

Abstract
Background and Purpose
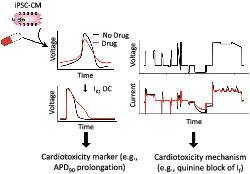
Before advancing to clinical trials, new drugs are screened for their pro‐arrhythmic potential using a method that is overly conservative and provides limited mechanistic insight. The shortcomings of this approach can lead to the mis‐classification of beneficial drugs as pro‐arrhythmic.
Experimental Approach
An in silico–in vitro pipeline was developed to circumvent these shortcomings. A computational human induced pluripotent stem cell‐derived cardiomyocyte (iPSC‐CM) model was used as part of a genetic algorithm to design experiments, specifically electrophysiological voltage clamp (VC) protocols, to identify which of several cardiac ion channels were blocked during in vitro drug studies. Such VC data, along with dynamically clamped action potentials (AP), were acquired from iPSC‐CMs before and after treatment with a control solution or a low‐ (verapamil), intermediate‐ (cisapride or quinine) or high‐risk (quinidine) drug.
Key Results
Significant AP prolongation (a pro‐arrhythmia marker) was seen in response to quinidine and quinine. The VC protocol identified block of IKr (a source of arrhythmias) by all strong IKr blockers, including cisapride, quinidine and quinine. The protocol also detected block of ICaL by verapamil and Ito by quinidine. Further demonstrating the power of the approach, the VC data uncovered a previously unidentified If block by quinine, which was confirmed with experiments using a HEK‐293 expression system and automated patch‐clamp.
Conclusion and Implications
We developed an in silico–in vitro pipeline that simultaneously identifies pro‐arrhythmia risk and mechanism of ion channel‐blocking drugs. The approach offers a new tool for evaluating cardiotoxicity during preclinical drug screening.
Keywords: arrhythmias, cardiotoxicity, computer simulation, induced pluripotent stem cells, ion channels, preclinical drug evaluation
Abbreviations
- AP
action potential
- APD20
action potential duration at 20% of repolarization
- APD90
action potential duration at 90% of repolarization
- CiPA
Comprehensive in Vitro Pro‐arrhythmia Assay
- EFPC
effective free plasma concentration
- ICaL
L‐type Ca2+ current
- If
funny current
- IK1
inward rectifier K+ current
- IKr
rapid delayed rectifier K+ current
- IKs
slow delayed rectifier K+ current
- INa
Na+ current
- iPSC‐CM
induced pluripotent stem cell‐derived cardiomyocytes
- Ito
transient outward K+ current
- VC
voltage clamp
What is already known
Human induced stem cell‐derived cardiomyocytes (iPSC‐CMs) are used to screen for drug cardiotoxicity.
iPSC‐CMs are heterogeneous, complicating the ability to link experimental cardiotoxicity markers to drug mechanism.
What does this study add
A model‐driven, optimized, experimental protocol to detect drug cardiotoxicity markers and mechanism during iPSC‐CM experiments.
A previously unreported block of cardiac funny current by quinine.
What is the clinical significance
A novel approach to screening for drug cardiotoxicity using human‐derived cells.
Quinine strongly blocks funny current above 3× its effective free plasma concentration.
1. INTRODUCTION
The use of in vitro assays of the rapid delayed rectifier K+ current (IKr ) by regulatory agencies and pharmaceutical companies prevents the approval of lethal pro‐arrhythmic drugs (Anonymous, 2005). However, the high sensitivity of such assays comes at the cost of low specificity (De Bruin et al., 2005; Gintant, 2011; Hancox et al., 2008) and have been shown to incorrectly label safe and effective therapies, such as verapamil and ranolazine, as pro‐arrhythmic (Crumb et al., 2016; Johannesen et al., 2014).
To address these specificity shortcomings, the Comprehensive in Vitro Pro‐arrhythmia Assay (CiPA) initiative was started in 2013 to guide the development of more accurate preclinical tests (Sager et al., 2014). The initiative recommended a three‐step drug screening approach that includes (1) quantifying drug effects on the major cardiac ionic currents using expression system cells, (2) integrating these effects into in silico models and using simulations to evaluate a drug's pro‐arrhythmic potential and (3) validating simulations with human‐derived induced pluripotent stem cell‐derived cardiomyocytes (iPSC‐CMs) and human ECG studies.
Over the last decade, drug effects on the action potential (AP) duration or field potential duration in iPSC‐CMs have become a common metric to evaluate drug risk (Blinova et al., 2018; Navarrete et al., 2013). iPSC‐CMs, however, are an imperfect model of adult physiology, with an immature phenotype, high degree of heterogeneity and depolarized maximum diastolic potential (Goversen, van der Heyden, et al., 2018). These features make it difficult to record consistent and reliable measures of pro‐arrhythmia risk. Furthermore, the heterogeneity of these cells limits the ability to validate in silico simulations and can produce discrepancies between experimental and computational results (Paci et al., 2021a).
Dynamic clamp of a synthetic inward rectifier potassium current (IK1 ) into iPSC‐CMs is a well‐established method to improve the apparent electrophysiological maturity of cells for drug studies. The IK1 model current is calculated in real time and injected into iPSC‐CMs to prevent spontaneous beating and establish a stable maximum diastolic potential below ‐65 mV (Fabbri et al., 2019; Quach et al., 2018). When paced from this hyperpolarized resting membrane potential, cells have a more consistent, and adult‐like AP phenotype, making drug‐induced AP changes easier to interpret (Goversen, Becker, et al., 2018; Li et al., 2019).
In this study, we developed a pipeline that uses IK1 dynamic clamp and a novel voltage clamp (VC) optimization approach to determine both the pro‐arrhythmia risk and mechanism of action for a drug, from iPSC‐CM experiments. We start with in vitro iPSC‐CM studies to determine drug block that we then confirm with expression system dose–response experiments. During the in vitro iPSC‐CM studies conducted here, we acquired optimized VC data, along with IK1 dynamically clamped APs, before and after application of a CiPA‐labelled low‐ (verapamil), intermediate‐ (cisapride) or high‐risk (quinidine) drug or quinine, which is also known to be pro‐arrhythmic (Colatsky et al., 2016; Woosley et al. n.d.). In addition to correctly identifying all three ion channels that were expected to be strongly blocked (>30%) by at least one of these drugs, the protocol also identified a previously unreported block of funny current (If ) by quinine. This approach has potential as a new cardiotoxicity screening tool that can increase specificity, while also providing information about the underlying mechanism.
2. METHODS
2.1. Pipeline design
Figure 1 displays the steps of a cardiotoxicity screening pipeline that we developed and validated in this study. The first step in the pipeline is to use an in silico iPSC‐CM model‐guided genetic algorithm (GA) to design a VC protocol that isolates individual currents (Step 1). While the VC protocol could, in principle, be designed to isolate any of the ionic currents present in the in silico model, in this study we focused on seven currents that are most associated with AP morphology: IK1, If, slow (IKs ) and delayed (IKr) rectifier potassium, L‐type calcium (ICaL ), sodium (INa ) and transient outward (Ito ). Optimized VC, as well as spontaneous and IK1 dynamic clamp and paced AP data, is acquired from a patient‐derived iPSC‐CM before and after drug application (Step 2). The IK1 dynamic clamp data are used to measure surrogate markers of cardiotoxicity (Step 3), while the optimized VC data are used to identify ion channel targets (Step 4). Dose–response data are then acquired for the identified targets using expression line cells (Step 5). For example, in this study we acquired the dose–response data for quinine block of HCN1, which further validated our findings on the unreported block of If by quinine.
FIGURE 1.
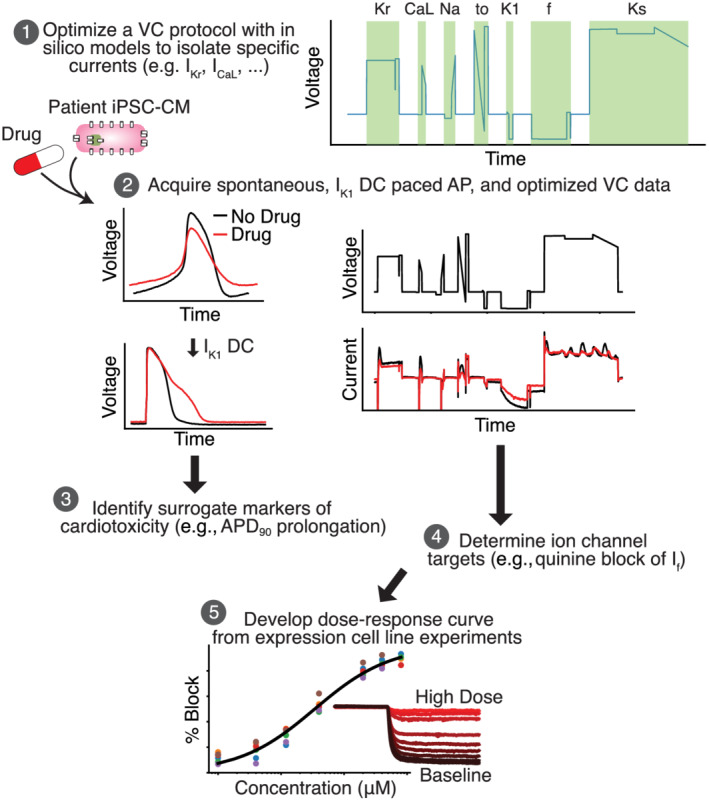
An in silico–in vitro pipeline to determine drug cardiotoxicity risk and mechanism. Step 1, the Kernik–Clancy model with experimental artefacts is used to develop a voltage clamp (VC) protocol that is specifically designed to isolate currents. Step 2, spontaneous, IK1 dynamic clamp and paced action potentials (AP), and optimized VC data are acquired from a patient‐derived induced pluripotent stem cell‐derived cardiomyocyte (iPSC‐CM) before and after drug application. Step 3, the change in IK1 dynamic clamp and paced AP data from pre‐ to post‐drug application is used to identify AP prolongation, a surrogate marker of cardiotoxicity. Step 4, changes in VC data are used to determine the ion channels targeted by a drug. Step 5, after identifying the ion channel targeted by a drug, a dose–response curve is developed for each of these ion channels using expression line cells.
2.2. Voltage clamp protocol optimization
The baseline Kernik–Clancy iPSC‐CM model was used in this study (Kernik et al., 2019). Experimental artefacts, such as seal leak, series resistance compensation and voltage offset, were included in our model simulations following the simplified artefact model from Lei et al. (2020). Taking experimental artefacts into account produced better fits of ion channel models to experimental data, while adjusting fewer parameters (Lei et al., 2020). The effects of these patch‐clamp artefacts are particularly pronounced within the first few milliseconds after a voltage step (Figure S1). Prior to the VC protocol optimization, the model was run to steady state, and then simulated under voltage clamp at −80 mV for 20 s.
