

PURPOSE
Multidisciplinary molecular tumor boards (MTBs) interpret next-generation sequencing reports and help oncologists determine best therapeutic options; however, there is a paucity of data regarding their clinical utility. The purpose of this study was to determine if MTB-directed therapy improves progression-free survival (PFS) over immediately prior therapy in patients with advanced cancer.
METHODS
This single-arm, prospective phase II clinical trial enrolled patients with advanced cancer with an actionable mutation who received MTB-recommended targeted therapy between January 1, 2017, and October 31, 2020. MTB-recommended both on-label (level 1 evidence) and off-label (evidence levels 2 and 3) therapies. Of the 93 enrolled patients, 43 were treated frontline and 50 received second-line or greater-line therapy. The primary outcome was the probability of patients treated with second-line or greater-line MTB-directed therapy who achieved a PFS ratio ≥ 1.3 (PFS on MTB-directed therapy divided by PFS on the patient's immediately prior therapy). Secondary outcomes included PFS for patients treated frontline and overall survival and adverse effects for the entire study population.
RESULTS
The most common disease sites were lung (35 of 93, 38%), gynecologic (17 of 93, 18%), GI (16 of 93, 17%), and head and neck (7 of 93, 8%). The Kaplan-Meier estimate of the probability of PFS ratio ≥ 1.3 was 0.59 (95% CI, 0.47 to 0.75) for patients treated with second-line or greater-line MTB-directed therapy. The median PFS was 449 (range 42-1,125) days for patients treated frontline. The median overall survival was 768 (range 22-1,240) days. There were four nontreatment-related deaths.
CONCLUSION
When treated with MTB-directed therapy, most patients experienced improved PFS compared with immediately prior treatment. MTB-directed targeted therapy may be a strategy to improve outcomes for patients with advanced cancer.
INTRODUCTION
Oncology care is shifting toward individualized driver mutation–directed cancer treatment. The growing number of companion diagnostic tests, next-generation sequencing (NGS) tests, and targeted therapies approved by the US Food and Drug Administration (FDA) is the evidence of the focus on these precision medicine initiatives.1
CONTEXT
Key Objective
Do patients with advanced cancer experience survival benefit from molecular tumor board (MTB)–directed targeted therapy?
Knowledge Generated
In this prospective phase II clinical trial, most patients treated with second-line or greater-line MTB-directed therapy experienced a superior progression-free survival compared with their most recent line of therapy. Patients treated with frontline MTB-directed therapy also experienced benefit.
Relevance
These findings demonstrate that patients with advanced cancer with targetable mutations experience improved progression-free survival when treated with MTB-directed therapy.
Although many targeted therapies are more efficacious and tolerable than cytotoxic chemotherapy,2-4 clinical use remains challenging because of the complexity of applying NGS results to patient care.5,6 To overcome this challenge, many institutions have implemented multidisciplinary molecular tumor boards (MTBs)7-9 to prescribe optimal targeted therapies. Furthermore, the availability of an MTB increases oncologists' willingness to use NGS tests.10
Although many institutions have implemented MTBs, data supporting their clinical utility are limited. A recent systematic review concluded that MTBs appear to improve clinical outcomes for patients with cancer; however, most of the existing literature is retrospective, observational, and underpowered for efficacy.11 Two prospective studies demonstrated improvement in survival indices for patients with a wide variety of tumor types12 and in patients with non–small-cell lung cancer (NSCLC).13
We report results from a phase II clinical trial of MTB-assisted care. We assessed the probability of patients receiving second-line or greater-line MTB-directed therapy that achieved a progression-free survival (PFS) ratio ≥ 1.3 compared with immediately prior therapy.
METHODS
Study Design
The Markey Cancer Center (MCC) MTB therapy trial (ClinicalTrials.gov identifier: NCT03089554) is a single-institution, open-label, and single-arm prospective clinical trial. Patients were followed from time of consent until death.
Patient data were collected and reported using Research Electronic Data Capture (Nashville, TN)14,15 hosted at the University of Kentucky and the OnCore Database (Advarra Inc, Columbia, MD), which is housed on secure servers maintained by the MCC Cancer Informatics Shared Resource Facility.
Ethical Considerations
The study was performed in accordance with the US FDA International Conference on Harmonization Guidelines for Good Clinical Practice and Declaration of Helsinki. The Protocol was approved by the University of Kentucky Institutional Review Board (IRB approval #48018). Each patient provided written informed consent before trial participation. This trial was regularly monitored by the MCC Data and Safety Monitoring Committee, and all adverse event data were recorded.
Study Population
Patients with cancer who had undergone NGS genomic testing, had an actionable mutation, no curative therapy options, and received an MTB-recommended therapy were eligible for inclusion. Initially, patients were required to have received prior cancer therapy; however, as genomic testing moved into the frontline setting, the Protocol was amended to include patients without prior therapy. A list of full eligibility criteria is available in the Protocol.
Study Intervention
Somatic profiling.
Before enrollment, patients underwent physician's choice somatic tumoral tissue or circulating tumoral deoxyribonucleic acid NGS testing at a commercial Clinical Laboratory Improvement Amendments–certified laboratory.
Tumor mutational burden (TMB), defined as the total number of somatic mutations per megabase (muts/Mb) of DNA, was classified as low (1-5 muts/Mb), intermediate (6-9 muts/Mb), or high (≥ 10 muts/Mb) on the basis of accepted ranges at study inception.16 Biomarker status, including immunohistochemical hormone receptor, microsatellite instability (MSI), programmed death-1 (PD-1)/programmed death-ligand 1 (PD-L1) expression, and loss of heterozygosity (LOH) scores were reviewed when available.
MTB structure and review.
