

Abstract
Autophagy is an evolutionarily conserved cellular process in which macromolecules undergo lysosomal degradation. It fulfills essential roles in quality controlling cellular constituents and in energy homeostasis. Basal autophagy is also widely accepted to provide a protective role in aging and aging‐related disorders, and its decline with age might precipitate the onset of a variety of diseases. In this review, we discuss the role of basal autophagy in maintaining homeostasis, in part through the maintenance of stem cell populations and the prevention of cellular senescence. We also consider how stress‐induced senescence, for example, during oncogene activation and in premalignant disease, might rely on autophagy, and the possibility that the age‐associated decline in autophagy might promote tumour development through a variety of mechanisms. Ultimately, evidence suggests that autophagy is required for malignant cancer progression in a number of settings. Thus, autophagy appears to be tumour‐suppressive during the early stages of tumorigenesis and tumour‐promoting at later stages.
Keywords: aging, autophagy, cancer, senescence
In this review, we discuss the role of basal autophagy in homeostasis through the maintenance of stem cell populations and the prevention of cellular senescence. We discuss how senescence, for example, during oncogene activation and in premalignant disease, may rely on autophagy, and how the age‐associated decline in autophagy could promote tumour development.
Abbreviations
- AMPK
AMP‐activated protein kinase
- APOE
apolipoprotein E
- ATG5
autophagy‐related gene 5
- ATG7
autophagy‐related gene 7
- ATG8
autophagy‐related gene 8
- BCL2
B‐cell lymphoma 2
- BECN1
Beclin‐1
- BUBR1
BUB1 mitotic checkpoint serine/threonine kinase B
- CCFs
cytoplasmic chromatin fragments
- CDK
cyclin dependent kinase
- DNMT1
DNA methyltransferase 1
- eIF5A
eukaryotic translation initiation factor 5A‐1
- FOXO
forkhead box protein O
- GATA4
GATA binding protein 4
- HSC
hematopoietic stem cell
- HSC70
heat shock cognate 71 kDa protein
- IL6
interleukin 6
- IL8
interleukin 8
- LAMP2A
lysosome‐associated membrane protein 2, variant A
- LC3
microtubule‐associated proteins 1A/1B light chain 3B
- mTORC1
mammalian target of rapamycin complex 1
- p62/SQSTM1
sequestosome‐1
- PAX7
paired box protein Pax‐7
- SAHF
senescence associated heterochromatin fragments
- SASP
senescence associated secretory phenotype
- SA‐β‐gal
senescence associated Beta‐galactosidase
- SIRT1
sirtuin 1
- TFEB
transcription factor EB
1. Introduction
Autophagy is a highly conserved process in which cellular components – from lipids and proteins, to large aggregates and organelles – are delivered to lysosomes for degradation. Autophagy was initially considered to be a broad non‐selective process, partly because autophagic activity can be dramatically upregulated during times of nutrient and of other cellular stresses, when intracellular components are mobilized and broken down to maintain energy homeostasis. It is now clear that this nutrient stress‐induced engagement concealed the remarkable complexity that exists with regard to cargo selection [1].
In addition to this bulk versus selective paradigm shift, we have also come to appreciate that autophagy is not simply one process in mammals, but three distinct processes (Fig. 1): macroautophagy, chaperone mediated autophagy, microautophagy. During macroautophagy, cellular components are sequestered and isolated into a double‐membrane‐bound vesicle (the autophagosome) before fusing with a lysosome, forming an autolysosome, leading to the breakdown of the vesicle contents. Chaperone‐mediated autophagy is a process in which proteins with a KFERQ‐like targeting motif can be recognized by the chaperone HSC70 before subsequent binding to the lysosomal membrane protein LAMP2A. This binding and ensuing formation of a translocation complex enable the transport of proteins across the lysosomal membrane where they are subsequently degraded [2]. Finally, during microautophagy cellular material is directly transported to the lysosome without the need for autophagosome biogenesis. Instead, late‐endosomes, or lysosomes, directly engulf cellular components before they are eventually broken down [3].
Fig. 1.
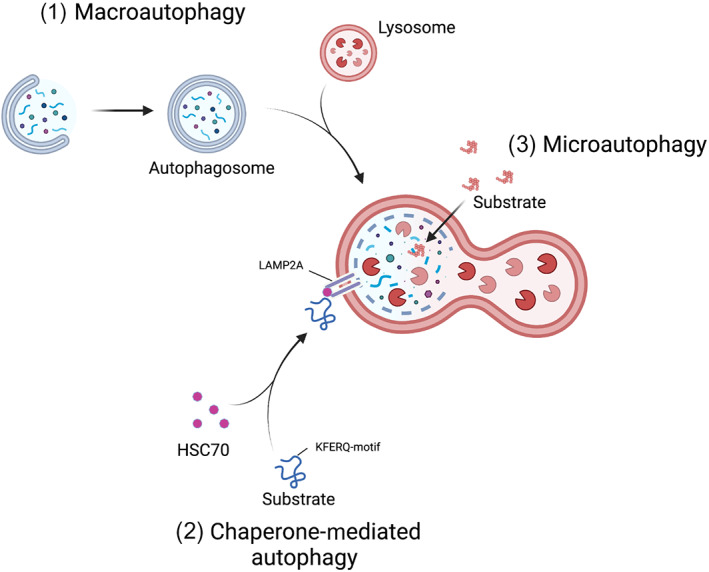
Three subtypes of autophagy. (1) During macroautophagy, cellular components are sequestered and isolated into a double‐membrane‐bound vesicle before fusing with a lysosome, forming an autolysosome, and leading to the breakdown of the vesicle contents. (2) During chaperone‐mediated autophagy proteins with a KFERQ‐like targeting motif are bound by the chaperone HSC70. The substrate‐HSC70 complex binds to the lysosomal membrane protein LAMP2A resulting in the translocation of proteins across the lysosomal membrane where they are subsequently degraded. (3) During microautophagy, cellular material is directly transported to late endosomes or directly to the lysosome for degradation through invagination of the vesicle membranes.
As the bulk of research has centred on macroautophagy, the term macroautophagy and autophagy are used interchangeably. However, significant evidence is accumulating that chaperone‐mediated autophagy has equally important implications in health and disease with regards to aging and cancer, and one must assume that the same is true of microautophagy. Here we focus primarily on macroautophagy, referring to it as simply autophagy due to its greater research focus in the community, however, our knowledge of the interplay of the three autophagic processes, and how they act in concert is poorly understood. If we are to understand the complex role autophagy has in mammalian cancer and aging, we are surely going to need to better balance our perspective on this.
In this review, we take a critical look at the data that underpins our understanding of when autophagy is modulated during aging and in the context of senescence and cancer. There is a complex interplay between these processes, autophagy is altered in aging, senescence, and cancer, equally its modulation can affect all three. In particular, we discuss the evidence that autophagy modulation through dietary, pharmalological, or genetic means from mammalian models may affect health and life span with a particular focus on stem cell functionality and cellular senescence. Simultaneously, we also place this information into the context of tumorigenesis where autophagy reactivation commonly occurs and is believed to promote tumor cell fitness. Finally, we argue that despite the clear links that exist between autophagy in aging, senescence, and in tumor development, our understanding of how altered autophagic flux influences these processes remains unclear. To ascertain this, we need more granular fundamental information about how and when autophagic flux changes, as well as more nuanced models that recapitulate these alterations, as opposed to blanket statements and an over‐reliance on complete knockout models.
2. Autophagy and aging
A central paradigm of both aging and autophagy research is that, with increasing age, there is a corresponding decrease in the rate of autophagic activity, at least in some tissues [4]. As autophagy is at the nexus of protein homeostasis, organelle turnover, and cellular metabolism, it is unsurprising that a swathe of evidence exists across species, both genetic and pharmacological, highlighting how the loss of autophagy is enough to drive cellular, tissue, and organismal dysfunction with detrimental effects on health and lifespan; by contrast boosting, autophagy has the opposite effects [5, 6, 7].
