

Abstract
Post-translational modifications (PTMs) create vast structural and functional diversity of proteins, ultimately modulating protein function and degradation, influencing cellular signaling, and regulating transcription. The combinatorial patterns of PTMs increase the heterogeneity of proteins and further mediates their interactions. Advances in mass spectrometry-based proteomics have resulted in identification of thousands of proteins and allowed characterization of numerous types and sites of PTMs. Examination of intact proteins, termed the top-down approach, offers the potential to map protein sequences and localize multiple PTMs on each protein, providing the most comprehensive cataloging of proteoforms. This review describes some of the dividends of using mass spectrometry to analyze intact proteins and showcases innovative strategies that have enhanced the promise of top-down proteomics for exploring the impact of combinatorial PTMs in unsurpassed detail.
Keywords: Proteomics, Top-down, Ion activation, Internal ions, Ion-ion reactions, Proteoform
Introduction
The construction of proteins by the ribosome results in thousands of protein sequences with unique function-dependent structures. Following translation, proteins are modulated by addition of post-translational modifications (PTMs) as well as further shaped by the key processes of folding and assembly into multimeric macromolecules to create functional structures. Many of the PTMs are reversible and dynamic, offering a means to regulate protein activity and subcellular localization while also contributing to dysfunction of pivotal biological processes in diseases [1–3]. The vast array of PTMs, each endowing proteins with different chemical properties, result in an immense diversity in the proteome with different modified proteins known as proteoforms [4–6]. Hundreds of types of PTMs are known, ranging from common ones such as phosphorylation, glycosylation and acetylation, to less common and even rare ones like sumoylation, cholesterolyation, and S-nitrosylation, among others [3]. Identification and localization of PTMs is a challenging task, exacerbated by the broad dynamic range of proteins, both in terms of sizes and abundance, in addition to the variations in lability of the PTMs and other chemical properties that influence the ability to detect proteoforms [1,2].
Mass spectrometry has proven to be one of the most versatile and powerful methods for qualitative and quantitative identification of proteins and their interactions with other molecules [7–9]. Numerous advances in sample preparation, fractionation, enrichment, MS/MS methods, quantification strategies, and data acquisition and processing methods have contributed to the ability to profile thousands of proteins and decipher variations in PTMs in a high throughput manner [10–15]. There have also been monumental inroads in functional characterization of proteins based on mass spectrometry approaches [16]. Mapping PTMs is made even more challenging by the fact that individual proteins may harbor a combination of modifications that further regulate their functions and activities as well as interactions with other proteins [17–19]. The combinatorial pattern of modifications affords an extraordinary way to fine-tune protein function and govern downstream signals while at the same time introducing far more heterogeneity into the potential protein repertoire. Understanding the co-dependence of PTMs is an enormous unsolved problem that demands even more advanced analytical strategies for accurately deciphering combinatorial modifications and their functional outcomes and elucidating how PTMs influence structure. This perspective focuses on the newest mass spectrometry-based strategies to advance the characterization of PTMs, particularly emphasizing those methods that have the greatest potential for mapping combinatorial PTMs.
Overview of the proteomics workflow
The major workflows for protein identification by mass spectrometry are categorized as “bottom-up” and “top-down” (with the intermediate “middle-down” strategy often considered a sub-category of bottom-up method) (Figure 1). Conventional “bottom-up” proteomics involves enzymatic digestion of proteins into peptides prior to LC-MS/MS analysis. Bioinformatics software is utilized to stitch together the original proteins based on matching the identified peptides to a proteome-scale database.
Figure 1.
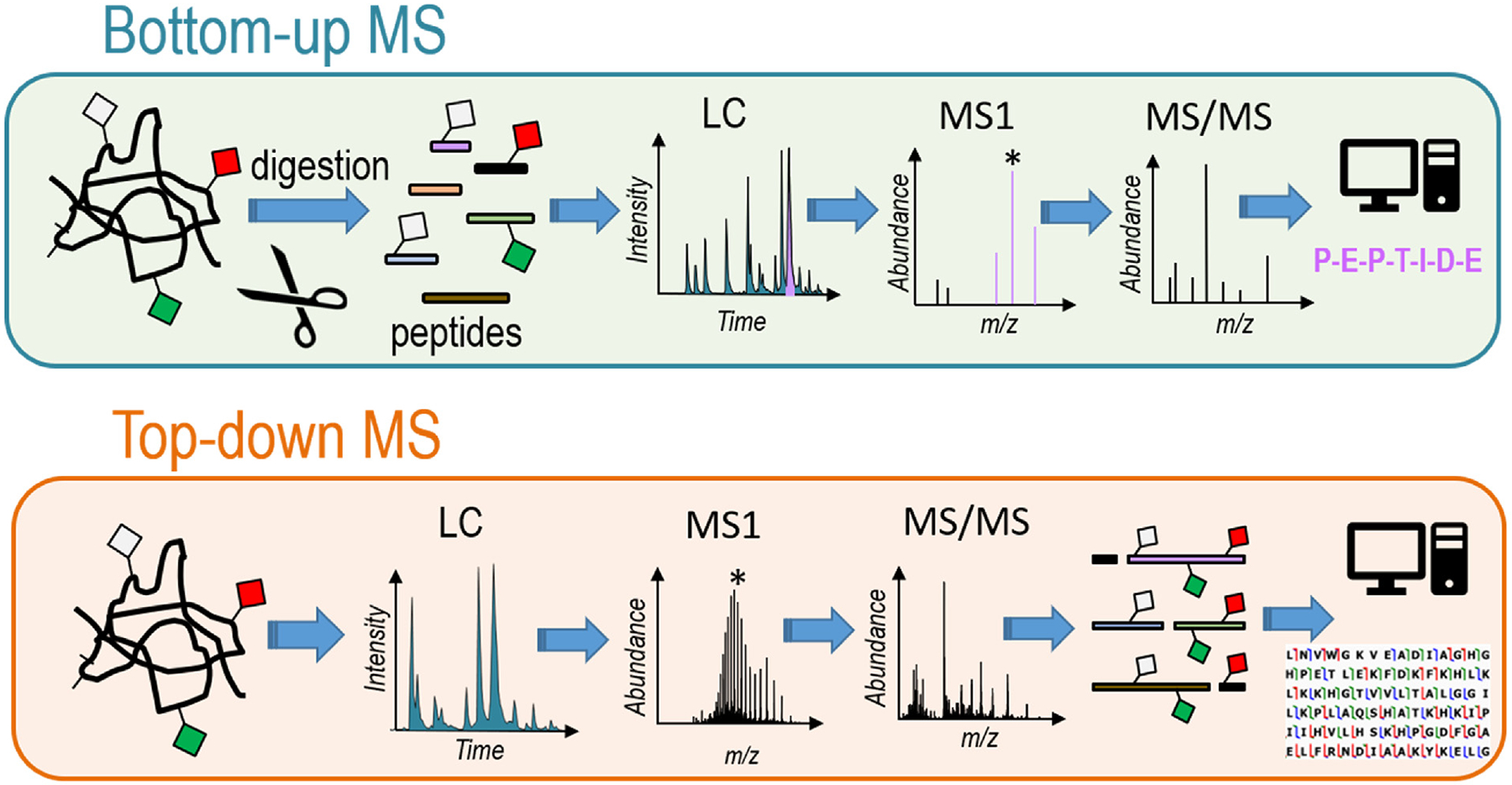
Schematic representation of bottom-up and top-down strategies. Bottom-up methods include a proteolytic digestion step undertaken on a mixture of proteins of interest, and the resulting mixture of peptides is separated and analyzed by LC-MS/MS in which MS1 and MS/MS spectra are acquired for the eluting peptides, then evaluated using database search methods to identify proteins. Top-down approaches examine intact proteins in which a mixture of proteins is separated and analyzed by LC-MS/MS in which MS1 and MS/MS spectra are acquired for the intact proteins as they elute, thus maintaining the entire context of modifications for each proteoform.
Bottom-up proteomics, allowing analysis of thousands of peptides in a single run, has become a routine procedure owing to robust nanoscale separation methods, high performance MS/MS instrumentation, and development of sophisticated bioinformatics tools designed for analysis of LC-MS data [7–9]. Bottom-up proteomics have advanced tremendously owing to improvements in enrichment methods, the widespread availability of high resolution/high accuracy mass spectrometers, and innovative data acquisition methods that enable more peptide identifications. These bottom-up methods excel for quantitative applications as well as for achieving unprecedented levels of protein identifications. Localizing specific PTMs has also advanced considerably owing to development of innovative fractionation and enrichment methods. However, the ability to map multiple PTMs, mutations, truncations, and polymorphisms of individual proteins is impeded by the analysis of peptides which cover only short sequence sections, often leaving gaps and blurring the context of combinatorial modifications.
The alternative workflow, top-down proteomics, offers several distinct advantages as well as notable challenges. The most compelling advantage is the potential to uniquely characterize each protein and its modifications (known as a proteoform) in its entirety [20–26]. A top-down approach uses the intact mass to identify differing proteoforms based on exact molecular compositions and then subsequent fragmentation (MS/MS) to confirm sequences and localize modifications. This method has resulted in identification of thousands of proteoforms from complex biological samples, such as the over 5000 proteoforms reported from human H1299 cells [27] and close to 2000 proteoforms from human fibroblasts with over 400 above 30 kDa [28]. This top down workflow has also been adapted for clinical-type human samples, such as human tissues and human peripheral blood mononuclear cells (from blood draws) to facilitate more translational applications [29–36]. Efforts to further expand the number of proteins and proteoforms identified range exploration of methods to improve solubility of certain classes of proteins [23]; development of more strategic enrichment, fractionation and separation methods to capture low abundance proteoforms and mitigate sample complexity [37–40]; and design of elevated informatics to provide more robust, user-friendly data processing tools [41,42]. Many of these challenges and concepts have been addressed in recent reviews or perspectives [20–26].
