

Abstract
Objective:
To determine the sensitivity and specificity of α-synuclein seed amplification assay (αSyn-SAA) in antemortem and postmortem CSF of autopsy-confirmed patients with different distributions of pathological αSyn, co-pathologies, and clinical diagnoses.
Methods:
αSyn-SAA was used to test antemortem CSF samples from 119 subjects with a variety of clinical syndromes and standardized neuropathological examinations from OHSU and UCSD (56 additional postmortem CSF samples available). The αSyn-SAA was also applied to frontal cortex and amygdala homogenates. Sensitivity and specificity were compared across distributions of αSyn-pathology. Clinical data and co-pathologies were compared across αSyn-SAA positive and negative groups.
Results:
Fifty-three individuals without and 66 with αSyn-pathology (neocortical (n=38), limbic (n=7), and amygdala-predominant (n=21)) were included. There was a sensitivity of 97.8% and specificity of 98.1% of the αSyn-SAA to identify patients with limbic/neocortical pathology from antemortem CSF. Sensitivity to detect amygdala-predominant pathology was only 14.3%. Postmortem CSF and brain tissue αSyn-SAA analyses also showed higher assay positivity in samples from limbic/neocortical cases.
Interpretation:
CSF αSyn-SAA reliably identifies αSyn seeds in patients with diffuse αSyn-pathology in the context of co-pathology and non-LBD diagnoses. The analysis of brain homogenates suggests that pathological αSyn in amygdala might differ from pathological αSyn in frontal cortex. αSyn-SAA might facilitate the differential diagnosis of dementias with mixed pathologies.
Introduction
Aggregated α-synuclein (αSyn) is the main component of cytoplasmic inclusions called Lewy bodies (LB) and Lewy neurites, which are the defining pathological features of Lewy body diseases (LBD), including Parkinson’s disease (PD) and dementia with Lewy bodies (DLB)1, 2. In addition, αSyn-laden LBs are found in the brains of as many as 50–60% of sporadic Alzheimer’s disease (AD) cases3–7, 96% in familial PSEN1 cases8, and in 10–20% of normal elders9, 10. AD cases with αSyn-pathology (sometimes called AD Lewy Body variant, AD-LBV) present relevant clinical differences compared to AD without αSyn-pathology, such as lower age of onset, lower age of death, more severe delusions, hallucinations, aberrant motor function, and sleep disorders7, 11. Similarly, co-incidental AD pathology in DLB may lower the likelihood of patients manifesting certain core features like visual hallucinations12. αSyn-pathology in AD cases affects amygdala, limbic and can affect neocortical areas with sparing of the brainstem and recent neuropathological studies in AD cases with amygdala-predominant αSyn-pathology found different αSyn truncations and modifications compared to limbic and neocortical αSyn-pathology found in PD or DLB13–15.
To date, neuropathological assessment at autopsy remains the gold standard to diagnose LBDs and in vivo αSyn biomarkers have been an unmet need. Recently, αSyn Seed Amplification Assays (αSyn-SAAs) (also known as protein misfolding cyclic amplification (PMCA) and real time quaking induced conversion (RT-QuIC)), have been adapted to detect misfolded αSyn aggregates (αSyn seeds) in CSF and peripheral tissues with remarkable diagnostic accuracy16–21. αSyn-SAA in CSF of clinically and in some cases pathologically confirmed PD and DLB cases has shown impressive results, with several independent groups reporting sensitivities and specificities near or above 90%16–18, 22–29. However, αSyn-SAA performance in neuropathologically validated cohorts with varying distribution of αSyn pathologies, co-pathologies, and non-LBD diagnoses has not been evaluated. Thus, it remains unknown if different types of αSyn-pathology distributions produce differences in seeding activity. A few studies have reported detection of αSyn seeds in CSF from clinically diagnosed AD patients (5/14 or 36% in one report17 and 0/16 in another18) and from patients clinically diagnosed with AD who were pathologically confirmed to have DLB (11/17 or 65%) or incidental Lewy bodies (2/13 or 15%)16. Despite the low number of cases, these results suggest that current assays may have different sensitivities, which may depend on αSyn-pathology distribution, co-pathologies, and/or pathological αSyn species.
In this multi-center study, we evaluated the capability of αSyn-SAA to detect αSyn seeds in antemortem and postmortem CSF samples as well as brain tissue of patients who underwent autopsy and neuropathological analyses. We compared the αSyn-SAA results to clinical and neuropathological data to determine sensitivity, specificity, clinical, and pathological correlations of this assay across different distributions of αSyn-pathology in the context of co-pathology and non-LBD diagnoses.
Methods
Patient Selection
eIRB 725 of Oregon Health and Science University ADRC gave ethical approval for this work. IRB 170957 of University of California San Diego ADRC gave ethical approval for this work. Informed consent was obtained from each subject for the retrieval of biological samples.
Participants in brain aging studies from the Oregon Alzheimer’s Disease Center (OADC) (n=57) and University of California San Diego Shiley-Marcos Alzheimer’s Disease Research Center (UCSD-ADRC) (n=62) who had 1) CSF collection during life, and 2) subsequent brain autopsy (n=119) were included in the study. All subjects had an annual battery of clinical, neuropsychologic, and other cognitive assessments, as described by the National Alzheimer’s Coordinating Center (NACC)30, including Mini-Mental State exam (MMSE), and Unified Parkinson’s Disease Rating Scale Part III (UPDRS). Blood was drawn for the determination of APOE genotype. Clinical diagnoses, assigned at the time of CSF collection, included AD (n=75), DLB (n=9), PD (n=4), mild cognitive impairment (MCI, n=11), other dementia (n=13, including frontotemporal dementia (n=10), mixed dementia (n=1), and “other dementia” (n=2)), and cognitively normal controls (n=7). Clinical diagnoses were assigned by a multidisciplinary consensus conference at each site. Pathologically, the cohort included patients with AD pathology (n=43), AD with αSyn-pathology (n=59), αSyn-pathology in isolation (n=3), progressive supranuclear palsy (n=2), corticobasal degeneration (n=2), FTLD TDP-43 (n=2), vascular disease in isolation (n=2), normal subjects (n=2), and patients with a mix of AD and other tauopathies (n=4) (Table 1). Cases were grouped by αSyn-pathology distribution as below. Patient-level information can be found in Supplemental Table 1.
Table 1.
αS-SAA positivity as a function of pathology diagnosis.
