

Introduction
Although most cases of metastatic non–small-cell lung cancer (NSCLC) harboring EGFR (ex19del or L858R) mutations respond to the EGFR tyrosine kinase inhibitor (TKI) osimertinib, they inevitably become resistant to this drug. The extent of progression-free survival depends on intrinsic or acquired on-target/off-target mechanisms of osimertinib resistance.1-3 In this respect, oncogenic fusions of receptor tyrosine kinases, detectable in 1%-10% of cases of acquired osimertinib resistance, are particularly relevant, as several of them are druggable by specific TKIs.3 Yet, although osimertinib-resistant cases with acquired ALK or ROS1 fusions have shown objective response (OR) to well-established ALK/ROS1-TKIs,4,5 the treatment and outcome of patients acquiring RET fusions during treatment with osimertinib are more difficult to envisage because of variable and rare fusion partners and limited experience with RET-TKIs in this setting. In this study, we describe a patient with disseminated EGFR-mutated (EGFR+) NSCLC, who progressed on second-line osimertinib acquiring ANK3-RET fusion (not reported before in this setting) together with MDM2 amplification and PTEN mutation. Despite these significant genomic coalterations, the patient has been displaying sustained ongoing OR to the osimertinib-pralsetinib combination for more than 12 months, providing clinical evidence for ANK3-RET fusion as important, effectively targetable mechanism of osimertinib resistance.
Case Report
The patient, a 62-year-old White female, never-smoker, underwent intracranial surgery for suspected pituitary adenoma. Histopathological assessment of the pituitary lesion and targeted next-generation sequencing (NGS) analysis of single nucleotide variants (SNVs), indels, and copy number variations (CNVs) across 161 unique cancer-associated genes using the Oncomine Comprehensive Assay v3 (Thermo Fisher Scientific, Waltham, MA) revealed instead metastatic pulmonary adenocarcinoma (immunohistochemically CK7+/TTF1+) with the EGFR ex19del p.E746_A750delELREA and negative programmed death ligand-1-expression. Moreover, total-body positron emission tomography-computed tomography (PET-CT) scan detected a tumor in the left lung's upper lobe and multiple metastases in both lungs, mediastinal lymph nodes, and L2 vertebra, corresponding to T3N2M1c. Figure 1 summarizes the systemic treatments used for the patient.
FIG 1.

Tumor evolution illustrating multiclonality of the disease during progression and adopted treatments. As the patient is still responding to osimertinib-pralsetinib combination, the future molecular state of the disease remains to be explored (symbol ?). NGS, next-generation sequencing; NSCLC, non–small-cell lung cancer.
Stereotactic radiosurgery against the pituitary metastasis was followed by systemic erlotinib treatment. Progression of primary tumor and mediastinal lymph nodal metastases was observed 15 months later. Lymph nodal biopsy and DNA-NGS analysis revealed the original EGFR ex19del and a new EGFR mutation p.T790M. The patient started second-line osimertinib 80 mg once daily. Plasma cell-free DNA (cfDNA) testing (Oncomine Lung cfDNA NGS assay; Thermo Fisher Scientific) at 4, 7, and 13 months of this treatment showed no EGFR mutations. However, two liver metastases were observed after 15 months. DNA-NGS analysis of the biopsy from one of them showed persistent EGFR ex19del and p.T790M mutations, which concomitantly reappeared in plasma cfDNA. Additionally, two putative resistance mechanisms, MDM2 amplification (19 gene copies) and the missense PTEN substitution p.C124S (oncogenic mutation in PTEN phosphatase catalytic domain, according to COSMIC6 and OncoKB databases7), were identified in this metastasis. Hence, no further targeted treatment options were deemed possible. The patient received three cycles of carboplatin-pemetrexed while continuing osimertinib but could not tolerate more chemotherapy because of bone marrow toxicity. However, supplementary NGS analysis of RNA from the hepatic metastasis using the Archer FusionPlex Lung kit (ArcherDX Inc, Boulder, CO) showed ANK3-RET fusion on chromosome 10q with break points at positions chr10:61994446 and chr10:43612032, respectively (Fig 2). The Archer Analysis software v.6.2.1 predicted the fusion to be in frame and detected it in 71.5% of reads. According to COSMIC annotations,6 both break points are outside the two genes' coding sequence (RET: chr10:43077027-43130351, positive strand; ANK3: chr10:60026298-60389730, negative strand). Furthermore, other genes are present on chromosome 10q between ANK3 and RET, suggesting that the ANK3-RET fusion was intergenic and comprised ANK3 exon 1-8 and RET exon 12-21 with completely preserved RET-kinase domain.
