

SUMMARY
Fungal infections in the central nervous system (CNS) cause high morbidity and mortality. The frequency of CNS mycosis has increased over the last two decades as more individuals go through immunocompromised conditions for various reasons. Nevertheless, options for clinical interventions for CNS mycosis are still limited. Thus, there is an urgent need to understand the host-pathogen interaction mechanisms in CNS mycosis for developing novel treatments. Although the CNS has been regarded as an immune-privileged site, recent studies demonstrate the critical involvement of immune responses elicited by CNS-resident and CNS-infiltrated cells during fungal infections. In this review, we discuss mechanisms of fungal invasion in the CNS, fungal pathogen detection by CNS-resident cells (microglia, astrocytes, oligodendrocytes, neurons), roles of CNS-infiltrated leukocytes, and host immune responses. We consider that understanding host immune responses in the CNS is crucial for endeavors to develop treatments for CNS mycosis.
Keywords: Fungal, Toll-like Receptors/Pattern Recognition Receptors, Neuroimmunology, microglia, astrocytes, neurons
1 |. INTRODUCTION
Incidences of invasive opportunistic fungal infections have risen dramatically over the last two decades and are expected to continuously rise because of increasing numbers of individuals with impaired immunity due to HIV/AIDS, genetic disorders, aging, and immunosuppressive therapies. Accordingly, clinical cases of fungal infections in the CNS are also increasing. CNS mycoses are particularly severe due to limited treatment options, causing high morbidity and mortality. Over 5 million fungal species are estimated to exist, and about 70,000 fungal species have been described1. Of these, only a few hundred are known to cause disease in humans2. A small number of fungi are known to have neurotropism, and such fungal genera include but are not limited to Cryptococcus, Candida, Aspergillus, Mucor, Rhizopus, Blastomyces, Coccidioides, and Histoplasma. Even with this relatively limited variety of known fungal pathogens, CNS mycosis is difficult to treat and causes devastating outcomes to patients.
When fungal clearance fails at the initial infection site, neurotropic fungi disseminate and enter the CNS through the bloodstream3. Although the blood-brain barrier (BBB) restricts the entrance of fungi into the CNS parenchyma, fungi use various tactics to breach the barrier. The host response will then depend on CNS-resident cells to recognize the presence of fungi that have infiltrated through the BBB. Host CNS-resident cells indeed express a wide range of pattern-recognition receptors (PRRs). Yet, fungi also use stealthy approaches to evade host detection. Even when detection of invading fungi is successful, immune responses in the CNS require tight and masterful regulation to limit collateral damages to CNS-resident cells. Concomitantly, leukocytes from the circulation are also recruited to the infected CNS, further challenging the fine-tuning of host immune responses. This review intends to highlight the urgency of basic and mechanistic understanding of CNS immune response in the context of fungal infections.
2 |. FUNGAL PATHOGENS THAT INFECT THE CNS
Candida, Cryptococcus, and Aspergillus are the three major fungal genera that typically cause fungal infections and disseminate to the CNS3–6. However, other fungi can also infect the CNS. These include non-Aspergillus molds, including fungi in the Mucorales order, and dimorphic fungi, such as Blastomyces, Coccidioides, and Histoplasma3,6,7.
2.1 |. Candida species
Candida is a genus of yeast fungi within the phylum Ascomycota and is the most common infectious fungi. Globally, it is estimated that there are ~750,000 cases of invasive candidiasis per year, with up to 40% mortality, depending on patients’ predisposing factors5. Candida species can be harmless commensals located on mucosal surfaces, mainly in the gastrointestinal, respiratory, reproductive tracts, and the skin. Yet, they can be pathogenic when the mucosal or skin barriers are breached or compromised. Candida shows morphologies of yeast, pseudohyphae, or true hyphae during infection8. The majority of Candida-related CNS infections are caused by hematogenous spread of Candida albicans and cause meningoencephalitis4. In addition to disseminated Candida infection (candidemia), Candida CNS infections can be caused by neurosurgery of contaminated surgical devices7. Less commonly, Candida infection can cause chronic meningitis, brain abscesses, vasculitis with cerebral infarctions, spinal infections, ventriculitis, and mycotic aneurysms.
2.2 |. Cryptococcus species
Cryptococcus is a genus of Basidiomycota fungi. They are yeast-like fungi but can also exhibit a hyphal state. However, they are not traditionally thought to be dimorphic since hyphal production typically occurs during the mating process. Cryptococcus yeasts are surrounded by a thick “capsule” consisting of polysaccharides, such as glucuronoxylomannan (GXM)9. The capsule enables Cryptococcus to evade immune recognition10. Currently, the global incidence of cryptococcal meningitis is about 223,100 cases per year and causes 15% of AIDS-related deaths globally5,11. Cryptococcus neoformans, an opportunistic fungus, is the most common species among the Cryptococcus genera. C. gattii is an endemic species, which infects immunocompetent individuals. An outbreak of C. gattii was reported two decades ago in the US Pacific Northwest12. Meningitis and meningoencephalitis are typical features of CNS cryptococcosis, most often caused by C. neoformans, and less commonly by C. gattii. CNS cryptococcosis often arises from a primary lung infection. Cryptococcus infection is a common complication of untreated HIV infection13, which can cause immune reconstitution inflammatory syndrome (IRIS) upon immune reconstitution by HIV antiretroviral therapy14,15.
2.3 |. Aspergillus species
Aspergillus fungi belong to Ascomycota, like Candida, but Aspergillus are conidial fungi, which go through asexual reproduction by forming conidia. Because Aspergillus conidia are airborne; lung infection through inhalation is the most common etiology of the infection16. Invasive aspergillosis is severe and aggressive. It is estimated that as many as 10 million individuals are at risk for invasive aspergillosis globally, with more than 300,000 cases reported yearly with an associated mortality range of 30–80%5,17. Aspergillus fumigatus typically causes CNS infections through hematogenous spread from the primary site of infection, mainly the lung, or from a contiguous anatomical site such as the paranasal sinuses4. Although Aspergillus meningitis is relatively rare, cases are found more frequently in immunocompetent than immunocompromised patients18.
2.4 |. Mucorales order
Mucorales, including Rhizopus and Mucor, are mold fungi that belong to the phylum Mucormycota, considered to be phylogenetically more primitive than Basidiomycota and Ascomycota. Globally, the prevalence of mucormycosis is greater than 10,000 cases5 with mortality rates of up to 95%19. The most common clinical presentation is rhino-orbital cerebral mucormycosis (ROCM), which are mainly caused by Rhizopus oryzae20. Although mucormycosis is relatively rare, immunocompromised individuals, as in hospital outbreaks, and diabetic patients are at high risk20. COVID-19-associated mucormycosis recently reported from India also raised a grave concern21. Routes of infection are inhalation, open wounds, and ingestion of contaminated food22. Cerebral mucormycosis is considered to be the most aggressive CNS fungal infection and uniformly fatal7. Nevertheless, how our immune system responds to Mucorales infections is still largely unknown.
2.5 |. Other neurotropic fungi (including dimorphic fungi)
Despite being less common than the fungi described above, other fungal pathogens can infect the CNS. Depending on the growth conditions, they include dimorphic endemic fungi that grow as yeast or as mold23. The group of fungi, including Blastomyces dermatitidis, Coccidioides spp., and Histoplasma capsulatum, can cause meningoencephalitis, hydrocephalus, brain or epidural abscesses, and spinal cord lesions6. Other groups of fungi that could infect the CNS are molds other than Aspergillus and Mucorales (including Fusarium, Scedosporium species, Dematiaceous molds)7 and yeast fungi other than Candida and Cryptococcus (including Sporothrix)24.
3 |. MECHANISMS OF CNS FUNGAL INVASION
Fungal infiltration into the CNS parenchyma predominantly occurs when host immune responses are inadequate in clearing pathogens at the primary infection site. To establish CNS infection, fungi need to penetrate the BBB, which consists of four different cell types: endothelial cells, pericytes, astrocytes, and microglia. The BBB exists at the interface of blood vessels and interstitial fluid in the brain and spinal cord, limiting microorganisms from entering the CNS. Pathogenic fungi secrete various molecules that induce host cell death, damaging host barriers. Fungi can also “hitchhike” host immune cells, i.e., taking advantage of host cells as carriers, to invade the CNS.
In this section, we discuss mechanisms of fungal dissemination by the three major CNS-infecting fungal genera, Cryptococcus, Candida, and Aspergillus. There are three proposed models of fungal BBB penetration; transcellular migration (transcytosis), paracellular migration, and the Trojan Horse model. Reports have demonstrated that at least some species in Cryptococcus, Candida, and Aspergillus employ these dissemination methods to infiltrate the CNS.
3.1 |. Transcellular migration (transcytosis)
Transcellular migration (termed transcytosis) is achieved by two different mechanisms. One is absorptive-mediated transcytosis (AMT), which does not require ligand-receptor binding but utilizes charge interactions25. The other mechanism is receptor-ligand mediated transcytosis (RMT), which requires specific binding between microbial ligand and a host cell receptor25. Transcytosis is not limited to CNS entry. Fungi take advantage of host cell transcytosis to exit the initial site of infection and cross the barriers to enter the bloodstream or lymphatics.
The transversal crossing of C. neoformans through brain microvascular endothelial cells (BMECs) is the essential step for developing cryptococcal meningoencephalitis (CM). Adhesion and transcytosis of cryptococci through human BMECs are observed, even with an acapusular mutant of C. neoformans26. As an example of RMT, hyaluronic acid (HA) on C. neoformans binds CD44 and ‘receptor of hyaluronan-mediated motility’ on human BMECs27,28 (Fig. 1A). Indeed, CD44-deficient mice show a reduced fungal burden of C. neoformans in the brain28. For cryptococci to attach to the brain endothelium, C. neoformans also secretes a metalloprotease, Mpr129. To assess the potency of Mpr1 in fungal dissemination, a study used Saccharomyces cerevisiae, which does not express Mpr1 and does not invade human BMECs30. Artificial expression of Mpr1 in S. cerevisiae successfully allowed the fungus to pass through the brain endothelium29. Because S. cerevisiae does not express HA, the result suggests that Mpr1 plays a critical role in transcytosis29. Another study showed that host Annexin A2 and its synergistic partner S100A10 mediate C. neoformans transcytosis31. In addition, C. neoformans releases microvesicles (termed “virulence bags”), containing polysaccharides, lipids, and cytoplasmic proteins32. The microvesicle contents fuse with host cells, as observed in human BMEC culture and in vivo mouse model of CNS infection32. Such microvesicles are also considered to be involved in the adhesion of C. neoformans to host BMECs. Furthermore, when C. neoformans crosses human BMEC monolayers, the host cell actin cytoskeleton is rearranged without affecting the integrity of the endothelial barrier26,33. Thus, without causing endothelial barrier disruption, C. neoformans can stealthily disseminate into the CNS without triggering host antifungal responses. Yet, a study suggested that C. neoformans ultimately induces endothelial cell necrosis34.
Figure 1. Mechanisms of fungal brain invasion.
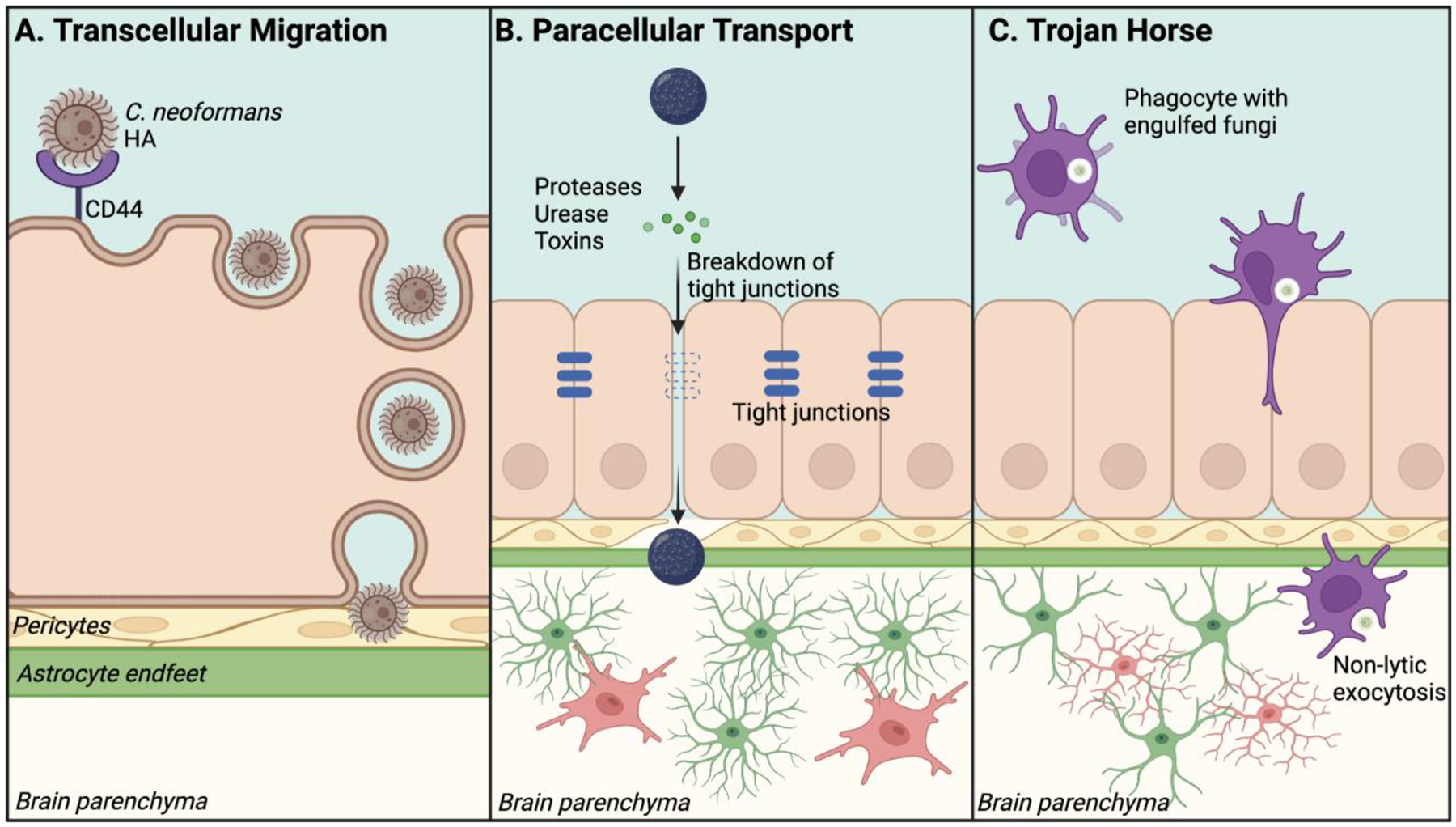
(A) Transcellular Migration: Interaction between a fungus and host barrier cells induces endocytosis of a fungus. Then, the fungus exits from host barrier cells and crosses the BBB. Shown is an example that HA in C. neoformans is detected by CD44 on BMEC to initiate receptor-ligand mediated transcytosis (RMT). (B) Paracellular Transport: Molecules secreted by fungi (e.g., proteases, urease) disrupt tight junctions between barrier cells to breach the BBB integrity. This allows the fungus to migrate across the endothelial layer into the brain parenchyma. (C) Trojan horse: Peripheral phagocytes uptake fungal spores or yeasts and migrate to the brain. Spores or yeasts are released from phagocytes once phagocytes migrate into the CNS. Created with BioRender.com
C. albicans express fungal invasins to achieve brain invasion. One invasin is Als3, which binds a heat shock protein, gp96, expressed specifically on the surface of brain endothelial cells and promotes Candida uptake by the host cells35. Als3 also interacts with human cadherins to aid Candida endocytosis, as described in a study using human umbilical vein endothelial cells and human oral epithelial lines36. Epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) cooperatively induce endocytosis of C. albicans in an Als3-dependent manner37, which complements the cadherin-dependent mechanism for endocytosis. These studies suggested Als3 as a critical molecule for Candida transcytosis. Independent of the yeast-mediated mechanism, the hyphal form of C. albicans also contributes to the loss of BBB integrity in disseminated candidasis38. C. albicans can pass through the BBB by developing pseudo-hyphae, a virulence mechanism that does not elicit host cell morphological changes and enables the fungus to adhere, invade, and transcytose across human BMEC without affecting monolayer integrity30.
Transcytosis of A. fumigatus across the BBB and infiltration into the CNS remains to be addressed. Yet, it is reported that sialic acid-specific lectin on A. fumigatus conidia adheres to fibrinogen or laminin on host cells39. Additionally, galactosaminogalactan, a cell wall-associated polysaccharide of A. fumigatus, has been identified as an adhesin40. Endocytosis of A. fumigatus conidia was identified in the A549 human lung epithelial cell line, human umbilical vein endothelial cells, and the J774 murine macrophage line41. Conidia internalization is promoted by fungal gliotoxin, which activates the actin cytoskeleton of host cells42. This mechanism is implicated in A. fumigatus transcytosis because gliotoxin alters the BBB integrity, at least ex vivo, without disrupting tight junction complexes43. Once A. fumigatus gains access to the brain, conidia germinate and establish CNS infection.
3.2 |. Paracellular transport
In paracellular transport, microbes pass through barriers between epithelial cells by disrupting tight junctions without damaging host cells (Fig. 1B). Like transcytosis, paracellular CNS penetration also requires the attachment of fungi to the host cells before they are transferred.
C. neoformans employs paracellular transport44,45, although paracellular transport is likely a minor route for migration across the BBB34. Ureases, cryptococcal virulence factors46, compromises barrier cell tight junction integrity47. The approach is also used by C. albicans, which proteolytically degrades host E-cadherin to pass through epithelial cells, as reported in a study using oral epithelial cells48. To target E-cadherin, C. neoformans uses host IL-33, which is highly expressed during pulmonary cryptococcosis49. The study demonstrated that E-cadherin downregulation depends on IL-3349.
