

Abstract
The administration of allogeneic natural killer (NK) cells following a lymphodepleting chemotherapy regimen is emerging as a well-tolerated therapeutic approach in the management of various malignancies. Contrary to the expected complications of allogeneic T cell therapy, there remains no evidence of graft-versus-host disease (GVHD) mediated by NK cells in numerous clinical trials. On the contrary, preclinical and clinical studies suggest that NK cells do not induce GVHD and in fact may prevent its development following allogeneic hematopoietic cell transplantation (HCT). In this study, we sought to determine the maximum tolerated dose of non-HLA-matched donor NK cells derived from peripheral blood and ex vivo expanded using a novel feeder cell platform. In a single-center Phase I clinical trial using a 3 × 3 design, 9 subjects each received 2 infusions of NK cells 2 weeks apart following a preparative regimen of cyclophosphamide (60 mg/kg i.v.) and fludarabine (25 mg/m2/day i.v for 5 days). No exogenous cytokines were administered. NK cells were administered at 3 dose levels: 1 × 107/kg, 2.5 × 107/kg, and 5 × 107/kg. Three subjects had myelodysplastic syndrome (MDS) or acute myelogenous leukemia (AML), and the other 6 subjects had colorectal carcinoma. Recipients were monitored over a 4-week period for GVHD as well as other adverse events and for persistence of donor NK cells in systemic circulation. Disease assessment was started at 28 days following the first NK cell infusion and continued until postinfusion day 100 or disease progression. In all 9 study subjects, there was no occurrence of GVHD and no dose-limiting toxicities that would warrant cohort expansion at any of the 3 planned cell dose levels. Low-level donor NK cell persistence was observed up to 4 weeks after the first NK cell infusion at all dose levels. The best observed response was a complete response with incomplete platelet recovery in a MDS subject who experienced disease relapse after prior allogeneic HCT. Other responses were stable disease in 1 subject with MDS and 2 subjects with colorectal cancer up to postinfusion day 100. This off-the-shelf, third-party NK cell product can be administered safely without inducing GVHD and exhibits in vivo persistence promoted by preparative lymphodepletion alone. The observed clinical responses could be enhanced by administration of exogenous cytokine support, as well as complementary approaches that promote NK cell function in the tumor microenvironment.
Keywords: Universal donor NK cells, NK cell therapy
INTRODUCTION
Adoptive natural killer (NK) cell therapy has demonstrated the potential for effective cancer immunotherapy in various malignancies with particular potential in hematologic malignancies and several solid tumors [1–4]. Several clinical trial reports have shown that infusions of HLA-haploidentical donor NK cells are well tolerated without inducing graft-versus-host disease (GVHD), cytokine release syndrome (CRS), or immune effector cell-related neurotoxicity syndrome [5–12]. In contrast, CRS and immune effector cell-related neurotoxicity syndrome are significant complications of autologous T cell therapies, whereas GVHD is mediated by allogeneic T cell therapies. As such, improving the application and efficacy of NK cell therapy remains of significant interest.
Breakthroughs in ex vivo NK cell expansion have allowed the generation of large NK cell doses [13–17]. This is an important development as higher doses have been associated with in vivo persistence and possibly efficacy [1–3]. Until recently, allogeneic NK cell adoptive therapies primarily have used NK cells derived from partially HLA-matched cord blood or from peripheral blood of HLA-haploidentical (haplo) donors [1–12]. In these models, the enormous costs (both logistical and financial) of cell manufacturing for each subject, as well as the often-limited availability of HLA-compatible donors for each intended subject, make generalizing this approach challenging.
We designed and completed a clinical trial to test the safety of ex vivo expanded “universal donor” NK cells in conjunction with lymphodepleting chemotherapy to enhance the survival and persistence of the allogeneic cells. For this trial, the NK cells were expanded ex vivo using a feeder cell line, called NKF, that was generated at our institution through the expression of membrane-bound IL-21 (mbIL-21) on the acute myelogenous leukemia (AML) cell line OCI-AML3 [18]. This feeder cell line has been demonstrated to lead to robust expansion (thousands of fold) of primary NK cells when irradiated NKF cells are cocultured with NK cells. The NK cell products generated using this feeder cell platform demonstrate high cytotoxic activity against a broad panel of cancer cell lines both in vitro and in murine xenografts [18]. To support the manufacture of NK cells under large-scale good manufacturing practice conditions for use in clinical studies, a master cell bank of NKF cells was generated.
The primary objective of the present clinical trial was to demonstrate that escalating doses of off-the-shelf NK cell products generated on the NKF expansion platform from third-party adult donors can be infused without inducing GVHD or other significant toxicities. A secondary goal of the study was to examine whether the proposed lymphocyte-depleting preparative regimen would prevent immediate rejection of the HLA-mismatched NK cell product, thereby allowing for a demonstrable antitumor effect.
