

Abstract
Telomere maintenance is essential for maintaining genome integrity in both normal and cancer cells. Without functional telomeres, chromosomes lose their protective structure and undergo fusion and breakage events that drive further genome instability, including cell arrest or death. One means by which this loss can be overcome in stem cells and cancer cells is via re‐addition of G‐rich telomeric repeats by the telomerase reverse transcriptase (TERT). During aging of somatic tissues, however, insufficient telomerase expression leads to a proliferative arrest called replicative senescence, which is triggered when telomeres reach a critically short threshold that induces a DNA damage response. Cancer cells express telomerase but do not entirely escape telomere instability as they often possess short telomeres; hence there is often selection for genetic alterations in the TERT promoter that result in increased telomerase expression. In this review, we discuss our current understanding of the consequences of telomere instability in cancer and aging, and outline the opportunities and challenges that lie ahead in exploiting the reliance of cells on telomere maintenance for preserving genome stability.
Keywords: aging, cancer, genome instability, senescence, telomeres, telomerase reverse transcriptases
Normal cells and stem cells rely on genome integrity to survive repeated cell divisions. In aging and cancer, the inability to maintain telomeres can lead to telomere erosion and a DNA damage response (DDR). Telomerase insufficiency induces phenotypes that can overlap between aged cells and cancer cells, or that can even further drive age‐associated cancer (e.g. the senescence‐associated secretory phenotype, or SASP).
Abbreviations
- ALT
alternative lengthening of telomeres
- AML
acute myeloid leukemia
- ATR
ataxia telangiectasia and Rad3‐related kinase
- ATRA
all‐trans retinoic acid
- AZT
azidothymidine (antiretroviral drug used to treat HIV)
- BRAF
serine/threonine‐protein kinase B‐raf
- BRCA1
breast cancer type 1 susceptibility protein
- BRCA2
breast cancer type 2 susceptibility protein
- c16orf72
chromosome 16 open reading frame 72
- CpG island
regions in the genome that contains repeated CG dinucleotides
- CTC1
CST complex subunit CTC1
- DDR
DNA damage response
- Dnmt3b
DNA methyltransferase 3B
- EGFR
epidermal growth factor receptor
- ETS
erythroid transformation specific
- EV
extracellular vesicles
- FDA
Food and Drug Administration
- H3K27
histone H3, lysine 27
- hTR
human telomerase RNA component
- kbp
kilobase pairs
- mESC
murine embryonic stem cells
- miRNA
micro RNAs
- nt
nucleotides
- NSCLC
non small cell lung cancer
- p21
cyclin‐dependent kinase inhibitor 1 or CDK‐interacting protein 1
- p53
cellular tumor antigen p53
- POT1
protection of telomeres protein 1
- PRC2
polycomb repressive complex 2
- RAP1
telomeric repeat‐binding factor 2‐interacting protein 1 or repressor activator protein 1
- Rb
retinoblastoma‐associated protein
- REV7
mitotic spindle assembly checkpoint protein MAD2B
- SASP
senescence‐associated secretory phenotype
- SHLD1
shieldin complex subunit 1
- SHLD2
shieldin complex subunit 2
- SHLD3
shieldin complex subunit 3
- STN1
CST complex subunit STN1
- TAPR1
telomere attrition and P53 response 1 protein (alias for c16orf72)
- TEN1
CST complex subunit TEN1
- TERT
telomerase reverse transcriptase
- THOR
TERT Hypermethylated Oncological Region
- TIMP
tissue inhibitors of matrix metalloproteinases
- TIN2
TRF1 interacting nuclear factor 2 protein
- TPP1
telomere protection protein 1, encoded by adrenocortical dysplasia homolog ACD
- TRF1
telomeric repeat binding factor 1 protein
- TRF2
telomeric repeat binding factor 2 protein
- VEGF
vascular endothelial growth factor
- WT
wildtype
1. Introduction
Telomeres are nucleoprotein complexes at the ends of chromosomes that protect cells from the loss of genetic information and from chromosomal fusions and other abnormalities caused by the untimely activation of the DNA damage response. In most eukaroytes, telomeric DNA is comprised of long tracts of a conserved, G‐rich repeat that is represented by the general sequence Tx(Ax)Gx(C). In humans and mice, telomeres consist of 5′‐TTAGGG‐3′ repeats that vary in length between < 20 kbp in humans and can exceed 50 kbp in mice [1, 2]. The conserved nature of telomeres suggests that they play an essential role that originated in our primitive ancestors [3, 4, 5, 6].