2.1. Lessons from model organisms
Historically some of the earliest data is leveraged from studies in lower organisms where the loss of autophagy has been shown to decrease life span and health span [8, 9], whilst overexpression of the autophagy machinery has been shown to promote longevity in some cases [10, 11]. Additionally, genetic perturbations that result in life‐span extension, also known as long‐lived mutants, have been found to be dependent on an intact autophagy system [12, 13, 14]. As loss of autophagy through genetic models or RNAi has been shown to reverse these longevity phenotypes in both Caenorhabditis elegans and Drosophila melanogaster, it suggests that autophagy may act as a central process in longevity across species [15, 16].
Traditionally, it has been a challenge to study the effects of systemic autophagy loss on longevity in mammalian models, because of the neonatal lethality that occurs in constitutive knockout models of key autophagy genes, and the rapid, severe, neurological phenotypes encountered upon whole‐body ablation of autophagy in the adult mouse [17, 18]. Consequently, much of the available evidence has been limited to tissue‐specific knockouts of essential autophagy genes. However, these are also complicated as the resultant phenotypes are often an amalgamation of both developmental and homeostasis effects. Recently, two models wherein autophagic activity is constitutively higher throughout life leading to health‐ and life‐span extension [19, 20], and two models wherein autophagy can be dynamically inhibited and restored [21], have provided fresh evidence that in the mammalian setting, autophagy acts causally at the interface with longevity.
Despite recent advances in our understanding of autophagy, our knowledge of how it is perturbed during aging and in disease states, particularly in cancer development, remains rudimentary. In C. elegans expression of a tandem reporter (Box 1) enabling autophagic activity to be determined not only confirmed that autophagic flux decreased upon entering old age, but uncovered evidence of tissue‐specific and differential age‐associated trajectories of autophagy activity, suggesting hitherto underappreciated spatial and temporal kinetics [22]. Further reinforcing the notion that we are still yet to truly understand how autophagy perturbations impact upon health and disease across the life course, in post‐reproductive C. elegans, life‐span can be extended through the inhibition of genes involved in the early stages of autophagy [23]. This seemingly paradoxical result is due to the alleviation of age‐associated dysfunctional autophagy which would otherwise result in neurodegeneration and shorten organismal life span [23].
Box 1. Glossary.
Atg8/LC3 tandem reporters
Tandem LC3 reporters exploit the change in pH that accompanies lysosomal fusion with autophagosomes, and the different pH stability of fluorophores, e.g., GFP and mRFP. LC3 puncta mark autophagosome formation, in this case being both GFP and mRFP positive. However, during fusion with lysosomes, the GFP signal is quenched and only the acid‐resistant mRFP can be visualized. As such, the ratio of GFP/RFP positive, and RFP positive alone enables a more quantitative analysis of autophagic flux.
In mammalian models, tandem Atg8/LC3 reporter mice [18, 24, 25, 26, 27] have also been developed and helped to deconvolute the hierarchy of autophagic regulation by AMPK‐mTORC1 signaling the kidney [24], as well as improving our understanding the role of autophagy and its key modulators during reperfusion injury in the kidney and heart [25, 26, 27]. It appears that selective forms of autophagy also display tissue and age‐associated complexity in mammalian systems. This at least appears the case for mitophagy wherein single or tandem tagged mitochondria have been used to uncover developmentally regulated mitophagy processes and in cell‐type‐specific activities [28]. A reduction in mitophagy can also be found in particular regions of the brain with aging as well as in Parkinson's disease, Huntington's disease, and as a result of a high‐fat diet [29, 30]. However, whilst these data are insightful, there is limited data on pan‐tissue analysis in wild‐type, or genetically perturbed mammalian models, during natural aging, with age‐related disease states, or during different stages of cancer development.
It has been noted that a number of autophagy and lysosomal‐associated genes and proteins decrease with age [10, 31, 32, 33, 34], and that the increased expression of autophagy/lysosomal master regulator transcription factors, such as TFEB and FOXO, can promote health‐ and life‐span in an autophagy‐dependent manner [35, 36, 37]. Interestingly, Zhang et al. [38] reported that the TFEB protein expression is maintained by the unique hypusinated translation factor eIF5A and that this post‐translational modification of eIF5A is mediated by an endogenous polyamine spermidine. Antiaging and autophagy‐inducing effects of spermidine (or other natural polyamines) and age‐associated decline of the natural polyamines have been noted: the antiaging effect of spermidine appears to be autophagy‐dependent [39]. Furthermore, the reported negative regulator of autophagy, Rubicon, is increased in aged tissue across species and its reduction is associated with life‐span extension in C. elegans and D. melanogaster, as well as the amelioration of aging phenotypes in mice [40]. Although the entire picture is still vague, these studies provide some mechanistic insights into the age‐associated autophagy decline.
2.2. Autophagy and stem cells
Several reports have, nonetheless, causally linked a reduction in autophagy with aging through a reduction in stem cell capacity. Skeletal muscle stem cells (also known as satellite cells) from geriatric humans and mice display heightened levels of the autophagy adaptor p62/Sqstm1, and reduced autophagic flux in comparison to satellite cells isolated from young counterparts, correlating with a decrease in function and the induction of senescence [41]. This loss of functionality is purportedly linked in part to organelle quality control (specifically mitochondria) and genetic or pharmacological promotion of autophagy in geriatric satellite cells appears to restore functionality to some degree [41]. Furthermore, the loss of autophagy from genetically engineered mouse models leads to the disruption of both muscle fiber structure, function, and satellite cell number supporting a causal role of autophagy in maintaining tissue and cellular functionality [21, 41, 42].
Additionally, autophagy seems to have a role in hematopoietic stem cell (HSC) fate determination through the control of metabolic and epigenetic modifications [43, 44], again, in part linked to mitochondrial turnover. HSCs isolated from aged mice are reported to form distinct populations based upon autophagy activity, with approximately a third maintaining high autophagy levels and a similar regenerative capacity to HSCs isolated from young mice. In contrast, the remaining autophagy low, aged HSCs displayed a reduced regenerative capacity and a pro‐myeloid skewing, phenotypes characterizing aged blood. In support of these findings the loss of autophagy in genetically engineered mouse models is associated with the premature acquisition of aged blood phenotypes, including a myeloid skewing due to altered epigenetic priming of the HSCs [44]. Similarly, the loss of chaperone‐mediated autophagy with age is associated with a shift in fatty acid metabolism in HSCs and a loss of function, which can be reversed with pharmacological activators [45]. Such data suggest that the decline of autophagy with age may limit the capacity of stem cell populations to regenerate tissue damage, which over time will negatively impact both tissue structure and function.
Several of these studies have also provided evidence that boosting autophagy through pharmacological or genetic means can restore the functionality of these stem cell populations. This is in keeping with the myriad of papers that report positive health‐ and life‐span benefits through the pharmacological promotion of autophagy in mouse models, and suggests that autophagy may be a bona fide therapeutic target to extend organismal health span [5, 46]. This idea is further supported by genetic evidence from mouse models where autophagy has been constitutively promoted throughout life. Transgenic mice overexpressing Atg5 live longer, in part attributed to an enhanced metabolic state, including an improved insulin sensitivity and reduced adipose accumulation with age [19]. Life span is also extended in Becn1 F121A/F121A knockin mice, in which a substitution mutation in the BH3 domain of Beclin1 increases autophagic flux by disrupting its interaction with the negative regulator Bcl2. This knockin allele also rescues premature aging in the Klotho hypomorphic mouse model [20]. However, it remains to be seen in these settings how tissue and stem cell populations change with increasing age, and whether these benefits are due to global autophagy promotion or the benefit can be attributed to alterations in tissue or cell‐type specific populations. Additionally, whilst boosting autophagic activity from birth appears to promote health and life span, this is almost certainly through prevention of macromolecular damage forming.