Electron microscopy captured that A. fumigatus conidia, which are not engulfed by host immune cells, can form extracellular hyphae without lysing host cells50.A. fumigatus produces proteases that disrupt the actin cytoskeleton in A549 cells51. A. fumigatus also produces mycotoxins, such as gliotoxin42, which are implicated in epithelium modifications during infection through paracellular transport, as well as transcytosis.
3.3 |. Trojan Horse Mechanism
Phagocytosis is a crucial mechanism in the clearance of pathogens. Yet paradoxically, phagocytic cells can become vehicles for fungal dissemination by serving as “Trojan Horses”52,53 (Fig. 1C), because phagocytic host cells can pass through the BBB under certain conditions. As previously described, C. neoformans has a thick polysaccharide capsule, which can protect yeasts against reactive oxygen species (ROS) generated by host cells54. The capsule enables C. neoformans to survive within the phagosome of human and mouse mononuclear phagocytes55–57. Studies have demonstrated that monocytes containing C. neoformans are present in the perivascular space of the brain vasculature of infected mice53,58 and transmigrate endothelial cell layers for C. neoformans to gain access to the brain59. Depletion of monocytes or neutrophils with clodronate liposomes or Ly6G antibody, respectively, reduces cryptococcal burdens in brains of infected mice53. Once C. neoformans crosses the BBB, the fungus exits macrophages60 and neutrophils61 via non-lytic exocytosis without triggering host cell death-associated inflammation60,61. Less is known about Candida and Aspergillus dissemination through the Trojan Horse mechanisms. However, a recent in vivo zebrafish study supported the Trojan Horse mechanism by demonstrating dissemination of C. albicans yeasts by using neutrophils as vehicles to invade the CNS62. A. fumigatus can also survive within phagocytes41, possibly due to the release of mycotoxins inhibiting macrophage oxidative burst63. Thus, Candida and Aspergillus may use the Trojan Horse mechanism, but further investigation is necessary to determine if these fungi use this mechanism to infiltrate the CNS.
4 |. PATHOGEN DETECTION BY CNS-RESIDENT CELLS
In this section, we describe the capacity of CNS-resident cells to detect pathogens through PRRs. Although peripheral innate immune cells express PRRs, studies have shown that CNS-resident cells also express a variety of PRRs and respond to PRR signaling, including the production of chemokines and cytokines. PRRs recognize pathogen-associated molecular patterns (PAMPs), but PRRs also detect damage-associated molecular patterns (DAMPs), which are released by host cells that are injured, stressed, or died due to inflammation and tissue insults. This section focuses on PRR expression and responses by CNS-resident cells (Table 1) in fungal and other microbial infections. In some cases, we also described PRR signaling activated by endogenous molecules.
Table 1.
PRR Expression in CNS-Resident Cells
| Stimulators/Ligands Described | CNS-Resident Cell | References | |
|---|---|---|---|
| Toll-like Receptors (TLRs) | |||
| TLR1 Dimerizes with TLR2 |
Triacyl lipopeptides α-synuclein |
Microglia | 72–75 |
| Astrocytes | 75,165,166 | ||
| Neurons | 202 | ||
| TLR2 | Peptidoglycan (PGN) Lipopeptides Lipoteichoic acid (LTA) Pam3CSK4 Zymosan |
Microglia | 72–75 |
| Astrocytes | 172 | ||
| Oligodendrocytes | 227,228 | ||
| Neurons | 204,209,210 | ||
| TLR3 | poly(I:C) | Microglia | 72–75,96,97 |
| Astrocytes | 75,165,166 | ||
| Oligodendrocytes | 228 | ||
| Neurons | 206,211,212 | ||
| TLR4 | Lipopolysaccharide (LPS) Neuraminidase (NA) 4-hydroxynonenal (HNE) |
Microglia | 72–75,86,88,89 |
| Astrocytes | 75,165,166,168,172 | ||
| Oligodendrocytes | 229 | ||
| Neurons | 204,210 | ||
| TLR5 | Flagellin | Microglia | 72–75,106–109 |
| Astrocytes | 75,165,166,172 | ||
| Neurons | 106,211,212 | ||
| TLR6 Dimerizes with TLR2 |
Diacyl lipopeptide | Microglia | 72–75 |
| Astrocytes | 75,165,166 | ||
| Neurons | 202 | ||
| TLR7 | ssRNA Imidazoquinolinone R848 Imiquimod |
Microglia | 72–76,100,101 |
| Astrocytes | 75,165,166,181 | ||
| Neurons | 205 | ||
| TLR8 | ssRNA Imidazoquinolinone R848 |
Microglia | 72–75 |
| Astrocytes | 75,165,166 | ||
| Neurons | 213 | ||
| TLR9 | CpG-DNA | Microglia | 72–75,214 |
| Astrocytes | 75,165,166,172,181 | ||
| Neurons | 214 | ||
| TLR10 (human) | Not determined | Microglia | 74,75 |
| TLR11 (murine) | Uropathogenic bacteria Profilin-like molecule from T. gondii |
Microglia | 72,73 |
| Astrocytes | 75,165,166,184 | ||
| Neurons | 184 | ||
| TLR12 (murine) | Not determined | Microglia | 72,73 |
| Astrocytes | 75,165,166 | ||
| TLR13 (murine) | Not determined | Microglia | 72,73 |
| Astrocytes | 75,165,166 | ||
| Neurons | 73 | ||
| C-type Lectin Receptors (CLRs) | |||
| DCIR | Not determined | Microglia | 285 |
| DC-SIGN | Mannose-containing glycoproteins Fucose Myelin/MOG |
Microglia | 116 |
| Mincle | SAP130 α-mannose |
Microglia | 286 |
| Neurons | 215,216 | ||
| Mannose Binding Lectin (MBL) | Mannose Fucose N-acetylglucosamine Amyloid β peptides |
Microglia | 187 |
| Astrocytes | 187 | ||
| Oligodendrocytes | 187 | ||
| Neurons | 186,187 | ||
| Mannose Receptor (MR) | Mannose Fucose N-acetylglucosamine |
Microglia | 287 |
| Astrocytes | 186 | ||
| Neurons | 186 | ||
| Dectin-1 | β-glucans Zymosan |
Microglia | 113,124,127 |
| NOD-like Receptors (NLRs) | |||
| NOD1 | Diaminopimelic acid (DAP) | Microglia | 137,138 |
| Astrocytes | 188 | ||
| Neurons | 188,217 | ||
| NOD2 | Muramyl dipeptide (MDP) | Microglia | 137,138 |
| Astrocytes | 188 | ||
| NLRC4 (IPAF) | Palmitate | Microglia | 147 |
| Astrocytes | 146 | ||
| Neurons | 219 | ||
| NLRC5 (NOD4) | Not determined | Microglia | 147 |
| Astrocytes | 288 | ||
| Neurons | 288 | ||
| NLRX1 (NOD5) | Not determined | Neurons | 221 |
| NLRP1 | Not determined | Microglia | 144 |
| Neurons | 218–220 | ||
| NLRP2 | ATP | Astrocytes | 190 |
| NLRP3 | Lysophosphatidylcholine (LPC) Nigericin |
Microglia | 144–147 |
| Astrocytes | 146 | ||
| RIG-I-like Receptors (RLRs) | |||
| RIG-I | poly(I:C) |
Microglia | 148 |
| Astrocytes | 195–197 | ||
| Neurons | 208,224,225 | ||
| MDA5 | poly(I:C) | Microglia | 148 |
| Astrocytes | 195–198 | ||
| Neurons | 208,224 | ||
| AIM2-like Receptors (ALRs) and other PRRs | |||
| AIM2 | Poly(dA:dT) dsDNA |
Microglia | 151,152,223 |
| Astrocytes | 145,223 | ||
| Neurons | 219,222,223 | ||
| cGAS-STING | dsDNA | Microglia | 154 |
4.1 |. Microglia
Microglia are CNS-resident macrophage-like cells and makeup 5–12% of brain cells in mice64 and 0.5–16.6% of brain cells in humans65. Microglia are involved in various stages of CNS biology, such as CNS development, homeostasis, and neuroinflammation. Microglia also serve as CNS sentinel cells to detect infections with highly developed phagocytic activity as macrophage-like cells. For the origin of microglia, monocytes from the circulation were previously considered to differentiate into microglia in the CNS. However, microglia are ontogenically distinct from other mononuclear phagocytes, including peripheral macrophages, and do not arise from hematopoietic stem cells in the bone marrow66–68. A parabiosis approach clarified that microglia progenitors are not recruited from the circulation68. Instead, in mice, microglia arise from the yolk sac at embryonic day 8.5–9.5 and migrate to the brain before the closure of the BBB66,67. Microglia self-renew after being seeded in the brain and are maintained throughout life independently of the hematopoietic system66,67. Unlike other macrophages, microglia resist high-dose irradiation69,70. Yet, like peripheral macrophages, microglia express a broad range of PRRs, detect pathogens, participate in antigen presentation, and secrete various soluble molecules to communicate with other cells in the CNS71. As CNS-resident macrophage-like cells, microglia are the best-studied CNS-resident cells regarding PRRs (Table 1; Fig. 2A). However, a limited number of previous studies used in vivo microglia-specific mutant systems. Therefore, it is possible that some PRRs, which were previously identified in microglial tissue culture, might not be functional in vivo, although this issue also applies to CNS-resident cells other than microglia.
Figure 2. Pattern Recognition Receptors identified in CNS-resident cells.
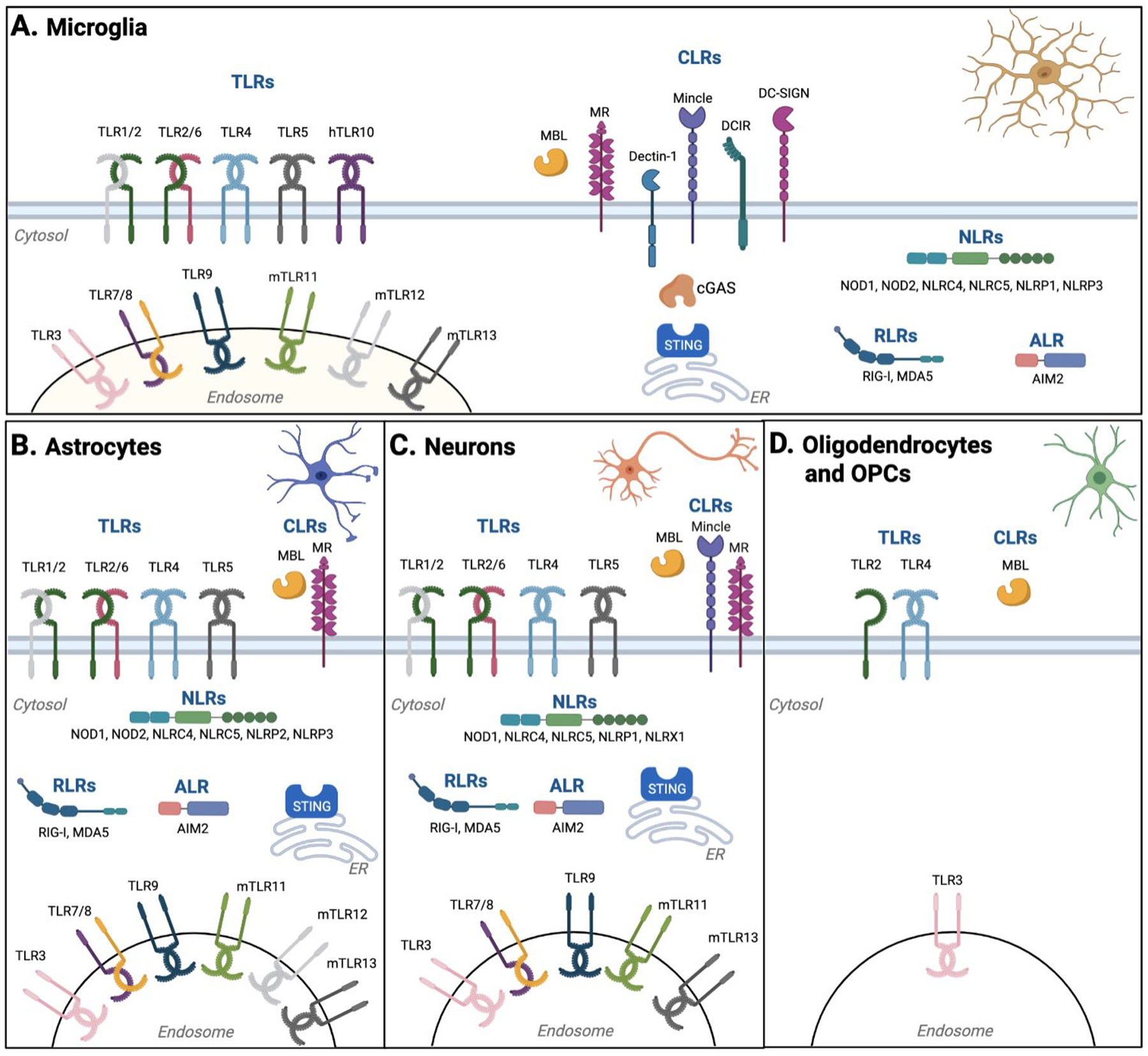
CNS-resident cells express a variety of PRRs to detect DAMPs and PAMPs. (A) Microglia, as CNS-resident macrophage-like cells, express an extensive repertoire of PRRs, including TLRs, CLRs, NLRs, RLRs, and ALR. (B, C) Compared to microglia, less variety of PRRs, particularly CLRs, was reported in astrocytes (B) and neurons (C). (D) A limited variety of PRRs in oligodendrocytes (and oligodendrocyte progenitor cells) have been reported. Created with BioRender.com.
4.1.1: TLRs in microglia
Microglia constitutively express Toll-like receptors (TLRs), such as TLR1–9, TLR11–13 in mice72,73 and TLR1–10 in humans74,75, together with their adaptor proteins. Expression levels of various TLRs depend on the localization of microglia, age of hosts, and the condition of the CNS, i.e., setting of neurodegenerative diseases or infections72,73,75,76. Microglial TLRs have been studied as inducers of proinflammatory responses against microbial pathogens. The functions of microglial TLRs have become a focus of intensive research. Together with C-type lectin receptors (CLRs), TLRs are critical for host fungal recognition. Studies on peripheral macrophages have revealed that fungal PAMPs are recognized by multiple TLRs (i.e., TLR2/1, TLR4, TLR3, TLR2/6, TLR7, and TLR9)77. Among these TLRs, TLR2, 4, and 9 have been most intensively studied as detectors of fungal pathogens, including species of Candida, Cryptococcus, and Aspergillus78–85, although further microglia-specific assessments of these TLRs will merit better understanding of CNS fungal infections.
Cell surface TLRs, including TLR4, are receptors that use Myeloid Differentiation primary response 88 (MyD88) as a key adaptor protein, which initiates activation of the transcription factors, mainly NFκB. TLR4 signaling induces the expression of proinflammatory cytokines72. In addition to well-known lipopolysaccharide (LPS), microglial TLR4 signaling is activated with neuraminidase (NA), a component of the envelope and membrane of some viruses and bacteria, respectively86. Because C. albicans also expresses NA87, microglial TLR4 may also be involved in Candida detection. An ex vivo study showed that TLR4 stimulation with LPS induces proinflammatory gene expression, such as Il1b, Il6, and Nos2, in primary microglia, although the same concentration of peptidoglycan, stimulating TLR2, induced expression of these genes to a slightly lesser extent72. More recent studies implicated enhanced microglial pyroptosis in response to TLR4 stimulation during spinal cord injury88. TLR4 inhibition induced microglia to have an “M2” phenotype89, characterized by the expression of Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) and IL-10, which may promote angiogenesis and prevent neuronal death90. TLR2 is also a cell surface TLR and forms a heterodimer with either TLR1 or TLR6. TLR2/1 and TLR2/6 detect triacyl lipopeptides and diacyl lipopeptides, respectively. TLR2/1 and TLR2/6 heterodimers on mouse macrophages detect GXM, a capsule component of C. neoformans91. However, A. fumigatus is detected only by TLR2/1 on human macrophages, despite mouse macrophages detecting A. fumigatus through both TLR2/1 and TLR2/692. For detecting CNS endogenous molecules, microglial TLR2 detects neuron-derived α-synuclein to enhance neuroinflammation93, where the involvement of TLR2/1 heterodimer is suggested94. Nevertheless, preferential formation of TLR2/1 or TLR2/6 and specific outcomes of a heterodimer have been largely unexplored in microglia.
TLR3, TLR7, TLR8, and TLR9 are localized in the endosome, recognize nucleic acids, and are extensively studied in eliciting antiviral responses through type-I interferon (IFN-I) production95. TLR3 detects double-stranded RNA (dsRNA) and depends on TRIF for downstream signaling as endosomal TLR4 does, while other endosomal TLRs require MyD88 for eliciting their signaling. Tlr3 mRNA is constitutively expressed in microglia96. Primary microglia in culture respond to TLR3 stimulation with poly(I:C), by inducing gene expression, including Tlr3 and Ifnb, and secreting Tumor Necrosis Factor alpha (TNFα), IL-6, and IL-12p4097. Microglia in Tlr3−/− mice did not undergo microgliosis, an intensive reaction of microglia to pathogenic insults, after a poly(I:C) intracerebroventricular (i.c.v.) injection, while in wild-type mice, the treatment induced microgliosis97. This study strongly suggested that TLR3 on microglia is biologically functional in vivo97. Unexpectedly, Tlr3−/− mice are more resistant than wild-type mice to encephalitic viral infections by West Nile virus (WNV) and a specific setting of Theiler’s murine encephalomyelitis virus infections96,98. The detrimental role of TLR3 in WNV infection, at least, could be explained by TLR3-dependent inflammatory response, which allows CNS infiltration of WNV and neuronal injury96. Although the role of microglial TLR3 is not known in fungal infections, dsRNA from A. fumigatus conidia activates lung epithelial cells through TLR399. Thus, TLR3 on microglia may also detect A. fumigatus conidia. However, it is unknown if microglial TLR3 is a beneficial or detrimental PRR in fungal infections.
