

Abstract
Histone variants represent chromatin components that diversify the structure and function of the genome. The variants of H2A, primarily H2A.X, H2A.Z and macroH2A, are well-established participants in DNA damage response (DDR) pathways, which function to protect the integrity of the genome. Through their deposition, post-translational modifications and unique protein interaction networks, these variants guard DNA from endogenous threats including replication stress and genome fragility as well as from DNA lesions inflicted by exogenous sources. A growing body of work is now providing a clearer picture on the involvement and mechanistic basis of H2A variant contribution to genome integrity. Beyond their well-documented involvement in gene regulation, we review here how histone H2A variants promote genome stability and how alterations in these pathways contribute to human diseases including cancer.
Keywords: H2A variants, Histones, H2AX, macroH2A, Genome integrity, DNA repair
1. Introduction
1.1. Chromatin and histone variants
Genetic information stored in nuclear DNA of eukaryotes must be compacted to fit into the volume of the nucleus while also being accessible to DNA-templated processes including transcription, replication and DNA repair. This is accomplished by packaging and organizing DNA into chromatin, the basic unit being the nucleosome. Nucleosomes consist of an octamer of histone proteins, which are assembled into higher order structures through linker histones, RNA, and various chromatin interacting proteins. In addition to the core histones H2A, H2B, H3 and H4 that make up the majority of nucleosomes in mammals, variants of core histones also exist that can replace their core histone counterparts and create nucleosomes with altered structural and functional capabilities. In humans, variants of all core histones have been identified, with H2A variants being the most diverse and numerous (Fig. 1a). Of note, there are many copies of each core histone including 17 individual histone genes alone that encode H2A. Surprisingly, even within these 17 genes there is sequence variation of up to several amino acids [1]. H2A histone variants consist of 11 genes that, unlike core histones, can contain introns (ex. H2A.Z.2 and macroH2A.1; Fig. 1b) and encode for at least 13 unique proteins due to alternative splicing. Differences in sequence identity among these variants also range broadly compared to H2A, from only a few amino acids to as little as ~18-% identity. Core histones are expressed mainly in S-phase while expression of histone variants occurs throughout the cell cycle, making their deposition and regulation independent of DNA replication. Analysis of the expression of these variants has revealed tissue and cellular heterogeneity, which also has been shown to be altered in some diseases including cancer [2].
Fig. 1.
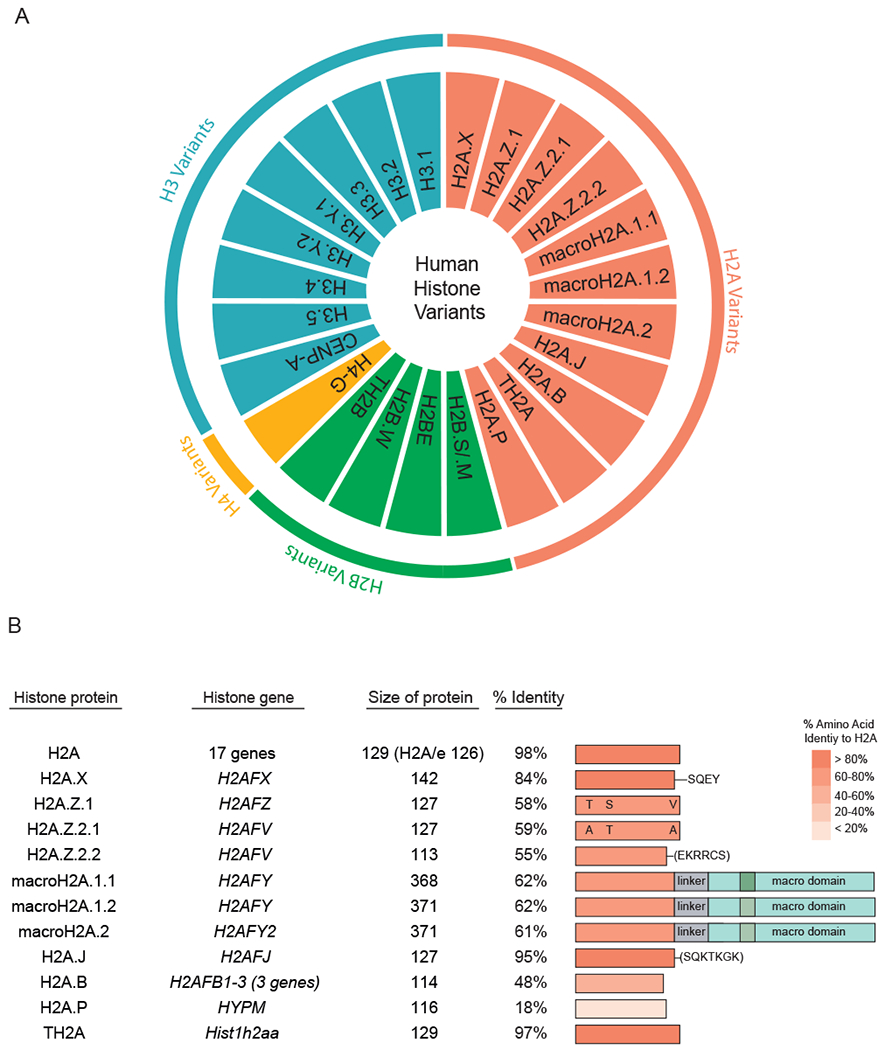
Human histone variants. (a) Complete list of human histone variants. Histone H2A variants consist of the highest number of variants (11) while histone H4 has only one variant reported to date in human cells. (b) Human histone H2A variants. Comparison of histone H2A variants with core H2A genes. Number of genes, gene names, protein size in amino acids and percent identity of variants to core H2A, along with key features of each variant, are provided. Note that H2A.Z.2.1 and H2A.Z.2.2, as well as macroH2A.1.1 and macroH2A.1.2, are splice variants of the same gene.
1.2. Post-translational modifications
The presence of numerous histone variants allows for an incredible potential for nucleosome diversity that can configure and fine-tune chromatin structure and function. Post-translational modifications (PTMs) of histone variants provide yet another level of complexity and diversity (Fig. 2). While writer enzymes have been defined for several of these PTMs (see Fig. 2), many remain poorly characterized and their biological function and regulatory mechanisms undefined. PTMs including phosphorylation, acetylation, methylation, ubiquitylation, SUMOylation and PARylation have been shown to decorate histone variants, which can provide binding sites for chromatin reader proteins that allow these factors to exert their function in a locally and often temporally controlled manner [2, 3]. This is equally true for the binding of DNA repair factors to PTM-mediated signals surrounding damaged DNA, which significantly impact chromatin and DNA repair processes [3, 4].
Fig. 2.
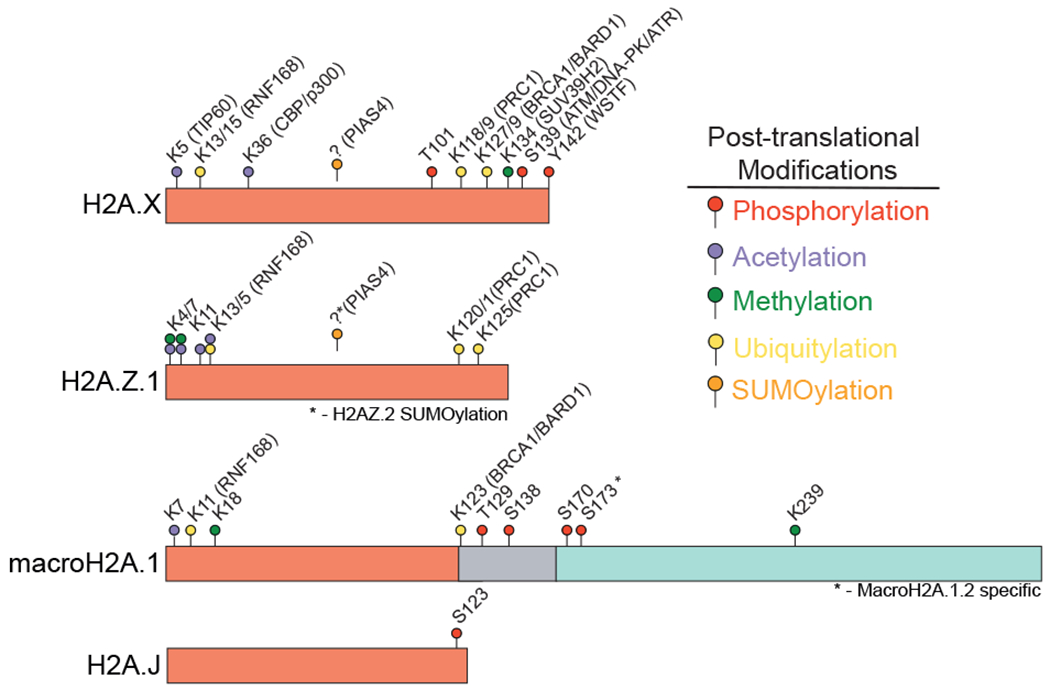
Post-translational modifications of histone H2A variants. Key PTMs of mammalian histone H2A variants with their modified residue (if known) are shown. Examples of histone writer enzymes for specific PTMs are included. Asterisks indicate variant specific modifications among closely related variants. Please note the list of histone H2A variant PTMs and their writer enzymes is not comprehensive.
1.3. Histone H2A variants in genome integrity and disease
While histone variants play well-established roles in regulating chromatin during transcription and development, which have been comprehensively reviewed elsewhere [2], emerging evidence reveals their important contribution for maintaining genome integrity. Here we focus our discussion on the involvement of mammalian histone H2A variants and their PTMs in DNA damage response pathways that respond to endogenous and exogenous sources of DNA damage, including during DNA double-strand break (DSB) repair (Fig. 2). Given that genome instability is a hallmark or enabling defect of several human diseases such as cancer, neurodegeneration and premature aging [5], we will further discuss how recent disease-related H2A variant biology may relate to their function in genome maintenance.
The complexity and necessity of maintaining the integrity of the genome is perhaps best characterized in the context of DSB repair, which represent dangerous lesions that if left unrepaired, can result in mutations and cell death. The DNA damage response (DDR) counters DSBs through a series of tightly controlled, sequential events that initiate with DNA end sensing, poly(ADP-ribose) polymerase (PARP) activation and concomitant chromatin relaxation, which is followed by the recruitment of the DDR kinases ATM, ATR and / or DNA-PKcs as well as a number of chromatin modifiers and remodelers that together recruit and activate downstream repair effector proteins [6–8]. The precise nature of these factors depends on the type of DSB repair, which in turn depends on the cell cycle phase as well as the location of the DNA lesion and has been summarized in excellent detail in several recent reviews [9–13]. In brief, there are two main DSB repair pathways: nonhomologous end-joining (NHEJ) which mediates the ligation of broken ends in a template-independent manner and homologous recombination (HR), which facilitates error-free repair using a homologous, undamaged DNA template [10, 12, 14–16]. Alternative end joining (Alt-EJ), which includes Polymerase theta mediated end-joining (TMEJ), represents a third, less frequently used group of DSB repair pathways that are normally suppressed by HR or NHEJ [12, 17]. Alt-EJ repairs DNA ends using small 3 - 25 bp patches of homology and is thus also error prone by nature, and often referred to as microhomology-mediated end joining (MMEJ) [10]. Both HR and Alt-EJ depend on DNA end resection, a process involving various endo- and exonuclease activities, such as MRE11 of the DNA end sensing MRE11-NBS1-RAD50 (MRN) complex, the 5’ to 3’ exonuclease EXO1, and others. In the case of HR, long-range resection is further supported by CtIP and the BRCA tumor suppressor genes [10, 17]. Resected single-stranded DNA (ssDNA) is coated with the ssDNA binding protein RPA, which is replaced by the RAD51 recombinase that forms filaments involved in homology search and strand invasion. The activity of BRCA1 and subsequent HR initiation is counteracted by 53BP1 in a manner that is tightly controlled by the underlying chromatin environment and thereby modulates the choice of repair pathway that acts on each individual DSB (see below and [18–21]).
Defects in DSB repair pathways and many of the underlying repair factors result in genome instability that can involve point mutations, small insertions and deletions, as well as larger chromosomal aberrations and translocations, all of which have been associated with human pathologies [5]. Repair pathway imbalance can further result in pathway-specific vulnerabilities, which has been exploited as a powerful strategy to develop new targeted therapies to treat cancer, a concept first used to develop PARP inhibitors (PARPi) to target HR-deficient tumors [22, 23]. In the following, we will discuss how H2A variants participate in DSB repair through their ability to alter the DNA lesion-surrounding chromatin context in a way that directly impacts chromatin accessibility, sensing of the DNA lesion, repair factor recruitment and, ultimately, optimal DNA repair. As defects in H2A variant function have been associated with a number of human pathologies, we further discuss the potential involvement of their genome repair activities in these diseases, which may be distinct from or complementary to their well-known functions in gene regulation and development.