The University of Kentucky MCC MTB serves as a consulting service where physicians submit cases for review. MTB participants include medical and gynecologic oncologists, genetic counselors, pharmacists, nurses, pathologists, radiologists, and basic scientists. Redacted patient history, histopathology, radiology, and genomic results are discussed. Each genomic pathogenic or likely pathogenic alteration is reviewed to determine its function, oncogenicity, availability of any targeted therapeutics, or clinical trials. MTB recommendations may include physician's choice standard therapy, clinical trial referral, targeted therapy with an FDA-approved on-label or off-label therapy, or additional testing, including recommendations for germline testing.
If a targeted therapeutic recommendation is made, on-label options are prioritized, followed by clinical trials and off-label options with the highest level of evidence. All recommendations are provided to the treating oncologist in a letter that includes evidence levels. The University of Kentucky Healthcare Pharmacy and Therapeutics Committee developed a consensus guideline for grading evidence, which is available in the Data Supplement. Level 1 evidence included FDA-approved indications and National Comprehensive Cancer Network guideline–recommended therapies. Evidence level 2 included drugs with an FDA-approved indication for the specific mutation in another disease type and phase II or III clinical trial evidence for activity in the disease of interest. Evidence level 3 included FDA-approved drugs for the specific mutation in another disease type with case report or phase I clinical trial evidence for activity in the disease of interest. Preclinical evidence or evidence of pathway inhibition was considered category 4 and not recommended.
Outcome Measures
The prespecified primary end point was the probability of patients treated with second-line or greater-line MTB-directed therapy who achieved a growth modulation index (GMI) ≥ 1.3. GMI is calculated as PFS2/PFS1, where PFS2 is PFS on MTB-directed therapy and PFS1 is PFS on the patient's immediately prior therapy. Von Hoff et al17 previously used this definition to define clinical benefit. PFS was defined as the time interval between the treatment start date and the date of clinical or radiographic disease progression as documented by the treating physician, date of death, or date of last follow-up, whichever occurred first. As PFS is expected to decrease with each subsequent therapy, an improvement in subsequent PFS suggests clinical benefit.18
Prespecified secondary outcomes included overall survival for all patients and treatment-related adverse events. PFS for patients treated with frontline MTB-directed therapy was an additional outcome but was not prespecified because of the Protocol amendment. Documentation of treatment-related adverse events included event duration, severity, temporal relationship to the receipt of study therapy, and if event resulted in study withdrawal. Toxicities were graded according to Common Terminology Criteria for Adverse Events version 5.0.19
Statistical Analysis
Sample size was calculated on the basis of research results from the study by Von Hoff et al.17 In this study, the null hypothesis was that ≤ 15% of patients had PFS2/PFS1 ≥1.3 and final results indicated that 27% of patients experienced PFS2/PFS1 ≥1.3.17 The alternative hypothesis for the present study was that ≥ 27% of patients had GMI ≥ 1.3 versus a null hypothesis of ≤ 15% of patients had GMI ≥ 1.3.17 To obtain 85% power with a 5% significance level, 93 patients were required, with one interim analysis planned on enrollment of 37 patients on the basis of a two-stage Simon's design.20,21 Stopping bound for futility was set at less than or equal to six patients with GMI ≥ 1.3.
Patient demographic, disease, treatment, and molecular characteristics were assessed with descriptive statistics. Separate analyses were performed for patients treated with MTB-directed second or greater line of therapy (cohort 1) and patients treated with MTB-directed frontline therapy (cohort 2). Since ignoring censoring of PFS2 using a simple proportion could result in an underestimated probability P = P(GMI > 1.3), the probability P = P(GMI > 1.3) was assessed by the Kaplan-Meier method.22 For cohort 2, the Kaplan-Meier method was used to evaluate PFS with 95% binomial CIs.
RESULTS
Patients
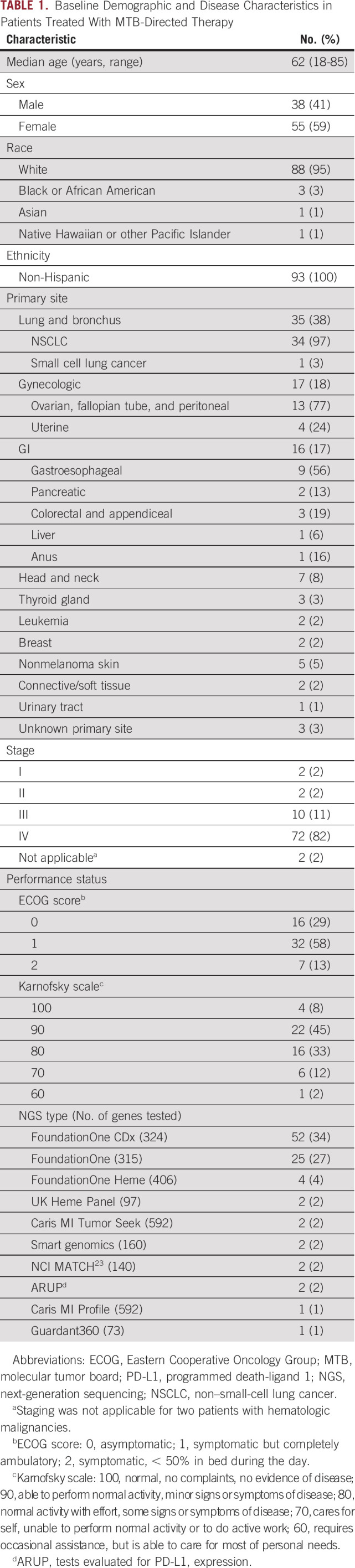
Between January 1, 2017, and October 31, 2020, 679 patients underwent MTB review. Of the 591 patients with actionable mutations, 98 were enrolled in this study. Before starting MTB-directed therapy, five patients were determined to be ineligible; 93 received the intervention (Fig 1). Accrual was stopped once the planned number of patients was enrolled. The median patient age was 63 (range 18-85) years; most were female (58 of 93, 62%), non-Hispanic (92 of 93, 99%), and White (88 of 93, 95%). Demographic and disease characteristics are summarized in Table 1. The most common disease sites were lung (35 of 93, 38%), gynecologic (17 of 93, 18%), GI (16 of 93, 17%), and head and neck (7 of 93, 8%).