Functions of other TLRs in microglia are still largely unexplored in fungal infection settings. Here, we briefly describe previous studies on roles of the microglial TLRs. The following studies were not performed in the context of fungal infections, but the findings might be extrapolated to possible outcomes when these TLRs are stimulated by fungi. TLR7 detects single-stranded RNA (ssRNA), and its expression has been observed in human76 and mouse microglia100 in numerous neurodegenerative diseases. In vivo administration of R848, a TLR7 agonist, induces acute sickness associated with CNS proinflammatory gene expression and microglia morphology changes101; thus, TLR7 is functional in microglia in vivo. TLR8 also recognizes ssRNA and is structurally related to TLR7. TLR8 is expressed in microglia72, but its function has yet to be addressed. TLR9 detects microbial and endogenous dsDNA. Previous studies suggested that TLR9 stimulation has substantial impacts on the CNS102–105, but it is still unknown if and how microglial TLR9 is involved in fungal infections. Additionally, although it is clear that TLR9 is expressed and functional in microglia, experimental systems to clarify the involvement of microglia-specific TLR9, such as using TLR9 conditional knockout mice, will clarify the implication of TLR9 stimulation in microglia in vivo. Other microglial TLRs reported include TLR5, which detects bacterial flagellin. TLR5 expression has been described in human and mouse microglia72,76, and its functional relevance has been explored in CNS disorders such as neuropathic pain, stroke, and Alzheimer’s disease (AD)106–108. Ex vivo microglial TLR5 stimulation with flagellin results in the release of proinflammatory cytokines, promoting chemotactic microglial migration, and enhancement of phagocytosis109. TLR10 is a human receptor, and its expression and function in microglia remain a subject for further research. TLR11–13 are murine TLRs, and their expression is elevated during murine neurocysticercosis, a parasitic infection that causes neurologic syndromes such as epileptic seizures73,110,111, but their functions in microglia remain unknown.
4.1.2: CLRs in microglia
Microglia also highly express a variety of CLRs, expressed primarily by myeloid cells (Table 1; Fig. 2A). In peripheral immune cells, dectin-1, dectin-2, MCL/dectin-3, Mincle, mannose receptor (MR), and Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) have been studied as detectors of fungal pathogens112. Among them, microglia express dectin-1, Mincle, MR, and DC-SIGN113–116. Dectin-2 does not appear to be expressed at detectable levels in human or murine microglia117. Dectin-1, encoded by the Clec7a gene, was the first CLR identified in macrophages to detect fungi118,119 through detection of β-glucans, cell wall components of fungi, some bacteria, and plants120. Dectin-1 on macrophages collaborates with TLR2 and MR for heightened fungal sensing121. Although dectin-1 is not required for detecting C. neoformans122, it is crucial to recognize Candida and Aspergillus118,123. Clec7a−/− mice infected with C. albicans showed increased fungal loads in the brain124. Thus, dectin-1 limits brain fungal loads in C. albicans infection but not in infections by C. glabrata, C. tropicalis, and C. parapsilosis124, suggesting that the role of dectin-1 in Candida is specific to species. Interestingly, a study showed that β-glucan stimulation is not sufficient for substantial production of cytokines or chemokines by microglia, although ROS generation and phagocytosis are actively induced125. In particular, the involvement of vav guanine nucleotide exchange factor 1 (Vav1) and phosphatidylinositol 3-kinase (PI3K) was identified in ROS generation by microglia126.
Dectin-1 is a critical receptor in eliciting host responses against fungal infections. Therefore, this section is intended to discuss microglial dectin-1 in neuropathological settings other than fungal infections. Clarifying if dectin-1 signaling in microglia shares common outcomes and mechanisms between CNS mycosis and CNS sterile inflammation will merit further studies. Recent studies have revealed the involvement of dectin-1 on microglia in various sterile and non-homeostatic settings. Microglia, in mice at least, have no or very low expression of dectin-1 during homeostasis, but its expression is significantly upregulated during neurodevelopment and under CNS pathological conditions113,127–130 (more details described in our previous review article131), and its expression is tightly upregulated in the spinal cord of mice with experimental autoimmune encephalomyelitis (EAE)113 and in the brain of aged APP-PS1 mice with β-amyloid accumulation127. Indeed, recent studies reported subsets of microglia, including microglial neurodegenerative phenotype (MGnD) and disease-associated microglia (DAM), highly express Clec7a in the brains of mice and humans in neuroinflammatory and neurodegenerative diseases127–129. Dectin-1-expressing microglia are associated with β-amyloid plaques and diffuse neuritic dystrophy in the cortex of AD model mice127. In addition, Clec7a upregulation was also identified in early postnatal proliferative region-associated microglia (PAM) during sterile CNS development130. PAM exhibit amoeboid morphology, are metabolically active, and phagocytose newly formed oligodendrocytes130. Thus, dectin-1 is highly expressed in microglia during pathological and neurodevelopmental settings independent of microbial infections131. Dectin-1 could function as an immunoregulatory receptor by detecting endogenous molecules in non-infectious settings113,132, and the immunoregulatory role of dectin-1 might become simultaneously active with the canonical proinflammatory role of dectin-1 during CNS fungal infections.
DC-SIGN in peripheral antigen-presenting cells recognizes mannose-containing glycoproteins on the surface of pathogens, such as C. albicans, Mycobacterium tuberculosis, and Helicobacter pylori133. Human microglia also express DC-SIGN116. Other studies demonstrated that DC-SIGN serves as a co-receptor for several viruses134,135. Unstimulated microglia show little to no expression of DC-SIGN, but DC-SIGN is detected in primary human microglia stimulated ex vivo116,136. DC-SIGN on dendritic cells (DCs) and microglia recognizes myelin, which is rich in carbohydrates and mediates internalization and endolysosomal routing of large myelin particles116. Costimulation of DC-SIGN and TLR4 enhances DC IL-10 production upon detecting myelin oligodendrocyte glycoprotein (MOG)116. The IL-10-producing immunoregulatory effect by DCs was abrogated when myelin had less fucosylated glycans present116. Thus, DC-SIGN may also play an immunoregulatory role in CNS fungal infection.
4.1.3: NLR in microglia
The nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) are intracellular sensors. The NOD subfamily of NLRs include NOD1 and NOD2, which recognize peptidoglycan motifs found mainly in bacteria. Expression of NOD1 and NOD2 was identified in microglia137,138. In pulmonary aspergillosis, NOD1 is detrimental to hosts by limiting host defense against Aspergillus139, but NOD2 is beneficial for host resistance140. NOD2 is also critical for the inflammatory response against neurotropic bacteria141,142.
The Nucleotide-binding oligomerization domain, Leucine-rich Repeat and Pyrin domain containing (NLRP) subfamily consists of intracellular sensors that mediate inflammasome activation by sensing a wide range of PAMPs and DAMPs. In particular, NLR Family Pyrin Domain Containing 3 (NLRP3) and NLR Family CARD Domain Containing 4 (NLRC4) inflammasomes detect Candida, Cryptococcus, Aspergillus, and Paracoccidioides (reviewed in143). Microglia express components necessary for the formation of inflammasomes, including NLRP1, NLRP3, pro-caspase-1, and ASC144. Expression of inflammasome components alone does not necessarily mean inflammasomes are activated or functional, because polymerization of inflammasome components is essential for inflammasome activation. For example, our lab recently reported that inflammasomes are not activated in microglia in the spinal cord during EAE145, although expression of NLRP3 inflammasome components is detected in microglia. Due to the technical limitation in detecting active inflammasomes in the CNS, most previous studies stopped short of demonstrating in vivo or in situ inflammasome activation in microglia and other CNS cells. Instead, many studies described the expression of inflammasome components or inflammasome activation ex vivo. For example, the NLRP3 inflammasome is activated ex vivo in microglia in response to lysophosphatidylcholine (LPC), a molecule associated with neurodegeneration and demyelination146. In a model of ischemia, NLRC5 is upregulated in microglia and directly binds to NLRP3 and NLRC4 in a nucleotide-binding domain-dependent manner to cooperatively drive microglial pyroptosis147.
4.1.4: RLRs, and other PRRs in microglia
RIG-I-like receptors (RLRs) are cytosolic RNA sensors, which trigger the production of IFN-I. The RLR family is composed of three members: retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2). Isolated mouse microglia constitutively express RIG-I and MDA5 transcripts and proteins148. Although RLRs have been extensively studied in the context of antiviral immunity, the involvement of RLRs in antifungal immunity has only recently been reported. One report using mouse macrophages and human PBMCs has shown that the yeast-to-hyphae transition of C. albicans induces expression of IFIH1, encoding for MDA5149. The study also showed that MDA5-deficient splenocytes failed to recognize C. albicans hyphae, resulting in reduced IFNβ expression149, suggesting recognition of C. albicans hyphae by MDA5. Furthermore, patients suffering from chronic mucocutaneous candidiasis have lower levels of IFIH1, suggesting the involvement of MDA5 in systemic candidiasis149. Based on the constitutive expression of MDA5 in microglia148, the involvement of microglial MDA5 in CNS candidiasis is expected.
AIM2-like receptors (ALRs) usually contain an N-terminal pyrin (PYD) domain, and one or two HIN domains. Absent In Melanoma 2 (AIM2) is a cytoplasmic DNA sensor, which forms an inflammasome upon detecting exogenous and endogenous dsDNA. Although the study focused on DCs in mice, A. fumigatus induces the formation of the AIM2 inflammasome complex, which also includes NLRP3 and caspase-8, in addition to canonical inflammasome components, ASC and caspase-1150. With this mechanism, the AIM2 inflammasome protects hosts from pulmonary aspergillosis150. Microglia express AIM2, and stimulation with poly(dA:dT) induces IL-1β and IL-18 release from microglia151, suggesting that the AIM2 inflammasome can be activated in microglia ex vivo. A more recent study showed that AIM2 in microglia limits EAE in an inflammasome-independent mechanism by negatively regulating cyclic GMP-AMP synthase (cGAS) and the DNA-dependent protein kinase (DNA-PK) pathways152. The study also demonstrated in an EAE setting that AIM2-deficient microglia induce the expression of genes related to immune responses against virus infections152. Thus, the study suggested that AIM2 in microglia negatively regulates inflammatory immune responses without forming an inflammasome. It is unknown if the inflammasome-independent AIM2 in microglia is functional in CNS fungal infections.
The cGAS- Stimulator of Interferon Genes (STING) pathway senses cytosolic dsDNA and induces IFN-I production by IRF3 activation. Although the study focused on human corneal epithelial cells (HCECs) and mouse corneas, cGAS-STING contributes to the progression of A. fumigatus keratitis153. Yet, it is not known which source of DNA, host or A. fumigatus, stimulates the cGAS-STING pathway. Another study identified the cGAS-STING pathway activation in Iba1-positive cells in the brain of mice in the middle cerebral artery occlusion (MCAO) model154, suggesting the possible presence of the cGAS-STING pathway in microglia. Using a microglia BV2 cell line, the study also demonstrated the activation of cGAS-STING to unspecified DAMPs154. Taken together, it is plausible that cGAS-STING activation in microglia occurs during CNS infection, but this possibility remains to be addressed.
4.2 |. Astrocytes
Arising from primary neural stem cells, astrocytes are the most abundant cell population in the CNS and contribute to CNS functions in health and disease. During CNS homeostasis, astrocytes are critical cells for the formation and maintenance of the BBB; controlling extracellular fluid, ions, and neurotransmitters; providing energy substrates to neurons; modulating blood flow; and regulating drainage of interstitial fluid155–158. Astrocytes also have roles in synapse development and plasticity159, and interact with various cells in the CNS. Despite significantly fewer reports on pathogen sensing by astrocytes than microglia, previous studies demonstrated the involvement of astrocytes in shaping the immunological milieu upon microbial infections and neurodegeneration113,160,161. In these pathological conditions, astrocytes undergo morphological and functional changes in a process known as reactive astrogliosis, in which the most common hallmarks include upregulation of glial fibrillary acidic protein (GFAP), the main component of astrocyte intermediate filaments and hypertrophy of astroglial processes162. During this process, astrocytes can form a glial scar around lesions during trauma or infection163. Additionally, astrocytes produce cytokines, chemokines, and growth factors, although astrocytes express less variation of PRRs than microglia (Table 1; Fig. 2B). Technical challenges in astrocyte handling, such as obtaining a highly pure astrocyte population and their susceptibility in ex vivo environments, hinder functional analyses of astrocytes. Their functions during fungal infections remain largely unknown. At least, the formation of an astrocytic border around lesions has been demonstrated in several mouse models of fungal infections28,164, although direct interactions between astrocytes’ PRRs and fungi have not been elucidated.
4.2.1: TLRs in astrocytes
Roles of astrocytic TLRs in fungal infections have yet to be explored, and limited information is available. In this subsection, we discuss previous findings, which provide knowledge to be considered when the roles of astrocytic TLRs in fungal infections are studied. The majority of TLR studies with astrocytes were performed ex vivo and focused on TLR expression or release of cytokines and chemokines by astrocytes in response to canonical TLR ligands. TLR expression under the homeostatic condition is reported in humans (TLR1–5, TLR9) and mice (TLR1–9 and TLR11–13) astrocytes75,165,166 (Table 1). As discussed below, astrocytes can substantially upregulate TLR expression levels; thus, TLRs in astrocytes may work secondary to PAMP detection by microglia, which are equipped with relatively high levels of TLRs without cell stimulation167.
TLR3 expression in human astrocytes is induced with various stimuli, including agonists of TLR3 and TLR4 and cytokines, such as TNFα, IL-6, IFNγ, and IL-12168. In contrast, IL-10, an anti-inflammatory cytokine, does not induce TLR3 expression168. After TLR3 stimulation, the expression of neuroprotective factors and anti-inflammatory cytokines, such as IL-10, are upregulated, and proinflammatory cytokine expression, such as IL-12p40 and IL-23, is downregulated in a human astrocyte tissue culture setting168. This results in inhibition of astrocyte growth, promotion of endothelial cell growth, and enhanced neuronal survival168. In an in vivo brain ischemia mouse model by MCAO, TLR3 agonist poly(I:C) attenuated reactive astrogliosis, reduced brain infarction volume, and improved neurological function169. These studies suggested that TLR3 in astrocytes could serve as an immunoregulatory receptor. However, another study indicated that TLR3 stimulation with poly(I:C) induced protein expression of TLR2, IL-6, C-C Motif Chemokine Ligand 5 (CCL5), Intercellular Adhesion Molecule 1 (ICAM-1), and Vascular Cell Adhesion Molecule 1 (VCAM-1) by primary mouse astrocytes in tissue culture170, suggesting TLR3 stimulation enhanced inflammation. The same study also demonstrated that TLR3-stimulated astrocytes slightly triggered antigen-specific CD4+ T cell activation ex vivo based on the evaluation of T cell proliferation and IFNγ production170. A more recent study indicated that human astrocytes treated with IFNγ (although TLR3 agonist was not tested), enhanced the expression of MHC class I and II, as well as co-stimulatory molecules (CD80/86/40)171. These studies suggest the involvement of TLR3 in modulating the CNS environment. However, further study is required to determine the involvement of astrocytic TLR3 in antigen-presentation in vivo as new antifungal host responses.
An early study using primary mouse astrocytes showed expression of Tlr2, 4, 5, and 9 and demonstrated that their gene expression levels could also be induced172. Various stimuli can induce TLR2 expression on astrocytes. For example, PGN, flagellin, and CpG-DNA highly induce Tlr2 expression in mouse astrocytes ex vivo172. Bacterial infection with Brucella melitensis also enhanced TLR2 expression in the white matter of rhesus macaques173. TLR2 on astrocytes recognizes Streptococcus suis, which causes meningitis, and induces expression of TLR2, TLR6 (but not TLR1), and a downstream proinflammatory response174,175. Another study showed that TLR2 expression on astrocytes depends on TNFα and NFκB pathways176. Multiple studies using primary astrocyte tissue culture demonstrated functional outcomes of TLR2 signaling on astrocytes. For example, stimulation of TLR2 with lipoteichoic acid induces the expression of matrix metalloproteinase 9 (MMP-9) in rat astrocytes177. Ex vivo treatment of mouse primary astrocytes with PGN triggers expression of TNFα, C-X-C Motif Chemokine Ligand 2 (CXCL2), and CCL4, and the involvement of TLR2 in the response was confirmed with Tlr2−/− astrocytes176. However, priming may be required for TLR2-mediated functions in astrocytes because TLR2 signaling needed pre-treatment of astrocytes with proinflammatory cytokines, such as TNFα, IFNγ, and IL-1β, for overt TLR2-mediated responses167. The study used primary mouse astrocytes in tissue culture167. Sensitization of TLR2 can also be achieved with conditioned media from microglia culture167, suggesting that astrocytes require crosstalk with microglia or CNS-infiltrated inflammatory cells to efficiently exert outcomes of TLR2 stimulation. For astrocytic TLR2 signaling activation, astrocyte priming may be necessary to induce TLR2 expression to a certain level.