An optimized VC protocol was designed for each of seven currents (IKr, ICaL, INa, Ito, IK1, If and IKs). Custom Python code that included the DEAP Python package was developed to implement the VC optimization GA (Fortin et al., 2012). The GA had 200 individual protocols per generation and 50 generations. Each protocol in the GA had a set of four voltage segments. Each segment could be either a step or ramp between 5 and 1000 ms long and was constrained to voltages between −120 and 60 mV.
To evaluate the fitness of a VC protocol, the Kernik–Clancy model with experimental artefacts was clamped, and the percent contribution (C[t]) of the target current was calculated at every timepoint.
In this equation, Ix is the target current. The denominator is the sum of the absolute values for all currents, including ionic currents and pipette leak (from the artefact equations). The best possible contribution score is equal to one and represents when the target current contributes all the current observed during a patch‐clamp experiment. We calculated the average contribution, C(t), over a 10‐ms window at each time point and used the highest contribution window value as the fitness score for the protocols.
2.3. Combining VC protocols
The protocol with the highest current contribution for each of the seven ionic currents (Figures S2–S8) was combined into one large protocol. Before combining the protocols, they were systematically shortened using a two‐step process. First, the portion of the protocol >50 ms after the maximum current contribution window, identified by the fitness function, was removed. Then, 10‐ms segments were incrementally removed from the beginning of the protocol, while ensuring the max current contribution did not decrease by more than 5%. The seven shortened protocols were connected by 500‐ms holding steps at −80 mV. The 500‐ms duration was chosen for two reasons: (1) it is long enough for many ion channel state variables to reach steady state and (2) the maximum contribution of each current did not decrease by more than 10% for any of the currents (Table S1).
To validate that the VC protocol isolates current during the same time windows in a cell with different conductance and kinetic parameters, the optimized VC protocol was applied to the Paci iPSC‐CM model (Paci et al., 2018) with experimental artefacts. The VC protocol was also applied to the Kernik–Clancy model with extracellular Ca2+ concentration set to 1.2 mM, a value within the normal physiological range, but different from our extracellular patch‐clamp solution. We found little difference in the amount of current isolation under these two conditions (Table S2).
2.4. iPSC‐CM experiments
Frozen vials of iPSC‐CMs were thawed and cultured as a monolayer in one well of a 6‐well plate precoated with 1% Matrigel and supplemented with RPMI media (Fisher/Corning 10–040‐CM) containing 5% FBS (Gibco 16000069) and 2% B27 (Gibco A1895601). Cells were placed in an incubator at 37°C, 5% CO2 and 85% humidity for 48 h. When replating, cells were lifted with 1‐ml Accutase (Corning A6964), and the enzymatic reaction was blocked with DMEM/F12 (Gibco 10565‐042) plus 5% FBS (Burridge et al., 2014). Cells were diluted to 100,000 cells ml−1 and re‐distributed to 124 sterile 8‐mm coverslips precoated with 1% Matrigel. RPMI media was replaced every other day. Cells were patched from days 5 to 15 after thaw. Each cell in this study was acquired from a different coverslip that had been independently cultured for >4 days in a 24‐well plate. Each cell was treated with drugs independently of all other cells in the study to produce independent experimental samples. Cells from five different vials were used in this study (see further details in Section 2.6 below).
Perforated patch‐clamp experiments were conducted using borosilicate glass pipettes pulled to a resistance of 2–4 M using a flaming/brown micropipette puller (Model P‐1000; Sutter Instrument, Novato, CA). The pipettes were filled with intracellular solution containing 10‐mM NaCl, 130‐mM KCl, 1‐mM MgCl2, 10‐mM CaCl2, 5.5‐mM dextrose and 10‐mM HEPES. Amphotericin B was used as the perforating agent. Amphotericin B allows only monovalent ions to pass through the cell membrane, so a high intrapipette calcium concentration was used as a quality control step. In the case of an unintended rupture, calcium will diffuse into the cell and cause calcium‐induced toxicity, which will terminate the experiment. The pipette tip was first dipped into intracellular solution with no amphotericin B for 2–5 s. Then, the pipette was backfilled with intracellular solution containing 0.44‐mM amphotericin B. The coverslips containing iPSC‐CMs were placed in the bath and constantly perfused with an extracellular solution at 35–37°C containing 137‐mM NaCl, 5.4‐mM KCl, 1‐mM MgSO4, 2‐mM CaCl2, 10‐mM dextrose and 10‐mM HEPES.
Patch‐clamp measurements were made at 10 kHz by a patch‐clamp amplifier (Model 2400; A‐M Systems, Sequim, WA) controlled by the Real Time eXperiment Interface (RTXI; http://rtxi.org) to send commands to the amplifier via the data acquisition card (PCI‐6025E; National Instruments, Austin, TX). After immersing the pipette into the extracellular solution, voltage was set to zero, and voltage offset in our recordings was assumed to be equal to the liquid junction potential of −2.8 mV. After contact with a cell was made and a seal of greater than 300 M was established, we waited for the access resistance to decrease below 40 M before starting experiments. A series resistance of 9–50 M was obtained for all experiments, and series resistance compensation was set to 70%. The 70% compensation was chosen because larger values caused oscillations during the recordings.
Spontaneous, IK1 dynamic clamp and VC data were acquired before and after drug application. Once access was gained, spontaneous behaviour was acquired for >10 s. Dynamic clamp IK1 data were acquired using a custom RTXI module that implemented the Ishihara et al. model (Ishihara et al., 2009). A recent in silico study showed that the Ishihara model has properties that are optimal for use in IK1 dynamic clamp drug studies of iPSC‐CMs (Fabbri et al., 2019). With this module, we injected 0.5× the baseline amount of Ishihara IK1. If spontaneous behaviour was not suppressed, and the resting membrane potential was above −65 mV, we incrementally increased the injected current by 0.25× or 0.5× of the baseline amount of Ishihara IK1 until these requirements were met. Resting at this hyperpolarized potential allows sodium channels to recover, resulting in APs with faster upstrokes and larger amplitudes, more closely resembling adult ventricular APs. The amount of IK1 dynamically clamped into a cell will influence its APD, which is why we chose to inject the minimum amount to suppress automaticity. The amount of IK1 dynamically clamped into each cell can be found in the data repository linked from the project GitHub page. The amount of IK1 injected into these cells (0.5–3×) is similar to modelled results with the Kernik–Clancy. After dynamic clamp data were acquired, the amplifier was switched to voltage clamp mode, and compensation of capacitance and access resistance was carried out. The cell was then clamped with the optimized VC protocol.
Following VC acquisition, the perfusion system was switched to an external solution containing either 0.1% of dimethyl sulfoxide (DMSO), or one of the following drugs: cisapride monohydrate at 250 nM, verapamil hydrochloride at 150 nM, quinidine at 2.7 μM or quinine at 12 μM. Drug solutions were prepared daily, by dissolving in DMSO before dilution in external solution. The DMSO concentration was <0.1% for all drug solutions. The drug perfusion system was run for >5 min, while square pulses were applied to observe changes in the current response. Once the perfusion system had run for >5 min and changes in the current response had stabilized, spontaneous, IK1 dynamic clamp and VC data were acquired.
2.5. HEK‐HCN1 experiments
Human embryonic kidney cells 293 with tetracycline‐inducible expression of human hyperpolarization‐gated cyclic nucleotide‐sensitive cation channel 1 (HEK‐HCN1) were cultured and maintained according to a protocol from Charles River. One frozen vial of cells was thawed in prewarm DMEM/F12 media plus 10% FBS and 100‐units ml−1 Penicillin/Streptomycin (Life Technologies 15,140) and placed in 37°C, 5% CO2 and 85% humidity incubator overnight. The media was replaced with selection media containing 0.005‐mg·ml−1 Blasticidin (InvivoGen ant‐bl‐5b) and 0.1‐mg·ml−1 Zeocin (InvivoGen ant‐zn‐5b), and the cells were sub‐cultured if they were at ~75% confluency.
To induce expression of HCN1 channels, cells were cultured at 37ºC in DMEM/F12 media plus 1.5‐μg·ml−1 tetracycline, 2 days before the experiment. To prepare the cells for use in the Nanion Patchliner automated patch system, they were rinsed twice with 5‐ml Hanks balanced salt solution (HBSS; Life Technologies 14175) and lifted with 2‐ml Accutase. The enzymatic reaction was blocked using DMEM/F12 media, and the cell solution was centrifuged at 1163xg for 2 min. Supernatant was discarded and the cell pellet was mixed with FBS‐free DMEM/F12 media plus 15‐mM HEPES (pH 7.3) and extracellular solution. The cell mix solution was placed in a 4°C fridge for 10 min before use.
HEK‐HCN1 current was recorded using an automated patch clamp system (Patchliner Quattro, Nanion Technologies GmbH), sampling at 25 kHz. Whole cell patch clamp was performed at room temperature (21‐24ºC) using standard medium resistance NPC‐16 chips (1.8–3 MΩ) after getting a GΩ seal. Series resistance was compensated at 80% for VC recordings.
If current–voltage (I‐V) traces were recorded starting from a holding potential of −20 mV. Cells were stepped to a potential between −20 and −120 mV for 3500 ms, decreasing by 10‐mV steps, and then stepped to −50 mV for 500 ms to acquire the tail current. Maximum currents were calculated as the average current over the last 800 ms of the hyperpolarizing step. Maximum tail currents were calculated as the average between 8 and 16 ms after the step to −50 mV.
To measure the dose–response of HCN1 current treated with quinine, peak currents were measured by stepping from a holding potential of −20 to −110 mV for 3500 ms. These data were acquired under each of the following conditions, in this order: no drug, 0.5% DMSO in external (the maximum DMSO concentration), and then 1, 4, 12, 40, 200, 400 and 800 μM quinine in external. Maximum currents were calculated as the average current over the last 800 ms of the hyperpolarizing step. These traces were acquired three or four times at each dose. The last two traces at each dose, which were acquired >40 s after drug application, were used for analysis. Each data point plotted in Figure 7c is an average of the currents from these two traces.
FIGURE 7.
