site for surgical biopsy, if required.
18F-fluorodeoxyglucose-positron emission tomography-CT
Whereas 18F-fluorodeoxyglucose-positron emission tomography-CT is useful for the evaluation of the extent of multisystem LCH and multifocal bone disease, its role in the assessment of isolated PLCH needs to be better defined [21, 22]. Pulmonary nodules can be hypermetabolic and they cannot be distinguished from malignant disease [21, 22]. Thick-walled cysts can occasionally also demonstrate high metabolic uptake, but they are not predictive of the natural disease course or the response of PLCH to treatment in a small series [21, 22].
Lung function
Pulmonary function abnormalities are variable, and depend on both the predominant anatomical lesions and the extent of cystic lesions on HRCT. Obstructive, restrictive and mixed patterns have been described. In the early stages, pulmonary function testing may be normal in ∼10% of patients [13]. The most common abnormality is a reduction of DLCO, which is observed in 80–90% of cases and is primarily a reflection of pulmonary vascular dysfunction [13, 14, 23].
The typical functional profile combines a reduction in VC, normal or increased RV, preserved TLC and increased or normal RV/TLC ratio (air trapping). Obstructive dysfunction with airflow limitation is observed in a sizeable proportion of patients, particularly in those with advanced cystic disease. A restrictive ventilatory defect (defined by TLC <80% predicted) is present in a minority of cases [13]. Patients with a predominantly nodular HRCT pattern usually have minimal lung function alterations with an isolated reduction in DLCO. The extent of cystic changes in HRCT is correlated with lung function abnormalities, including airflow obstruction [15, 24].
Blood gas levels at rest remain normal for a long time, but exertion may cause alveolar arterial oxygen gradient elevation and hypoxaemia [25, 26]. 6MWT distance may be impaired in patients with advanced disease [16].
Bronchoscopy and BAL
The macroscopic appearance of the bronchial tree is normal or shows only nonspecific inflammation related to smoking. Bronchial mucosa biopsy specimens do not confirm the diagnosis of PLCH, but are helpful for ruling out an alternative diagnosis in atypical cases.
Transbronchial lung biopsies may be diagnostic for PLCH in expert centres with variable diagnostic yields (15–40%) due to the focal nature of histological lesions. Multiple specimens should be obtained and examined extensively using immunohistochemical techniques [27–29].
BAL usually reflects smoking exposure and shows an increased number of cells recovered, with a marked predominance of alveolar macrophages. Differential BAL fluid cell counts may show a moderate nonspecific increase in eosinophils. The proportion of alveolar lymphocytes is normal or diminished, with a decreased CD4/CD8 ratio related to smoking.
The identification of ≥5% CD1a+ cells in BAL was observed only in PLCH, but has a low sensitivity [27, 30, 31]. False-positive results are common and the specimens must be examined by an experienced cytologist. Thus, BAL rarely establishes a definite diagnosis for PLCH in adults, but rather provides additional orientation by showing macrophage alveolitis. It is useful to differentiate PLCH from interstitial lung diseases with more characteristic BAL findings, such as sarcoidosis in patients with predominant nodular lesions on HRCT. BAL also allows lung infections to be ruled out, particularly in patients with constitutional symptoms.
Routine laboratory tests are usually normal. A mild increase in peripheral neutrophil counts and blood inflammatory markers unrelated to patient smoking consumption may be observed [32]. Blood eosinophil counts are normal. In the rare patients who have lymphopenia, other diagnoses should be ruled out, such as HIV infection [32].
Diagnosis of PLCH
Although transbronchial lung biopsies may show LCH granulomas, a definitive diagnosis of PLCH is most commonly obtained through video-assisted thoracoscopic surgical biopsy guided by HRCT findings. The biopsy specimens should ideally be taken from nodular areas of lung HRCT. The histological hallmark of PLCH is the accumulation of CD1a+/CD207+ cells organised into loosely formed granulomas, which are preferentially located in and destroy the wall of distal bronchioles [33, 34]. Large numbers of lymphocytes and inflammatory cells, specifically macrophages and eosinophils, are also present in early LCH lesions [33, 34].
With the progression of lesions, the numbers of CD1a+ cells decrease and are subsequently replaced by either fibrosis in the form of a characteristic stellate scar or a cystic cavity surrounded by a fibrous ring. However, CD1a+ cells may still be observed in cystic forms on HRCT and inflammatory cells may persist even inside thin-walled cysts [35, 36].
The indication for a lung biopsy must be determined in each individual case, with a careful evaluation of risks and benefits of the procedure. In a young adult smoker with mild or no symptoms and a typical nodulocystic HRCT pattern, a presumptive diagnosis is acceptable with a close follow-up. Conversely, a surgical lung biopsy is performed during surgical pleurodesis in patients with recurrent or persistent pneumothorax. In contrast, the biopsy of an extrathoracic lesion, such as a lesion in bone or skin, may confirm the diagnosis if the pulmonary manifestations are consistent with PLCH. In patients with extensive cystic lesions and impaired lung function, the risk of a surgical lung biopsy should be balanced with the need for a definitive diagnosis.