FIG 2.
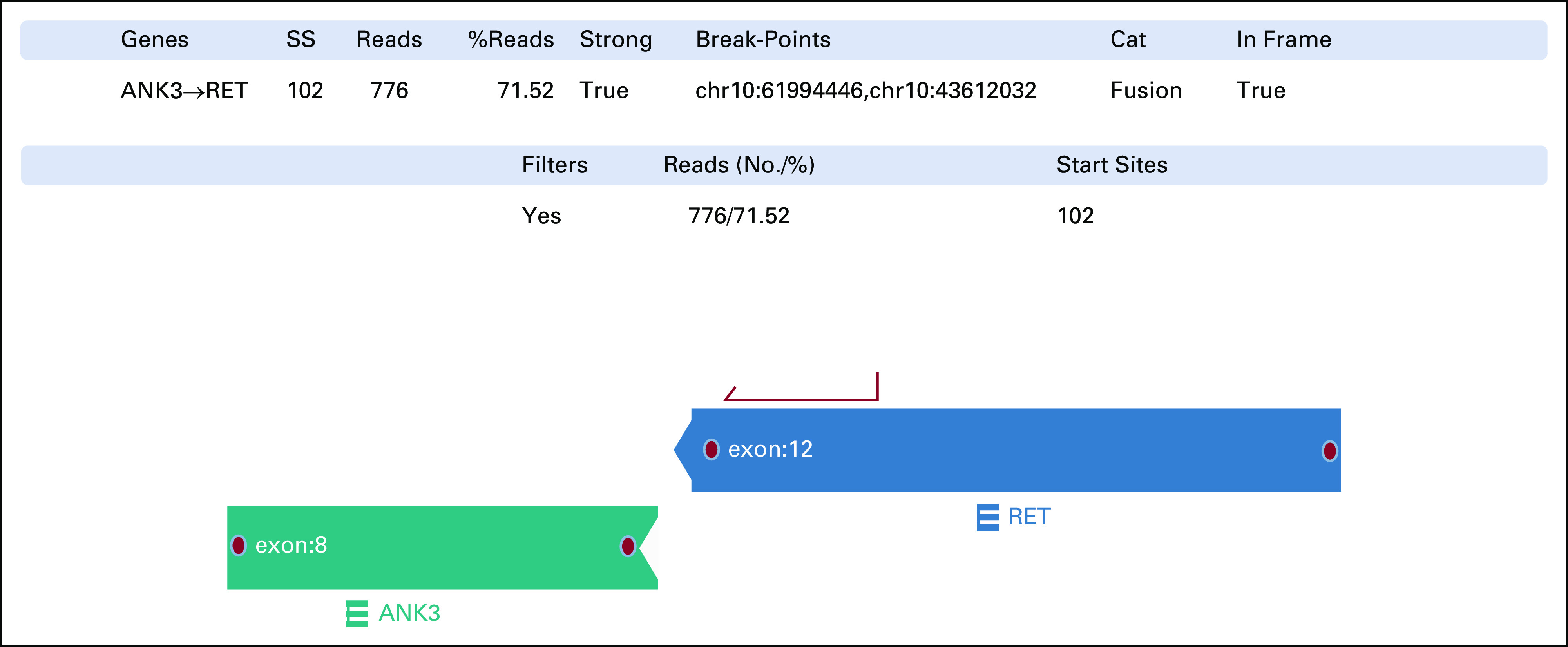
Intergenic ANK3-RET fusion on chromosome 10q (break point chr10:61994446, chr10:43612032) identified by RNA-NGS with an Archer FusionPlex Lung kit. SS, start sites.