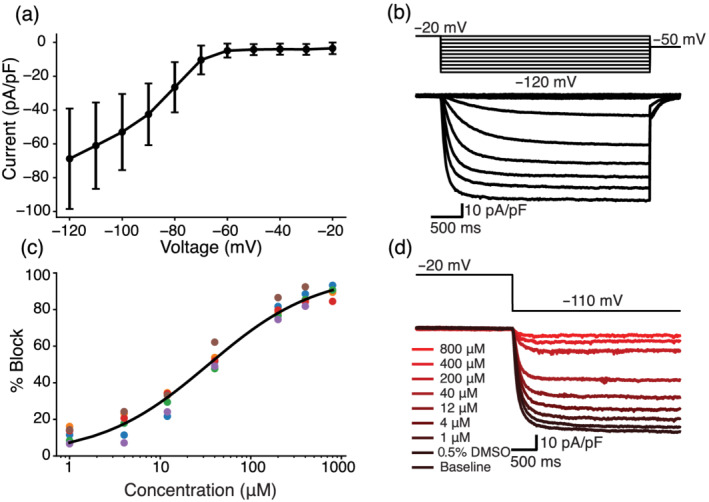
Dose–response curve shows quinine block of funny current in HEK‐293 cells stably expressing HCN1. (a) IV curve with averages and errors calculated from six HEK‐HCN1 cells. (b) Representative HEK‐HCN1 current–voltage traces. (c) Dose–response curve fit to pharmacology data from six HEK‐HCN1 cells. (d) Traces generated from a representative cell by clamping at −20 mV for 3000 ms, and then stepping to −110 mV for 3500 ms at all tested concentrations
In all experiment, cells were measured using extracellular solution with the following concentrations (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 D‐glucose monohydrate, 10 HEPES, pH adjusted to 7.4 with NaOH and 298 mOsm. The intracellular solution had the following concentrations (in mM): 10 EGTA, 10 HEPES, 10 KCl, 10 NaCl, 110 KF, pH 7.2 with KOH, 280 mOsm.
2.6. Data analysis and statistics
The data and statistical analyses in this paper comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All results are presented as mean ± SEM. All cells that lasted until the end of an experiment and had a seal greater than 300 M before perforation and access below 50 M were included in this study. In total, 40 iPSC‐CMs were analysed in this study. We compared changes in AP features and VC current traces from iPSC‐CM data acquired before and after application of cisapride (n = 6), verapamil (n = 9), quinidine (n = 6), quinine (n = 9) or a DMSO control solution (n = 10).
For each drug, a single iPSC‐CM vial was thawed and experiments were conducted with either the drug of interest or a DMSO control solution between 5 and 15 days after the thaw. On a given day, all trials were conducted with either the DMSO control or drug solution. Information about patched cells was not used to select the perfusion solution (e.g., randomization). Data were analysed after day 15, and cells that did not meet our quality control thresholds for seal and access resistance were removed. An additional vial was thawed if n < 6. A single thaw was enough to meet these standards for verapamil, quinidine and quinine. Two vials were required for cisapride. The DMSO control group includes cells from all the thawed vials. The experimenter was not blinded to the drugs used. This was not required, as the analysis and quality control thresholds were established a priori, with one exception (see discussion of IKr segment). Statistical tests were conducted after all data was collected.
Significant differences between the DMSO and each drug group were calculated using the SciPy unpaired t‐test function in Python, with significance indicating P < 0.05. Significance was calculated to compare IK1 dynamic clamp AP prolongation. For the VC data, significance was calculated over the seven regions that the Kernik–Clancy model identified as maximizing the isolation IKr, INa, ICaL, Ito, IK1, If and IKs. The goal was to identify time windows over which ionic currents were isolated and use the occurrence of current reduction in individual cells within those windows to identify drug block. Confidence intervals are set to 95% for each point plot. All statistical analyses were performed using the raw experimental data. For presentation in figures, data were smoothed with a 0.4‐ms moving average.
A functional t test (Keser, 2014) was used to determine the time windows when current response changes to drug treatment differed from responses to DMSO treatment. We used the following steps to develop a null distribution, conduct a t test at every time point and determine windows of significant difference between DMSO control and drug groups:
For both the control and drug groups, we calculated the change in current at every time point from pre‐ to post‐treatment.
We developed a null distribution by completing the following step 200 times: we combined and randomly shuffled individuals from the drug and control groups, and then redistributed them into two distinct groups. We calculated a T statistic (T(t)) at every time point.
We found the t value at the 95th quantile of the null distribution and used it as the threshold for determining significant differences between our control and drug groups. In other words, the control and drug groups were labelled significantly different at a time when the T value comparing these two groups was greater than the T value at the 95th percentile of the null distribution.
The windows plotted in Figures 5, 6 and S10–S13 show where there is a significant difference between the drug and DMSO groups that lasts for more than 1 ms. The functional t test calculations were completed with custom Python code using the SciPy unpaired t test function.
FIGURE 5.
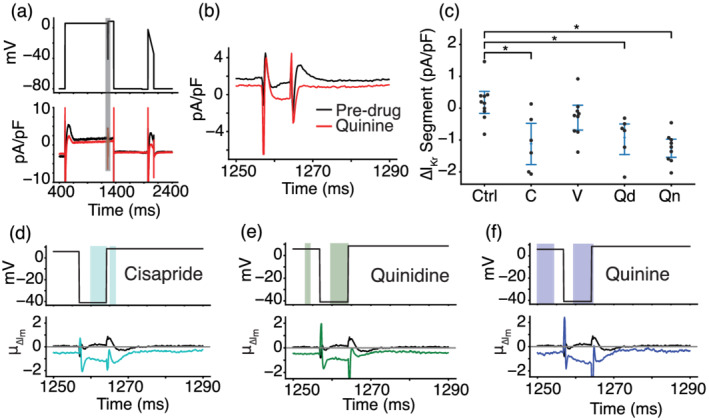
Optimized voltage clamp (VC) protocol correctly identifies IKr as a target of cisapride, quinidine and quinine. (a) Representative cell shows the effect of quinine on the current response during the segment of the VC protocol designed to isolate IKr. The black trace is pre‐drug, and the red trace is post‐drug. (b) The current trace during the segment meant to isolate IKr (shaded grey in panel a). (c) Cells treated with cisapride (C), quinidine (Qd) and quinine (Qn) showed a decrease in total current during the VC segment designed to isolate IKr. At the concentrations used in this experiment, cisapride, quinidine and quinine should block 95%, 89% and 72% of IKr, respectively. *P< 0.05, significantly different as indicated. (d–f) Functional t tests show a significant difference in the average change in current during the IKr‐isolating segment when comparing cells treated with DMSO to cells treated with cisapride (d), quinidine (e) and quinine (f). Verapamil was excluded because there was no significant difference during this segment of the protocol.
FIGURE 6.
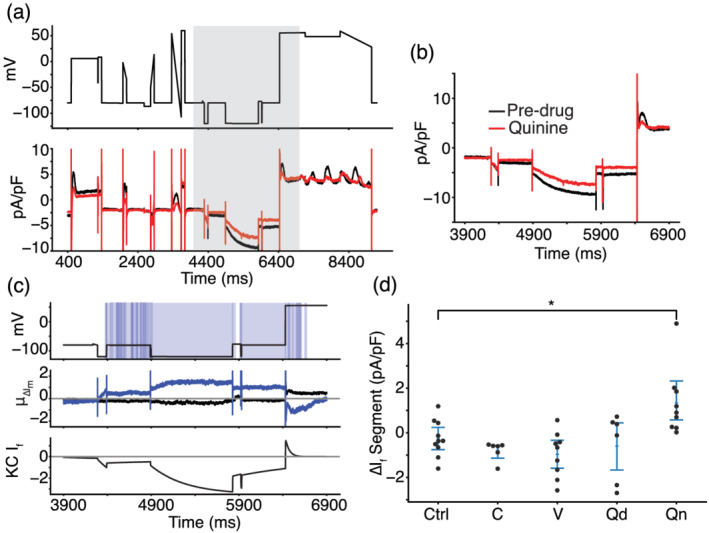
Voltage clamp (VC) protocol identifies funny current as a target of quinine. (a) Representative cell treated with quinine shows a reduction in inward current when the cell is clamped to a hyperpolarized potential of −120 mV, before 6000 ms. After 6000 ms, outward current is reduced as If reverses in direction before inactivating. (b) The current trace during and around the segment meant to isolate If (shaded grey in panel a). (c) Functional t test shows a significant difference between quinine‐treated cells and control cells throughout the period of the protocol when funny current would be active (c, top). The average difference between quinine‐treated and control cells is between 0.8 and 1.2 pA/pF throughout this period (c, middle). The Kernik–Clancy induced pluripotent stem cell‐derived cardiomyocyte (iPSC‐CM) model funny current becomes active during the period when quinine‐treated cells show a significant change in current and turns off just before the P value increases above 0.05 (c, bottom). (d) Cells treated with quinine (Qn) show a decrease in total current during the VC segment designed to isolate If. *P< 0.05, significantly different as indicated.
2.7. Materials
Cisapride monohydrate and quinine were obtained from Sigma Aldrich (Saint Louis, MO). Verapamil hydrochloride was obtained from MP Biomedicals (Solon, OH). Quinidine was obtained from Tocris (Bristol, UK). Frozen stocks of human iPSC‐CMs from a healthy individual (SCVI‐480CM) were obtained from Joseph C. Wu, MD, PhD, at the Stanford Cardiovascular Institute Biobank. The iPSC‐CM line was derived from an African‐American female donor and was approved by Stanford University Human Subjects Research Institutional Review Board. The cells were differentiated to cardiomyocytes as described previously (Burridge et al., 2014; Churko et al., 2013). HEK‐HCN1 (CT6114) cells were obtained from Charles River (Wilmington, MA).
2.8. Code and data availability
All code has been made publicly available on GitHub at: https://github.com/Christini-Lab/vc-optimization-cardiotoxicity. All the data presented in this manuscript has been made publicly available through Cornell eCommons.
2.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander et al., 2021).
3. RESULTS
3.1. Optimizing a VC protocol to isolate individual currents for drug cardiotoxicity screening
We used a GA to optimize VC protocols that maximize the current contribution for each of seven ionic currents: IKr, ICaL, INa, Ito, IK1, IKs and If (Figures 2a and S2–S8). The VC protocols were systematically shortened and then combined into a single protocol that contained the segments that maximized the isolation of each ionic current (Figure 2b). The resulting protocol (~9 s) is short enough to be run multiple times in each cell under control conditions and after drug application.
FIGURE 2.