The differential diagnoses to consider vary according to nodulocystic or isolated cystic patterns in lung HRCT and have been recently reviewed [10, 37, 38].
Outcome and prognosis
The natural history of the disease is variable and it is difficult to predict in an individual patient. Partial or complete resolution of lung HRCT abnormalities may occur without treatment, particularly after smoking cessation. Up to 40% of patients may experience a significant (≥15%) decline of FEV1 or DLCO during the first 2 years after diagnosis [13]. At 5 years, ∼50% of patients will experience impaired pulmonary function over time and some will develop obstructive lung disease [15]. In some patients, despite regression of the disease, pulmonary function continues to deteriorate due to smoking-related COPD. Finally, in 10–20% of patients, early severe manifestations are present at diagnosis and they evolve to progressive respiratory failure with chronic cor pulmonale [14, 39].
Retrospective studies have found that altered lung function parameters, particularly FEV1 and DLCO, could predict worse outcomes [14, 15, 39]. However, only pulmonary hypertension is a reliable factor of a pejorative prognosis in an individual patient [16, 40, 41]. In a recent multicentre prospective study, the persistence of smoking was associated with an increased risk of lung function deterioration [13].
Pregnancy does not appear to modify the course of PLCH, but certain precautions, such as a caesarean section, are required for women with diffuse cystic lesions and impaired pulmonary function due to the risk of pneumothorax during labour.
In two retrospective studies, the median survival of patients with PLCH was found to be significantly shorter compared with individuals of the same sex and age, but mortality in these studies is probably overestimated due to the number of patients lost to follow-up [14, 39]. In addition to the association with lymphoma, particularly Hodgkin's lymphoma, increased incidences of primary lung cancer (related to continued smoking) as well as various other malignancies have also been reported [14, 42–44].
Initial evaluation and follow-up
At the diagnosis of PLCH, a thorough history and a comprehensive physical examination (including ear/nose/throat) and stomatology are essential to search for extrapulmonary LCH involvement. It is recommended that a skeletal radiograph survey be performed, including a dental panoramic radiograph [7]. However, if patients are meticulously and clinically evaluated for the absence of extrathoracic LCH manifestations, the systematic search for bone involvement seems uninformative [32].
A complete blood count, blood chemistry analysis (including total protein, electrolyte, creatinine, bilirubin, alanine aminotransferase, aspartic aminotransferase, alkaline phosphatase, γ-glutamyl transpeptidase, C-reactive protein and fibrinogen levels) and protein electrophoresis are systematically performed [7, 32]. They can rarely reveal LCH-related abnormalities (such as cholestasis in liver LCH involvement) and are useful for distinguishing alternative diagnoses or for patient therapeutic management [32, 45, 46]. Morning urine osmolality is a screening test for suspected diabetes insipidus [7].
Follow-up with a physical examination, chest radiography and lung function tests should be performed periodically for PLCH. Patients with recently diagnosed PLCH may experience early progression of their disease [13]. It is recommended that all patients undergo follow-up every 3–6 months for the first year after diagnosis [7, 8, 13]. Systematic sequential chest CT scans add little information [13, 15]. In practice, we perform a serial lung HRCT only in those patients who exhibit changes in their clinical, functional or chest radiography status during follow-up. Doppler echocardiography is indicated in patients with unexplained dyspnoea or isolated/disproportionate decrease in DLCO to search for pulmonary hypertension, which needs to be confirmed by cardiac catheterisation [16, 40, 47].
Long-term follow-up is recommended and may detect respiratory dysfunction [14, 15] or a rare relapse with recurrent nodule formation [48].
Pathogenesis
The cell-specific gene expression signature in Langerin (CD207)+ cells within LCH lesions has shown that these cells are more consistent with immature myeloid dendritic cell precursors than Langerhans cells [49, 50].
Very few LCH cells proliferate in the lesions [51, 52]. The accumulation of CD1a+ cells in granulomas around small airways mainly results from recruitment in response to various soluble factors (particularly granulocyte–macrophage colony-stimulating factor (GM-CSF) and CCL20 chemokine) of circulating peripheral blood myeloid haematopoietic precursor cells, which then differentiate in the tissues that are involved [53–56]. Additionally, the CD1a+ cells in PLCH granulomas appear to be less sensitive to apoptosis [57].
A specific characteristic of PLCH granulomas is their capacity to destroy and remodel surrounding tissues. CD1a+ cells of PLCH granulomas express, to varying degrees, membrane maturation markers (especially costimulation molecules) similar to those that are present on the surface of dendritic cells after exposure to pathogens or activating cytokines [58]. CD1a+ cells extracted from bone LCH granulomas appear, however, to be functionally impaired [59]. An important proportion of the T-lymphocytes that infiltrate LCH granulomas are regulatory T-lymphocytes [51]. Various metalloproteinases have been identified in LCH granulomas and could account for LCH-induced tissue destruction [49, 50, 60]. Recently, the activation of the Notch1 signalling pathway has been shown to be at least partly responsible for the specific profile of LCH cells [49].