TLR4 on astrocytes appears to be ready to detect ligands because of its constitutive expression. Tlr4 mRNA expression levels without stimulation are already higher than those of Tlr1, Tlr2, and Tlr3 in human astrocytes168. A study showed that LPS stimulation of primary murine astrocytes induced gene expression of Tnf, Il1b, Il6, Cxcl1, Cxcl2, Ccl2, Ccl7, and Ccl12 as early as 2 hours after stimulation178, suggesting that TLR4 was present on astrocytes and able to detect LPS178. Other studies also showed functional TLR4 on astrocytes without pre-treatment. For example, LPS stimulation of murine astrocytes activates NFκB, MAPKs, and JAK/STAT signaling pathways179, and enhances the expression of inducible Nitric Oxide Synthase (iNOS), IL-6, CCL2, ICAM1, and VCAM1170. Yet, earlier studies showed conflicting results. One study concluded that astrocytes (and oligodendrocytes) do not express Tlr4 mRNA, despite the positive Tlr4 detection in microglia, using primary cells from newborn rat forebrains without stimulation180. The study180 was published in 2002; therefore, the detection of cDNA was performed on an agarose gel, rather than quantitative PCR. Thus, it is possible that detection was not sensitive enough to detect Tlr4 transcripts. In contrast, another study published in 2003 also used agarose gels and clearly showed the Tlr4 cDNA band, although astrocyte stimulation was necessary for visualizing solid Tlr4 cDNA bands172. A different study showed a very low TLR4 protein expression level in resting mouse astrocytes when detected by flow cytometry170. Here, it is possible that TLR4 detection by flow cytometry might have been technically challenging if the TLR4 antibody was not sensitive, and the absence of a positive TLR4 staining control prevented evaluation of the TLR4 antibody sensitivity170. Mouse astrocytes show only mild induction of Tlr4 gene activation with various stimulations, such as LPS, poly(I:C), PGN, flagellin, and CpG-DNA170,172. The mild induction of Tlr4 is a substantial contrast to that of Tlr2 and Tlr3170. Tlr4 gene expression in human primary astrocytes is even more resistant to stimulation with inflammatory cytokines, such as IL-1β, TNFα, and IFNγ168. These basal TLR4 expression levels may be low enough to be missed, and Tlr4 expression is relatively resistant for upregulation. However, it appears that astrocytes can express functional TLR4.
Expression of TLR7 and TLR9 in astrocytes is inducible by ZIKA virus infection181. TLR7 agonist, imiquimod, reactivates astrocyte and induces production of proinflammatory cytokines and chemokines, including IFNβ, TNF, CCL2, and CXCL10182. Ex vivo TLR9 stimulation of mouse astrocytes with its ligand, CpG-DNA, upregulates the expression of cathelin-related antimicrobial peptide183, suggesting that TLR9 signaling is functional in astrocytes. Although ligands and functions for TLR11–13 in astrocytes remain to be addressed, toxoplasmosis in mice upregulates the expression of TLR11 in astrocytes184.
4.2.2: CLRs in astrocytes
CLR expression by astrocytes appears to be limited. For example, the expression of “Aplec” genes (the “Antigen-Presenting lectin-like receptor gene complex” and including a cluster of C-type lectin (CLEC) receptor gene loci), including Clec4e (encoding Mincle), was not detected in rat astrocytes, while microglia express them when stimulated185. To the best of our knowledge, no reports showed astrocytic dectin-1 and other major CLRs, involved in fungal detection. Yet, MR is highly expressed in astrocytes in one week-old neonatal mice and decreases its expression level to maintain low levels throughout adulthood186. Mannose-binding lectin (MBL) is detected in astrocytes of post-mortem brain tissue of patients with HIV encephalitis187. These studies suggest that astrocytes may not be equipped to directly detect fungal infections through CLRs, although our understanding of CLRs in astrocytes is still not sufficient for this conclusion.
4.2.3: NLRs and ALRs in astrocytes
Astrocytes are known to express some NLRs and AIM2 inflammasome components. For NLRs, NOD1 and NOD2 are expressed in astrocytes188. NOD2 in retinal astrocytes has been shown to synergize with TLR2 for the recruitment of pathogenic T cells during intraocular inflammation141,142,189. NLRP3 and NLRC4 inflammasomes are activated in LPC-stimulated mouse primary astrocytes ex vivo, and resulting IL-1β secretion was reported146, suggesting that astrocytes could behave as inflammatory cells through these inflammasomes. Similarly, ex vivo treatment of mouse primary astrocytes with nigericin, an NLRP3 inflammasome stimulator, also allows IL-1β secretion, although poly(dA:dT)/liposome, an AIM2 inflammasomes stimulator, fails to secrete detectable levels of IL-1β145. Human primary astrocytes express NLRP2, which can be activated by adenosine triphosphate (ATP) as a DAMP, and process pro-caspase-1 and pro-IL-1β190. Based on these studies, astrocytes appear to have the capacity to activate various inflammasomes, including NLRs.
Our lab recently reported that astrocytes can also have activated AIM2 inflammasome in the spinal cord during a late stage of EAE145. Unexpectedly, astrocytes are the main CNS cell type exhibiting AIM2 inflammasome “activation”, and neither microglia nor CNS-infiltrated myeloid cells are sources of any active inflammasomes during EAE145. Also, the NLRP3 inflammasome is not activated in astrocytes, contrasting to the NLRP3 inflammasome activation and its pathogenic role in EAE in peripheral myeloid cells145,191–193. Interestingly, mouse primary astrocytes with active AIM2 inflammasome release little IL-1β due to globally limited expression of inflammasome components145. The function of the AIM2 inflammasome in astrocytes is not inflammatory based on limited IL-1β production by astrocytes and more severe EAE in Aim2−/− mice145,152,194. Yet, the biological role of active AIM2 inflammasome in astrocytes during EAE is still unknown. Taken together, the roles of activated inflammasomes in astrocytes in vivo are still largely elusive, particularly due to its generally low expression levels of inflammasome components145. Nevertheless, NLR inflammasomes in astrocytes appear to induce inflammation, while the AIM2 inflammasome in astrocytes may regulate inflammation, or at least its activation is not inflammatory.
4.2.4: RLRs in astrocytes
Reports have shown that RIG-I and MDA5 are expressed in rat, human, and murine astrocytes195–197. While many studies focus on understanding their roles in CNS injury, few reports address their functionality during infections. Cultured astrocytes in a stretch injury model showed elevated RIG-I and MDA5 expression, leading to the production of IFN-I and an increase in GFAP and vimentin, hallmarks of reactive astrogliosis195. RIG-I expression in astrocytes is also inducible by hypoxia and infection by vesicular stomatitis virus (VSV)196,197. In the VSV study, primary human astrocytes produce inflammatory cytokines and neurotoxic mediators197. In mouse astrocytes, cytosolic poly(I:C) complexed with Lipofectamine activate MDA5, but not RIG-I, and induces the release of IFNβ and IL-6198.
Astrocytes have functional STING, although the setting in a report did not directly sense cytoplasmic DNA from the astrocyte nucleus199. Rather, STING was activated by cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), transferred from brain metastatic cancer cells through gap junction channels with connexin-43 (Cx43) through cell-cell contact199. In addition, the astroglial STING pathway activation elicited the production of IFNα and TNF199. This intriguing study suggests a possible role of astrocytes as secondary sensors of DNA damage in other CNS-resident cells, such as injured neurons, as well as injured astrocytes, known to be physically connected with healthy astrocytes through Cx43200. The cGAMP-STING signaling in bystander cells is also reported with murine embryonic fibroblasts (MEFs) during virus infection201. In this case, volume-regulated anion channels (VRACs) play a role in cell-to-cell transmission of cGAMP to enhance antiviral IFN-I response201.
Although astrocytes are not well equipped with various PRRs, compared to microglia with their extensive PRR repertoire, further studies are merited to identify the roles of astrocytes as a primary or secondary cell type to sense infections or even as amplifiers or regulators of infection-triggered cell responses by CNS immune sentinels.
4.3 |. Neurons
Neurons are fundamental cells in the CNS and peripheral nervous system (PNS) responsible for the relay of information throughout the body. Some neuronal PRRs are used to detect infections, but a majority of previous studies focused on the involvement of PRRs in neurons in the light of neuronal death and growth (Table 1; Fig. 2C).
4.3.1: TLRs in neurons
Neuronal TLRs have been studied mainly in the context of neurodegenerative diseases and neurodevelopment. Several studies reported the expression of TLR1–9 in neurons in the CNS and the PNS166,202–207. Interestingly, some neuronal TLRs may function through glycogen synthase kinase 3β (GSK3β), jun-N-terminal kinase (JNK), and PI3K/protein kinase B (AKT) pathways, rather than the canonical NFκB pathway204,208,209. Expression of TLR2 and TLR4 in neurons is increased in response to IFNγ stimulation and energy deprivation, as well as in response to ischemic brain injury204. The study also reported that TLR2 and TLR4 in neurons enhance proapoptotic signaling, rendering neurons vulnerable to ischemic death204. TLR4 in neurons may have a distinct role or different expression pattern from TLR2 in neurons in AD. For example, only TLR4 expression was increased in neurons when exposed to amyloid β (Aβ) peptide, despite both TLR4 and TLR2 being upregulated in cultured neurons upon exposure to hydoxynonenal (HNE), an AD-related byproduct210. TLR4 is also responsible for JNK phosphorylation and caspase-3 cleavage, resulting in neuronal death, upon HNE treatment210. This suggests that TLR4 signaling in neurons may be detrimental during AD; but the functional consequence of TLR2 in neurons remains undefined in this setting. Nevertheless, neuronal TLR2 is required for opioid-induced neuronal death via the activation of the GSK3β signaling pathways209, as well as glucose deprivation-induced cell death204. Thus, TLR2 and TLR4 signaling are generally neurotoxic. If fungi stimulate neuronal TLR2 and TLR4, it may be detrimental to neurons rather than eliciting antifungal responses.
TLR3 and TLR5 in primary sensory neurons are involved in controlling itch and pain211,212. For example, a TLR3 agonist induces action potentials in dorsal root ganglion (DRG) neurons and elicits scratching in wild-type mice but not in TLR3-deficient mice206. TLR5 in DRG neurons was suggested to be a target to treat neuropathic pain through the blockade of Aβ-fibers, which are implicated in mechanical allodynia106. These studies show that TLR3 and TLR5 regulate neuronal excitability and synaptic transmission.
TLR7 is expressed in neurons, and in vivo neuronal cell death is induced by ssRNA40, an oligoribonucleotide from HIV205. TLR8 in neurons is a negative regulator of neurite growth and an inducer of neuronal apoptosis213. The study also reported that stimulating TLR8 in neurons with R848 (also a TLR7 agonist) downregulates phosphorylated IκB without impacting NFκB p65 activation213. TLR9 in spinal cord neurons is considered involved in the excitotoxic death of neurons, at least partly due to stress in the endoplasmic reticulum214. Another study showed that some neurons in mice express TLR11 after encephalitic Toxoplasma gondii infection184, but the role of TLR11 is unknown.
4.3.2: CLRs in neurons
Spinal cord neurons express low levels of Mincle and induce Mincle expression in response to peripheral nerve injury (PNI). Mincle signaling in neurons appears to induce TNFα production and allodynia induced by PNI215,216. Neurons also express MR under certain conditions186, but its functional relevance in neurons remains undetermined. MBL is expressed in human neurons with increased expression during HIV-encephalitis, but the function of MBL is still not clear187. The roles of CLRs during fungal infections remain unaddressed.
4.3.3: NLRs and ALRs in neurons
NOD1 is highly expressed in neurons of developing mouse brain, and it may function to recognize microbiota-derived PGN that cross the BBB during brain development188. Rat cortical neurons also highly express NOD1 by an ex vivo oxygen-glucose deprivation and reperfusion treatment217. Among this PRR category, neurons have been relatively well studied in the light of inflammasomes. Neurons from naïve mice express detectable levels of NLRP1, caspase-1, and ASC proteins218. Human cortical brain neurons also showed the expression of NLRP1, caspase-1, ASC, NLRC4 (IPAF), and AIM2, although NLRP3 expression was not identified219. The study demonstrated that ex vivo treatment of human neurons with muramyl dipeptide (MDP), flagellin, and poly(dA:dT) activated caspase-1 in neurons, suggesting functional NLRP1, NLRC4, and AIM2 inflammasomes219. Furthermore, IL-1β release from neurons by NLRP1 inflammasome activation induced neuroinflammation, leading to caspase-6-mediated axonal degeneration and Aβ42 overproduction219. An earlier ex vivo study showed NLRP1 overexpression in rat primary cerebellar granule neurons led to apoptosis, accompanied by caspase-3 activation, resulting in neuron injury220. In contrast, a non-inflammasome forming NLR, NLR Family Member X1 (NLRX1), protects neuron-like N2A cells from necrosis221.
AIM2 expression has also been reported in human neuron cultures219. The AIM2 inflammasome is activated in rat embryonic cortical neurons, based on the detection of pro-caspase-1 processing, along with the formation of “ASC specks,” which are microscopically detectable signals reflecting the inflammasome polymer complex formation222. A more recent study indicated the detection of Aim2 transcripts in neurons, along with astrocytes and microglia, during brain development in mice223. AIM2 total knockout mice demonstrated increased DNA damage accumulation, which was also identified in neuron-specific caspase-1 knockouts223; thus, ASC specks detected in the brain development appear to be activated by AIM2 inflammasome in neurons. Indeed, we identified activated AIM2 inflammasome was also detected in motor neurons expressing Choline Acetyltransferase (ChAT) during a late stage of EAE, although the levels of activated AIM2 inflammasome in neurons are significantly lower than those in astrocytes145.
4.3.4: RLRs and other PRRs in neurons
Neurons express RLRs in various mammalian models but mainly in the context of viral infections208,224–226. Expression of RIG-I and MDA5 in cultured human and rat primary neurons increases in response to IFN-I; and brain infection by simian immunodeficiency virus also enhances the expression of RIG-I and MDA5 in neurons of macaques208,224. Furthermore, RIG-I signaling in neurons induces inflammatory molecules, including IL-6, IL-12p70, CCL2, CXCL10, and TNFα, upon Japanese encephalitis virus (JEV) infection225. A later study showed that RIG-I and STING interact to elicit immunological responses involving IFN-I226. Together, these reports suggest that neurons are sensitive to detecting viruses through RLRs and elicit antiviral immune responses. However, the roles of neuronal RLRs in the context of fungal infections are still unknown.
4.4 |. Oligodendrocytes and Oligodendrocyte Precursor Cells (OPCs)
Oligodendrocytes are responsible for maintaining and generating the myelin sheath that surrounds axons. They also support neurons by producing neurotrophic factors, which maintain axonal integrity and propagate the action potential along axons. Due to their importance in protecting neurons, oligodendrocytes have become a target of research in neurodegenerative disorders.
To the best of our knowledge, reports on PRR expression in oligodendrocytes and OPCs are limited to a few TLRs (Table 1; Fig. 2D). Among TLRs, oligodendrocytes express TLR2, and the expression is upregulated in MS lesions227. TLR2 stimulation by its agonists inhibits OPC maturation, while agonists of other TLRs do not227. Expression of TLR2, and TLR3 to a lesser extent, was also identified in primary rat oligodendrocytes culture228. TLR3 agonist, poly(I:C), induces apoptosis in oligodendrocytes, while zymosan promotes survival, differentiation, and myelin-like membrane formation in oligodendrocytes228. Although the study describes zymosan as “a TLR2 agonist”228, zymosan is a yeast cell wall derivative and stimulates dectin-1 together with TLR2 as well. Thus, dectin-1 signaling may also be involved in the outcomes in zymosan-treated oligodendrocytes, although dectin-1 was not discussed228. TLR4 was also identified on OPCs and triggers apoptosis of OPCs with Hsp60, a DAMP released by microglia229, through the TLR4-MyD88-NFκB signaling pathway229. Another study showed that LPS-mediated TLR4 stimulation is detrimental and toxic for oligodendrocytes in culture180.
Information on CLRs, NLRs, RLRs, and ALRs in oligodendrocytes and OPCs is limited, except for the study on zymosan-treated oligodendrocytes as mentioned above, although the possible involvement of dectin-1 was not addressed228. Other studies showed that oligodendrocytes do not express MR186, but MBL is a CLR expressed by oligodendrocytes of the frontal cortex of HIV-1 infected brains, although the role MBL in oligodendrocytes remains unknown187.
5 |. RECRUITMENT OF BLOOD-BORNE IMMUNE CELLS TO THE CNS DURING FUNGAL INFECTIONS
Recruitment of immune cells from circulation is essential for fungal clearance in the CNS. Having CNS-infiltrated cells in the CNS adds a broader variety of PRRs and effector functions, including the secretion of cytokines and chemokines, as well as phagocytosis and killing of fungi. However, heightened immunity by CNS-infiltrated cells could cause irreparable damage to the CNS, due to relatively poor regenerative capacity of CNS-resident cells. In addition, as discussed in Section 3, recruited immune cells can contribute to fungal dissemination as vehicles of the Trojan Horse mechanism. Here, we discuss peripheral immune cells recruited to the CNS during fungal infections and their contribution to fungal clearance and CNS pathology.