FIG 1.
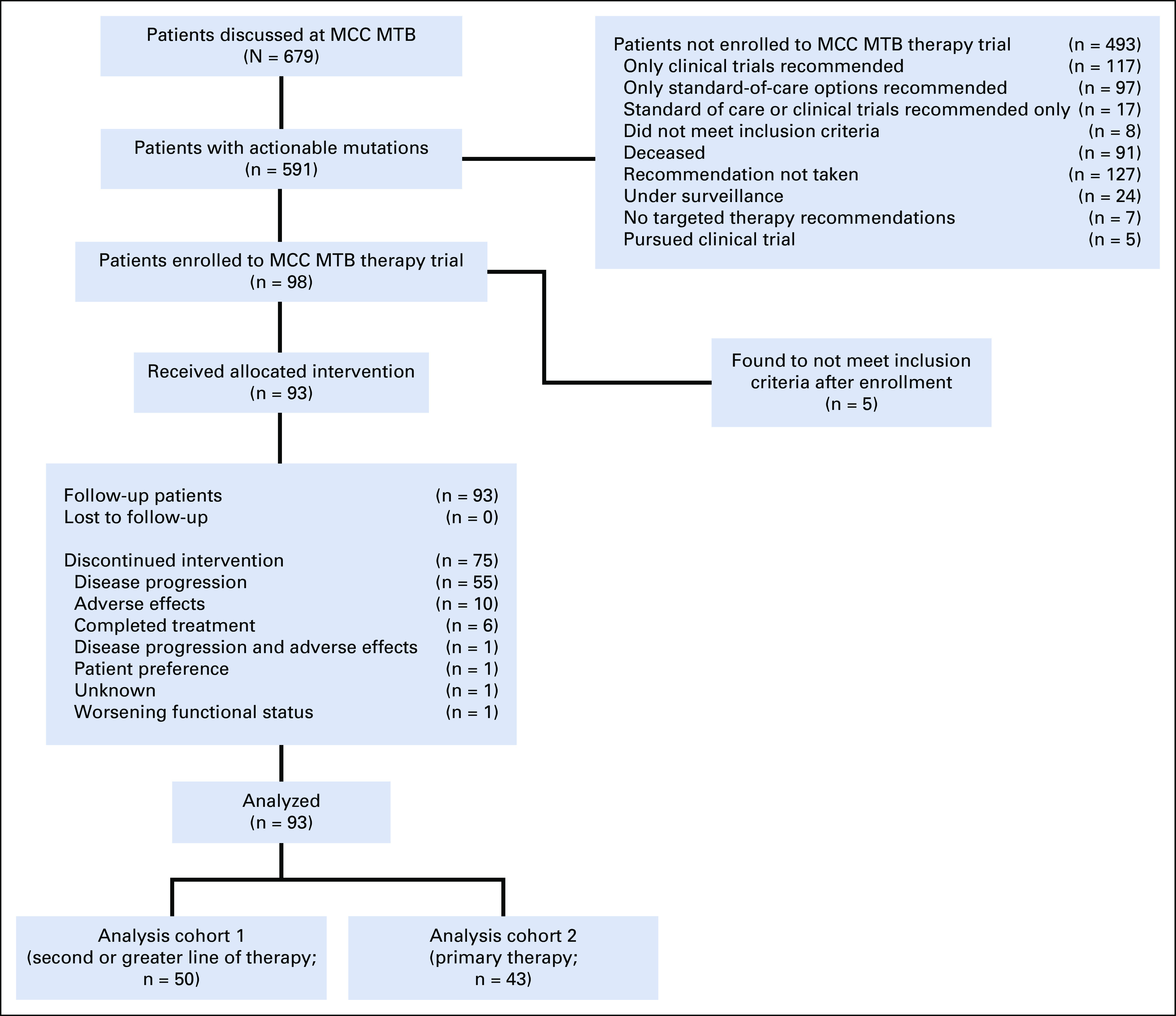
Study flow diagram. Flow diagram shows identification of patients who underwent MTB review and subsequent enrollment in this trial. MCC, Markey Cancer Center; MTB, molecular tumor board.
TABLE 1.
Baseline Demographic and Disease Characteristics in Patients Treated With MTB-Directed Therapy
Cohort 1 (n = 50) included advanced ovarian, NSCLC, and head and neck cancers treated with second-line or greater-line MTB-directed therapy. Cohort 2 (n = 43) included patients with NSCLC, gastroesophageal, head and neck, nonmelanoma skin, and other cancers treated with first-line MTB-directed therapy. Primary tumor sites, targets, treatments, level of evidence, and individual patient PFS outcomes are detailed in the Data Supplement (cohort 124 and cohort 2).
Molecular Profiling
Treatment targets were identified using commercial (n = 89), in-house (n = 2), or trial-specific23 (n = 2) genomic testing and are detailed in Table 1. Tissue NGS was performed for 92 (99%) patients, and blood circulating tumoral deoxyribonucleic acid was performed for one patient. Ninety-four percent (87 of 93) of patients were tested for at least one immunotherapy biomarker, including TMB (n = 81), MSI (n = 83), and/or PD-1/PD-L1 expression (n = 38).
The most common genetic alterations included those in TP53 (64 of 93, 69%), CDKN2A (23 of 93, 25%), CDKN2B (17 of 93, 18%), and EGFR (11 of 93 12%). Among tumors evaluated for TMB, 24% (19 of 81) were high, 41% (33 of 81) were intermediate, and 36% (29 of 81) were low. MSI was present in 10% (8 of 83) of tumors tested, and 76% (29 of 38) of tumors evaluated for PD-L1 expression (≥ 1% combined positive score) were positive.