The strong link between smoking and PLCH highly suggests that tobacco plays a role in the pathogenesis of PLCH. The role for smoking in triggering PLCH is highlighted by the finding that children with extrapulmonary LCH who subsequently develop PLCH during adolescence or adulthood are often smokers [61]. Smoking induces accumulation of CD1a+ cells in the lungs in healthy smokers as well as in COPD patients [62–64]. It stimulates local production of cytokines, which are important for the recruitment and differentiation of dendritic cells (particularly tumour necrosis factor-α, GM-CSF, transforming growth factor-β and CCL20), and these cytokines are expressed in PLCH lesions [56, 65–67]. Osteopontin also appears to play a role in PLCH because large quantities of osteopontin are present in the BAL of patients with PLCH compared with control smokers [68]. Tobacco smoke also promotes the survival of dendritic cells through anti-apoptotic mechanisms [63].
An important breakthrough in the understanding of LCH was the recent identification of a recurrent BRAFV600E mutation in ∼50% of LCH lesions [17], including PLCH lesions [69–72]. The presence of this mutation can be determined both by molecular genotyping and immunohistochemistry with paraffin-embedded specimens.
The BRAFV600E mutation is present in various types of cancers, including malignant melanomas and almost all cases of hairy cell leukaemia [73, 74]. However, BRAFV600E somatic mutation does not necessarily mean that LCH is a malignant disease because this mutation was also observed in benign nevi [75]. Interestingly, in children with severe systemic forms of LCH, the BRAFV600E mutation may also be detected in circulating and sometimes bone marrow precursors of dendritic cells [76].
The mitogen-activated protein kinase (MAPK) pathway is activated in LCH lesions regardless of the presence or not of the BRAFV600E mutation and may be activated by mutations in other key factors of this signalling cascade [17]. Similar to other forms of LCH, MAP2K1 (MEK1) mutations were observed in a subset of BRAF-wild-type PLCH lesions [69, 77–79]. Recently, activating NRASQ61K/R mutations have been described in a substantial subset of PLCH lesions, whereas these mutations were only exceptionally reported in other forms of LCH (figure 5) [69]. These NRAS mutations occurred concurrently with BRAFV600E mutations in most cases and both mutations were carried by different cell clones [69].
FIGURE 5.
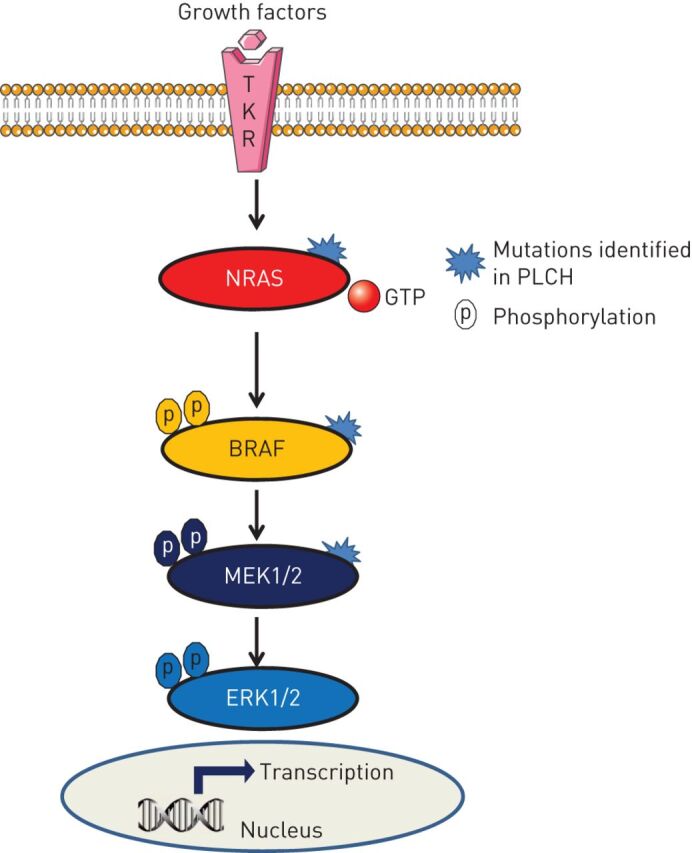
The mitogen-activated protein kinase (MAPK) pathway showing mutations identified in pulmonary Langerhans cell histiocytosis (PLCH). TKR: tyrosine kinase receptor; GTP: guanosine triphosphate; MEK: MAPK kinase; ERK: extracellular signal-regulated kinase. Under physiological conditions, MAPK is activated through binding growth factors to the TKR. The phosphorylation of downstream factors ultimately results in the translocation of phosphorylated ERK1/2 into the nucleus and the activation of transcription for genes involved in cell activation. The MAPK pathway is constitutively activated downstream of each mutated factor.