One impressive example illustrating the power of the top-down approach is illustrated in Figure 2 for a bis-phosphorylated, N-acetylated cardiac troponin proteoform, cTnI, which is one key protein in the cardiac troponin complex that regulates contraction and relaxation of cardiac muscle [36]. The phosphorylation patterns modulate cardiac contractility and thus are critical harbingers of heart disease. After enrichment and purification, cardiac troponin proteoforms from cardiac tissue were characterized by a combination of two MS/MS methods, electron capture dissociation (ECD) and collisionally activated dissociation (CAD), using Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometry to localize the PTMs based systematic analysis of the fragment ion assignments via high accuracy measurements and isotope patterns [36].
Figure 2.
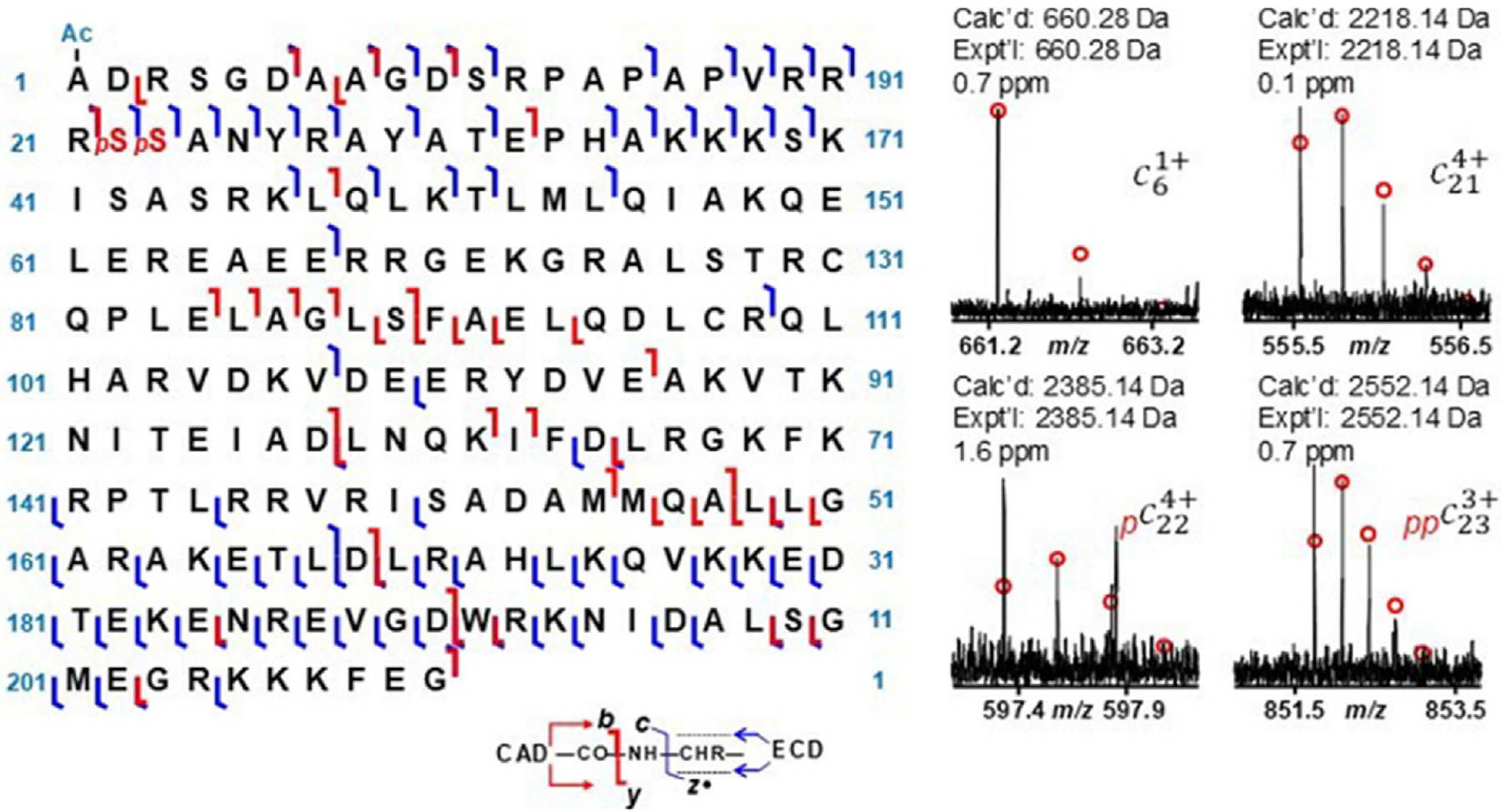
Sequence map showing backbone cleavages caused by ECD (resulting in c/z fragment ions) and CAD (yielding b/y fragment ions) of bis-phosphorylated swine cTnI proteoform (with Met excision and N-terminal acetylation) and isotopic profiles of four c ions including c6 ion with acetylation, c21 with no phosphorylation, c22 with one phosphorylation, and c23 with two phosphorylations which localized phosphorylation sites at Ser22 and Ser 23. The red “p” represents phosphorylation; Ac, acetylation. Reproduced from [36].
Owing to unsolved limitations in instrumentation necessary to routinely analyze larger proteins (typically ones greater than 30–40 kDa) or those with low abundances, top-down methods have yet to surpass bottom-up methods in terms of the breadth and throughput of proteome analysis. However, the top-down strategy allows multiple modifications to be pinpointed on individual proteins if the fragmentation patterns are sufficiently detailed, and this access to mapping combinatorial patterns of modifications is arguably the most powerful attribute that remains inaccessible by any bottom-up proteomics approach. Capitalizing on and magnifying this asset requires the ability to generate the richest fragmentation patterns of proteins and fully dissect those fragmentation patterns to maximize the information content. The following sections will consider the evolution of ion activation methods utilized for characterization of intact proteins as well as auxiliary methods to glean more information from the fragmentation patterns.
Ion activation and MS/MS for intact proteins
An essential keystone in characterizing proteins is the production of extensive series of fragment ions that allow confirmation of sequence and PTM information. The MS/MS aspect arguably poses one of the most significant challenges for several reasons. A protein must be activated with sufficient energy to generate meaningful fragment ions that map the sequence and bracket modifications. Attaining this goal requires generation of hundreds of fragment ions, and confident assignment requires valid isotopic patterns and high mass accuracy. MS/MS spectra with insufficient assigned fragment ions may allow protein identification but not comprehensive characterization of sequence variations or site-specific localization of PTMs.
Collisional-based methods (collision-induced dissociation (CID) or collisionally activated dissociation (CAD)) remain the mostly popular activation methods in tandem mass spectrometry and mesh well with high throughput workflows [43,44]. CID has been used for analysis of intact proteins, allowing identification of proteins via database searches that match the fragment ions to in silico-generated collections of fragment ions for each protein. The relatively low energy deposition of collisional activation, a feature that is more pronounced for larger proteins, the dependence on mobile protons to facilitate the mechanisms of fragmentation, and the notable preferential backbone cleavages that occur adjacent to specific amino acids (such as Pro, Asp, Glu) often results in limited sequence coverage [45–47]. These factors have contributed to the development of other activation methods, ones that enable more extensive fragmentation of proteins.
Electron-based activation methods (electron capture dissociation (ECD) and electron transfer dissociation (ETD)) are currently the most widely used for top-down analysis [48–55]. In general, these methods have yielded higher levels of sequence coverage than collisional activation methods for intact proteins, with the significant benefit of preserving labile PTMs which facilitates site localization. The scope of ECD, originally restricted to FTICR systems, has been extended to other platforms owing to the development of a modular electromagnetostatic cell that enables ECD on both Orbitrap and TOF platforms [56–59].
Ultraviolet photodissociation (UVPD) has offered another alternative to collisional activation for analysis of intact proteins [60,61]. The peptide bond in proteins exhibits strong absorption around 190 nm, making it a chromophore well-suited for photoactivation using 193 nm [62–66] or 213 nm [67–69] photons (two wavelengths that have been utilized for UVPD of proteins) and resulting in high energy deposition (6.3 eV or 5.9 eV per photon, respectively). Because UV photo-absorption results in excitation of electrons to excited electronic states, the fragmentation pathways observed for proteins are rather unique in that the most prominent fragment ions are typically a/x ions originating from cleavage of the C – Cα backbone bond, in contrast to the cleavage of C-N amide bonds that are dominant for collisional activation (b/y ions) or N – Cα bonds for electron activation (c/z ions). Proteins may dissociate directly from the excited states or after internal conversion to the ground electronic state along with intra-molecular vibrational energy re-distribution, leading to the diverse array of product ions that are the hallmark of UVPD [60]. An equally important facet of UVPD is the preservation of labile PTMs, facilitating their localization (akin to electron activation methods and in contrast to collisional activation).