METHODS
NK Cell Manufacture
Healthy adult volunteers (age ≥18 years) identified by the Hematopoietic Cell Biorepository Core of the Case Comprehensive Cancer Center provided informed consent in accordance with an Institutional Review Board-approved protocol. Preferential selection was for NK cell donors classified as the killer immunoglobulin-like receptor (KIR) B haplotype with ≥2 activating KIR genes [19,20]. This KIR genotyping was performed using the Invitrogen KIR SSP Genotyping Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions by the HLA laboratory at University Hospitals Cleveland Medical Center. Donor eligibility determination included a physical examination and a Donor Heath Questionnaire for evidence of cardiovascular and communicable disease, as well as screening for infectious disease markers according to 21CFR1271.50 using blood tests performed in a Clinical Laboratory Improvement Amendments-certified laboratory (Indiana Blood Center, Indianapolis, IN). Two donors were used for this clinical study, including 1 donor for the first dose level and a second donor for the next 2 dose levels. Nonmobilized mononuclear cells were harvested from a 2-blood volume leukapheresis procedure. This apheresis mononuclear cell product was subsequently depleted of CD3+ lymphocytes using the CliniMACS Plus system (Miltenyi Biotech, Bergisch Gladbach, Germany) according to the manufacturer’s protocol. The final CD3+-depleted product was plated in G-REX flasks (Wilson Wolf Manufacturing, St Paul, MN) at a starting density of approximately 5 × 107/L NK cells in coculture with gamma-irradiated (90 Gy; Cesium-137 irradiator; CIS BIO International, Paris, France) NKF feeder cells. The NKF cells were plated at a ratio of 10 irradiated NKF cells to 1 NK cell on day 1, followed by subsequent weekly feeder additions at a 5 NKF-to-1 NK cell ratio. The cell culture medium was supplemented with 100 U/mL IL-2 (Miltenyi Biotech) at a minimum of every 2 days and during media changes. NK cells were harvested from culture after 2 to 3 weeks of culture and cryopreserved in aliquots of approximately 5 × 107/mL NK cells with 5% dimethyl sulfoxide (Cryoserv; Merit Pharmaceutical, Los Angeles, CA) in human serum albumin (Grifols Biologics, Los Angeles, CA). Cryostore freezing bags (OriGen Biomedical, Austin, TX) containing product aliquots were subjected to controlled-rate freezing to a temperature of −100 °C before transfer to liquid nitrogen storage.
Study Design
This Phase I study used a “3 + 3” design cell dose escalation in which 3 subjects were enrolled sequentially to each of 3 planned NK cell dose levels: 1 × 107/kg, 2.5 × 107/kg, and 5 × 107/kg. The study was approved by the Institutional Review Board of University Hospitals Cleveland Medical Center under an Investigational New Drug application reviewed by the US Food and Drug Administration, and all subjects provided written informed consent. Patients age ≥18 years were eligible for the study if they had relapsed/refractory metastatic colorectal adenocarcinoma, sarcoma, or any hematologic malignancy. Patients with hematologic malignancies who had undergone previous allogeneic hematopoietic cell transplantation (HCT) were eligible if they were beyond 1 year from transplantation and did not have active GVHD requiring systemic therapy.
Study Treatment
Study subjects first received a lymphocyte-depleting chemotherapy regimen consisting of i.v. cyclophosphamide 60 mg/kg as a single dose (T-7), followed by i.v. fludarabine 25 mg/m2 per day for 5 days (T-6 to T-2). At 48 hours after the last fludarabine infusion (T-0), study subjects received NK cell infusions at the corresponding dose level, followed by a second dose repeated 2 weeks later. The second NK cell infusion was administered without repeating the chemotherapy regimen. No exogenous cytokines or hematopoietic growth factors were administered.
Preparation for NK Cell Infusion
Selected units were thawed in a water bath at 37 °C according to institutional policy with <10 minutes in the water bath until a slushy consistency was observed before further processing (dilution, wash, and final formulation). For the first cell dose level (1 × 107/kg), sufficient cryopreserved aliquots to meet 1.5 to 2 times the intended cell dose were thawed the day before the planned infusion and maintained overnight in NK expansion medium supplemented with rhIL-2 (1000 U/mL). On the morning of infusion, NK cells were washed twice, and the calculated cell dose was resuspended into a final formulation of 5 to 200 × 106/mL NK cells in Plasma-Lyte A (Baxter Healthcare, Deerfield, IL) with 2.5% human serum albumin. For the second and third dose levels, the overnight culture step was omitted. Instead, on the morning of infusion sufficient cryopreserved aliquots to meet 1.5 times the intended cell dose were thawed and washed before resuspension of the calculated cell dose. Release criteria were viability >60% (by 7-AAD flow cytometry) when overnight culture was performed or >70% if the product was infused immediately after thawing, CD3+CD56− content <5 × 105/kg, CD19+ B cell content <1%, and negative endotoxin and Gram stain. Products were distributed in Luer Lock syringes at the first 2 cell dose levels and in infusion bags at the third dose level.
Patient Monitoring
All study subjects received both NK cell infusions while hospitalized on the Hematopoietic Cell Transplant Service and were discharged to home on the day after the first infusion and a at minimum of 4 hours after the second infusion. Outpatient follow-up included physical examination for symptoms or signs of GVHD, laboratory monitoring of hematologic and biochemistry profiles, as well as blood product transfusions when indicated. Blood samples were obtained at T+1, +3, +5, +7, +14, +21, and +28 days for correlative assessments of serum and mononuclear cells. Adverse events occurring up to 28 days after the first NK cell infusion were considered dose-limiting toxicities (DLTs) if they met prespecified criteria: grade II-IV acute GVHD (graded according to the 1994 Consensus Conference on Acute GVHD Grading) and grade 3-4 nonhematologic toxicities (graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events, version 4.03) attributed to the NK cell product. Because cytopenia is expected following the lymphodepleting regimen, a hematologic DLT was defined as profound cytopenia (absolute neutrophil count ≤500/μL, platelet count ≤20,000/μL, and hemoglobin level <7 g/dL) persisting for 28 days after the start of chemotherapy if different than baseline values. Disease assessments were obtained as appropriate for each diagnosis at 28 days and 3 months after the first NK cell infusion.
