

Abstract
Background
Glycogen storage disease type 1a (GSD1a) is a rare autosomal recessive metabolic disorder characterized by hypoglycaemia, growth retardation, lactic acidosis, hepatomegaly, hyperlipidemia, and nephromegaly. GSD1a is caused by a mutation in the G6PC gene encoding glucose-6-phosphatase (G6Pase); an enzyme that catalyses the hydrolysis of glucose-6-phosphate (G6P) to phosphate and glucose.
Objective
To elaborate on the clinical findings, biochemical data, molecular genetic analysis, and short-term prognosis of 13 GSD1a patients in Malaysia.
Methods
The information about 13 clinically classified GSD1a patients was retrospectively studied. The G6PC mutation analysis was performed by PCR-DNA sequencing.
Results
Patients were presented with hepatomegaly (92%), hypoglycaemia (38%), poor weight gain (23%), and short stature (15%). Mutation analysis revealed nine heterozygous mutations; eight previously reported mutations (c.155 A > T, c.209 G > A, c.226 A > T, c.248 G > A, c.648 G > T, c.706 T > A, c.1022 T > A, c.262delG) and a novel mutation (c.325 T > C). The most common mutation found in Malaysian patients was c.648 G > T in ten patients (77%) of mostly Malay ethnicity, followed by c.248 G > A in 4 patients of Chinese ethnicity (30%). A novel missense mutation (c.325 T > C) was predicted to be disease-causing by various in silico software.
Conclusions
The establishment of G6PC molecular genetic testing will enable the detection of presymptomatic patients, assisting in genetic counselling while avoiding the invasive methods of liver biopsy.
1. Introduction
Glycogen storage diseases (GSD) are a group of metabolic disorders of glycogen metabolism. GSD mostly affects the liver, skeletal muscles, heart, and sometimes the central nervous system [1]. There are more than 12 different types, and they are classified based on the deficient enzymes and affected tissues [2]. The most common type is GSD type 1a, representing about 80% of GSD1 patients. GSD1a was first described by Von Gierke in 1929. It is a recessively inherited metabolic disorder with a prevalence of one in 100,000 live births [3]. There are limited prevalence data for GSD in the Malaysian population; however, available data from the national referral centre at Genetic Clinic Hospital Kuala Lumpur from 1998–2021, suggests that the majority of patients have GSD1a (47.6%) as well (Figure 1).
Figure 1.
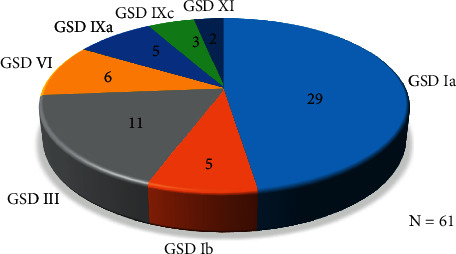
Number of patients diagnosed at the National Referral Centre, Genetic Clinic, Hospital Kuala Lumpur, Malaysia, from 1998–2021.
GSD1a (MIM #232220) is caused by the deficiency of glucose-6-phosphatase (G6Pase), an enzyme which catalyses the hydrolysis of glucose-6-phosphate (G6P) to phosphate and glucose. Deficiency of G6Pase causes an increase of G6P in the cytoplasm and triggers alternative metabolic pathways, thus leading to the accumulation of glycogen in glucose-generating organs, including the liver, kidney, and small intestine. G6Pase is a hydrophobic protein located in the endoplasmic reticulum containing 357 amino acids and nine transmembrane helix structures with its NH- and COOH- termini facing the ER lumen and cell cytoplasm, respectively. This enzyme is encoded by G6PC gene (OMIM #613742) located on chromosome 17q21, spanning about 12.5kb region and contains 5 exons [4].
The initial diagnosis of GSD1a is based on the clinical presentation and biochemical analysis such as hepatomegaly, hypoglycaemia, lactic acidosis, hypercholesterolemia, hypertriglyceridaemia, and hyperuricaemia which are usually manifested in the infantile period. The confirmation of diagnosis can be made either by measuring the G6Pase activity in liver biopsy tissue or by G6PC gene sequencing. Genetic analysis of the G6PC gene is preferred as it is less invasive compared to liver biopsy and it also facilitates genetic counselling.