The widespread availability of high performance mass spectrometers has allow successful adoption of the top-down approach for solving a wide range of biological problems. A few specific representative (but by no means comprehensive) examples are briefly summarized here to illustrate the growing impact of proteoform analysis by top-down MS/MS methods [70–84]. CID and ETD were used to facilitate differentiation of hemoglobin variants in blood, pinpointing single amino acid differences and providing diagnostic markers for thalassemia screening [72]. ECD and CID were used to identify variations in profiles of sarcomeric proteins extracted from septal myectomy tissues from patients with hypertrophic cardiomyopathy, revealing alterations in splicing and phosphorylation patterns among FHL2, ALP-H, elfin, cypher-5, cypher-6, and calsarcin-1 [75]. Histone proteoforms (H2A, H2B, H3, H4) extracted from CD8 T cells obtained after in vivo influenza infection of mice were characterized in detail using ETD and CID, unveiling numerous increases in PTMs linked to transcriptional activation and correlating with the stage of T cell differentiation [76]. The heterogeneity of O-glycan proteoforms of the spike protein receptor-binding domain of severe acute respiratory syndrome coronavirus 2 (SAR-CoV-2) was determined using ECD and CID, identifying eight O-glycoforms as well as a new 2-fucosylated glycan [81]. A large scale top-down study reported 30,000 proteoforms expressed from 1690 human genes spanning 21 cell types from human blood and bone marrow, representing an impressive compilation of a blood proteoform atlas [83].
Although these examples have highlighted the exciting attributes of the top-down workflow for protein characterization, particularly in the context of elucidating proteoforms, the compelling fruits of top-down methods for complete characterization of combinatorial PTMs have not been fully harvested. As observed for all activation methods for characterization of intact proteins, performance (primarily in terms of sequence coverage) falters for the mid-section of proteins and tends to degrade with the size of the protein. A number of factors contribute to the deterioration in performance, but the root cause largely originates with the decreasing signal-to-noise as the precursor ion decomposes into a great array of fragment ions (sub-division of the finite ion current) and the inability to assign many fragment ions. The latter problem arises from congestion of the MS/MS spectra containing hundreds of fragment ions in multiple charge states, resulting in overlap in the isotope patterns that are essential for confirming fragment ion identities. In addition, assignment of fragment ions is typically achieved by matching accurate m/z values to in silico product ions referenced to the N-terminus or C-terminus of each protein of interest. Those fragment ions that don’t retain the N- or C-terminus, classified as “internal ions”, are generally discarded. Owing to these reasons, typical MS/MS spectra of proteins may reveal hundreds of assignable fragment ions, in addition to an even greater array of non-assigned ions. The loss of information content is particularly critical for advancing the ability to characterize combinatorial patterns of PTMs which rely on comprehensive fragmentation to localize sites. Four inroads are briefly described to underscore some of the ongoing efforts to expand the opportunities of top-down methods for solving the combinatorial PTM puzzle.
Hybrid MS/MS methods
Notable gains in performance are obtained by combining ion activation methods, either in tandem or by integrating different MS/MS strategies [85–91]. For example, electron, collisional, and photoactivation methods can be combined in several formats, performed simultaneously to create a broader range of fragment ions or in series to maximize conversion of non-dissociated precursors into informative fragment ions. Combining different activation methods may also be used to decrease the density of fragment ions by dispersing them among a greater range of charge states (encompassing a broader m/z range). For example, ECD has been combined with UVPD (ECuvPD) or collisional activation (EChcD) in order to increase the sequence coverage of subunits of monoclonal antibodies (mAbs) as well as intact mAbs, including mapping the key complementarity determining regions [91]. 213 nm UVPD has been integrated with ETD and EThcD to expand the coverage of monoclonal IgG1, illustrating the reciprocity of orthogonal activation methods [88]. In another study, 266 nm UVPD was used to preferentially cleave disulfide bonds in combination with ECD for characterization of the resulting proteins [86]. These types of hybrid MS/MS methods are generally straight-forward to implement and have motivated other efforts to glean insight from top-down spectra.
Ion-Ion reactions
A hugely promising frontier for extending the performance of top-down methods entails the integration of ion-ion reactions into the top-down workflow [92]. Ion-ion reactions allow manipulation of charge states of ions based on fast and efficient charge reduction processes, primarily via gas-phase proton transfer charge reduction (PTCR) reactions as originally introduced ~25 years ago [93] and since expanded to other types of reactions [92]. For proteins dispersed among a broad range of charge states, PTCR may be used to consolidate them into single charge states and significantly increase the signal-to-noise ratios [93–95]. Alternatively, PTCR may be used to shift fragment ions to lower charge states, thereby decreasing spectral crowding in regions of the MS/MS spectra that are densely congested with ions of similar m/z values [97–101]. As illustrated in Figure 3, the fragmentation pattern generated by UVPD of carbonic anhydrase (25+ charge state) shown in the uppermost trace is dense and packed with fragment ions, many with overlapping isotope patterns that impede determination of molecular compositions and prevent ion assignment [98]. The ability to alleviate overlap in isotopic distributions of ions is critically important for effective MS/MS analysis and identification of proteoforms. By subjecting m/z-selected slices of the fragment ion population to reactions with a proton-scavenging reagent, fragment ions are shifted to higher m/z regions to alleviate congestion, as shown in the four lower traces in Figure 3. Significant improvements in proteoform analysis have been obtained by incorporating PTCR, either to fractionate very congested regions of MS1 spectra or to disperse fragments over a broader m/z landscape in MS/MS analysis in top-down workflows, on high performance Orbitrap or Fourier transform ion cyclotron resonance mass spectrometers [94–101]. The impressive performance gains by strategic manipulation and concentration of charge states in the gas phase offers one of the most compelling opportunities for advancing proteoform analysis.
Figure 3.
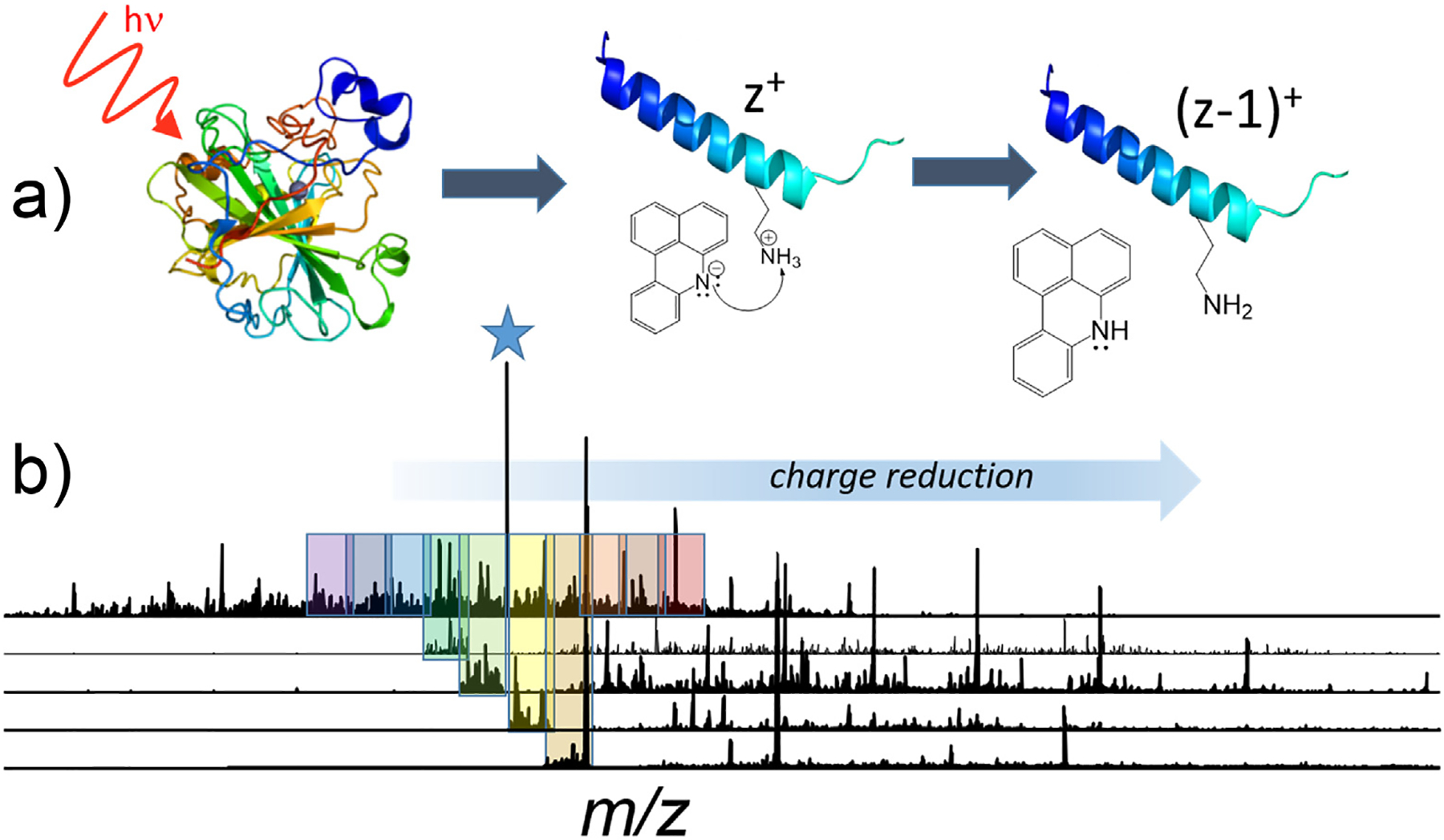
Proton transfer charge reduction (PTCR) reactions may be undertaken on fragment ions produced from top-down MS/MS characterization of proteins. A) UVPD of a protein generates many fragment ions (F) in a variety of charge states. PTCR using a suitable proton-scavenging reagent ion reduce the charge states of fragment ions (charge state z+), thus shifting them to lower m/z values (i.e., retaining the same mass m (aside from loss of a proton) and a lower charge state (z-1)+) and consequently dispersing the fragment ions over a broader m/z range. B) A protein, such as carbonic anhydrase (25+), creates many fragment ions upon UVPD. PTCR reactions undertaken on the groups of fragment ions outlined in the shaded boxes alleviates congestion in the MS/MS spectra and allows confident assignment of a much larger portion of the fragment ions. Adapted from [98].