Recently, a significant expression of the programmed cell death PD-1/PDL-1 immune checkpoint inhibitors and T-regulatory cells was shown to be present in the micro-environment of LCH lesions, and these markers were correlated with the presence of the BRAFV600E mutation [80–82].
The correlation of MAPK mutations with clinical presentation and prognosis of PLCH is currently being evaluated. In children with LCH, the BRAFV600E mutation is associated with an increased risk of recurrence in systemic LCH, a high-risk disease (with risk organs) with increased resistance to first-line therapy [76, 83].
Taken together, LCH is currently considered an inflammatory myeloid neoplasm with variable clinical expression [84]. Smoking probably plays a triggering role in the development of lung localisation. Another exogenous cofactor, such as viral infection, has been suspected but has not yet been identified [85].
Treatment
As the natural history of PLCH is mostly unpredictable in the individual patient and the disease may spontaneously resolve, the first step for patient management is observation with a close follow-up [13]. Smoking cessation is essential in all patients and they may need to be enrolled in individualised programmes. It is the only required intervention in a significant proportion of patients [86]. Combined inhaled corticosteroid and long-acting β2-agonist therapy may provide benefit to patients with wheezing and obstructive lung disease [13]. Pneumothorax management may be particularly difficult and requires almost constant drainage. Surgical pleurodesis is indicated in cases of recurrence, and is often needed for cases of a single large pneumothorax because of a frequent and prolonged air leak and a high rate of recurrence with a conservative approach [87]. Ideally, these patients should be referred to centres with expertise in these procedures. Pleurectomy is best avoided for patients who may be candidates for lung transplantation in the future, but pleurectomy is occasionally inevitable because of recurring pneumothoraces.
Lower respiratory tract infection is a common cause of deterioration for PLCH and should be promptly treated. An annual vaccination against influenza and an antipneumococcal vaccine are recommended for patients with impaired lung function.
Systemic treatment is considered for symptomatic patients with impaired lung function.
Oral corticosteroids are prescribed by some physicians in patients with progressive disease [88], although there is no controlled proof of their efficacy in stabilising or inducing disease remission. Vinblastine, which is the cornerstone treatment of LCH in children [6, 89], has limited efficacy in PLCH [90].
Cladribine (2-chlorodeoxyadenosine), which is a purine nucleoside analogue and is directly toxic for lymphocyte and monocyte cells, has been used as a second-line treatment in multisystem LCH [91, 92] and has been reported to induce remission or improve lung disease in several PLCH cases [18, 93–96]. As this treatment is highly immunosuppressive (inducing prolonged lymphopenia), prolonged infection prophylaxis of Pneumocystis jirovecii and herpes viruses should also be included in the treatment. The efficacy and tolerance of this promising treatment for symptomatic forms of PLCH with impaired pulmonary function is currently being evaluated in a phase II clinical trial (Clinicaltrials.gov: identifier NCT01473797).
Hypoxaemia is treated with supplemental oxygen. Symptomatic treatment is often needed in cases of pulmonary hypertension. The role of vasodilator therapy for the treatment PLCH/pulmonary hypertension is not well established, and should be reserved for centres with expertise in both vascular and advanced lung disease [16, 97, 98]. Patients with advanced PLCH benefit from lung transplantation [41]. Disease relapse in the transplanted lungs has been described, especially for patients with pre-operative extrapulmonary manifestations and patients who resume smoking following transplantation [41, 99].
Perspectives
As a result of the progress made in recent years, it is possible to provide recommendations for the management of patients with PLCH in clinical practice. Smoking cessation (and weaning the patient off other addictive substances) remains an important challenge that, at best, can be achieved through individualised programmes and psychological support whenever necessary. Patient associations are very helpful for patients who are often discouraged when coping with this orphan disease. Another important goal is the search for predictive factors that can be reliably used at the individual level. Better knowledge of the pathogenesis of LCH has led to the use of targeted therapies, including BRAF or MEK (MAPK kinase) inhibitors, in patients with severe disease. Cooperative efforts are needed to rigorously evaluate the efficacy and tolerance of these treatments due to their potentially severe side-effects.
Disclosures
A. Tazi ERR-0070-2017_Tazi (1.2MB, pdf)
Acknowledgements
The authors thank M. Mao and E. Savariau (Service de Pneumologie, Assistance Publique-Hôpitaux de Paris, Hôpital Saint-Louis, Paris, France) for their technical assistance.
Authorship contributions: G. Lorillon collected data for clinical situations and drafted the manuscript; A. Tazi conceived the design and edited the manuscript. Both authors approved the final version of the manuscript and take responsibility for the content of this review.
Footnotes
Conflict of interest: Disclosures can be found alongside this article at err.ersjournals.com
Provenance: Commissioned article, peer reviewed.