| αS-SAA positivity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pathology | n | αSyn Pathology | Antemortem | Postmortem | Frontal | Amygdala | ||||
| AD | 26 | Negative | 4% | 1/26 | 20% | 3/14 | - | - | - | - |
| AD + VD | 5 | Negative | 0% | 0/5 | 0% | 0/3 | - | - | - | - |
| AD + VD + HS | 2 | Negative | 0% | 0/2 | 0% | 0/2 | - | - | - | - |
| AD + VD + AA | 1 | Negative | 0% | 0/1 | - | - | - | - | - | - |
| AD + AA | 3 | Negative | 0% | 0/3 | 0% | 0/1 | 0% | 0/1 | 0% | 0/1 |
| AD + HS | 1 | Negative | 0% | 0/1 | 0% | 0/1 | - | - | - | - |
| AD + HS + LMN Encephalitis | 1 | Negative | 0% | 0/1 | - | - | - | - | - | - |
| AD + Pick’s disease | 1 | Negative | 0% | 0/1 | - | - | - | - | - | - |
| AD + PART | 1 | Negative | 0% | 0/1 | - | - | - | - | - | - |
| AD + PART + METS | 1 | Negative | 0% | 0/1 | 0% | 0/1 | - | - | - | - |
| AD + VD + AA + ARTAG | 1 | Negative | 0% | 0/1 | 0% | 0/1 | 0% | 0/1 | 0% | 0/1 |
| CBD + VD + AA | 1 | Negative | 0% | 0/1 | - | - | 100%* | 1/1 | 0% | 0/1 |
| PSP | 2 | Negative | 0% | 0/2 | - | - | - | - | - | - |
| CBD | 1 | Negative | 0% | 0/1 | 0% | 0/1 | - | - | - | - |
| FTLD-TDP43 | 1 | Negative | 0% | 0/1 | 0% | 0/1 | - | - | - | - |
| FTLD-Tau | 1 | Negative | 0% | 0/1 | - | - | - | - | - | - |
| VD | 2 | Negative | 0% | 0/2 | - | - | 0% | 0/1 | 0% | 0/1 |
| Normal | 2 | Negative | 0% | 0/2 | 0% | 0/1 | - | - | - | - |
| AD + αSyn-Path | 26 | Neocortical/Limbic | 100% | 26/26 | 91% | 10/11 | 100% | 4/4 | 100% | 4/4 |
| AD + αSyn-Path | 7 | Amygdala-predominant | 14% | 1/7 | - | - | 50% | 1/2 | 100% | 1/1 |
| AD + VD + αSyn-Path | 6 | Neocortical/Limbic | 83% | 5/6 | 75% | 3/4 | 100% | 1/1 | 100% | 1/1 |
| AD + VD + αSyn-Path | 4 | Amygdala-predominant | 0% | 0/4 | 100% | 4/4 | 100%* | 1/1 | 100% | 1/1 |
| AD + AA + αSyn-Path | 2 | Neocortical | 100% | 2/2 | 100% | 2/2 | 100% | 1/1 | 100% | 1/1 |
| AD + AA + αSyn-Path | 3 | Amygdala-predominant | 0% | 0/3 | 50% | 1/2 | 0% | 0/2 | 50% | 1/2 |
| AD + AA + FTLD-TDP43 + αSyn-Path | 2 | Neocortical/Limbic | 100% | 2/2 | 100% | 1/1 | 100% | 1/1 | 100% | 1/1 |
| AD + AA + FTLD-TDP43 + αSyn-Path | 3 | Amygdala-predominant | 0% | 0/3 | 33% | 1/3 | 0% | 0/3 | 33% | 1/3 |
| AD + HS + αSyn-Path | 4 | Neocortical/Limbic | 100% | 4/4 | 100% | 1/1 | - | - | - | - |
| AD + Pick’s disease + αSyn-Path | 1 | Amygdala-predominant | 0% | 0/1 | - | - | - | - | - | - |
| AD + ARTAG + αSyn-Path | 1 | Neocortical | 100% | 1/1 | 100% | 1/1 | 100% | 1/1 | 100% | 1/1 |
| AD + VD + PSP + αSyn-Path | 1 | Amygdala-predominant | 100% | 1/1 | - | - | 0% | 0/1 | 100%* | 1/1 |
| AD + Infarcts + αSyn-Path | 1 | Amygdala-predominant | 100% | 1/1 | - | - | - | - | - | - |
| PSP + CBD + HS + αSyn-Path | 1 | Neocortical | 100% | 1/1 | - | - | - | - | - | - |
| FTLD-TDP43 + αSyn-Path | 1 | Amygdala-predominant | 0% | 0/1 | 0% | 0/1 | 0% | 0/1 | 0% | 0/1 |
| αSyn-Path | 3 | Neocortical | 100% | 3/3 | - | - | - | - | - | - |
AD:Alzheimer’s disease, VD: Vascular disease, HS: hippocampal sclerosis, AA: Includes amyloid angyopathy, leptomeningial congophilic angiopathy, and lepto/parenchymal congophilic angiopathy, LMN Encephalitis: Limbic Microglial Nodular Encephalitis , PART: Primary-Age Related Tauopathy, METS: Micrometastases, ARTAG: Aging-related tau astrogliopathy, CBD: corticobasal degeneration, PSP: progressive supranuclear palsy, FTLD-TDP43: Frontotemporal lobe degeneration with TDP43 pathology, and αSyn-Path: Includes neocortical, limbic, and amygdala predominant αSyn pathology.
2/3 wells were positive.
CSF Analysis
CSF was collected for all 119 cases by lumbar puncture in the morning fasting condition according to a standardized protocol31. A subset of patients (n=56) had additional CSF samples obtained at the time of brain removal. CSF specimens were divided into 0.5 ml aliquots and stored at -80°C. Antemortem CSF collection occurred 1–15 years prior to autopsy (17.6% in 0–2yr, 46.3% in 2–5yr, 18.5% in 5–8yr, 6.7% in 8–10yr, 10.9% in 10–16yr). Antemortem CSF was analyzed for Aβ40, Aβ42, t-tau and p-tau (Lumipulse, Fujirebio at both sites).
CSF samples were initially analyzed by the endpoint qualitative version of the αSyn-SAA that has been validated for clinical use under CLIA/CAP certifications (clinical assay, SYNTap™). Each sample was analyzed in triplicate (40µL CSF per well) in a 96-well plate (COSTAR, cat# 3603) with a final volume reaction of 200µL). The reaction mixture consisted of 0.3mg/mL rec-αSyn (Amprion, cat# S2020) in 100mM PIPES pH 6.50, 500mM NaCl, 10µM ThT, and a 2.5mm borosilicate glass bead per well. Plates were sealed using an Optical Adhesive Film (ThermoFisher, cat# 4311971) and shaken at 800rpm with orbital shaking for 1min every 29min of quiescent incubation in a TIMIX 5 shaker (Edmund Buehler) placed in an incubator set to 37°C. Bottom fluorescence readings at 490nm were performed using a BMG FLUOstar Omega. This clinical version of the assay was performed according to standard operational procedures in agreement with CLIA regulation. CSF samples were deemed “Detected” or “Not Detected” based on a preestablished threshold for the median maximum fluorescence of the triplicate. The research and development (R&D) kinetic αSyn-SAA was utilized to analyze CSF samples and brain tissues. The methods of the kinetic αSyn-SAA have been reported in detail elsewhere22, 23. Briefly, CSF samples and brain homogenates (BHs) were evaluated in triplicates (40µL/well) in a 96-well plate (COSTAR 96, cat# 3916), in a reaction mix consisting of 0.3mg/ml rec-αSyn (Amprion, cat# S2021), 100mM PIPES pH 6.50 (Sigma, cat# 80635), 500mM NaCl (Lonza, cat# 51202), 10µM ThT (Sigma, cat# T3516), and a 3/32-inch BSA-blocked Si3N4 bead (Tsubaki Nakashima). This assay was performed in a BMG FLUOstar Omega shaker/reader with orbital shaking at 800rpm for 1min and 29min of quiescent incubation at 37°C. Fluorescence at 490nm was measured every 30min for accurate estimation of kinetic parameters. The assay outcomes of the R&D kinetic assay are positive, inconclusive, or negative, based on a probabilistic algorithm that uses maximum fluorescence and kinetic parameters22. Maximum fluorescence (Fmax, RFU) was the highest fluorescence reading within the length of the assay. A 4-parameter fit (Mars, BMG) was fit to estimate the slope (RFU/h) and the time to reach 50% of the Fmax (T50, h) of each replicate/well. The time to threshold (TTT, h) was determined with a user defined formula (Mars, BMG); threshold was set to 5,000 RFU. Scientists performing the assay were blinded to the clinical or pathological diagnoses associated with the samples.
Brain Tissue Analysis
In a subset of patients (n=22), 500mg samples of frozen brain tissue from the middle frontal cortex and amygdala were provided for αSyn-SAA. Cases included those without αSyn-pathology (n=4), amygdala-predominant αSyn-pathology (n=10), and limbic/neocortical αSyn-pathology (n=8). All frozen samples were provided from the UCSD-ADRC.
Frontal cortex and amygdala samples were homogenized to 10% w/v in 1XPBS (Cytiva, cat# SH30256.02) with cOmplete Mini EDTA-free protease inhibitor cocktail (Roche, cat# 11836170001). Approximately 100µg of brain sample was homogenized in 1.5mL tubes preloaded with 1mm zirconium beads (cat# 11079110zx) in a MP FastPrep 24 homogenizer. Two rounds of homogenization were performed for all samples (15s at 4m/s and 30s at 6m/s). If additional homogenization was needed, samples were chilled on ice for 5min in between additional homogenization rounds at 6m/s for 30s. BHs were centrifuged at 800xg for 1 minute to remove cellular debris. Supernatants were collected, vortexed, aliquoted, and stored at -80°C until αSyn-SAA analysis. BH aliquots were 10-fold serially diluted in synthetic CSF (Amprion, cat# S2022) up to 10-9 and analyzed in triplicates. Results for 10-8 dilution are shown to avoid negativity by over-dilution.
Neuropathological Assessments
Neuropathological assessments were performed in a standardized manner with various pathologies assessed using hematoxylin and eosin staining and immunohistochemistry directed against tau, amyloid-β, α-synuclein, and TDP-43 species as appropriate and pathological diagnoses were assigned by expert neuropathologists32–35. MSA cases were excluded from this study given the known altered kinetics on αSyn-SAA assays compared to PD and DLB cases36. Alzheimer’s disease neuropathological change was assigned according to NACC guidelines after Braak tau stage, CERAD stage, and Thal phase was determined33, 37. Distribution of LRP was determined via α-synuclein immunohistochemistry staining (OADC: αSyn MJFR1, Abcam; UCSD-ADRC: pSer129 αSyn 81A, Biolegend Laboratories) using slices from pons and/or midbrain, hippocampus, amygdala, and neocortical areas including temporal cortex and/or middle frontal cortex and the following staging definitions were applied: Neocortical: midbrain+ pons+ hippocampus+ amygdala+ neocortex+; Limbic: midbrain+ pons+ hippocampus+ amygdala+ neocortex-; Amygdala-predominant: midbrain- pons- hippocampus+/- amygdala+ neocortex-38.