Correlative Analyses
NK cells derived from 3 manufacturing runs were assessed phenotypically either fresh (at harvest) or post-thaw following cryopreservation by flow cytometry (Navios EX flow cytometer; Beckman Coulter, Indianapolis, IN). The following antibodies were used: CD159a-FITC, CD335-p3, CD314-PE vio615, CD158a_h-PC5.5, CD158b1_b2-pc7, CD158e1_e2-APC, CD16 APC-750, CD94-PB (all from Beckman Coulter).
Cytokine levels in patient plasma samples were measured using Luminex multiplex cytokine kits (R&D Systems) following the manufacturer’s protocol for the following: IL-1, IL-2, IL-4, IL-6, IL-7, IL-8, IL-10, IL-12, IL-15, IL-18, GM-CSF, and IFN-γ. All statistical comparisons were analyzed using Student’s t test.
RESULTS
Product Manufacture
For the first manufacturing run intended for dose level 1, the CD3-depleted apheresis product containing 0.4 × 109 NK cells was seeded into culture as described in Methods. After 2 weeks of culture 14.2 × 109 NK cells were harvested (36-fold expansion). For the subsequent manufacturing run, 0.76 × 109 NK cells were seeded, and a 152-fold expansion was observed over 3 weeks. For this run, a partial harvest was performed after week 2 because we had reach our maximal manufacturing capacity, and a portion of the cells were maintained in culture until week 3. There was a 31.6-fold NK cell fold expansion yielding 24 × 109 NK cells in the first 2 weeks of culture and a 4.8-fold expansion in week 3 yielding an additional 54.2 × 109 NK cells. Given the high product availability, NK cells from the second manufacturing run were administered to all 6 study subjects at dose levels 2 and 3.
Among the 18 NK cell product infusions administered during the study, the median viability of the final infused products was 77.15% (range, 70% to 90.3%). The final cryopreserved/infused products were highly enriched for NK cells (median, 98.03%) and maximally depleted of B cells (median, 0.05%) and T cells (median, 1.06%) (Figure 1A). Phenotypic analysis showed that post-thaw NK cell products retained similar proportions of cells expressing NKG2D, NKG2A, NKp46, and NKG2C as the freshly cultured product at harvest (Figure 1B). In comparison, CD16-expressing cells were decreased post-thaw to a statistically significant degree (P = .0295). Expression levels of corresponding KIRs were unchanged after cryopreservation (data not shown). Overnight stimulation of post-thaw NK cells with high-dose IL-2 at dose level 1 did not change these observed differences significantly.
Figure 1.
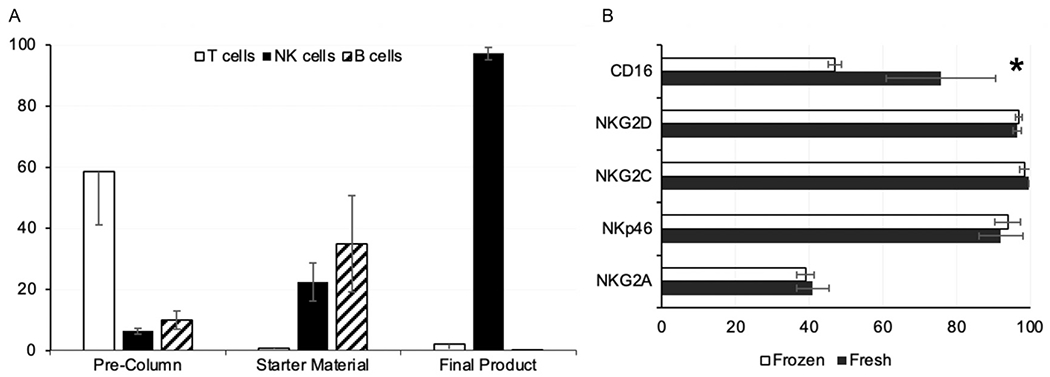
Phenotypic analyses of NK cell products manufactured from 3 donors. (A) Presented as percentages are the T cell, NK cell, and B cell content of PBMCs obtained from donor leukapheresis (precolumn), CD3-depleted PBMCs at start of cell culture (starter material), and final NK cell product harvested after 2 to 3 weeks of culture. (B) NK cell products were analyzed for retention of various functional receptors at thaw following cryopreservation (frozen) compared with the same products when harvested from culture (fresh). The observed difference in CD16+ cell population was statistically significant. *P = .029. Values shown are percentages.