To date, more than 135 unique mutations have been reported in the HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), most of which were missense types, followed by deletion and/or insertion and splicing. In the different ethnic groups, specific mutations were found at high frequencies, such as Arg83Cys in Jewish (98%) and Caucasian (33%), Arg83His in Chinese, and c.648 G > T in East Asians (Japanese 91%, Korean 86.2%, and Chinese 54%) [5–7]. In the present study, we report the clinical, biochemical, molecular analysis, and short-term prognosis of 13 GSD1a patients in Malaysia.
2. Materials and Methods
2.1. Patient and Sample Collection
Thirteen patients with a clinical diagnosis of GSD 1a who have not had a molecular analysis were referred to the Molecular Diagnostic Unit, Institute of Medical Research for molecular investigation. Patients' medical records were retrospectively reviewed for medical history, clinical examination, and laboratory study results. Standard deviations for height were calculated using standard growth charts from the World Health Organization (WHO) [8]. Routine biochemistry was performed in the clinical laboratory in a Hospital in Kuala Lumpur and the laboratory's reference ranges were provided. Descriptive statistics, including means, minimum, and maximum, were calculated. All statistical analyses were performed using Microsoft Excel. Parents of the affected child were guided to sign the consent form for genetic testing. Approximately 2.5 to 5 ml of peripheral blood in EDTA tubes were taken from patients for molecular genetic analysis of the G6PC gene.
2.2. Polymerase Chain Reaction
Genomic DNA was extracted using Chemagic Prepito D (Perkin Elmer) and both the quantity and quality of extracted DNA were measured using a NanoDrop ND-1000 Spectrophotometer. Six sets of primers were designed in-house to amplify five coding exons and flanking intronic sequences of the G6PC gene including splice sites (Supplement Table 1). PCR was performed in a 50 µl volume containing 50 ng genomic DNA, 0.1 U Taq DNA polymerase, 1X PCR buffer with MgSO4, 1 µmol of each primer and 0.2 mM of 10 mM dNTP mix. Amplification was performed using a touchdown PCR protocol as described by [9].
2.3. DNA Sequencing and Variant Analysis
Purification of PCR products and Sanger sequencing were performed as described previously [10]. Sequencing results were aligned to the reference sequence of the G6PC gene (NM_000151.3) using the SeqScape software v.3.0 (Applied Biosystem) to identify DNA variants. All variants identified were sought in the following database: Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php), Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), and Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org).
Novel variants were further checked using variant data from 100 genomes of the Singaporean Malays retrieved from the Singapore Sequencing Malay Project (SSMP) (http://phg.nus.edu.sg/StatGen/public_html/SSMP/SSMP_index.html) [11]. Several in silico tools were used to predict the pathogenicity of novel missense mutations by using MutationTaster (http://www.mutationtaster.org) [12], VarSome (https://varsome.com/) [13], and CADD (https://cadd.gs.washington.edu/).
2.4. Protein Structure Analysis
The crystal structure for G6Pase is currently not available on the Protein Data Bank (PDB), therefore we used the structure predicted by AlphaFold-2 (AF-2) for this analysis [14]. The PDB file was retrieved from the UniProt database (https://www.uniprot.org/uniprotkb/P35575/entry#structure) and the impact of novel missense mutations on protein structure was predicted by using Missense3D (http://missense3d.bc.ic.ac.uk/∼missense3d/) [15]. Next, we performed in silico mutagenesis using FoldX to predict the impact of the novel missense mutation on the thermodynamic stability of the protein structure [16]. The PDB file was first repaired using the FoldX RepairPDB command, and the repaired PDB was subjected to mutagenesis using the BuildModel command. We used the same criteria described by Caswell et al. [17] to interpret the change in free energy of the mutated structure compared to the wild-type structure. Both protein structures (containing native or mutated residues) were visually inspected using PyMOL.