In support of the central role of autophagy in aging, whole‐body inhibition of autophagy (except for the brain) through the expression of a doxycycline‐inducible shRNA targeting Atg5 (Atg5i mice), also drives the development of macroscopic premature aging phenotypes such as graying, kyphosis, loss of muscle mass, and a shortening of life span in two closely related models [21]. These models differ in only their targeting sequence and the degree to which they inhibit autophagy; Atg5i mice provide a robust knockdown phenocopying a knockout in many tissues, meanwhile, Atg5i‐2 mice are hypomorphic, and despite a build‐up of p62/Sqstm1 lack the gross phenotypes associated with Atg5i and Atg5 knockout mice. Upon loss of autophagy markers of aging such as inflammatory cytokines, telomere‐associated DNA damage, and markers of senescence were also found elevated across various tissues [21]. When autophagy was restored, through doxycycline withdrawal and the re‐expression of Atg5, this led to a striking reacquisition of organismal health and life span. However, it should be noted that this rejuvenation is incomplete as evidence of molecular damage and markers of senescence remain elevated, whilst life span was still reduced in comparison to control mice (in which shAtg5 was not expressed). This suggests that some of the damage induced by loss of Atg5, and its resultant effects such as senescence induction, may be irreversible and might continue to contribute to organismal age‐related decline.
3. Autophagy and senescence
Cellular senescence is a distinct cellular state activated in response to stress (such as DNA damage, telomere attrition, mitochondrial dysfunction, and high oncogenic signaling), as well as present in normal physiological settings, such as during embryogenesis and wound healing [47, 48]. Phenotypically, it is characterized by a stable inhibition of proliferation, often accompanied by a high lysosomal mass (as shown by staining for senescence‐associated beta‐galactosidase activity, SA‐β‐gal), and the acquisition of a distinct secretory phenotype. This later feature, known as the senescence‐associated secretory phenotype (the SASP), is believed to underpin the functional role of senescence in diverse biology from biological patterning, wound healing, and immunosurveillance, to tumor promotion through the expression and release of a wide array of cytokines and signaling molecules that act both locally and systemically [49, 50, 51]. Due to its stable cell cycle arrest, senescence is considered to be a tumor‐suppressive program, and markers of senescence are often found upregulated in premalignant lesions [52]. Conversely, senescent cells through the SASP may also have deleterious effects through shaping the tumorigenic microenvironment and supporting tumor progression [53]. To understand the functional relevance of senescence in vivo, it is important to collectively consider these two aspects of senescence: stable exit from the cell cycle (cell‐autonomous), and the capability to modulate surrounding tissues (non‐cell‐autonomous).
3.1. Basal autophagy
While senescence, particularly that caused by telomere shortening, has been implicated in organismal aging, the causative relationship between these processes was not clear until relatively recently [54, 55]. A series of mouse studies in 2006 showed an age‐associated accumulation of cells expressing p16 (a cyclin‐dependent kinase inhibitor functionally linked to senescence) in HSCs, neural progenitors, and pancreatic β‐cells. Importantly, the decline of self‐renewal potential of these cells was in part dependent on p16 expression [56, 57, 58]. These studies collectively reinforced the idea that senescence in tissue stem cells or progenitor cells limits a tissue's regenerative capacity, thus enhancing age‐associated tissue degeneration [59]. This is reminiscent of the key role of autophagy in the maintenance of stem cell regenerative potential in HSCs and muscle satellite cells, as described above [43, 44, 60]. While senescence and autophagy have not been experimentally linked in HSCs in the context of aging, their functional association has been reported in muscle stem cells. In geriatric mice, the regenerative capacity of satellite cells declines partly through the development of senescence in these cells [60]. In a subsequent study, the same group showed that Atg7 depletion in Pax7‐positive satellite cells induced senescence, and that geriatric senescence could be prevented in these cells by treating them with rapamycin or spermidine [41]. These results further indicate that an intimate relationship exists between organismal age, cumulative and degenerative alterations in organs, and the decline of cellular self‐renewal capacity, where basal autophagy actively serves to maintain cellular fitness, thus preventing senescence [53, 61].
In addition to the cell‐autonomous aspect of senescence, emerging evidence suggests its non‐cell‐autonomous activities contribute to aging. In an elegantly devised mouse model, in which p16‐expressing (and thus likely senescent) cells were induced to apoptose, the enforced elimination of these cells attenuated age‐associated deterioration in several organs in both progeroid mice (with a BubR1 hypomorphic genetic background) and naturally aging mice, even in later life [62, 63]. While the exact mechanism of how senescent cells locally (in diverse albeit not all tissues) or even systemically promote the aging process remains to be elucidated, it has been attributed to the accumulating and persistent non‐cell‐autonomous activities of senescent cells, represented by the SASP [54, 55, 64]. How autophagy is involved in this scenario appears to be highly complex and is less well explored. However, as described below, abundant evidence indicates that senescence (both its cell‐autonomous and non‐cell‐autonomous aspects) and autophagy are stress‐induced processes.
3.2. Stress‐induced autophagy
Considering the role of basal autophagy in metabolic and energy homeostasis and in cytoprotection, it is not surprising that a reduction of basal autophagy induces some types of senescence, which itself is a highly heterogenous state [48, 53]. In addition to the in vivo studies described above (for example, in the Atg5i model and in satellite cells), autophagy inhibition has also been shown to promote senescence in cell culture models [65, 66]. But it remains unclear whether or not the age‐associated decline of autophagy activity is the major cause of an increased senescence load with age.
Autophagy is also activated in response to diverse cellular stress, including oncogenic stress and stress caused by chemotherapeutic reagents, both of which can also induce senescence. It was also shown in human fibroblasts that, during replicative senescence, induced through telomere attrition and resulting in persistent DNA damage response, there is an adaptive shift from the proteasomal system towards the autophagic pathway [67]. Indeed, the hallmark of senescence, SA‐β‐gal activity represents elevated lysosomal mass and function, likely to be reflecting autolysosomes at least in some contexts [68, 69, 70].
The functional relevance of autophagy in senescence appears to be highly diverse and context‐dependent. It is conceivable that autophagy activation may promote cell survival under stress conditions [71] but abundant evidence indicates that autophagy modulates specific senescence effectors at multiple levels, which are not necessarily linear. We have previously shown that autophagy is activated during human fibroblast senescence and that it promotes the SASP through mTOR signaling [70, 72]. While mTOR, which can promote the SASP at both post‐transcription (mRNA stability) and mRNA translation levels [73, 74] negatively controls autophagy upstream at autophagosome formation, amino acids, (outputs of autolysosomal degradation) activate mTOR at the surface of (auto)lysosomes [75, 76]. Interestingly, while starvation typically inhibits mTOR and activates autophagy, prolonged starvation reactivates mTOR through autolysosomal degradation products [77]. As such, a spatially regulated feedback module between mTOR and lysosomes coordinates anabolic (mTOR signaling) and catabolic (autophagy) response, allowing their simultaneous activation [70, 78].
Autophagy is also involved in modulating the senescence phenotype, including the SASP, through specific protein degradation. For example, Kang et al. [79] have reported that the transcription factor GATA4 is both a substrate of p62‐mediated selective autophagy and a positive regulator of the inflammatory SASP. Interestingly, they showed that transient autophagy inhibition, which stabilizes GATA4 and thereby promotes the SASP, induces senescence more efficiently compared to the continuous inhibition of autophagy, proposing that global and selective autophagy have opposite effects, promoting and mitigating senescence, respectively [79].
The selectivity of autophagy cargo also appears to alter during senescence. Lamin B1, a major component of the nuclear envelope, is downregulated during senescence, at least in part through selective autophagy degradation via APOE and LC3 binding [80, 81, 82]. Downregulation of Lamin B1, a widely recognized senescence feature [83, 84, 85, 86, 87, 88], has been functionally associated with other senescence features, including the formation of senescence‐associated heterochromatin foci (SAHFs) [87] and cytoplasmic chromatin fragments (CCFs) [82, 89]. CCFs are recognized by the cytosolic DNA‐sensing cGAS–STING pathway to activate the inflammatory SASP [90, 91, 92, 93]. In addition, a recent study identified that the cGAS–STING pathway can also activate autophagy in the context of replicative crisis, in which excessive autophagy can be detrimental [94]. Although these observations indicate a positive feedback loop between autophagy and the cGAS–STING pathway, autophagy can also degrade cytosolic DNA species [95, 96, 97], providing an additional layer of complexity in the SASP regulation and cellular viability.