Internal Ions
As underscored in the examples above, improvements in mass accuracy and resolution, ion activation methods, and search algorithms have elevated the characterization of intact proteins by top-down approaches. However, careful curation of MS/MS spectra reveal that numerous fragment ions are not assigned. Many of these unassigned ions correspond to internal fragment ions, products that do not contain either the N- or C-termini of proteins [102–109]. Internal ions are often ignored owing to the substantial computational demands to search for them and the corresponding high rate of false positives related to the potential to match the mass of an internal ion to others with the same elemental composition that are not uniquely anchored to the N- or C-terminus of a protein. The recent introduction of the ClipsMS (Comprehensive Localization of Internal Protein Sequences) algorithm facilitates searches and assignment of internal fragments [107]. One graphical example of the striking array of internal fragment ions identified upon electron ionization dissociation of myoglobin (16+ charge state) is shown in Figure 4, indicating numerous fragment ions that span sections of the protein [105]. Since these internal ions are not anchored by the N- or C-terminus, these ions are typically not assigned in most database search methods. As reported in a series of recent studies [103–109], assignment of internal ions offers a means to increase the coverages and localization of PTMs, particularly in the mid-sections of proteins which are frequently less thoroughly mapped by top-down MS/MS strategies.
Figure 4.
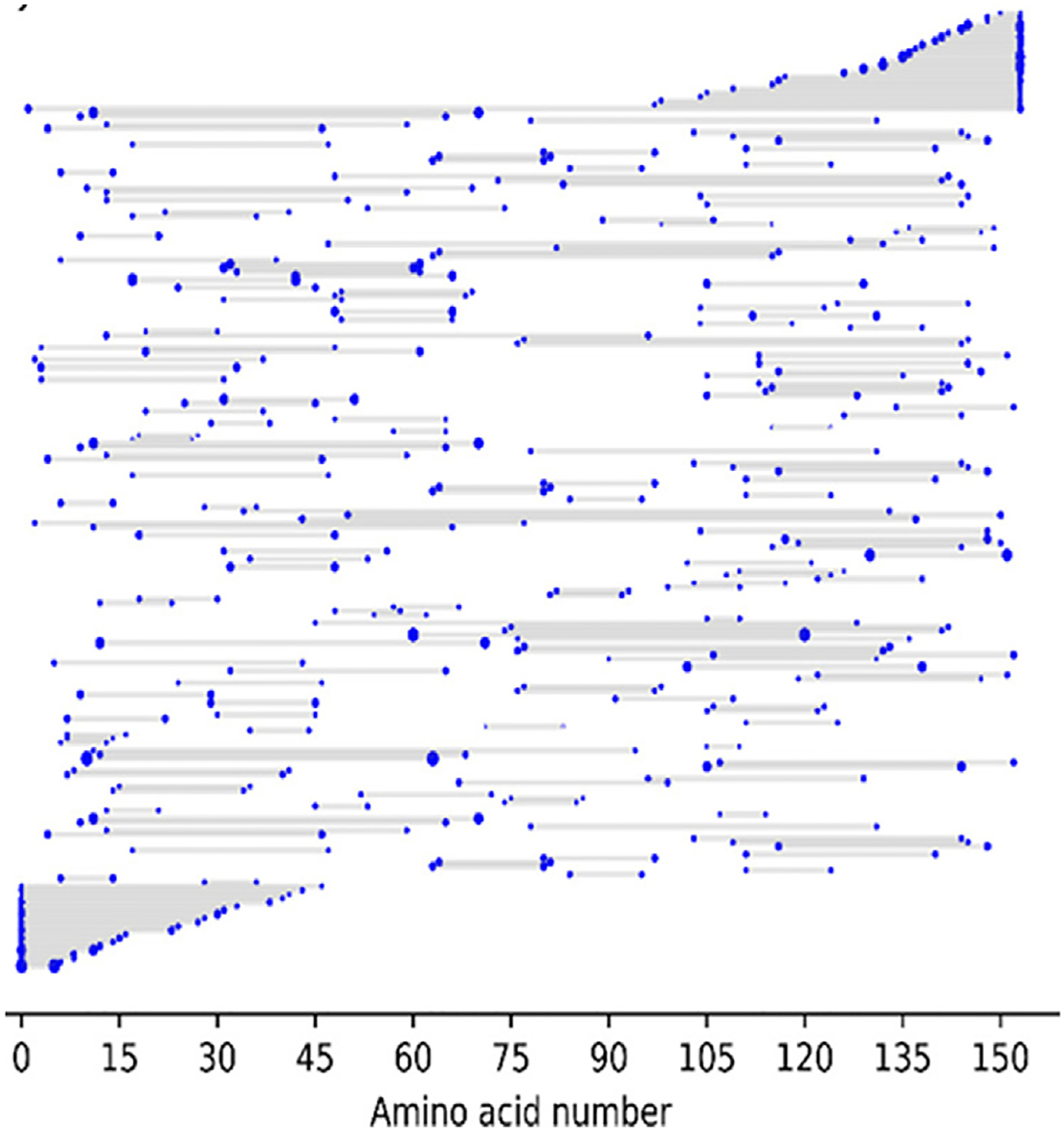
Electron ionization dissociation of myoglobin (16+ charge state) results in production of a large array of fragment ions originating from cleavages of the protein backbone The fragment location map indicates the region of the protein sequence covered by terminal and internal fragments. The solid gray contoured regions represent those fragment ions that include the N-terminus (lower gray contour) or C- terminus (upper gray contour) of the protein. The numerous horizontal bars in the middle region of the map represent the segments spanned by internal ions, indicating many different sizes of internal ions, none of which contain the N-terminal or C-terminal amino acid. Reproduced from [108].
Dissecting protein complexes
Advances in top-down methods have inspired efforts to use mass spectrometry to characterize protein complexes. This growing field, “native mass spectrometry”, utilizes electrospray ionization to transfer intact protein assemblies into the gas phase in a manner that preserves quaternary structures [26,110–112]. High resolution mass spectra can reveal the stoichiometries of complexes, and subsequent activation can disassemble the complexes in a manner that reveals the arrangement of subunits (Figure 5) [113–118]. For example, the fast, high energy deposition of surface-induced dissociation (SID) causes disassembly of protein complexes without significant unfolding and in a manner that preferentially disrupts the weakest interfaces, thus providing direct insight about subunit connectivity [119,120]. The possibility of analyzing supramolecular complexes, disassembling them into their constituent proteins, and then characterizing each protein in a step-wise top-down scheme offers a compelling opportunity to directly explore the compositions of protein assemblies in a mode that harmonizes proteomics and structural biology. In this scenario, the ability to characterize PTMs of the individual proteins in the complexes has the potential to allow construction of structure/functional relationships at an unprecedented level of detail.
Figure 5.
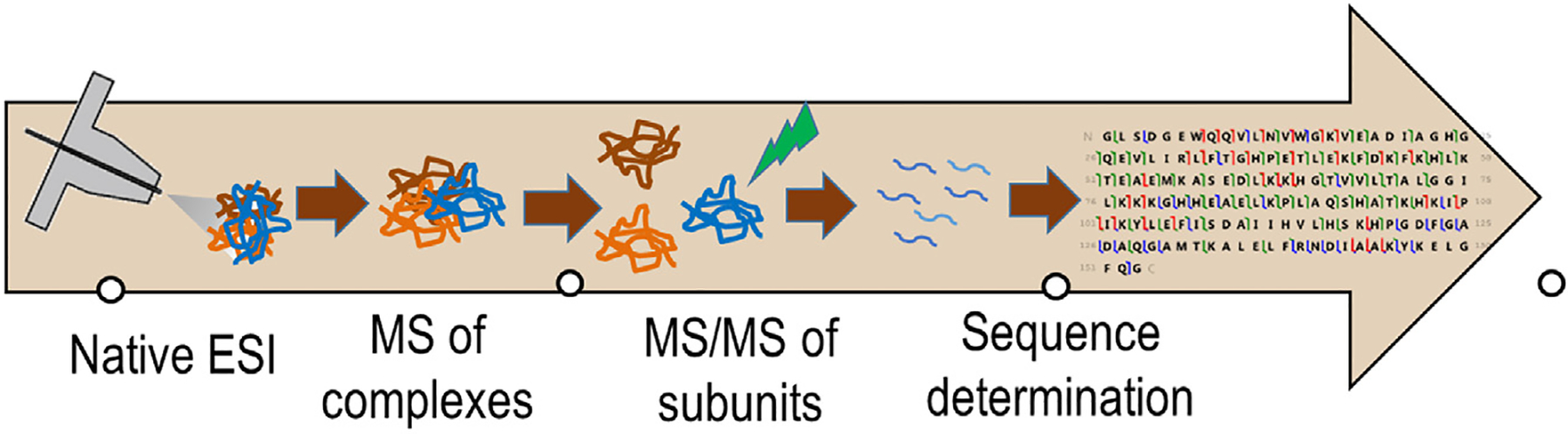
Intact protein complexes transferred to the gas phase are disassembled into protein subunits and characterized by MS/MS to allow identification of the proteins within the complexes.