3. Results
3.1. Clinical Analysis
The clinical features, biochemical and G6PC mutations of 13 Malaysian patients from twelve unrelated families with GSD1a are summarized in Table 1. Nine were males (69%) and four were females (31%). The median age of diagnosis was 13 months old (range 9 months to 16 years old). All presented with hepatomegaly except for patient 13 (92%). Other clinical features on presentation were hypoglycaemia (5/13, 38%), poor weight gain (3/13, 23%), and short stature (2/13, 15%). Motor delay, epistaxis, splenomegaly, and gouty arthritis were found in one patient, respectively (8%). Biochemical features included raised hyperlactataemia (mean 4.5 mmol/L, range 1.8–13.4 mmol/L), hypertriglyceridaemia (mean 11 mmol/L, range 5.7–24.6 mmol/L), hyperuricaemia (mean 503 mmol/L, range 228–866 mmol/L), raised alanine aminotransferase (mean 168 U/L, range 29–351 U/L), and raised aspartate aminotransferase (mean 223 U/L, range 87–357 U/L).
Table 1.
Clinical features, biochemical and the G6PC mutations in GSD1a Malaysian patients.
| Pt No | Sex/Ethnicity | Age onset | Age of dx | Initial clinical manifestation | Lactate, mmol/L (ref <2) | Triglyceride, mmol/L (ref <1.7) | Uric acid, µmol/L (ref <400) | ALT/AST, IU/L (ref <42/<51) | Nucleotide changes | Protein changes | Exon | Mutation reported |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1# | F/Chinese | 7m | 9m | Hepatomegaly, hypoglycaemia | 5.5 | 11 | 687 | 129/148 | c.248 G > A c.1022 T > A |
p.Arg83His p.Ile341Asn |
2 5 |
Hwu (1995) Lee (1996) |
| 2∗# | M/Malay | 3y 8 m | 3y 8 m | Hepatomegaly | 2.6 | 9.1 | 569 | 309/275 | c.648 G > T c.706 T > A |
p.Leu216 = p.Trp236Arg |
5 5 |
Kajihara [18] Lei [19] |
| 3 | M/Chinese | 8m | 2y 2m | Hepatomegaly, poor weight gain, epistaxis | 4.2 | 24.6 | 560 | 351/357 | c.262delG c.648 G > T |
p.Val88Phefs∗14 p.Leu216 = |
2 5 |
Lam (1998) Kajihara [18] |
| 4 | F/Chinese | ND | 6y | Hepatomegaly, short stature, poor weight gain | ND | ND | ND | ND | c.209 G > A c.248 G > A |
p.Trp70∗ p.Arg83His |
1 2 |
Trioche (1999) Hwu (1995) |
| 5 | M/Malay | 12m | 12m | Hepatomegaly, hypoglycaemia | ND | ND | 500 | ND | c.155 A > T c.325 T > C |
p.His52Leu p.Cys109Arg |
1 2 |
Rahman [20] This study |
| 6# | F/Malay | 12m | 13m | Hepatomegaly | 13.4 | 11.4 | 405 | 89/158 | c.226 A > T c.648 G > T |
p.Lys76∗ p.Leu216 = |
1 5 |
Rahman [20] Kajihara [18] |
| 7 | M/Chinese | 10m | 12m | Hepatomegaly, hypoglycaemia | ND | ND | ND | ND | c.248 G > A c.648 G > T |
p.Arg83His p.Leu216 = |
1 5 |
Hwu (1995) Kajihara [18] |
| 8# | M/Malay | 6m | 9m | Hepatomegaly, hypoglycaemia, poor weight gain | 5.5 | 13 | 451 | 229/335 | Homo c.648 G > T | p.(Leu216 = ) | 5 | Kajihara [18] |
| 9# | M/Chinese | 15m | 2y 10m | Hepatomegaly | 4.2 | 7.1 | 269 | 181/149 | c.248 G > A c.648 G > T |
p.(Arg83His) p.(Leu216 = ) | 2 5 |
Hwu (1995) Kajihara [18] |
| 10# | M/Malay | 9m | 9m | Hepatomegaly, splenomegaly, motor delay | 3.3 | 6.6 | 228 | 156/275 | Homo c.648 G > T | p.(Leu216 = ) | 5 | Kajihara [18] |
| 11 | M/Malay | ND | 4y | Hepatomegaly, hypoglycaemia | ND | ND | ND | ND | Homo c.648 G > T | p.(Leu216 = ) | 5 | Kajihara [18] |
| 12 | F/Malay | 5m | 10m | hepatomegaly | 8 | 11.2 | ND | 39/87 |
c.325 T > C c.648 G > T |
p.(Cys109Arg)
p.(Leu216 = ) |
2 5 |
This study
Kajihara [18] |
| 13∗ | M/Malay | 13y | 16y | Short stature, gouty arthritis | 1.8 | 5.7 | 866 | 29/ND | c.648 G > T c.706 T > A |
p.Leu216 = p.Trp236Arg |
5 5 |
Kajihara [18] Lei [19] |
Abbreviations. ALT, alanine transaminase; AST, aspartate transaminase; Dx, diagnosis; F female; M, male; m, months; ND, not determined; Pt, patient; y, years; ∗ siblings, #Inherited from parents.