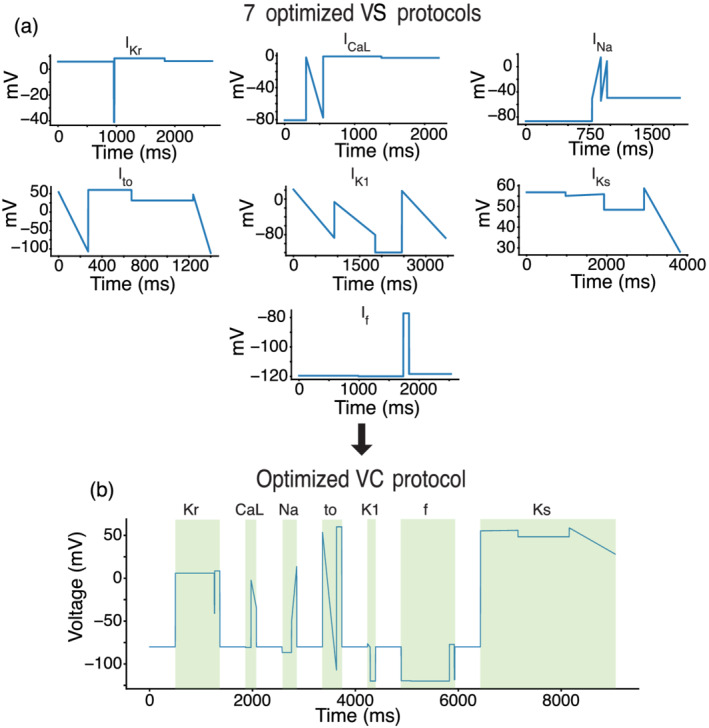
A genetic algorithm (GA) optimized a voltage clamp (VC) protocol to isolate individual current contributions in an in silico induced pluripotent stem cell‐derived cardiomyocyte (iPSC‐CM) model. (a) The GA was used to develop individual VC protocols for seven of the most predominant ionic currents in cardiac cells. (b) These seven protocols were systematically shortened and joined together by a −80‐mV holding step to produce an optimized VC protocol that isolates each of the seven currents.
We validated the VC protocol by applying it to a different iPSC‐CM model (Paci et al., 2018) and comparing the windows of maximum current (Figure S9). This validation step provided us with confidence that the VC protocol could isolate currents during the same time windows from cells with different conductances and kinetics.
3.2. Synthetic maturation of iPSC‐CMs by IK1 dynamic clamp improves interpretability of iPSC‐CM AP data
The iPSC‐CMs displayed a heterogeneous spontaneous phenotype (Figure 3a), which is consistent with previous work on single‐cell iPSC‐CMs (Garg et al., 2019). To recover consistent AP data from all cells, we injected a synthetic IK1 model current (Ishihara et al., 2009) and paced the cells at 1 Hz (Figure 3b). These data are consistent with previous work showing that IK1 dynamic clamp can reduce (although not eliminate) cell‐to‐cell heterogeneity while eliciting more adult‐like AP behaviour from iPSC‐CMs (Li et al., 2019). The IK1 dynamic clamp and paced APs (n = 40) had an average resting membrane potential of −74.2 ± 2.8 mV (Figure 3c) and action potential duration at 90% repolarization of 142.0 ± 48.3 ms (Figure 3d).
FIGURE 3.
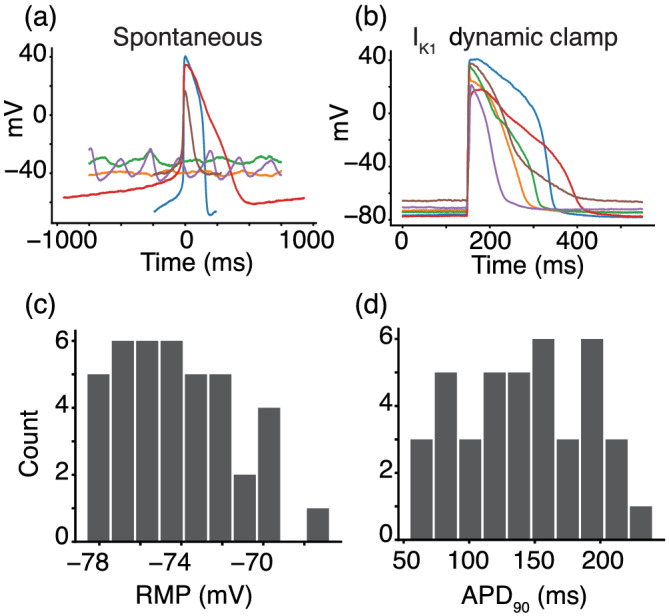
Synthetic maturation of induced pluripotent stem cell‐derived cardiomyocytes (iPSC‐CMs) with dynamically clamped IK1 ensures consistent action potentials (APs) under 1‐Hz pacing conditions. (a) Spontaneous behaviour from a subpopulation of six cells (each with a different coloured trace) that indicates the level of heterogeneity and inconsistent presence of APs in the cell population. (b) IK1 dynamic clamp applied to the same cells makes them appear more mature when paced at 1 Hz. (c and d) Histograms for the IK1 dynamically clamped and paced AP resting membrane potential (−74.2 ± 2.8 mV) and action potential duration at 90% repolarization (142.0 ± 48.3 ms) for the 40 cells in this study
3.3. IK1 dynamic clamp AP data identifies surrogate markers of cardiotoxicity
We compared changes in AP features for iPSC‐CM data acquired before and after application of cisapride (n = 6), verapamil (n = 9), quinidine (n = 6), quinine (n = 9) or a DMSO control solution (n = 10). Table 1 shows the percent block of each cardiac ion channel by these drugs based on previously published results (Crumb et al., 2016). Cisapride is a CiPA‐labelled intermediate‐risk drug and blocks IKr specifically at the concentration used in this study (50× the effective free plasma concentration, EFPC). The cell in Figure 4a did not show AP prolongation after cisapride treatment, which was the case for four of six cells. Verapamil is a low‐risk drug that moderately blocks ICaL and lightly blocks IKr at the concentrations used in this study (3xEFPC). Figure 4b shows a verapamil‐treated cell that displays slight shortening during the plateau phase, which is characteristic of ICaL block. Quinidine and quinine are both pro‐arrhythmic and block multiple ion channels with a strong affinity for IKr at concentrations used in this study (3xEFPC). Both example quinidine‐ (Figure 4c) and quinine‐treated (Figure 4d) cells show AP prolongation characteristic of such IKr block.
TABLE 1.
The optimized VC protocol correctly identifies strong drug blocks
| Drug | IKr | ICaL | INa | Ito | IK1 | If | IKs |
|---|---|---|---|---|---|---|---|
| Cisapride (125 nM) | 95%* | 1% | 2% | 13% | 5% | ?? | 2% |
| Verapamil (150 nM) | 21% | 39%* | <1% | 1% | 3% | ?? | 3% |
| Quinidine (2529 nM) | 89%* | 16% | 10% | 43%* | 1% | ?? | 27% |
| Quinine (12,000 nM) | 72%* | 29% | 28% | 15% | <1% | 32%* † | 20% |
Note: All percent block data, except quinine block of If, is taken from Crumb et al. (2016). The quinine block of If is from the HEK‐HCN1 experiments in this study. The optimized voltage clamp (VC) protocol qualitatively identifies drug blocks that Crumb et al. showed were greater than 30% and identifies funny current as a target of quinine (†).
P < 0.05, significantly greater than 30% block.
FIGURE 4.
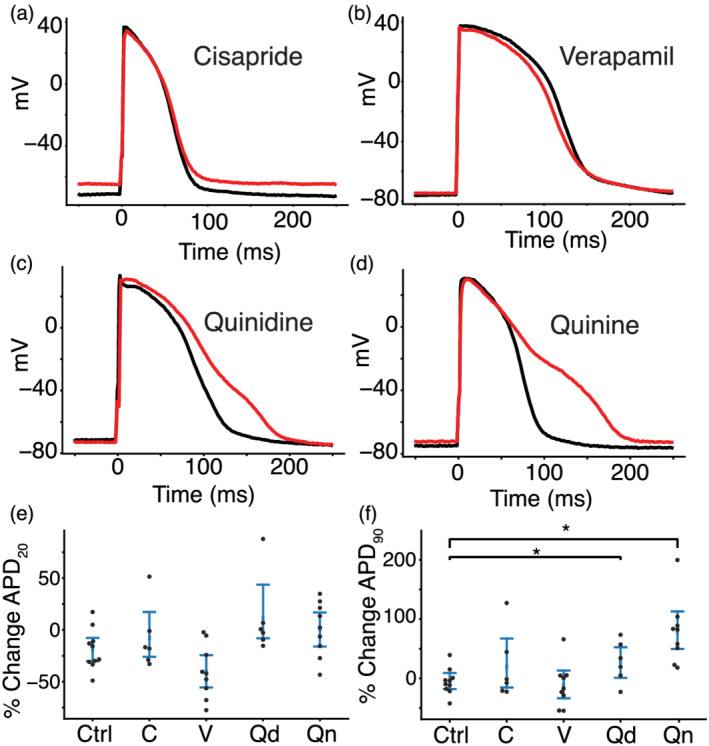
Drug treatment resulted in action potential (AP) morphology changes consistent with the effects caused by drug block of the expected currents. Four cells with dynamically clamped IK1 and paced at 1 Hz before (black) and after (red) treatment with cisapride (a), verapamil (b), quinidine (c) and quinine (d). (e) Percent change in APD20 and (f) APD90 for drugs treated with cisapride ( C), verapamil (V), quinidine (Qd), quinine (Qn) or a DMSO control solution (Ctrl). No drugs caused a significant change in APD20 compared to control cells. Quinidine and quinine showed significant prolongation when compared to control cells. *P< 0.05, significantly different as indicated.
When compared to control, verapamil‐treated cells showed shortening in the action potential duration at 20% of repolarization (APD20), but the difference was not statistically significant (Figure 4e). Cells that were treated with quinidine and quinine showed a significant prolongation in the action potential duration at 90% of repolarization (APD90) when compared to control cells (Figure 4f). Because APD90 prolongation is an established pro‐arrhythmia risk indicator, these findings agree with the CiPA classification or known pro‐arrhythmia risk for verapamil (low risk), quinidine (high risk), and quinine (conditional risk) at 3× their EFPC. Cisapride‐treated cells did not show a significant change in APD90, despite it being a stronger IKr blocker than quinidine and quinine at the concentrations used in this study. This may be because quinidine and quinine block other outward potassium currents, including IKs, at the concentrations used in this study. Such block of both dominant repolarizing potassium currents (IKr and IKs) may cause greater and more consistent AP prolongation in such a heterogeneous iPSC‐CM population.
3.4. The optimized VC protocol qualitatively identifies drugs that block greater than 30% of an ionic current
To identify the ion channel targets of each drug (i.e., mechanism), we compared the average change in VC responses from each drug treatment group to the DMSO control group. Because the protocol was designed to isolate specific ionic currents during brief windows of the protocol, changes in each of these seven segments could reveal ionic mechanism.
Figure 5a shows a representative example of a cell that was treated with quinine. The zoomed‐in panel in Figure 5b shows the portion of the VC protocol that maximized the current contribution of IKr relative to the total current. This inset shows a decrease in the total current present after treatment with quinine. The difference is particularly pronounced from 1260 to 1270 ms, the segment where IKr is predicted by the model to have a large relative contribution compared to the other ionic currents present. According to our modelling work, 1265–1270 ms is predicted to have the largest amount of IKr, although the experimental data from this region was highly variable due to variations in access resistance among cells. We chose, instead, to focus on the 1260–1265 ms window, because it has the second largest IKr current isolation and is a few milliseconds after a voltage step, so access resistance has less of an effect on this region. Figure 5c shows the change in current during the window from 1260 to 1265 ms for all cells in this study.