Study Enrollment
Subjects were accrued between June 2018 and April 2021. Fifteen subjects were screened and consented to enroll on the study; however, 6 of these subjects did not receive treatment according to the protocol. Five of these 6 subjects were deemed ineligible to proceed owing to acute clinical events related to disease progression before the start of lymphodepleting chemotherapy. One subject developed high-grade fever with transient hypotension shortly after the start of cyclophosphamide therapy (T-7), which was attributed to a catheter-related bacteremia from a preexisting implanted device. Chemotherapy was discontinued, and the subject was withdrawn from the study and replaced with another subject at the same dose level.
Subject Characteristics
Of the 9 study subjects, 6 had metastatic colon/rectal carcinoma that progressed after 3 or more lines of chemotherapy including 5-fluorouracil-based combination regimens (Table 1). Sites of disease metastases were predominantly lungs and liver. Two of the remaining 3 subjects had high-grade myelodysplastic syndrome (MDS) with circulating blasts and high tumor burden following several lines of treatment, including an allogeneic HCT (subject 8). The third subject (subject 12) had recurrent AML presenting with extramedullary disease in the form of myeloid sarcoma of the breast. At the time of enrollment, this subject had experienced another breast cancer relapse after her second haploidentical donor HCT.
Table 1.
Patient Characteristics, Treatment Profiles, and Outcomes (Clinical and Correlative) of the 9 Study Subjects Who Received NK Cell Products in 3 Dose Level Cohorts.
| NK Cell Dose | 1 × 107/kg | 2.5 × 107/kg | 5 × 107/kg | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Subject ID | #1 | #4 | #5 | #7 | #8 | #12 | #13 | #14 | #15 |
| Gender, Age (years) | M, 73 | F, 61 | M, 66 | F, 59 | F, 33 | F, 23 | M, 56 | F, 55 | F, 61 |
| Diagnosis | MDS | Colorectal | Colon | Colon Ca | High Risk MDS, post SCT relapse | Myeloid sarcoma (s/p 2nd SCT) | Colon Ca | Rectal Ca | Rectal Ca |
| Met sites | N/A | multiple | lungs | multiple | n/a | breast | multiple | Lungs | Liver, lungs |
| HLA Match with NK cell donar | 4/10 | 3/10 | 2/10 | 3/10 | 1/10 | 1/10 | 3/10 | 2/10 | 4/10 |
| KIR-L Pairs: HvG GvH | Mismatch (B) Match | Mismatch (C) Match | Match Match | Mismatch (B) Mismatch (C) | Mismatch (B) Mismatch (C) | Mismatch (B) Match | Match Match | Match Match | Mismatch (B) Match |
| NK Cell Dates | 06/01/18 06/15/18 | 12/20/18 01/03/19 | 02/14/19 02/28/19 | 11/21/19 12/05/19 | 04/30/20 05/14/20 | 11/19/20 12/03/20 | 01/12/21 01/26/21 | 03/16/21 03/30/21 | 05/11/21 05/25/21 |
| D28 donor STR chimerism | N/A | 2% (8% on +7) | 1% (4% on +7) | 4% (14% on +7) | 2% (N/A on +7) | 2% (N/A on +7) | N/A | 3% (20% on +8) | 2% (7% on +7) |
| GVHD (Y/N) | N | N | N | N | N | N | N | N | N |
| DSA (HLA Class I and II) | N* | N | N* | N* | N* | N* | N (Day 21) | N* | N |
| Day 28 | Stable | Progression | Stable | Progression | Stable disease; Increased SCT donor chimerism | Stable | Progression | Stable (mixed response) | Progression |
| Day 100 | Progression | Hospice | Stable | Hospice | CRi | N/A (out of state clinical trial) | Death | Progression | Other clinical trial |
MDS (Myelodysplastic syndrome); SCT (allogeneic hematopoietic cell transplant); N/A (not assessable); NK cell (natural killer cell); D28 (Day 28); STR (short tandem repeat); GVHD (graft versus host disease); DSA (donor specific antibodies); HLA (human leukocyte antigen): HLA-A, -B, -C, DRB1, DQB1; KIR-L (killer cell immunoglobulin-like receptor ligand) Pairs reporting if recipient (H) and NK cell donor (G) were KIR-ligand Matched or Mismatched. Direction of NK cell alloreactivity predicted by KIR-Ligand status is shown as recipient versus donor (HvG) or donor versus recipient (GvH). The mismatched KIR-ligand epitope is shown if HLA-Bw4/6 (B) or HLA-C1/C2 (C)
Donor STR chimerism on Day 7 was not interpretable (designated N/A) on day +7 if lymphocytes were not detectable on complete blood count
Subjects were observed to have antibodies to HLA Class I and/or II antigens that were not present in their respective NK cell donors
Study Treatment
All 9 study subjects received the complete cyclophosphamide and fludarabine conditioning regimen according to protocol. Cell products obtained from the first manufacturing run were administered to all 3 subjects at dose level 1. All 6 subjects at dose levels 2 and 3 received products from a different manufacturing run that used a second donor. Because no DLTs were observed, no cohort expansion was required at any cell dose level. All 9 subjects received 2 NK cell infusions as planned. The treatment profiles of the 9 subjects who completed the study treatment are presented in Table 1. All 9 subjects were HLA-disparate with the NK cell donors at a match range of 1 to 4 out of 10 (HLA-A, -B, C, -DR, and -DQ). Mismatches in KIR ligands were observed in the recipient-versus-donor direction in 6 subjects (1 HLA-C1/C2 and 5 HLA-Bw4/6 epitopes) and in the donor-versus-host direction in 2 subjects (both HLA C1/C2 epitopes) (Table 1).