Statistical Analysis
Clinical and pathological differences between the OADC and UCSD-ADRC cohorts were assessed to determine the necessity for stratification by site. All DLB and PD patients were from UCSD. Sensitivity, specificity, and predictive values were calculated via chi-squared test with 95% confidence intervals calculated using the hybrid Wilson-Brown method. Differences in kinetic parameters were analyzed by one-way ANOVA with Tukey’s multiple comparisons test or unpaired t-test. Prior to testing group differences, all outcome variables were assessed for normality. For normally distributed continuous variables, we used the General Linear Model (GLM) to test whether there were group differences in the outcome variables (age at death, onset of cognitive symptoms, and MMSE decline rate). For non-normally distributed continuous variables (UPDRS at lumbar puncture, MMSE at lumbar puncture, UPDRS at most recent visit, MMSE at most recent visit, CDR at most recent visit, lumbar puncture to autopsy interval, CSF Aβ40, Aβ42, t-tau, and p-tau, disease duration, postmortem interval), we used a Kruskal-Wallis test (more than two groups) or a Wilcoxon rank-sum test (two groups) to test for group differences. Post hoc pairwise comparisons were tested using the Dwass, Steel, Critchlow-Fligner Method. We used chi-square tests or fisher’s exact tests to test for group differences when outcome variables were categorical (biological sex, early-onset status, neuropathology diagnosis, clinical diagnosis, NACC variables: Thal phase for amyloid plaques, Braak stage for neurofibrillary degeneration, density of neocortical neuritic plaques, NIA-AA Alzheimer’s disease neuropathologic change (ADNC), density of diffuse plaques, cerebral amyloid angiopathy, arteriosclerosis, APOE status. For the following variables, we had data from both the OADC and UCSD-ADRC cohorts: onset of cognitive symptoms, disease duration, age at death, rate of MMSE decline, MMSE at lumbar puncture, most recent MMSE score, interval between lumbar puncture and autopsy, postmortem interval, biological sex, clinical diagnosis, Thal phase, Braak tau stage, Cerad stage, ADNC, APOE genotype, CSF Aβ40, Aβ42, t-tau, and p-tau. UPDRS score at lumbar puncture was only collected at UCSD. Statistical significance was set at p < 0.05.
Results
Neuropathological αSyn analysis and comparison
The neuropathological analysis of the 119 subjects revealed αSyn-pathology in the brains of 66 (55%) cases. Of the 66 patients with αSyn-pathology, 38 showed neocortical stage αSyn-pathology, 7 showed limbic stage αSyn-pathology, and 21 showed amygdala-predominant αSyn-pathology. Rates of AD pathology was high across the cohort. 40/53 (75%) of cases without αSyn-pathology had intermediate or high degrees of AD neuropathological change, as did 19/21 (90%) cases with amygdala predominant αSyn-pathology and 39/45 (87%) cases limbic or neocortical disease (Table 1). These rates were not statistically significant across the αSyn driven categories (χ2=3.3, p=0.2). The cases that did not have significant AD neuropathological change composed a variety of tauopathies, TDP-43-opathies, and vascular disease (Table 1, Supplemental Table 1). No significant difference in Aβ40, Aβ42, Aβ42/40 ratio, t-tau, and p-tau in antemortem CSF were observed between the αSyn-pathology groups within institution (Supplemental Table 2).
Using a Kruskal-Wallis test, we compared patients within αSyn distribution groups (none, amygdala-predominant, limbic/neocortical) on several standardized clinical and pathological variables to determine if there were important group differences. UPDRS part III scores were significantly different between αSyn groups at lumbar puncture (X2=21.59, p<0.0001, Supplemental Table 2) and at last visit prior to death (X2=14.93, p=0.0006, Supplemental Table 2). Post-hoc analyses showed that the limbic/neocortical group had higher UPDRS part III scores at lumbar puncture than those without αSyn-pathology and the amygdala-predominant αSyn group (Wilcoxon z=-3.71, p=0.0006 and Wilcoxon z=-3.44, p=0.002 respectively). The limbic/neocortical group also had higher UPDRS III scores at last visit prior to death compared to the amygdala-predominant group (Wilcoxon z=-3.70, p=0.0007). The majority of patients diagnosed with DLB (8/9) and PD (4/4) showed limbic/neocortical αSyn while 16/21 patients with amygdala predominant αSyn had a clinical diagnosis of AD (X2=28, p=0.002, Supplemental Table 2, Supplemental Table 1). Lastly, male sex was over-represented across the three αSyn distribution groups (X2=6.94, p=0.03, Supplemental Table 2).
Sensitivity and specificity of the αSyn-SAA using CSF samples
A total of 119 antemortem CSF samples were analyzed with the clinical αSyn-SAA. All but 1 of the 53 patients without αSyn-pathology were negative by the clinical αSyn-SAA and thus, the specificity for the clinical assay in this cohort was 98.1% (95% CI 90.1% to 99.9%) (Table 2). Of the 66 individuals with αSyn-pathology, 47 were found positive by the clinical αSyn-SAA; Neuropathological analysis is the gold standard to which αSyn-SAA results were compared to. Thus, samples with positive αSyn-SAA results from patients with pathological αSyn found at autopsy were called true-positives, while samples with negative αSyn-SAA results from patients without αSyn-pathology were called true-negatives. The overall sensitivity of the assay to detect αSyn-pathology in any form was 71.2% (95% CI 59.4% to 80.7%). However, significant differences were observed when stratifying sensitivity analysis by pathological αSyn distribution. αSyn-SAA had sensitivity of 97.8% (95% CI 88.4% to 99.9%) in detecting αSyn seeds in limbic/neocortical pathology, but only 14.3% (95% CI 5.0% to 34.6%) in detecting amygdala-predominant αSyn-pathology (Table 2).
Table 2.
Sensitivity, specificity, and predictive values for antemortem and postmortem CSF αSyn-SAA against αSyn-pathology.
| ANTEMORTEM (n=119) | |
|---|---|
|
| |
| Variable | Value, % (95% CI) |
| Sensitivity | 71.2 (59.4–80.7) |
| Limbic/Neocortical | 97.8 (88.4–99.9) |
| Amygdala | 14.3 (5.0–34.6) |
|
| |
| Specificity | 98.1 (90.1–99.9) |
|
| |
| Positive predictive value | 97.9 (89.1–99.9) |
|
| |
| Negative predictive value | 73.2 (62.0–82.2) |
|
| |
| POSTMORTEM (n=56) | |
|
| |
| Variable | Value, % (95% CI) |
|
| |
| Sensitivity | 80.0 (62.7–90.5) |
| Limbic/Neocortical | 90.0 (69.9–98.2) |
| Amygdala | 60.0 (31.3–83.2) |
|
| |
| Specificity | 88.5 (71.0–96.0) |
|
| |
| Positive predictive value | 88.9 (71.9–96.2) |
|
| |
| Negative predictive value | 79.3 (61.6–90.2) |
Fifty six of the 119 patients had postmortem CSF for clinical αSyn-SAA analysis, 26 had no αSyn-pathology and 30 had αSyn-pathology at autopsy (limbic/neocortical n=20, amygdala-predominant n=10). Of the 26 patients without αSyn-pathology, 23 were found negative by the αSyn-SAA, for an estimated specificity of 88.5% (95% CI 71.0% to 96.0%) (Supplemental Table 2). Of the 30 individuals with αSyn-pathology, 24 were found positive by αSyn-SAA; thus, the sensitivity for the combined cohort was 80% (95% CI 62.7% to 90.5%). Similarly, when stratified by αSyn distribution, the clinical αSyn-SAA in postmortem CSF had sensitivity of 90% (95% CI 69.9% to 98.2%) to detect individuals with limbic or neocortical αSyn, but sensitivity of only 60% (95% CI 31.3% to 83.2%), to detect amygdala-predominant αSyn (Table 2). Despite a decrease in sensitivity of the αSyn-SAA between antemortem and postmortem CSF samples, there was no significant difference in postmortem interval between patients that tested positive or negative using postmortem CSF in both limbic/neocortical (p=0.45) and amygdala-predominant groups (p=0.12).
Of the 56 individuals with both antemortem and postmortem CSF, 46 (82.1%) showed concordant αSyn-SAA results, 9 (16.1%) changed from negative results antemortem to positive results on the postmortem assay, and 1 (1.8%) changed from positive to negative. Interestingly, changes between antemortem and postmortem CSF αSyn-SAA results was significantly higher in amygdala-predominant cases (6/10, all negative to positive) than in limbic/neocortical cases (1/20) (X2=28.49, p<0.0001).