Senescence‐associated nuclear lamina breakdown can be partly explained by a unique type of nuclear autophagy [98]. LC3 is a key protein involved in autophagy vesicle formation (thus often utilized as a marker of autophagosomes and autolysosomes puncta), but it also plays a role in the recruitment of autophagy cargos to autophagosomes [99, 100]. Interestingly, LC3 is also distributed in the nuclei and interacts with Lamin B1 at the basal state. During oncogene‐induced senescence in human fibroblast, the LC3‐Lamin B1 complex is recruited to autophagosomes and degraded at the cytoplasm [82]. This mechanism is not limited to Lamin B1. SIRT1, a conserved NAD+‐dependent deacetylase (substrates include histones and non‐histone proteins, such as p53 [101, 102, 103, 104], is involved in diverse biological processes, including senescence and aging [105, 106]. SIRT1 is known to be post‐transcriptionally downregulated during senescence, and its ectopic expression of SIRT1 delays replicative senescence in human fibroblasts [107, 108]. Similar to Lamin B1, Xu et al. [109] have shown that SIRT1 binds LC3 at the nuclei in a basal condition, and upon senescence induction, their interaction is enhanced and the complex is degraded in the cytoplasm through autophagy. This autophagy‐dependent SIRT1 downregulation was also seen in hematopoietic tissues during aging in both humans and mice [109]. LC3 has been previously shown to be a nuclear substrate of SIRT1: the deacetylation of LC3 promotes its exportation to the cytoplasm [110]. The study by Xu et al. is particularly relevant in senescence and aging as it indicates that SIRT1 is also a substrate of LC3‐mediated selective autophagy. Notably, SIRT1 appears to negatively regulate the expression of IL6 and IL8, encoding major SASP factors, through direct binding to their promoter regions [111], providing an additional mechanistic link between autophagy and the SASP. Finally, the maintenance DNA methyltransferase, DNMT1 has also been shown to be degraded by autophagy during senescence induced by CDK inhibitors in cancer cells [112]. DNMT1 is also repressed during replicative senescence [113, 114], and that its depletion accelerates senescence in human fibroblasts [113].
Combined, autophagy activation can modulate the phenotype through multiple paths. To understand the phenotypic impact of autophagy, it is crucial to consider the balance between basal and stress‐induced autophagy as well as between bulk and selective autophagy (Fig. 2). Some of the specific nuclear autophagy cargos are involved in the control of both epigenetic and secretory programs during senescence (e.g., Lamin B1 and SIRT1), reinforcing the idea that autophagy is a focal point on which cell‐autonomous and non‐cell‐autonomous regulation of senescence converges.
Fig. 2.
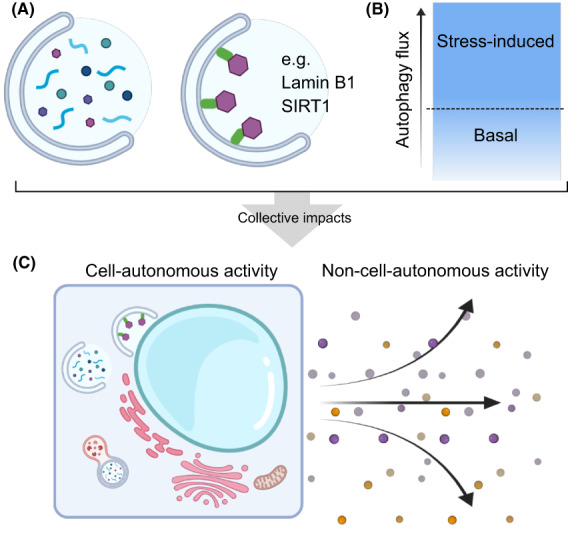
Basal and stress‐induced autophagy in senescence. (A) The collective effect of autophagy is both the bulk selective and nonselective degradation of cellular components. (B) During stress induction which may result in senescence, such as telomere attrition or oncogene activation, autophagy can be up‐regulated to maintain the cell‐autonomous and the non‐cell‐autonomous functionalities of a cell (as shown in C), i.e., proliferative arrest, metabolism, secretory outputs.
4. Autophagy and tumorigenesis
During tumor development, the role of autophagy is complex and appears dependent upon the stage and tissue of origin. On one hand, autophagy is considered essential for cellular health and its loss may otherwise lead to the accumulation of pro‐tumor damage; as such, its promotion is believed to help prevent the development of this initial damage. Consistently, increased basal autophagy in Becn1 F121A/F121A knockin mice not only extends lifespan but also diminishes age‐associated spontaneous tumorigenesis in comparison to littermate controls [20]. Meanwhile, as tumor formation is a microcosm of evolution, and with autophagy being a pro‐fitness and cytoprotective program, many end‐stage tumors are believed to be dependent on an intact, or enhanced autophagy process. As such dynamic fluctuations in autophagy may occur during the progression of the disease (Fig. 3).
Fig. 3.
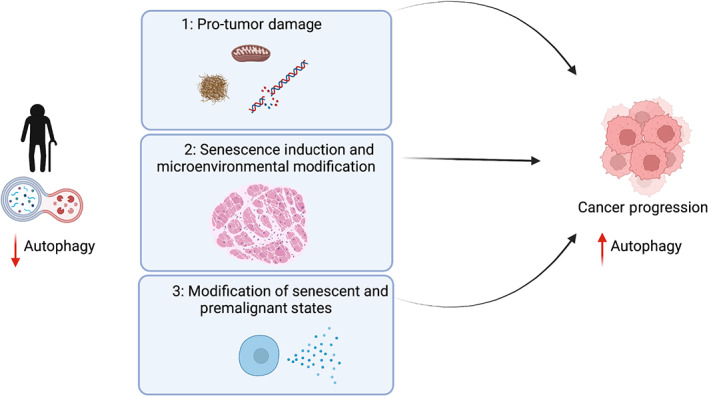
Dynamic fluctuations in autophagy promote cancer progression. With an increasing age and a subsequent decrease in autophagic flux, early tumor development can be promoted through several mechanisms. (1) Through the creation of pro‐tumor damage at the level of protein, organelle homeostasis, and/or DNA damage; (2) the induction of cellular senescence which can generate a pro‐tumorigenic microenvironment; (3) modification of precancerous cellular states accelerating the transition to malignant cancer.
In the liver, the accumulation of the autophagy receptor Sqstm1/p62 is considered a key initiating event in the development of malignant hepatocellular carcinoma [115]. Congruently, the loss of autophagy in either mice with whole liver deletion of Atg7 (Atg7 flox/flox ; Alb‐Cre) or whole‐body mosaic deletion of Atg5 (Atg5 f/f ; CAG‐Cre) results in hepatic adenoma development, and loss of Sqstm1/p62 severely impedes this [116]. Similarly, the loss of chaperone‐mediated autophagy through Lamp2A knockout (Lamp2A flox/flox ; Alb‐Cre) also promotes the development of hepatocellular adenomas with increasing age [117]. Yet whilst the loss of autophagic activity can drive the initial formation of tumorigenesis, this is limited to benign adenomas, and no malignant tumors arise [116].
Similarly, in lung models of autophagy inhibition, the loss of either Atg7 or Atg5 appears to significantly impact the development of aggressive disease in K‐rasG12D driven models, yet with some notable differences. In the case of Atg7 knockout, tumors appear to develop at a similar rate in the early stages of disease regardless of autophagy status. However, whilst autophagy competent tumors develop into malignant tumors, Atg7 deficiency forces tumors to develop towards benign oncocytomas [118]. Of note, concomitant loss of Lkb1 with K‐rasG12D activation is associated with a requirement for an intact autophagy system, and its loss severely impacts tumorigenesis, suggesting tumor genotype and timing of mutational acquisition may also dictate the role of autophagy in disease progression [119]. In the Atg5 knockout model, the loss of autophagy reduces tumor burden and progression to adenocarcinoma in the lung. Interestingly, the authors also noted that the initial transition from hyperplasia to adenoma in autophagy‐deficient mice was higher, suggesting that the loss of Atg5 in this model promotes the early stages of tumor development, whilst blocking the later stages [120]. In keeping with this, in K‐rasG12D driven models of pancreatic cancer development, autophagy inhibition (via simultaneous deletion of Atg7 or Atg5) has shown to increase the incidence of early premalignant lesion development [121, 122]. However, despite this increase in early grade pancreatic intraepithelial neoplasia (PanIN) lesions, these rarely transition to malignant tumors except in the p53 knockout background.