Adverse Events
Although donor NK cells were demonstrated to persist in circulation for up to 28 days after initial administration, no study subject developed acute GVHD (Table 1). Of note, subject 12 had completed systemic management of chronic GVHD from 2 previous allogeneic HCTs at 6 months before enrollment. This subject did not develop acute GVHD or recurrent chronic GVHD following NK cell administration.
There were no significant NK cell infusion-related events; 1 subject developed transient chills at 1 hour after the first NK cell infusion, and the second infusion involved no complications. In all study subjects, hematologic recovery (ie, absolute neutrophil count >500/μL and platelet count >20,000/μL without transfusion support or return to pretreatment baseline) was observed between days 11 and 14 following the first NK cell infusion. Excluding cytopenias, the most common adverse event observed was fatigue, seen in all subjects, with onset typically 5 to 6 days after the start of the lymphocyte-depleting chemotherapy regimen (Table 2). Other events observed in >30% of subjects were nausea (56%), headache (56%) and transaminitis (44%), all of which were grade 1 to 2 only (Table 2). All observed events were expected events attributable to the lymphocyte-depleting chemotherapy regimen.
Table 2.
Observed Nonhematologic Adverse Events in First 28 Days after NK Cell Infusion.
| Adverse Event | Number (%) | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|---|
| Fatigue | 9 (100%) | 7 | 0 | 2 | 0 |
| Headache | 5 (56%) | 4 | 1 | 0 | 0 |
| Nausea | 5 (56%) | 2 | 3 | 0 | 0 |
| Transaminitis | 4 (44%) | 3 | 1 | 0 | 0 |
| Hypoalbuminemia | 3 (33%) | 1 | 2 | 0 | 0 |
| Anorexia / weight loss | 3 (33%) | 1 | 2 | 0 | 0 |
| Constipation | 3 (33%) | 3 | 0 | 0 | 0 |
| Pedal edema | 2 (22%) | 0 | 2 | 0 | 0 |
| Pruritus | 2 (22%) | 1 | 1 | 0 | 0 |
| Petechiae | 2 (22%) | 2 | 0 | 0 | 0 |
| Tachycardia | 2 (22%) | 2 | 0 | 0 | 0 |
| Neutropenic fever | 2 (22%) | 1 | 0 | 1 | 0 |
| Hyperglycemia | 2 (22%) | 1 | 1 | 0 | 0 |
| Dysgeusia | 2 (22%) | 2 | 0 | 0 | 0 |
| Alopecia | 2 (22%) | 1 | 1 | 0 | 0 |
| Dyspnea | 2 (22%) | 1 | 1 | 0 | 0 |
| Orthostasis | 2 (22%) | 1 | 1 | 0 | 0 |
| Rash | 1 (11%) | 1 | 0 | 0 | 0 |
| Diarrhea | 1 (11%) | 1 | 0 | 0 | 0 |
| Hypokalemia | 1 (11%) | 1 | 0 | 0 | 0 |
| Hypophosphatemia | 1 (11%) | 1 | 0 | 0 | 0 |
| Hypomagnesemia | 1 (11%) | 0 | 1 | 0 | 0 |
| Hypocalcemia | 1 (11%) | 1 | 0 | 0 | 0 |
| Prolonged INR | 1 (11%) | 1 | 0 | 0 | 0 |
| Vomiting | 1 (11%) | 1 | 0 | 0 | 0 |
| Cough | 1 (11%) | 0 | 1 | 0 | 0 |
| Catheter infection | 1 (11%) | 0 | 0 | 1 | 0 |
| Dysuria | 1 (11%) | 1 | 0 | 0 | 0 |
| Dehydration | 1 (11%) | 1 | 0 | 0 | 0 |
| Pain (Musculoskeletal) | 1 (11%) | 0 | 1 | 0 | 0 |
| Oral mucositis | 1 (11%) | 0 | 1 | 0 | 0 |
| Colitis | 1 (11%) | 0 | 0 | 1 | 0 |
| Pleural effusion | 1 (11%) | 0 | 1 | 0 | 0 |
| Visual hallucinations | 1 (11%) | 1 | 0 | 0 | 0 |
| Abdominal pain | 1 (11%) | 0 | 0 | 1 | 0 |
No study subjects exhibited clinical evidence of CRS. In 7 subjects with adequately evaluable samples, only IL-15 and IL-7 showed significant increases over baseline levels up to 28 days after the first infusion (Figure 2). There were no observable differences in the profiles of IL-2, IL-6, IL-10, IL-8 (Figure 2), IL-1, IL-4, IL-12, GM-CSF, or IFN-γ (data not shown).
Figure 2.