2. H2A.X
2.1. γH2A.X in genome maintenance
The histone H2A variant H2A.X, which comprises ~ 10% of chromatin-associated H2A species, was first described in 1980 by West and Bonner, who used two dimensional histone gel electrophoresis to separate and identify histone H2A variants [24]. Several years later, the same group realized the involvement of H2A.X in the DDR, when they determined that H2A.X from mammalian cells and mice was phosphorylated on serine 139 within the SQEY motif comprising the 4 most C-terminal amino acids of this variant. This modified protein species was termed “γH2A.X” from its discovery following DNA damage induction by ionizing gamma-radiation (IR) [25]. Interestingly, the SQ motif located two amino acids from the end of the histone protein is invariant across species, being found in animals, plants, fungi and even protists [26]. For example, in fission and budding yeast, the core histone H2A is a homolog of H2A.X and contains the SQ motif. Using a specific antibody raised against the S139-phosphorylated H2A.X peptide, immunofluorescence of irradiated mammalian cells and other species revealed discrete nuclear foci of γH2A.X representing megabases of modified chromatin that marked individual DSBs (Fig. 3a) [21]. The ability of γH2A.X to identify unrepaired DNA lesions in cells was a landmark finding that remains the standard technique used by most researchers to label DNA lesions in cells, which is reflected in the number of citations on H2A.X and γH2A.X that continue to be published to this day (Fig. 3b).
Fig. 3.
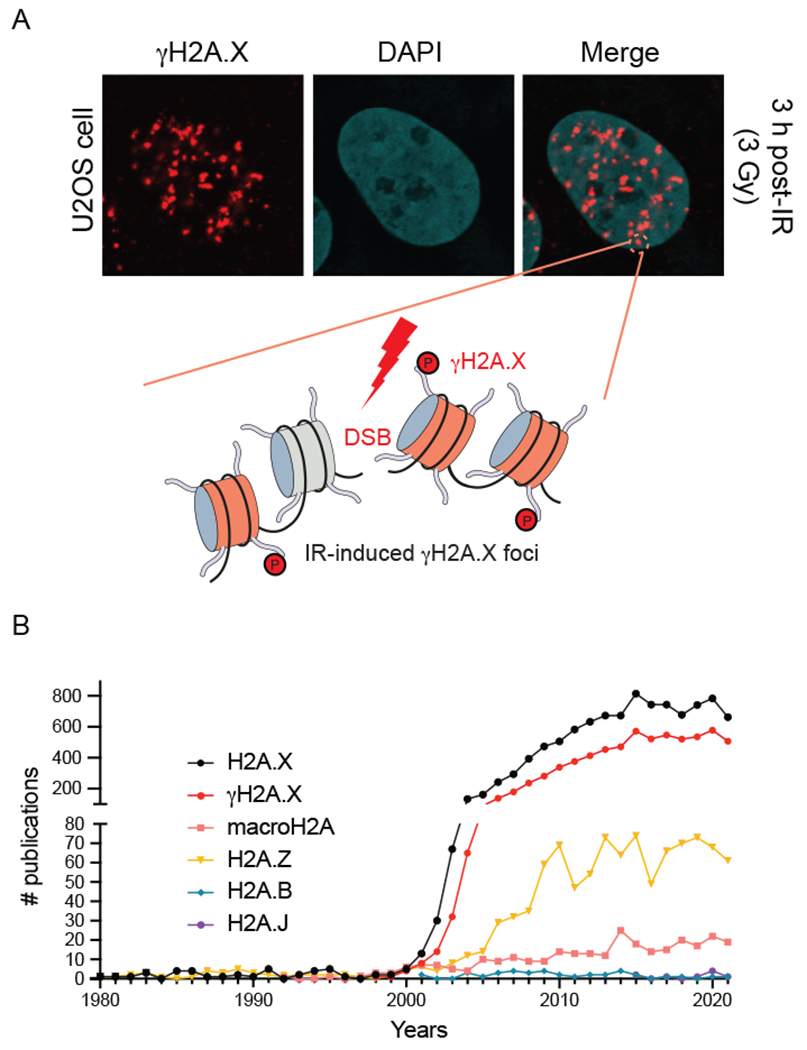
Characterization of γH2A.X and histone H2A variant publications. (a) Ionizing radiation-induced γH2A.X foci. Human U2OS cancer cells treated with IR and analyzed by immunofluorescence with a γH2A.X specific antibody (image provided by Doohyung Lee, Miller lab, UT Austin). Each focus represents γH2AX signal surrounding an individual DNA double-strand break. γH2AX-modified nucleosomes are highlighted in red and unmodified nucleosomes are in gray. (b) Analysis of publications involving histone H2A variants. H2A.X is the most highly studied histone H2A variants, with the majority of publications involving γH2A.X.
After its initial discovery, γH2A.X was shown to co-localize with several DNA repair proteins including RAD50 of the MRN complex, BRCA1 and RAD51 [28]. Several other groups rapidly identified additional factors that localized to γH2A.X-marked DSBs including ATM, 53BP1 and MDC1 (reviewed in [29]). Using various phospho-H2A.X peptide probes to mimic γH2A.X, MDC1 and 53BP1 were shown to directly bind to H2A.X phosphorylated on S139, identifying these factors as “readers” of γH2A.X within chromatin-surrounding DNA break sites [30–33]. The identification of the kinase(s) responsible for γH2A.X was first hinted at by the observation that treatment with the PI-3 kinase inhibitor wortmannin prevented damage-induced γH2A.X [28]. The ataxia-telangiectasia mutated (ATM) kinase, a phosphoinositide 3 kinase-related protein kinase (PI3KK), was shown to directly phosphorylate H2A.X on S139 within its preferred SQ/TQ motif in vitro, and cells deficient for ATM resulted in defective γH2A.X formation [34]. The related PI3KKs DNA-PK and ATR are also capable of phosphorylating γH2A.X [35, 36]. Upon DDR activation, MDC1 becomes a platform for the recruitment of additional proteins to break sites through both phospho-specific and other interactions (reviewed in [6]). These include NBS1 of the MRN complex [37–40], ATM [41], and the E3 ubiquitin ligase RNF8 [42–44]. The ability of MDC1 to bind both γH2A.X and MRN/ATM provides a mechanism whereby this signal can spread on chromatin across several 100 kb to create the visible foci observed by immunofluorescence in cells. The γH2A.X-MDC1 axis plays a pivotal role in blocking CtIP-mediated DNA end resection in G1 phase lymphocytes to promote V(D)J recombination and genome stability [45]. Thus, γH2A.X serves as a molecular amplifier and platform that acts to recruit and retain DDR factors to DSB-surrounding chromatin to promote their signaling, processing and repair.
The physiological relevance of γH2A.X was first established by the ability to eliminate H2A.X in mice and mammalian cells. The Nussenzweig and Alt labs generated H2A.X null mice, which helped define the physiological role of this variant in suppressing genome instability [46, 47]. Indeed, although H2A.X loss was not essential for viability, H2A.X knockout (KO) mice were born ~30% smaller than their WT littermates and displayed sensitivity to IR. Embryonic fibroblasts derived from H2A.X KO mice grew poorly in culture and displayed premature senescence, consistent with the observation that these cells contained increased levels of endogenous DNA damage. Importantly, the DNA damage-induced foci of repair proteins including 53BP1 and BRCA1 that colocalized with γH2A.X were impaired in H2A.X KO cells as well as in cells complemented with H2A.X containing a non-phosphorylatable S139A mutation, demonstrating that the phosphorylation of H2A.X was specifically required to mount the chromatin response to DSBs. Similar results were observed in mouse embryonic stem cells and human cells lacking H2A.X [48, 49], suggesting a conserved function for H2A.X in DNA repair across mammalian species and cell types.
2.2. PTMs beyond mammalian γH2A.X
As described above and highlighted in Fig. 2, H2A.X is decorated by other PTMs in addition to phosphorylation on S139 and these play additional roles in DNA damage signaling and repair. Once RNF8 is recruited to damage sites, in addition to H2A and H2A.X, it ubiquitinates linker histone H1 with K63-linked ubiquitin chains that are recognized by an additional E3 ligase RNF168, which is required for 53BP1 and BRCA1 recruitment to DNA lesions [50]. RNF168 mono-ubiquitylates both H2A and H2A.X on K15 [51], a mark read by the ubiquitin-dependent recruitment (UDR) domain of 53BP1 [52]. More recently, several groups reported that the obligate E3 ligase partner of the heterodimer complex containing BRCA1 contained a ubiquitin binding domain that also reads K15ub within H2A and possibly H2A.X [53–55]. These studies suggest an elegant competition mechanism between BRCA1 and 53BP1 that regulates DSB repair pathway choice.
Additional PTMs on H2A.X that fine-tune the chromatin response to DSBs have been recently reviewed [3, 20]. For example, H2A.X is sumoylated by PIAS4 (Fig. 2; [49]), which along with another SUMO E3 ligase PIAS1, regulate 53BP1 and BRCA1 dynamics at break sites [56, 57]. The terminal tyrosine of H2A.X is phosphorylated by the non-canonical, bromodomain protein WSTF and dephosphorylated by EYA1 [58, 59]. This mark has been proposed to collaborate with γH2A.X to regulate an apoptotic DNA repair pathway choice, including through a mechanism involving the binding of these marks by the Fe65-JNK1 complex [58]. This mark is also read by the SH2 domain of GRB2, which helps enforce the recruitment of MRE11 to break sites and subsequent HR repair [60]. H2AX has several additional phosphorylations, including on T101, which is induced by DNA damage and contributes to radiation resistance [61]. H2AX is acetylated on K5 and K36 by TIP60 and CBP/p300 respectively, which aids in promoting IR-resistance and HR repair [61, 62]. Methylation of H2A.X on K134 by SUV39H2 promotes γH2A.X formation and DSB repair [63]. PRC1-mediated ubiquitination on K119 is conserved between H2A and H2A.X and has been implicated in DSB repair [64, 65], Poly (ADP-ribose) modification (PARylation) of E141 on H2A.X promotes base excision repair (BER) through the recruitment of NEIL3 glycosylase to oxidative DNA lesions [66]. NEIL3 was shown to bind directly to PAR chains, including ADP-ribosylated H2AX, through its C-terminal GRF zinc-finger domains. Remarkably, E141 PARylation appears to prevent aberrant phosphorylation of nearby S139 (γH2A.X to counteract an erroneous accumulation of DSB repair factors at single-stranded DNA lesions, supporting a regulatory role for H2A.X beyond DSB repair [66]. Together, these studies highlight the importance and complex nature of modifications on H2A.X that are associated with the DDR. However, we still know little about the coordinated regulation of these marks in a spatiotemporal manner and how this relates mechanistically to DNA damage signaling and repair events that are established by γH2A.X Despite its discovery over 20 years ago, future studies are clearly needed to fully comprehend how γH2A.X and its other modifications orchestrate genome maintenance during the DDR and in other biological contexts. Of note, many of the modifications discussed above may not be restricted to H2A.X or H2A. Whether and how other H2A variants are modified, and how such modifications may affect the DDR, remains to be determined.
ATM-mediated phosphorylation of H2A.X is conserved in plants, which express an additional histone H2A variant H2A.W that is also phosphorylated on an SQ residue by ATM [67]. While H2A.X appears to function in euchromatin in plants, H2A.W is localized to heterochromatin where it facilitates genome maintenance pathways including DSB repair, transposon silencing and DNA methylation [67–69]. These studies highlight that while general genome maintenance pathways involving histone H2A variants are conserved from mammals to plants, additional diversification of these pathways has occurred. We refer readers to several excellent reviews focusing on histone variants in plants for additional details [70, 71].
2.3. Anatomy and function of γH2A.X domains
Although the above studies established the functional importance of γH2A.X in DNA damage signaling and repair, the physical nature of γH2A.X damage-induced foci has only recently been investigated. Early work used various γH2A.X chromatin immunoprecipitation (ChIP) techniques to determine that this signal emanated from break sites at senescent telomeres, as well as RAG- and restriction enzyme-induced, site-specific DSBs [72–74]. Of note, these studies observed a non-uniform and asymmetric distribution of γH2A.X spreading from breaks. Using super resolution microscopy, γH2A.X foci induced by IR were further resolved to identify multiple “nano-foci” of γH2A.X associated with each individual DSB with an approximate size of 75 kb, a structure observed as a single focus by traditional microscopy [75]. Notably, these clustered γH2A.X foci were shown to be in close proximity to CTCF, a chromosomal architectural protein involved in organizing topologically associated domains (TADs) and chromosome loops. Similar results were obtained in a study using genome-wide Hi-C chromatin contact maps to map γH2AX propagation as it relates to the spatial organization of TADs and other genomic interactions [76]. Depletion of CTCF resulted in radio-sensitization and a diminution of γH2A.X nano-foci, which strongly suggested that chromatin architecture dictates γH2A.X formation and its function in chromatin-based responses to DNA damage [75]. Related to these findings, super resolution microscopy approaches allowed for the detection of 53BP1 and its effector RIF1 at break-associated topological domains [77]. Cells deficient for 53BP1 or RIF, but not core DSB factors, were unable to maintain these 3-dimensional chromatin domains at DSBs. Finally, using human DIvA (DSB inducible via AsiSI) cells coupled with 4C-Seq and Hi-C, the Legube lab demonstrated that TADs establish γH2A.X-53BP1 domains surrounding DSBs through a loop extrusion mechanism involving CTCF-anchored ATM and cohesins [78]. Thus, the 3D genome organization in cells regulates the establishment of γH2A.X revealing a vital interplay between nuclear architecture and genome integrity. These findings also suggest that variations in genome organization across individual cells, cell types or pathological states may contribute to the engagement, use and efficiency of DNA repair mechanisms that impact genome maintenance [11].