Follow-up data (Table 2) were available for 11 patients with a mean age of 9.4 years (median 6 years). Patient 4, who defaulted treatment at 17 years old, died at 20 years old due to sepsis. Dietary advice was frequent complex carbohydrates for all patients, and 9/11 patients (81.8%) took uncooked corn starch with doses ranging from 0.3–1.6 g/kg/feed. No patient used overnight perfusion feed in our cohort. Of the 11 patients with follow-up, seven (63.6%) had short stature, and all (100%) had hyperlactataemia and hypertriglyceridaemia. Seven patients (63.6%) took Allopurinol, an oral xanthine oxidase inhibitor, but despite that, four of them had plasma uric acid above the reference range (>400 µmol/L) at the last follow-up. Two (18.2%) had multiple focal liver lesions on ultrasonography, but none underwent liver biopsy, and serum alpha-fetoprotein was not raised.
Table 2.
Follow-up, treatment, and outcome of GSD1a Malaysians patients.
| Pt No | Age at last follow-up (yrs) | Short stature (<2SD for age and sex) | Uncooked corn starch (g/kg/feed) | Allopurinol use | Blood lactate, mmol/L (ref <2) | Uric acid, µmol/L (ref <400) | Triglyceride, mmol/L (ref <1.7) | Proteinuria (ref <3.5 mg/mmol creat) | Multiple liver focal lesions (ultrasonography) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | N | 1.3 | y | 6.2 | 344 | 5.3 | N | N |
| 2 | 12 | Y | 0.3 | y | 3.56 | 500 | 15.2 | Y | N |
| 3 | 3 | N | 1 | Y | 4.81 | 531 | 13.7 | N | N |
| 4 | 17 | Y | N | N | 17.6 | 443 | 8.5 | ND | Y |
| 5 | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| 6 | 6 | Y | 1.6 | y | 10.9 | 150 | 21.5 | N | N |
| 7 | 25 | Y | N | Y | 3.67 | 318 | 8.3 | N | Y |
| 8 | 2 | Y | 1.3 | N | 3.1 | 300 | 6.4 | N | N |
| 9 | 3 | N | 0.7 | N | 4.1 | 247 | 5.7 | N | N |
| 10 | 1 | N | 0.6 | N | 4.8 | 279 | 3.6 | N | N |
| 11 | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| 12 | 12 | Y | 1 | Y | 6.6 | 540 | 8.6 | N | N |
| 13 | 18 | Y | 0.5 | Y | 5.2 | 570 | 3.3 | Y | N |
Abbreviations. N, no; ND, not determined; SD, standard deviation; Y, yes.
3.2. G6PC Genotyping
Nine mutations were identified in 13 patients (Table 1). These included five missense mutations (His52Leu, Arg83His, Cys109Arg, Trp236Arg, and Ile341Asn), two nonsense mutations (Trp70∗ and Lys76∗), a splice site mutation (c.648 G > T) and a frameshift mutation (c.262delG). Schematic of nine mutations spanned all exons except exon 3 and 4 were identified in G6PC gene are shown at Figure 2. Two recurrent mutations (c.648 G > T and c.Arg83His) were identified in 13 and 4 alleles of the total mutant alleles in our patient cohort, respectively. Carrier status of the parents was confirmed for patients 1, 2, 6, 8, 9, and 10, whereas others were not available for carrier testing. A novel mutation, Cys109Arg, found in this study was not detected in 100 genomes of Singaporean Malay. The novel Cys109Arg was one of the compound heterozygous mutations exhibited in Patients 5 and 12, whereby the c.325 T > C change replaced cysteine with arginine at codon 109 of the G6Pase protein. MutationTaster predicts c.325 T > C to be disease causing and CADD score was 26.9 (deleterious) whereas VarSome classified the variant as uncertain significance/likely pathogenic based on evidence of one moderate (PM2); that is not present in the population database (gnomAD) and two supporting evidence; (PP2) missense variants in G6PC is the common mechanism of the disease and (PP3) 11 pathogenic computational verdict as deleterious effect on the gene.