Cells treated with cisapride, quinidine and quinine all showed significant reductions in current during this segment compared to control cells. Cells treated with verapamil, a weaker IKr blocker at the concentration used here (21%), did not show a significant difference in current during this segment. These data suggest that the VC protocol can detect strong IKr block during the model‐designed IKr isolation segment but fails to detect weak block of IKr current.
In addition to focusing on the model‐identified segments of the protocol, we also performed a functional t test to compare the average change in total current between control and drug‐treated cells at every timepoint during the VC protocol.
Figure 5d–f shows the segment of the VC protocol where IKr should be isolated. The bottom panel shows the average change in membrane current (μ∆Im) at each timepoint for cells treated with cisapride (Figure 5d, cyan), quinidine (Figure 5e, green) or quinine (Figure 5f, blue). The black line in each of these panels shows the average change in cells treated with a DMSO control solution. The shaded window on the top of each panel shows the time points where there is a significant difference between control and treatment groups. The functional t test identifies a significant difference in the currents between 1260 and 1265 ms for all three of these IKr blockers. There was no significant difference between DMSO and verapamil during this period. We plotted these significance windows over the entire VC protocol and compared them to individual currents from the Kernik–Clancy model (Figures S10–S13). Figures S10, S12, and S13 show significant differences throughout the regions where the Kernik–Clancy IKr current is present.
We conducted the same statistical analysis, as shown in Figure 5c, to determine changes during the isolating segments for each of the other currents. These isolating segments correspond to the time windows of the VC protocol that maximized the isolation of each current in the Kernik–Clancy model (Figure S9). The boxes marked with (*) in Table 1 indicate currents that were identified as blocked based on significant changes in current during the corresponding model‐identified segment. In addition to the IKr blocks noted above, the VC protocol correctly identified verapamil block of ICaL and quinidine block of Ito. The VC protocol did not identify any reductions in INa, IK1 or IKs, which were all weakly blocked by at least one of these drugs. Importantly, all blocks of more than 30% were correctly identified, and there were no currents that were incorrectly identified as being blocked. Taken with dynamic clamp AP data, these results indicate that the optimized VC protocol can be used to identify underlying currents responsible for changes in AP morphology.
3.5. The optimized VC protocol identifies a previously unreported quinine block of If
As Table 1 indicates, quinine‐treated iPSC‐CMs revealed a previously unreported block of If. A representative cell trace shows, after quinine treatment, a net positive shift in membrane current throughout the region where the Kernik–Clancy model predicts If is present (Figure 6a,b). This positive shift in membrane current is consistent with If block. The areas of significant difference in this part of the protocol closely align with the Kernik–Clancy simulated If current (Figure 6c). This holds largely true both for when If is predicted to conduct an inward current (between 4400 and 6400 ms) and an outward current (between 6400 and 6600 ms). When we consider only the segment of the protocol where If current isolation is maximized, we find a significant increase in total current due to reduction of negative If, when compared to control cells (Figure 6d).
To verify the finding that quinine blocks If, we used a HEK‐293 cell line with tetracycline‐inducible expression of HCN1, the ion channel that conducts If. We chose the HCN1 isoform because it was recently shown to be present at high densities in iPSC‐CMs (Giannetti et al., 2021). Figure 7a,b show that these HCN1 cells have typical current–voltage If behaviour. We acquired dose–response data under baseline external conditions, after treatment with a DMSO control solution and at seven concentrations of quinine (Figure 7c,d). There was no significant difference between the traces acquired before and after the addition of a 0.5% DMSO control solution. The best fit Hill equation curve has an IC50 of 34.2 μM and Hill coefficient of 0.72. At 12 μM, which was the concentration used in the iPSC‐CM study, the estimated block is 32.0%. Overall, this dose–response data confirms the findings from our iPSC‐CM study, that quinine blocks If at 3× the EFPC.
4. DISCUSSION
In this study, we demonstrated the potential of a drug screening pipeline that uses iPSC‐CMs to simultaneously identify drug cardiotoxicity markers and mechanism. The pipeline relies on a novel optimization algorithm to produce VC protocols that we can use to identify multi‐channel drug block that provide a mechanistic insight into AP morphology changes. We found that quinidine and quinine, both pro‐arrhythmic drugs, caused AP prolongation. From the VC data, we correctly identified the four ion channels that were blocked by more than 30%. The VC protocol also uncovered a previously unreported block of If by quinine in iPSC‐CMs. A similar block was recently demonstrated in iPSC‐derived neurons (Zou et al., 2018). We confirmed these findings with a dose–response study on an HCN1 expression system and found that quinine blocked the pore domain of If by 32% at the concentration used in this experiment.
4.1. Optimizing VC protocols for iPSC‐CM drug experiments
Traditional VC protocols are developed manually and typically designed to fit and/or measure specific model parameters (Beattie et al., 2018; Groenendaal et al., 2015; Hobbs & Hooper, 2008). The current study is a departure from this approach in two ways: (1) the protocols were designed automatically using a model‐guided optimization and (2) the resulting protocols were specifically designed to identify drug targets, not to improve fits. Due to the conserved nature of ion channels across various tissues, this new approach to protocol design can be extended to toxicity applications of other electrically excitable cells, such as neurons.
The success of such a VC protocol optimization requires reasonable agreement between the computational model (e.g., Kernik–Clancy) and in vitro cells (e.g., iPSC‐CMs from Stanford Biobank). While the model in this study was able to create VC protocols that could detect strong blocks of ionic current, the heterogeneity within the iPSC‐CM population (Figure 3) indicates that the model does not fully capture the dynamics of the cells used here. Despite this discrepancy, the success of our in vitro experiments validates that the Kernik–Clancy model kinetics are close enough descriptive of the iPSC‐CMs we used to identify when currents are isolated during voltage clamp. This novel application of the model to optimize experiments differs from previous uses of iPSC‐CM models, which have largely focused on predicting drug cardiotoxicity (Gong & Sobie, 2018; Jæger et al., 2021; Tveito et al., 2018) and screening channel mutations (Kernik et al., 2020) through in silico simulations. To the best of our knowledge, this is the first work that shows how in silico models can be used to improve the design, and therefore the effects, of cardiotoxicity drug studies. Additionally, we believe the accuracy of the VC protocol optimization approach will improve with models developed to closely reflect the dynamics of the cell lines used in the cardiotoxicity studies.
4.2. Generating VC protocols that take advantage of the unique gating kinetics for each channel
The GA was designed to isolate individual currents, thereby producing protocols (Figure 2a) that exploit the unique gating kinetics for each ion channel. For example, the IKr protocol (Figure S5) takes advantage of the fast‐inactivation, slow‐activation gating that is characteristic of this channel. The initial step to just above 0 mV inactivates the channel while the activation gate opens. The step to −40 mV opens the inactivation gate, allowing current to flow through the channel before the slower activation gate closes. The final step, back to more depolarized than 0 mV, increases the driving force, which provides a brief window (about 5 ms) where the activation gate is open, the inactivation gate is open, and there is a large driving force pushing potassium out of the cell.
The other protocols also identified dynamic ranges that highlight each channel's unique kinetics. The IKs (Figure S7) and If (Figure S8) protocols settle into positive (>50 mV) and negative (<−110 mV) extremes where most other channels are closed, but these channels are open and remain open. The IK1 protocol (Figure S6) isolates its current at the same potential as If, but is maximized before If has a chance to open. The INa protocol (Figure S2) steps to a hyperpolarized potential (−87 mV) to fully open the inactivation gate and then jumps to a ramp that is depolarized enough to activate the channel (−50 mV), while minimizing the activation of other ion channels (e.g., ICaL). The ICaL and Ito channels have fast activation and slow inactivation kinetics—these protocols take advantage of this by stepping to potentials that will open their channels but minimize the contribution from the other currents.
4.3. Screening for drug cardiotoxicity using the novel VC protocol and iPSC‐CMs
The existence of overly conservative cardiotoxicity screening guidelines points to the need to develop new methods that improve specificity and provide insight into the mechanism of drug block. In recent years, high‐throughput iPSC‐CM screening approaches that rely on surrogate markers of cardiotoxicity risk have improved prediction accuracy and the ability to evaluate drugs at scale (Bedut et al., 2016; Lu et al., 2019; Pioner et al., 2019). Expression system cell lines and molecular‐dynamic simulations have supplemented these findings by providing detailed single‐channel mechanisms of drug action (Demarco et al., 2020; Yang et al., 2020). However, there have been few methods that provide both measures of cardiotoxicity and mechanism from the same iPSC‐CM cells.
One recent approach fit an iPSC‐CM model to voltage and fluorescent calcium AP data acquired before and after drug application (Jæger et al., 2021; Tveito et al., 2018). Because this approach only considers spontaneous AP and conduction velocity data, successful estimates of current block is limited to the currents that are sensitive to changes in these data such as IKr, ICaL and INa.
Here, we built upon this work by developing an approach that provides both surrogate measures of drug cardiotoxicity and identification of drug block for seven cardiac ion channels. Our in vitro AP results showed significant prolongation of quinidine‐ and quinine‐treated cells, which indicates their pro‐arrhythmic potential at 3xEFPC. The VC protocol identified strong block of outward potassium currents by both quinidine (IKr and Ito) and quinine (IKr and If) which may explain such AP prolongation. Cisapride did not cause significant prolongation; however, the VC protocol correctly identified its block of IKr. This result indicates the value that the VC protocol has in detecting pro‐arrhythmic mechanisms in the absence of cardiotoxic markers. If such a discrepant finding were identified from this pipeline, we would still label the drug as likely to be pro‐arrhythmic, based on block of IKr.
If is not often linked to repolarization in iPSC‐CMs. However, we think it is likely to be playing a role in the cells from this study. If appears to be a relatively large current in these cells (Figure 6). Therefore, even if a small fraction of the channels are turned on during AP repolarization, as was recently shown in mouse sinoatrial node myocytes (Peters et al., 2021), If block could lead to AP prolongation. Additionally, the cells in this study had relatively short APs (Figure 3d), so repolarizing If would be active for a greater proportion of the AP duration, before it turns off. However, this finding needs to be further validated.
4.4. Limitations and future directions
This study has several limitations that will need to be addressed before this pipeline can be broadly implemented. First, before this approach can be used at scale, it should be validated with a high‐throughput automated patch‐clamp system. With this type of system, data could be acquired 10–100× faster at multiple drug concentrations and by operators who do not need the specialized patch‐clamp skill. Such studies would produce a greater quantity of data with consistent experimental artefact parameters (Goversen, Becker, et al., 2018), resulting in less variability and greater power for statistical analyses.