5.1 |. Neutrophils
Neutrophils serve as the first line of defense against fungal infections and can eliminate pathogens through various mechanisms, including phagocytosis, secretion of antimicrobial factors, generation of oxidative bursts, and release of neutrophil extracellular traps (NETs)230. In the absence of neutrophils, a condition known as neutropenia, individuals face a higher risk of developing invasive candidiasis231 and aspergillosis232. In contrast, neutrophil-mediated control of fungal infections is known to cause immunopathology, at least in the peripheral organs233,234
During disseminated candidiasis, neutrophils are recruited and accumulate in the mouse brain early during infection235. The number of neutrophils recruited in the brain peaks at 4 days after C. albicans infection, and the kinetics of neutrophil counts in the brain contrasts with a steady increase in the kidney up to day 7235. Thus, even in the same mouse, the brain appears to limit neutrophil infiltration to a certain point. Recruitment of neutrophils to the CNS during invasive candidiasis relies on Caspase Recruitment Domain Family Member 9 (CARD9) in microglia236, an adaptor protein downstream of Syk-mediated CLR signaling. CARD9 in neutrophils is also required to produce chemokines that further recruit monocytes and additional neutrophils236. Neutrophils also play a role to limit the transition from the hyphal form to the yeast form237.
Cryptococcus infection models using mice238–240 and zebrafish241 showed that neutrophils are the first cells to interact with Cryptococcus in the brain vasculature. Complement C5a receptor signaling is critical for neutrophil recruitment to the brain vasculature, in which neutrophils are the primary cell type to clear C. neoformans240. The clearance of C. neoformans in the brain microvasculature by neutrophils is a crucial step for host protection; thus, increasing neutrophil recruitment to clear arrested cryptococci in the brain microvasculature was proposed as a therapeutic approach to improve cryptococcal meningoencephalitis240. Complement C3 is also critically involved in the recognition and clearance of C. neoformans in the brain238.
Neutrophil recruitment to the CNS during aspergillosis needs further investigation. Yet, it is reported that neutrophils, recruited to the lung242 and eye243, mediate the clearance of conidia244 and restrict hyphal growth243. Based on the studies, it is possible that neutrophils also exert anti-Aspergillus activities in the brain.
5.2 |. Monocytes
Monocytes, which travel through the blood to tissues where they become macrophages or dendritic cells (DCs), are crucial for controlling fungal infections in both mice and humans. (DCs were identified in the cerebrospinal fluid (CSF) of patients with severe cryptococcal meningoencephalitis245, but limited information on brain DCs during CNS mycosis is available.) Monocytes also exert effector functions, such as phagocytosis and secretion of cytokines and chemokines246,247. Inflammatory monocytes express CCR2, a key chemokine receptor for monocyte migration. CCR2, together with C-X3-C Motif Chemokine Receptor 1 (CX3CR1), is critical for monocyte trafficking along inflamed vessels and subsequent accumulation in the brain248. Although the study is on pulmonary fungal infections, depletion of CCR2+ cells in mice reduced host survival and fungal clearance in C. albicans249. The study also suggested that the antifungal activity of CCR2+ cells is exerted in the first 48 hours after infection249. In a mouse pulmonary aspergillosis model, CCR2+Ly6C+ monocytes and their derivatives are required for antifungal CD4+ T cell responses in the lung250. More studies describe monocytes in the CNS in C. neoformans infection. Monocytes are recruited and accumulate in the brain vasculature hours after systemic administration of C. neoformans251. In this context, monocytes internalize C. neoformans and are taken advantage of as “Trojan Horses” by the fungus to cross the BBB and enter the brain parynchema52. Thus, monocytes are generally antifungal, but they are used as vehicles for fungal CNS dissemination.
5.3 |. T cells
The protective role of T cells, particularly CD4+ T cells, is well demonstrated in fungal infections. Yet, a limited number of studies described T cells infiltrated in the CNS during fungal brain infections. Among the previous studies, Cryptococcus brain infection is the most well documented among all the fungal infections in the brain, probably due to the urgency to understand prevalent CNS cryptococcosis in immunocompromised individuals, such as HIV/AIDS patients with low CD4+ T cell counts252. In comparison to CD4+ T cells, CD8+ T cells are rather involved in low inocula brain infection by C. neoformans; and CD8+ T cells are relatively inefficient in clearing the fungus253. We also demonstrated that CD8+ T cells also do not induce cryptococcal IRIS (C-IRIS), which is induced by CD4+ T cells14. Another ex vivo study showed that CD4+ T cells interact with microglia, as possible antigen-presenting cells, better than CD8+ T cells253. These studies suggest a substantially less impact of CD8+ T cells in CNS cryptococcosis than CD4+ T cells. Nevertheless, CD4+ T cell transfer into immunocompromised mice with low degree cryptococcosis did not reduce brain fungal burdens14, implicating that brain-infiltrated CD4+ T cells may also not necessarily be efficient to fight against C. neoformans. However, our study showed that brain-infiltrated CD4+ T cells are pathogenic enough to elicit lethal outcomes in C-IRIS, with a majority of T helper cells are Th1 cells14. Here, IFNγ exacerbates C-IRIS14. Now, multiple questions remain on brain-infiltrated T cells during fungal infections. For example, which conditions make T cells in the CNS protective or pathogenic during brain fungal infection? Why are CD8+ T cells less potent than CD4+ T cells in providing either protective or detrimental outcomes in CNS fungal infections? Roles of brain-infiltrated T cells in fungal infections other than cryptococcosis are still largely elusive and merit further investigation.
5.4 |. Other CNS-infiltrated leukocytes
There is limited information on CNS-infiltrated leukocytes other than those mentioned above. Natural killer (NK) cells kill Cryptococcus by a perforin-mediated process254 and show intrathecal expansion and activation during Cryptococcus infection245. Brains from patients with cryptococcosis showed NK cells present within cryptococcomas255. B cells are also recruited to the CNS245 and reduce Cryptococcus dissemination to the brain256. In a mouse model of invasive candidiasis, a study demonstrated a wide range of leukocytes (including NK cells, NKT cells, γδT cells, B cells, CD4+ and CD8+ T cells, and DCs) detected in the brain235.
6 |. HOST RESPONSES AGAINST FUNGAL PATHOGENS IN THE CNS
Immune responses in the CNS require tight and masterful regulation to limit collateral damage. Hyperactive inflammatory responses within the CNS are detrimental as they may lead to irreparable tissue damage that can leave long-term and possibly irreversible effects on the host. The CNS has historically been regarded as ‘immune-privileged’, isolated from the peripheral immune system by the BBB. Although a limited number of circulating immune cells are known to patrol the CNS, microglia and perivascular macrophages reside within the CNS257 and are considered the first line of CNS defense against invading pathogens. Additionally, recent advances in neuroimmunology have presented that CNS-resident cells modulate immune responses in the CNS258. However, technical limitations remain in elucidating CNS-resident cells in vivo responses due to the difficulty in reflecting the biological functions of the cells using ex vivo experimental approaches. This section discusses the mechanistic and physiological outcomes of host CNS-resident cells upon detection of fungal pathogens.
6.1 |. CNS immune responses against Candida
Although most Candida infections are seen in mucosal surfaces and on the skin, CNS Candida infections do occur, particularly in the meninges in humans. The bloodstream carries disseminated Candida to all organs, including the CNS. Candida CNS infiltration starts with traversing the BBB259, as described in Section 3.
Once Candida infiltrates the brain, CNS-resident cells respond. Microglia are currently the best-studied CNS-resident cells for their responses and functions. The first article investigating microglia using a mouse model of Candida infection was published nearly 30 years ago. In this study, intracerebral transfer of the BV2 microglial cell line to syngeneic mice inhibited fungal growth in the brain, suggesting the overall protective role of microglia during Candida infection260. A host protective role of microglia could be attributed to their capacity to phagocytose pathogens and serve a sentinel role, by microglia promptly detecting pathogens and producing chemokines and cytokines to recruit inflammatory cells from circulation. Indeed, microglia express CXCL1 upon detection of candidalysin to recruit neutrophils for Candida clearance and release a proinflammatory cytokine IL-1β through NLRP3 inflammasome activation261. In microglia, the production of both CXCL1 and IL-1β depends on CARD9261. CXCL1 is a critical chemokine for neutrophil recruitment to efficiently clear fungus; and astrocytes assist microglial CXCL1 production by providing additional signals to microglia producing CXCL1 in response to candidalysin261. The involvement of astrocytes in the sentinel role of microglia is intriguing because it suggests the active participation of astrocytes at the early stage of antifungal immune responses. At this point, it is unknown if direct contact between microglia and astrocytes is required or if some soluble factors mediate the crosstalk to enhance CXCL1 production by microglia.
As described above, a major role of microglia, at least in Candida infection, is sensing the infection and sending a signal to recruit neutrophils in the brain. The sentinel function of microglia is reminiscent of tissue-resident macrophages in the peripheral organs, such as alveolar macrophages, which secrete chemokines CXCL1 and CXCL2 faster than other cell types during pulmonary cryptococcosis, as our lab previously reported262. We also demonstrated that tissue-resident macrophages produce CXCL1 and CXCL2 more efficiently than blood-borne macrophages for neutrophil recruitment upon C. albicans detection263. Here, tissue-resident macrophages are poised to induce NFκB activation and secrete CXCL1 and CXCL2 by autophagic digestion of A20 upon detecting Candida263. Indeed, microglia in naive adult mice also highly express TNF Alpha Induced Protein 3 (TNFAIP3/A20)264. Thus, microglia may also use autophagic A20 degradation as an approach to fulfill their role as CNS immune sentinels.
Candida infection in the CNS also promotes the accumulation of activated microglia, astrocytes, and cleaved Aβ peptide around yeast aggregates and forming fungal-induced glial granulomas164. Aβ has antimicrobial activities265,266 and efficiently aids the clearance of Candida by enhancing microglial phagocytosis and antifungal activity164. However, Aβ is also the key mediator of Alzheimer’s disease pathology267. Therefore, further investigation is warranted to understand the long-term consequence of fungal infections in the brain and their possible connection to the induction of neurodegenerative diseases in later life.
6.2 |. CNS immune responses against Cryptococcus
Dissemination of Cryptococcus into the CNS can result in the development of CM268. Although CM was initially described over a century ago269, immune responses of CNS-resident cells to Cryptococcus remain poorly understood. Cryptococcus forms a large polysaccharide capsule that hides components of the yeast cell wall9, resulting in limited and delayed recognition by host immune cells270. Studies using BV2 microglial cells demonstrated that acapsular mutant Cryptococcus yeasts were more susceptible to phagocytosis and clearance than encapsulated yeasts271. However, both acapusular mutant and encapsulated yeasts similarly fail to elicit TNFα and IL-1β expression by BV2271. Interestingly, the addition of either acapusular or encapsulated yeasts to BV2 cell culture inhibits LPS-mediated TNFα expression271. Furthermore, Cryptococcus capsule components, GXM and galactoxylomannan (GalXM), also inhibit LPS-mediated TNFα expression271, suggesting that the Cryptococcus capsule can inhibit TLR4 signaling, although it is not clear why acapusular mutant yeasts also inhibited the TLR4 signaling. Here, an antibody against GXM opsonizes C. neoformans, enhancing microglial phagocytosis, limiting Cryptococcus growth272, and inducing chemokine expression of CCL3 (MIP-1α) and CCL4 (MIP-1β)273. The data highlights the critical involvement of the Cryptococcal capsule in hindering microglia effector functions. At the same time, something other than capsule components appears to be involved in limiting cytokine expression upon detection of C. neoformans. Another study suggested that microglia are, unexpectedly, relatively slow to respond in a mouse model of CM274. Based on the flow cytometry data, seven days post-infection (dpi) was still too early for microglia to upregulate the expression of activation markers, CD11c and major histocompatibility complex class II (MHCII), and the upregulation of the two molecules was found on 14-dpi274. Additionally, the production of cytokines and chemokines, including TNFα, CCL3, and CCL4, required at least 21-dpi, and some molecules took more than 30-dpi for significant upregulation274.
C. neoformans induces immune reconstitution inflammatory syndrome (IRIS), a pathological condition whereby a recovering immune system paradoxically worsens the patient’s condition by excessively responding to a previously acquired C. neoformans infection. IRIS can also be associated with Candida and Aspergillus, but Cryptococcus-associated IRIS (C-IRIS) is most prevalent, and a serious concern, for HIV/AIDS patients. We used a mouse model of C-IRIS, mimicking human C-IRIS observed in immunocompromised patients, and demonstrated that the combination of previous subclinical C. neoformans infection and immune reconstitution by CD4+ T cells led to brain swelling by dysregulation of aquaporin-4 expression in astrocytes14. Microglia activation was also reported in mouse C-IRIS275. In the mouse C-IRIS model, CD4+ T, particularly Th1 cells, are involved in the lethal outcomes, but CD8+ T cells are not14. Although it is unknown why CD8+ T cells do not elicit the lethal C-IRIS outcomes, another study suggested less involvement of CD8+ T cells compared to CD4+ T cells in controlling Cryptococcus infection in the presence of microglia253. The study also indicated a lesser magnitude of MHC-I upregulation than MHC-II upregulation in an IFNγ-stimulated BV2 microglial cell line253. These studies suggest the participation of IFNγ and CD4+ T cells in controlling CNS cryptococcosis, but IFNγ and CD4+ T cells could cause collateral damage in the CNS, as observed in C-IRIS14,275.
Astrocytes also undergo structural and functional changes during CNS mycosis, i.e., astrogliosis. Astrogliosis is associated with destructive parenchymal cryptococcomas in humans268. In patients with CM, astrocytes are the primary source of CXCL10276, one of the most highly expressed chemokines in the CSF245. Human astrocytoma cells have also been demonstrated to engulf cryptocci277. We currently do not fully understand the functions of reactive astrocytes during in vivo Cryptococcus infection. Yet, astrocytes may be involved in limiting CM by nitric oxide production278 and possible CD4+ T cell activation by upregulation of human leukocyte antigen (HLA) class II, as demonstrated with astrocytoma cells277. The involvement of neurons and oligodendrocytes in CNS cryptococcosis is not known.
6.3 |. CNS immune responses against Aspergillus
Although Aspergillus infects the brain and forms brain abscesses in immunocompromised individuals279, limited reports are available for host cell responses in CNS aspergillosis. A. flavus infects the microglia cell line CHME-3 and induces expression of various cytokines, Tlr genes, and Mmp genes280. In particular, Il17, Tlr7, Tlr9, and Mmp9 expression is highly upregulated, and the magnitude of gene expression by A. flavus stimulation is generally more significant than that by C. albicans stimulation280. Secreted factors from A. fumigatus, A. flavus, A. terreus, and A. niger. are toxic to primary astrocytes and an astrocyte cell line, but neurons appear to be more sensitive to Aspergillus-secreted toxins281. As demonstrated with monocytes and macrophages, conidia and hyphae of Aspergillus potently activate the host complement system to opsonize the fungus and support phagocytosis and killing282. Cerebral aspergillosis induces the expression of complement proteins (C1q, C3, and C4) by neurons, oligodendrocytes, and CNS-infiltrated myeloid cells. However, unexpectedly, microglia only showed a marginal increase in complement protein expression283. The complement system can enhance various proinflammatory biological functions in the brain, such as cell stimulation and induction of chemotaxis284. At the same time, it can lead to collateral damages in the brain with loss of neurons and oligodendrocytes284.
7 |. CONCLUSIONS
CNS mycoses are emerging as major global health concerns, causing morbidity and mortality, especially in immunocompromised individuals. Although the CNS has been deemed immune-privileged, recent data demonstrate that CNS-resident cell types, particularly glial cells, can detect and respond to invading pathogenic fungi via PRRs and initiate immune responses during infection. Also, immune cell recruitment in the CNS from the circulation during CNS fungal infections was demonstrated. Animal in vivo mycosis models on peripheral organs have provided considerable information, but our knowledge of CNS mycosis is still behind. For example, little is known about what happens to host cells and fungi in the CNS after detecting fungal invasion. Multiple factors contribute to the paucity of our knowledge in CNS mycosis, despite the unacceptable morbidity and mortality rate. The CNS environment is unique: It involves neuro-immune communications and requires tightly regulated immune responses to avoid irreparable damage. In addition, the CNS environment and structure make it technically challenging to assess host cellular responses. New technologies in studying cells in the CNS greatly merit expanding our understanding of CNS mycosis to overcome the challenges.
ACKNOWLEDGEMENTS
We apologize to the authors whose publications could not be cited in the scope of this review. This work was supported by Burroughs Wellcome Fund Graduate Diversity Enrichment Program Grant #1020286 (to EYR), National Institutes of Health R01-AI088100, R01-NS120417, R01-AI160737 (to MLS), and National Multiple Sclerosis Society (NMSS) Research Grant RG-4536B2/1 (to MLS). We appreciate Ms. Miranda Lumbreras and Mr. Devon DiPalma for editorial suggestions and proofreading.
Funding information:
Burroughs Wellcome Fund Graduate Diversity Enrichment Program Grant: 1020286
National Institutes of Health: R01-AI088100, R01-NS120417, R01-AI160737
National Multiple Sclerosis Society: RG-4536B2/1
Footnotes
CONFLICTS OF INTEREST
All authors declare they have no conflicts of interest to disclose.