Tumoral genomics, MTB-recommended targets, and received treatments are detailed in Figure 2. The most common targets were high TMB (26 of 93, 28%), PD-L1 expression (12 of 93, 13%), intermediate TMB (12 of 93, 13%), and ERBB2 amplifications or mutations (11 of 93, 12%).
FIG 2.
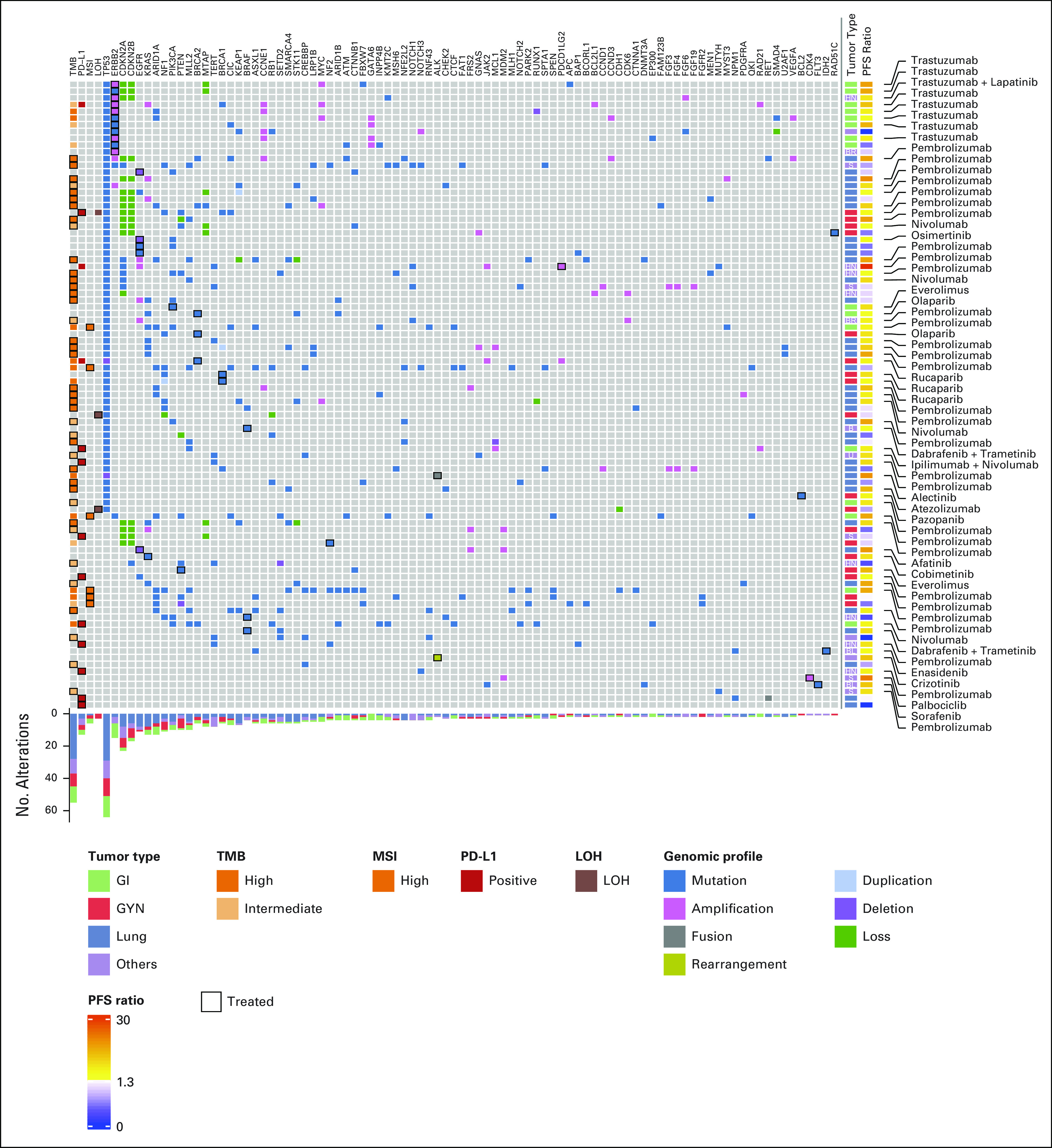
Genomic profiles, recommendations, and MTB-directed targeted therapy, and outcomes of patients enrolled in this trial. Genes and biomarkers are displayed in columns. Rows demonstrate individual tumor profiles, targeted therapy, and PFS ratio for each patient. GYN, gynecologic; LOH, loss of heterozygosity; MSI, microsatellite instability; MTB, molecular tumor board; PD-L1: programmed death-ligand 1; PFS, progression-free survival; TMB, tumor mutational burden.
Treatments
Immune checkpoint inhibitors were recommended for 59% (55 of 93) of participants, targeted high TMB for 47% (26 of 55), PD-L1 expression for 24% (13 of 55), intermediate TMB for 22% (12 of 55), and MSI for 9% (5 of 55). Immune checkpoint inhibitor therapy was recommended for one patient with PD-1 ligand 2 amplification (1 of 55, 2%) whose tumor also demonstrated PD-L1 positivity.
Small molecule inhibitors were recommended for 29% (27 of 93) of patients. Tyrosine kinase inhibitors (n = 10) included osimertinib (n = 4) and afatinib (n = 1), which targeted EGFR mutations; alectinib targeted an ALK fusion (n = 1); crizotinib targeted an ALK rearrangement (n = 1); pazopanib targeted a BCL2 rearrangement (n = 1); sorafenib targeted a FLT3 mutation, and lapatinib was used in combination with trastuzumab to target an ERBB2 amplification (n = 1). Poly-ADP ribose polymerase (PARP) inhibitors were used in eight patients, which targeted BRCA2 (olaparib, n = 2; rucaparib, n = 1), BRCA1 (rucaparib, n = 2), and RAD51C (rucaparib, n = 1) mutations and elevated LOH scores (niraparib, n = 1; rucaparib, n = 1). Mitogen-activated protein kinase (MEK) inhibitors targeted a KRAS mutation (cobimetinib, n = 1) and were used in combination with BRAF inhibitors (dabrafenib and trametinib, n = 3) to target BRAF V600E mutations. The mechanistic target of rapamycin inhibitor everolimus targeted an NF2 mutation (n = 1) and a PTEN mutation (n = 1). Enasidenib, an isocitrate dehydrogenase-2 (IDH2) inhibitor, targeted an IDH2 mutation (n = 1), and palbociclib, a cyclin dependent kinase 4/6 (CDK4/6) inhibitor, targeted a CDK4 amplification (n = 1).