116 antemortem (51 no αSyn-pathology, 44 limbic/neocortical αSyn-pathology, 21 amygdala-predominant αSyn-pathology) and 33 postmortem (11 no αSyn-pathology, 15 limbic/neocortical αSyn-pathology, 7 amygdala-predominant αSyn-pathology) CSF samples were also analyzed by a research kinetic αSyn-SAA to accurately estimate kinetic parameters and further characterize seeding activity in these samples. Fewer samples were run using this assay because some samples had been exhausted in the previous analysis. The kinetic assay provides a diagnostic output based on a probabilistic algorithm, which deems samples as “negative”, “positive”, or “inconclusive. The kinetic αSyn-SAA “negative” and “positive” determinations were consistent with the CLIA-regulated version of the assay for the antemortem and postmortem analyzed in parallel (data not shown). Fmax was analyzed between groups, with no αSyn-pathology (p<0.0001, q=20.42, DF=113) and amygdala-predominant αSyn-pathology (p<0.0001, q=14.07, DF=113) groups having significantly lower Fmax than individuals with neocortical or limbic αSyn-pathology on antemortem CSF, most likely caused by the abundance of “negative” samples (Figure 1A). Representative raw kinetic graphs are shown in Figure 1B. There were kinetic differences in the seed amplification of amygdala-predominant cases compared to neocortical/limbic cases (TTT (p=0.0007) and T50 (p=0.0002)) where amygdala-predominant cases had slower seeding activity. However, the small number of amygdala-predominant αSyn-SAA positive cases (n=3) precludes reliable conclusions. There were no significant differences in kinetic parameters between αSyn-pathology groups using postmortem CSF in the kinetic αSyn-SAA (data not shown).
Figure 1. Kinetic parameters of Research SAA stratified by alpha-synuclein distribution.
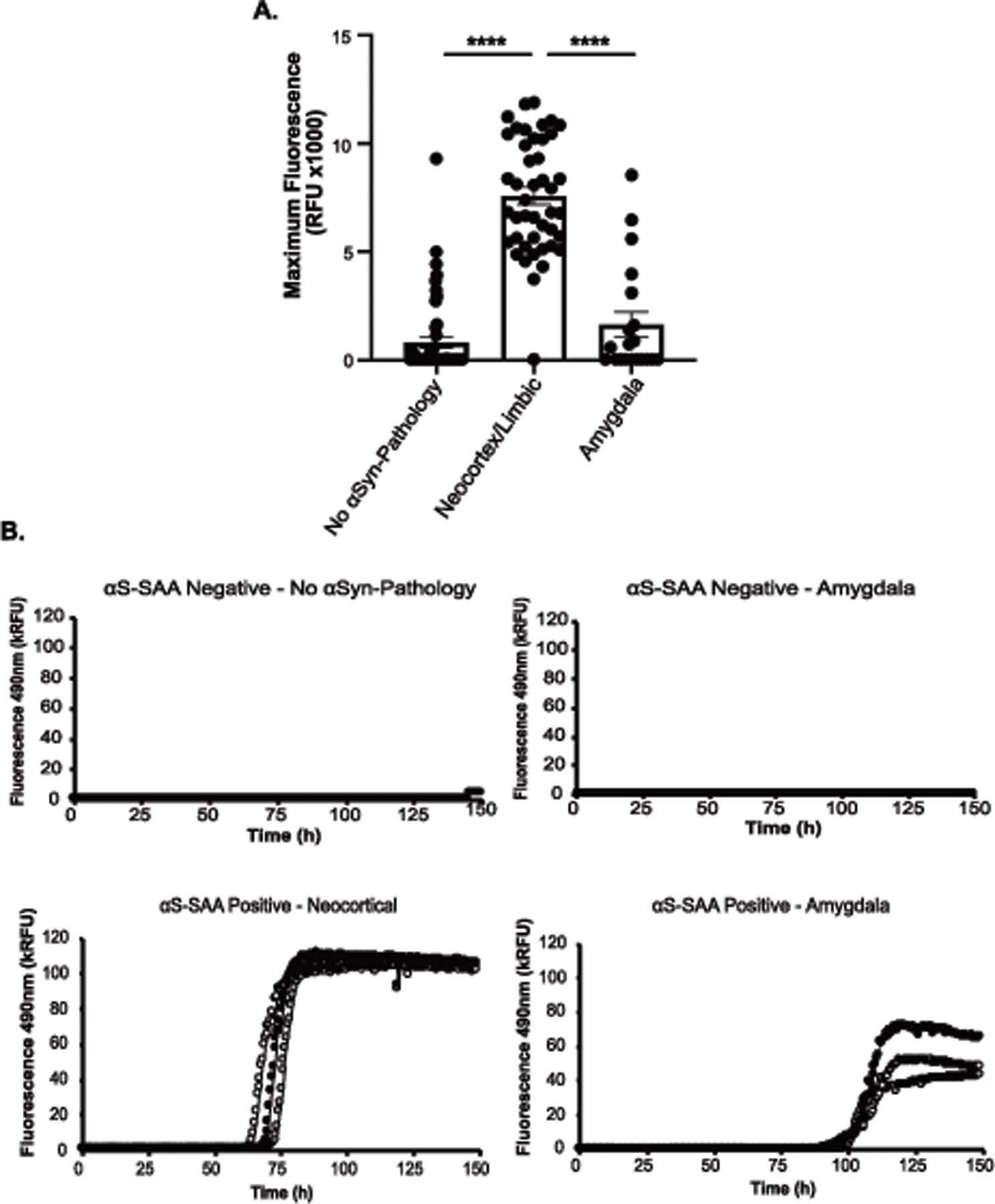
A) Maximum Fluorescence Signal from R/D αSyn-SAA using antemortem CSF between no αSyn-pathology (n=51), Neocortex/Limbic (n=44), and Amygdala-predominant (n=21) groups. B) Representative figures of raw kinetic data from the Research SAA using antemortem CSF. Included are “negative” samples that are from no αSyn-pathology and amygdala-predominant individuals, and “positive” samples that are from neocortical and amygdala-predominant individuals. Statistical analysis using one-way ANOVA with Tukey’s multiple comparisons post hoc (A). Error bars represent Standard Error of the Mean (SEM).
To investigate the potential effects of AD co-pathology on likelihood of αSyn-SAA seeding activity, we compared antemortem CSF αSyn-SAA results to CERAD scores (C0/C1 v C2/C3), Braak tau stage (B0/B1 v B2/B3) and Amyloid-β Thal Phase (A0/A1 v A2/A3) for cases with pathological αSyn (Figure 2). There were no significant associations between the likelihood of αSyn-SAA positivity and CERAD score (p=0.7), Thal phase (p>0.9), and by Braak tau stage (p>0.9) (Figure 2). We also evaluated the effect of proteins associated to AD biomarkers in in CSF as they could interfere with the amplification process in the assay. No significant differences were found in levels of Aβ40, Aβ42, Aβ42/40 ratio, t-tau, and p-tau, between limbic/neocortical cases and amygdala-predominant cases as a function of αSyn-SAA result within institution (data not shown). Overall, αSyn-SAA positivity or lack thereof is not associated with the presence of AD co-pathology or commonly used AD CSF biomarkers.
Figure 2. Differences in neuropathology scores between synuclein-pathology groups as a function of SAA result.