Retrospective NGS of RNA isolated from the pre-osimertinib biopsies (diagnostic pituitary biopsy and tumor rebiopsy at first intrathoracic progression) did not show the ANK3-RET fusion. This confirmed that the latter had been acquired during osimertinib treatment and could explain progression, as other RET fusions have been reported as mechanisms of acquired resistance to EGFR-TKIs.4,8,9 Thus, RET-TKI treatment with pralsetinib 400 mg once daily combined with continuation of osimertinib 80 mg once daily was initiated. After 2 weeks, this combination was shortly paused because of interstitial pneumonia, which completely resolved on steroid therapy. The patient resumed the combined TKI treatment at reduced dose (osimertinib 40 mg once daily, pralsetinib 200 mg once daily) without experiencing additional adverse events. The first assessment performed 1 month later revealed a mixed response, with regression of the primary tumor, disappearance of one liver metastasis, and growth of the other one (Figs 3A-3C v Figs 3D-3F). At the second assessment 4 weeks later, further reduction of primary tumor and regression of both liver metastases were noticed (Figs 3G-3I). Stable disease (SD) in the chest and abdomen was observed after 5 months of treatment (Figs 3J-3L). The following assessments showed complete response in abdomen and slight progression of single pleural lesions, which were biopsied and displayed the PTEN mutation and MDM2 amplification (34 gene copies), but not the pralsetinib-targeted RET fusion. Consequently, to control the pleural lesions, osimertinib dose was reincreased to 80 mg once daily, while pralsetinib 200 mg once daily was maintained. The last PET-CT, so far, performed after 12 months of treatment displayed persisting abdominal complete response and thoracic SD (data not shown). Accordingly, sequential plasma cfDNA did not show reappearance of EGFR mutations during the combination treatment.
FIG 3.

Response to combined treatment with osimertinib and pralsetinib. Before treatment: (A) lung tumor in left upper lobe (5.1 cm; arrow), (B) liver metastasis (1.7 cm; arrow), and (C) residual liver metastasis (nonmeasurable; arrow). First assessment: (D) lung tumor in left upper lobe (4.3 cm; arrow), (E) liver metastasis (1.1 cm; arrow), and (F) residual liver metastasis (4.6 cm; arrow). Second assessment: (G) lung tumor in left upper lobe (4.0 cm; arrow), (H) liver metastasis (nonmeasurable; arrow), and (I) residual liver metastasis (3.8 cm; arrow). Third assessment: (J) lung tumor in left upper lobe (3.9 cm; arrow), (K) liver metastasis (nonvisible), and (L) residual liver metastasis (3.8 cm; arrow).
The study was conducted according to the Declaration of Helsinki's guidelines and approved by the Danish Tumor Board Phase I and Lung Team Tumor Board (20200122) at Department of Oncology, Rigshospitalet, University of Copenhagen. Written informed consent for this report was obtained from the patient.
Discussion
Our finding of acquired ANK3-RET fusion as druggable mechanism of osimertinib resistance underlines the importance of including RNA-NGS for gene fusions in clinical practice to find resistance mechanisms and new targeted treatment options. Indeed, the use of complementary RNA-NGS in a prospective genomic profiling of 2,522 lung adenocarcinomas revealed 14% additional actionable gene fusions and METex14 alterations that had not been detected by DNA-NGS.10 Since detecting RET fusions in plasma cfDNA is challenging, tumor rebiopsies, whenever feasible, should be performed in the setting of EGFR-TKI resistance to identify acquired fusions by RNA-NGS and histologic transformation. Progression on osimertinib due to acquired RET fusions has previously been reported and treated with different modalities, including continuing osimertinib beyond progression,4,11 replacing it with the RET-targeting multi-TKI cabozantinib,12 or combining osimertinib with RET-TKI (Table 1).9,13,14 Although RET fusions develop more frequently during treatment with osimertinib than earlier-generation EGFR-TKIs,4,11 a large-scale multicancer study identified patients with NSCLC acquiring CCDC6-RET (n = 10), NCOA4-RET (n = 4), or TRIM24-RET (n = 1) fusions at progression on first/second-generation EGFR-TKI, without though reporting any combined EGFR-TKI-RET-TKI therapy for these patients.15 In another study including 3,505 EGFR+ NSCLCs, six cases acquiring RET fusions with CCDC6, NCOA4, or TRIM24 during treatment with erlotinib or afatinib were detected.16 One of these cases harboring NCOA4-RET fusion as resistance mechanism to afatinib responded with SD for 7 months