Conclusions
Numerous innovative advances in mass spectrometry technologies have accelerated the understanding of the interplay between protein structure and function. Integral to extending the depth of the structure/function synergy is the ability to exhaustively map combinatorial patterns of post-translational modifications. The development of new strategies aimed at extracting more information from fragmentation patterns of intact proteins have cemented the dividends of the top-down approach, now being applied to increasingly complex biological problems such as ribosomes, proteasomes and viruses [26]. Moreover, transformative breakthroughs in the development of multiplexed individual ion/charge detection mass spectrometry methods offer the potential for unsurpassed gains in sensitivity and harvesting information from complex samples [121–123]. The next step requires integrating the established methods and upcoming breakthroughs to enable high throughput analysis of all proteins and assemblies in cells, further capitalizing on the emerging field of single-cell proteomics [124,125]. This will require inroads in separation methods compatible with native mass spectrometry and even more sophisticated data analysis for a systems biology approach to understanding structure, function, interactions, and assembly.
Acknowledgements
Funding from the National Institutes of Health [R35GM139658] and the Robert A. Welch Foundation [F-1155] is acknowledged by JSB. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- CAD
collisionally activated dissociation
- ECD
electron capture dissociation
- ETD
electron transfer dissociation
- FT-ICR
Fourier-transform ion cyclotron resonance
- HCD
higher energy collisional dissociation
- PTM
post-translational modification
- UVPD
ultraviolet photodissociation
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
- 1.Conibear AC: Deciphering protein post-translational modifications using chemical biology tools. Nat Rev Chem 2020, 4: 674–695. [DOI] [PubMed] [Google Scholar]
- 2.Xu Y, Chou K-C: Recent progress in predicting post-translational modification sites in proteins. Curr Top Med Chem 2016, 16:591–603. [DOI] [PubMed] [Google Scholar]
- 3.Millar AH, Heazlewood JL, Giglione C, Holdsworth JH, Bachair A, Schulze WX: The scope, functions and dynamics of post-translational protein modifications. Annu Rev Plant Biol 2019, 70:119–151. [DOI] [PubMed] [Google Scholar]
- 4.Smith LM, Kelleher NL, Proteomics CTD: Proteoform: a single term describing protein complexity. Nat Methods 2013, 10: 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith LM, Kelleher NL: Proteoforms as the next proteomics currency. Science 2018, 359:1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adhikari S, Nice EC, Deutsch EW, Lane L, Omenn GS, Pennington SR, Paik YK, Overall CM, Corrales FJ, Cristea IM, Van Eyk JE, Uhlén M, Lindskog C, Chan DW, Bairoch A, Waddington JC, Justice JL, LaBaer J, Rodriguez H, He F, Kostrzewa M, Ping P, Gundry RL, Stewart P, Srivastava S, Srivastava S, Nogueira FCS, Domont GB, Vandenbrouck Y, Lam MPY, Wennersten S, Vizcaino JA, Wilkins M, Schwenk JM, Lundberg E, Bandeira N, Marko-Varga G, Weintraub ST, Pineau C, Kusebauch U, Moritz RL, Ahn SB, Palmblad M, Snyder MP, Aebersold R, Baker MS: A high-stringency blue-print of the human proteome. Nat Commun 2020, 11:5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha Ankit, Mann Matthias: A beginner’s guide to mass spectrometry–based proteomics. Biochemist 2020, 42:64–69. [Google Scholar]
- 8.Macklin A, Khan S, Kislinger T: Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin Proteonomics 2020, 17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yates JR III: The revolution and evolution of shotgun proteomics for large-scale proteome analysis. J Am Chem Soc 2013, 135:1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doll S, Burlingame AL: Mass spectrometry-based detection and assignment of protein posttranslational modifications. ACS Chem Biol 2015, 10:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mnatsakanyan R, Shema G, Basik M, Batist G, Borchers CH, Sickmann A, Zahedi RP: Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expet Rev Proteonomics 2018, 15:515–535. [DOI] [PubMed] [Google Scholar]
- 12.Dunphy K, Dowling P, Bazou D, O’Gorman P: Current methods of post-translational modification analysis and their applications in blood cancers. Cancers 2021, 13:1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Udeshi ND, Mani DC, Satpathy S, Fereshetian S, Gasser JA, Svinkina T, Olive ME, Ebert BL, Mertins P, Carr SA: Rapid and deep-scale ubiquitylation profiling for biology and translational research. Nat Commun 2020, 11:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musiani D, Massignani E, Cuomo A, Yadav A, Bonaldi T: Biochemical and computational approaches for the large-scale analysis of protein arginine methylation by mass spectrometry. Curr Protein Pept Sci 2020, 21:725–739. [DOI] [PubMed] [Google Scholar]
- 15.Gabriele C, Prestagiacomo LE, Cuda G, Gaspari M: Mass spectrometry-based glycoproteomics and prostate cancer. Int J Mol Sci 2021, 22:5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Neill JR : An overview of mass spectrometry-based methods for functional proteomics. Methods Mol Biol 2019, 1871:179–196. [DOI] [PubMed] [Google Scholar]
- 17.Leutert M, Entwisle SW, Villen J: Decoding post-translation modification crosstalk with proteomics. Mol Cell Proteomics 2021, 20, 100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venne AS, Kollipara L, Zahedi RP: The next level of complexity: crosstalk of posttranslational modifications. Proteomics 2014, 14:513–524. [DOI] [PubMed] [Google Scholar]
- 19.Dai Vu L, Gevaert K, De Smet I: Protein language: post-translational modifications talking to each other. Trends Plant Sci 2018, 23:1068–1080. [DOI] [PubMed] [Google Scholar]
- 20.Gregorich ZR, Ge Y: Top-down proteomics in health and disease: challenges and opportunities. Proteomics 2014, 14: 1195–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toby TK, Fornelli L, Kelleher NL: Progress in top-down proteomics and the analysis of proteoforms. Annu Rev Anal Chem 2016, 9:499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou M, Lantz C, Brown KA, Ge Y, Paša-Tolić L, Loo JA, Lermyte F: Higher-order structural characterisation of native proteins and complexes by top-down mass spectrometry. Chem Sci 2020, 11:12918–12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melby JA, Roberts DS, Larson EJ, Brown KA, Bayne EF, Jin S, Ge Y: Novel strategies to address the challenges in top-down proteomics. J Am Soc Mass Spectrom 2021, 32:1278–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fornelli L, Toby TK: Characterization of large intact protein ions by mass spectrometry: what directions should we follow? Biochim Biophys Acta Protein Proteonomics 2022, 1870, 140758. [DOI] [PubMed] [Google Scholar]
- 25.Tucholski T, Ge Y: Fourier-transform ion cyclotron resonance mass spectrometry for characterizating proteoforms. Mass Spectrom Rev 2022, 41:158–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamara S, den Boer MA, Heck AJR, High-resolution native mass spectrometry, Chem Rev Article ASAP, DOI: 10.1021/acs.chemrev.1c00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Catherman AD, Durbin KR, Ahlf DR, Early BP, Fellers RT, Tran JC, Thomas PM, Kelleher NL: Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol Cell Proteomics 2013, 12:3465–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fornelli L, Durbin KR, Fellers RT, Early BP, Greer jB, LeDuc RD, Copton PD, Kelleher NL: Advancing top-down analysis of the human proteome using a benchtop quadrupole-orbitrap mass spectrometer. J Proteome Res 2017, 16:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iavarone F, Melis M, Platania G, Cabras T, Manconi B, Petruzzelli R, Cordaro M, Siracusano A, Faa G, Messana I, Zanasi M: Characterization of salivary proteins of schizophrenic and bipolar disorder patients by top-down proteomics. J Proteonomics 2014, 103:15–22. [DOI] [PubMed] [Google Scholar]
- 30.Kellie JF, Higgs RE, Ryder JW, Major A, Beach TG, Adler CH, Merchant K, Knierman MD: Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci Rep 2014, 4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martelli C, Iavarone F, D’Angelo L, Arba M, Vincenzoni F, Inserra I, Delfino D, Rossetti DV, Caretto M, Massimi L, Tamburrini G: Integrated proteomic platforms for the comparative characterization of medulloblastoma and pilocytic astrocytoma pediatric brain tumors: a preliminary study. Mol Biosyst 2015, 11:1668–1683. [DOI] [PubMed] [Google Scholar]
- 32.Durbin KR, Fornelli L, Fellers RT, Doubleday PF, Narita M, Kelleher NL: Quantitation and identification of thousands of human proteoforms below 30 kDa. J Proteome Res 2016, 15: 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. *.Lin Z, Wei L, Cai W, Zhu Y, Tucholski T, Mitchell SD, Guo W, Ford SP, Diffee GM, Ge Y: Simultaneous quantification of protein expression and modifications by top-down targeted proteomics: a case of the sarcomeric subproteome. Mol Cell Proteomics 2019, 18:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]; Integrated top-down LCMS/MS for characterization of PTMs of sarcomeric protin isoforms.
- 34. *.Toby TK, Fornelli L, Srzentić K, DeHart CJ, Levitsky J,Friedewald J, Kelleher NL: A comprehensive pipeline for translational top-down proteomics from a single blood draw. Nat Protoc 2019, 14:119–152. [DOI] [PMC free article] [PubMed] [Google Scholar]; Detailed protocol for high throughput top-down characterization of blood mononuclear cell proteoforms.