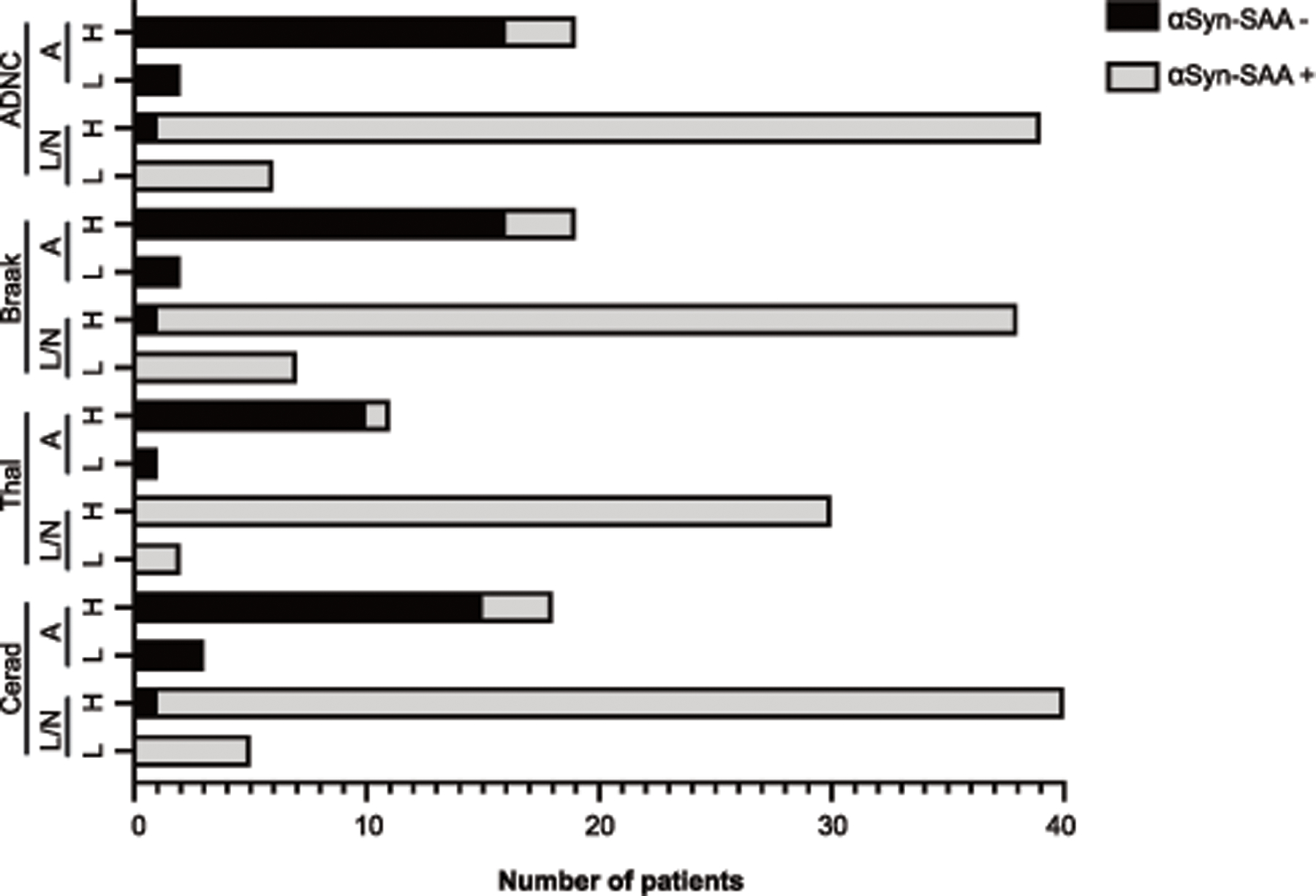
Bars represent the distribution of SAA positive or SAA negative within high (“H”) or low (“L”) categorization of ADNC, Braak, Thal, and Cerad neuropathological staging. Patients are further classified by limbic/neocortical (“L/N”) or amygdala-predominant (“A”) groups. Statistical analysis using Fisher’s exact test within synuclein-pathology group.
Comparisons of subjects with positive vs negative CSF αSyn-SAA results
UPDRS part III scores at the time of lumbar puncture were significantly lower in the antemortem false negative group compared to the true positive group (Z=-3.12, p=0.002), considering pathological analysis as gold standard. The interval between lumbar puncture and death was significantly different between true positive and false negative groups, with the false negative group having on average a longer interval than the true positive group (Z=2.09, p=0.04, Figure 3A). The two groups also differed in the distribution of αSyn-pathology (X2=48.69, p<0.0001); 94.7% of the false negatives fell into the amygdala-predominant group, while 93.6% of the true positives fell into the limbic/neocortical group. Similarly, in postmortem CSF, 66.7% of false negatives were in the amygdala-predominant group and 75% of true positives were in the limbic/neocortical αSyn group (X2=3.75, p=0.05) (Figure 3B).
Figure 3. Clinical and pathological differences between True Positive and False Negative.
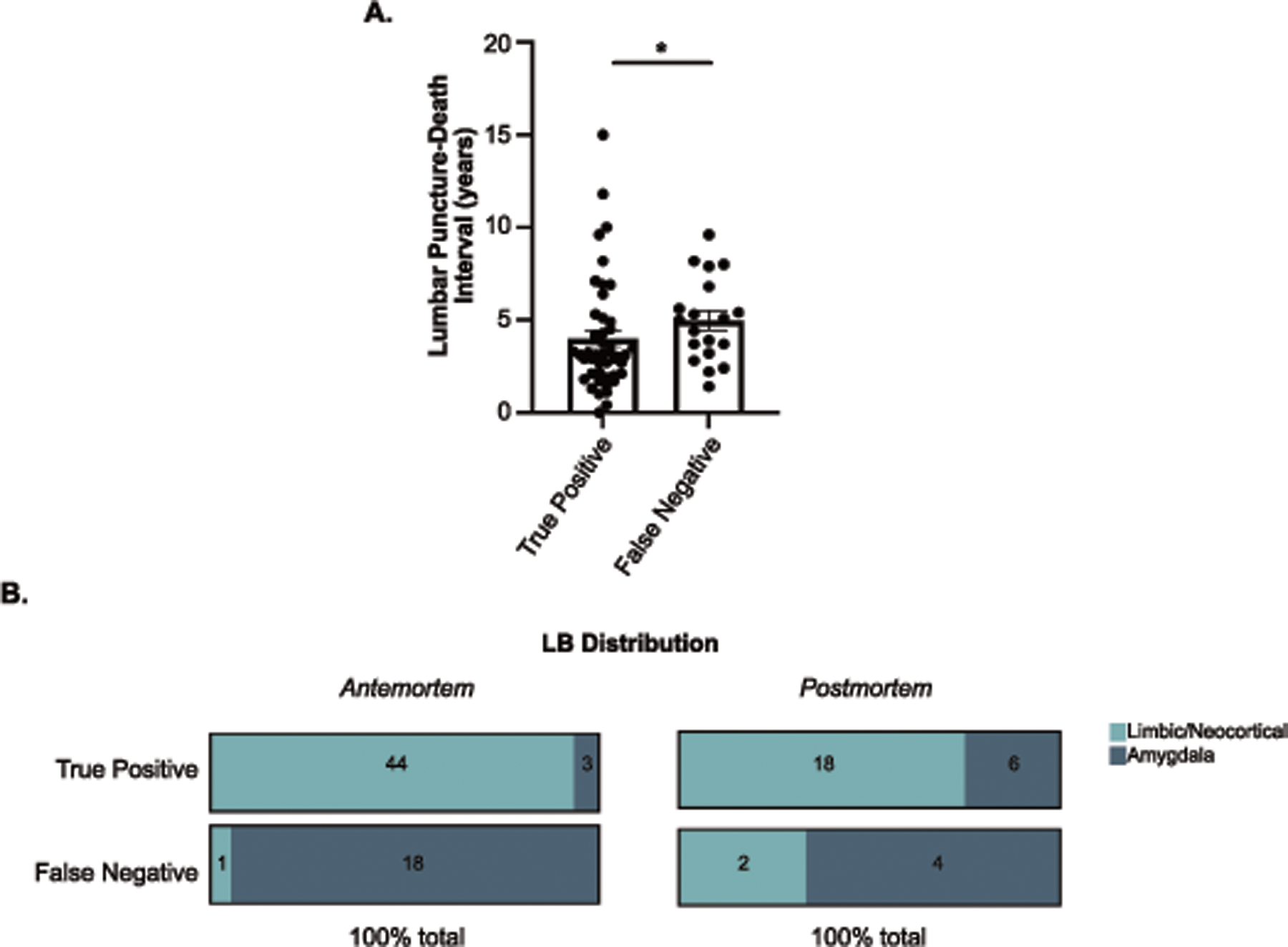
A) Interval in years from lumbar puncture to death between True Positives (n=47) and False Negative (n=19) groups. B) Distribution of Neocortex/Limbic and Amygdala-predominant LRP in True Positive and False Negative groups for antemortem and postmortem CSF analysis. Number of patients in each category is indicated on the bar. Statistical analysis using Wilcoxon rank-sum test with post hoc pairwise comparisons from Dwass, Steel, Critchlow-Fligner method (A) or chi-square (B). Error bars represent Standard Error of the Mean (SEM).
Clinical significance of incidental synuclein pathology
Lastly, we explored how clinical diagnosis related to clinical αSyn-SAA performance, in order to better understand whether subtle clinical predictors were present among patients without a diagnosis of a synucleinopathy whose antemortem CSF tested positive by αSyn-SAA. In this analysis, we examined all patients who were clinically diagnosed with AD, without concomitant PD or DLB, and whose antemortem CSF αSyn-SAA results were positive versus negative. There was a significant difference in patient biological sex, where αSyn-SAA-positive patients had a significantly greater proportion of males (23/29, 79.3%) compared to αSyn-SAA-negative patients (25/46, 54.3%) (X2=7.84, p=0.005). Clinically diagnosed AD patients with positive αSyn-SAA CSF had higher UPDRS part III scores (6.71 +/- 8.6) than those with negative αSyn-SAA CSF (1.82 +/- 4.92) at most recent visit prior to death (Z=2.53, p=0.01).