Trastuzumab was used in 11 (11 of 93, 12%) patients and targeted an ERBB2 amplification (n = 9), point mutation (n = 1), and positive human epidermal growth factor receptor 2 immunohistochemistry (n = 1).
On-label therapies with level 1 evidence were recommended for 72% (67 of 93) of patients (cohort 1: 27 of 50; cohort 2: 40 of 43). Seven patients were treated with level 2–recommended therapies, and 19 were treated with level 3–recommended therapies. In cohort 1, five level 2 recommendations included an ERBB2-amplified salivary gland adenocarcinoma with trastuzumab and lapatinib, a BRCA2-mutated pancreatic cancer with a PARP inhibitor, a PD-L1–positive nonmelanoma skin cancer with pembrolizumab, and a neuroendocrine lung cancer with nivolumab. Before the site-agnostic FDA approval of pembrolizumab for high TMB,25 one patient with anaplastic thyroid carcinoma in cohort 1 and one patient with squamous cell carcinoma arising from an ovarian teratoma in cohort 2 were treated with pembrolizumab for this indication. One patient with a BRAF V600E–mutated head and neck sarcoma was treated with BRAF and MEK inhibitors.
Level 3 recommendations (n = 19) were common in cohort 1 (n = 18) and included treatment of a PTEN-mutated ovarian cancer (n = 1), an NF2-mutated ovarian cancer (n = 1), and a PIK3CA-mutated cholangiocarcinoma (n = 1) with mechanistic target of rapamycin inhibitors; an ALK-rearranged mesothelioma with an ALK inhibitor (n = 1)24; a BRAF V600E–mutated anaplastic thyroid cancer with BRAF and MEK inhibitors (n = 1); a KRAS-mutated ovarian cancer with a MEK inhibitor (n = 1); and a CDK4-amplified liposarcoma with a CDK4/6 inhibitor (n = 1). Trastuzumab targeted an ERBB2-mutated neuroendocrine tumor (n = 1). Immune checkpoint inhibitors were recommended as a level 3 recommendation for PD-L1 positivity in head and neck mesothelioma (n = 1) and ovarian cancer (n = 1) and targeted intermediate TMB in appendiceal (n = 1), large cell neuroendocrine ovarian (n = 1), breast (n = 1), Hurthle cell thyroid (n = 1) cancers and retroperitoneal (n = 1) and head and neck (n = 1) sarcomas. Pembrolizumab was recommended for an MSI endometrial carcinoma (n = 1) before the FDA site-agnostic approval,26 and niraparib was recommended for an ovarian cancer with an elevated LOH score (n = 1) as a level 3 recommendation before this FDA approval.27 In cohort 2, one nonmelanoma skin cancer with an intermediate TMB was treated with pembrolizumab.
Efficacy and Survival
As of April 15, 2021, the probability of patients treated with second-line or greater-line MTB-directed therapy achieving GMI ≥ 1.3 was 0.54 (27 of 50); 95% CI, 0.40 to 0.68; the null hypothesis of 0.15 was rejected (P < .0001). A Kaplan-Meier estimate of P(GMI ≥ 1.3), which accounted for censoring, was 0.59 (95% CI, 0.47 to 0.75; Fig 3A). The median baseline PFS1 for this cohort was 145 (range 35-1767) days, the median PFS2 was 186 (range 22-1,240) days, and the median GMI was 2.053 as estimated by the Kaplan-Meier method. Individual patient PFS2/PFS1 ratios are illustrated in Figures 3B and 4.
FIG 3.

Growth modulation index among patients treated with second-line or greater-line MTB-directed therapy (cohort 1; n = 50). (A) GMI analysis for 50 patients who had prior therapy PFS data. Larger GMI values represent higher PFS2/PFS1 ratios, indicating superior PFS on MTB-directed therapy (PFS2). Survival probability indicates the probability of obtaining or exceeding a particular GMI. A GMI ≥ 1.3 suggests the benefit of subsequent therapy. The simple proportion of patients experiencing GMI ≥ 1.3 (27 of 50) is 0.54. To provide an accurate estimation, the Kaplan-Meier method accounts for censoring of PFS2 and is 0.59. The Hall-Wellner band indicates the 95% CI, 0.466 to 0.746. (B) Individual patients' time to progression (days) on prior therapy (x axis) and MTB-directed therapy (y axis) is delineated by a circle. The blue line indicates GMI = 1.3. Patients to the left of the blue line demonstrated the most benefit from MTB-directed therapy with some long-term survivors. The red line indicates GMI = 1.0. Patients with GMI = 1.0 conferred the same benefit from MTB-directed therapy as they with their most recent therapy. GMI, growth modulation index; MTB, molecular tumor board; PFS, progression-free survival; TTP, time to progression.
FIG 4.

Waterfall plot detailing individual PFS ratio (PFS2/PFS1) values for patients treated with second-line or greater-line MTB-directed therapy (cohort 1; n = 50). A larger PFS2/PFS1 value indicates a greater benefit of MTB-directed therapy. MTB, molecular tumor board; PFS, progression-free survival.