to afatinib-cabozantinib combination therapy.16 In a group of 41 patients progressing on second-line osimertinib, two with acquired CCDC6-RET and one with NCOA4-RET fusion were identified and treated with the osimertinib-pralsetinib combination obtaining an ongoing OR of 4 months.9 Also combining osimertinib with another RET-TKI, selpercatinib, was effective for a group of EGFR+ NSCLC patients with acquired NCOA4-RET, CCDC6-RET, KIF5B-RET, or RUFY2-RET fusions, as the ORR was 50% and median duration of treatment 7.4 months.14 Our case, after progressing on second-line osimertinib, acquired a RET fusion with ANK3, an unreported partner in this setting. After combining osimertinib with pralsetinib, we have obtained an ongoing OR that so far has been maintained for more than 12 months, which, to our knowledge, is the longest reported until now with osimertinib-RET-TKI combinations (Table 1).13,14 The efficacy of these combinations against osimertinib resistance linked to ANK3-RET fusion needs confirmation in additional cases. Nonetheless, the observations summarized in Table 1 strongly indicate that combining osimertinib with a RET-TKI may counteract osimertinib resistance mediated by different RET fusions. It is also noteworthy that, after initial interruption of the osimertinib-pralsetinib therapy at standard dose because of pulmonary toxicity, we have been able to safely resume and continue the combination at reduced dose of both drugs for 9 months, still obtaining a durable OR. Afterward, we have reincreased osimertinib to standard dose to control the disease in the chest without causing toxicity. These results may be relevant for patients with acquired resistance mechanisms requiring dual targeted therapy with the risk of overlapping toxicity, as they may still benefit, at least for some time, from a dose reduction of the combined drugs.
TABLE 1.
Summary of Reported Cases Demonstrating Feasibility of Combined EGFR Tyrosine Kinase Inhibitor Plus RET Tyrosine Kinase Inhibitor Treatment for EGFR+ Non–Small-Cell Lung Cancer Patients With Acquired RET Fusions

As DNA-NGS identified MDM2 amplification and PTEN mutation as putative mechanisms of osimertinib resistance and our patient's permanent steroid replacement for hypophysis insufficiency as well as EGFR+ NSCLC were contraindications for immunotherapy,17 only chemotherapy could be offered to her. However, complementary RNA-NGS revealed the acquired ANK3-RET fusion, allowing further targeted treatment with combined osimertinib-pralsetinib. Hitherto, 48 different RET fusions have been reported in NSCLC,18 both as de novo cancer drivers in EGFR wild-type cases and as acquired mechanisms of resistance to osimertinib and other EGFR-TKIs.1,3,9,18,19 Notably, de novo ANK3-RET fusion has previously been identified in EGFR wild-type NSCLC, although without any information on response to RET-TKIs.20,21 To our knowledge, this is the first report of this fusion as acquired osimertinib-resistance mechanism that can be effectively counteracted by combining pralsetinib with osimertinib continuation. Our report also adds clinically relevant information to the list of intergenic RET rearrangements in NSCLC.18
The PTEN mutation and MDM2 amplification co-occurring in our patient may contribute to TKI resistance.1 The PTEN substitution has not been described before under osimertinib treatment and may function as parallel resistance mechanism alongside the RET fusion, as the PTEN tumor suppressor inhibits the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin pathway. The concomitant high-level MDM2 amplification may also promote osimertinib resistance, as MDM2 functions as an E3 ubiquitin ligase targeting the p53 tumor suppressor for proteolysis and antagonizing its transcriptional activity.22 Indeed, coexisting MDM2 amplification is associated with worse outcome in EGFR-TKI–treated EGFR+ NSCLC.23 Neither PTEN mutation nor MDM2 amplification were found in the diagnostic biopsy or the first rebiopsy (Fig 1); thus, we considered them as acquired TKI-resistance mechanisms that, together with the ANK3-RET fusion, reflect the clonal heterogeneity of advanced EGFR+ NSCLC at progression on TKIs.24 Despite preclinical attempts to target MDM2 overexpression and PTEN inactivation,25,26 these alterations remain undruggable in clinical practice. Yet, despite such coalterations, our patient's disseminated NSCLC has been exhibiting a prolonged, ongoing, clinically meaningful response to the pralsetinib-osimertinib combination, further revealing the ANK3-RET fusion as crucial, targetable mechanism of osimertinib resistance.