Detection of αSyn seeds from frontal cortex and amygdala brain samples
We next analyzed a subset of patients (n=22) from the UCSD-ADRC cohort who had frozen brain tissue available for analysis, including 4 no αSyn-pathology, 10 amygdala-predominant, and 8 limbic/neocortical cases. In both brain regions, the 4 patients without αSyn-pathology were negative by the αSyn-SAA, consistent with the results for antemortem CSF in both kinetic and clinical assays (Table 3). In agreement with the high sensitivity in CSF for limbic/neocortical cases, seeding activity was detected in both frontal cortex and amygdala of all 8 analyzed cases. However, there was a significant decrease in seeding activity in both frontal cortex and amygdala of the amygdala-predominant cases. Of the 10 amygdala-predominant cases, 4 cases showed no seeding activity in both frontal cortex and amygdala. There were 2 cases with seeding activity detected in the amygdala, with one of them showing 2/3 wells positive in frontal cortex.
Table 3.
Patient categorization from brain homogenate samples.
| Case Information | Brain Tissue | AM CSF |
PM CSF | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Sex | Age at onset | Age at death | Primary Pathology | Thal Phase | CERAD Stage | Braak Stage | ADNC | LRP Classification | Frontal Cortex | Amygdala | LP to death (y) | Result | Result |
| 1 | Male | NA | 84 | Normal | A0 | C0 | I | Not | None | − | − | 1.8 | − | |
| 2 | Male | 65 | 71 | CBD | A3 | C1 | I | Low | None | 2/3 | − | 2.9 | − | |
| 3 | Female | 65 | 76 | AD | A3 | C3 | VI | High | None | − | − | 4.7 | − | − |
| 4 | Female | 58 | 66 | AD | A3 | C3 | VI | High | None | − | − | 3.0 | − | − |
|
| ||||||||||||||
| 5 | Female | 72 | 84 | AD | A3 | C2 | VI | High | Amygdala | − | 2.7 | − | ||
| 6 | Female | 83 | 90 | AD + PSP | A3 | C2 | VI | High | Amygdala | − | 2/3 | 0.9 | + | |
| 7 | Female | 84 | 91 | AD | A3 | C3 | VI | High | Amygdala | 2/3 | + | 4.6 | − | |
| 8 | Male | 75 | 84 | AD | A3 | C2 | VI | High | Amygdala | 2/3 | + | 4.9 | − | + |
| 9 | Female | 56 | 66 | FTLD TDP-43 | A0 | C0 | I | Not | Amygdala | − | − | 3.8 | − | − |
| 10 | Male | 69 | 76 | AD | A3 | C3 | VI | High | Amygdala | − | − | 5.5 | − | − |
| 11 | Male | 55 | 73 | AD | A3 | C3 | V | High | Amygdala | − | − | 1.9 | − | − |
| 12 | Male | 77 | 86 | AD | A3 | C3 | V | High | Amygdala | − | + | 5.3 | − | + |
| 13 | Female | 79 | 87 | AD | A3 | C3 | IV | Intermediate | Amygdala | − | + | 5.3 | − | + |
| 14 | Female | 90 | 100 | AD | A3 | C3 | V | High | Amygdala | − | − | 8.0 | − | − |
|
| ||||||||||||||
| 15 | Male | 54 | 67 | LBD | A1 | C2 | I | Low | Limbic/Neocortical | + | + | 8.1 | + | |
| 16 | Male | 72 | 81 | LBD | A3 | C2 | V | High | Limbic/Neocortical | + | + | 3.7 | + | |
| 17 | Male | 59 | 71 | LBD | A3 | C2 | IV | Intermediate | Limbic/Neocortical | + | + | 3.5 | + | |
| 18 | Male | 63 | 71 | LBD | A3 | C2 | III | Intermediate | Limbic/Neocortical | + | + | 1.2 | + | |
| 19 | Male | 66 | 71 | LBD | A3 | C3 | VI | High | Limbic/Neocortical | + | + | 1.7 | + | + |
| 20 | Male | 62 | 73 | LBD | A3 | C1 | II | Low | Limbic/Neocortical | + | + | 6.8 | + | + |
| 21 | Male | 52 | 72 | LBD | A2 | C2 | V | Intermediate | Limbic/Neocortical | + | + | 9.3 | + | + |
| 22 | Female | 51 | 59 | LBD | A3 | C3 | VI | High | Limbic/Neocortical | + | + | 2.7 | + | + |
Abbreviations: AM: Antemortem, PM: Postmortem, ADNC: Alzheimer’s disease neuropathological change NA: not applicable. LRP: Lewy Related Pathology, CSF: cerebrospinal fluid. CBD: corticobasal degeneration, AD: Alzheimer’s disease, PSP: progressive supranuclear Palsy, FTLD TDP-43: frontotemporal lobar degeneration TAR DNA-binding protein 43, LBD: Lewy-body disease. Inconclusive cases have 2/3 replicate wells that were positive. Brain tissue samples were analyzed at 10-8 dilution. Positive results indicate 3/3 replicates were positive and negative results indicate 0/3 replicates were positive. Amygdala tissue could not be obtained for case 5.
Within no αSyn-pathology and limbic/neocortical groups, there was 100% concordance between brain homogenate results and CSF results. Of the 5 patients with amygdala-predominant αSyn-pathology that also tested positive on the αSyn-SAA using amygdala brain tissue, 3 also had some seeding activity on the αSyn-SAA using either antemortem or postmortem CSF (Table 3). Overall, the assay detected higher seeding activity in amygdala tissue in amygdala-predominant cases, while neocortical cases presented high levels of seeding activity in both brain regions.
Discussion
Although there have been large strides in the understanding of the molecular basis of synucleinopathies, in vivo methods for detecting αSyn are still limited. Misfolded αSyn aggregation likely begins years to decades before the onset of symptoms, allowing for the potential ability to identify patients in the earliest stages of their diseases. The development of a sensitive and specific diagnostic tool for synucleinopathies would allow for early diagnosis of patients where often there is the highest level of clinical uncertainty and when disease modifying therapies are of the greatest potential use39. Thus, αSyn-related biomarkers remain a crucial need to the field. Several publications have shown promising results for αSyn-SAAs performed in academic laboratories17, 24, but the performance of the assay within a regulated CLIA environment, and against pathology-confirmed samples, has been a gap. Moreover, the knowledge of whether current generations of αSyn-SAAs can detect pathological αSyn in patients with other pathologies and with clinical diagnoses other than PD or DLB, is crucial to understanding the range of their diagnostic utility. αSyn-SAA offers the ability to identify αSyn seeds in living patients and studies have focused largely on cases with clinical DLB, PD, and MSA and where performed, autopsy was used as a validation of the clinical diagnosis. However, these assays offer the potential ability to identify patients with αSyn-pathology who may not exhibit a ‘synucleinopathy phenotype’. One factor that can complicate diagnosis is the presence of AD co-pathology which affect clinical expression particularly in PD and DLB12, 32, 40–44. Furthermore, in AD, αSyn-pathology pathology in AD-LBV is common and also associated with worse prognosis and specific clinical features7, 11. The use of αSyn-SAA assays to help characterize patients in terms of their αSyn-pathology is immediately clinically applicable and potentially valuable in clinical trials to recruit homogenous populations; but detailed studies in well-characterized pathologically validated cohorts has been needed to understand how the current αSyn-SAA assay can be applied. We used pathologically driven categories of αSyn pathology, independent of clinical diagnosis, in a cohort of patients with high degrees of co-pathology to assess the performance of the αSyn-SAA assay. In these cases, the use of such a biomarker could prove useful in identifying αSyn pathology that was not necessarily suspected.
Our results add to the previous reports that αSyn-SAAs can robustly detect αSyn seeds in limbic/neocortical stage αSyn-pathology, but also show decreased sensitivity in detecting αSyn seeds in amygdala-predominant cases. An additional unique feature to this study is the number of subjects with postmortem CSF, providing a proximal time point to the autopsy assessment. Classification using postmortem CSF showed a sensitivity of 80% and specificity of 88.5%, however when stratified by pathology distribution, again the assay performed significantly better in detecting limbic/neocortical than amygdala-predominant αSyn-pathology. Lastly, we also observed decreased seeding activity from amygdala-predominant cases when assaying frozen brain tissue from frontal cortex and amygdala.