Second, the VC protocol does not provide information about drug effects on channel kinetics and is designed to identify pore block of channels in a limited dynamic range. Designing an algorithm to tease apart such effects is a non‐trivial extension of this project, and would need a target objective that is likely to be very different from the one used in this study.
Third, the cisapride‐treated iPSC‐CMs did not show significant prolongation, despite the VC protocol correctly identifying a strong block of IKr. Unlike cisapride, the quinidine‐ and quinine‐treated cells showed significant AP prolongation. This discrepancy would occur if IKr were not the dominant repolarizing ionic current in the cells used in this study, as quinidine and quinine block other potassium currents in addition to IKr. Simultaneous block of these currents would deplete the repolarization reserve and cause more consistent AP prolongation from such a heterogeneous population.
Fourth, the iPSC‐CMs often produced an oscillatory current trace when held at large positive voltages, which is probably caused by calcium overload that can occur at high potentials. This oscillatory pattern could decrease the sensitivity of the protocol to determine strong IKs‐blockers, which we plan to test in the future.
Fifth, while we have shown that the protocol can identify strong block of four ion channels (IKr, ICaL, If and Ito) with no false positives, further validation of the approach is needed using drugs that block the remaining ion channels known to exist in iPSC‐CMs (INa, IK1 and IKs). Because the VC protocol was designed using the same model and GA, we are hopeful that it will work with these ion channels as well.
Sixth, we did not account for the effect that cell‐to‐cell variation and changes in series resistance have on the interpretation of current responses. Such variations can make it difficult to interpret current traces within 1 ms after a voltage step. For example, we measured changes from the second most‐isolated IKr segment, because the peak occurred just after stepping to a new voltage. In the future, this issue may be avoided by adjusting the GA fitness function to exclude contribution calculations just after voltage steps. Such an approach could be validated with a set of iPSC‐CM models that approximate the heterogeneity seen in experiments. One could simulate this heterogeneity by adjusting conductances and developing hybrid populations with currents from both the Paci and Kernik–Clancy models, as suggested in Paci et al. (2021b).
Finally, we need to reproduce this study with cells from multiple donors. We do not expect donor demographics to have a meaningful effect on the success of this approach, as iPSC‐CMs currently do not exhibit the physiological differences between female and male adult cardiomyocytes well (Huo et al., 2019). However, patient‐to‐patient variations in ion channel expression levels could affect the extent of pre‐ to post‐drug electrophysiological changes.
In conclusion, we have outlined in this study, a new pipeline for determining drug cardiotoxicity and underlying mechanism by applying a novel VC protocol and IK1 dynamic clamp to iPSC‐CMs. This pipeline correctly identified the ion channels that were strongly blocked by each drug and identified pro‐arrhythmic markers (AP prolongation) or mechanism (IKr block) for the intermediate‐ and high‐risk drugs tested in this study. We believe that this cardiotoxicity pipeline could have far‐reaching effects on how drugs are screened and could ultimately increase the number of safe and effective drugs available to patients.
CONFLICT OF INTEREST
The authors declare no competing interest.
AUTHOR CONTRIBUTIONS
A.C., T.K.M. and D.C. designed the study; A.C. and S.W. conducted experiments; A.C. and D.K. developed the optimization code; A.C. performed computational studies and statistical tests; A.C., S.W., T.K.M. and D.C. wrote the paper.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, and as recommended by funding agencies, publishers and other organiations engaged with supporting research.
Supporting information
Table S1: Percentage of current isolated
Table S2: Simulated effect of extracellular calcium concentration on current isolation with the optimized VC protocol.
Figure S1: The effect of experimental artefact on VC data designed to activate sodium channels. The experimental artefact used in this simulation included a voltage offset of −2.8 mV, seal resistance of 1 GΩ, and access resistance of 20 MΩ. The top panel shows the voltage experienced by the cell (dashed blue) compared to the command voltage (black). The voltage offset shifts the membrane voltage negative by 2.8 mV, which has little effect on the current response. The relatively high access resistance is what causes the gradual slope upwards from the starting voltage of −80 mV to the ending voltage of −30 mV. This gradual slope in the membrane voltage leads to a delayed and reduced peak current (bottom) response.
Figure S2: The optimized protocol for INa with Kernik‐Clancy and Paci current response. A, Optimized INa protocol. The total current response (C) and INa contribution (E) for both the Kernik‐Clancy and Paci models. B, The portion of the protocol shaded in A, which includes the maximum INa contribution. The total current response (D) and INa contribution (F) for the portion of the protocol displayed in B.
Figure S3: The optimized protocol for Ito with Kernik‐Clancy and Paci current responses.
Figure S4: The optimized protocol for ICaL with Kernik‐Clancy and Paci current response.
Figure S5: The optimized protocol for IKr with Kernik‐Clancy and Paci current responses.
Figure S6: The optimized protocol for IK1 with Kernik‐Clancy and Paci current responses.
Figure S7: The optimized protocol for IKs with Kernik‐Clancy and Paci current responses.
Figure S8: The optimized protocol for If with Kernik‐Clancy and Paci current responses.
Figure S9: The timepoints of maximum current isolation for the Paci and Kernik‐Clancy models. Five (IK1, Ito, IKr, IKs, and INa) of the seven currents in the Paci model were isolated within 10 ms of when they were isolated in the Kernik‐Clancy model. These time windows are highlighted grey in the top panel. The maximum ICaL isolation in the Paci model occurs far from where the current is maximized in the Kernik‐Clancy model. However, the Paci model had a current isolation within 5% of its maximum during the Kernik‐Clancy window. The timepoints for If also differed between the two models. However, these timepoints are near one another and have similar voltage dynamics, indicating that the Kernik‐Clancy timepoint is likely generalizable for these currents. The remaining panels display the percent contribution of each of the seven currents for the Kernik‐Clancy and Paci models throughout the protocol.
Figure S10: Differences in cell response to cisapride vs. DMSO. The VC protocol (panel 1), Kernik‐Clancy simulated change in membrane current after cisapride treatment (panel 2), average change in experimental cell response from pre‐ to post‐drug application for both DMSO and cisapride (panel 3), and the Kernik‐Clancy IKr response to the VC protocol (panel 4). The blue overlays indicate where there is a significant difference (p < .05) between the average cisapride and DMSO responses. We expected cisapride to strongly and specifically block IKr. The bottom panel shows that most of the areas that are significantly different occur when IKr is present in the Kernik‐Clancy model.
Figure S11: Differences in cell response to verapamil vs. DMSO. This figure shows the VC protocol (panel 1), Kernik‐Clancy simulated change in membrane current after verapamil treatment (panel 2), average change in experimental cell response from pre‐ to post‐drug application for both DMSO and verapamil (panel 3), and the Kernik‐Clancy ICaL response to the VC protocol. The red overlays indicate where there is a significant difference (p < .05) between the average verapamil and DMSO responses. At the concentration tested, we expect verapamil to block ~40% of ICaL and ~20% of IKr. The bottom panel shows that most of the areas that are significantly different occur when the ICaL is present in the Kernik‐Clancy model. There are two brief windows that the functional t‐test identifies after 4,000 ms, that are not likely IKr or ICaL.
Figure S12: Differences in cell response to quinidine vs. DMSO. This figure shows the VC protocol (panel 1), Kernik‐Clancy simulated change in membrane current after quinidine treatment (panel 2), average change in experimental cell response from pre‐ to post‐drug application for both DMSO and quinidine (panel 3), and the Kernik‐Clancy IKr (panel 4), Ito (panel 5), and IKs (panel 6) responses to the VC protocol. The green overlays indicate where there is a significant difference (p < .05) between the average quinidine and DMSO responses. At the concentration tested, we expect quinidine to block ~89% of IKr, ~43% of Ito, and ~27% of IKs. The significance windows overlap very well with the Kernik‐Clancy IKr, Ito, and IKs currents. This is to be expected, as quinidine is known to be a strong and general blocker of potassium currents.
Figure S13: Differences in cell response to quinine vs. DMSO. This figure shows the VC protocol, Kernik‐Clancy simulated change in membrane current after quinine treatment (panel 2), average change in drug response from pre‐ to post‐drug application for both DMSO and quinidine (panel 3), and the Kernik‐Clancy IKr (panel 4), If (panel 5), and ICaL (panel 6) responses to the VC protocol. The blue overlays indicate where there is a significant difference (p < .05) between the average quinine and DMSO responses. At the concentration tested, we expect quinine to block ~72% of IKr and ~29% of ICaL. During the experiments, we noticed a likely block of If with quinine treatment. In Figure 7, we show how we calculate a block of ~32% of If by quinine at this concentration using a HEK‐HCN1 cell line. The significance windows overlap very well with the Kernik‐Clancy IKr, ICaL, and If currents.
Figure S14: Max current vs voltage for HCN1 tail current. The max conductance‐voltage curve was found by stepping to ‐50 mV after the channels had been activated with a depolarizing step. The max tail current values in this plot indicate that most, if not all, funny current channels are open when stepping to voltages below ‐100 mV.
ACKNOWLEDGEMENTS
We thank Dr. Drew Tilley for his experimental insights and expertise. We thank Dr. Sumanta Basu for consulting on the functional t test and Dr. Henry Sutanto for feedback on the manuscript. Please note that this manuscript has also been posted as a preprint on bioRxiv: https://www.biorxiv.org/content/10.1101/2021.09.10.459625. Research reported in this publication was supported by the National Heart, Lung And Blood Institute of the National Institutes of Health under Award Number F31HL154655 (to A.C.) and U01HL136297 (to D.J.C.).
Clark, A. P. , Wei, S. , Kalola, D. , Krogh‐Madsen, T. , & Christini, D. J. (2022). An in silico–in vitro pipeline for drug cardiotoxicity screening identifies ionic pro‐arrhythmia mechanisms. British Journal of Pharmacology, 179(20), 4829–4843. 10.1111/bph.15915
Funding information National Heart, Lung, and Blood Institute, Grant/Award Numbers: U01HL136297, F31HL154655
DATA AVAILABILITY STATEMENT
Data that support the findings of this study are available through eCommons at https://doi.org/10.7298/c883-s773.
REFERENCES
- Alexander, S. P. , Mathie, A. , Peters, J. A. , Veale, E. L. , Striessnig, J. , Kelly, E. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Aldrich, R. W. , Attali, B. , Baggetta, A. M. , Becirovic, E. , Biel, M. , Bill, R. M. , Catterall, W. A. , … Zhu, M. (2021). The concise guide to PHARMACOLOGY 2021/22: Ion channels. British Journal of Pharmacology, 178(Suppl), S157–S245. [DOI] [PubMed] [Google Scholar]
- Anonymous . (2005). ICH S7B Note for Guidance on the Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals. Int. Conf. Harmon. Tech. Requir. Regist. Pharm. Hum. Use.