REFERENCES
- 1.Hawksworth DL, Lucking R. Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol Spectr. 2017;5(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kohler JR, Casadevall A, Perfect J. The Spectrum of Fungi That Infects Humans. Cold Spring Harbor Perspectives in Medicine. 2015;5(1):a019273–a019273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panackal AA, Williamson PR Fungal Infections of the Central Nervous System. Continuum. 2015;21 (6):1662–1678. [DOI] [PubMed] [Google Scholar]
- 4.Kullberg BJ, Arendrup MC. Invasive Candidiasis. N Engl J Med. 2015;373(15):1445–1456. [DOI] [PubMed] [Google Scholar]
- 5.Bongomin F, Gago S, Oladele RO, Denning DW. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J Fungi (Basel). 2017;3(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz S, Kontoyiannis DP, Harrison T, Ruhnke M. Advances in the diagnosis and treatment of fungal infections of the CNS. Lancet Neurol. 2018;17(4):362–372. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy M, Rosengart A, Schuetz AN, Kontoyiannis DP, Walsh TJ. Mold infections of the central nervous system. N Engl J Med. 2014;371(2):150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobsen ID, Wilson D, Wachtler B, Brunke S, Naglik JR, Hube B. Candida albicans dimorphism as a therapeutic target. Expert Rev Anti Infect Ther. 2012;10(1):85–93. [DOI] [PubMed] [Google Scholar]
- 9.Zaragoza O, Rodrigues ML, De Jesus M, Frases S, Dadachova E, Casadevall A. The capsule of the fungal pathogen Cryptococcus neoformans. Adv Appl Microbiol. 2009;68:133–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Decote-Ricardo D, LaRocque-de-Freitas IF, Rocha JDB, et al. Immunomodulatory Role of Capsular Polysaccharides Constituents of Cryptococcus neoformans. Front Med (Lausanne). 2019;6:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajasingham R, Smith RM, Park BJ, et al. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis. 2017;17(8):873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris JR, Lockhart SR, Sondermeyer G, et al. Cryptococcus gattii infections in multiple states outside the US Pacific Northwest. Emerg Infect Dis. 2013;19(10):1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis JA, Horn DL, Marr KA, Fishman JA. Central nervous system involvement in cryptococcal infection in individuals after solid organ transplantation or with AIDS. Transplant Infectious Disease. 2009;11(5):432–437. [DOI] [PubMed] [Google Scholar]
- 14.Khaw YM, Aggarwal N, Barclay WE, Kang E, Inoue M, Shinohara ML. Th1-Dependent Cryptococcus-Associated Immune Reconstitution Inflammatory Syndrome Model With Brain Damage. Front Immunol. 2020;11:529219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aggarwal N, Barclay W, Shinohara ML. Understanding mechanisms underlying the pathology of immune reconstitution inflammatory syndrome (IRIS) by using animal models. Curr Clin Microbiol Rep. 2018;5(3):201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kosmidis C, Denning DW. The clinical spectrum of pulmonary aspergillosis. Thorax. 2015;70(3):270–277. [DOI] [PubMed] [Google Scholar]
- 17.Lin SJ, Schranz J, Teutsch SM. Aspergillosis case-fatality rate: systematic review of the literature. Clin Infect Dis. 2001;32(3):358–366. [DOI] [PubMed] [Google Scholar]
- 18.Antinori S, Corbellino M, Meroni L, et al. Aspergillus meningitis: a rare clinical manifestation of central nervous system aspergillosis. Case report and review of 92 cases. J Infect. 2013;66(3):218–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petrikkos G, Skiada A, Lortholary O, Roilides E, Walsh TJ, Kontoyiannis DP. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54 Suppl 1(suppl_1):S23–34. [DOI] [PubMed] [Google Scholar]
- 20.Chikley A, Ben-Ami R, Kontoyiannis DP. Mucormycosis of the Central Nervous System. J Fungi (Basel). 2019;5(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raut A, Huy NT. Rising incidence of mucormycosis in patients with COVID-19: another challenge for India amidst the second wave? Lancet Respir Med. 2021;9(8):e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roden MM, Zaoutis TE, Buchanan WL, et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin Infect Dis. 2005;41(5):634–653. [DOI] [PubMed] [Google Scholar]
- 23.Lockhart SR, Toda M, Benedict K, Caceres DH, Litvintseva AP. Endemic and Other Dimorphic Mycoses in The Americas. J Fungi (Basel). 2021;7(2):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nathan CL, Emmert BE, Nelson E, Berger JR. CNS fungal infections: A review. J Neurol Sci. 2021;422:117325. [DOI] [PubMed] [Google Scholar]
- 25.Jones AR, Shusta EV. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24(9):1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen SHM, Stins MF, Huang SH, et al. Cryptococcus neoformans induces alterations in the cytoskeleton of human brain microvascular endothelial cells. J Med Microbiol. 2003;52(Pt 11):961–970. [DOI] [PubMed] [Google Scholar]
- 27.Jong A, Wu CH, Prasadarao NV, et al. Invasion of Cryptococcus neoformans into human brain microvascular endothelial cells requires protein kinase C-alpha activation. Cell Microbiol. 2008;10(9):1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jong A, Wu CH, Gonzales-Gomez I, et al. Hyaluronic acid receptor CD44 deficiency is associated with decreased Cryptococcus neoformans brain infection. J Biol Chem. 2012;287(19):15298–15306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vu K, Tham R, Uhrig JP, et al. Invasion of the central nervous system by Cryptococcus neoformans requires a secreted fungal metalloprotease. mBio. 2014;5(3):e01101–01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jong AY, Stins MF, Huang SH, Chen SH, Kim KS. Traversal of Candida albicans across human blood-brain barrier in vitro. Infect Immun. 2001;69(7):4536–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang W, Fa ZZ, Xie Q, et al. Complex Roles of Annexin A2 in Host Blood-Brain Barrier Invasion by Cryptococcus neoformans. CNS Neurosci Ther. 2017;23(4):291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang SH, Wu CH, Chang YC, Kwon-Chung KJ, Brown RJ, Jong A. Cryptococcus neoformans-derived microvesicles enhance the pathogenesis of fungal brain infection. PLoS One. 2012;7(11):e48570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang YC, Stins MF, McCaffery MJ, et al. Cryptococcal yeast cells invade the central nervous system via transcellular penetration of the blood-brain barrier. Infect Immun. 2004;72(9):4985–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vu K, Eigenheer RA, Phinney BS, Gelli A. Cryptococcus neoformans promotes its transmigration into the central nervous system by inducing molecular and cellular changes in brain endothelial cells. Infect Immun. 2013;81(9):3139–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Mittal R, Solis NV, Prasadarao NV, Filler SG. Mechanisms of Candida albicans trafficking to the brain. PLoS Pathog. 2011;7(10):e1002305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phan QT, Myers CL, Fu Y, et al. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 2007;5(3):e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu W, Phan QT, Boontheung P, Solis NV, Loo JA, Filler SG. EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc Natl Acad Sci U S A. 2012;109(35):14194–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Navarathna DH, Munasinghe J, Lizak MJ, Nayak D, McGavern DB, Roberts DD. MRI confirms loss of blood-brain barrier integrity in a mouse model of disseminated candidiasis. NMR Biomed. 2013;26(9):1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouchara JP, Sanchez M, Chevailler A, et al. Sialic acid-dependent recognition of laminin and fibrinogen by Aspergillus fumigatus conidia. Infect Immun. 1997;65(7):2717–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gravelat FN, Beauvais A, Liu H, et al. Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal beta-glucan from the immune system. PLoS Pathog. 2013;9(8):e1003575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wasylnka JA, Moore MM. Uptake of Aspergillus fumigatus Conidia by phagocytic and nonphagocytic cells in vitro: quantitation using strains expressing green fluorescent protein. Infect Immun. 2002;70(6):3156–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang C, Chen F, Liu X, et al. Gliotoxin Induces Cofilin Phosphorylation to Promote Actin Cytoskeleton Dynamics and Internalization of Aspergillus fumigatus Into Type II Human Pneumocyte Cells. Front Microbiol. 2019;10:1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel R, Hossain MA, German N, Al-Ahmad AJ. Gliotoxin penetrates and impairs the integrity of the human blood-brain barrier in vitro. Mycotoxin Res. 2018;34(4):257–268. [DOI] [PubMed] [Google Scholar]
- 44.Olszewski MA, Noverr MC, Chen GH, et al. Urease expression by Cryptococcus neoformans promotes microvascular sequestration, thereby enhancing central nervous system invasion. Am J Pathol. 2004;164(5):1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charlier C, Chretien F, Baudrimont M, Mordelet E, Lortholary O, Dromer F. Capsule structure changes associated with Cryptococcus neoformans crossing of the blood-brain barrier. Am J Pathol. 2005;166(2):421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cox GM, Mukherjee J, Cole GT, Casadevall A, Perfect JR. Urease as a virulence factor in experimental cryptococcosis. Infect Immun. 2000;68(2):443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh A, Panting RJ, Varma A, et al. Factors required for activation of urease as a virulence determinant in Cryptococcus neoformans. mBio. 2013;4(3):e00220–00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villar CC, Kashleva H, Nobile CJ, Mitchell AP, Dongari-Bagtzoglou A. Mucosal tissue invasion by Candida albicans is associated with E-cadherin degradation, mediated by transcription factor Rim101p and protease Sap5p. Infect Immun. 2007;75(5):2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heyen L, Muller U, Siegemund S, et al. Lung epithelium is the major source of IL-33 and is regulated by IL-33-dependent and IL-33-independent mechanisms in pulmonary cryptococcosis. Pathog Dis. 2016;74(7):ftw086. [DOI] [PubMed] [Google Scholar]
- 50.DeHart DJ, Agwu DE, Julian NC, Washburn RG. Binding and germination of Aspergillus fumigatus conidia on cultured A549 pneumocytes. J Infect Dis. 1997;175(1):146–150. [DOI] [PubMed] [Google Scholar]
- 51.Kogan TV, Jadoun J, Mittelman L, Hirschberg K, Osherov N. Involvement of secreted Aspergillus fumigatus proteases in disruption of the actin fiber cytoskeleton and loss of focal adhesion sites in infected A549 lung pneumocytes. J Infect Dis. 2004;189(11):1965–1973. [DOI] [PubMed] [Google Scholar]
- 52.Charlier C, Nielsen K, Daou S, Brigitte M, Chretien F, Dromer F. Evidence of a role for monocytes in dissemination and brain invasion by Cryptococcus neoformans. Infect Immun. 2009;77(1):120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaufman-Francis K, Djordjevic JT, Juillard PG, et al. The Early Innate Immune Response to, and Phagocyte-Dependent Entry of, Cryptococcus neoformans Map to the Perivascular Space of Cortical Post-Capillary Venules in Neurocryptococcosis. Am J Pathol. 2018;188(7):1653–1665. [DOI] [PubMed] [Google Scholar]
- 54.Zaragoza O, Chrisman CJ, Castelli MV, et al. Capsule enlargement in Cryptococcus neoformans confers resistance to oxidative stress suggesting a mechanism for intracellular survival. Cell Microbiol. 2008;10(10):2043–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levitz SM, Harrison TS, Tabuni A, Liu X. Chloroquine induces human mononuclear phagocytes to inhibit and kill Cryptococcus neoformans by a mechanism independent of iron deprivation. J Clin Invest. 1997;100(6):1640–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levitz SM, Nong SH, Seetoo KF, Harrison TS, Speizer RA, Simons ER. Cryptococcus neoformans resides in an acidic phagolysosome of human macrophages. Infect Immun. 1999;67(2):885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Santiago-Tirado FH, Onken MD, Cooper JA, Klein RS, Doering TL. Trojan Horse Transit Contributes to Blood-Brain Barrier Crossing of a Eukaryotic Pathogen. mBio. 2017;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chretien F, Lortholary O, Kansau I, Neuville S, Gray F, Dromer F. Pathogenesis of cerebral Cryptococcus neoformans infection after fungemia. J Infect Dis. 2002;186(4):522–530. [DOI] [PubMed] [Google Scholar]
- 59.Sorrell TC, Juillard PG, Djordjevic JT, et al. Cryptococcal transmigration across a model brain blood-barrier: evidence of the Trojan horse mechanism and differences between Cryptococcus neoformans var. grubii strain H99 and Cryptococcus gattii strain R265. Microbes Infect. 2016;18(1):57–67. [DOI] [PubMed] [Google Scholar]
- 60.Ma H, Croudace JE, Lammas DA, May RC. Expulsion of live pathogenic yeast by macrophages. Curr Biol. 2006;16(21):2156–2160. [DOI] [PubMed] [Google Scholar]
- 61.Yang X, Wang H, Hu F, Chen X, Zhang M. Nonlytic exocytosis of Cryptococcus neoformans from neutrophils in the brain vasculature. Cell Commun Signal. 2019;17(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scherer AK, Blair BA, Park J, Seman BG, Kelley JB, Wheeler RT. Redundant Trojan horse and endothelial-circulatory mechanisms for host-mediated spread of Candida albicans yeast. PLoS Pathog. 2020;16(8):e1008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mitchell CG, Slight J, Donaldson K. Diffusible component from the spore surface of the fungus Aspergillus fumigatus which inhibits the macrophage oxidative burst is distinct from gliotoxin and other hyphal toxins. Thorax. 1997;52(9):796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. [DOI] [PubMed] [Google Scholar]
- 65.Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathologica. 2001;101(3):249–255. [DOI] [PubMed] [Google Scholar]
- 66.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoeffel G, Chen J, Lavin Y, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. 2015;42(4):665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–1543. [DOI] [PubMed] [Google Scholar]
- 69.Han W, Umekawa T, Zhou K, et al. Cranial irradiation induces transient microglia accumulation, followed by long-lasting inflammation and loss of microglia. Oncotarget. 2016;7(50):82305–82323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Menzel F, Kaiser N, Haehnel S, et al. Impact of X-irradiation on microglia. Glia. 2018;66(1):15–33. [DOI] [PubMed] [Google Scholar]
- 71.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. [DOI] [PubMed] [Google Scholar]
- 72.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173(6):3916–3924. [DOI] [PubMed] [Google Scholar]
- 73.Mishra BB, Gundra UM, Teale JM. Expression and distribution of Toll-like receptors 11–13 in the brain during murine neurocysticercosis. J Neuroinflammation. 2008;5(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verma R, Kim JY. 1,25-Dihydroxyvitamin D3 Facilitates M2 Polarization and Upregulates TLR10 Expression on Human Microglial Cells. Neuroimmunomodulation. 2016;23(2):75–80. [DOI] [PubMed] [Google Scholar]
- 75.Jack CS, Arbour N, Manusow J, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175(7):4320–4330. [DOI] [PubMed] [Google Scholar]
- 76.Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61(11):1013–1021. [DOI] [PubMed] [Google Scholar]
- 77.Bourgeois C, Kuchler K. Fungal pathogens-a sweet and sour treat for toll-like receptors. Front Cell Infect Microbiol. 2012;2:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shoham S, Huang C, Chen JM, Golenbock DT, Levitz SM. Toll-like receptor 4 mediates intracellular signaling without TNF-alpha release in response to Cryptococcus neoformans polysaccharide capsule. J Immunol. 2001;166(7):4620–4626. [DOI] [PubMed] [Google Scholar]
- 79.Shah VB, Williams DL, Keshvara L. beta-Glucan attenuates TLR2- and TLR4-mediated cytokine production by microglia. Neurosci Lett. 2009;458(3):111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang JE, Warris A, Ellingsen EA, et al. Involvement of CD14 and toll-like receptors in activation of human monocytes by Aspergillus fumigatus hyphae. Infect Immun. 2001;69(4):2402–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koutsouras GW, Ramos RL, Martinez LR. Role of microglia in fungal infections of the central nervous system. Virulence. 2017;8(6):705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Netea MG, Gow NA, Joosten LA, Verschueren I, van der Meer JW, Kullberg BJ. Variable recognition of Candida albicans strains by TLR4 and lectin recognition receptors. Med Mycol. 2010;48(7):897–903. [DOI] [PubMed] [Google Scholar]
- 83.Netea MG, Gow NA, Munro CA, et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest. 2006;116(6):1642–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Netea MG, Van der Graaf C, Van der Meer JW, Kullberg BJ. Recognition of fungal pathogens by Toll-like receptors. Eur J Clin Microbiol Infect Dis. 2004;23(9):672–676. [DOI] [PubMed] [Google Scholar]
- 85.Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, Kullberg BJ. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis. 2002;185(10):1483–1489. [DOI] [PubMed] [Google Scholar]
- 86.Fernandez-Arjona MDM, Grondona JM, Fernandez-Llebrez P, Lopez-Avalos MD. Microglial activation by microbial neuraminidase through TLR2 and TLR4 receptors. J Neuroinflammation. 2019;16(1):245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Royal GC Jr., Nandedkar AK, Sampson CC, Faggett T. Neuraminidase production by Candida albicans. J Natl Med Assoc. 1984;76(2):143–145. [PMC free article] [PubMed] [Google Scholar]
- 88.Xu S, Wang J, Jiang J, et al. TLR4 promotes microglial pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway after spinal cord injury. Cell Death Dis. 2020;11(8):693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cui W, Sun C, Ma Y, Wang S, Wang X, Zhang Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front Neurosci. 2020;14:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Businaro R, Corsi M, Asprino R, et al. Modulation of Inflammation as a Way of Delaying Alzheimer’s Disease Progression: The Diet’s Role. Curr Alzheimer Res. 2018;15(4):363–380. [DOI] [PubMed] [Google Scholar]
- 91.Fonseca FL, Nohara LL, Cordero RJ, et al. Immunomodulatory effects of serotype B glucuronoxylomannan from Cryptococcus gattii correlate with polysaccharide diameter. Infect Immun. 2010;78(9):3861–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rubino I, Coste A, Le Roy D, et al. Species-specific recognition of Aspergillus fumigatus by Toll-like receptor 1 and Toll-like receptor 6. J Infect Dis. 2012;205(6):944–954. [DOI] [PubMed] [Google Scholar]