References
- 1.Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage–dendritic cell lineages. Blood 2016; 127: 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weitzman S, Egeler RM. Langerhans cell histiocytosis: update for the pediatrician. Curr Opin Pediatr 2008; 20: 23–29. [DOI] [PubMed] [Google Scholar]
- 3.Aricò M, Girschikofsky M, Généreau T, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer 2003; 39: 2341–2348. [DOI] [PubMed] [Google Scholar]
- 4.Aricò M, Nichols K, Whitlock JA, et al. Familial clustering of Langerhans cell histiocytosis. Br J Haematol 1999; 107: 883–888. [DOI] [PubMed] [Google Scholar]
- 5.Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer 2008; 51: 71–75. [DOI] [PubMed] [Google Scholar]
- 6.Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 2013; 60: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis 2013; 8: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suri H, Yi ES, Nowakowski GS, et al. Pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis 2012; 7: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tazi A. Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J 2006; 27: 1272–1285. [DOI] [PubMed] [Google Scholar]
- 10.Gupta N, Vassallo R, Wikenheiser-Brokamp KA, et al. Diffuse cystic lung disease. Part I. Am J Respir Crit Care Med 2015; 191: 1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomeer M, Demedts M, Vandeurzen K. Registration of interstitial lung diseases by 20 centres of respiratory medicine in Flanders. Acta Clin Belg 2001; 56: 163–172. [DOI] [PubMed] [Google Scholar]
- 12.Gaensler E, Carrington C. Open biopsy for chronic diffuse infiltrative lung disease: clinical, reontgenographic, and physiological correlations in 502 patients. Ann Thor Surg 1980; 30: 411–426. [DOI] [PubMed] [Google Scholar]
- 13.Tazi A, de Margerie C, Naccache JM, et al. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis 2015; 10: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vassallo R, Ryu JH, Schroeder DR, et al. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med 2002; 346: 484–490. [DOI] [PubMed] [Google Scholar]
- 15.Tazi A, Marc K, Dominique S, et al. Serial computed tomography and lung function testing in pulmonary Langerhans’ cell histiocytosis. Eur Respir J 2012; 40: 905–912. [DOI] [PubMed] [Google Scholar]
- 16.Le Pavec J, Lorillon G, Jais X, et al. Pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension: clinical characteristics and impact of pulmonary arterial hypertension therapies. Chest 2012; 142: 1150–1157. [DOI] [PubMed] [Google Scholar]
- 17.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010; 116: 1919–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lorillon G, Bergeron A, Detourmignies L, et al. Cladribine is effective against cystic pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med 2012; 186: 930–932. [DOI] [PubMed] [Google Scholar]
- 19.Vassallo R, Jensen EA, Colby TV, et al. The overlap between respiratory bronchiolitis and desquamative interstitial pneumonia in pulmonary Langerhans cell histiocytosis: high-resolution CT, histologic, and functional correlations. Chest 2003; 124: 1199–1205. [DOI] [PubMed] [Google Scholar]
- 20.Seely JM, Salahudeen S Sr, Cadaval-Goncalves AT, et al. Pulmonary Langerhans cell histiocytosis: a comparative study of computed tomography in children and adults. J Thorac Imaging 2012; 27: 65–70. [DOI] [PubMed] [Google Scholar]
- 21.Obert J, Vercellino L, Van Der Gucht A, et al. 18F-fluorodeoxyglucose positron emission tomography-computed tomography in the management of adult multisystem Langerhans cell histiocytosis. Eur J Nucl Med Mol Imaging 2017; 44: 598–610. [DOI] [PubMed] [Google Scholar]
- 22.Krajicek BJ, Ryu JH, Hartman TE, et al. Abnormal fluorodeoxyglucose PET in pulmonary Langerhans cell histiocytosis. Chest 2009; 135: 1542–1549. [DOI] [PubMed] [Google Scholar]
- 23.Crausman RS, King TE Jr. Pulmonary vascular involvement in pulmonary histiocytosis X. Chest 1997; 112: 1714. [DOI] [PubMed] [Google Scholar]
- 24.Canuet M, Kessler R, Jeung MY, et al. Correlation between high-resolution computed tomography findings and lung function in pulmonary Langerhans cell histiocytosis. Respiration 2007; 74: 640–646. [DOI] [PubMed] [Google Scholar]
- 25.Rolland-Debord C, Fry S, Giovannelli J, et al. Physiologic determinants of exercise capacity in pulmonary Langerhans cell histiocytosis: a multidimensional analysis. PLoS One 2017; 12: e0170035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crausman RS, Jennings CA, Tuder RM, et al. Pulmonary histiocytosis X: pulmonary function and exercise pathophysiology. Am J Respir Crit Care Med 1996; 153: 426–435. [DOI] [PubMed] [Google Scholar]