Edyta M. Urbanska
Honoraria: AstraZeneca, Roche, Takeda
Consulting or Advisory Role: Amgen, Pfizer, Novartis
Research Funding: Pfizer
Travel, Accommodations, Expenses: Takeda
Jens B. Sørensen
Honoraria: AstraZeneca, Roche, Takeda
Consulting or Advisory Role: AstraZeneca, Pfizer, Roche, Takeda
Linea C. Melchior
Honoraria: Takeda
Eric Santoni-Rugiu
Honoraria: Roche (Inst), Amgen (Inst), Takeda Science Foundation (Inst), AstraZeneca (Inst), Bristol Myers Squibb Foundation (Inst)
Consulting or Advisory Role: Bayer (Inst), Roche (Inst), Takeda, Amgen (Inst)
Travel, Accommodations, Expenses: Takeda
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the virtual IASLC 2021 World Conference on Lung Cancer, September 8-14, 2021 (Suppl; abstr FP46.02). Part of the data also presented as a preprint (doi: 10.20944/preprints202111.0052.v1).
DATA SHARING STATEMENT
The data from the study will be made available by the authors on request.
AUTHOR CONTRIBUTIONS
Conception and design: Edyta M. Urbanska, Jens B. Sørensen, Eric Santoni-Rugiu
Provision of study materials or patients: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Edyta M. Urbanska
Honoraria: AstraZeneca, Roche, Takeda
Consulting or Advisory Role: Amgen, Pfizer, Novartis
Research Funding: Pfizer
Travel, Accommodations, Expenses: Takeda
Jens B. Sørensen
Honoraria: AstraZeneca, Roche, Takeda
Consulting or Advisory Role: AstraZeneca, Pfizer, Roche, Takeda
Linea C. Melchior
Honoraria: Takeda
Eric Santoni-Rugiu
Honoraria: Roche (Inst), Amgen (Inst), Takeda Science Foundation (Inst), AstraZeneca (Inst), Bristol Myers Squibb Foundation (Inst)
Consulting or Advisory Role: Bayer (Inst), Roche (Inst), Takeda, Amgen (Inst)
Travel, Accommodations, Expenses: Takeda
No other potential conflicts of interest were reported.
REFERENCES
- 1. Santoni-Rugiu E, Melchior LC, Urbanska EM, et al. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: Differences and similarities with acquired resistance. Cancers (Basel) 2019;1:923. doi: 10.3390/cancers11070923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382:41–50. doi: 10.1056/NEJMoa1913662. [DOI] [PubMed] [Google Scholar]
- 3. Leonetti A, Sharma S, Minari R, et al. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121:725–737. doi: 10.1038/s41416-019-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Offin M, Somwar R, Rekhtman N, et al. Acquired ALK and RET gene fusions as mechanisms of resistance to Osimertinib in EGFR-mutant lung cancers. JCO Precis Oncol. 2018;2:1–12. doi: 10.1200/PO.18.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zeng L, Yang N, Zhang Y. GOPC-ROS1 rearrangement as an acquired resistance mechanism to osimertinib and responding to crizotinib combined treatments in lung adenocarcinoma. J Thorac Oncol. 2018;13:e114–e116. doi: 10.1016/j.jtho.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 6.COSMIC, Catalogue of Somatic Mutations in Cancer. https://cancer.sanger.ac.uk/cosmic [Google Scholar]
- 7.OncoKB, MSK's Precision Oncology Knowledge Base. An FDA-Recognized Human Genetic Variant Database. https://www.oncokb.org [Google Scholar]
- 8. Klempner SJ, Bazhenova LA, Braiteh FS, et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC-patients who has progressed on first- or second-generation EGFR-TKI. Lung Cancer. 2015;89:357–359. doi: 10.1016/j.lungcan.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 9. Piotrowska Z, Isozaki H, Lennerz JK, et al. Landscape of acquired resistance to Osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with Osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018;8:1529–1539. doi: 10.1158/2159-8290.CD-18-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Benayed R, Offin M, Mullaney K, et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden. Clin Cancer Res. 2019;1:4712–4722. doi: 10.1158/1078-0432.CCR-19-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu H, Shen J, Xiang J, et al. Characterization of acquired receptor tyrosine–kinase fusions as mechanisms of resistance to EGFR tyrosine–kinase inhibitors. Cancer Manag Res. 2019;11:6343–6351. doi: 10.2147/CMAR.S197337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chae YK, Viveiros P, Heleno CT, et al. Use of cabozantinib in a patient with EGFR-mutated non–small-cell lung cancer harboring acquired CCDC6-RET fusion. JCO Precis Oncol. 