- 93.Kim C, Lee HJ, Masliah E, Lee SJ. Non-cell-autonomous Neurotoxicity of alpha-synuclein Through Microglial Toll-like Receptor 2. Exp Neurobiol. 2016;25(3):113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fiebich BL, Batista CRA, Saliba SW, Yousif NM, de Oliveira ACP. Role of Microglia TLRs in Neurodegeneration. Front Cell Neurosci. 2018;12:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lester SN, Li K. Toll-like receptors in antiviral innate immunity. J Mol Biol. 2014;426(6):1246–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10(12):1366–1373. [DOI] [PubMed] [Google Scholar]
- 97.Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. Microglia recognize double-stranded RNA via TLR3. J Immunol. 2006;176(6):3804–3812. [DOI] [PubMed] [Google Scholar]
- 98.Jin YH, Kaneyama T, Kang MH, Kang HS, Koh CS, Kim BS. TLR3 signaling is either protective or pathogenic for the development of Theiler’s virus-induced demyelinating disease depending on the time of viral infection. J Neuroinflammation. 2011;8:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beisswenger C, Hess C, Bals R. Aspergillus fumigatus conidia induce interferon-beta signalling in respiratory epithelial cells. Eur Respir J. 2012;39(2):411–418. [DOI] [PubMed] [Google Scholar]
- 100.Beraud D, Twomey M, Bloom B, et al. alpha-Synuclein Alters Toll-Like Receptor Expression. Front Neurosci. 2011;5:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Michaelis KA, Norgard MA, Levasseur PR, et al. Persistent Toll-like receptor 7 stimulation induces behavioral and molecular innate immune tolerance. Brain Behav Immun. 2019;82:338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tauber SC, Ebert S, Weishaupt JH, Reich A, Nau R, Gerber J. Stimulation of Toll-like receptor 9 by chronic intraventricular unmethylated cytosine-guanine DNA infusion causes neuroinflammation and impaired spatial memory. J Neuropathol Exp Neurol. 2009;68(10):1116–1124. [DOI] [PubMed] [Google Scholar]
- 103.Maatouk L, Compagnion AC, Sauvage MC, et al. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat Commun. 2018;9(1):2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Scholtzova H, Kascsak RJ, Bates KA, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29(6):1846–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Scholtzova H, Chianchiano P, Pan J, et al. Amyloid beta and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol Commun. 2014;2(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu ZZ, Kim YH, Bang S, et al. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat Med. 2015;21(11):1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chakrabarty P, Li A, Ladd TB, et al. TLR5 decoy receptor as a novel anti-amyloid therapeutic for Alzheimer’s disease. J Exp Med. 2018;215(9):2247–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herrera-Rivero M, Santarelli F, Brosseron F, Kummer MP, Heneka MT. Dysregulation of TLR5 and TAM Ligands in the Alzheimer’s Brain as Contributors to Disease Progression. Mol Neurobiol. 2019;56(9):6539–6550. [DOI] [PubMed] [Google Scholar]
- 109.Ifuku M, Hinkelmann L, Kuhrt LD, et al. Activation of Toll-like receptor 5 in microglia modulates their function and triggers neuronal injury. Acta Neuropathol Commun. 2020;8(1):159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang D, Zhang G, Hayden MS, et al. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303(5663):1522–1526. [DOI] [PubMed] [Google Scholar]
- 111.Yarovinsky F, Zhang D, Andersen JF, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308(5728):1626–1629. [DOI] [PubMed] [Google Scholar]
- 112.Goyal S, Castrillon-Betancur JC, Klaile E, Slevogt H. The Interaction of Human Pathogenic Fungi With C-Type Lectin Receptors. Front Immunol. 2018;9:1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deerhake ME, Danzaki K, Inoue M, et al. Dectin-1 limits autoimmune neuroinflammation and promotes myeloid cell-astrocyte crosstalk via Card9-independent expression of Oncostatin M. Immunity. 2021;54(3):484–498 e488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Burudi EM, Riese S, Stahl PD, Regnier-Vigouroux A. Identification and functional characterization of the mannose receptor in astrocytes. Glia. 1999;25(1):44–55. [DOI] [PubMed] [Google Scholar]
- 115.Ishikawa A, Miyake Y, Kobayashi K, et al. Essential roles of C-type lectin Mincle in induction of neuropathic pain in mice. Sci Rep. 2019;9(1):872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Garcia-Vallejo JJ, Ilarregui JM, Kalay H, et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J Exp Med. 2014;211(7):1465–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Taylor PR, Reid DM, Heinsbroek SE, Brown GD, Gordon S, Wong SY. Dectin-2 is predominantly myeloid restricted and exhibits unique activation-dependent expression on maturing inflammatory monocytes elicited in vivo. Eur J Immunol. 2005;35(7):2163–2174. [DOI] [PubMed] [Google Scholar]
- 118.Taylor PR, Tsoni SV, Willment JA, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol. 2007;8(1):31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Saijo S, Fujikado N, Furuta T, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol. 2007;8(1):39–46. [DOI] [PubMed] [Google Scholar]
- 120.Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413(6851):36–37. [DOI] [PubMed] [Google Scholar]
- 121.Inoue M, Moriwaki Y, Arikawa T, et al. Cutting edge: critical role of intracellular osteopontin in antifungal innate immune responses. J Immunol. 2011;186(1):19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nakamura K, Kinjo T, Saijo S, et al. Dectin-1 is not required for the host defense to Cryptococcus neoformans. Microbiol Immunol. 2007;51(11):1115–1119. [DOI] [PubMed] [Google Scholar]
- 123.Mezger M, Kneitz S, Wozniok I, Kurzai O, Einsele H, Loeffler J. Proinflammatory response of immature human dendritic cells is mediated by dectin-1 after exposure to Aspergillus fumigatus germ tubes. J Infect Dis. 2008;197(6):924–931. [DOI] [PubMed] [Google Scholar]
- 124.Thompson A, Griffiths JS, Walker L, et al. Dependence on Dectin-1 Varies With Multiple Candida Species. Front Microbiol. 2019;10:1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shah VB, Huang Y, Keshwara R, Ozment-Skelton T, Williams DL, Keshvara L. Beta-glucan activates microglia without inducing cytokine production in Dectin-1-dependent manner. J Immunol. 2008;180(5):2777–2785. [DOI] [PubMed] [Google Scholar]
- 126.Shah VB, Ozment-Skelton TR, Williams DL, Keshvara L. Vav1 and PI3K are required for phagocytosis of beta-glucan and subsequent superoxide generation by microglia. Mol Immunol. 2009;46(8–9):1845–1853. [DOI] [PubMed] [Google Scholar]
- 127.Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47(3):566–581 e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Keren-Shaul H, Spinrad A, Weiner A, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169(7):1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- 129.Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell. 2018;173(5):1073–1081. [DOI] [PubMed] [Google Scholar]
- 130.Li Q, Cheng Z, Zhou L, et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron. 2019;101(2):207–223 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Deerhake ME, Shinohara ML. Emerging roles of Dectin-1 in noninfectious settings and in the CNS. Trends Immunol. 2021;42(10):891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bode K, Bujupi F, Link C, et al. Dectin-1 Binding to Annexins on Apoptotic Cells Induces Peripheral Immune Tolerance via NADPH Oxidase-2. Cell Rep. 2019;29(13):4435–4446 e4439. [DOI] [PubMed] [Google Scholar]
- 133.Appelmelk BJ, van Die I, van Vliet SJ, Vandenbroucke-Grauls CM, Geijtenbeek TB, van Kooyk Y. Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol. 2003;170(4):1635–1639. [DOI] [PubMed] [Google Scholar]
- 134.Soilleux EJ, Morris LS, Leslie G, et al. Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J Leukoc Biol. 2002;71(3):445–457. [PubMed] [Google Scholar]
- 135.Curtis BM, Scharnowske S, Watson AJ. Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc Natl Acad Sci U S A. 1992;89(17):8356–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lambert C, Desbarats J, Arbour N, et al. Dendritic cell differentiation signals induce anti-inflammatory properties in human adult microglia. J Immunol. 2008;181(12):8288–8297. [DOI] [PubMed] [Google Scholar]
- 137.Wang M, Ye X, Hu J, et al. NOD1/RIP2 signalling enhances the microglia-driven inflammatory response and undergoes crosstalk with inflammatory cytokines to exacerbate brain damage following intracerebral haemorrhage in mice. J Neuroinflammation. 2020;17(1):364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chauhan VS, Sterka DG Jr., Furr SR, Young AB, Marriott I. NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia. 2009;57(4):414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gresnigt MS, Jaeger M, Subbarao Malireddi RK, et al. The Absence of NOD1 Enhances Killing of Aspergillus fumigatus Through Modulation of Dectin-1 Expression. Front Immunol. 2017;8:1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gresnigt MS, Cunha C, Jaeger M, et al. Genetic deficiency of NOD2 confers resistance to invasive aspergillosis. Nat Commun. 2018;9(1):2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu X, Chauhan VS, Young AB, Marriott I. NOD2 mediates inflammatory responses of primary murine glia to Streptococcus pneumoniae. Glia. 2010;58(7):839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu X, Chauhan VS, Marriott I. NOD2 contributes to the inflammatory responses of primary murine microglia and astrocytes to Staphylococcus aureus. Neurosci Lett. 2010;474(2):93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tavares AH, Burgel PH, Bocca AL. Turning Up the Heat: Inflammasome Activation by Fungal Pathogens. PLoS Pathog. 2015;11(7):e1004948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Burm SM, Zuiderwijk-Sick EA, t Jong AE, et al. Inflammasome-induced IL-1beta secretion in microglia is characterized by delayed kinetics and is only partially dependent on inflammatory caspases. J Neurosci. 2015;35(2):678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Barclay WE, Aggarwal N, Deerhake ME, et al. The AIM2 inflammasome is activated in astrocytes during the late phase of EAE. JCI Insight. 2022;7(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. 2017;214(5):1351–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Deng Y, Fu Y, Sheng L, et al. The Regulatory NOD-Like Receptor NLRC5 Promotes Ganglion Cell Death in Ischemic Retinopathy by Inducing Microglial Pyroptosis. Front Cell Dev Biol. 2021;9:669696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Furr SR, Chauhan VS, Sterka D Jr., Grdzelishvili V, Marriott I. Characterization of retinoic acid-inducible gene-I expression in primary murine glia following exposure to vesicular stomatitis virus. J Neurovirol. 2008;14(6):503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jaeger M, van der Lee R, Cheng SC, et al. The RIG-I-like helicase receptor MDA5 (IFIH1) is involved in the host defense against Candida infections. Eur J Clin Microbiol Infect Dis. 2015;34(5):963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Karki R, Man SM, Malireddi RKS, et al. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe. 2015;17(3):357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gustin A, Kirchmeyer M, Koncina E, et al. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS One. 2015;10(6):e0130624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ma C, Li S, Hu Y, et al. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J Exp Med. 2021;218(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Han F, Guo H, Wang L, et al. The cGAS-STING signaling pathway contributes to the inflammatory response and autophagy in Aspergillus fumigatus keratitis. Exp Eye Res. 2021;202:108366. [DOI] [PubMed] [Google Scholar]
- 154.Jiang GL, Yang XL, Zhou HJ, et al. cGAS knockdown promotes microglial M2 polarization to alleviate neuroinflammation by inhibiting cGAS-STING signaling pathway in cerebral ischemic stroke. Brain Res Bull. 2021;171:183–195. [DOI] [PubMed] [Google Scholar]
- 155.MacVicar BA, Newman EA. Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect Biol. 2015;7(5):a020388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rossi D, Volterra A. Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res Bull. 2009;80(4–5):224–232. [DOI] [PubMed] [Google Scholar]
- 157.Turner DA, Adamson DC. Neuronal-astrocyte metabolic interactions: understanding the transition into abnormal astrocytoma metabolism. J Neuropathol Exp Neurol. 2011;70(3):167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Salman MM, Kitchen P, Halsey A, et al. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vainchtein ID, Molofsky AV. Astrocytes and Microglia: In Sickness and in Health. Trends in Neurosciences. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Still KM, Batista SJ, O’Brien CA, et al. Astrocytes promote a protective immune response to brain Toxoplasma gondii infection via IL-33-ST2 signaling. PLoS Pathog. 2020;16(10):e1009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity. 2017;46(6):957–967. [DOI] [PubMed] [Google Scholar]
- 163.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32(12):638–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wu Y, Du S, Johnson JL, et al. Microglia and amyloid precursor protein coordinate control of transient Candida cerebritis with memory deficits. Nat Commun. 2019;10(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Carty M, Bowie AG. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol. 2011;81(7):825–837. [DOI] [PubMed] [Google Scholar]
- 166.Mishra BB, Mishra PK, Teale JM. Expression and distribution of Toll-like receptors in the brain during murine neurocysticercosis. J Neuroimmunol. 2006;181(1–2):46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Henn A, Kirner S, Leist M. TLR2 hypersensitivity of astrocytes as functional consequence of previous inflammatory episodes. J Immunol. 2011;186(5):3237–3247. [DOI] [PubMed] [Google Scholar]
- 168.Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53(7):688–695. [DOI] [PubMed] [Google Scholar]
- 169.Li Y, Xu XL, Zhao D, et al. TLR3 ligand Poly IC Attenuates Reactive Astrogliosis and Improves Recovery of Rats after Focal Cerebral Ischemia. CNS Neurosci Ther. 2015;21(11):905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49(3):360–374. [DOI] [PubMed] [Google Scholar]
- 171.Rostami J, Fotaki G, Sirois J, et al. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J Neuroinflammation. 2020;17(1):119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43(3):281–291. [DOI] [PubMed] [Google Scholar]
- 173.Lee KM, Chiu KB, Sansing HA, et al. Aerosol-induced brucellosis increases TLR-2 expression and increased complexity in the microanatomy of astroglia in rhesus macaques. Front Cell Infect Microbiol. 2013;3:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Graveline R, Segura M, Radzioch D, Gottschalk M. TLR2-dependent recognition of Streptococcus suis is modulated by the presence of capsular polysaccharide which modifies macrophage responsiveness. Int Immunol. 2007;19(4):375–389. [DOI] [PubMed] [Google Scholar]
- 175.Zheng H, Punaro MC, Segura M, et al. Toll-like receptor 2 is partially involved in the activation of murine astrocytes by Streptococcus suis, an important zoonotic agent of meningitis. J Neuroimmunol. 2011;234(1–2):71–83. [DOI] [PubMed] [Google Scholar]
- 176.Phulwani NK, Esen N, Syed MM, Kielian T. TLR2 expression in astrocytes is induced by TNF-alpha- and NF-kappa B-dependent pathways. J Immunol. 2008;181(6):3841–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hsieh HL, Wang HH, Wu CY, Tung WH, Yang CM. Lipoteichoic acid induces matrix metalloproteinase-9 expression via transactivation of PDGF receptors and NF-kappaB activation in rat brain astrocytes. Neurotox Res. 2010;17(4):344–359. [DOI] [PubMed] [Google Scholar]
- 178.Rodgers KR, Lin Y, Langan TJ, Iwakura Y, Chou RC. Innate Immune Functions of Astrocytes are Dependent Upon Tumor Necrosis Factor-Alpha. Sci Rep. 2020;10(1):7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59(2):242–255. [DOI] [PubMed] [Google Scholar]
- 180.Lehnardt S, Lachance C, Patrizi S, et al. The Toll-Like Receptor TLR4 Is Necessary for Lipopolysaccharide-Induced Oligodendrocyte Injury in the CNS. J Neurosci. 2002;22(7):2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hamel R, Ferraris P, Wichit S, et al. African and Asian Zika virus strains differentially induce early antiviral responses in primary human astrocytes. Infect Genet Evol. 2017;49:134–137. [DOI] [PubMed] [Google Scholar]
- 182.Butchi NB, Pourciau S, Du M, Morgan TW, Peterson KE. Analysis of the neuroinflammatory response to TLR7 stimulation in the brain: comparison of multiple TLR7 and/or TLR8 agonists. J Immunol. 2008;180(11):7604–7612. [DOI] [PubMed] [Google Scholar]
- 183.Brandenburg LO, Jansen S, Albrecht LJ, et al. CpG oligodeoxynucleotides induce the expression of the antimicrobial peptide cathelicidin in glial cells. J Neuroimmunol. 2013;255(1–2):18–31. [DOI] [PubMed] [Google Scholar]
- 184.Atmaca HT, Kul O, Karakus E, Terzi OS, Canpolat S, Anteplioglu T. Astrocytes, microglia/macrophages, and neurons expressing Toll-like receptor 11 contribute to innate immunity against encephalitic Toxoplasma gondii infection. Neuroscience. 2014;269:184–191. [DOI] [PubMed] [Google Scholar]
- 185.Lindblom RP, Aeinehband S, Parsa R, et al. Genetic variability in the rat Aplec C-type lectin gene cluster regulates lymphocyte trafficking and motor neuron survival after traumatic nerve root injury. J Neuroinflammation. 2013;10:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Burudi EM, Regnier-Vigouroux A. Regional and cellular expression of the mannose receptor in the post-natal developing mouse brain. Cell Tissue Res. 2001;303(3):307–317. [DOI] [PubMed] [Google Scholar]
- 187.Singh KK, Nathamu S, Adame A, et al. Expression of mannose binding lectin in HIV-1-infected brain: implications for HIV-related neuronal damage and neuroAIDS. Neurobehav HIV Med. 2011;3:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Arentsen T, Qian Y, Gkotzis S, et al. The bacterial peptidoglycan-sensing molecule Pglyrp2 modulates brain development and behavior. Mol Psychiatry. 2017;22(2):257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jiang G, Sun D, Kaplan HJ, Shao H. Retinal astrocytes pretreated with NOD2 and TLR2 ligands activate uveitogenic T cells. PLoS One. 2012;7(7):e40510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61(7):1113–1121. [DOI] [PubMed] [Google Scholar]