- 35. *.Fulcher JM, Makaju A, Moore RJ, Zhou M, Bennett DA, De Jager PL, Qian WJ, Paša-Tolić L, Petyuk VA: Enhancing top-down proteomics of brain tissue with FAIMS. J Proteome Res 2021, 20:2780–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]; High-field asymmetric waveform ion mobility spectrometry (FAIMS) facilitates gas-phase fractionation of proteions prior to top-down analysis of proteoforms from Alzherimer’s disease brain tissue.
- 36. *.Tiambeng TN, Tucholski T, Wu Z, Zhu Y, Mitchell SD, Roberts DS, Jin Y, Ge Y: Analysis of cardiac troponin proteoforms by top-down mass spectrometry. Methods Enzymol 2019, 626:347–374. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive localization of PTMs of cardiac troponin proteins by FTICR-MS by combining ECD and CAD information.
- 37.Brown KA, Tucholski T, Alpert AJ, Eken C, Wesemann L, Kyrvasilis A, Jin S, Ge Y: Top-down proteomics of endogenous membrane proteins enabled by cloud point enrichment and multidimensional liquid chromatography–mass spectrometry. Anal Chem 2020, 92:15726–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. *.Jooß K, Schachner LF, Watson R, Gillespie ZB, Howard SA, Cheek MA, Meiners MJ, Sobh A, Licht JD, Keogh MC, Kelleher NL: Separation and characterization of endogenous nucleosomes by native capillary zone electrophoresis-top-down mass spectrometry. Anal Chem 2021, 93:5151–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]; Capillary electrophoresis combined with top-down mass spectrometry allows separation and PTM characterization of histones from whole nucleosomes.
- 39. *.Gerbasi VR, Melani RD, Abbatiello SE, Belford MW, Huget R, McGee JP, Dayhoff D, Thomas PM, Kelleher NL: Deeper protein identification using field asymmetric ion mobility spectrometry in top-down proteomics. Anal Chem 2021, 93:6323–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]; Addition of high-field asymmetric waveform ion mobility spectrometry (FAIMS) capability significantly increases the number of identified proteins and assigned proteoforms in top-down analysis of complex mixtures.
- 40.Fulcher JM, Makaju Am, Moore RJ, Zhou M, Bennett DA, De Jager PL, Qian WJ, Pasa-Tolic L, Petyuk VA: Enhancing top-down proteomics of brain tissue with FAIMS. J Proteome Res 2021, 20:2780–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeHart CJ, Fellers RT, Fornelli L, Kelleher NL, Thomas PM: Bioinformatics analysis of top-down mass spectrometry data with ProSight lite. Methods Mol Biol 2017, 1558:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, He B, Kou Q, Wang Z, Wu S, Liu Y, Feng W, Liu X: Evaluation of top-down mass spectral identification with homologous protein sequences. BMC Bioinf 2018, 19:494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wells JM, McLuckey SA: Collision-induced dissociation (CID) of peptides and proteins. Methods Enzymol 2005, 402: 148–185. [DOI] [PubMed] [Google Scholar]
- 44.Macias L, Santos I, Brodbelt JS: Ion activation for peptides and proteins. Anal Chem 2020, 92:227–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haverland NA, Skinner OS, Fellers RT, Tariq AA, Early BP, LeDuc RD, Fornelli L, Compton PD: Kelleher NL defining gas-phase fragmentation propensities of intact proteins during native top-down mass spectrometry. J Am Soc Mass Spectrom 2017, 28:1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. *.Ives AN, Su T, Durbin KR, Early BP, Dos Santos Seckler H, Fellers RT, LeDuc RD, Schachner LF, Patrie SM, Kelleher NL: Using 10,000 fragment ions to inform scoring in native top-down proteomics. J Am Soc Mass Spectrom 2020, 31: 1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]; Patterns of fragment ions were compiled for over 150 proteins subjected to higher-energy collisional dissociation to establish trends in backbone cleavages in top-down analysis.
- 47.Cobb JS, Easterling ML, Agar JN: Structural characterization of intact proteins is enhanced by prevalent fragmentation pathways rarely observed for peptides. J Am Soc Mass Spectrom 2010, 21:949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riley NM, Westphall MS, Coon JJ: Activated ion-electron transfer dissociation enables comprehensive top-down protein fragmentation. J Proteome Res 2017, 16:2653–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fornelli L, Ayoub D, Aizikov K, Liu X, Damoc E, Pevzner PA, Makarov A, Beck A, Tsybin YO: Top-down analysis of immunoglobulin G isotypes 1 and 2 with electron transfer dissociation on a high-field Orbitrap mass spectrometer. J Proteonomics 2017, 159:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riley NM, Coon JJ: The role of electron transfer dissociation in modern proteomics. Anal Chem 2018, 90:40–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lermyte F, Valkenborg D, Loo JA, Sobott F: Radical solutions: principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure. Mass Spectrom Rev 2018, 37:750–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rush MJP, Riley NM, Westphall MS, Coon JJ: Top-down characterization of proteins with intact disulfide bonds using activated-ion electron transfer dissociation. Anal Chem 2018, 90:8946–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riley NM, Westphall MS, Coon JJ: Sequencing larger intact proteins (30–70 kDa) with activated ion electron transfer dissociation. J Am Soc Mass Spectrom 2018, 29:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. *.McCool EN, Lodge JM, Basharat AR, Liu X, Coon JJ, Sun L: Capillary zone electrophoresis-tandem mass spectrometry with activated ion electron transfer dissociation for large-scale top-down proteomics. J Am Soc Mass Spectrom 2019, 30:2470–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]; Capillary zone electrophoresis was integrated with top-down analysis using a combined infrared photoactivation/electron transfer dissociation method (activated ion electron transfer dissociation) for high throughput top-down proteomics of E. coli cells.
- 55. *.Lodge JM, Schauer KL, Brademan DR, Riley NM, Shishkova E, Westphall MS, Coon JJ: Top-down characterization of an intact monoclonal antibody using activated ion electron transfer dissociation. Anal Chem 2020, 92:10246–10251. [DOI] [PMC free article] [PubMed] [Google Scholar]; Activated ion electron transfer dissociation enhances fragmentation of disulfide-bound regions of lintact monoclonal antibodies.
- 56.Fort KL, Cramer CN, Voinov VG, Vasil’ev YV, Lopez NI, Beckman JS, Heck AJR: Exploring ECD on a benchtop Q exactive Orbitrap mass spectrometer. J Proteome Res 2018, 17:926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaw JB, Malhan N, Vasil’ev YV, Lopez NI, Makarov A, Beckman JS, Voinov VG: Sequencing grade tandem mass spectrometry for top-down proteomics using hybrid electron capture dissociation methods in a benchtop Orbitrap mass spectrometer. Anal Chem 2018, 90:10819–10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. *.Beckman JS, Voinov VG, Hare M, Sturgeon D, Vasil’ev Y, Oppenheimer D, Shaw JB, Wu S, Glaskin R, Klein C, Schwarzer C, Stafford G: Improved protein and PTM characterization with a practical electron-based fragmentation on Q-TOF instruments. J Am Soc Mass Spectrom 2021, 32: 2081–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]; Modular electromagnetostatic cell allows implementation of ECD on a Q-TOF platform for top-down characterization of proteoforms.
- 59.Shaw JB, Cooper-Shepherd DA, Hewitt D, Wildgoose JL, Beckman JS, Langridge JI, Voinov VG: Enhanced top-down protein characterization with electron capture dissociation and cyclic ion mobility spectrometry. Anal Chem 2022, 94: 3888–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brodbelt JS, Morrison L, Santos I: Ultraviolet photodissociation mass spectrometry for analysis of biological molecules. Chem Rev 2020, 120:3328–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hendricks NG, Julian RR: Leveraging ultraviolet photodissociation and spectroscopy to investigate peptide and protein three-dimensional structure with mass spectrometry. Analyst 2016, 141:4534–4540. [DOI] [PubMed] [Google Scholar]
- 62.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS: Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J Am Chem Soc 2013, 135: 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cleland TP, DeHart CJ, Fellers RT, VanNispen AJ, Greer JB, LeDuc RD, Parker WR, Thomas PM, Kelleher NL, Brodbelt JS: High throughput analysis of intact human proteins using UVPD and HCD on an Orbitrap mass spectrometer. J Proteome Res 2017, 16:2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greer SM, Brodbelt JS: Top-down characterization of heavily modified histones using 193 nm ultraviolet photodissociation (UVPD) mass spectrometry. J Proteome Res 2018, 17: 1138–1145. [DOI] [PubMed] [Google Scholar]
- 65. *.Sipe SN, Patrick JW, Laganowsky A, Brodbelt JS: Enhanced characterization of membrane protein complexes by ultraviolet photodissociation mass spectrometry. Anal Chem 2020, 92:899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]; UVPD provides high levels of sequence coverage and localization of PTMs for membrane proteins.