In addition to being modified upon DNA damage, H2A.X has also been shown to be incorporated de novo at sites of UVC-induced and replication damage in a mechanism dependent on the histone chaperone FACT [79]. Conversely, H2A.Z was observed to be removed from break sites, consistent with other studies (see below). This work highlights a potential role for H2A.X deposition directly at break sites in supporting DDR factor recruitment and repair functions at damage sites, beyond its initial phosphorylation. It is worth noting that H2A.X has been reported to promote replication fork stability, and H2A.X deficiency results in sensitivity to replication stress [80, 81]. This process may further involve H2A variant exchanges at stalled forks, given the γH2A.X-dependent deposition of the replication-stress protective macroH2A1 variant at stalled replication forks (see below and [82]). Therefore, the addition and removal of various histone variants likely acts to shape a repair-competent chromatin environment for both the repair of DNA lesions and the epigenetic maintenance of the surrounding chromatin. It is important to note that the initial recruitment of DNA damage response factors is independent of H2A.X [83]. Consistent with this notion, H2AX-independent repair pathways have also been identified. These can involve MDC1 damage recruitment through its interactions with the nucleosome acidic patch, an interaction hub known for coordinating epigenetic and DDR factor chromatin interactions, which promotes 53BP1 damage recognition and DNA repair in the absence of H2A.X [84]. Altogether, γH2A.X acts as a robust marker of DNA damage and H2A.X is an important facilitator of the chromatin response to DNA damage through its ability to promote the assembly of DNA damage signaling and repair proteins at DNA breaks.
2.4. H2A.X in disease
Although H2A.X is best known for its role in DNA damage signaling and repair, studies have begun to establish the function of this variant in other biological processes. As these have been reviewed extensively, we point readers to these more comprehensive reviews and highlight here a few key studies as examples of the multifunctional nature of H2A.X in mammals [85– 87]. Early work using mouse models of H2A.X loss revealed a tumor suppressor function of H2A.X. Indeed, mice with either one or both copies of H2A.X removed in a p53 mutant background developed tumors and unstable genomes including translocations, establishing H2A.X as haploinsufficient for tumor suppression [46, 88]. Intriguingly, human H2A.X maps to a region frequently altered in several cancers including leukemias, raising the possibility that H2A.X is involved in human cancers [46, 85]. Loss of H2AX results in increased cancer cell migration and invasion, consistent with the notion that H2A.X is involved in the epithelial-mesenchymal transition (EMT) [89]. While loss of H2A.X did not result in increased metastasis in an in vivo model in this study, re-expression of H2A.X resulted in increased tumorigenesis, highlighting a complex relationship between H2A.X and cancer. γH2A.X has also been used extensively to identify DNA damage sites in many cancer cell and tissue samples under normal conditions and following treatments with therapeutic drugs including DNA damaging agents ([90]; reviewed in [91]). This provides a robust readout of DNA damage for analysis of genotoxicity as well as cytotoxicity as apoptotic cells also induce γH2A.X Furthermore, γH2A.X is being evaluated as a potential sensitive and early detection biomarker for cancer and other damage related pathologies including aging, given the high frequency and involvement of genome instability in these diseases.
In addition to cancer, H2A.X has been shown to be involved in other pathologies. H2A.X deficiency in mice results in defective mitochondrial morphology and function, implicating H2A.X in metabolism [92]. Interestingly, H2A.X KO mice display defects in neurological function and behavior that is linked to altered oxidative stress responses [93]. Similar defects have been associated with ATM deficiency in mice and patients with Ataxia-Telangectasia, pointing to a central role for DDR defects in common neurodegenerative disorders. We refer the reader to a recent comprehensive review on this topic . Combined loss of ATM and H2A.X results in embryonic lethality in mice, revealing a genetic dependency for both of these genes during embryogenesis [94]. H2A.X protein levels are reduced in cells exposed to persistent oxidative stress, which alters DNA integrity pathways under such conditions [95]. Together with its role as a modulator of BER [66], this observation has important implications for H2A.X regulation and functions in human pathologies where oxidative stress is implicated, including cancer, aging, neurodegeneration, metabolic disorders and others (reviewed in [96]). Functional roles in meiosis, development, stem cell biology and cell cycle control have also been reported (reviewed in [87]). In many cases, it is unclear how these H2A.X functions are orchestrated, and if they are related to its role in DNA damage signaling and repair. While potential reader proteins of γH2A.X and other PTMs on this variant may participate, little is known about bona fide effector proteins, genome localization, chromatin structure and function involving H2A.X in these biological processes. In many cases, a role for H2A.X in gene regulation rather than in signaling has not been ruled out. Although many questions remain, these studies emphasize the multifunctional and pivotal roles of H2A.X in not only in the DDR but also in other biological processes.
3. H2A.Z
3.1. H2A.Z and genome integrity
The histone H2A variant H2A.Z is encoded by two genes whose protein products, H2A.Z.1 and H2A.Z.2, differ by only 3 amino acids (Fig. 1b). H2A.Z.2 is found in two splice variants with H2A.Z.2.2 containing a truncated and variable C-terminus [97]. Unlike other H2A variants, H2A.Z is essential in mice, where it regulates development and stem cell functions [98]. H2A.Z is known to play multiple roles in transcription, including gene expression, RNA Polymerase II (Pol II) dynamics, enhancer function and 3D genome organization (reviewed in [99]). Not surprisingly, this variant has been implicated in other DNA-related processes including DNA repair, replication, cell cycle and chromosome segregation, and cellular senescence . Many of these pathways are linked to human pathologies and accordingly, H2A.Z has been implicated in cancer, metabolism and neurodegeneration [2, 99]. Given the diverse functions of H2A.Z, it is not unexpected that this variant is also highly modified by PTMs that participate in these underlying biological processes (Fig. 2) [2]. Structurally, H2A.Z incorporation results in nucleosome destabilization when compared to H2A and other variants, suggesting that this H2A.Z property may be important for its unique functions [97, 100, 101]. A detailed overview of H2A.Z biology can be found in a recent review by Colino-Sanguino et al. [99]. We will therefore limit this section to a brief synopsis of key operations performed by H2A.Z in genome maintenance.
H2A.Z is an established contributor to genome integrity in mammalian cells. Early studies revealed that H2A.Z was incorporated at DSBs by the p400 remodeler where it promoted chromatin opening that supported the localization of DNA repair factors Ku70/80 and BRCA1 [102]. Moreover, the FRRUC ubiquitin ligase complex facilitates H2A.Z loading at DNA breaks in a H2A119ub and PARP-dependent manner [103]. In addition to the repair process per se, H2A.Z loading also promotes break-proximal transcriptional repression, a function governed by several chromatin modifying and binding proteins including PRC1, PRC2 and others [104–106]. Interestingly, H2A.Z recruitment to breaks was found to be transient, involving its active removal by the chaperone Anp32E and chromatin remodeler INO80, which in turn was demonstrated to support DSB repair [107, 108]. Remarkably, in the context of HR, depletion of H2A.Z restored the repair defects in Anp32E and INO80 deficient cells, pointing to H2A.Z removal as the main function of these factors in HR. Whether this function is exclusively break-proximal or involves gene regulation or chromatin organization at sites other than DNA lesions is not known.
Although these studies have established mammalian H2A.Z as a contributor to DSB repair, several additional questions remain. It is unclear if H2A.Z plays a structural role in regulating chromatin dynamics at DNA breaks that facilitate repair, or if effector proteins that bind to H2A.Z also participate in repair. An attractive model is that the removal of H2A.Z from breaks may allow other H2A variants to be incorporated at sites of damage, for example macroH2A variants which show delayed accumulation at DSBs [109]. The involvement of mammalian H2A.Z in genome integrity pathways in addition to DSB repair are poorly understood. In yeast, loss of H2A.Z (Htz1) in replication checkpoint mutants results in genome instability and replication stress [110]. In mammalian cells, H2A.Z is enriched at replication origins and is required for a subset of early origins to be activated [111, 112]. Interestingly, H2A.Z was shown to promote H4K20me2 through its association with the methyltransferase SUV420H1 [112]. This histone mark is one of the signals read by 53BP1 for its recruitment to chromatin and function at DSBs [113]. Thus, although H2A.Z is undoubtedly involved in genome integrity, more information is needed to mechanistically define these roles and how they may relate to human deficiencies that potentially involve H2A.Z impairment.
4. macroH2A
4.1. macroH2A genes and functions
Macro-histones are unique among H2A variants in that they possess a large extranucleosomal domain consisting of a disordered linker region and a macrodomain (Fig. 1b). Macrodomains fold into a globular mixed α-helix and β-sheet structure containing a deep groove, which serves as a potential ligand-binding pocket for various forms of ADP-ribose (ADPR), including mono-ADPR, poly-ADPR (PAR), poly(A), and O-acetyl-ADP-ribose (OAADPR). Substantial sequence variation between different macrodomains may explain their ligand-selectivity. While macrodomains are highly evolutionarily conserved, macrodomain-containing histones are predominantly found in vertebrates and notably absent in common model organisms such as yeast and flies. Macro-histones exclusively involve the H2A core histone, and much has been learned in recent years about mammalian macroH2A.1 (reviewed in [2, 114]). Of emerging importance are the distinct biological roles of its two alternatively spliced macroH2A.1 isoforms, macroH2A1.1 and macroH2A.1.2 (Fig. 1b; [115]). The two isoforms differ in 33 aa within the macrodomain, which results in a macroH2A.1.1-specific ability to bind ADPR derivatives, while no ligands have been identified to date for the macrodomain of macroH2A.1.2. Significantly less is known about the independently encoded macroH2A.2 gene, which, like macroH2A.1.2, is unable to bind ADPR [116].
Macro-histones were originally identified as epigenetic modulators of gene expression, regulating developmental and/or senescence-associated transcription programs (see [2, 114] for a comprehensive discussion of this aspect of macroH2A1 function). Reminiscent of its alternatively spliced isoforms, macroH2A1 was shown to localize to two functionally distinct chromatin subtypes marked either by the repressive H3K27me3 mark or by H2B acetylations [117, 118]. However, a formal link between macroH2A.1 splicing and chromatin state remains to be established. Genome-wide macroH2A.1 mapping studies consistently show low to modest enrichment across large chromatin domains, often coinciding with H3K27me3, and to a lesser extent H3K9me3. Association with repressive chromatin marks was shown to facilitate the interaction of heterochromatic regions with lamin-associated domains (LADs), pointing to a role for macroH2A.1 in nuclear organization. Consistent with this, macroH2A.1 loss resulted in perinuclear heterochromatin loss and nuclear membrane deformation [119], which are generally associated with increased DNA damage and genome instability [120, 121]. Extending their contribution to genome maintenance, recent work points to a central and perhaps functionally related role for macroH2A.1.1 and macroH2A.1.2 in DNA repair, while defects in either variant have been linked to increased DNA damage accumulation, altered cell growth, and genomic aberrations [115]. Best understood to date is the role of macroH2A.1 proteins in DSB repair, although increasing evidence points to additional roles in the resolution of other types of DNA lesions. While transcriptional control of repair-relevant genes by macroH2A.1 cannot be discounted, it is the direct recruitment to or presence of macroH2A.1 at DNA lesions that appears to be critical for its DNA repair function(s). In the following, we review recent advances in our understanding of the often splice variant-specific roles of macroH2A.1 in mammalian DNA repair, their impact on genomic and overall nuclear integrity, and discuss possible implications for human pathologies where appropriate.
4.2. DSB repair and macroH2A
The first evidence for macroH2A.1 accumulation at a DNA lesion dates back over a decade, when the macroH2A.1.1 variant was found to accumulate at sites of laser-induced DNA damage (Fig. 4a) [122]. Recruitment of macroH2A.1.1 was dependent on both PARP1 and the ADPR-binding residues in the macroH2A.1.1 macrodomain, suggesting that this process occurs via and/or in response to PAR accumulation at sites of DNA damage. A similar phenomenon was observed at enzymatically induced DSBs [123]. In both cases, macroH2A.1.1 was found to affect DNA damage signaling and repair. While macroH2A.1.1 overexpression resulted in impaired recruitment of the NHEJ effectors Ku70/Ku80 and a concomitant delay in DNA damage resolution [122], macroH2A.1.1 knockdown caused a reduction in 53BP1 recruitment and a moderate increase in NHEJ efficiency [123]. Thus, increased and decreased macroH2A.1.1 levels appear to have a similar impact on the NHEJ process, although the mechanistic basis for this observation remains to be determined. More recently, macroH2A.1.1 was shown to promote repair via Alt-EJ in a splice variant-specific knockout mouse model [124]. Both Ku70/Ku80 and 53BP1 oppose Alt-EJ, consistent with the notion that macroH2A.1.1 may contribute to this repair pathway at least in part by affecting the recruitment of NHEJ effectors. In addition, macroH2A.1.1 was shown to interact with two key Alt-EJ mediators, PARP1 [125, 126] and Ligase 3, in a PARP1 activity-dependent manner that likely involves ADPR binding [124].