- Beattie, K. A. , Hill, A. P. , Bardenet, R. , Cui, Y. , Vandenberg, J. I. , Gavaghan, D. J. , de Boer, T. P. , & Mirams, G. R. (2018). Sinusoidal voltage protocols for rapid characterisation of ion channel kinetics. The Journal of Physiology, 596, 1813–1828. 10.1113/JP275733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedut, S. , Seminatore‐Nole, C. , Lamamy, V. , Caignard, S. , Boutin, J. A. , Nosjean, O. , Stephan, J. P. , & Coge, F. (2016). High‐throughput drug profiling with voltage‐and calcium‐sensitive fluorescent probes in human iPSC‐derived cardiomyocytes. American Journal of Physiology‐Heart and Circulatory, 311, H44–H53. 10.1152/ajpheart.00793.2015 [DOI] [PubMed] [Google Scholar]
- Blinova, K. , Dang, Q. , Millard, D. , Smith, G. , Pierson, J. , Guo, L. , Brock, M. , Lu, H. R. , Kraushaar, U. , Zeng, H. , Shi, H. , Zhang, X. , Sawada, K. , Osada, T. , Kanda, Y. , Sekino, Y. , Pang, L. , Feaster, T. K. , Kettenhofen, R. , … Gintant, G. (2018). International multisite study of human‐induced pluripotent stem cell‐derived cardiomyocytes for drug Proarrhythmic potential assessment. Cell Reports, 24, 3582–3592. 10.1016/j.celrep.2018.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge, P. W. , Matsa, E. , Shukla, P. , Lin, Z. C. , Churko, J. M. , Ebert, A. D. , Lan, F. , Diecke, S. , Huber, B. , Mordwinkin, N. M. , Plews, J. R. , Abilez, O. J. , Cui, B. , Gold, J. D. , & Wu, J. C. (2014). Chemically defined and small molecule‐based generation of human cardiomyocytes. Nature Methods, 11, 855–860. 10.1038/nmeth.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churko, J. M. , Burridge, P. W. , & Wu, J. C. (2013). Cellular Cardiomyoplasty: Methods and protocols. Methods in Molecular Biology., 1036, 81–88. 10.1007/978-1-62703-511-8_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colatsky, T. , Fermini, B. , Gintant, G. , Pierson, J. B. , Sager, P. , Sekino, Y. , Strauss, D. G. , & Stockbridge, N. (2016). The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative — Update on progress. Journal of Pharmacological and Toxicological Methods, 81, 15–20. 10.1016/j.vascn.2016.06.002 [DOI] [PubMed] [Google Scholar]
- Crumb, W. J. , Vicente, J. , Johannesen, L. , & Strauss, D. G. (2016). An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. Journal of Pharmacological and Toxicological Methods, 81, 251–262. 10.1016/j.vascn.2016.03.009 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruin, M. L. , Pettersson, M. , Meyboom, R. H. B. , Hoes, A. W. , & Leufkens, H. G. M. (2005). Anti‐HERG activity and the risk of drug‐induced arrhythmias and sudden death. European Heart Journal, 26, 590–597. 10.1093/eurheartj/ehi092 [DOI] [PubMed] [Google Scholar]
- Demarco, K. R. , Yang, P.‐C. , Singh, V. , Furutani, K. , Dawson, J. , Jeng, M.‐T. , Fettinger, J. C. , Bekker, S. , Ngo, V. A. , Noskov, S. Y. , Yarov‐Yarovoy, V. , Sack, J. T. , Wulff, H. , Clancy, C. E. , & Vorobyov, I. (2020). Molecular determinants of pro‐arrhythmia proclivity of d‐ and l‐ sotalol via a multi‐scale modeling pipeline. Journal of Molecular and Cellular Cardiology, 115800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri, A. , Goversen, B. , Vos, M. A. , van Veen, T. A. B. , & de Boer, T. P. (2019). Required GK1 to suppress automaticity of iPSC‐CMs depends strongly on IK1 model structure. Biophysical Journal, 117, 2303–2315. 10.1016/j.bpj.2019.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin, F. A. , De Rainville, F. M. , Gardner, M. A. , Parizeau, M. , & Gagńe, C. (2012). DEAP: Evolutionary algorithms made easy. Journal of Machine Learning Research, 13, 2171–2175. [Google Scholar]
- Garg, P. , Oikonomopoulos, A. , Chen, H. , Li, Y. , Lam, C. K. , Sallam, K. , Perez, M. , Lux, R. L. , Sanguinetti, M. C. , & Wu, J. C. (2019). Genome editing and induced pluripotent stem cells in cardiac Channelopathy. Journal of the American College of Cardiology, 72, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannetti, F. , Benzoni, P. , Campostrini, G. , Milanesi, R. , Bucchi, A. , Baruscotti, M. , Dell'Era, P. , Rossini, A. , & Barbuti, A. (2021). A detailed characterization of the hyperpolarization‐activated ‘funny’ current (if) in human‐induced pluripotent stem cell (iPSC)‐derived cardiomyocytes with pacemaker activity. Pflügers Archiv, 473, 1009–1021. 10.1007/s00424-021-02571-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gintant, G. (2011). An evaluation of hERG current assay performance: Translating preclinical safety studies to clinical QT prolongation. Pharmacology & Therapeutics, 129, 109–119. 10.1016/j.pharmthera.2010.08.008 [DOI] [PubMed] [Google Scholar]
- Gong, J. Q. X. , & Sobie, E. A. (2018). Population‐based mechanistic modeling allows for quantitative predictions of drug responses across cell types. NPJ Systems Biology and Applications, 4, 11. 10.1038/s41540-018-0047-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goversen, B. , Becker, N. , Stoelzle‐Feix, S. , Obergrussberger, A. , Vos, M. A. , van Veen, T. A. B. , Fertig, N. , & de Boer, T. P. (2018). A hybrid model for safety pharmacology on an automated patch clamp platform: Using dynamic clamp to join iPSC‐derived cardiomyocytes and simulations of Ik1 ion channels in real‐time. Frontiers in Physiology, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goversen, B. , van der Heyden, M. A. G. , van Veen, T. A. B. , & de Boer, T. P. (2018). The immature electrophysiological phenotype of iPSC‐CMs still hampers in vitro drug screening: Special focus on IK1. Pharmacology & Therapeutics, 183, 127–136. 10.1016/j.pharmthera.2017.10.001 [DOI] [PubMed] [Google Scholar]
- Groenendaal, W. , Ortega, F. A. , Kherlopian, A. R. , Zygmunt, A. C. , Krogh‐Madsen, T. , & Christini, D. J. (2015). Cell‐specific cardiac electrophysiology models. PLoS Computational Biology, 11, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancox, J. C. , McPate, M. J. , El Harchi, A. , & Zhang, Y. H. (2008). The hERG potassium channel and hERG screening for drug‐induced torsades de pointes. Pharmacology & Therapeutics, 119, 118–132. 10.1016/j.pharmthera.2008.05.009 [DOI] [PubMed] [Google Scholar]
- Hobbs, K. H. , & Hooper, S. L. (2008). Using complicated, wide dynamic range driving to develop models of single neurons in single recording sessions. Journal of Neurophysiology, 99, 1871–1883. 10.1152/jn.00032.2008 [DOI] [PubMed] [Google Scholar]
- Huo, J. , Wei, F. , Cai, C. , Lyn‐Cook, B. , & Pang, L. (2019). Sex‐related differences in drug‐induced QT prolongation and Torsades de pointes: A new model system with human iPSC‐CMs. Toxicological Sciences, 167, 360–374. 10.1093/toxsci/kfy239 [DOI] [PubMed] [Google Scholar]
- Ishihara, K. , Sarai, N. , Asakura, K. , Noma, A. , & Matsuoka, S. (2009). Role of Mg2+ block of the inward rectifier K+ current in cardiac repolarization reserve: A quantitative simulation. Journal of Molecular and Cellular Cardiology, 47, 76–84. 10.1016/j.yjmcc.2009.03.008 [DOI] [PubMed] [Google Scholar]
- Jæger, K. H. , Charwat, V. , Wall, S. , Healy, K. E. , & Tveito, A. (2021). Identifying drug response by combining measurements of the membrane potential, the cytosolic calcium concentration, and the extracellular potential in microphysiological systems. Frontiers in Pharmacology, 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannesen, L. , Vicente, J. , Mason, J. W. , Sanabria, C. , Waite‐Labott, K. , Hong, M. , Guo, P. , Lin, J. , Sørensen, J. S. , Galeotti, L. , Florian, J. , Ugander, M. , Stockbridge, N. , & Strauss, D. G. (2014). Differentiating drug‐induced multichannel block on the electrocardiogram: Randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clinical Pharmacology and Therapeutics, 96, 549–558. 10.1038/clpt.2014.155 [DOI] [PubMed] [Google Scholar]
- Kernik, D. C. , Morotti, S. , Di Wu, H. , Garg, P. , Duff, H. J. , Kurokawa, J. , Jalife, J. , Wu, J. C. , Grandi, E. , & Clancy, C. E. (2019). A computational model of induced pluripotent stem‐cell derived cardiomyocytes incorporating experimental variability from multiple data sources. The Journal of Physiology, 597, 4533–4564. 10.1113/JP277724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernik, D. C. , Yang, P. C. , Kurokawa, J. , Wu, J. C. , & Clancy, C. E. (2020). A computational model of induced pluripotent stem‐cell derived cardiomyocytes for high throughput risk stratification of KCNQ1 genetic variants. PLoS Computational Biology, 16, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keser, I. K. (2014). Comparing two mean humidity curves using functional t‐tests: Turkey case. Electronic Journal of Applied Statistical Analysis, 7, 254–278. [Google Scholar]
- Lei, C. L. , Clerx, M. , Whittaker, D. G. , Gavaghan, D. J. , de Boer, T. P. , & Mirams, G. R. (2020). Accounting for variability in ion current recordings using a mathematical model of artefacts in voltage‐clamp experiments. Philosophical Transactions. Series a, Mathematical, Physoical and Engineering Sciences, 378, 20190348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Luo, X. , Ulbricht, Y. , Wagner, M. , Piorkowski, C. , El‐Armouche, A. , & Guan, K. (2019). Establishment of an automated patch‐clamp platform for electrophysiological and pharmacological evaluation of hiPSC‐CMs. Stem Cell Research, 41, 101662. 10.1016/j.scr.2019.101662 [DOI] [PubMed] [Google Scholar]