The lower sensitivity of CSF αSyn-SAA to detect αSyn seeds in amygdala-predominant pathology may represent assay dependence on degree of brain αSyn “burden”. Alternatively, negative αSyn-SAA CSF samples in the amygdala-predominant group could be explained by localized brain pathology that does not enter the CSF. However, direct analysis of amygdala homogenate from amygdala-predominant cases showed low detection, suggesting less seeding activity by these particular αSyn species. Recent studies have found that αSyn species in amygdala-predominant pathology found in AD may have different immunohistochemical properties than PD or DLB patients with limbic and neocortical αSyn-pathology13–15. It is plausible that these amygdala-predominant αSyn seeds have lower rates of amplification due to unique conformation or post-translational modifications of these αSyn species. Currently, there is no method to quantify αSyn seeds in a sample, thus, it is not possible to determine if αSyn seeds were extracted with similar efficiencies from amygdala and frontal lobe tissues. Lower concentrations in the amygdala homogenates could explain negative results. However, we found positivity in dilutions up to 10-9 in some cases which is higher than previously shown in the literature (not shown), suggesting the homogenization protocol did not artificially decrease the amount of αSyn seeds. The small number of amygdala-predominant cases who had seeding activity had slower time-to-threshold and T50 values than limbic/neocortical cases (TTT (p=0.0007) and T50 (p=0.0002)). This is potentially of interest given that in vitro models have shown that lower levels of synthetic αSyn seeds take longer to amplify in αSyn-SAA17, 18. However, future studies of larger cohorts will be needed to confirm these preliminary observations.
Since αSyn-pathology commonly coexists in AD and may be associated with faster clinical progression45, identifying this pathology with a biomarker would improve clinical monitoring and create options for clinical trials targeting αSyn in these patients. If amygdala-predominant type αSyn-pathology is an early stage or precursor of more widespread concomitant LB pathology in AD, then detecting its presence through biomarkers such as αSyn-SAA would be useful. However, the effect of amygdala-predominant αSyn-pathology in AD appears to have less clinical impact in some cases or may take years to convert to a more widespread seeding. Further work is needed to determine why the seeding potential of amygdala-predominant αSyn-pathology is lower in some cases, or whether different types of αSyn-SAAs could provide detection of this pathology. We also report for the first time that αSyn seeds can be amplified from postmortem CSF samples. This is relevant because it could offer some insights when antemortem CSF samples are negative but there is detectable brain pathological αSyn upon neuropathological analysis. In these cases, positive postmortem CSF results could indicate that the αSyn pathological process started after antemortem CSF collection or that the disease process was too early at the time of antemortem CSF collection. However, we observed a reduction in sensitivity when testing postmortem CSF from neocortical/limbic cases, driven by 2 samples that were negative. Since we observed an increase in sensitivity when analyzing amygdala-predominant postmortem CSF, αSyn seed degradation or overall CSF instability is unlikely to explain the difference. Preliminary observations suggest that brain debris or cellular breakdown products could contaminate the sample during postmortem CSF collection, which effects could be minimized at least partially by centrifugation. Nevertheless, the instability of αSyn seeds and other CSF components in postmortem CSF and their potential effects on αSyn-SAA have not been systematically studied and require further exploration.
The assay’s ability to identify clinically unexpected synuclein pathology is an area of great potential. Our results indicate that 27/75 (36%) of the clinically diagnosed AD patients had αSyn aggregates in their antemortem CSF and were later autopsy-confirmed to have limbic/neocortical LB disease. DLB can be misdiagnosed as AD during life, and the presence of moderate to severe AD-related tau pathology is associated with a lower likelihood of visual hallucinations and cognitive fluctuations, and worse performance on tests of episodic memory and naming in DLB patients, meaning that it is more challenging to diagnose these patients with mixed pathologies accurately32, 44, 46.
Clinically, our cohort included only 4 PD and 9 DLB cases, and pathologically, there were no cases with brainstem-only αSyn-pathology which are limitations of the study. Our study adds valuable new information about the accuracy of αSyn-SAA in the context of co-pathology and non-LBD diagnoses. In another study, CSF from 4 cases with incidental αSyn-pathology in brainstem-only have been analyzed by αSyn-SAA25. Three of these cases were positive, suggesting that brainstem pathological αSyn shares propagation features with limbic and neocortical rather than amygdala-predominant pathological αSyn. Since brainstem-only pathological αSyn is an early event, these results are consistent detection of αSyn seeds in CSF of prodromal PD cases, like isolated REM sleep behavior disorder (iRBD)25, 29, 47. Finally, other minor weaknesses include potential differences in interpretation of the NACC guidelines between the two institutions and the impossibility to determine if patients with αSyn-SAA negative antemortem CSF and pathological αSyn upon autopsy represent true false negatives or if the pathology developed after antemortem CSF collection. Additionally, the limbic/neocortical group was skewed towards male participants. This is congruent with numerous studies identifying a sex-link for risk of synucleinopathy48–50. Larger numbers of cases with additional distributions of pathological αSyn, particularly brainstem-only and olfactory-only, should be further investigated to get a full picture of the relationship between brain pathology and CSF αSyn-SAA positivity. Lastly, further work is needed to fully interrogate differences in the seeding activity between pathological αSyn from different brain regions. It is unknown if the differences reflect the conformation of the seeds (strains), interactions with co-localized co-pathology, or perhaps brain region specific components (proteins, lipids, polysaccharides, nucleic acids, etc.) that may have an effect of the αSyn-SAA. Our data suggest that AD co-pathology is unlikely to explain the differences based on CSF measures, CERAD scores, Braak-tau stages, and Thal phases.
In this large, multicentered autopsy-validated cohort of patients with a variety of stages of αSyn-pathology, our results indicate that the αSyn-SAA is highly predictive of neocortical or limbic αSyn-pathology in aging patients for whom αSyn-pathology is not clinically suspected. This feature makes αSyn-SAA a diagnostic tool with great potential for clinical trials aiming to initiate interventions early in the disease process or to select-out patients with co-incidental αSyn-pathology. However, there was substantially lower sensitivity to detect amygdala-predominant αSyn pathology in brain tissue and CSF, which may have distinct biochemical properties and seeding potential that reduces detection in current generation of αSyn-SAAs.
Supplementary Material
Summary for Social Media If Published.
Twitter handles of the authors: @edwardwilsoniii
Alpha-synuclein seed amplification assays (αSyn-SAAs) are highly accurate detecting αSyn seeds in CSF from LBD cohorts. Accuracy in patients with co-pathologies and non-LBD clinical diagnoses is not well known. αSyn-SAA results have not been correlated with the distribution of αSyn-pathology in the brain.
Here we examine the ability of the αSyn-SAA to detect αSyn seeds in a multicenter cohort of autopsy-validated cases with different αSyn-pathology distributions. Antemortem CSF, and when available postmortem CSF, amygdala and frontal cortex tissues were tested by αSyn-SAA.
High accuracy of the αSyn-SAA is confirmed in limbic and neocortical αSyn-pathology cases. Lower detection is observed in cases with amygdala-predominant αSyn-pathology. Similar results were found in brain tissues. These results indicate differences between frontal cortex and amygdala pathological αSyn.
αSyn-SAAs present high sensitivity and specificity for the identification of cases with limbic and neocortical αSyn-pathology, even in the presence of co-pathology and independent of clinical diagnosis. αSyn-SAA could assist in the differential diagnosis in dementias with mixed pathology, especially AD, DLB, and AD-LBV.
Acknowledgments
We thank the patients who participated in this research, their families, and the investigators and staff at the OHSU Layton Aging and Alzheimer’s Disease Research Center and Oregon VA Parkinson’s Disease Research Education and Clinical Care Center; UCSD Shiley Marcos Alzheimer’s Disease Research Center; Robin Guariglia and Nora Mattek for compilation of clinical and genetic data; Babett Lind for CSF sample collection. Additional thanks to the individuals at Amprion Inc. involved in this project: Frank Espin, John Middleton, Russell M. Lebovitz, and Karen MacLeod.
This study was funded in part by the Alzheimer Disease Center Clinical Core at Oregon Health and Science University (PI: Kaye; eIRB 725; supported by NIH P30 AG008017, P30 AG066518) as well as the National Center for Advancing Translational Sciences (National Institutes of Health, Grant Award Number UL1TR002369). Amprion’s efforts were funded in part by the Alzheimer’s Drug Discovery Foundation (ADDF) Diagnostics Accelerator, as well as by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (Award Number U44NS111672). Moriah R. Arnold is funded through the Medical Scientist Training Program of Oregon Health & Science University (T32 GM 109835). Dr. Coughlin is funded by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (Award Number NS120038) and the National Institute on Aging (Award Number AG062429). Dr. Galasko is funded by the UCSD Shiley-Marcos ADRC (AG062429), and by the DLB Research Center of Excellence award from the Lewy Body Dementia Association. The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review of approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Potential Conflicts of Interest
Dr. Concha, Ms. Farris, and Mr. Ma are inventors on several patents related to PMCA technology (SAA) and are associated to Amprion Inc, a biotech company focused on the commercial utilization of SAA for diagnosis. All other authors have no conflicts of interest to disclose.