Fig. 4.
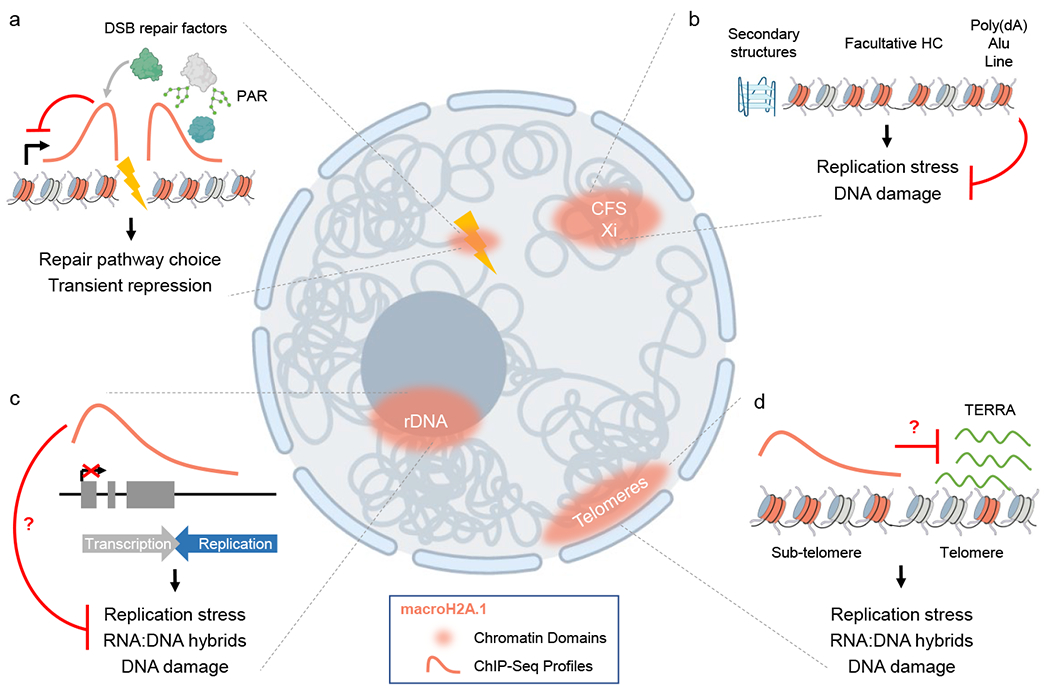
Involvement of macroH2A.1 in genome maintenance. (a-d) macroH2A.1 accumulation on chromatin coincides with DNA damage-susceptibility. (a) DSBs promote macroH2A.1 accumulation to modulate DSB repair factor engagement and transcription repression at breaks, which in the case of the macroH2A.1.1 variant can involve PARylation. (b) CFSs contain difficult to replicate elements including G4 structures, heterochromatin and repetitive DNA, which are often enriched for macroH2A.1 that in turn protects from replication stress and DNA damage. (c) rRNA-encoding rDNA repeat segments are enriched for macroH2A.1, which suppresses rDNA transcription and may protect from transcription:replication conflicts and resulting RNA:DNA hybrids. (d) MacroH2A.1 enrichment at subtelomeric DNA counteracts replication stress and DNA damage formation. Whether or not macroH2A.1 suppresses TERRA transcripts and subsequent RNA:DNA hybrids remains to be determined. macroH2A.1-containing nucleosomes are in light red, red lines depict macroH2A.1 enrichment and distribution based on ChIP.
Like macroH2A.1.1, macroH2A1.2 is recruited to sites of DSBs, albeit with delayed kinetics and in a manner dependent on ATM signaling rather than PARP1 activation [109]. MacroH2A.1.2 was found to be required for optimal BRCA1 recruitment and consequently promoted HR, suggesting that the two splice variants may support distinct DSB repair outcomes. While depletion of either variant had a similar, modest positive effect on NHEJ [109, 123], opposite effects were indeed observed for both HR and Alt-EJ upon variant-specific macroH2A.1 depletion [109, 124]. These findings support the view that macroH2A.1 variants regulate DNA repair pathway choice specifically at resected DNA ends, the obligate intermediates for both HR and Alt-EJ. Given that macroH2A.1.2 facilitates end resection via the BRCA1/CtIP axis [109], it is tempting to speculate that its absence results in limited resection, which impairs HR but not Alt-EJ [127]. MacroH2A.1.1, in turn, facilitates recruitment of the Alt-EJ machinery when macroH2A.1.2 abundance is low and resection and/or HR factor recruitment is suboptimal. While it is plausible that competition amongst macroH2A.1 variants during repair of resected ends occurs in a genome-site specific manner, a more general effect at the level of overall protein abundance cannot be excluded. The implications of these findings for genome integrity and ultimately malignant transformation are discussed in the context of fragile DNA elements below.
The presence of macroH2A.1 at DSBs does not only affect repair factor recruitment, but further has a significant impact on the DSB-surrounding chromatin environment, although the two processes are likely closely intertwined. Both macroH2A.1.1 and macroH2A.1.2 have been shown to promote a dynamic condensation of break proximal chromatin [109, 122], consistent with their established role in repressive chromatin formation [2]. If and how this structural change in break-proximal chromatin contributes to repair outcomes remains to be established. Of note, chromatin decondensation through histone deacetylase inhibition resulted in impaired BRCA1 retention, similar to the effect of decondensation observed upon macroH2A.1.2 depletion [109]. However, increased acetylation of specific histone residues that interfere with 53BP1 chromatin association has been linked to an increase in BRCA1 recruitment [128]. Together, these findings point to a complex role for chromatin dynamics in repair factor assembly and disassembly that is likely directly affected by the DSB-surrounding chromatin environment.
Consistent with a preference for open chromatin, HR has been shown to be preferentially active at transcribed loci, likely to avoid mutagenic repair of transcribing genic regions [129]. A set of recent observations involving macroH2A.1 among other factors, may help reconcile the seemingly conflicting requirements for active and repressive chromatin in the context of HR. The bromodomain-containing and hence acetyl-histone-binding ZMYND8 protein is recruited to breaks in actively transcribed genes, which in turn initiates the recruitment of the NuRD (nucleosome remodeling and histone deacetylation) complex and establishment of a repressive chromatin environment at genic DSBs [130–133]. ZMYND8/NuRD accumulation is further dependent on PAR-mediated recruitment of the histone demethylase KDM5A and concomitant H3K4 demethylation [131, 134]. Of note, optimal KDM5A recruitment also required the presence of macroH2A.1.2 [134], suggesting a need for the dynamic exchange of active and repressive chromatin marks to facilitate an HR-permissive repair environment. Consistent with this, DBSs were shown to induce sequential expansion and recondensation of chromatin, the latter of which being dependent on macroH2A.1.2 [109, 135]. Although not formally demonstrated, H2A.Z may account for the former, which could at least in part explain its role on HR (see above). Dynamic chromatin condensation further provides a plausible mechanistic basis for previously reported DSB-induced transcriptional silencing thought to help restrict gene expression until repair is completed [104–106]. Indeed, depletion of KDM5A, ZMYND8 or macroH2A.1.2 each results in defective break-proximal transcriptional silencing as well as impaired HR [20, 130, 131, 134]. If repression of DSB-proximal transcription is a prerequisite for HR or an auxiliary event to insulate the repair environment from active RNA polymerase encounters remains to be established.
It should be noted that whether or not macroH2A.1 variants are incorporated into break-proximal nucleosomes is an ongoing debate. While PARP1-dependent macroH2A.1.1 accumulation at DSBs does not appear to involve its nucleosome deposition [122, 123], recent work in the context of replication stress-associated DNA damage points to chromatin incorporation of macroH2A.1.2 [82]. However, no definitive histone chaperones have been identified for either variant. Moreover, given that both PAR and intrinsically disordered domains can promote liquid:liquid phase separation, it is possible that macroH2A.1 variant accumulation may reflect an aspect of the recently reported liquid condensate formation at DSB sites through either their disordered linker region or macroH2A.1.1:PAR binding (Fig. 4a; [11, 136, 137]). While a role for macroH2A.1.1 in HR remains to be determined, early PAR/macroH2A.1.1 accumulation may reflect an effort to restrain HR until cells encounter a more HR-permissive environment. A similar role has recently been proposed for phase separation of DNA lesions encountered in mitosis [11, 138]. Irrespective of the precise nature of macroH2A.1 variant accumulation at DSBs, the body of work discussed here underlines the importance of macroH2A not only in DSB repair but also for the coordination of the dynamic structural changes to chromatin that are associated with this process.
4.3. Involvement of macroH2A In ssDNA lesion repair?
In contrast to DSB repair, little is known about how macroH2A.1 affects the repair of ssDNA breaks, although the prominent role for PARP1 in the cellular response to ssDNA breaks points to involvement of at least the PAR-binding macroH2A.1.1 variant. In support of the latter, macroH2A.1.1 was recently shown to protect cells from oxidative DNA damage in a manner dependent in part on the activity of PARP1 [126]. Depletion of macroH2A.1.1 resulted in a pronounced increase in necrotic cell death, which was attributed to defective PAR chain stabilization by macroH2A.1.1, a feature that has been linked to excessive nuclear NAD+ consumption and metabolic homeostasis [126, 139]. Moreover, macroH2A.1 was able to facilitate the repair of the predominant oxidative DNA lesion 8-oxo-guanine, although the underlying mechanism remains unclear [126]. Support for a role in BER, the predominant repair pathway for 8-oxo-guanine, comes from the recent observation that macroH2A.1.1 can bind Ligase 3, which in addition to Alt-EJ is a key effector of BER [124]. Of note, macroH2A.1.2 depletion also results in an increase in susceptibility to oxidative stress, albeit in a PARP1-independent manner [126]. It will be interesting to determine if the latter is due to DSBs resulting from excessive oxidative damage, or if it reflects a bona fide role for macroH2A.1.2 in BER, perhaps complementary to or synergistic with that of macroH2A.1.1. Lastly, it will be imperative to assess if macro-histones, as well as other variants, are involved in additional ssDNA repair processes.
4.4. macroH2A and replication stress
Both ssDNA and dsDNA breaks are a hallmark of DNA replication stress. Of note, macroH2A.1 mapping efforts across a range of mammalian cell lines and tissues has revealed preferential macroH2A.1 enrichment at expansive, replication stress-susceptible genomic regions, including common fragile sites (CFSs), the inactive X chromosome, telomeres, and nucleolar ribosomal DNA (rDNA); (Fig. 4b–d; reviewed in [114]). Moreover, macroH2A.1 protects at least a subset of these regions from DNA damage, which has been most comprehensively studied at CFSs [82]. CFSs are genomic regions rich in secondary DNA structures such as G-quadruplexes, stem loop-prone short and long interspersed nuclear elements (SINEs/LINEs), RNA:DNA hybridforming regions and densely packed heterochromatin, all of which pose obstacles to DNA polymerases [140]. Stalling of the replication machinery results in the accumulation of under-replicated ssDNA regions, which activates a DNA damage checkpoint response involving ATR and CHK1 kinases to delay replication until the resulting damage can be resolved or repaired [141]. Consistent with its role at DSBs [109], macroH2A.1.2 facilitates the recruitment of the replication fork-protective BRCA1 protein to stalled replication forks [82]. MacroH2A.1.2 depletion conversely results in the accumulation of various hallmarks of replication stress, including phosphorylation of the ssDNA sensor and ATR activator RPA, accumulation of FANCD2, a marker of under-replicated CFSs, impaired RAD51 loading and concomitant replication fork degradation, suggesting HR as the main effector of macroH2A.1-mediated fork protection [82, 142]. Of note, CFS-associated DNA damage appears to be a driver of macroH2A.1 deposition, which was proposed to help shape a protective CFS chromatin environment for subsequent cell divisions (Fig. 4b) [82]. Both the histone chaperone FACT and the LSH2/HELLS chromatin remodeler have been implicated in this process [82, 142]. Since LSH2/HELLS is frequently mutated in Immunodeficiency Centromeric Instability Facial Anomalies (ICF) 4 syndrome [143], and given the epistatic function of macroH2A.1 and LSH2 in response to replication stress, it will be interesting to investigate if macroH2A.1 is similarly implicated in this severe human disease with symptoms including immunodeficiency, neurologic defects, and reduced growth [142, 144].
It is worth noting that ChIP-Seq analyses with splice variant-specific antibodies revealed similar enrichment patterns for both macroH2A.1.1 and macroH2A.1.2 [82, 145]. While replication stress has been proposed as a driver for CFS-specific accumulation of macroH2A.1.2, the underlying principles that govern macroH2A.1.1 deposition at these sites remain to be established. Their genomic overlap suggests that much like DNA methylation, existing macroH2A.1 molecules may serve as a bookmark for subsequent macroH2A.1 deposition, irrespective of the isoform. Consistent with this, LSH2 was recently shown to facilitate incorporation of two distinct macroH2A variants, macroH2A.1 and macroH2A.2 [146]. While macroH2A.1.1 does not appear to be directly implicated in the replication stress response [82], it may mediate a complementary genome-protective effect through its ability to inhibit basal poly-ADP ribose polymerase 1 (PARP-1) activity and concomitant nuclear NAD+ consumption [139].