Combined these data suggest that the loss of autophagy may provide initial evolutionary momentum, yet its continued absence is either insufficient alone to promote, or indeed may be detrimental in of itself to malignant conversion. This idea was tested in the Atg5i mouse model, in which autophagy can be dynamically regulated through doxycycline‐inducible expression of sh‐Atg5. Atg5i mice in which autophagy was inhibited for ~ 4 months displayed signs of premature aging with a reduction in health status. When autophagy was subsequently re‐established in these mice, these phenotypes were rescued as was their life span [21]. However, these mice also displayed an increase in age‐associated spontaneous tumor formation in comparison with control mice (where autophagy was intact all the time), thus transient systemic inhibition of autophagy appears to increase the tumorigenic risk. Of note, continuous autophagy inhibition in Atg5i mice was not associated with tumor development, although their premature death due to accelerated aging precluded this assessment. Nevertheless, given that malignant tumors failed to form in long‐lived mosaic Atg5 knockout mice [116], the transient inhibition of autophagy appears to increase the risk of cancer relative to the continuous inhibition of autophagy.
These findings support the theory that autophagy has phase‐dependent roles during tumor development, but what is the mechanism for ‘rejuvenation‐induced’ tumorigenesis, as seen in the Atg5i mice? It is conceivable that reduced autophagy leads to damage accumulation, either proteomic or (epi)genomic, which can be pro‐tumorigenic. Yet without sufficient autophagy, cellular fitness is below the threshold required to support malignant transformation. This idea is largely biased towards the cell‐autonomous aspect of the tumor origin. Notably, it has been shown that short‐term autophagy deficiency in mouse livers causes acute liver damage, which is mostly ‘cured’ by autophagy restoration [123]. However, this autophagy restoration also promotes liver fibrosis. Hepatic stellate cells are a major component of the fibrosis reaction, and autophagy inhibition prevents their function, as such the reactivation of autophagy may enable these cells remodel the tissue microenvironment [123, 124, 125]. Therefore, even though macroscopic restoration occurs in Atg5i mice, the local and tissue microenvironment could remain different. Indeed, as mentioned earlier, Atg5i mice also retain heightened levels of senescent cells after autophagy restoration. As such the outstanding question is whether those residual senescent cells support and shape tumorigenic microenvironment and/or can those pretumoral senescent cells with persistent DNA damage somehow occasionally regain the proliferative capacity with increased cellular fitness after autophagy restoration.
It is also clear that microenvironmental autophagy has an important role in end‐stage tumors as well as potentially during the initial development of cancer. In pancreatic cancer, it is reported that tumor cells promote autophagy in pancreatic stellate cells, which in turn supports tumor growth through metabolic support [126]. Similarly autophagy loss in hepatic stellate cells appears to reduced hepatocellular carcinoma growth, partly through loss of GDF15 expression [127]. Furthermore, several reports have also implicated hepatic autophagy in modulating systemic effects of tumor growth, either through maintaining circulating amino acid (arginine) [128] or inflammatory cytokine levels [129]. However, as these effects may be due to the dramatic effect of complete autophagy loss in the liver, exemplified by gross hepatomegaly and widespread hepatocyte cell death, it is not known to what degree these effects may be recapitulated during aging, when autophagic activity is decreased but not completely abrogated.
In this vein it should also be noted that the complete knockout models of autophagy represent extreme examples that do not fully recapitulate the natural biogenesis of disease states. Interestingly, human cancers are not associated with biallelic inactivating mutations in genes essential for autophagic activity. For example, early reports have described enhanced spontaneous tumor formation in mice with only one functional copy of the Beclin1 gene [130, 131], which is often lost in a heterozygous manner in human breast, ovarian, and prostate cancer [132, 133, 134]. Although the proximity of Beclin1 to the BRCA1 tumor suppressor gene renders the interpretation difficult particularly in these tumor types [135], such a haplo‐insufficient tumor‐suppressive activity was also observed in the Atg5+/− mouse models for pancreatic cancer [136] and AML [137].
As such we must assume that, whilst our current genetic mouse models lend important insights into disease progression, more nuanced models in which autophagic activity (or even adaptor proteins and cargo) can be titrated, and do not profoundly introduce tissue architecture artifacts, are required. As examples, autophagy deletion in the liver is associated with extreme hepatomegaly and liver damage [123], whilst in the pancreas, its loss is associated with the development of pancreatic atrophy [121, 138]. Such phenotypes may impact the progression of disease, not necessarily in a way that mirrors the etiology of human disease with regards to autophagic activity and aging. In this regard, the Atg5i models are promising. Although the original Atg5i mice exhibited a highly efficient knockdown of Atg5, thus developing a phenotype reminiscent of complete knockouts, the second Atg5i model (Atg5i‐2), in which Atg5 is downregulated using a weaker shRNA and which produces milder phenotype with, e.g., no obvious hepatomegaly [21], still displays premature aging phenotypes. However, tumor susceptibility in the Atg5i‐2 remains to be evaluated and this model may be of greater interest in future studies. Similarly, the recently developed HyD‐LIR flox/flox mouse model, wherein Atg8‐dependent selective autophagy can be impaired, due to expression of a synthetic gene that acts as a competitive inhibitor to p62/sqstm1, offers a remarkable opportunity to study aspects of autophagy loss without the caveats that accompany the complete knockout of essential autophagy genes [139, 140].
5. Perspectives and conclusions
Advancing age represents the single greatest risk factor for cancer development. As organisms age, the rate at which autophagy occurs decreases. Indeed, it is clear that the loss of autophagy can lead to cellular dysfunction and can perturb stem cell populations in vivo, limiting their regenerative potential and reducing the homeostatic capacity of a tissue [41, 44]. As autophagy is associated with metabolism, mitochondrial homeostasis, reactive oxygen species generation, turnover of toxic and tumor‐promoting aggregates, as well as homeostasis of other organelles, it is likely that this age‐associated decrease in autophagy impacts some or all of these processes at once. However, to what degree each is compromised during aging and is functionally relevant to age‐associated decline is difficult to disentangle, and almost certainly cell‐type‐specific.
Importantly, the loss of basal autophagy appears also to be associated with the acquisition of cellular senescence [65, 141]. As mammals age, and concomitant with a decrease in autophagy, there is also an increase in the senescent burden, or the amount of senescent cells residing in tissues. The historical viewpoint, that senescence is primarily a cell autonomous process, has long since been expanded by the realization that senescent cells modify their surrounding local microenvironment, and have systemic effects on an organism [50, 53]. Yet whether the loss of autophagy is a major driver of senescence in naturally aged systems, or is limited to particular populations (i.e., stem cells with low proliferative indexes) remains to be investigated, as does the phenotype of this form of senescence. It is now widely accepted that senescence itself is a heterogenous cellular state that may depend on the cell of origin, and on the type of stress that led to its induction. As autophagy has been linked to the maintenance of senescence, metabolism, and secretion, it is tempting to speculate that this form of senescence will be distinct with unique effects on the local milieu [65, 70, 72, 142].
Senescence is also considered to be a barrier to tumorigenesis in a number of settings, and senescence markers are often found to be upregulated during the premalignant stages of cancer development in vivo [52]. As such the intersection of cancer development, particularly at these early stages of tumor formation, with an age‐associated decrease in autophagy represents a potential source of synergy for tumor promotion. How a reduction in autophagy, as opposed to its complete loss, in these early lesions may impinge upon senescent and premalignant biology is still relatively unexplored in vivo. Moreover, reduced levels of autophagy may lead to tumor progression through cell autonomous or nonautonomous mechanisms. In addition to its typical nonselective degradation, cargo selectivity also plays a key role in downstream phenotypes, such as stress‐induced senescence [72, 80, 82]. Selective autophagy, as exemplified by nuclear autophagy, appears to have a distinct layer of regulation, and the net effect of the bulk autophagy and fine‐tuned selective autophagy not only shape the intrinsic cellular phenotype but also surrounding tissue microenvironments by modulating non‐cell‐autonomous activities (e.g., the SASP). To‐date, only a handful of specific (nuclear autophagy) cargos that are associated with senescence have been characterized. The future identification of such cargos would enrich our understanding of the crosstalk between senescence and autophagy and their relevance to aging and cancer, and might provide new opportunities to intervene in aging and age‐associated disorders.