We compared survival outcomes for patients treated with second-line or greater-line MTB-directed therapy by evidence level (level 1 v level 2 or 3 evidence; Data Supplement). The Kaplan-Meier estimate of P(GMI ≥ 1.3) was 0.61 (95% CI, 0.39 to 0.77), and the median PFS ratio was 2.053 (95% CI, 0.85 to 2.88) for patients treated with therapies with level 1 evidence. The Kaplan-Meier estimate of P(GMI ≥ 1.3) was 0.57 (95% CI, 0.34 to 0.74), and the median PFS ratio was 1.551 (95% CI, 0.54 to 2.56) for patients treated with therapies with level 2 or 3 evidence. There was no difference between these groups (log-rank test; P = .54), suggesting that benefit of MTB-directed therapy was similar regardless of the evidence level.
In cohort 2, the Kaplan-Meier estimate of the median PFS on MTB-directed frontline therapy was 449 (range 42-1,125) days (Data Supplement). In the entire cohort, the median overall survival on MTB-directed therapy was 768 (range 22-1,240) days (Data Supplement).
Exceptional responses were noted in both cohorts. One patient with head and neck squamous cell carcinoma treated with pembrolizumab experienced a PFS2/PFS1 of 31.79. Although the indication for this on-label therapy was PDCD1LG2 amplification, which is predictive of clinical response in this disease,28 this patient was subsequently tested for PD-L1 expression, which was also positive. One patient with a PTEN-mutated ovarian cancer experienced a PFS2/PFS1 ratio of 5.12 on (evidence level 3) everolimus, consistent with a previous report suggesting benefit in this population.29 Before the FDA site-agnostic approval of pembrolizumab for high TMB,25 one patient with squamous cell carcinoma arising from an ovarian teratoma was treated with frontline MTB-directed (evidence level 2) pembrolizumab for a TMB of 25 and experienced a PFS of 880 days.
Poor responses were also noted for patients treated with on-label therapies. These included a PD-L1–positive NSCLC treated with pembrolizumab (PFS2/PFS1 = 0.02), an ERBB2-amplified esophageal adenocarcinoma treated with trastuzumab (PFS2/PFS1 = 0.36), an MSI endometrial cancer treated with pembrolizumab (PFS2/PFS1 = 0.37), and two ovarian cancers treated with PARP inhibitors for a RAD51C mutation (PFS2/PFS1 = 0.39) and an elevated LOH score (PFS2/PFS1 = 0.54).
Safety
A summary of adverse event data is available in the Data Supplement. All-grade adverse events occurred in 23% of patients (21 of 93). Grade 3 events were experienced by 17% (16 of 93) and included GI (14 events, nine patients) and respiratory, thoracic, and mediastinal (nine events, five patients) disorders. Grade 4 events occurred in two patients (2 of 93, 2%) and included neutropenia (1 of 93, 1%) and nonfatal respiratory failure (1 of 93, 1%). While on study, two patients died of progressive disease and two patients died of sepsis. One patient died of sepsis related to aspiration pneumonia 5 days after pembrolizumab administration. A separate patient died of sepsis resulting from a chronic pelvic abscess 15 days after nivolumab administration. Neither of the deaths were determined to be drug-related on independent reviews by the study investigators and treating physicians.
DISCUSSION
This prospective phase II trial demonstrated a survival benefit of MTB-directed therapy over immediately prior therapy. The probability of patients treated with MTB-directed second-line or greater-line therapy experiencing GMI ≥ 1.3 was 0.59. These results are superior to those previously reported by Von Hoff et al,17 where 27% (18 of 66) of patients demonstrated a PFS ratio ≥ 1.317 and comparable with those reported by Radovich et al,30 which showed that 43% (19 of 44) of patients achieved a PFS ratio ≥ 1.3. Differences are likely related to increased availability of targeted therapies, including immune checkpoint inhibitors. The patients most likely to achieve a PFS ratio ≥ 1.3 in these publications were breast and colon cancers17 and sarcomas.30 Although our study enrolled few patients with sarcoma and breast cancer, 8 of 9 (89%) patients with GI cancer, 4 of 5 (80%) patients with head and neck cancer, and 8 of 14 (57%) patients with gynecologic cancer experienced PFS2/PFS1 ≥1.3. This demonstrates the benefit of MTB-directed care across a wide variety of tumor types.
Personalized cancer therapies are quickly becoming approved for multiple molecular targets and primary disease sites. This results in more difficult decisions for patients whose tumors harbor more than one actionable mutation. MTBs may help oncologists choose the most efficacious therapy option, especially when multiple options are available. Although a benefit of the MTB is identifying off-label therapies, oncologists are more likely to use NGS testing to support on-label therapies.10 The present study demonstrates similar evidence of benefit for patients treated with on-label and off-label therapies, which supports the continuous practice of MTB off-label recommendations.
Although most MTB studies include few or no patients treated with up-front targeted therapy, patients treated with frontline MTB-directed therapy in this study experienced a median PFS of 14.9 months (449 days). Sixty percent (26 of 43) of patients in this cohort had advanced or metastatic NSCLC, where NGS sequencing and targeted therapy are frontline.31 Our findings echo the report of Koopman et al,32 describing a median PFS equal to 6.3 months in patients with NSCLC receiving MTB-directed frontline therapy. Although 60% of patients treated with frontline MTB-directed therapy in our study had a diagnosis of NSCLC, the other 40% with varying histologies also benefitted.
Frequency of adverse events was consistent with previous MTB studies,33 and toxicities were as expected for the anticancer agents used in the study.
A strength of this study is its prospective, interventional design. As most previous MTB-related studies are observational, we provide evidence of benefit of MTB-directed care. The statistical design with the use of the novel GMI end point and the Kaplan-Meier method to adjust for censoring is another strength as it compares MTB-directed therapy with the patients' immediately prior therapy, allowing the patient to serve as their own control. Of the 139 patients whom the MTB identified to be eligible for at least one clinical trial, five patients pursued these opportunities. It is important to note that these patients were excluded from this analysis as we felt that it was unethical to count their outcomes in two clinical trials.