The balanced presence and/or activity of both macroH2A.1 variants appears to be essential for genome stability at the replication stress-susceptible inactive X chromosome (Xi), which is significantly enriched for all macroH2A variants [114]. Consistent with its role at CFSs, macroH2A.1.2 loss resulted in a pronounced increase in both aneuploidy and micronuclei formation, which can be attributed to an accumulation of under-replicated DNA causing DNA fragility and DSBs upon entering mitosis [147]. However, while macroH2A.1.2 loss was the likely cause for the underlying lesions, it was their aberrant, Alt-EJ-dependent repair supported through macroH2A.1.1 that accounted for anaphase bridge formation and associated chromosomal aberrations [124]. Supporting the physiological relevance of this finding, mice deficient in macroH2A.1.2 have increased female-biased lethality observed as early as embryonic day 9.5, while mice that lack both macroH2A.1.2 and the Alt-EJ-promoting macroH2A.1.1 variant show no overt survival defects [124, 148].
Although chromosomal defects upon macroH2A.1.2 loss were most pronounced at the macroH2A.1-rich Xi, lower levels of aneuploidy could be observed on autosomes, suggesting that CFSs across the genome may be prone to similar defective repair when macroH2A.1 splicing is perturbed. Aberrant macroH2A.1 splicing is a frequent feature of most cancers and has been linked to poor patient survival [2, 115]. It is, thus, worth considering that macroH2A.1 splice variant profiles may reflect DSB repair potential and therapy responsiveness, given that both HR and Alt-EJ have been identified as cancer-specific vulnerabilities [22, 149–151]. In support of this notion, macroH2A.1 appears to alter tumor progression in a splice-variant and cancer-type dependent manner. Importantly, the impact of macroH2A.1 on cancer progression is likely to reflect a combination of its gene regulatory and genome maintenance functions (reviewed in [2, 114]).
4.5. Protection from replication:transcription conflicts by macroH2A?
CFSs are not the only genomic regions where genome integrity is impacted by macroH2A.1. Both telomeric and ribosomal DNA (rDNA) are enriched for macroH2A.1 and display functional defects upon macroH2A1 loss (Fig. 4c–d; [114]). Notably, at both loci, transcription poses a major source of genome instability, pointing to a common, genome-protective role of macroH2A.1-associated repressive chromatin [152]. In the case of the rDNA, rRNA-encoding genes exist in several hundred copies spread in clusters over 5 chromosomes, which aggregate in the nucleolus. Only a subset of rDNA repeats is expressed at any given time, and the silencing of the remaining rDNA units is at least in part mediated by macroH2A.1 [119, 153]. To prevent transcription:replication conflicts in active rDNA units, evolutionarily conserved replication fork barriers (RFB) separate the rRNA encoding region from the rDNA intergenic spacer region, where DNA replication initiates [154]. Spontaneous DNA breaks have recently been detected precisely at this barrier, even in the absence of added replication stress [155]. Notably, the separation of the rRNA-coding region and the intergenic spacer is recapitulated in the pattern of macroH2A.1 accumulation, which is restricted to the former (Fig. 4c; [153]). While it remains to be determined whether and how macroH2A.1 mediates protection from replication:transcription conflicts, several recent findings support this notion: first, macroH2A.1 was shown to coordinate replication and transcription processes during human papilloma virus replication in a process that appears to involve their spatial separation within viral replication foci [156]; second, macroH2A.1 has been implicated in the suppression of replication stress at telomeres [157], which is at least in part attributed to the transcription of telomeric repeat containing RNA (TERRA) (Fig. 4d; [158, 159]), third, TERRA-associated transcription:replication conflicts and the resulting formation of RNA:DNA hybrid structures can be resolved by BRCA1 [159], raising the possibility that macroH2A.1.2-mediated BRCA1 recruitment may help protect from these often detrimental, aberrant structures. Finally, telomeric RNA:DNA hybrids are particularly abundant in tumor cells undergoing Alternative Lengthening of Telomeres, which were shown to depend on macroH2A.1.2 for efficient tumor growth [157]. It is worth noting that an involvement of macroH2A.1 and/or its alternative splicing in RNA:DNA hybrid resolution more generally could have a significant impact on a range of RNA:DNA hybrid-associated pathologies [160–162], although no evidence for such a function exists to date.
4.6. macroH2A.1 PTMs in repair
Like all H2A variants, macroH2A.1 is subject to PTMs, which may affect its function in the repair processes described above (see Fig. 2 for a summary of known/validated macroH2A.1 PTMs). In support of the latter, macroH2A.1-ubiquitination has been linked to the DDR-associated E3 ligases RNF168 and BRCA1/BARD1 [163, 164]. RNF168 mediates K11Ub of macroH2A.1 in proximity to an alpha1 extension helix region that is conserved among H2A variants [163], and an analogous N-terminal Ub modification is observed at K15 on H2A.Z [163, 165]. Whether or not these modifications are induced in response to DNA damage remains to be determined, as does their impact on DNA repair. However, ubiquitination of the equivalent K residues on H2A plays an important role in the control of 53BP1 and BRCA1/BARD1 repair factor recruitment, directing the choice between HR and NHEJ [20, 52–55, 166]. In further support of a link to HR, BRCA1/BARD1 mediates macroH2A.1 ubiquitination at K123, although the functional relevance of this modification remains to be fully understood [164]. Of potential relevance for macroH2A.1-mediated Alt-EJ and Xi instability [124], phosphorylation of S137 was found to be enriched in mitosis and excluded from the inactive X chromosome [167]. Other macroH2A.1 phosphorylations may further be relevant for splice variant-specific macroH2A.1 functions, as T129 and S170 in the common macroH2A.1 linker region were found to be preferentially modified in macroH2A.1.1 and macroH2A.1.2, respectively [168]. What regulates this differential phosphorylation, and whether or not it contributes to the distinct repair outcomes associated with each variant, remains to be determined.
Altogether, macroH2A.1 plays a central role in maintaining nuclear integrity both in response to defined DNA lesions and at genomic regions that are prone to DNA damage. Remarkably, these protective functions involve several, possibly complementary functional and/or structural aspects of macroH2A.1 biology, including its genomic location, PTMs and the distinct biological roles of macroH2A.1 splice variants.
5. Mechanisms of genome integrity by other H2A variants
5.1. H2A.J
The H2A histone variant H2A.J is encoded by the H2AFJ gene in humans and is highly similar to core H2A, with a difference of approximately 5 amino acids only (Fig. 1b). This includes the presence of an SQ motif in the C-terminus that, like H2A.X, is phosphorylated after DNA damage [169]. H2A.J was first linked to DNA damage when its expression was observed to increase 10-fold in both replicative and DNA-damage induced senescent human fibroblasts [170]. H2A.J was shown to be required for the induction of immune and inflammatory genes, including cytokines associated with senescent-associated secretory phenotype (SASP). The ability of H2A.J to promote inflammatory genes may be contributed to its weakened interaction with linker histone H1. This is in line with the differences between H2A and H2A.J in the C-terminus where H2A binds H1. It has been proposed that phosphorylation of H2A.J could further weaken these contacts with H1 and DNA resulting in increased gene expression of immune and inflammation genes following DNA damage [169]. Of note, macroH2A.1 variants have also been implicated in senescence and the associated SASP phenotype [171]. If and how H2A.J relates to or affects macroH2A.1 function may be a worthwhile subject for future investigation. Further supporting a role in senescence and aging, aged tissues in mouse and human, as well as prolonged irradiation of mouse tissues, resulted in increased expression of H2A.J. In addition, gene amplification and overexpression of H2A.J has been reported in a subset of cancers, consistent with its involvement in DNA damage-associated human pathologies [170]. However, while irradiation increases the expression of H2A.J and leads to its phosphorylation, how these changes contribute to its role in genome maintenance remains unclear. H2A.J phosphorylation was observed only after high levels of radiation in mouse cells and H2A.J localization at breaks was not observed after acute irradiation in human cells [169, 172]. Thus, several questions remain for the potential involvement of H2A.J in genome integrity pathways. The identity of the kinase(s) involved in DNA damage-dependent phosphorylation of this variant on S123 are unknown, as is the localization and presence of this modification in human cells. Potential readers of this damage-induced mark are as-yet-unidentified and roles for H2A.J beyond those reported for gene regulation in response to damage remain undetermined. It will be informative to perform additional experiments to address these questions as they may reveal the function of this histone variant in damage-associated human diseases including cancer, aging and potentially neurodegeneration, a condition where H2A.J has not yet been studied. A recently reported H2A.J knockout mouse is viable and fertile and may provide an important next step to dissecting the physiological impact of H2A.J deficiency, particularly under conditions of stress [173].
5.2. H2A.B
H2A.B (also referred to as H2A.BbD) is another histone H2A variant implicated in DNA damage responses. This H2A variant is only 48% identical to H2A and was first identified as being excluded from the inactive X-chromosome in female human cells, suggesting a role in gene regulation [174]. Later work showed that H2A.B nucleosomes display reduced spacing compared to H2A and localize to active genes [175]. Depletion of H2A.B resulted in alterations in gene expression and defects in splicing. H2A.B is further likely to be actively involved in the response to DNA damage, as overexpressed, GFP-tagged H2A.B localizes to sites of laser microirradiation and replication. Reduced S-phase progression and defective UV damage repair was observed upon overexpression of H2A.B [176]. Proteomic analysis of this variant revealed an association with replication proteins, which is in line with H2A.B being involved in DNA replication and repair. Thus, several histone H2A variants in addition to H2A.X, H2A.Z and macroH2A also participate in genome integrity pathways. These variants remain insufficiently studied and several important questions remain. Neither do we know what cellular context these variants function in, nor are the molecular machinery regulating the dynamics, potential modifications and interaction partners identified or any disease relevance defined in most cases. Addressing these questions will likely provide important insights into the collective role that histone H2A variants play in genome integrity.
6. Conclusions and Future Directions
H2A variants come in many varieties and account for the majority of the nucleosome diversity observed in mammalian cells (Fig. 1a). Remarkably, all H2A variants studied in-depth to date show some involvement in genome maintenance pathways, underlining the importance of the chromatin environment for genomic integrity. While several H2A variants, like H2A.X, H2A.Z and more recently macroH2A.1, have been studied extensively, the relevance of variants such as H2A.J and H2A.B is only emerging. Moreover, changes between annotated H2A variants and core H2A are sometimes limited to a few amino acids, analogous to the variability observed between core H2A encoding genes, which draws into question what defines a histone variant. It will be interesting to determine if coding sequence variation across the many core H2A genes translates into functional differences, as observed for example between H2A.J and H2A.
Building on what we have learned to date, several key questions are emerging that will require attention if we want to truly understand how diversity in H2A histones, and nucleosome composition more generally, can be leveraged to understand repair pathways, genome integrity, and ultimately human diseases and their respective therapies. For one, the often pronounced, differential impact of H2A variants on DNA repair outcome and genome stability raises the question of what controls their incorporation and dynamic exchange in the context of distinct DNA lesions and chromatin states. Chaperones of key modulators of DNA repair such as macroH2A.1 remain to be uncovered, and the contribution of H2A variant exchange to repair kinetics and progression is only beginning to be understood. Is nucleosome incorporation even critical in all cases or are many of these functions channeled through pre-existing variant deposition as well as the numerous PTMs identified for H2A variants? Moreover, at least for macroH2A.1, chromatin association rather than nucleosome incorporation may be sufficient for their function in repair. What is the impact of non-break-proximal H2A variants on DNA repair? Recent insights into the importance of three-dimensional chromatin configuration for DSB repair suggest that TAD formation, chromatin looping and possibly phase separation can define repair outcome [11]. These processes are likely to be modulated by H2A variant nucleosomes, even when they are not directly recruited to or present at DNA lesions. It will further be important to functionally dissect the relevance for TADs and other three-dimensional structures for the DNA repair process itself. Manipulation of TAD-associated histone variants, such as H2A.X, may present a promising approach to address this issue. Moreover, there are many H2A variant PTMs with yet to be defined contribution to DNA repair. Given the significant impact that small changes in H2A variant modifications can have on repair outcomes and genome integrity, a wealth of additional information can be expected from further studies of PTM dynamics and the underlying modifiers and readers.
Finally, disease relevance of H2A variants remains an underexplored area of investigation. Linking H2A variants and their modifications and/or modifiers to disease-associated mutations should be a focus of future research. This will greatly benefit from the wealth of information that is being generated through various (single-cell) omics efforts to characterize human tissues, such as the Human Cell Atlas and the cancer-focused Human Tumor Atlas Network and Clinical Proteomic Tumor Analysis consortium [177–179]. Emphasis should also be put on regulators of alternative splicing, which may be of particular relevance for macroH2A.1 and H2A.Z physiology and function. Ultimately, a greater understanding of how H2A variants participate in the various DNA-templated processes including transcription, replication and DNA repair will be required to define the contribution that these epigenetic regulators have in normal physiology and disease. This information has the potential to be leveraged for new therapeutic strategies and tools such as biomarkers for human diseases, including those impacted by the loss of genome integrity such as aging, neurodegeneration and cancer.