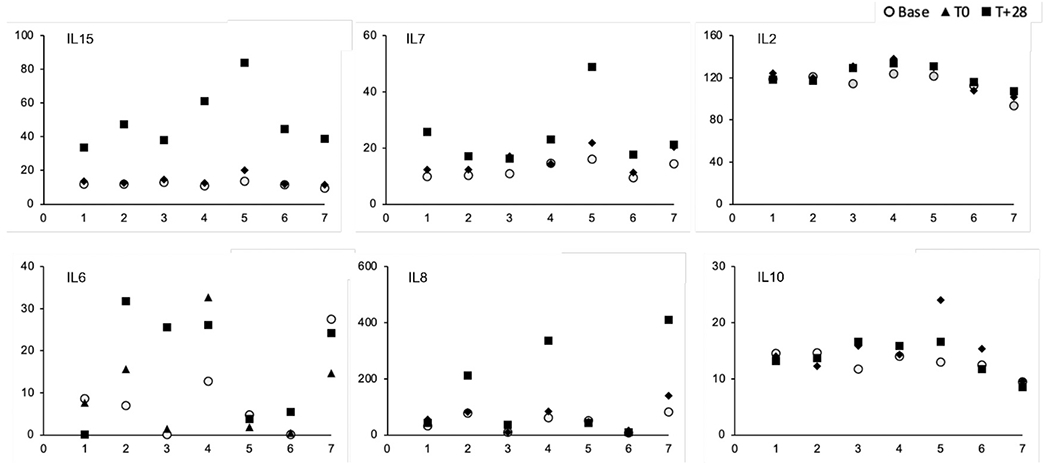
Serum cytokine assays obtained from 7 study subjects before (baseline; circles); on the day of (T0; triangles), and 28 days after (T+28; squares) the first NK cell infusion. All cytokine levels are in pg/mL (y-axis); individual subjects are on the x-axis.
Persistence of Donor-Derived NK Cells
Eight study subjects were tested for circulating donor NK cells by short tandem repeat chimerism at multiple time points up to 28 days after the first NK cell infusion. In 5 subjects, peak NK cell donor chimerism was observed at day 7 following the first NK cell infusion, ranging from 4% to 20% of peripheral blood mononuclear cells (PBMCs). Donor NK cells constituted 1% to 3% of circulating PBMCs at 28 days after the first NK cell infusion (Table 1).
Development of Donor-Directed Anti-HLA Antibodies
As of 28 days after the first NK cell infusion, no study subject was observed to have antibodies directed against NK cell donor class I or II HLA antigens. Six subjects were found to have antibodies against nondonor class I and/or II antigens at this time point.
Clinical Outcomes
At the first NK cell dose level, the best observed responses were stable disease in the high-risk MDS patient and a colon cancer patient with lung metastases (Table 1). The MDS patient (subject 1) had unchanged dysplasia and blast percentage in the marrow at day 28; however, circulating blast burden after hematologic recovery was decreased by 50%. This subject also had a decreased transfusion requirement for 90 days before developing disease progression. The third colon cancer patient (subject 5) who had progressively enlarging lung metastases on 2 computed tomography scans obtained every 3 months before study enrollment subsequently had stable disease until day 100.
At the second NK cell dose level, the best observed response was in subject 8, who relapsed with MDS at 9 months after undergoing allogeneic HCT and subsequently had persistent disease following hypomethylating agent therapy before study enrollment. At 28 days after the first NK cell infusion, although marrow dysplasia and low-level blasts (<5%) remained evident, a rise in transplant donor chimerism was observed. Without additional treatment, full donor chimerism and complete remission with incomplete platelet recovery occurred 4 weeks later. The patient subsequently underwent a second allogeneic HCT to consolidate this remission and had sustained complete remission at 28 days post-transplantation (100 days after NK cell infusion). This patient remained disease-free at 15 months following study treatment and at 12 months post-transplantation. At postinfusion day 28, subject 12 was found to have no change in size of the extramedullary AML in the breast and no new sites of disease.
At NK cell dose level 3, the only observed response was in subject 14 (rectal carcinoma), in whom a 25% decrease in size was observed in some lung metastases, whereas others were unchanged at 28 days after her first NK cell infusion. This study subject also had the highest NK cell donor chimerism at all time points assessed: 20% of circulating PBMCs at day 7 and 3% still circulating as of day 28. Disease progression was subsequently observed on her day 100 imaging. All other study subjects had progressive disease by their day 28 assessment and proceeded to subsequent therapies except subjects 4 and 13, who transitioned to hospice care.
DISCUSSION
In this first human study of ex vivo-expanded, off-the-shelf universal donor NK cells derived from peripheral blood and administered following lymphocyte depletion, we demonstrate an absence of GVHD, including in HCT recipients. Even in the absence of exogenous cytokine administration, we see significant in vivo persistence of these HLA-disparate NK cell products, as well as signals of therapeutic effect in both AML/MDS and colon cancer.
The absence of GVHD in all study subjects in this trial is very striking and lends credence to the concept of universal-donor, off-the-shelf platforms for adoptive NK cell therapy. This is consistent with findings from the few published clinical trials of NK cells derived from HLA-mismatched cord blood and adult donors also without evidence of GVHD [21,22]. One important benefit of the HLA-mismatched donor approach is that it increases the likelihood of NK cell alloreactivity, thereby enhancing “graft-versus-tumor” effect [23,24]. This alloreactivity is mediated by a discordance between inhibitory KIRs on donor NK cells and their ligands, epitopes of HLA class I antigens, on recipient tumor cells. Donor KIR versus recipient KIR-ligand (KIR-L) mismatch is associated with improved disease-free survival and reduced GVHD following allogeneic HCT [23–26]. The 2 subjects with the highest dose of donor KIR/recipient KIR-L incompatible NK cells in a clinical trial published by Yang et al [21]. also had the longest progression-free survival.