Figure 2.
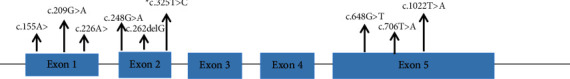
Schematic of nine mutations spanned all exons except exon 3 and 4 were identified in G6PC gene. Mutation c.648 G > T is the most common mutation present in 13 alleles followed with c.248 G > T in four alleles. Novel mutation is labelled with ∗.
The Cys109 residue is located in an extended, unstructured loop region of G6Pase which has a high predicted Local Distance Difference Test (pLDDT) score of 92.99 (Figure 3). This score exceeds the recommended threshold of 70 for the AF-2 model, therefore structural analysis performed using this model is likely to generate reliable predictions [17]. Missense3D predicted that replacement of Cys with Arg would abolish the disulphide bond formed between Cys109 and residue Cys254. This is supported by in silico mutagenesis analysis by FoldX that predicted the missense change to severely destabilise the protein structure.
Figure 3.
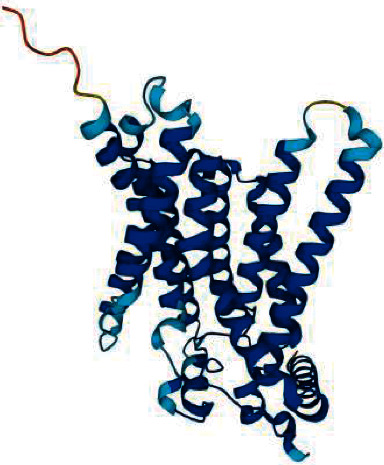
Structure of G6Pase predicted by AlphaFold-2. Dark blue indicates a predicted Local Distance Difference Test (pLDDT) score above 90 and light blue indicates a pLDDT score of between 70 and 90.
4. Discussion
In this paper, we presented the clinical, biochemical, and molecular findings of 13 GSD1a patients from Malaysia. The spectrum of mutations identified in our patients were similar to HGMD, where missense changes were the most common type of mutation in the G6PC gene.
The most common mutation in our cohort was c.648 G > T, which was found in ten patients, followed by c.248 G > T that was identified in four patients. The c.648 G > T mutation was common in patients of Asian ancestry as reported in patients from Japanese (91%) [18], South Korean (75%) [21], Chinese ethnicity (54%) [22], and Malay ethnicity (78%) [20]. Our findings supported this observation that the c.648 G > T mutation was prevalent among patients of Malay origin. This variant was shown to alter splicing by producing an aberrant transcript that eliminated 91 nucleotides resulting in an altered reading frame and premature termination Kajihara (1995). The c.248 G > A mutation was identified in four unrelated Chinese patients, which was also in agreement with the high prevalence of this mutation among patients of Chinese origin [20].
Both nonsense mutations (Trp70∗ and Lys76∗) and a frameshift mutation (c.262delG) were predicted to create a premature stop codon. A nonsense mutation occurring 11 amino acids from the carboxyl terminus was devoid of enzymatic activity [19] and, because of this, the shorter enzymes produced by the above-mentioned mutations could also result in loss of function. More importantly, Lys76 was one of the active site residues in G6Pase and substitution with asparagine was shown to abolish enzyme activity [23].