The promotion of autophagy throughout life has been shown to extend life and health span, with a reduction in spontaneous tumor formation in mammalian systems [20]. Whilst the genetic promotion of autophagy can restore cellular and tissue functionality, as well as extend or recapture health and life span, there is evidence that damage remains, including senescent cells, and may continue to have detrimental effects [19, 20, 21, 123]. Indeed, ‘rejuvenation’ via the re‐establishment of autophagy in Atg5i mice is accompanied by increased tumorigenesis in the presence of residual senescence cells. These persistent senescent cells may represent early tumor formation, or may promote tumor development through non‐cell autonomous activities. To‐date therapies targeting senescent cells for destruction with senolytics and therapeutics‐boosting autophagy have only been considered individually. However, this raises the question of whether senolytic compounds may synergize with autophagy augmentation to promote a more holistic organismal rejuvenation therapy.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by a Cancer Research UK Cambridge Institute Core Grant (C9545/A29580) to MN and the UKRI Biotechnology and Biological Science Research Council (BB/T013486/1) to LDC and MN. Figures created with Biorender.com.
Contributor Information
Liam D. Cassidy, Email: liam.cassidy@cruk.cam.ac.uk.
Masashi Narita, Email: masashi.narita@cruk.cam.ac.uk.
References
- 1. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–42. 10.1038/s41556-018-0037-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaushik S, Cuervo AM. The coming of age of chaperone‐mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365–81. 10.1038/s41580-018-0001-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schuck S. Microautophagy – distinct molecular mechanisms handle cargoes of many sizes. J Cell Sci. 2020;133(17):jcs246322. 10.1242/jcs.246322 [DOI] [PubMed] [Google Scholar]
- 4. Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24(12):604–12. 10.1016/j.tig.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125(1):85–93. 10.1172/JCI73946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–93. 10.1038/s41580-018-0033-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. López‐Otín C, Galluzzi L, Freije JMP, Madeo F, Kroemer G. Metabolic control of longevity. Cell. 2016;166(4):802–21. 10.1016/j.cell.2016.07.031 [DOI] [PubMed] [Google Scholar]
- 8. Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7‐dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21(23):3061–6. 10.1101/gad.1600707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hars ES, Qi H, Ryazanov AG, Jin S, Cai L, Hu C, et al. Autophagy regulates ageing in C. elegans . Autophagy. 2007;3(2):93–5. [DOI] [PubMed] [Google Scholar]
- 10. Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4(2):176–84. 10.4161/auto.5269 [DOI] [PubMed] [Google Scholar]
- 11. Ulgherait M, Rana A, Rera M, Graniel J, Walker DW. AMPK modulates tissue and organismal aging in a non‐cell‐autonomous manner. Cell Rep. 2014;8(6):1767–80. 10.1016/j.celrep.2014.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meléndez A, Tallóczy Z, Seaman M, Eskelinen E‐L, Hall DH, Levine B. Autophagy genes are essential for dauer development and life‐span extension in C. elegans . Science. 2003;301(5638):1387–91. 10.1126/science.1087782 [DOI] [PubMed] [Google Scholar]
- 13. Lapierre LR, Gelino S, Meléndez A, Hansen M. Autophagy and lipid metabolism coordinately modulate life span in germline‐less C. elegans . Curr Biol. 2011;21(18):1507–14. 10.1016/j.cub.2011.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans . PLoS Genet. 2008;4(2):e24. 10.1371/journal.pgen.0040024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tóth ML, Sigmond T, Borsos E, Barna J, Erdélyi P, Takacs‐Vellai K, et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans . Autophagy. 2008;4(3):330–8. 10.4161/auto.5618 [DOI] [PubMed] [Google Scholar]
- 16. Jia K, Levine B. Autophagy is required for dietary restriction‐mediated life span extension in C. elegans . Autophagy. 2007;3(6):597–9. [DOI] [PubMed] [Google Scholar]
- 17. Karsli‐Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–27. 10.1158/2159-8290.CD-14-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuma A, Komatsu M, Mizushima N. Autophagy‐monitoring and autophagy‐deficient mice. Autophagy. 2017;13:1619–28. 10.1080/15548627.2017.1343770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pyo J‐O, Yoo S‐M, Ahn H‐H, Nah J, Hong S‐H, Kam T‐I, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. 10.1038/ncomms3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, et al. Disruption of the beclin 1‐BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;125:85. 10.1038/s41586-018-0162-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cassidy LD, Young ARJ, Young CNJ, Soilleux EJ, Fielder E, Weigand BM, et al. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat Commun. 2020;11(1):307–12. 10.1038/s41467-019-14187-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang JT, Kumsta C, Hellman AB, Adams LM, Hansen M. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. Elife. 2017;6:e18459. 10.7554/eLife.18459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilhelm T, Byrne J, Medina R, Kolundžić E, Geisinger J, Hajduskova M, et al. Neuronal inhibition of the autophagy nucleation complex extends life span in post‐reproductive C. elegans . Genes Dev. 2017;31(15):1561–72. 10.1101/gad.301648.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bork T, Liang W, Yamahara K, Lee P, Tian Z, Liu S, et al. Podocytes maintain high basal levels of autophagy independent of mtor signaling. Autophagy. 2020;16(11):1932–48. 10.1080/15548627.2019.1705007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li L, Wang ZV, Hill JA, Lin F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol. 2014;25(2):305–15. 10.1681/ASN.2013040374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125(25):3170–81. 10.1161/CIRCULATIONAHA.111.041814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li L, Kang H, Zhang Q, D'Agati VD, Al‐Awqati Q, Lin F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J Clin Invest. 2019;129(6):2374–89. 10.1172/JCI122256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McWilliams TG, Prescott AR, Allen GFG, Tamjar J, Munson MJ, Thomson C, et al. mito‐QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol. 2016;214(3):333–45. 10.1083/jcb.201603039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katayama H, Hama H, Nagasawa K, Kurokawa H, Sugiyama M, Ando R, et al. Visualizing and modulating mitophagy for therapeutic studies of neurodegeneration. Cell. 2020;181(5):1176–87.e16. 10.1016/j.cell.2020.04.025 [DOI] [PubMed] [Google Scholar]
- 30. Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, et al. Measuring in vivo mitophagy. Mol Cell. 2015;60(4):685–96. 10.1016/j.molcel.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, et al. Genome‐wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci USA. 2010;107(32):14164–9. 10.1073/pnas.1009485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, et al. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281(20):14474–85. 10.1074/jbc.M600364200 [DOI] [PubMed] [Google Scholar]
- 33. Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014;8(5):1509–21. 10.1016/j.celrep.2014.07.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, Sahu S, et al. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012;13(3):258–65. 10.1038/embor.2011.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu C‐C, Visvikis O, Chang JT, et al. The TFEB orthologue HLH‐30 regulates autophagy and modulates longevity in Caenorhabditis elegans . Nat Commun. 2013;4(1):2267–8. 10.1038/ncomms3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Demontis F, Perrimon N. FOXO/4E‐BP signaling in Drosophila muscles regulates organism‐wide proteostasis during aging. Cell. 2010;143(5):813–25. 10.1016/j.cell.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–33. 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang H, Alsaleh G, Feltham J, Sun Y, Napolitano G, Riffelmacher T, et al. Polyamines control eIF5A hypusination, TFEB translation, and autophagy to reverse B cell senescence. Mol Cell. 2019;76(1):110–25.e9. 10.1016/j.molcel.2019.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Green DR. Polyamines and aging: a CLEAR connection? Mol Cell. 2019;76(1):5–7. 10.1016/j.molcel.2019.09.003 [DOI] [PubMed] [Google Scholar]
- 40. Nakamura S, Oba M, Suzuki M, Takahashi A, Yamamuro T, Fujiwara M, et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat Commun. 2019;10(1):847–11. 10.1038/s41467-019-08729-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. García‐Prat L, Martínez‐Vicente M, Perdiguero E, Ortet L, Rodríguez‐Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529(7584):37–42. 10.1038/nature16187 [DOI] [PubMed] [Google Scholar]