- 66.Mehaffey MR, Lee J, Jun J, Lanzillottti MB, Escobar EE, Morgenstern KR, Georgious G, Brodbelt JS: Mapping a conformational epitope of hemagglutinin a using native mass spectrometry and ultraviolet photodissociation. Anal Chem 2020, 92:11869–11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fornelli L, Srzentić K, Huguet R, Mullen C, Sharma S, Zabrouskov V, Fellers RT, Durbin KR, Compton PD, Kelleher NL: Accurate sequence analysis of a monoclonal antibody by top-down and middle-down Orbitrap mass spectrometry applying multiple ion activation techniques. Anal Chem 2018, 90:8421–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCool EN, Chen D, Li W, Liu Y, Sun L: Capillary zone electrophoresis-tandem mass spectrometry using ultraviolet photodissociation (213 nm) for large-scale top-down proteomics. Anal Methods 2019, 11:2855–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. *.Fornelli L, Srzentić K, Toby TK, Doubleday PF, Huguet R, Mullen C, Melani RD, Dos Santos Seckler H, DeHart CJ, Weisbrod CR, Durbin KR, Greer JB, Early BP, Fellers RT, Zabrouskov V, Thomas PM, Compton PD, Kelleher NL: Thorough performance evaluation of 213 nm ultraviolet photodissociation for top-down proteomics. Mol Cell Proteomics 2020, 19:405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]; Performance of 213 nm UVPD for characterization of intact proteins is benchmarked.
- 70.Wei L, Gregorich ZR, Lin Z, Cai W, Jin Y, McKiernan SH, McIlwain S, Aiken JM, Moss RL, Diffee GM, Ge Y: Novel sarcopenia-related alterations in sarcomeric protein post-translational modifications (PTMs) in skeletal muscles identified by top-down proteomics. Mol Cell Proteomics 2018, 17: 134–145. ECD FTICR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. *.Wilkins JT, Seckler HS, Rink J, Compton PD, Fornelli L, Thaxton CS, LeDuc R, Jacobs D, Doubleday PF, Sniderman A, Lloyd-Jones DM, Kelleher NL: Spectrum of apolipoprotein AI and apolipoprotein AII proteoforms and their associations with indices of cardiometabolic health: the CARDIA study. J Am Heart Assoc 2021, 10, e019890. [DOI] [PMC free article] [PubMed] [Google Scholar]; Extensive characterization of PTMs of apolipoproteins from serum samples by top-down analysis.
- 72. *.He Lidong, Rockwood Alan L, Agarwal Archana M, Anderson Lissa C, Weisbrod Chad R, Hendrickson Christopher L, Marshall Alan G: Diagnosis of hemoglobinopathy and β-thalassemia by 21 tesla fourier transform ion cyclotron resonance mass spectrometry and tandem mass spectrometry of hemoglobin from blood. Clin Chem 2019, 65:986–994. [DOI] [PubMed] [Google Scholar]; Top-down analysis of hemoglobin variants from blood allowed diagnosis of β-thalassemia.
- 73.Lin Z, Wei L, Cai W, Zhu Y, Tucholski T, Mitchell SD, Guo W, Ford SP, Diffee GM, Ge Y: Simultaneous quantification of protein expression and modifications by top-down targeted proteomics: a case of the sarcomeric subproteome. Mol Cell Proteomics 2019, 18:594–605. CID. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jin Y, Lin Z, Xu Q, Fu C, Zhang Z, Zhang Q, Pritts WA, Ge Y: Comprehensive characterization of monoclonal antibody by Fourier transform ion cyclotron resonance mass spectrometry. MAbs 2019, 11:106–115. ECD CID. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. *.Tucholski T, Cai W, Gregorich ZR, Bayne EF, Mitchell SD, McIlwain SJ, de Lange WJ, Wrobbel M, Karp H, Hite Z, Vikhorev PG, Marston SB, Lal S, Li A, Dos Remedios C, Kohmoto T, Hermsen J, Ralphe JC, Kamp TJ, Moss RL, Ge Y: Distinct hypertrophic cardiomyopathy genotypes result in convergent sarcomeric proteoform profiles revealed by top-down proteomics. Proc Natl Acad Sci U S A 2020, 117: 24691–24700. [DOI] [PMC free article] [PubMed] [Google Scholar]; Top-down characterization of sarcomeric proteoforms from heart tissue confirms signatures of hypertrophic cardiomyopathy.
- 76. *.Rezinciuc S, Tian Z, Wu S, Hengel S, Pasa-Tolic L, Smallwood HS: Mapping influenza-induced posttranslational modifications on histones from CD8+ T cells. Viruses 2020, 12:1409. [DOI] [PMC free article] [PubMed] [Google Scholar]; Histones from CD8 T cells reveal altered patterns of PTMs after in vivo influenza infection.
- 77.Williams JP, Morrison LJ, Brown JM, Beckman JS, Voinov VG, Lermyte F: Top-down characterization of denatured proteins and native protein complexes using electron capture dissociation implemented within a modified ion mobility-mass spectrometer. Anal Chem 2020, 92:3674–3681. ECD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. *.Adams LM, DeHart CJ, Kelleher NL: Precise characterization of KRAS4B proteoforms by combining immunoprecipitation with top-down mass spectrometry. Methods Mol Biol 2021, 2262:47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]; Detailed protocol for top-down analysis of KRAS4B proteoforms from cancer cell lines and tissue samples.
- 79.Greisch JF, den Boer MA, Lai SH, Gallagher K, Bondt A, Commandeur J, Heck AJR: Extending native top-down electron capture dissociation to MDa immunoglobulin complexes provides useful sequence tags covering their critical variable complementarity-determining regions. Anal Chem 2021, 93: 16068–16075. [DOI] [PMC free article] [PubMed] [Google Scholar]; Top-down analysis of MDa immunoglobulins via ECD provides insight into complementarity-determining regions.
- 80.Jeanne Dit Fouque K, Kaplan D, Voinov VG, Holck FHV, Jensen ON, Fernandez-Lima F: Proteoform differentiation using tandem trapped ion mobility, electron capture dissociation, and ToF mass spectrometry. Anal Chem 2021, 93: 9575–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. *.Roberts DS, Mann M, Melby JA, Larson EJ, Zhu Y, Brasier AR, Jin S, Ge Y: Structural O-glycoform heterogeneity of the SARS-CoV-2 spike protein receptor-binding domain revealed by top-down mass spectrometry. J Am Chem Soc 2021, 143: 12014–12024. [DOI] [PMC free article] [PubMed] [Google Scholar]; High resolution elucidation of O-glycan proteoforms of S protein receptor binding domain of severe acute respiratory syndrome coronavirus 2
- 82.Fulcher JM, Makaju A, Moore RJ, Zhou M, Bennett DA, De Jager PL, Qian WJ, Paša-Tolić L, Petyuk VA: Enhancing top-down proteomics of brain tissue with FAIMS. J Proteome Res 2021, 20:2780–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. *.Melani RD, Gerbasi VR, Anderson LC, Sikora JW, Toby TK, Hutton JE, Butcher DS, Negrao F, Seckler HS, Srzenti K, Fornelli L, Camarillo JM, LeDuc RD, Cesnik AJ, Lundberg E, Greer JB, Fellers RT, Robey MT, DeHart CJ, Forte E, Hendrickson CL, Abbatiello SE, Thomas PM, Kojaji AI, Levitsky J, Kelleher NL: The Blood Proteoform Atlas: a reference map of proteoforms in human hematopoietic cells. Science 2022, 375:411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]; Top-down analysis maps 30,000 proteoforms from 1690 human genes covering 21 cell types and plasma from human blood and bone marrow.
- 84.Shaw JB, Cooper-Shepherd DA, Hewitt D, Wildgoose JL, Beckman JS, Langridge JI, Voinov VG: Enhanced top-down protein characterization with electron capture dissociation and cyclic ion mobility spectrometry. Anal Chem 2022, 94: 3888–3896. ECD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cannon JR, Holden DD, Brodbelt JS: Hybridizing ultraviolet photodissociation with electron transfer dissociation for intact protein characterization. Anal Chem 2014, 86: 10970–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wongkongkathep P, Li H, Zhang X, Loo RR, Julian RR, Loo JA: Enhancing protein disulfide bond cleavage by UV excitation and electron capture dissociation for top-down mass spectrometry. Int J Mass Spectrom 2015, 390:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shliaha PV, Gibb S, Gorshkov V, Jespersen MS, Anderson GR, Bailey D, Schwartz J, Eliuk S, Schwammle V, Jensen ON: Maximizing sequence coverage in top-down proteomics by automated multimodal gas-phase protein fragmentation. Anal Chem 2018, 90:12519–12526. [DOI] [PubMed] [Google Scholar]
- 88.Fornelli L, Srzentić K, Huguet R, Mullen C, Sharma S, Zabrouskov V, Fellers RT, Durbin KR, Compton PD, Kelleher NL: Accurate sequence analysis of a monoclonal antibody by top-down and middle-down Orbitrap mass spectrometry applying multiple ion activation techniques. Anal Chem 2018, 90:8421–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Riley NM, Sikora JW, Seckler HS, Greer JB, Fellers RT, LeDuc RD, Westphall MS, Thomas PM, Kelleher NL, Coon JJ: The value of activated ion electron transfer dissociation for high-throughput top-down characterization of intact proteins. Anal Chem 2018, 90:8553–8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.He L, Anderson LC, Barnidge DR, Murray DL, Dasari S, Dispenzieri A, Hendrickson CL, Marshall AG: Classification of plasma cell disorders by 21 tesla fourier transform ion cyclotron resonance top-down and middle-down MS/MS analysis of monoclonal immunoglobulin light chains in human serum. Anal Chem 2019, 91:3263–3269. [DOI] [PubMed] [Google Scholar]
- 91.Shaw JB, Liu W, Vasil Ev YV, Bracken CC, Malhan N, Guthals A, Beckman JS, Voinov VG: Direct determination of antibody chain pairing by top-down and middle-down mass spectrometry using electron capture dissociation and ultraviolet photodissociation. Anal Chem 2020, 92:766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Foreman DJ, McLuckey SA: Recent developments in gas-phase ion/ion reactions for analytical mass spectrometry. Anal Chem 2020, 92:252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stephenson JL, McLuckey SA: Ion/ion proton transfer reactions for protein mixture analysis. Anal Chem 1996, 68: 4026–4032. [DOI] [PubMed] [Google Scholar]
- 94.Anderson LC, Karch KR, Ugrin SA, Coradin M, English AM, Sidoli S, Shabanowitz J, Garcia BA, Hunt DF: Analyses of histone proteoforms using front-end electron transfer dissociation-enabled Orbitrap instruments. Mol Cell Proteomics 2016, 15:975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Holden DD, McGee W, Brodbelt JS: Integration of ultraviolet photodissociation with proton transfer reactions and ion parking for analysis of intact proteins. Anal Chem 2016, 88: 1008–1016. [DOI] [PubMed] [Google Scholar]
- 96. *.Ugrin SA, English AM, Syka JEP, Bai DL, Anderson LC, Shabanowitz J, Hunt DF: Ion-ion proton transfer and parallel ion parking for the analysis of mixtures of intact proteins on a modified Orbitrap mass analyzer. J Am Soc Mass Spectrom 2019, 30:2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]; Proton transfer reactions and parallel ion parking are used to consolidate multiple charge states of proteins into single charge states to enhance signal intensity in top-down analysis.