- Lu, H. R. , Zeng, H. , Kettenhofen, R. , Guo, L. , Kopljar, I. , van Ammel, K. , Tekle, F. , Teisman, A. , Zhai, J. , Clouse, H. , Pierson, J. , Furniss, M. , Lagrutta, A. , Sannajust, F. , & Gallacher, D. J. (2019). Assessing drug‐induced long QT and Proarrhythmic risk using human stem‐cell‐derived cardiomyocytes in a Ca2+ imaging assay: Evaluation of 28 CiPA compounds at three test sites. Toxicological Sciences, 170, 345–356. 10.1093/toxsci/kfz102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete, E. G. , Liang, P. , Lan, F. , Sanchez‐Freire, V. , Simmons, C. , Gong, T. , Sharma, A. , Burridge, P. W. , Patlolla, B. , Lee, A. S. , Wu, H. , Beygui, R. E. , Wu, S. M. , Robbins, R. C. , Bers, D. M. , & Wu, J. C. (2013). Screening drug‐induced arrhythmia using human induced pluripotent stem cell‐derived cardiomyocytes and low‐impedance microelectrode arrays. Circulation, 128, S3–S13. 10.1161/CIRCULATIONAHA.112.000570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paci, M. , Koivumäki, J. T. , Lu, H. R. , Gallacher, D. J. , Passini, E. , & Rodriguez, B. (2021a). Comparison of the simulated response of three in silico human stem cell‐derived cardiomyocytes models and in vitro data under 15 drug actions. Frontiers Pharmacology, 12, 1–16. 10.3389/fphar.2021.604713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paci, M. , Koivumäki, J. T. , Lu, H. R. , Gallacher, D. J. , Passini, E. , & Rodriguez, B. (2021b). Comparison of the simulated response of three in silico human stem cell‐derived cardiomyocytes models and in vitro data under 15 drug actions. Frontiers in Pharmacology, 12, 1–16. 10.3389/fphar.2021.604713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paci, M. , Pölönen, R. P. , Cori, D. , Penttinen, K. , Aalto‐Setälä, K. , Severi, S. , & Hyttinen, J. (2018). Automatic optimization of an in silico model of human iPSC derived cardiomyocytes recapitulating calcium handling abnormalities. Frontiers in Physiology, 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, C. H. , Liu, P. W. , Morotti, S. , Gantz, S. C. , Grandi, E. , Bean, P. , & Proenza, C. (2021). Bi‐directional flow of the funny current (I f) during the pacemaking cycle in murine sinoatrial node myocytes. Proceedings of the National Academy of Sciences of the United States of America, 118(28), e2104668118. 10.1073/pnas.2104668118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioner, J. M. , Santini, L. , Palandri, C. , Martella, D. , Lupi, F. , Langione, M. , Querceto, S. , Grandinetti, B. , Balducci, V. , Benzoni, P. , Landi, S. , Barbuti, A. , Ferrarese Lupi, F. , Boarino, L. , Sartiani, L. , Tesi, C. , Mack, D. L. , Regnier, M. , Cerbai, E. , … Coppini, R. (2019). Optical investigation of action potential and calcium handling maturation of hiPSC‐cardiomyocytes on biomimetic substrates (Vol. 20) (p. 20). Int. J. Mol. 10.3390/ijms20153799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach, B. , Krogh‐Madsen, T. , Entcheva, E. , & Christini, D. J. (2018). Light‐activated dynamic clamp using iPSC‐derived cardiomyocytes. Biophysical Journal, 115, 2206–2217. 10.1016/j.bpj.2018.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager, P. T. , Gintant, G. , Turner, J. R. , Pettit, S. , & Stockbridge, N. (2014). Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the cardiac safety research consortium. American Heart Journal, 167, 292–300. 10.1016/j.ahj.2013.11.004 [DOI] [PubMed] [Google Scholar]
- Tveito, A. , Jæger, K. H. , Huebsch, N. , Charrez, B. , Edwards, A. G. , Wall, S. , & Healy, K. E. (2018). Inversion and computational maturation of drug response using human stem cell derived cardiomyocytes in microphysiological systems. Scientific Reports, 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woosley, R. , Heise, C. , Gallo, T. , Tate, J. , Woosley, D. , & Ka, R. (n.d.) www.CredibleMeds.org, QTdrugs List.
- Yang, P. C. , Demarco, K. R. , Aghasafari, P. , Jeng, M. T. , Dawson, J. R. D. , Bekker, S. , Noskov, S. Y. , Yarov‐Yarovoy, V. , Vorobyov, I. , & Clancy, C. E. (2020). A computational pipeline to predict cardiotoxicity: From the atom to the rhythm. Circulation Research, 126, 947–964. 10.1161/CIRCRESAHA.119.316404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, L. , Xue, Y. , Jones, M. , Heinbockel, T. , Ying, M. , & Zhan, X. (2018). The effects of quinine on neurophysiological properties of dopaminergic neurons. Neurotoxicity Research, 34, 62–73. 10.1007/s12640-017-9855-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Percentage of current isolated
Table S2: Simulated effect of extracellular calcium concentration on current isolation with the optimized VC protocol.
Figure S1: The effect of experimental artefact on VC data designed to activate sodium channels. The experimental artefact used in this simulation included a voltage offset of −2.8 mV, seal resistance of 1 GΩ, and access resistance of 20 MΩ. The top panel shows the voltage experienced by the cell (dashed blue) compared to the command voltage (black). The voltage offset shifts the membrane voltage negative by 2.8 mV, which has little effect on the current response. The relatively high access resistance is what causes the gradual slope upwards from the starting voltage of −80 mV to the ending voltage of −30 mV. This gradual slope in the membrane voltage leads to a delayed and reduced peak current (bottom) response.
Figure S2: The optimized protocol for INa with Kernik‐Clancy and Paci current response. A, Optimized INa protocol. The total current response (C) and INa contribution (E) for both the Kernik‐Clancy and Paci models. B, The portion of the protocol shaded in A, which includes the maximum INa contribution. The total current response (D) and INa contribution (F) for the portion of the protocol displayed in B.
Figure S3: The optimized protocol for Ito with Kernik‐Clancy and Paci current responses.
Figure S4: The optimized protocol for ICaL with Kernik‐Clancy and Paci current response.
Figure S5: The optimized protocol for IKr with Kernik‐Clancy and Paci current responses.
Figure S6: The optimized protocol for IK1 with Kernik‐Clancy and Paci current responses.
Figure S7: The optimized protocol for IKs with Kernik‐Clancy and Paci current responses.
Figure S8: The optimized protocol for If with Kernik‐Clancy and Paci current responses.
Figure S9: The timepoints of maximum current isolation for the Paci and Kernik‐Clancy models. Five (IK1, Ito, IKr, IKs, and INa) of the seven currents in the Paci model were isolated within 10 ms of when they were isolated in the Kernik‐Clancy model. These time windows are highlighted grey in the top panel. The maximum ICaL isolation in the Paci model occurs far from where the current is maximized in the Kernik‐Clancy model. However, the Paci model had a current isolation within 5% of its maximum during the Kernik‐Clancy window. The timepoints for If also differed between the two models. However, these timepoints are near one another and have similar voltage dynamics, indicating that the Kernik‐Clancy timepoint is likely generalizable for these currents. The remaining panels display the percent contribution of each of the seven currents for the Kernik‐Clancy and Paci models throughout the protocol.
Figure S10: Differences in cell response to cisapride vs. DMSO. The VC protocol (panel 1), Kernik‐Clancy simulated change in membrane current after cisapride treatment (panel 2), average change in experimental cell response from pre‐ to post‐drug application for both DMSO and cisapride (panel 3), and the Kernik‐Clancy IKr response to the VC protocol (panel 4). The blue overlays indicate where there is a significant difference (p < .05) between the average cisapride and DMSO responses. We expected cisapride to strongly and specifically block IKr. The bottom panel shows that most of the areas that are significantly different occur when IKr is present in the Kernik‐Clancy model.
Figure S11: Differences in cell response to verapamil vs. DMSO. This figure shows the VC protocol (panel 1), Kernik‐Clancy simulated change in membrane current after verapamil treatment (panel 2), average change in experimental cell response from pre‐ to post‐drug application for both DMSO and verapamil (panel 3), and the Kernik‐Clancy ICaL response to the VC protocol. The red overlays indicate where there is a significant difference (p < .05) between the average verapamil and DMSO responses. At the concentration tested, we expect verapamil to block ~40% of ICaL and ~20% of IKr. The bottom panel shows that most of the areas that are significantly different occur when the ICaL is present in the Kernik‐Clancy model. There are two brief windows that the functional t‐test identifies after 4,000 ms, that are not likely IKr or ICaL.
Figure S12: Differences in cell response to quinidine vs. DMSO. This figure shows the VC protocol (panel 1), Kernik‐Clancy simulated change in membrane current after quinidine treatment (panel 2), average change in experimental cell response from pre‐ to post‐drug application for both DMSO and quinidine (panel 3), and the Kernik‐Clancy IKr (panel 4), Ito (panel 5), and IKs (panel 6) responses to the VC protocol. The green overlays indicate where there is a significant difference (p < .05) between the average quinidine and DMSO responses. At the concentration tested, we expect quinidine to block ~89% of IKr, ~43% of Ito, and ~27% of IKs. The significance windows overlap very well with the Kernik‐Clancy IKr, Ito, and IKs currents. This is to be expected, as quinidine is known to be a strong and general blocker of potassium currents.
Figure S13: Differences in cell response to quinine vs. DMSO. This figure shows the VC protocol, Kernik‐Clancy simulated change in membrane current after quinine treatment (panel 2), average change in drug response from pre‐ to post‐drug application for both DMSO and quinidine (panel 3), and the Kernik‐Clancy IKr (panel 4), If (panel 5), and ICaL (panel 6) responses to the VC protocol. The blue overlays indicate where there is a significant difference (p < .05) between the average quinine and DMSO responses. At the concentration tested, we expect quinine to block ~72% of IKr and ~29% of ICaL. During the experiments, we noticed a likely block of If with quinine treatment. In Figure 7, we show how we calculate a block of ~32% of If by quinine at this concentration using a HEK‐HCN1 cell line. The significance windows overlap very well with the Kernik‐Clancy IKr, ICaL, and If currents.
Figure S14: Max current vs voltage for HCN1 tail current. The max conductance‐voltage curve was found by stepping to ‐50 mV after the channels had been activated with a depolarizing step. The max tail current values in this plot indicate that most, if not all, funny current channels are open when stepping to voltages below ‐100 mV.
Data Availability Statement
All code has been made publicly available on GitHub at: https://github.com/Christini-Lab/vc-optimization-cardiotoxicity. All the data presented in this manuscript has been made publicly available through Cornell eCommons.
Data that support the findings of this study are available through eCommons at https://doi.org/10.7298/c883-s773.