- 191.Inoue M, Chen PH, Siecinski S, et al. An interferon-beta-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat Neurosci. 2016;19(12):1599–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109(26):10480–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Inoue M, Williams KL, Oliver T, et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal. 2012;5(225):ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chou WC, Guo Z, Guo H, et al. AIM2 in regulatory T cells restrains autoimmune diseases. Nature. 2021;591(7849):300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.de Rivero Vaccari JP, Minkiewicz J, Wang X, et al. Astrogliosis involves activation of retinoic acid-inducible gene-like signaling in the innate immune response after spinal cord injury. Glia. 2012;60(3):414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li L, Yang R, Feng M, et al. Rig-I is involved in inflammation through the IPS-1/TRAF(6) pathway in astrocytes under chemical hypoxia. Neurosci Lett. 2018;672:46–52. [DOI] [PubMed] [Google Scholar]
- 197.Furr SR, Moerdyk-Schauwecker M, Grdzelishvili VZ, Marriott I. RIG-I mediates nonsegmented negative-sense RNA virus-induced inflammatory immune responses of primary human astrocytes. Glia. 2010;58(13):1620–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.De Miranda J, Yaddanapudi K, Hornig M, Lipkin WI. Astrocytes recognize intracellular polyinosinic-polycytidylic acid via MDA-5. FASEB J. 2009;23(4):1064–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chen Q, Boire A, Jin X, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533(7604):493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Liang Z, Wang X, Hao Y, et al. The Multifaceted Role of Astrocyte Connexin 43 in Ischemic Stroke Through Forming Hemichannels and Gap Junctions. Front Neurol. 2020;11:703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhou C, Chen X, Planells-Cases R, et al. Transfer of cGAMP into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-viral Immunity. Immunity. 2020;52(5):767–781 e766. [DOI] [PubMed] [Google Scholar]
- 202.Goethals S, Ydens E, Timmerman V, Janssens S. Toll-like receptor expression in the peripheral nerve. Glia. 2010;58(14):1701–1709. [DOI] [PubMed] [Google Scholar]
- 203.Barajon I, Serrao G, Arnaboldi F, et al. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J Histochem Cytochem. 2009;57(11):1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104(34):13798–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lehmann SM, Rosenberger K, Kruger C, et al. Extracellularly delivered single-stranded viral RNA causes neurodegeneration dependent on TLR7. J Immunol. 2012;189(3):1448–1458. [DOI] [PubMed] [Google Scholar]
- 206.Liu T, Berta T, Xu ZZ, et al. TLR3 deficiency impairs spinal cord synaptic transmission, central sensitization, and pruritus in mice. J Clin Invest. 2012;122(6):2195–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cameron JS, Alexopoulou L, Sloane JA, et al. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci. 2007;27(47):13033–13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Peltier DC, Simms A, Farmer JR, Miller DJ. Human neuronal cells possess functional cytoplasmic and TLR-mediated innate immune pathways influenced by phosphatidylinositol-3 kinase signaling. J Immunol. 2010;184(12):7010–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Li Y, Li H, Zhang Y, et al. Toll-like receptor 2 is required for opioids-induced neuronal apoptosis. Biochem Biophys Res Commun. 2010;391(1):426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tang SC, Lathia JD, Selvaraj PK, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol. 2008;213(1):114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Toll-like receptor 7 mediates pruritus. Nat Neurosci. 2010;13(12):1460–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull. 2012;28(2):131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ma Y, Li J, Chiu I, et al. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175(2):209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Acioglu C, Mirabelli E, Baykal AT, et al. Toll like receptor 9 antagonism modulates spinal cord neuronal function and survival: Direct versus astrocyte-mediated mechanisms. Brain Behav Immun. 2016;56:310–324. [DOI] [PubMed] [Google Scholar]
- 215.Suzuki Y, Nakano Y, Mishiro K, et al. Involvement of Mincle and Syk in the changes to innate immunity after ischemic stroke. Sci Rep. 2013;3(1):3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yang J, Lee HG, Cho S, et al. Activation of spinal macrophage-inducible C-type lectin induces mechanical allodynia and microglial activation in rats. Neurosci Lett. 2019;690:42–47. [DOI] [PubMed] [Google Scholar]
- 217.Ma X, Zhang W, Xu C, et al. Nucleotide-binding oligomerization domain protein 1 enhances oxygen-glucose deprivation and reperfusion injury in cortical neurons via activation of endoplasmic reticulum stress-mediated autophagy. Exp Mol Pathol. 2020;117:104525. [DOI] [PubMed] [Google Scholar]
- 218.Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab. 2009;29(3):534–544. [DOI] [PubMed] [Google Scholar]
- 219.Kaushal V, Dye R, Pakavathkumar P, et al. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015;22(10):1676–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Liu F, Lo CF, Ning X, et al. Expression of NALP1 in cerebellar granule neurons stimulates apoptosis. Cell Signal. 2004;16(9):1013–1021. [DOI] [PubMed] [Google Scholar]
- 221.Imbeault E, Mahvelati TM, Braun R, Gris P, Gris D. Nlrx1 regulates neuronal cell death. Mol Brain. 2014;7(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Adamczak SE, de Rivero Vaccari JP, Dale G, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab. 2014;34(4):621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lammert CR, Frost EL, Bellinger CE, et al. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature. 2020;580(7805):647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Co JG, Witwer KW, Gama L, Zink MC, Clements JE. Induction of innate immune responses by SIV in vivo and in vitro: differential expression and function of RIG-I and MDA5. J Infect Dis. 2011;204(7):1104–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Nazmi A, Dutta K, Basu A. RIG-I mediates innate immune response in mouse neurons following Japanese encephalitis virus infection. PLoS One. 2011;6(6):e21761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Nazmi A, Mukhopadhyay R, Dutta K, Basu A. STING mediates neuronal innate immune response following Japanese encephalitis virus infection. Sci Rep. 2012;2(1):347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A. 2010;107(25):11555–11560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bsibsi M, Nomden A, van Noort JM, Baron W. Toll-like receptors 2 and 3 agonists differentially affect oligodendrocyte survival, differentiation, and myelin membrane formation. J Neurosci Res. 2012;90(2):388–398. [DOI] [PubMed] [Google Scholar]
- 229.Li Y, Zhang R, Hou X, et al. Microglia activation triggers oligodendrocyte precursor cells apoptosis via HSP60. Mol Med Rep. 2017;16(1):603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. [DOI] [PubMed] [Google Scholar]
- 231.Uzun O, Ascioglu S, Anaissie EJ, Rex JH. Risk factors and predictors of outcome in patients with cancer and breakthrough candidemia. Clin Infect Dis. 2001;32(12):1713–1717. [DOI] [PubMed] [Google Scholar]
- 232.Muhlemann K, Wenger C, Zenhausern R, Tauber MG. Risk factors for invasive aspergillosis in neutropenic patients with hematologic malignancies. Leukemia. 2005;19(4):545–550. [DOI] [PubMed] [Google Scholar]
- 233.Lee EKS, Gillrie MR, Li L, et al. Leukotriene B4-Mediated Neutrophil Recruitment Causes Pulmonary Capillaritis during Lethal Fungal Sepsis. Cell Host Microbe. 2018;23(1):121–133 e124. [DOI] [PubMed] [Google Scholar]
- 234.Lionakis MS, Fischer BG, Lim JK, et al. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog. 2012;8(8):e1002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lionakis MS, Lim JK, Lee CC, Murphy PM. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun. 2011;3(2):180–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Drummond RA, Collar AL, Swamydas M, et al. CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System. PLoS Pathog. 2015;11(12):e1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wilson D, Thewes S, Zakikhany K, et al. Identifying infection-associated genes of Candida albicans in the postgenomic era. FEMS Yeast Res. 2009;9(5):688–700. [DOI] [PubMed] [Google Scholar]
- 238.Zhang M, Sun D, Liu G, Wu H, Zhou H, Shi M. Real-time in vivo imaging reveals the ability of neutrophils to remove Cryptococcus neoformans directly from the brain vasculature. J Leukoc Biol. 2016;99(3):467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Buchanan KL, Doyle HA. Requirement for CD4 + T Lymphocytes in Host Resistance against Cryptococcus neoformans in the Central Nervous System of Immunized Mice. Infection and Immunity. 2000;68(2):456–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sun D, Zhang M, Liu G, et al. Intravascular clearance of disseminating Cryptococcus neoformans in the brain can be improved by enhancing neutrophil recruitment in mice. Eur J Immunol. 2016;46(7):1704–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Davis JM, Huang M, Botts MR, Hull CM, Huttenlocher A. A Zebrafish Model of Cryptococcal Infection Reveals Roles for Macrophages, Endothelial Cells, and Neutrophils in the Establishment and Control of Sustained Fungemia. Infect Immun. 2016;84(10):3047–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Caffrey AK, Lehmann MM, Zickovich JM, et al. IL-1alpha signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog. 2015;11(1):e1004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Clark HL, Jhingran A, Sun Y, et al. Zinc and Manganese Chelation by Neutrophil S100A8/A9 (Calprotectin) Limits Extracellular Aspergillus fumigatus Hyphal Growth and Corneal Infection. J Immunol. 2016;196(1):336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Shlezinger N, Irmer H, Dhingra S, et al. Sterilizing immunity in the lung relies on targeting fungal apoptosis-like programmed cell death. Science. 2017;357(6355):1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Panackal AA, Wuest SC, Lin YC, et al. Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis. PLoS Pathog. 2015;11(5):e1004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Loeffler J, Haddad Z, Bonin M, et al. Interaction analyses of human monocytes co-cultured with different forms of Aspergillus fumigatus. J Med Microbiol. 2009;58(Pt 1):49–58. [DOI] [PubMed] [Google Scholar]
- 247.Dominguez-Andres J, Feo-Lucas L, Minguito de la Escalera M, Gonzalez L, Lopez-Bravo M, Ardavin C. Inflammatory Ly6C(high) Monocytes Protect against Candidiasis through IL-15-Driven NK Cell/Neutrophil Activation. Immunity. 2017;46(6):1059–1072 e1054. [DOI] [PubMed] [Google Scholar]
- 248.Prinz M, Priller J. Tickets to the brain: role of CCR2 and CX3CR1 in myeloid cell entry in the CNS. J Neuroimmunol. 2010;224(1–2):80–84. [DOI] [PubMed] [Google Scholar]
- 249.Ngo LY, Kasahara S, Kumasaka DK, Knoblaugh SE, Jhingran A, Hohl TM. Inflammatory monocytes mediate early and organ-specific innate defense during systemic candidiasis. J Infect Dis. 2014;209(1):109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Hohl TM, Rivera A, Lipuma L, et al. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. 2009;6(5):470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sun D, Zhang M, Sun P, et al. VCAM1/VLA4 interaction mediates Ly6Clow monocyte recruitment to the brain in a TNFR signaling dependent manner during fungal infection. PLoS Pathog. 2020;16(2):e1008361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis and therapy. Nat Rev Neurol. 2017;13(1):13–24. [DOI] [PubMed] [Google Scholar]
- 253.Aguirre K, Crowe J, Haas A, Smith J. Resistance to Cryptococcus neoformans infection in the absence of CD4+ T cells. Med Mycol. 2004;42(1):15–25. [PubMed] [Google Scholar]
- 254.Li SS, Kyei SK, Timm-McCann M, et al. The NK receptor NKp30 mediates direct fungal recognition and killing and is diminished in NK cells from HIV-infected patients. Cell Host Microbe. 2013;14(4):387–397. [DOI] [PubMed] [Google Scholar]
- 255.Islam A, Li SS, Oykhman P, et al. An acidic microenvironment increases NK cell killing of Cryptococcus neoformans and Cryptococcus gattii by enhancing perforin degranulation. PLoS Pathog. 2013;9(7):e1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Dufaud C, Rivera J, Rohatgi S, Pirofski LA. Naive B cells reduce fungal dissemination in Cryptococcus neoformans infected Rag1(−/−) mice. Virulence. 2018;9(1):173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Goldmann T, Wieghofer P, Jordao MJ, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17(7):797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Colombo E, Farina C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016;37(9):608–620. [DOI] [PubMed] [Google Scholar]
- 259.Sheppard DC, Filler SG. Host cell invasion by medically important fungi. Cold Spring Harb Perspect Med. 2014;5(1):a019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Blasi E, Mazzolla R, Barluzzi R, Mosci P, Bartoli A, Bistoni F. Intracerebral transfer of an in vitro established microglial cell line: local induction of a protective state against lethal challenge with Candida albicans. J Neuroimmunol. 1991;32(3):249–257. [DOI] [PubMed] [Google Scholar]
- 261.Drummond RA, Swamydas M, Oikonomou V, et al. CARD9(+) microglia promote antifungal immunity via IL-1beta- and CXCL1-mediated neutrophil recruitment. Nat Immunol. 2019;20(5):559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Xu-Vanpala S, Deerhake ME, Wheaton JD, et al. Functional heterogeneity of alveolar macrophage population based on expression of CXCL2. Sci Immunol. 2020;5(50). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun. 2015;6:5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Voet S, Mc Guire C, Hagemeyer N, et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun. 2018;9(1):2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Spitzer P, Condic M, Herrmann M, et al. Amyloidogenic amyloid-beta-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci Rep. 2016;6(1):32228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron. 2004;43(5):605–608. [DOI] [PubMed] [Google Scholar]
- 268.Lee SC, Dickson DW, Casadevall A. Pathology of cryptococcal meningoencephalitis: analysis of 27 patients with pathogenetic implications. Hum Pathol. 1996;27(8):839–847. [DOI] [PubMed] [Google Scholar]
- 269.Stott KE, Loyse A, Jarvis JN, et al. Cryptococcal meningoencephalitis: time for action. Lancet Infect Dis. 2021;21(9):e259–e271. [DOI] [PubMed] [Google Scholar]
- 270.Vecchiarelli A Immunoregulation by capsular components ofCryptococcus neoformans. Medical Mycology. 2000;38(6):407–417. [DOI] [PubMed] [Google Scholar]
- 271.Barluzzi R, Brozzetti A, Delfino D, Bistoni F, Blasi E. Role of the capsule in microglial cell—Cryptococcus neoformansinteraction: impairment of antifungal activity but not of secretory functions. Medical Mycology. 1998;36(4):189–197. [PubMed] [Google Scholar]
- 272.Lee SC, Kress Y, Dickson DW, Casadevall A. Human microglia mediate anti-Cryptococcus neoformans activity in the presence of specific antibody. J Neuroimmunol. 1995;62(1):43–52. [DOI] [PubMed] [Google Scholar]
- 273.Goldman D, Song X, Kitai R, Casadevall A, Zhao ML, Lee SC. Cryptococcus neoformans induces macrophage inflammatory protein 1alpha (MIP-1alpha) and MIP-1beta in human microglia: role of specific antibody and soluble capsular polysaccharide. Infect Immun. 2001;69(3):1808–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Neal LM, Xing E, Xu J, et al. CD4(+) T Cells Orchestrate Lethal Immune Pathology despite Fungal Clearance during Cryptococcus neoformans Meningoencephalitis. mBio. 2017;8(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Eschke M, Piehler D, Schulze B, et al. A novel experimental model of Cryptococcus neoformans-related immune reconstitution inflammatory syndrome (IRIS) provides insights into pathogenesis. Eur J Immunol. 2015;45(12):3339–3350. [DOI] [PubMed] [Google Scholar]
- 276.Xu J, Neal LM, Ganguly A, et al. Chemokine receptor CXCR3 is required for lethal brain pathology but not pathogen clearance during cryptococcal meningoencephalitis. Sci Adv. 2020;6(25):eaba2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Olave MC, Vargas-Zambrano JC, Celis AM, Castaneda E, Gonzalez JM. Infective capacity of Cryptococcus neoformans and Cryptococcus gattii in a human astrocytoma cell line. Mycoses. 2017;60(7):447–453. [DOI] [PubMed] [Google Scholar]
- 278.Lee SC, Dickson DW, Brosnan CF, Casadevall A. Human astrocytes inhibit Cryptococcus neoformans growth by a nitric oxide-mediated mechanism. J Exp Med. 1994;180(1):365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Almutairi BM, Nguyen TB, Jansen GH, Asseri AH. Invasive aspergillosis of the brain: radiologic-pathologic correlation. Radiographics. 2009;29(2):375–379. [DOI] [PubMed] [Google Scholar]
- 280.Gandhi J, Naik P, Kaur I, Kumar A, Joseph J. Microglial Response to Aspergillus flavus and Candida albicans: Implications in Endophthalmitis. J Fungi (Basel). 2020;6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Speth C, Rambach G, Lass-Florl C, et al. Culture supernatants of patient-derived Aspergillus isolates have toxic and lytic activity towards neurons and glial cells. FEMS Immunol Med Microbiol. 2000;29(4):303–313. [DOI] [PubMed] [Google Scholar]
- 282.Rambach G, Hagleitner M, Mohsenipour I, et al. Antifungal activity of the local complement system in cerebral aspergillosis. Microbes Infect. 2005;7(13):1285–1295. [DOI] [PubMed] [Google Scholar]
- 283.Rambach G, Maier H, Vago G, et al. Complement induction and complement evasion in patients with cerebral aspergillosis. Microbes Infect. 2008;10(14–15):1567–1576. [DOI] [PubMed] [Google Scholar]
- 284.Speth C, Dierich MP, Gasque P. Neuroinvasion by pathogens: a key role of the complement system. Mol Immunol. 2002;38(9):669–679. [DOI] [PubMed] [Google Scholar]
- 285.Hickman SE, Kingery ND, Ohsumi TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16(12):1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Suzuki Y, Nakano Y, Mishiro K, et al. Involvement of Mincle and Syk in the changes to innate immunity after ischemic stroke. Sci Rep. 2013;3:3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Burudi EME, Riese S, Stahl PD, Regnier-Vigouroux A. Identification and functional characterization of the mannose receptor in astrocytes. Glia. 1999;25(1):44–55. [DOI] [PubMed] [Google Scholar]
- 288.Li P, Shen Y, Cui P, et al. Neuronal NLRC5 regulates MHC class I expression in Neuro-2a cells and also during hippocampal development. J Neurochem. 2020;152(2):182–194. [DOI] [PubMed] [Google Scholar]