Limitations include the possibility of selection bias as this was a single-arm study that only enrolled patients with an actionable mutation and received MTB-directed therapy; therefore, benefit is confined to patients with targetable mutations. This study was conducted at a single-institution tertiary referral center with a largely rural, Appalachian population and may not be generalizable to other populations. Furthermore, this was an unblinded, nonrandomized study with clinical disease assessments and PFS calculated by the study investigators. This could result in less precise assessment and bias in ascertaining PFS; however, patients were usually managed by the same treating oncologist for both regimens, and measurement error is likely similar between the MTB-directed and immediately prior regimens. To minimize bias, study investigators used predefined criteria to assess PFS.
Finally, because of the markedly better-than-anticipated outcomes demonstrated in the planned interim analysis, only 50 patients were enrolled into cohort 1 despite initial sample size calculations requiring 93 patients to detect GMI ≥ 1.3 in 27% of patients. As a result, an IRB-approved Protocol modification permitted enrollment of patients treated with frontline MTB-directed therapy into a second cohort to total 93 patients.
In conclusion, this prospective study demonstrates that patients with advanced cancer with targetable mutations experience survival benefit when receiving MTB-directed care. Additional trials in disease-specific populations and with randomized designs are warranted.
Abigail Anderson
Employment: Quest Diagnostics (I)
Reema A. Patel
Honoraria: Onc Live
Research Funding: Ipsen (Inst), Genentech (Inst)
Travel, Accommodations, Expenses: Genentech
Susanne M. Arnold
Research Funding: AstraZeneca (Inst), Genentech (Inst), Merck Sharp & Dohme (Inst), Nektar (Inst), Exelixis (Inst), Ohio State University (Inst), National Cancer Institute (Inst), Kura Oncology (Inst), Regeneron (Inst), LabCorp (Inst), Syneos Health (Inst), AbbVie (Inst)
Jill M. Kolesar
Stock and Other Ownership Interests: Helix Diagnostics
Consulting or Advisory Role: The Jackson Laboratory
Research Funding: ArtemiLife
Patents, Royalties, Other Intellectual Property: Patent pending for a cell-based therapy derived from human macrophages
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented orally at the Society of Gynecologic Oncology Annual Winter Meeting, Olympic Valley, CA, January 27-29, 2022, and as a poster at the Society of Gynecologic Oncology Annual Meeting, Phoenix, AZ, March 18-21, 2022.
SUPPORT
Supported by the National Cancer Institute at the National Institutes of Health, which provided support for the Biostatistics and Bioinformatics Shared Resource Facility, Biospecimen and Tissue Procurement Shared Resource, and the Cancer Research Informatics Shared Resource Facilities of the University of Kentucky Markey Cancer Center (P30CA177558, B. Mark Evers).
CLINICAL TRIAL INFORMATION
R.W.M. and M.L.H. contributed equally to this work and are joint first authors.
DATA SHARING STATEMENT
Anonymized data sets may be available upon request. Requests for access to data may be obtained by contacting the corresponding author.
AUTHOR CONTRIBUTIONS
Conception and design: Rachel W. Miller, Susanne M. Arnold, Jill M. Kolesar
Financial support: Jill M. Kolesar
Administrative support: Rachel W. Miller, Mikayla Buchanan, Abigail Anderson, Derek B. Allison, Riham H. El Khouli, Reema A. Patel, John L. Villano, Susanne M. Arnold, Jill M. Kolesar
Provision of study materials or patients: Rachel W. Miller, Mikayla Buchanan, Abigail Anderson, Derek B. Allison, Riham H. El Khouli, Reema A. Patel, John L. Villano, Susanne M. Arnold, Jill M. Kolesar
Collection and assembly of data: All authors
Data analysis and interpretation: Megan L. Hutchcraft, Heidi L. Weiss, Jianrong Wu, Chi Wang, Jinpeng Liu, Rani Jayswal
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Abigail Anderson
Employment: Quest Diagnostics (I)
Reema A. Patel
Honoraria: Onc Live
Research Funding: Ipsen (Inst), Genentech (Inst)
Travel, Accommodations, Expenses: Genentech
Susanne M. Arnold
Research Funding: AstraZeneca (Inst), Genentech (Inst), Merck Sharp & Dohme (Inst), Nektar (Inst), Exelixis (Inst), Ohio State University (Inst), National Cancer Institute (Inst), Kura Oncology (Inst), Regeneron (Inst), LabCorp (Inst), Syneos Health (Inst), AbbVie (Inst)
Jill M. Kolesar
Stock and Other Ownership Interests: Helix Diagnostics
Consulting or Advisory Role: The Jackson Laboratory
Research Funding: ArtemiLife
Patents, Royalties, Other Intellectual Property: Patent pending for a cell-based therapy derived from human macrophages
No other potential conflicts of interest were reported.