It is also notable that none of the HCT recipients treated on this study developed GVHD after our NK cell product infusion. In another clinical trial reported by Shaffer et al., [11] GVHD was not observed up to 100 days following infusion of haploidentical donor NK cells and IL-2 in AML patients who relapsed at a median of 6.8 months after allogeneic HCT. Given our pilot use of unrelated donor products, we took the added precaution of excluding patients in the first year after allogeneic HCT and/or were receiving systemic treatment for GVHD at time of screening. Of note, 1 of our 2 post-HCT subjects had a history of chronic GVHD of the gut but was no longer requiring systemic therapy at 6 months before study enrollment.
Another future benefit of our study’s exclusion of familial and HLA-match from allogeneic NK cell donor selection is that this approach expands the pool of “ideal” donors selected for specific qualities, such as activating KIR profile. Grafts from HCT donors with a genotype that encodes a preponderance of activating KIRs (KIR B haplotypes) have been associated with improved clinical outcomes (disease-free survival, overall survival, and GVHD) following allogeneic HCT for AML [19,20,27]. Both NK cell donors in our study genotyped as KIR B-haplotype had a predicted KIR B content of “better.” Each donor had at least 2 activating KIRs (donor for dose level 1: KIR2DS1, KIR2DS2, KIR2DS5, KIR3DS5; donor for dose levels 2 and 3: KIR2DS2, KIR2DS3, KIR3DS1) in addition to the conserved KIR2DS4. Four of 11 healthy volunteers in the biorepository from which NK cell donors were selected genotyped as KIR B haplotype with similar activating KIR content profiles (>2).
Although the clinical benefit that we found despite the use of KIR B haplotype donors was minimal, all 3 MDS/AML patients in our study demonstrated some response: 2 with stable disease and 1 with CR with incomplete count recovery. As mentioned earlier, the post-transplantation treatment benefit of KIR B haplotype donors has been reported largely only in AML settings; however, from our present data, we cannot deduce the true impact of KIR B on colon cancer in the absence of other treatment conditions that may improve in vivo NK cell proliferation and function. As an example, we did not administer exogenous IL-2 or IL-15 to support these cells in vivo. In addition, the optimal cell doses and conditions that could permit adequate NK cell infiltration into solid tumor beds are not known. Despite these limitations, we did observe 2 of 6 colon cancer patients with stable disease or mixed response.
This trial also demonstrates that the lymphodepleting regimen of cyclophosphamide 60 mg/kg and fludarabine 125 mg/m2 adequately prevents immediate host rejection of HLA-disparate NK cells without causing prolonged hematologic toxicity or other significant adverse events. Previous studies that administered lower doses of cyclophosphamide and omission of fludarabine from the preparative regimen before infusion of haploidentical donor NK cells were associated with rapid disappearance of infused NK cells [11]. Only transient (≤4 days) circulation of third-party donor NK cells was observed in a study published by Yang et al [21]. of third-party NK cells in which no lymphodepleting regimen was given. Although the absence of GVHD in that study may be attributed to the host rejection of donor NK cells, our study demonstrates a persistence of circulating donor NK cells up to 4 weeks from the first infusion (2 weeks from the second infusion) still with an absence of GVHD.
Finally, our novel feeder cell-based ex vivo expansion platform for manufacturing our “universal donor” NK cell product also improved financial and logistic feasibility of clinical use. We successfully generated NK cell products from one 21-day manufacturing run per donor to infuse both projected cell doses for all study subjects. Cells generated from 1 donor were administered to all 6 subjects at the second and third dose levels, with residual products remaining in the inventory. Ongoing efforts are directed at improving cryopreservation techniques to ensure adequate post-thaw viability and function and to further enhance the manufacturing workflow.
One limitation of this study is that exogenous cytokines (IL-2 and IL-15) were not administered to support the in vivo function and proliferation of the adoptively transferred NK cells. This was a safety measure to ensure that adverse events that could be induced by the allogeneic, nonfamilial nature of the NK cell products would not be perpetuated by cytokine support. As the safety profile of these products—in particular, the absence of GVHD—demonstrates, subsequent studies that include exogenous cytokine support are planned.
Another limitation is the challenge of using cryopreserved NK cell products with subsequent cell loss and expected diminished function post-thaw. To compensate for this, we thawed and washed 1.5 to 2 times the intended cell dose to ensure that the infused cells were mostly viable. In addition, we observed attrition of CD16+ cells post-thaw even though other cytotoxicity/activating receptors were preserved. This diminishes the antibody-dependent cellular cytotoxicity potential of these cells. Studies to improve cryopreservation and thaw techniques as a way of optimizing the post-thaw product are necessary for this field.