References
- 1.Spillantini MG, Crowther RA, Jakes R et al. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 1998; 95 (11): 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baba M, Nakajo S, Tu PH et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 1998; 152 (4): 879–884. [PMC free article] [PubMed] [Google Scholar]
- 3.Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 2000; 10 (3): 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jellinger KA. Alpha-synuclein pathology in Parkinson’s and Alzheimer’s disease brain: incidence and topographic distribution--a pilot study. Acta Neuropathol 2003; 106 (3): 191–201. [DOI] [PubMed] [Google Scholar]
- 5.Kotzbauer PT, Trojanowsk JQ, Lee VM. Lewy body pathology in Alzheimer’s disease. J Mol Neurosci 2001; 17 (2): 225–232. [DOI] [PubMed] [Google Scholar]
- 6.Roudil J, Deramecourt V, Dufournet B et al. Influence of Lewy Pathology on Alzheimer’s Disease Phenotype: A Retrospective Clinico-Pathological Study. J Alzheimers Dis 2018; 63 (4): 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Twohig D, Nielsen HM. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener 2019; 14 (1): 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leverenz JB, Fishel MA, Peskind ER et al. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol 2006; 63 (3): 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchman AS, Shulman JM, Nag S et al. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann Neurol 2012; 71 (2): 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iacono D, Geraci-Erck M, Rabin ML et al. Parkinson disease and incidental Lewy body disease: Just a question of time? Neurology 2015; 85 (19): 1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung EJ, Babulal GM, Monsell SE et al. Clinical Features of Alzheimer Disease With and Without Lewy Bodies. JAMA Neurol 2015; 72 (7): 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merdes AR, Hansen LA, Jeste DV et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 2003; 60 (10): 1586–1590. [DOI] [PubMed] [Google Scholar]
- 13.Sorrentino ZA, Goodwin MS, Riffe CJ et al. Unique alpha-synuclein pathology within the amygdala in Lewy body dementia: implications for disease initiation and progression. Acta Neuropathol Commun 2019; 7 (1): 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Covell DJ, Robinson JL, Akhtar RS et al. Novel conformation-selective alpha-synuclein antibodies raised against different in vitro fibril forms show distinct patterns of Lewy pathology in Parkinson’s disease. Neuropathol Appl Neurobiol 2017; 43 (7): 604–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson PT, Abner EL, Patel E et al. The Amygdala as a Locus of Pathologic Misfolding in Neurodegenerative Diseases. J Neuropathol Exp Neurol 2018; 77 (1): 2–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairfoul G, McGuire LI, Pal S et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 2016; 3 (10): 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shahnawaz M, Tokuda T, Waragai M et al. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of alpha-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol 2017; 74 (2): 163–172. [DOI] [PubMed] [Google Scholar]
- 18.Groveman BR, Orru CD, Hughson AG et al. Rapid and ultra-sensitive quantitation of disease-associated alpha-synuclein seeds in brain and cerebrospinal fluid by alphaSyn RT-QuIC. Acta Neuropathol Commun 2018; 6 (1): 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manne S, Kondru N, Jin H et al. alpha-Synuclein real-time quaking-induced conversion in the submandibular glands of Parkinson’s disease patients. Mov Disord 2020; 35 (2): 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manne S, Kondru N, Jin H et al. Blinded RT-QuIC Analysis of alpha-Synuclein Biomarker in Skin Tissue From Parkinson’s Disease Patients. Mov Disord 2020; 35 (12): 2230–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Luca CMG, Elia AE, Portaleone SM et al. Efficient RT-QuIC seeding activity for alpha-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Transl Neurodegener 2019; 8: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Concha-Marambio L, Farris CM, Holguin B et al. Seed Amplification Assay to Diagnose Early Parkinson’s and Predict Dopaminergic Deficit Progression. Mov Disord 2021; 36 (10): 2444–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russo MJ, Orru CD, Concha-Marambio L et al. High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol Commun 2021; 9 (1): 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang UJ, Boehme AK, Fairfoul G et al. Comparative study of cerebrospinal fluid alpha-synuclein seeding aggregation assays for diagnosis of Parkinson’s disease. Mov Disord 2019; 34 (4): 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi M, Candelise N, Baiardi S et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol 2020; 140 (1): 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singer W, Schmeichel AM, Shahnawaz M et al. Alpha-Synuclein Oligomers and Neurofilament Light Chain in Spinal Fluid Differentiate Multiple System Atrophy from Lewy Body Synucleinopathies. Ann Neurol 2020; 88 (3): 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shahnawaz M, Bilkis T, Park IS. Amyloid beta cytotoxicity is enhanced or reduced depending on formation of amyloid beta oligomeric forms. Biotechnol Lett 2021; 43 (1): 165–175. [DOI] [PubMed] [Google Scholar]
- 28.Singer W, Schmeichel AM, Shahnawaz M et al. Alpha-Synuclein Oligomers and Neurofilament Light Chain Predict Phenoconversion of Pure Autonomic Failure. Ann Neurol 2021; 89 (6): 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poggiolini I, Erskine D, Vaikath NN et al. RT-QuIC Using C-Terminally Truncated alpha-Synuclein Forms Detects Differences in Seeding Propensity of Different Brain Regions from Synucleinopathies. Biomolecules 2021; 11 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weintraub S, Salmon D, Mercaldo N et al. The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord 2009; 23 (2): 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peskind ER, Riekse R, Quinn JF et al. Safety and acceptability of the research lumbar puncture. Alzheimer Dis Assoc Disord 2005; 19 (4): 220–225. [DOI] [PubMed] [Google Scholar]
- 32.McKeith IG, Boeve BF, Dickson DW et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017; 89 (1): 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Montine TJ, Phelps CH, Beach TG et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123 (1): 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dickson DW, Kouri N, Murray ME et al. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci 2011; 45 (3): 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cairns NJ, Bigio EH, Mackenzie IR et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007; 114 (1): 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shahnawaz M, Mukherjee A, Pritzkow S et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020; 578 (7794): 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirra SS, Heyman A, McKeel D et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991; 41 (4): 479–486. [DOI] [PubMed] [Google Scholar]
- 38.Leverenz JB, Hamilton R, Tsuang DW et al. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol 2008; 18 (2): 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adler CH, Beach TG, Hentz JG et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology 2014; 83 (5): 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irwin DJ, White MT, Toledo JB et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012; 72 (4): 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irwin DJ, Grossman M, Weintraub D et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 2017; 16 (1): 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coughlin D, Xie SX, Liang M et al. Cognitive and Pathological Influences of Tau Pathology in Lewy Body Disorders. Ann Neurol 2019; 85 (2): 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coughlin DG, Phillips JS, Roll E et al. Multimodal in vivo and postmortem assessments of tau in Lewy body disorders. Neurobiol Aging 2020; 96: 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peavy GM, Edland SD, Toole BM et al. Phenotypic differences based on staging of Alzheimer’s neuropathology in autopsy-confirmed dementia with Lewy bodies. Parkinsonism Relat Disord 2016; 31: 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malek-Ahmadi M, Beach TG, Zamrini E et al. Faster cognitive decline in dementia due to Alzheimer disease with clinically undiagnosed Lewy body disease. PLoS One 2019; 14 (6): e0217566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis 2006; 9 (3 Suppl): 417–423. [DOI] [PubMed] [Google Scholar]
- 47.Iranzo A, Fairfoul G, Ayudhaya ACN et al. Detection of alpha-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol 2021; 20 (3): 203–212. [DOI] [PubMed] [Google Scholar]
- 48.Jurado-Coronel JC, Cabezas R, Avila Rodriguez MF et al. Sex differences in Parkinson’s disease: Features on clinical symptoms, treatment outcome, sexual hormones and genetics. Front Neuroendocrinol 2018; 50: 18–30. [DOI] [PubMed] [Google Scholar]
- 49.Lubomski M, Louise Rushworth R, Lee W et al. Sex differences in Parkinson’s disease. J Clin Neurosci 2014; 21 (9): 1503–1506. [DOI] [PubMed] [Google Scholar]
- 50.Smith KM, Dahodwala N. Sex differences in Parkinson’s disease and other movement disorders. Exp Neurol 2014; 259: 44–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.