- 42. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10(6):507–15. 10.1016/j.cmet.2009.10.008 [DOI] [PubMed] [Google Scholar]
- 43. Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi‐Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208(3):455–67. 10.1084/jem.20101145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543(7644):205–10. 10.1038/nature21388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dong S, Wang Q, Kao Y‐R, Diaz A, Tasset I, Kaushik S, et al. Chaperone‐mediated autophagy sustains haematopoietic stem‐cell function. Nature. 2021;591(7848):117–23. 10.1038/s41586-020-03129-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–95. 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- 47. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28(2):99–114. 10.1101/gad.235184.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. 2019;179(4):813–27. 10.1016/j.cell.2019.10.005 [DOI] [PubMed] [Google Scholar]
- 49. Pérez‐Mancera PA, Young ARJ, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14(8):547–58. 10.1038/nrc3773 [DOI] [PubMed] [Google Scholar]
- 50. Muñoz‐Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–96. 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 51. Coppé J‐P, Desprez P‐Y, Krtolica A, Campisi J. The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5(1):99–118. 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642–2. 10.1038/436642a [DOI] [PubMed] [Google Scholar]
- 53. Chan ASL, Narita M. Short‐term gain, long‐term pain: the senescence life cycle and cancer. Genes Dev. 2019;33(3–4):127–43. 10.1101/gad.320937.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Childs BG, Gluscevic M, Baker DJ, Laberge R‐M, Marquess D, Dananberg J, et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16(10):718–35. 10.1038/nrd.2017.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tchkonia T, Zhu Y, Van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–72. 10.1172/JCI64098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem‐cell ageing modified by the cyclin‐dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421–6. 10.1038/nature05159 [DOI] [PubMed] [Google Scholar]
- 57. Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443(7110):448–52. 10.1038/nature05091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner‐Weir S, et al. p16INK4a induces an age‐dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–7. 10.1038/nature05092 [DOI] [PubMed] [Google Scholar]
- 59. Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8(9):703–13. 10.1038/nrm2241 [DOI] [PubMed] [Google Scholar]
- 60. Sousa‐Victor P, Gutarra S, García‐Prat L, Rodríguez‐Ubreva J, Ortet L, Ruiz‐Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506(7488):316–21. 10.1038/nature13013 [DOI] [PubMed] [Google Scholar]
- 61. Young ARJ, Cassidy LD, Narita M. Autophagy and senescence, converging roles in pathophysiology as seen through mouse models. Adv Cancer Res. 2021;150:113–45. 10.1016/bs.acr.2021.02.001 [DOI] [PubMed] [Google Scholar]
- 62. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature. 2011;479(7372):232–6. 10.1038/nature10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)‐positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–9. 10.1038/nature16932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van Deursen JM. Senolytic therapies for healthy longevity. Science. 2019;364(6441):636–7. 10.1126/science.aaw1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PloS One. 2011;6(8):e23367. 10.1371/journal.pone.0023367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He Y‐W, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci USA. 2012;109(33):13325–30. 10.1073/pnas.1120193109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009;28(7):889–901. 10.1038/emboj.2009.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence‐associated (beta)‐galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113(Pt 20):3613–22. 10.1242/jcs.113.20.3613 [DOI] [PubMed] [Google Scholar]
- 69. Gerland L‐M, Peyrol S, Lallemand C, Branche R, Magaud J‐P, Ffrench M. Association of increased autophagic inclusions labeled for beta‐galactosidase with fibroblastic aging. Exp Gerontol. 2003;38(8):887–95. 10.1016/s0531-5565(03)00132-3 [DOI] [PubMed] [Google Scholar]
- 70. Narita M, Young ARJ, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332(6032):966–70. 10.1126/science.1205407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Murrow L, Debnath J. Autophagy as a stress‐response and quality‐control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–37. 10.1146/annurev-pathol-020712-163918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. 10.1101/gad.519709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence‐associated secretory phenotype. Nat Cell Biol. 2015;17(9):1205–17. 10.1038/ncb3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Laberge R‐M, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–61. 10.1038/ncb3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zoncu R, Bar‐Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside‐out mechanism that requires the vacuolar H(+)‐ATPase. Science. 2011;334(6056):678–83. 10.1126/science.1207056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sancak Y, Bar‐Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator‐Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–6. 10.1038/nature09076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13(4):453–60. 10.1038/ncb2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612. 10.1126/science.aaa5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhao H, Ji Q, Wu Z, Wang S, Ren J, Yan K, et al. Destabilizing heterochromatin by APOE mediates senescence. Nat Aging. 2022;2(4):303–16. 10.1038/s43587-022-00186-z [DOI] [PubMed] [Google Scholar]
- 81. Lenain C, Gusyatiner O, Douma S, van den Broek B, Peeper DS. Autophagy‐mediated degradation of nuclear envelope proteins during oncogene‐induced senescence. Carcinogenesis. 2015;36(11):1263–74. 10.1093/carcin/bgv124 [DOI] [PubMed] [Google Scholar]
- 82. Dou Z, Xu C, Donahue G, Shimi T, Pan J‐A, Zhu J, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527(7576):105–9. 10.1038/nature15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shimi T, Butin‐Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, et al. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011;25(24):2579–93. 10.1101/gad.179515.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Barascu A, Le Chalony C, Pennarun G, Genet D, Imam N, Lopez B, et al. Oxidative stress induces an ATM‐independent senescence pathway through p38 MAPK‐mediated lamin B1 accumulation. EMBO J. 2012;31(5):1080–94. 10.1038/emboj.2011.492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Freund A, Laberge R‐M, Demaria M, Campisi J. Lamin B1 loss is a senescence‐associated biomarker. Mol Biol Cell. 2012;23(11):2066–75. 10.1091/mbc.E11-10-0884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dreesen O, Chojnowski A, Ong PF, Zhao TY, Common JE, Lunny D, et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J Cell Biol. 2013;200(5):605–17. 10.1083/jcb.201206121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young ARJ, et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013;27(16):1800–8. 10.1101/gad.217281.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, et al. Lamin B1 depletion in senescent cells triggers large‐scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27(16):1787–99. 10.1101/gad.223834.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, et al. Lysosome‐mediated processing of chromatin in senescence. J Cell Biol. 2013;202(1):129–43. 10.1083/jcb.201212110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114(23):E4612–20. 10.1073/pnas.1705499114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402–6. 10.1038/nature24050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Glück S, Guey B, Gulen MF, Wolter K, Kang T‐W, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061–70. 10.1038/ncb3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, et al. Mitochondria‐to‐nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020;34(5–6):428–45. 10.1101/gad.331272.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nassour J, Radford R, Correia A, Fusté JM, Schoell B, Jauch A, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. 2019;565(7741):659–63. 10.1038/s41586-019-0885-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Han X, Chen H, Gong H, Tang X, Huang N, Xu W, et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress‐induced senescence. J Biol Chem. 2020;295(14):4451–63. 10.1074/jbc.RA119.010734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Miller KN, Dasgupta N, Liu T, Adams PD, Vizioli MG. Cytoplasmic chromatin fragments‐from mechanisms to therapeutic potential. Elife. 2021;10:e63728. 10.7554/eLife.63728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rello‐Varona S, Lissa D, Shen S, Niso‐Santano M, Senovilla L, Mariño G, et al. Autophagic removal of micronuclei. Cell Cycle. 2012;11(1):170–6. 10.4161/cc.11.1.18564 [DOI] [PubMed] [Google Scholar]