Highlights.
Genome integrity relies on diverse histone H2A variants and their modifications
γH2A.X foci represent 3-D damage signaling frameworks formed by chromatin loops
macroH2A.1 is a splicing-regulated modulator of DSB repair pathway choice
H2A.J and H2A.B variants are emerging as effectors of DNA repair
H2A variant functions including genome integrity are linked with several human pathologies
Acknowledgements
We apologize to authors whose work was not included due to space limitations in this brief review. BioRender was used for parts of Fig. 4. The K.M.M. lab is supported in part through grants from the National Cancer Institute, National Institute of Health (N.I.H., CA198279 and CA201268) and the American Cancer Society (RSG-16-042-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interest statement
The authors declare no competing interests.
References
- [1].Pinto DMSF, The human canonical core histone catalogue, bioRxiv. [Google Scholar]
- [2].Buschbeck M, Hake SB, Variants of core histones and their roles in cell fate decisions, development and cancer, Nat Rev Mol Cell Biol 18(5) (2017) 299–314. [DOI] [PubMed] [Google Scholar]
- [3].Kim JJ, Lee SY, Miller KM, Preserving genome integrity and function: the DNA damage response and histone modifications, Crit Rev Biochem Mol Biol 54(3) (2019) 208–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dantuma NP, van Attikum H, Spatiotemporal regulation of posttranslational modifications in the DNA damage response, EMBO J 35(1) (2016) 6–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jackson SP, Bartek J, The DNA-damage response in human biology and disease, Nature 461(7267) (2009) 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ciccia A, Elledge SJ, The DNA damage response: making it safe to play with knives, Mol Cell 40(2) (2010) 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Price BD, D’Andrea AD, Chromatin remodeling at DNA double-strand breaks, Cell 152(6) (2013) 1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Smeenk G, van Attikum H, The chromatin response to DNA breaks: leaving a mark on genome integrity, Annu Rev Biochem 82 (2013) 55–80. [DOI] [PubMed] [Google Scholar]
- [9].Mitrentsi I, Yilmaz D, Soutoglou E, How to maintain the genome in nuclear space, Curr Opin Cell Biol 64 (2020) 58–66. [DOI] [PubMed] [Google Scholar]
- [10].Scully R, Panday A, Elango R, Willis NA, DNA double-strand break repair-pathway choice in somatic mammalian cells, Nat Rev Mol Cell Biol 20(11) (2019) 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sebastian R, Aladjem MI, Oberdoerffer P, Encounters in Three Dimensions: How Nuclear Topology Shapes Genome Integrity, Front Genet 12 (2021) 746380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhao B, Rothenberg E, Ramsden DA, Lieber MR, The molecular basis and disease relevance of non-homologous DNA end joining, Nat Rev Mol Cell Biol 21(12) (2020) 765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hustedt N, Durocher D, The control of DNA repair by the cell cycle, Nat Cell Biol 19(1) (2016) 1–9. [DOI] [PubMed] [Google Scholar]
- [14].Chang HHY, Pannunzio NR, Adachi N, Lieber MR, Non-homologous DNA end joining and alternative pathways to double-strand break repair, Nat Rev Mol Cell Biol 18(8) (2017) 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jasin M, Rothstein R, Repair of strand breaks by homologous recombination, Cold Spring Harb Perspect Biol 5(11) (2013) a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen CC, Feng W, Lim PX, Kass EM, Jasin M, Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer, Annu Rev Cancer Biol 2 (2018) 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brambati A, Barry RM, Sfeir A, DNA polymerase theta (Poltheta) - an error-prone polymerase necessary for genome stability, Curr Opin Genet Dev 60 (2020) 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Densham RM, Morris JR, Moving Mountains-The BRCA1 Promotion of DNA Resection, Front Mol Biosci 6 (2019) 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mirman Z, de Lange T, 53BP1: a dSb escort, Genes Dev 34(1-2) (2020) 7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sanchez A, Lee D, Kim DI, Miller KM, Making Connections: Integrative Signaling Mechanisms Coordinate DNA Break Repair in Chromatin, Front Genet 12 (2021) 747734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Setiaputra D, Durocher D, Shieldin - the protector of DNA ends, EMBO Rep 20(5) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lord CJ, Ashworth A, PARP inhibitors: Synthetic lethality in the clinic, Science 355(6330) (2017) 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].O’Neil NJ, Bailey ML, Hieter P, Synthetic lethality and cancer, Nat Rev Genet 18(10) (2017) 613–623. [DOI] [PubMed] [Google Scholar]
- [24].West MH, Bonner WM, Histone 2A, a heteromorphous family of eight protein species, Biochemistry 19(14) (1980) 3238–45. [DOI] [PubMed] [Google Scholar]
- [25].Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM, DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139, J Biol Chem 273(10) (1998) 5858–68. [DOI] [PubMed] [Google Scholar]
- [26].Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W, Histone H2A variants H2AX and H2AZ, Curr Opin Genet Dev 12(2) (2002) 162–9. [DOI] [PubMed] [Google Scholar]
- [27].Rogakou EP, Boon C, Redon C, Bonner WM, Megabase chromatin domains involved in DNA double-strand breaks in vivo, J Cell Biol 146(5) (1999) 905–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM, A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage, Curr Biol 10(15) (2000) 886–95. [DOI] [PubMed] [Google Scholar]
- [29].Fernandez-Capetillo O, Celeste A, Nussenzweig A, Focusing on foci: H2AX and the recruitment of DNA-damage response factors, Cell Cycle 2(5) (2003) 426–7. [PubMed] [Google Scholar]
- [30].Kleiner RE, Verma P, Molloy KR, Chait BT, Kapoor TM, Chemical proteomics reveals a gammaH2AX-53BP1 interaction in the DNA damage response, Nat Chem Biol 11(10) (2015) 807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ, MDC1 is a mediator of the mammalian DNA damage checkpoint, Nature 421(6926) (2003) 961–6. [DOI] [PubMed] [Google Scholar]
- [32].Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP, MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks, Cell 123(7) (2005) 1213–26. [DOI] [PubMed] [Google Scholar]
- [33].Ward IM, Minn K, Jorda KG, Chen J, Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX, J Biol Chem 278(22) (2003) 19579–82. [DOI] [PubMed] [Google Scholar]
- [34].Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ, ATM phosphorylates histone H2AX in response to DNA double-strand breaks, J Biol Chem 276(45) (2001) 42462–7. [DOI] [PubMed] [Google Scholar]
- [35].Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA, ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation, Cancer Res 64(7) (2004) 2390–6. [DOI] [PubMed] [Google Scholar]
- [36].Ward IM, Chen J, Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress, J Biol Chem 276(51) (2001) 47759–62. [DOI] [PubMed] [Google Scholar]
- [37].Chapman JR, Jackson SP, Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage, EMBO Rep 9(8) (2008) 795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Melander F, Bekker-Jensen S, Falck J, Bartek J, Mailand N, Lukas J, Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin, J Cell Biol 181(2) (2008) 213–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M, Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin, J Cell Biol 181(2) (2008) 227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wu L, Luo K, Lou Z, Chen J, MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks, Proc Natl Acad Sci U S A 105(32) (2008) 11200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J, MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals, Mol Cell 21(2) (2006) 187–200. [DOI] [PubMed] [Google Scholar]
- [42].Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J, RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly, Cell 131(5) (2007) 901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, Pelletier L, Jackson SP, Durocher D, Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase, Science 318(5856) (2007) 1637–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J, RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins, Cell 131(5) (2007) 887–900. [DOI] [PubMed] [Google Scholar]
- [45].Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH, Sleckman BP, H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes, Nature 469(7329) (2011) 245–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW, Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors, Cell 114(3) (2003) 359–70. [DOI] [PubMed] [Google Scholar]
- [47].Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A, Genomic instability in mice lacking histone H2AX, Science 296(5569) (2002) 922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW, Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX, Proc Natl Acad Sci U S A 99(12) (2002) 8173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chen WT, Alpert A, Leiter C, Gong F, Jackson SP, Miller KM, Systematic identification of functional residues in mammalian histone H2AX, Mol Cell Biol 33(1) (2013) 111–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, Mailand N, Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage, Nature 527(7578) (2015) 389–93. [DOI] [PubMed] [Google Scholar]
- [51].Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK, RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling, Cell 150(6) (2012) 1182–95. [DOI] [PubMed] [Google Scholar]
- [52].Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D, 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark, Nature 499(7456) (2013) 50–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Becker JR, Clifford G, Bonnet C, Groth A, Wilson MD, Chapman JR, BARD1 reads H2A lysine 15 ubiquitination to direct homologous recombination, Nature (2021). [DOI] [PubMed] [Google Scholar]
- [54].Dai L, Dai Y, Han J, Huang Y, Wang L, Huang J, Zhou Z, Structural insight into BRCA1-BARD1 complex recruitment to damaged chromatin, Mol Cell (2021). [DOI] [PubMed] [Google Scholar]
- [55].Hu Q, Botuyan MV, Zhao D, Cui G, Mer E, Mer G, Mechanisms of BRCA1-BARD1 nucleosome recognition and ubiquitylation, Nature (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP, Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks, Nature 462(7275) (2009) 935–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, Ng T, Solomon E, The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress, Nature 462(7275) (2009) 886–90. [DOI] [PubMed] [Google Scholar]
- [58].Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG, Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions, Nature 458(7238) (2009) 591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, Patel DJ, Elledge SJ, Allis CD, WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity, Nature 457(7225) (2009) 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ye Z, Xu S, Shi Y, Bacolla A, Syed A, Moiani D, Tsai CL, Shen Q, Peng G, Leonard PG, Jones DE, Wang B, Tainer JA, Ahmed Z, GRB2 enforces homology-directed repair initiation by MRE11, Sci Adv 7(32) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Xie A, Odate S, Chandramouly G, Scully R, H2AX post-translational modifications in the ionizing radiation response and homologous recombination, Cell Cycle 9(17) (2010) 3602–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jiang X, Xu Y, Price BD, Acetylation of H2AX on lysine 36 plays a key role in the DNA double-strand break repair pathway, FEBS Lett 584(13) (2010) 2926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sone K, Piao L, Nakakido M, Ueda K, Jenuwein T, Nakamura Y, Hamamoto R, Critical role of lysine 134 methylation on histone H2AX for gamma-H2AX production and DNA repair, Nat Commun 5 (2014) 5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pan MR, Peng G, Hung WC, Lin SY, Monoubiquitination of H2AX protein regulates DNA damage response signaling, J Biol Chem 286(32) (2011) 28599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wu CY, Kang HY, Yang WL, Wu J, Jeong YS, Wang J, Chan CH, Lee SW, Zhang X, Lamothe B, Campos AD, Darnay BG, Lin HK, Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response, J Biol Chem 286(35) (2011) 30806–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen Q, Bian C, Wang X, Liu X, Ahmad Kassab M, Yu Y, Yu X, ADP-ribosylation of histone variant H2AX promotes base excision repair, EMBO J 40(2) (2021) e104542.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lorkovic ZJ, Park C, Goiser M, Jiang D, Kurzbauer MT, Schlogelhofer P, Berger F, Compartmentalization of DNA Damage Response between Heterochromatin and Euchromatin Is Mediated by Distinct H2A Histone Variants, Curr Biol 27(8) (2017) 1192–1199. [DOI] [PubMed] [Google Scholar]
- [68].Bourguet P, Picard CL, Yelagandula R, Pelissier T, Lorkovic ZJ, Feng S, Pouch-Pelissier MN, Schmucker A, Jacobsen SE, Berger F, Mathieu O, The histone variant H2A.W and linker histone H1 co-regulate heterochromatin accessibility and DNA methylation, Nat Commun 12(1) (2021) 2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Osakabe A, Jamge B, Axelsson E, Montgomery SA, Akimcheva S, Kuehn AL, Pisupati R, Lorkovic ZJ, Yelagandula R, Kakutani T, Berger F, The chromatin remodeler DDM1 prevents transposon mobility through deposition of histone variant H2A.W, Nat Cell Biol 23(4) (2021) 391–400. [DOI] [PubMed] [Google Scholar]