- 27.Baqir M, Vassallo R, Maldonado F, et al. Utility of bronchoscopy in pulmonary Langerhans cell histiocytosis. J Bronchology Interv Pulmonol 2013; 20: 309–312. [DOI] [PubMed] [Google Scholar]
- 28.Houssini I, Tomashefski J, Cohen A. Transbronchial biopsy in patients with pulmonary eosinophilic granuloma. Arch Pathol Lab Med 1994; 118: 523–530. [PubMed] [Google Scholar]
- 29.Travis WD, Borok Z, Roum JH, et al. Pulmonary Langerhans cell granulomatosis (histiocytosis X). A clinicopathologic study of 48 cases. Am J Surg Pathol 1993; 17: 971–986. [DOI] [PubMed] [Google Scholar]
- 30.Harari S, Torre O, Cassandro R, et al. Bronchoscopic diagnosis of Langerhans cell histiocytosis and lymphangioleiomyomatosis. Respir Med 2012; 106: 1286–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tazi A, Soler P, Hance AJ. Adult pulmonary Langerhans’ cell histiocytosis. Thorax 2000; 55: 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tazi A, de Margerie-Mellon C, Vercellino L, et al. Extrathoracic investigation in adult patients with isolated pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis 2016; 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roden AC, Yi ES. Pulmonary Langerhans cell histiocytosis: an update from the pathologists’ perspective. Arch Pathol Lab Med 2016; 140: 230–240. [DOI] [PubMed] [Google Scholar]
- 34.Kambouchner M, Basset F, Marchal J, et al. Three-dimensional characterization of pathologic lesions in pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med 2002; 166: 1483–1490. [DOI] [PubMed] [Google Scholar]
- 35.Kim HJ, Lee KS, Johkoh T, et al. Pulmonary Langerhans cell histiocytosis in adults: high-resolution CT – pathology comparisons and evolutional changes at CT. Eur Radiol 2011; 21: 1406–1415. [DOI] [PubMed] [Google Scholar]
- 36.Soler P, Bergeron A, Kambouchner M, et al. Is high-resolution computed tomography a reliable tool to predict the histopathological activity of pulmonary Langerhans cell histiocytosis? Am J Respir Crit Care Med 2000; 162: 264–270. [DOI] [PubMed] [Google Scholar]
- 37.Raoof S, Bondalapati P, Vydyula R, et al. Cystic lung diseases: algorithmic approach. Chest 2016; 150: 945–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta N, Vassallo R, Wikenheiser-Brokamp KA, et al. Diffuse cystic lung disease. Part II. Am J Respir Crit Care Med 2015; 192: 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delobbe A, Durieu J, Duhamel A, et al. Determinants of survival in pulmonary Langerhans’ cell granulomatosis (histiocytosis X). Groupe d'Etude en Pathologie Interstitielle de la Societe de Pathologie Thoracique du Nord. Eur Respir J 1996; 9: 2002–2006. [DOI] [PubMed] [Google Scholar]
- 40.Chaowalit N, Pellikka PA, Decker PA, et al. Echocardiographic and clinical characteristics of pulmonary hypertension complicating pulmonary Langerhans cell histiocytosis. Mayo Clin Proc 2004; 79: 1269–1275. [DOI] [PubMed] [Google Scholar]
- 41.Dauriat G, Mal H, Thabut G, et al. Lung transplantation for pulmonary Langerhans’ cell histiocytosis: a multicenter analysis. Transplantation 2006; 81: 746–750. [DOI] [PubMed] [Google Scholar]
- 42.Feuillet S, Louis L, Bergeron A, et al. Pulmonary Langerhans cell histiocytosis associated with Hodgkin's lymphoma. Eur Respir Rev 2010; 19: 86–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egeler RM, Neglia JP, Arico M, et al. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am 1998; 12: 369–378. [DOI] [PubMed] [Google Scholar]
- 44.Sadoun D, Vaylet F, Valeyre D, et al. Bronchogenic carcinoma in patients with pulmonary histiocytosis X. Chest 1992; 101: 1610–1613. [DOI] [PubMed] [Google Scholar]
- 45.Araujo B, Costa F, Lopes J, et al. Adult Langerhans cell histiocytosis with hepatic and pulmonary involvement. Case Rep Radiol 2015; 2015: 536328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdallah M, Genereau T, Donadieu J, et al. Langerhans’ cell histiocytosis of the liver in adults. Clin Res Hepatol Gastroenterol 2011; 35: 475–481. [DOI] [PubMed] [Google Scholar]
- 47.Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30: 2493–2537. [DOI] [PubMed] [Google Scholar]
- 48.Tazi A, Montcelly L, Bergeron A, et al. Relapsing nodular lesions in the course of adult pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med 1998; 157: 2007–2010. [DOI] [PubMed] [Google Scholar]
- 49.Hutter C, Kauer M, Simonitsch-Klupp I, et al. Notch is active in Langerhans cell histiocytosis and confers pathognomonic features on dendritic cells. Blood 2013; 120: 5199–5208. [DOI] [PubMed] [Google Scholar]
- 50.Allen CE, Li L, Peters TL, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol 2010; 184: 4557–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Senechal B, Elain G, Jeziorski E, et al. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med 2007; 4: e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brabencova E, Tazi A, Lorenzato M, et al. Langerhans cells in Langerhans cell granulomatosis are not actively proliferating cells. Am J Pathol 1998; 152: 1143–1149. [PMC free article] [PubMed] [Google Scholar]