- 98. Luo M, Zhao X, Song Y, Cheng H, Zhou R. Nuclear autophagy: an evolutionarily conserved mechanism of nuclear degradation in the cytoplasm. Autophagy. 2016;12(11):1973–83. 10.1080/15548627.2016.1217381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41. 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 100. Klionsky DJ, Abdel‐Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. 2021;17:1–382. 10.1080/15548627.2020.1797280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD‐dependent histone deacetylase. Nature. 2000;403(6771):795–800. 10.1038/35001622 [DOI] [PubMed] [Google Scholar]
- 102. Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, et al. A phylogenetically conserved NAD+‐dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97(12):6658–63. 10.1073/pnas.97.12.6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107(2):137–48. 10.1016/s0092-8674(01)00524-4 [DOI] [PubMed] [Google Scholar]
- 104. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD‐dependent p53 deacetylase. Cell. 2001;107(2):149–59. 10.1016/s0092-8674(01)00527-x [DOI] [PubMed] [Google Scholar]
- 105. Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6(4):298–305. 10.1038/nrm1616 [DOI] [PubMed] [Google Scholar]
- 106. Yi J, Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta. 2010;1804(8):1684–9. 10.1016/j.bbapap.2010.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sasaki T, Maier B, Bartke A, Scrable H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell. 2006;5(5):413–22. 10.1111/j.1474-9726.2006.00235.x [DOI] [PubMed] [Google Scholar]
- 108. Huang J, Gan Q, Han L, Li J, Zhang H, Sun Y, et al. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PloS One. 2008;3(3):e1710. 10.1371/journal.pone.0001710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Xu C, Wang L, Fozouni P, Evjen G, Chandra V, Jiang J, et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol. 2020;22(10):1170–9. 10.1038/s41556-020-00579-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57(3):456–66. 10.1016/j.molcel.2014.12.013 [DOI] [PubMed] [Google Scholar]
- 111. Hayakawa T, Iwai M, Aoki S, Takimoto K, Maruyama M, Maruyama W, et al. SIRT1 suppresses the senescence‐associated secretory phenotype through epigenetic gene regulation. PloS One. 2015;10(1):e0116480. 10.1371/journal.pone.0116480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Acevedo M, Vernier M, Mignacca L, Lessard F, Huot G, Moiseeva O, et al. A CDK4/6‐dependent epigenetic mechanism protects cancer cells from PML‐induced senescence. Cancer Res. 2016;76(11):3252–64. 10.1158/0008-5472.CAN-15-2347 [DOI] [PubMed] [Google Scholar]
- 113. Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013;15(12):1495–506. 10.1038/ncb2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lopatina N, Haskell JF, Andrews LG, Poole JC, Saldanha S, Tollefsbol T. Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J Cell Biochem. 2002;84(2):324–34. 10.1002/jcb.10015 [DOI] [PubMed] [Google Scholar]
- 115. Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font‐Burgada J, et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC‐initiating cells. Cancer Cell. 2016;29(6):935–48. 10.1016/j.ccell.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy‐deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. 10.1101/gad.2016211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Schneider JL, Villarroya J, Diaz‐Carretero A, Patel B, Urbanska AM, Thi MM, et al. Loss of hepatic chaperone‐mediated autophagy accelerates proteostasis failure in aging. Aging Cell. 2015;14(2):249–64. 10.1111/acel.12310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Guo JY, Karsli‐Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K‐ras‐induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447–61. 10.1101/gad.219642.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bhatt V, Khayati K, Hu ZS, Lee A, Kamran W, Su X, et al. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1‐deficient Kras‐driven lung tumorigenesis. Genes Dev. 2019;33(3–4):150–65. 10.1101/gad.320481.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. 10.1038/ncomms4056 [DOI] [PubMed] [Google Scholar]
- 121. Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. 10.1038/nature12865 [DOI] [PubMed] [Google Scholar]
- 122. Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4(8):905–13. 10.1158/2159-8290.CD-14-0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cassidy LD, Young AR, Pérez‐Mancera PA, Nimmervoll B, Jaulim A, Chen H‐C, et al. A novel Atg5‐shRNA mouse model enables temporal control of autophagy in vivo. Autophagy. 2018;14:1256–66. 10.1080/15548627.2018.1458172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Thoen LFR, Guimarães ELM, Dollé L, Mannaerts I, Najimi M, Sokal E, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55(6):1353–60. 10.1016/j.jhep.2011.07.010 [DOI] [PubMed] [Google Scholar]
- 125. Hernández Gea V, Ghiassi Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–46. 10.1053/j.gastro.2011.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479–83. 10.1038/nature19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Myojin Y, Hikita H, Sugiyama M, Sasaki Y, Fukumoto K, Sakane S, et al. Hepatic stellate cells in hepatocellular carcinoma promote tumor growth via growth differentiation factor 15 production. Gastroenterology. 2021;160(5):1741–54.e16. 10.1053/j.gastro.2020.12.015 [DOI] [PubMed] [Google Scholar]
- 128. Poillet‐Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018;563(7732):569–73. 10.1038/s41586-018-0697-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Poillet‐Perez L, Sharp DW, Yang Y, Laddha SV, Ibrahim M, Bommareddy PK, et al. Autophagy promotes growth of tumors with high mutational burden by inhibiting a T‐cell immune response. Nat Cancer. 2020;1(9):923–34. 10.1038/s43018-020-00110-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20. 10.1172/JCI20039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100(25):15077–82. 10.1073/pnas.2436255100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Delaney JR, Patel CB, Willis KM, Haghighiabyaneh M, Axelrod J, Tancioni I, et al. Haploinsufficiency networks identify targetable patterns of allelic deficiency in low mutation ovarian cancer. Nat Commun. 2017;8(1):14423–11. 10.1038/ncomms14423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Delaney JR, Patel CB, Bapat J, Jones CM, Ramos‐Zapatero M, Ortell KK, et al. Autophagy gene haploinsufficiency drives chromosome instability, increases migration, and promotes early ovarian tumors. PLoS Genet. 2020;16(1):e1008558. 10.1371/journal.pgen.1008558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gao X, Zacharek A, Salkowski A, Grignon DJ, Sakr W, Porter AT, et al. Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer. Cancer Res. 1995;55(5):1002–5. [PubMed] [Google Scholar]
- 135. Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res. 2014;12(4):485–90. 10.1158/1541-7786.MCR-13-0614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Görgülü K, Diakopoulos KN, Ai J, Schoeps B, Kabacaoglu D, Karpathaki A‐F, et al. Levels of the autophagy‐related 5 protein affect progression and metastasis of pancreatic tumors in mice. Gastroenterology. 2019;156(1):203–17.e20. 10.1053/j.gastro.2018.09.053 [DOI] [PubMed] [Google Scholar]
- 137. Watson AS, Riffelmacher T, Stranks A, Williams O, De Boer J, Cain K, et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015;1(1):371. 10.1038/cddiscovery.2015.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Diakopoulos KN, Lesina M, Wörmann S, Song L, Aichler M, Schild L, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex‐ and nutrition‐dependent processes. Gastroenterology. 2015;148(3):626–38.e17. 10.1053/j.gastro.2014.12.003 [DOI] [PubMed] [Google Scholar]
- 139. Kageyama S, Gudmundsson SR, Sou Y‐S, Ichimura Y, Tamura N, Kazuno S, et al. p62/SQSTM1‐droplet serves as a platform for autophagosome formation and anti‐oxidative stress response. Nat Commun. 2021;12(1):16–6. 10.1038/s41467-020-20185-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lee Y‐K, Jun Y‐W, Choi H‐E, Huh YH, Kaang B‐K, Jang D‐J, et al. Development of LC3/GABARAP sensors containing a LIR and a hydrophobic domain to monitor autophagy. EMBO J. 2017;36(8):1100–16. 10.15252/embj.201696315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kwon Y, Kim JW, Jeoung JA, Kim M‐S, Kang C. Autophagy is pro‐senescence when seen in close‐up, but anti‐senescence in long‐shot. Mol Cells. 2017;40(9):607–12. 10.14348/molcells.2017.0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Jiang S, Dupont N, Castillo EF, Deretic V. Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun. 2013;5(5):471–9. 10.1159/000346707 [DOI] [PMC free article] [PubMed] [Google Scholar]