- [70].Foroozani M, Holder DH, Deal RB, Histone Variants in the Specialization of Plant Chromatin, Annu Rev Plant Biol (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lorkovic ZJ, Berger F, Heterochromatin and DNA damage repair: Use different histone variants and relax, Nucleus 8(6) (2017) 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G, High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome, EMBO J 29(8) (2010) 1446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Meier A, Fiegler H, Munoz P, Ellis P, Rigler D, Langford C, Blasco MA, Carter N, Jackson SP, Spreading of mammalian DNA-damage response factors studied by ChIP-chip at damaged telomeres, EMBO J 26(11) (2007) 2707–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Savic V, Yin B, Maas NL, Bredemeyer AL, Carpenter AC, Helmink BA, Yang-Iott KS, Sleckman BP, Bassing CH, Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin, Mol Cell 34(3) (2009) 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Natale F, Rapp A, Yu W, Maiser A, Harz H, Scholl A, Grulich S, Anton T, Horl D, Chen W, Durante M, Taucher-Scholz G, Leonhardt H, Cardoso MC, Identification of the elementary structural units of the DNA damage response, Nat Commun 8 (2017) 15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Collins PL, Purman C, Porter SI, Nganga V, Saini A, Hayer KE, Gurewitz GL, Sleckman BP, Bednarski JJ, Bassing CH, Oltz EM, DNA double-strand breaks induce H2Ax phosphorylation domains in a contact-dependent manner, Nat Commun 11(1) (2020) 3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ochs F, Karemore G, Miron E, Brown J, Sedlackova H, Rask MB, Lampe M, Buckle V, Schermelleh L, Lukas J, Lukas C, Stabilization of chromatin topology safeguards genome integrity, Nature 574(7779) (2019) 571–574. [DOI] [PubMed] [Google Scholar]
- [78].Arnould C, Rocher V, Finoux AL, Clouaire T, Li K, Zhou F, Caron P, Mangeot PE, Ricci EP, Mourad R, Haber JE, Noordermeer D, Legube G, Loop extrusion as a mechanism for formation of DNA damage repair foci, Nature 590(7847) (2021) 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Piquet S, Le Parc F, Bai SK, Chevallier O, Adam S, Polo SE, The Histone Chaperone FACT Coordinates H2A.X-Dependent Signaling and Repair of DNA Damage, Mol Cell 72(5) (2018) 888–901 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ, ATR and H2AX cooperate in maintaining genome stability under replication stress, J Biol Chem 284(9) (2009) 5994–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Chen BR, Quinet A, Byrum AK, Jackson J, Berti M, Thangavel S, Bredemeyer AL, Hindi I, Mosammaparast N, Tyler JK, Vindigni A, Sleckman BP, XLF and H2AX function in series to promote replication fork stability, J Cell Biol 218(7) (2019) 2113–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kim J, Sturgill D, Sebastian R, Khurana S, Tran AD, Edwards GB, Kruswick A, Burkett S, Hosogane EK, Hannon WW, Weyemi U, Bonner WM, Luger K, Oberdoerffer P, Replication Stress Shapes a Protective Chromatin Environment across Fragile Genomic Regions, Mol Cell 69(1) (2018) 36–47 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A, Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks, Nat Cell Biol 5(7) (2003) 675–9. [DOI] [PubMed] [Google Scholar]
- [84].Salguero I, Belotserkovskaya R, Coates J, Sczaniecka-Clift M, Demir M, Jhujh S, Wilson MD, Jackson SP, MDC1 PST-repeat region promotes histone H2AX-independent chromatin association and DNA damage tolerance, Nat Commun 10(1) (2019) 5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y, GammaH2AX and cancer, Nat Rev Cancer 8(12) (2008) 957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Palla VV, Karaolanis G, Katafigiotis I, Anastasiou I, Patapis P, Dimitroulis D, Perrea D, gamma-H2AX: Can it be established as a classical cancer prognostic factor?, Tumour Biol 39(3) (2017) 1010428317695931. [DOI] [PubMed] [Google Scholar]
- [87].Turinetto V, Giachino C, Multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions, Nucleic Acids Res 43(5) (2015) 2489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A, H2AX haploinsufficiency modifies genomic stability and tumor susceptibility, Cell 114(3) (2003) 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Weyemi U, Redon CE, Choudhuri R, Aziz T, Maeda D, Boufraqech M, Parekh PR, Sethi TK, Kasoji M, Abrams N, Merchant A, Rajapakse VN, Bonner WM, The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition, Nat Commun 7 (2016) 10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ji J, Zhang Y, Redon CE, Reinhold WC, Chen AP, Fogli LK, Holbeck SL, Parchment RE, Hollingshead M, Tomaszewski JE, Dudon Q, Pommier Y, Doroshow JH, Bonner WM, Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay, PLoS One 12(2) (2017) e0171582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Rahmanian N, Shokrzadeh M, Eskandani M, Recent advances in gammaH2AX biomarker-based genotoxicity assays: A marker of DNA damage and repair, DNA Repair (Amst) 108 (2021) 103243. [DOI] [PubMed] [Google Scholar]
- [92].Weyemi U, Paul BD, Bhattacharya D, Malla AP, Boufraqech M, Harraz MM, Bonner WM, Snyder SH, Histone H2AX promotes neuronal health by controlling mitochondrial homeostasis, Proc Natl Acad Sci U S A 116(15) (2019) 7471–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Weyemi U, Paul BD, Snowman AM, Jailwala P, Nussenzweig A, Bonner WM, Snyder SH, Histone H2AX deficiency causes neurobehavioral deficits and impaired redox homeostasis, Nat Commun 9(1) (2018) 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zha S, Sekiguchi J, Brush JW, Bassing CH, Alt FW, Complementary functions of ATM and H2AX in development and suppression of genomic instability, Proc Natl Acad Sci U S A 105(27) (2008) 9302–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Gruosso T, Mieulet V, Cardon M, Bourachot B, Kieffer Y, Devun F, Dubois T, Dutreix M, Vincent-Salomon A, Miller KM, Mechta-Grigoriou F, Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients, EMBO Mol Med 8(5) (2016) 527–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D, Abete P, Oxidative stress, aging, and diseases, Clin Interv Aging 13 (2018) 757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Bonisch C, Schneider K, Punzeler S, Wiedemann SM, Bielmeier C, Bocola M, Eberl HC, Kuegel W, Neumann J, Kremmer E, Leonhardt H, Mann M, Michaelis J, Schermelleh L, Hake SB, H2A.Z.2.2 is an alternatively spliced histone H2A.Z variant that causes severe nucleosome destabilization, Nucleic Acids Res 40(13) (2012) 5951–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Faast R, Thonglairoam V, Schulz TC, Beall J, Wells JR, Taylor H, Matthaei K, Rathjen PD, Tremethick DJ, Lyons I, Histone variant H2A.Z is required for early mammalian development, Curr Biol 11(15) (2001) 1183–7. [DOI] [PubMed] [Google Scholar]
- [99].Colino-Sanguino Y, Clark SJ, Valdes-Mora F, The H2A.Z-nuclesome code in mammals: emerging functions, Trends Genet (2021). [DOI] [PubMed] [Google Scholar]
- [100].Lewis TS, Sokolova V, Jung H, Ng H, Tan D, Structural basis of chromatin regulation by histone variant H2A.Z, Nucleic Acids Res 49(19) (2021) 11379–11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Suto RK, Clarkson MJ, Tremethick DJ, Luger K, Crystal structure of a nucleosome core particle containing the variant histone H2A.Z, Nat Struct Biol 7(12) (2000) 1121–4. [DOI] [PubMed] [Google Scholar]
- [102].Xu Y, Ayrapetov MK, Xu C, Gursoy-Yuzugullu O, Hu Y, Price BD, Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair, Mol Cell 48(5) (2012) 723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Rona G, Roberti D, Yin Y, Pagan JK, Homer H, Sassani E, Zeke A, Busino L, Rothenberg E, Pagano M, PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading, Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Caron P, van der Linden J, van Attikum H, Bon voyage: A transcriptional journey around DNA breaks, DNA Repair (Amst) 82 (2019) 102686. [DOI] [PubMed] [Google Scholar]
- [105].Puget N, Miller KM, Legube G, Non-canonical DNA/RNA structures during Transcription-Coupled Double-Strand Break Repair: Roadblocks or Bona fide repair intermediates?, DNA Repair (Amst) 81 (2019) 102661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Tan XY, Huen MSY, Perfecting DnA double-strand break repair on transcribed chromatin, Essays Biochem 64(5) (2020) 705–719. [DOI] [PubMed] [Google Scholar]
- [107].Alatwi HE, Downs JA, Removal of H2A.Z by INO80 promotes homologous recombination, EMBO Rep 16(8) (2015) 986–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Gursoy-Yuzugullu O, Ayrapetov MK, Price BD, Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair, Proc Natl Acad Sci U S A 112(24) (2015) 7507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Khurana S, Kruhlak MJ, Kim J, Tran AD, Liu J, Nyswaner K, Shi L, Jailwala P, Sung MH, Hakim O, Oberdoerffer P, A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance, Cell Rep 8(4) (2014) 1049–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Srivatsan A, Li BZ, Szakal B, Branzei D, Putnam CD, Kolodner RD, The Swr1 chromatin-remodeling complex prevents genome instability induced by replication fork progression defects, Nat Commun 9(1) (2018) 3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Cayrou C, Ballester B, Peiffer I, Fenouil R, Coulombe P, Andrau JC, van Helden J, Mechali M, The chromatin environment shapes DNA replication origin organization and defines origin classes, Genome Res 25(12) (2015) 1873–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Long H, Zhang L, Lv M, Wen Z, Zhang W, Chen X, Zhang P, Li T, Chang L, Jin C, Wu G, Wang X, Yang F, Pei J, Chen P, Margueron R, Deng H, Zhu M, Li G, H2A.Z facilitates licensing and activation of early replication origins, Nature 577(7791) (2020) 576–581. [DOI] [PubMed] [Google Scholar]
- [113].Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G, Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair, Cell 127(7) (2006) 1361–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Sun Z, Bernstein E, Histone variant macroH2A: from chromatin deposition to molecular function, Essays Biochem 63(1) (2019) 59–74. [DOI] [PubMed] [Google Scholar]
- [115].Kim J, Oberdoerffer P, Khurana S, The histone variant macroH2A1 is a splicing-modulated caretaker of genome integrity and tumor growth, Mol Cell Oncol 5(3) (2018) e1441629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kozlowski M, Corujo D, Hothorn M, Guberovic I, Mandemaker IK, Blessing C, Sporn J, Gutierrez-Triana A, Smith R, Portmann T, Treier M, Scheffzek K, Huet S, Timinszky G, Buschbeck M, Ladurner AG, MacroH2A histone variants limit chromatin plasticity through two distinct mechanisms, EMBO Rep 19(10) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chen H, Ruiz PD, Novikov L, Casill AD, Park JW, Gamble MJ, MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation, Nat Struct Mol Biol 21(11) (2014) 981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Ruiz PD, Gamble MJ, MacroH2A1 chromatin specification requires its docking domain and acetylation of H2B lysine 20, Nat Commun 9(1) (2018) 5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Douet J, Corujo D, Malinverni R, Renauld J, Sansoni V, Posavec Marjanovic M, Cantarino N, Valero V, Mongelard F, Bouvet P, Imhof A, Thiry M, Buschbeck M, MacroH2A histone variants maintain nuclear organization and heterochromatin architecture, J Cell Sci 130(9) (2017) 1570–1582. [DOI] [PubMed] [Google Scholar]
- [120].Martins F, Sousa J, Pereira CD, da Cruz ESOAB, Rebelo S, Nuclear envelope dysfunction and its contribution to the aging process, Aging Cell 19(5) (2020) e13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Pfeifer CR, Vashisth M, Xia Y, Discher DE, Nuclear failure DNA damage, and cell cycle disruption after migration through small pores: a brief review, Essays Biochem 63(5) (2019) 569–577. [DOI] [PubMed] [Google Scholar]
- [122].Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EH, Scheffzek K, Hottiger MO, Ladurner AG, A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation, Nat Struct Mol Biol 16(9) (2009) 923–9. [DOI] [PubMed] [Google Scholar]
- [123].Xu C, Xu Y, Gursoy-Yuzugullu O, Price BD, The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1, FEBS Lett 586(21) (2012) 3920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Sebastian R, Hosogane EK, Sun EG, Tran AD, Reinhold WC, Burkett S, Sturgill DM, Gudla PR, Pommier Y, Aladjem MI, Oberdoerffer P, Epigenetic Regulation of DNA Repair Pathway Choice by MacroH2A1 Splice Variants Ensures Genome Stability, Mol Cell 79(5) (2020) 836–845 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG, Splicing regulates NAD metabolite binding to histone macroH2A, Nat Struct Mol Biol 12(7) (2005) 624–5. [DOI] [PubMed] [Google Scholar]