The novel mutation c.325 T > C (p.Cys109Arg) has been identified in two different GSD1a patients in the heterozygous state. Furthermore, Cys109Arg is positioned at the luminal loop and is shown to play a crucial role in the catalytic activity of the enzyme. This has been demonstrated by Shieh et al. [24] and Angaroni et al. [25] as the mutation Thr108Ile and Glu110Lys located at the luminal loop were shown to have inactivated G6Pase activity. Another possibility as shown by in silico analysis is the potential for the missense mutation to destabilise the structure of G6Pase which also could severely impact enzymatic activity. Despite the absence of functional studies for our novel mutation, it was predicted to be pathogenic by in silico program and since the mutation was found in patients from ethnic Malay, the absence of mutations in the Singaporean Malay genome database showed that the new allele was extremely rare in Malay population.
To date, there is no clear genotype-phenotype relation for GSD1a, even though several studies have proposed that some mutations may be associated with certain phenotypes. Nevertheless, some studies have reported the relationship between homozygous c.648 G > T with the level of severity in hepatocellular carcinoma [26–28], but we have yet to determine this phenotype in our patients as they are still young. However, two of our homozygous c.648 G > T patients (Patient 8 and 10) showed a severe phenotype, presenting in infancy with hepatomegaly, hypoglycaemia, hyperlactataemia, and hypertriglyceridaemia. Unfortunately, clinical data was incomplete for the third homozygous patient (Patient 11).
The oldest GSD1a patient was diagnosed at 16 years old (patient 13), presenting with gouty arthritis and short stature, demonstrating again that hyperuricaemia in adolescence can be a presenting feature for GSD1a as previously reported [29]. His clinical presentation contrasts with his younger brother (patient (2), who had massive hepatomegaly by the age of 4 years old. There are similar reports of variable phenotypes among affected siblings [29–31], which suggests additional genetic and/or environmental modifying factors [32].
The diagnosis is complicated and challenging because GSD patients exhibit phenotypic heterogeneity. However, gene mutational analysis enables a noninvasive and accurate way of diagnosing type Ia patients. Hence, as prompt and accurate diagnosis is the most important point for the proper treatment of metabolic diseases, next generation sequencing (NGS) can provide the most accurate and cost- and time-efficient approach for the fast diagnosis of the disease as well as overcome the difficulties in analysing diseases with broad clinical and genetic heterogeneity.
5. Conclusion
The establishment of a molecular genetic testing service for the G6PC gene will allow the diagnosis of GSD1a patients and eliminate the need for a liver biopsy. Besides that, it also enables the detection of presymptomatic patients and assists in genetic counselling. In conclusion, we have characterized both the clinical and molecular aspects of patients with GSD1a in Malaysia. The novel mutation identified in this study will further expand the spectrum of pathogenic mutations associated with GSD1a.
Acknowledgments
The authors would like to thank the Director-General of Health of Malaysia for his permission to publish this article. Our special thanks to Director of Institute for Medical Research and the Head Centre of SDC for critical reading of the manuscript and valuable comments. The authors would like to extend our gratitude to all clinical and laboratory staff for their contributions in this study. This study was part of the diagnostic services offered by the Ministry of Health, Malaysia.
Abbreviations:
- GSD1a:
Glycogen storage disease 1a
- G6PC:
Glucose-6-phosphatase
- HGMD:
Human Gene Mutation Database.
Data Availability
The datasets used and analysed during the current study can be obtained from the corresponding author upon reasonable request.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and the 1964 Helsinki Declaration. This study has been approved by Medical Research and Ethics Committee Malaysia (MREC) NMRR ID-21-02060-WNI.
Consent
An informed consent to participate was obtained from all individual participants included in the study. A consent for publication was obtained from the parents of all patients for publication.
Conflicts of Interest
All authors declare that there are no conflicts of interest.
Authors' Contributions
SitiAishah AW carried out the genetic test, performed the data analysis, and drafted and revised the manuscript. Noraishah A and Leong HY assisted with data collection and revised the manuscript. KhairulNizam performed 3D protein prediction analysis and revised the manuscript. Ngu LH revised the manuscript. Yusnita Y supervised the research study, and drafted and revised the manuscript.
Supplementary Materials
Table 1 Primers for exon-specific sequencing of G6PC gene.