- 97.Huguet R, Mullen C, Srzentić K, Greer JB, Fellers RT, Zabrouskov V, Syka JEP, Kelleher NL, Fornelli L: Proton transfer charge reduction enables high-throughput top-down analysis of large proteoforms. Anal Chem 2019, 91: 15732–15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. *.Sanders JD, Mullen C, Watts E, Holden DD, Schwartz J,Brodbelt JS: Enhanced sequence coverage of large proteins by combining ultraviolet photodissociation with proton transfer reactions. Anal Chem 2020, 92:1041–1049. [DOI] [PubMed] [Google Scholar]; Sequence coverages of intact proteins were increased by combining UVPD and proton transfer reactions to alleviate congestion and uncover additional fragment ions.
- 99. *.Kline JT, Mullen C, Durbin KR, Oates RN, Huguet R, Syka JEP, Fornelli L: Sequential ion–ion reactions for enhanced gas-phase sequencing of large intact proteins in a tribrid Orbitrap mass spectrometer. J Am Soc Mass Spectrom 2021, 32: 2334–2345. [DOI] [PubMed] [Google Scholar]; Proton transfer reactions are used to reduce signal overlap in ETD and EThcD spectra for intact proteins.
- 100. *.Weisbrod CR, Anderson LC, Hendrickson CL, Schaffer LV, Shortreed MR, Smith LM, Shabanowitz J, Hunt DF: Advanced strategies for proton-transfer reactions coupled with parallel ion parking on a 21 T FT-ICR MS for intact protein analysis. Anal Chem 2021, 93:9119–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]; Significant gains in signal-to-noise ratios by concentrating multiple charge states of proteins into single charge states and also reduce congestion observed in ETD spectra.
- 101.Duselis EM, Panepinto MC, Syka JEP, Mullen C, D’Ippolito RA, English AM, Ugrin SA, Shabanowitz J, Hunt DF: Improved sequence analysis of intact proteins by parallel ion parking during electron transfer dissociation. Anal Chem 2021, 93: 15728–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Durbin KR, Skinner OS, Fellers RT, Kelleher NL: Analyzing internal fragmentation of electrosprayed ubiquitin ions during beam-type collisional dissociation. J Am Soc Mass Spectrom 2015, 26:782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen J, Shiyanov P, Green KB: Top-down mass spectrometry of intact phosphorylated b-casein: correlation between the precursor charge state and internal fragments. J Mass Spectrom 2019, 54:527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lyon YA, Riggs D, Fornelli L, Compton PD, Julian RR: The ups and downs of repeated cleavage and internal fragment production in top-down proteomics. J Am Soc Mass Spectrom 2018, 29:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. *.Zenaidee MA, Lantz C, Perkins T, Jung W, Loo RRO, Loo JA: Internal fragments generated by electron ionization dissociation enhance protein top-down mass spectrometry. J Am Soc Mass Spectrom 2020, 31:1896–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]; Internal ions may contribute up to 40% of mass spectral signals produced by electron ionization dissociation of intact proteins.
- 106.Schmitt ND, Berger JM, Conway JB, Agar JN: Increasing top-down mass spectrometry sequence coverage by an order of magnitude through optimized internal fragment generation and assignment. Anal Chem 2021, 93:6355–6362. [DOI] [PubMed] [Google Scholar]
- 107. *.Lantz C, Zenaidee MA, Wei B, Hemminger Z, Ogorzalek Loo RR, Loo JA: ClipsMS: an algorithm for analyzing internal fragments resulting from top-down mass spectrometry. J Proteome Res 2021, 20:1928–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]; Development of an algorithm to assign internal fragment ions in top-down mass spectra of proteins.
- 108. *.Zenaidee MA, Wei B, Lantz C, Lambeth TR, Diedrich JK, Ogorzalek Loo RR, Julian RR, Loo JA: Internal fragments generated from different top-down mass spectrometry fragmentation methods extend protein sequence coverage. J Am Soc Mass Spectrom 2021, 32:1752–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]; Combining internal and conventional terminal fragment ions provides greatest sequence coverage of proteins.
- 109.Wei B, Zenaidee MA, Lantz C, Ogorzalek Loo RR, Loo JA: Towards understanding the formation of internal fragments generated by collisionally activated dissociation for top-down mass spectrometry. Anal Chim Acta 2022, 1194, 339400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Robinson CV: Mass Spectrometry: from plasma proteins to mitochondrial membranes. Proc Natl Acad Sci U S A 2019, 116:2814–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barth M, Schmidt C: Native mass spectrometry-a valuable tool in structural biology. J Mass Spectrom 2020, 55:e4578. [DOI] [PubMed] [Google Scholar]
- 112.Li H, Nguyen HH, Ogorzalek Loo RR, Campuzano IDG, Loo JA: An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macroMol. complexes. Nat Chem 2018, 10:139–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA: Kelleher NL from protein complexes to subunit backbone fragments: a multi-stage approach to native mass spectrometry. Anal Chem 2013, 85:11163–11173. [DOI] [PubMed] [Google Scholar]
- 114.Ben-Nissan G, Belov ME, Morgenstern D, Levin Y, Dym O, Arkind G, Lipson C, Makarov AA, Sharon M: Triple-stage mass spectrometry unravels the heterogeneity of an endogenous protein complex. Anal Chem 2017, 89:4708–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mehaffey MR, Sanders JD, Holden DD, Nilsson CL, Brodbelt JS: Multi-stage ultraviolet photodissociation mass spectrometry to characterize single amino acid variants of human mitochondrial BCAT2. Anal Chem 2018, 90:9904–9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Skinner OS, Haverland NA, Fornelli L, Melani RD, Do Vale LHF, Seckler HS, Doubleday PF, Schachner LF, Srzentić K, Kelleher NL, Compton PD: Top-down characterization of endogenous protein complexes with native proteomics. Nat Chem Biol 2018, 14:36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. *.Mehaffey MR, Xia JQ, Brodbelt JS: Uniting native capillary electrophoresis and multistage ultraviolet photodissociation mass spectrometry for on-line separation and characterization of E. Coli ribosomal proteins and protein complexes. Anal Chem 2020, 92:15202–15211. [DOI] [PMC free article] [PubMed] [Google Scholar]; UVPD characterizes proteoforms in E. coli 70S ribosome using a multi-stage top-down MS/MS approach.
- 118.Greisch JF, den Boer MA, Lai SH, Gallagher K, Bondt A, Commandeur J, Heck AJR: Extending native top-down electron capture dissociation to MDa immunoglobulin complexes provides useful sequence tags covering their critical variable complementarity-determining regions. Anal Chem 2021, 93: 16068–16075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Harvey SR, O’Neale C, Schey KL, Wysocki VH: Native mass spectrometry and surface induced dissociation provide insight into the post-translational modifications of tetrameric AQP0 isolated from bovine eye lens. Anal Chem 2022, 94:1515–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Snyder DT, Harvey SR, Wysocki VH: Surface-induced dissociation mass spectrometry as a structural biology tool. Chem Rev 2022, 122:7442–7487, 10.1021/acs.chemrev.1c00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kafader JO, Durbin KR, Melani RD, Des Soye BJ, Schachner LF, Senko MW, Compton PD, Kelleher NL: Individual ion mass spectrometry enhances the sensitivity and sequence coverage of top-down mass spectrometry. J Proteome Res 2020, 19:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kafader JO, Melani RD, Durbin KR, Ikwuagwu B, Early BP, Fellers RT, Beu SC, Zabrouskov V, Makarov AA, Maze JT, Shinholt DL, Yip PF, Tullman-Ercek D, Senko MW, Compton PD, Kelleher NL: Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nat Methods 2020, 17:391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wörner TP, Snijder J, Bennett A, Agbandje-McKenna M, Makarov AA, Heck AJR: Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nat Methods 2020, 17:395–398. [DOI] [PubMed] [Google Scholar]
- 124.Kelly RT: Single-cell proteomics: progress and prospects. Mol Cell Proteomics 2020, 19:1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Slavov N: Single-cell protein analysis by mass spectrometry. Curr Opin Chem Biol 2021, 60:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]