2019;3:1–5. doi: 10.1200/PO.18.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rehman M, Kim C, Reuss JE, et al. Divergent RET- and BRAF-mediated resistance to osimertinib in EGFR-mutant NSCLC: A case report. JCO Precis Oncol. 2021;5:939–942. doi: 10.1200/PO.21.00083. [DOI] [PubMed] [Google Scholar]
- 14. Rotow J, Patel J, Hanley M, et al. Combination osimertinib plus selpercatinib for EGFR-mutant non-small cell lung cancer with acquired RET fusions. J Thorac Oncol. 2021;16(suppl):S230. abstr FP14.07. [Google Scholar]
- 15. Rich TA, Reckamp KL, Chae YK, et al. Analysis of cell-free DNA from 32,989 advanced cancers reveals novel co-occurring activating RET alterations and oncogenic signaling pathway aberrations. Clin Cancer Res. 2019;25:5832–5842. doi: 10.1158/1078-0432.CCR-18-4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schrock AB, Zhu VW, Hsieh W-S, et al. Receptor tyrosine kinase fusions and BRAF kinase fusions are rare but actionable resistance mechanisms to EGFR tyrosine kinase inhibitors. J Thorac Oncol. 2018;13:1312–1323. doi: 10.1016/j.jtho.2018.05.027. [DOI] [PubMed] [Google Scholar]
- 17. Melosky B, Juergens R, Hirsh V, et al. Amplifying outcomes: Checkpoint inhibitor combinations in first-line non-small cell lung cancer. Oncologist. 2020;25:64–77. doi: 10.1634/theoncologist.2019-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ou S-HI, Zhu VW. Catalog of 5' fusion partners in RET+ NSCLC Circa 2020. JTO Clin Res Rep. 2020;1:1–7. doi: 10.1016/j.jtocrr.2020.100037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoenfeld AJ, Chan JM, Kubota D, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res. 2020;1:2654–2663. doi: 10.1158/1078-0432.CCR-19-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu C, Dong XR, Zhao J, et al. Association of genetic and immuno-characteristics with clinical outcomes in patients with RET-rearranged non-small cell lung cancer: A retrospective multicenter study. J Hematol Oncol. 2020;13:37. doi: 10.1186/s13045-020-00866-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li B, Qu H, Zhang J, et al. Genomic characterization and outcome evaluation of kinome fusions in lung cancer revealed novel druggable fusions. NPJ Precis Oncol. 2021;5:81. doi: 10.1038/s41698-021-00221-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hou H, Sun D, Zhang X. The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int. 2019;19:216. doi: 10.1186/s12935-019-0937-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim Y, Lee B, Shim JH, et al. Concurrent genetic alterations predict the progression to target therapy in EGFR-mutated advanced NSCLC. J Thorac Oncol. 2019;14:193–202. doi: 10.1016/j.jtho.2018.10.150. [DOI] [PubMed] [Google Scholar]
- 24. Blakely CM, Watkins TBK, Wu W, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet. 2017;49:1693–1704. doi: 10.1038/ng.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang L, Zawacka-Pankau J. The p53/MDM2/MDMX-targeted therapies—A clinical synopsis. Cell Death Dis. 2020;11:237. doi: 10.1038/s41419-020-2445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gkountakos A, Sartori G, Falcone I, et al. PTEN in lung cancer: Dealing with the problem, building on new knowledge and turning the game around. Cancers (Basel) 2019;11:1141. doi: 10.3390/cancers11081141. [DOI] [PMC free article] [PubMed] [Google Scholar]