References
- 1.Koshy A., Ramaswamy K., Correa M., Rekha S. Glycogen storage disease: report of 17 cases from southern India. Indian Journal of Gastroenterology : Official Journal of the Indian Society of Gastroenterology . 2006;25:182–184. [PubMed] [Google Scholar]
- 2.Wolfsdorf J. I., Weinstein D. A. Glycogen storage disease. Reviews in Endocrine and Metabolic Disorders . 2003;4(1):95–102. doi: 10.1023/a:1021831621210. [DOI] [PubMed] [Google Scholar]
- 3.Mundy H., Lee P. J. The glycogen storage diseases. Current Paediatrics . 2004;14(5):407–413. doi: 10.1016/j.cupe.2004.05.004. [DOI] [Google Scholar]
- 4.Lei K. J., Shelly L. L., Pan C. J., Sidbury J. B., Chou J. Y. Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science . 1993;262(5133):580–583. doi: 10.1126/science.8211187. [DOI] [PubMed] [Google Scholar]
- 5.Chou Y., Jun B. C. M. Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nature Reviews Endocrinology . 2010;6(12):676–688. doi: 10.1038/nrendo.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosaline F., Piraud M., Boudjemline A. M., et al. Glucose-6 phosphatase deficiency. Orphanet Journal of Rare Diseases . 2011;20:6–27. doi: 10.1186/1750-1172-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y. M., Choi J. H., Lee B. H., Kim G. H., Kim K. M., Yoo H. W. Predominance of the c.648G > T G6PC gene mutation and late complications in Korean patients with glycogen storage disease type Ia. Orphanet Journal of Rare Diseases . 2020;15(1):p. 45. doi: 10.1186/s13023-020-1321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.World Health Organization. The WHO Child Growth Standards . http://www.who.int/childgrowth/standards/en/ [Google Scholar]
- 9.Azize N. A., Ngah W. Z., Othman Z., et al. Mutation analysis of glycine decarboxylase, aminomethyltransferase and glycine cleavage system protein-H genes in 13 unrelated families with glycine encephalopathy. Journal of Human Genetics . 2014;59(11):593–597. doi: 10.1038/jhg.2014.69. [DOI] [PubMed] [Google Scholar]
- 10.Abdul Wahab S. A., Yakob Y., Abdul Azize N. A., et al. Clinical and mutational analysis of theGCDHGene in Malaysian patients with glutaric aciduria type 1. BioMed Research International . 2016;2016:5. doi: 10.1155/2016/4074365.4074365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai-Ping W., Liu X., Peng C., Li R., Kar Seng S., Xu H. Deep whole-genome sequencing of 100 southeast Asian malays. The American Journal of Human Genetics . 2013;10(1):52–66. doi: 10.1016/j.ajhg.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwarz J. M., Cooper D. N., Schuelke M., Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nature Methods . 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 13.Sian E., Emma L. B., Alison C., et al. ACGS best practice guidelines for variant classification in rare disease 2020. 2020. https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf . [DOI] [PubMed]
- 14.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O. Highly accurate protein structure prediction with AlphaFold. Nature . 2021;596(7873):583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ittisoponpisan S., Islam S. A., Khanna T., Alhuzimi E., David A., Sternberg M. J. E. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? Journal of Molecular Biology . 2019;431(11):2197–2212. doi: 10.1016/j.jmb.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. The FoldX web server: an online force field. Nucleic Acids Research . 2005;33(Web Server issue):W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caswell R. C., Gunning A. C., Owens M. M. Assessing the clinical utility of protein structural analysis in genomic variant classification: experiences from a diagnostic laboratory. Genome Medicine . 2022;14(1):p. 77. doi: 10.1186/s13073-022-01082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Susumu K., Matsuhashi S., Yamamoto K., et al. Exon redefinition by a point mutation within exon 5 of the glucose-6-phosphatase gene is the major cause of glycogen storage disease type 1a in Japan. The American Journal of Human Genetics . 1995;57:549–555. doi: 10.1016/0270-9139(95)95224-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei K. J., Pan C. J., Shelly L. L., Liu J. L., Chou J. Y. Identification of mutations in the gene for glucose-6-phosphatase, the enzyme deficient in glycogen storage disease type 1a. Journal of Clinical Investigation . 1994;93(5):1994–1999. doi: 10.1172/jci117192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdul Rahman A. A., Abdullah I. S., Teh S. H., et al. Novel mutations of the G6PC gene in Malaysians with glycogen storage disease 1a (GSD1a) Malaysian Journal of Science . 2021;40(1):34–45. [Google Scholar]