In conclusion, the persistence of our off-the-shelf NK cell product in vivo, albeit at low levels, along with the demonstrated antitumor effect against both colon cancer and AML/MDS, provides evidence that this treatment approach can be refined to amplify therapeutic efficacy. Such refinements would include increasing the administered NK cell doses, providing optimized exogenous cytokine support or other complementary agents, and promoting tumor bed infiltration, particularly in solid malignancies [28–30]. We have demonstrated preclinically that inhibiting transforming growth factor-β, particularly in the metastatic colon cancer setting, would enhance the activity of our ex vivo expanded NK cell product in the tumor microenvironment [29]. Future iterations of this protocol would include coadministration of supporting cytokines (ie, IL-15 analogs) and transforming growth factor-β inhibitors to further explore this clinical potential.
ACKNOWLEDGMENTS
This work was supported by the dedicated clinical efforts of the Advanced Practice Provider team of the Bone Marrow Transplant program at Seidman Cancer Center and by investigations conducted by the University Hospitals Cleveland Medical Center (UHCMC) Flow Cytometry Core and by Thomas Prior and James Metcalf of the UHCMC Genetics Lab. The authors acknowledge Basabi Maitra and the Case Comprehensive Cancer Center Hematopoietic and Biorepository Core for processing and storage of correlative blood samples.
Financial disclosure:
Funding support was provided by the Adolescent and Young Adult Pilot Program UHCMC/Rainbow (pilot funding for development of the NKF master cell bank), the Case Comprehensive Cancer Center GI SPORE (Career Development Award and Pilot Funding for F.O. and D.W.), the UHCMC Clinical Research Center/Clinical Trials Unit (research coordination and Early Phase Clinical Research Awards for F.O.), and the Leukemia Lymphoma Society (Therapy Accelerated Program Award; D.W., PI; F.O. co-PI).
Footnotes
Conflict of interest statement: There are no conflicts of interest to report.
REFERENCES
- 1.Geller MA, Miller JS. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy. 2011;3:1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knorr DA, Bachanova V, Verneris MR, Miller JS. Clinical utility of natural killer cells in cancer therapy and transplantation. Semin Immunol. 2014;26:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller JS. Therapeutic applications: natural killer cells in the clinic. Hematology Am Soc Hematol Educ Program. 2013;2013:247–253. [DOI] [PubMed] [Google Scholar]
- 4.Locatelli F, Moretta F, Brescia L, Merli P. Natural killer cells in the treatment of high-risk acute leukaemia. Semin Immunol. 2014;26:173–179. [DOI] [PubMed] [Google Scholar]
- 5.Shi J, Tricot G, Szmania S, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol. 2008;143:641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubnitz JE, Inaba H, Ribeiro RC, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. 2010;28:955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon SR, Lee YS, Yang SH, et al. Generation of donor natural killer cells from CD34(+) progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: a feasibility study. Bone Marrow Transplant. 2010;45:1038–1046. [DOI] [PubMed] [Google Scholar]
- 8.Curti A, Ruggeri L, D’Addio A, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. 2011;118:3273–3279. [DOI] [PubMed] [Google Scholar]
- 9.Stern M, Passweg JR,, Meyer-Monard S, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant. 2013;48:433–438. [DOI] [PubMed] [Google Scholar]
- 10.Bachanova V, Cooley S, Defor TE, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123:3855–3863. ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaffer BC, Le Luduec JB, Forlenza C, et al. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2016;22:705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciurea SO, Schafer JR, Bassett R, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130:1857–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho D, Campana D. Expansion and activation of natural killer cells for cancer immunotherapy. Korean J Lab Med. 2009;29:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Childs RW, Berg M. Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology Am Soc Hematol. Educ Program 2013;2013:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szmania S, Lapteva N, Garg T, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother. 2015;38:24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Somanchi SS, Senyukov VV, Denman CJ, Lee DA. Expansion, purification, and functional assessment of human peripheral blood NK cells. J Vis Exp. 2011;2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah N, Martin-Antonio B, Yang H, et al. Antigen-presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS One. 2013;8:e76781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ojo EO, Sharma AA, Liu R, et al. Membrane-bound IL-21-based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci Rep. 2019;9:14916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooley S, Weisdorf DJ, Guethlein LA, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood. 2010;116:2411–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancusi A, Ruggeri L, Urbani E, et al. Haploidentical hematopoietic transplantation from KIR ligand-mismatched donors with activating KIRs reduces nonrelapse mortality. Blood. 2015;125:3173–3182. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Lim O, Kim TM, et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol Res. 2016;4:215–224. [DOI] [PubMed] [Google Scholar]
- 22.Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruggeri L, Capanni M, Casucci M, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94:333–339. [PubMed] [Google Scholar]
- 24.Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood. 2004;103:767–776. [DOI] [PubMed] [Google Scholar]
- 25.Giebel S, Locatelli F, Lamparelli T, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood. 2003;102:814–819. [DOI] [PubMed] [Google Scholar]
- 26.Willemze R, Rodrigues CA, Labopin M, et al. KIR-ligand incompatibility in the graft-versus-host direction improves outcomes after umbilical cord blood transplantation for acute leukemia. Leukemia. 2009;23:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venstrom JM, Pittari G, Gooley TA, et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med. 2012;367:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otegbeye F, Ojo E, Moreton S, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS One. 2018;13: e0191358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biederstädt A, Rezvani K. Engineering the next generation of CAR-NK immunotherapies. Int J Hematol. 2021;114:554–571. [DOI] [PMC free article] [PubMed] [Google Scholar]