- [126].Ruiz PD, Hamilton GA, Park JW, Gamble MJ, MacroH2A1 Regulation of Poly(ADP-Ribose) Synthesis and Stability Prevents Necrosis and Promotes DnA Repair, Mol Cell Biol 40(1) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Wang H, Xu X, Microhomology-mediated end joining: new players join the team, Cell Biosci 7 (2017) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA, Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination, Nat Struct Mol Biol 20(3) (2013) 317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, lacovoni JS, Daburon V, Miller KM, Jackson SP, Legube G, Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks, Nat Struct Mol Biol 21(4) (2014) 366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Gong F, Chiu LY, Cox B, Aymard F, Clouaire T, Leung JW, Cammarata M, Perez M, Agarwal P, Brodbelt JS, Legube G, Miller KM, Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination, Genes Dev 29(2) (2015) 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller KM, Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair, J Cell Biol 216(7) (2017) 1959–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Savitsky P, Krojer T, Fujisawa T, Lambert JP, Picaud S, Wang CY, Shanle EK, Krajewski K, Friedrichsen H, Kanapin A, Goding C, Schapira M, Samsonova A, Strahl BD, Gingras AC, Filippakopoulos P, Multivalent Histone and DNA Engagement by a PHD/BRd/PwWP Triple Reader Cassette Recruits ZMYND8 to K14ac-Rich Chromatin, Cell Rep 17(10) (2016) 2724–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Spruijt CG, Luijsterburg MS, Menafra R, Lindeboom RG, Jansen PW, Edupuganti RR, Baltissen MP, Wiegant WW, Voelker-Albert MC, Matarese F, Mensinga A, Poser I, Vos HR, Stunnenberg HG, van Attikum H, Vermeulen M, ZMYND8 Co-localizes with NuRD on Target Genes and Regulates Poly(ADP-Ribose)-Dependent Recruitment of GATAD2A/NuRD to Sites of DNA Damage, Cell Rep 17(3) (2016) 783–798. [DOI] [PubMed] [Google Scholar]
- [134].Kumbhar R, Sanchez A, Perren J, Gong F, Corujo D, Medina F, Devanathan SK, Xhemalce B, Matouschek A, Buschbeck M, Buck-Koehntop BA, Miller KM, Poly(ADP-ribose) binding and macroH2A mediate recruitment and functions of KDM5A at DNA lesions, J Cell Biol 220(7) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Burgess RC, Burman B, Kruhlak MJ, Misteli T, Activation of DnA damage response signaling by condensed chromatin, Cell Rep 9(5) (2014) 1703–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Fijen C, Rothenberg E, The evolving complexity of DNA damage foci: RNA, condensates and chromatin in DNA double-strand break repair, DNA Repair (Amst) 105 (2021) 103170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Spegg V, Altmeyer M, Biomolecular condensates at sites of DNA damage: More than just a phase, DNA Repair (Amst) 106 (2021) 103179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lezaja A, Altmeyer M, Inherited DNA lesions determine G1 duration in the next cell cycle, Cell Cycle 17(1) (2018) 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Posavec Marjanovic M, Hurtado-Bages S, Lassi M, Valero V, Malinverni R, Delage H, Navarro M, Corujo D, Guberovic I, Douet J, Gama-Perez P, Garcia-Roves PM, Ahel I, Ladurner AG, Yanes O, Bouvet P, Suelves M, Teperino R, Pospisilik JA, Buschbeck M, MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD(+) consumption, Nat Struct Mol Biol 24(11) (2017) 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Glover TW, Wilson TE, Arlt MF, Fragile sites in cancer: more than meets the eye, Nat Rev Cancer 17(8) (2017) 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Berti M, Cortez D, Lopes M, The plasticity of DNA replication forks in response to clinically relevant genotoxic stress, Nat Rev Mol Cell Biol 21(10) (2020) 633–651. [DOI] [PubMed] [Google Scholar]
- [142].Xu X, Ni K, He Y, Ren J, Sun C, Liu Y, Aladjem MI, Burkett S, Finney R, Ding X, Sharan SK, Muegge K, The epigenetic regulator LSH maintains fork protection and genomic stability via MacroH2A deposition and RAD51 filament formation, Nat Commun 12(1) (2021) 3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Thijssen PE, Ito Y, Grillo G, Wang J, Velasco G, Nitta H, Unoki M, Yoshihara M, Suyama M, Sun Y, Lemmers RJ, de Greef JC, Gennery A, Picco P, Kloeckener-Gruissem B, Gungor T, Reisli I, Picard C, Kebaili K, Roquelaure B, Iwai T, Kondo I, Kubota T, van Ostaijen-Ten Dam MM, van Tol MJ, Weemaes C, Francastel C, van der Maarel SM, Sasaki H, Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome, Nat Commun 6 (2015) 7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ni K, Ren J, Xu X, He Y, Finney R, Braun SMG, Hathaway NA, Crabtree GR, Muegge K, LSH mediates gene repression through macroH2A deposition, Nat Commun 11(1) (2020) 5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].L.H. Recoules A; Flavien Raynal F; Karasu N; Moutahir F; Bejjani F; Jariel-Encontre I; Cuvier O; Sexton T; Lavigne A; Bystricky K, The histone variant macroH2A1.1 regulates RNA Polymerase II paused genes within defined chromatin interaction landscapes, bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Ni K, Muegge K, LSH catalyzes ATP-driven exchange of histone variants macroH2A1 and macroH2A2, Nucleic Acids Res 49(14) (2021) 8024–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Bakhoum SF, Kabeche L, Compton DA, Powell SN, Bastians H, Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities, Trends Cancer 3(3) (2017) 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Pehrson JR, Changolkar LN, Costanzi C, Leu NA, Mice without macroH2A histone variants, Mol Cell Biol 34(24) (2014) 4523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, Yusufzai T, D’Andrea AD, Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair, Nature 518(7538) (2015) 258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A, Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination, Nature 518(7538) (2015) 254–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Schrempf A, Slyskova J, Loizou JI, Targeting the DNA Repair Enzyme Polymerase theta in Cancer Therapy, Trends Cancer 7(2) (2021) 98–111. [DOI] [PubMed] [Google Scholar]
- [152].Hall AC, Ostrowski LA, Pietrobon V, Mekhail K, Repetitive DNA loci and their modulation by the non-canonical nucleic acid structures R-loops and G-quadruplexes, Nucleus 8(2) (2017) 162–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Cong R, Das S, Douet J, Wong J, Buschbeck M, Mongelard F, Bouvet P, macroH2A1 histone variant represses rDNA transcription, Nucleic Acids Res 42(1) (2014) 181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Tsang E, Carr AM, Replication fork arrest, recombination and the maintenance of ribosomal DNA stability, DNA Repair (Amst) 7(10) (2008) 1613–23. [DOI] [PubMed] [Google Scholar]
- [155].Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, Stanlie A, Wu W, Wu X, Day A, Wong N, Yin M, Canela A, Fu H, Redon C, Pruitt SC, Jaszczyszyn Y, Aladjem MI, Aplan PD, Hyrien O, Nussenzweig A, Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse, Cell 174(5) (2018) 1127–1142 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Khurana S, Markowitz TE, Kabat J, McBride AA, Spatial and Functional Organization of Human Papillomavirus Replication Foci in the Productive Stage of Infection, mBio (2021) e0268421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Kim J, Sun C, Tran AD, Chin PJ, Ruiz PD, Wang K, Gibbons RJ, Gamble MJ, Liu Y, Oberdoerffer P, The macroH2A1.2 histone variant links ATRX loss to alternative telomere lengthening, Nat Struct Mol Biol 26(3) (2019) 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Silva B, Arora R, Bione S, Azzalin CM, TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells, Nat Commun 12(1) (2021) 3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Vohhodina J, Goehring LJ, Liu B, Kong Q, Botchkarev VV Jr., Huynh M, Liu Z, Abderazzaq FO, Clark AP, Ficarro SB, Marto JA, Hatchi E, Livingston DM, BRCA1 binds TERRA RNA and suppresses R-Loop-based telomeric DNA damage, Nat Commun 12(1) (2021) 3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Zong D, Oberdoerffer P, Batista PJ, Nussenzweig A, RNA: a double-edged sword in genome maintenance, Nat Rev Genet 21(11) (2020) 651–670. [DOI] [PubMed] [Google Scholar]
- [161].Groh M, Gromak N, Out of balance: R-loops in human disease, PLoS Genet 10(9) (2014) e1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Wells JP, White J, Stirling PC, R Loops and Their Composite Cancer Connections, Trends Cancer 5(10) (2019) 619–631. [DOI] [PubMed] [Google Scholar]
- [163].Kelliher JL, West KL, Gong Q, Leung JWC, Histone H2A variants alpha1-extension helix directs RNF168-mediated ubiquitination, Nat Commun 11(1) (2020) 2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Kim BJ, Chan DW, Jung SY, Chen Y, Qin J, Wang Y, The Histone Variant MacroH2A1 Is a BRCA1 Ubiquitin Ligase Substrate, Cell Rep 19(9) (2017) 1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].O’Connor HF, Lyon N, Leung JW, Agarwal P, Swaim CD, Miller KM, Huibregtse JM, Ubiquitin-Activated Interaction Traps (UBAITs) identify E3 ligase binding partners, EMBO Rep 16(12) (2015) 1699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Wilson MD, Benlekbir S, Fradet-Turcotte A, Sherker A, Julien JP, McEwan A, Noordermeer SM, Sicheri F, Rubinstein JL, Durocher D, The structural basis of modified nucleosome recognition by 53BP1, Nature 536(7614) (2016) 100–3. [DOI] [PubMed] [Google Scholar]
- [167].Bernstein E, Muratore-Schroeder TL, Diaz RL, Chow JC, Changolkar LN, Shabanowitz J, Heard E, Pehrson JR, Hunt DF, Allis CD, A phosphorylated subpopulation of the histone variant macroH2A1 is excluded from the inactive X chromosome and enriched during mitosis, Proc Natl Acad Sci U S A 105(5) (2008) 1533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Giallongo S, Lo Re O, Lochmanova G, Parca L, Petrizzelli F, Zdrahal Z, Mazza T, Vinciguerra M, Phosphorylation within Intrinsic Disordered Region Discriminates Histone Variant macroH2A1 Splicing Isoforms-macroH2A1.1 and macroH2A1.2, Biology (Basel) 10(7) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Adèle Mangelinck CC, Courbeyrette Régis, Ourarhni Khalid, Hamiche Ali, Redon Christophe, Bonner William M., van Dijk Erwin, Derbois Céline, Olaso Robert, Deleuze Jean-François, Fenaille François, Rübe Claudia E., Thuret Jean-Yves, Mann Carl, The H2A.J histone variant contributes to Interferon-Stimulated Gene expression in senescence by its weak interaction with H1 and the derepression of repeated DNA sequences, bioRxiv (2020). [Google Scholar]
- [170].Contrepois K, Coudereau C, Benayoun BA, Schuler N, Roux PF, Bischof O, Courbeyrette R, Carvalho C, Thuret JY, Ma Z, Derbois C, Nevers MC, Volland H, Redon CE, Bonner WM, Deleuze JF, Wiel C, Bernard D, Snyder MP, Rube CE, Olaso R, Fenaille F, Mann C, Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression, Nat Commun 8 (2017) 14995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ, MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype, Mol Cell 59(5) (2015) 719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Isermann A, Mann C, Rube CE, Histone Variant H2A.J Marks Persistent DNA Damage and Triggers the Secretory Phenotype in Radiation-Induced Senescence, Int J Mol Sci 21(23) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].C.E.S. Redon Z; Tewary G; Mangelinck A; Courbeyrette R; Thuret J-Y; Aladjem MI; Bonner WM; Rübe CE; Mann C , Histone Variant H2A.J Is Enriched in Luminal Epithelial Gland Cells, Genes 12(11) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Chadwick BP, Willard HF, A novel chromatin protein, distantly related to histone H2A, is largely excluded from the inactive X chromosome, J Cell Biol 152(2) (2001) 375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Tolstorukov MY, Goldman JA, Gilbert C, Ogryzko V, Kingston RE, Park PJ, Histone variant H2A.Bbd is associated with active transcription and mRNA processing in human cells, Mol Cell 47(4) (2012) 596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Sansoni V, Casas-Delucchi CS, Rajan M, Schmidt A, Bonisch C, Thomae AW, Staege MS, Hake SB, Cardoso MC, Imhof A, The histone variant H2A.Bbd is enriched at sites of DNA synthesis, Nucleic Acids Res 42(10) (2014) 6405–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Lindeboom RGH, Regev A, Teichmann SA, Towards a Human Cell Atlas: Taking Notes from the Past, Trends Genet 37(7) (2021) 625–630. [DOI] [PubMed] [Google Scholar]
- [178].Rozenblatt-Rosen O, Regev A, Oberdoerffer P, Nawy T, Hupalowska A, Rood JE, Ashenberg O, Cerami E, Coffey RJ, Demir E, Ding L, Esplin ED, Ford JM, Goecks J, Ghosh S, Gray JW, Guinney J, Hanlon SE, Hughes SK, Hwang ES, Iacobuzio-Donahue CA, Jane-Valbuena J, Johnson BE, Lau KS, Lively T, Mazzilli SA, Pe’er D, Santagata S, Shalek AK, Schapiro D, Snyder MP, Sorger PK, Spira AE, Srivastava S, Tan K, West RB, Williams EH, Human Tumor Atlas N, The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution, Cell 181(2) (2020) 236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Yang M, Petralia F, Li Z, Li H, Ma W, Song X, Kim S, Lee H, Yu H, Lee B, Bae S, Heo E, Kaczmarczyk J, Stepniak P, Warchol M, Yu T, Calinawan AP, Boutros PC, Payne SH, Reva B, Consortium N-C-D, Boja E, Rodriguez H, Stolovitzky G, Guan Y, Kang J, Wang P, Fenyo D, Saez-Rodriguez J, Community Assessment of the Predictability of Cancer Protein and Phosphoprotein Levels from Genomics and Transcriptomics, Cell Syst 11(2) (2020) 186–195 e9. [DOI] [PubMed] [Google Scholar]