- 21.Ki C. S., Han S. H., Kim H. J., et al. Mutation spectrum of the glucose-6-phosphatase gene and its implication in molecular diagnosis of Korean patients with glycogen storage disease type Ia. Clinical Genetics . 2004;65(6):487–489. doi: 10.1111/j.1399-0004.2004.00260.x. [DOI] [PubMed] [Google Scholar]
- 22.Chou Y., Mansfield B. C. Mutations in the glucose-6-phosphatase-α (G6PC) gene that cause type Ia glycogen storage disease. Human Mutation . 2008;29(7):921–930. doi: 10.1002/humu.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shieh S., Terzioglu T., Hiraiwa H., Marsh M., Pan C., Chou J. The molecular basis of glycogen storage disease type 1a. Journal of Biological Chemistry . 2002;15(7):5047–5053. doi: 10.1074/jbc.m110486200. [DOI] [PubMed] [Google Scholar]
- 24.Shieh S., Pan C.-J., Mansfield C., Chou J. A glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. Journal of Biological Chemistry . 2003;278(47):47098–47103. doi: 10.1074/jbc.m309472200. [DOI] [PubMed] [Google Scholar]
- 25.Angaroni J., de Kremer R., Argaraña E., et al. Glycogen storage disease type Ia in Argentina: two novel glucose-6-phosphatase mutations affecting protein stability. Molecular Genetics and Metabolism . 2004;83(3):276–279. doi: 10.1016/j.ymgme.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Matern D., Seydewitz H. H., Bali D., Lang C., Chen Y. T. Glycogen storage disease type I: diagnosis and phenotype/genotype correlation. European Journal of Pediatrics . 2002;161(1):S10–S19. doi: 10.1007/bf02679989. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura T., Ozawa T., Kawasaki T., Nakamura H., Sugimura H. Glucose-6-Phosphatase gene mutations in 20 adult Japanese patients with glycogen storage disease type 1a with reference to hepatic tumors. Journal of Gastroenterology and Hepatology . 2001;16(12):1402–1408. doi: 10.1046/j.1440-1746.2001.02645.x. [DOI] [PubMed] [Google Scholar]
- 28.Akanuma J., Nishigaki T., Fujii K., Matsubara Y., Inui K., Takahashi K. Glycogen storage disease type Ia: molecular diagnosis of 51 Japanese patients and characterization of splicing mutations by analysis of ectopically transcribed mRNA from lymphoblastoid cells. American Journal of Medical Genetics . 2000;91(2):107–112. [PubMed] [Google Scholar]
- 29.Shieh S., Lu Y.-H., Huang H., et al. Misdiagnosis as steatohepatitis in a family with mild glycogen storage disease type 1a. Gene . 2012;509(1):154–157. doi: 10.1016/j.gene.2012.07.057. [DOI] [PubMed] [Google Scholar]
- 30.Parvari R., Isam J., Moses S. W. Glycogen storage disease type 1a in three siblings with the G270V mutation. Journal of Inherited Metabolic Disease . 1999;22(2):149–154. doi: 10.1023/a:1005445802822. [DOI] [PubMed] [Google Scholar]
- 31.Rake J. P., ten Berge A. M., Visser G., et al. Identification of a novel mutation (867delA) in the glucose-6-phosphatase gene in two siblings with glycogen storage disease type Ia with different phenotypes. Human Mutation . 2000;15(4):p. 381. doi: 10.1002/(SICI)1098-1004(200004)15:4<381::AID-HUMU13>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 32.Peeks F., Steunenberg T. A. H., de Boer F., et al. Clinical and biochemical heterogeneity between patients with glycogen storage disease type IA: the added value of CUSUM for metabolic control. Journal of Inherited Metabolic Disease . 2017;40(5):695–702. doi: 10.1007/s10545-017-0039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1 Primers for exon-specific sequencing of G6PC gene.
Data Availability Statement
The datasets used and analysed during the current study can be obtained from the corresponding author upon reasonable request.