REFERENCES
- 1. Colomer R, Mondejar R, Romero-Laorden N, et al. When should we order a next generation sequencing test in a patient with cancer? EClinicalMedicine. 2020;25:100487. doi: 10.1016/j.eclinm.2020.100487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao TT, Xu H, Xu HM, et al. The efficacy and safety of targeted therapy with or without chemotherapy in advanced gastric cancer treatment: A network meta-analysis of well-designed randomized controlled trials. Gastric Cancer. 2018;21:361–371. doi: 10.1007/s10120-018-0813-2. [DOI] [PubMed] [Google Scholar]
- 3. Schwaederle M, Zhao M, Lee JJ, et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: A meta-analysis. JAMA Oncol. 2016;2:1452–1459. doi: 10.1001/jamaoncol.2016.2129. [DOI] [PubMed] [Google Scholar]
- 4. Jardim DL, Schwaederle M, Wei C, et al. Impact of a biomarker-based strategy on oncology drug development: A meta-analysis of clinical trials leading to FDA approval. J Natl Cancer Inst. 2015;107:djv253. doi: 10.1093/jnci/djv253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gray SW, Hicks-Courant K, Cronin A, et al. Physicians' attitudes about multiplex tumor genomic testing. J Clin Oncol. 2014;32:1317–1323. doi: 10.1200/JCO.2013.52.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kurzrock R, Colevas AD, Olszanski A, et al. NCCN oncology research program's investigator steering committee and NCCN best practices committee molecular profiling surveys. J Natl Compr Canc Netw. 2015;13:1337–1346. doi: 10.6004/jnccn.2015.0163. [DOI] [PubMed] [Google Scholar]
- 7. Bourret P, Cambrosio A. Genomic expertise in action: Molecular tumour boards and decision-making in precision oncology. Sociol Health Illn. 2019;41:1568–1584. doi: 10.1111/1467-9566.12970. [DOI] [PubMed] [Google Scholar]
- 8. Burkard ME, Deming DA, Parsons BM, et al. Implementation and clinical utility of an integrated academic-community regional molecular tumor board. JCO Precis Oncol. 2017 doi: 10.1200/PO.16.00022. 10.1200/PO.16.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walko C, Kiel PJ, Kolesar J. Precision medicine in oncology: New practice models and roles for oncology pharmacists. Am J Health Syst Pharm. 2016;73:1935–1942. doi: 10.2146/ajhp160211. [DOI] [PubMed] [Google Scholar]
- 10. Freedman AN, Klabunde CN, Wiant K, et al. Use of next-generation sequencing tests to guide cancer treatment: Results from a nationally representative survey of oncologists in the United States. JCO Precis Oncol. 2018;2:1–13. doi: 10.1200/PO.18.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Larson KL, Huang B, Weiss HL, et al. Clinical outcomes of molecular tumor boards: A systematic review. JCO Precis Oncol. 2021;5:1122–1132. doi: 10.1200/PO.20.00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kato S, Kim KH, Lim HJ, et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun. 2020;11:4965. doi: 10.1038/s41467-020-18613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang B, Chen Q, Allison D, et al. Molecular tumor board review and improved overall survival in non-small-cell lung cancer. JCO Precis Oncol. 2021;5:1530–1539. doi: 10.1200/PO.21.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harris PA, Taylor R, Minor BL, et al. The REDCap consortium: Building an international community of software platform partners. J Biomed Inform. 2019;95:103208. doi: 10.1016/j.jbi.2019.103208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Von Hoff DD, Stephenson JJ, Jr., Rosen P, et al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28:4877–4883. doi: 10.1200/JCO.2009.26.5983. [DOI] [PubMed] [Google Scholar]
- 18. Bailey CH, Jameson G, Sima C, et al. Progression-free survival decreases with each subsequent therapy in patients presenting for phase I clinical trials. J Cancer. 2012;3:7–13. doi: 10.7150/jca.3.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lichtman SM. Pharmacokinetics and pharmacodynamics in the elderly. Clin Adv Hematol Oncol. 2007;5:181–182. [PubMed] [Google Scholar]
- 20. Jung SH, Kim KM. On the estimation of the binomial probability in multistage clinical trials. Stat Med. 2004;23:881–896. doi: 10.1002/sim.1653. [DOI] [PubMed] [Google Scholar]
- 21. Koyama T, Chen H. Proper inference from Simon's two-stage designs. Stat Med. 2008;27:3145–3154. doi: 10.1002/sim.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu J, Chen L, Wei J, et al. Phase II trial design with growth modulation index as the primary endpoint. Pharm Stat. 2019;18:212–222. doi: 10.1002/pst.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Flaherty KT, Gray RJ, Chen AP, et al. Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH) J Clin Oncol. 2020;38:3883–3894. doi: 10.1200/JCO.19.03010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patel RA, Lin M, Harper MM, et al. Pediatric patient with peritoneal mesothelioma harboring ALK rearrangement. Curr Probl Cancer Case Rep. 2021;4:100074. [Google Scholar]
- 25.United States Food and Drug Administration . FDA Approves Pembrolizumab for Adults and Children With TMB-H Solid Tumors. 2020. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors [Google Scholar]
- 26.United States Food and Drug Administration . FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication. 2017. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication [Google Scholar]
- 27.United States Food and Drug Administration . FDA Approves Niraparib for HRD-Positive Advanced Ovarian Cancer. 2019. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-niraparib-hrd-positive-advanced-ovarian-cancer [Google Scholar]
- 28. Yearley JH, Gibson C, Yu N, et al. PD-L2 expression in human tumors: Relevance to anti-PD-1 therapy in cancer. Clin Cancer Res. 2017;23:3158–3167. doi: 10.1158/1078-0432.CCR-16-1761. [DOI] [PubMed] [Google Scholar]
- 29. Wheler JJ, Moulder SL, Naing A, et al. Anastrozole and everolimus in advanced gynecologic and breast malignancies: Activity and molecular alterations in the PI3K/AKT/mTOR pathway. Oncotarget. 2014;5:3029–3038. doi: 10.18632/oncotarget.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Radovich M, Kiel PJ, Nance SM, et al. Clinical benefit of a precision medicine based approach for guiding treatment of refractory cancers. Oncotarget. 2016;7:56491–56500. doi: 10.18632/oncotarget.10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ettinger DS, Wood DE, Aisner DL, et al. NCCN Clinical Practice Guidelines in Oncology: Non-small Cell Lung Cancer. Version 7.2021. 2021. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf [Google Scholar]
- 32. Koopman B, van der Wekken AJ, Elst A, et al. Relevance and effectiveness of molecular tumor board recommendations for patients with non–small-cell lung cancer with rare or complex mutational profiles. JCO Precis Oncol. 2020:393–410. doi: 10.1200/PO.20.00008. [DOI] [PubMed] [Google Scholar]
- 33. Réda M, Richard C, Bertaut A, et al. Implementation and use of whole exome sequencing for metastatic solid cancer. EBioMedicine. 2020;51:102624. doi: 10.1016/j.ebiom.2019.102624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data sets may be available upon request. Requests for access to data may be obtained by contacting the corresponding author.