- 53.Rolland A, Guyon L, Gill M, et al. Increased blood myeloid dendritic cells and dendritic cell-poietins in Langerhans cell histiocytosis. J Immunol 2005; 174: 3067–3071. [DOI] [PubMed] [Google Scholar]
- 54.Annels NE, Da Costa CE, Prins FA, et al. Aberrant chemokine receptor expression and chemokine production by Langerhans cells underlies the pathogenesis of Langerhans cell histiocytosis. J Exp Med 2003; 197: 1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caux C, Vanbervliet B, Massacrier C, et al. Regulation of dendritic cell recruitment by chemokines. Transplantation 2002; 73: S7–S11. [DOI] [PubMed] [Google Scholar]
- 56.Tazi A, Bonay M, Bergeron A, et al. Role of granulocyte-macrophage colony stimulating factor (GM-CSF) in the pathogenesis of adult pulmonary histiocytosis X. Thorax 1996; 51: 611–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marchal J, Kambouchner M, Tazi A, et al. Expression of apoptosis-regulatory proteins in lesions of pulmonary Langerhans cell histiocytosis. Histopathology 2004; 45: 20–28. [DOI] [PubMed] [Google Scholar]
- 58.Tazi A, Moreau J, Bergeron A, et al. Evidence that Langerhans cells in adult pulmonary Langerhans cell histiocytosis are mature dendritic cells: importance of the cytokine microenvironment. J Immunol 1999; 163: 3511–3515. [PubMed] [Google Scholar]
- 59.Geissmann F, Lepelletier Y, Fraitag S, et al. Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood 2001; 97: 1241–1248. [DOI] [PubMed] [Google Scholar]
- 60.Hayashi T, Rush WL, Travis WD, et al. Immunohistochemical study of matrix metalloproteinases and their tissue inhibitors in pulmonary Langerhans’ cell granulomatosis. Arch Pathol Lab Med 1997; 121: 930–937. [PubMed] [Google Scholar]
- 61.Bernstrand C, Cederlund K, Sandstedt B, et al. Pulmonary abnormalities at long-term follow-up of patients with Langerhans cell histiocytosis. Med Pediatr Oncol 2001; 36: 459–468. [DOI] [PubMed] [Google Scholar]
- 62.Van Pottelberge GR, Bracke KR, Demedts IK, et al. Selective accumulation of Langerhans-type dendritic cells in small airways of patients with COPD. Respir Res 2010; 11: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vassallo R, Walters PR, Lamont J, et al. Cigarette smoke promotes dendritic cell accumulation in COPD; a Lung Tissue Research Consortium study. Respir Res 2010; 11: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soler P, Moreau A, Basset F, et al. Cigarette smoking-induced changes in the number and differentiated state of pulmonary dendritic cells/Langerhans cells. Am Rev Respir Dis 1989; 139: 1112–1117. [DOI] [PubMed] [Google Scholar]
- 65.Hashimoto M, Yanagisawa H, Minagawa S, et al. TGF-beta-dependent dendritic cell chemokinesis in murine models of airway disease. J Immunol 2015; 195: 1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bracke KR, D'Hulst A I, Maes T, et al. Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient mice. J Immunol 2006; 177: 4350–4359. [DOI] [PubMed] [Google Scholar]
- 67.Reibman J, Hsu Y, Chen LC, et al. Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. Am J Respir Cell Mol Biol 2003; 28: 648–654. [DOI] [PubMed] [Google Scholar]
- 68.Prasse A, Stahl M, Schulz G, et al. Essential role of osteopontin in smoking-related interstitial lung diseases. Am J Pathol 2009; 174: 1683–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mourah S, How-Kit A, Meignin V, et al. Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J 2016; 47: 1785–1796. [DOI] [PubMed] [Google Scholar]
- 70.Chilosi M, Facchetti F, Caliò A, et al. Oncogene-induced senescence distinguishes indolent from aggressive forms of pulmonary and non-pulmonary Langerhans cell histiocytosis. Leuk Lymphoma 2014; 55: 2620–2626. [DOI] [PubMed] [Google Scholar]
- 71.Roden AC, Hu X, Kip S, et al. BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol 2014; 38: 548–551. [DOI] [PubMed] [Google Scholar]
- 72.Yousem SA, Dacic S, Nikiforov YE, et al. Pulmonary Langerhans cell histiocytosis: profiling of multifocal tumors using next-generation sequencing identifies concordant occurrence of BRAF V600E mutations. Chest 2013; 143: 1679–1684. [DOI] [PubMed] [Google Scholar]
- 73.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–954. [DOI] [PubMed] [Google Scholar]
- 74.Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011; 364: 2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet 2003; 33: 19–20. [DOI] [PubMed] [Google Scholar]
- 76.Berres ML, Lim KP, Peters T, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014; 211: 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kamionek M, Ahmadi Moghaddam P, Sakhdari A, et al. Mutually exclusive extracellular signal-regulated kinase pathway mutations are present in different stages of multi-focal pulmonary Langerhans cell histiocytosis supporting clonal nature of the disease. Histopathology 2016; 69: 499–509. [DOI] [PubMed] [Google Scholar]
- 78.Brown NA, Furtado LV, Betz BL, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood 2014; 124: 1655–1658. [DOI] [PubMed] [Google Scholar]
- 79.Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 2014; 124: 3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zeng K, Wang Z, Ohshima K, et al. BRAF V600E mutation correlates with suppressive tumor immune microenvironment and reduced disease-free survival in Langerhans cell histiocytosis. Oncoimmunology 2016; 5: e1185582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu J, Sun HH, Fletcher CD, et al. Expression of programmed cell death 1 ligands (PD-L1 and PD-L2) in histiocytic and dendritic cell disorders. Am J Surg Pathol 2016; 40: 443–453. [DOI] [PubMed] [Google Scholar]
- 82.Gatalica Z, Bilalovic N, Palazzo JP, et al. Disseminated histiocytoses biomarkers beyond BRAFV600E: frequent expression of PD-L1. Oncotarget 2015; 6: 19819–19825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Héritier S, Emile JF, Barkaoui MA, et al. BRAF mutation correlates with high-risk Langerhans cell histiocytosis and increased resistance to first-line therapy. J Clin Oncol 2016; 34: 3023–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to Histiocytosis X? Br J Haematol 2015; 169: 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jeziorski E, Senechal B, Molina TJ, et al. Herpes-virus infection in patients with Langerhans cell histiocytosis: a case-controlled sero-epidemiological study, and in situ analysis. PLoS One 2008; 3: e3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Elia D, Torre O, Cassandro R, et al. Pulmonary Langerhans cell histiocytosis: a comprehensive analysis of 40 patients and literature review. Eur J Intern Med 2015; 26: 351–356. [DOI] [PubMed] [Google Scholar]
- 87.Mendez JL, Nadrous HF, Vassallo R, et al. Pneumothorax in pulmonary Langerhans cell histiocytosis. Chest 2004; 125: 1028–1032. [DOI] [PubMed] [Google Scholar]
- 88.Schönfeld N, Frank W, Wenig S, et al. Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration 1993; 60: 38–44. [DOI] [PubMed] [Google Scholar]
- 89.Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood 2015; 126: 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tazi A, Lorillon G, Haroche J, et al. Vinblastine chemotherapy in adult patients with Langerhans cell histiocytosis: a multicenter retrospective study. Orphanet J Rare Dis 2017; 12: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saven A, Burian C. Cladribine activity in adult Langerhans-cell histiocytosis. Blood 1999; 93: 4125–4130. [PubMed] [Google Scholar]
- 92.Weitzman S, Braier J, Donadieu J, et al. 2′-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer 2009; 53: 1271–1276. [DOI] [PubMed] [Google Scholar]
- 93.Epaud R, Ducou Le Pointe H, Fasola S, et al. Cladribine improves lung cysts and pulmonary function in a child with histiocytosis. Eur Respir J 2015; 45: 831–833. [DOI] [PubMed] [Google Scholar]
- 94.Grobost V, Khouatra C, Lazor R, et al. Effectiveness of cladribine therapy in patients with pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis 2014; 9: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lazor R, Etienne-Mastroianni B, Khouatra C, et al. Progressive diffuse pulmonary Langerhans cell histiocytosis improved by cladribine chemotherapy. Thorax 2009; 64: 274–275. [DOI] [PubMed] [Google Scholar]
- 96.Aerni MR, Aubry MC, Myers JL, et al. Complete remission of nodular pulmonary Langerhans cell histiocytosis lesions induced by 2-chlorodeoxyadenosine in a non-smoker. Respir Med 2008; 102: 316–319. [DOI] [PubMed] [Google Scholar]
- 97.Nemoto K, Oh-Ishi S, Inui T, et al. Long-term improvement during tadalafil therapy in a patient with pulmonary hypertension secondary to pulmonary Langerhans cell histiocytosis. Respir Med Case Rep 2016; 18: 54–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.May A, Kane G, Yi E, et al. Dramatic and sustained responsiveness of pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension to vasodilator therapy. Respir Med Case Rep 2014; 14: 13–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yazicioglu A, Turkkan S, Demirag F, et al. Recurrence pattern of pulmonary Langerhans cell histiocytosis after lung transplantation: a case report. Transplant Proc 2016; 48: 3231–3233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Tazi ERR-0070-2017_Tazi (1.2MB, pdf)