Telomeric DNA repeats are double‐stranded but end with a single‐stranded, G‐rich 3′ overhang, typically 150 nucleotides (nt) in length. As this overhang contains the same sequence as telomeric DNA that is more distal to the chromosome terminus, it can loop back and invade the double‐stranded DNA region to form a displacement loop (D‐loop); when localized at the telomere, this structure is also called a telomere loop (T‐loop) [7, 8, 9, 10]. This unusual looped DNA structure serves as a docking site for shelterin, a six‐subunit complex (containing TRF1, TRF2, TIN2, RAP1, POT1 and TPP1) that binds to telomeres [11, 12]. Shelterin interacts together with other complexes, such as CST (CTC1, TEN1, STN1) [13, 14, 15, 16] and the multi‐subunit complex, shieldin (SHLD1, SHLD2, SHLD3, REV7) [17] to regulate telomere DNA replication and suppress inappropriate telomere processing or recombination [9, 18, 19, 20, 21]. In addition to structural protection there is a need for telomere replenishment because the 5′ terminus of each DNA molecule is incompletely replicated. This so‐called ‘end replication problem’, together with nucleolytic trimming of chromosome ends, leads to progressive loss of telomeric sequence during each round of DNA replication [22, 23, 24, 25].
Without a means to counterbalance this loss, inexorable telomere erosion results in a critically short threshold of telomere length that triggers a DNA damage response and cell cycle arrest (in normal cells that retain an intact DNA damage checkpoint) or cell death due to mitotic catastrophe. In human fibroblasts in culture, this response limits cellular lifespan – a phenomenon called replicative senescence or the Hayflick limit [26, 27]. In vivo, adult somatic cells from highly proliferative or minimally proliferative tissues often undergo telomere attrition at a similar rate [28]. In many eukaryotic cells, including stem cells and cancer cells, this loss is often counteracted by the expression of the telomerase reverse transcriptase (TERT) and its integral RNA template component hTR (encoded by TERC). This telomerase complex and its associated co‐factors are recruited to telomeres during S‐phase and serve to elongate the 3′ telomeric overhang [29, 30, 31]. Despite retaining low levels of telomerase activity, telomere erosion nonetheless occurs in most human stem and progenitor cells [32, 33].
Emerging evidence underscores that there are important similarities in how telomere integrity affects aging, normal stem cells (as discussed below), and cancer cells (as discussed later). In this review, our goal is to highlight these common themes and the opportunities and challenges that lie ahead in exploiting this information in the development of new cancer treatments.
2. Telomeres in aging and stem cells
Telomere shortening is one of the main hallmarks of aging and is commonly observed in most cell types [34]. Telomere shortening can occur through the well‐known end‐replication problem or as a result of independent stochastic events.
In aging, the impact of telomere erosion on human and organismal lifespan is an extant question. While telomere length decreases with age in all cell types, the average telomere length varies between species significantly and does not strictly correlate with lifespan [35, 36]. One such example is that of mice and humans; mice have longer telomeres than do humans but a shorter lifespan [2]. However, mice that lack sufficient levels of telomerase activity undergo telomere erosion and eventually reach an average length similar to some normal human tissues. As such, they provide a powerful genetic model in which to assess the role of eroded telomeres in aging and cancer [37, 38]. In marine animals, it is the body size of marine mammals, rather than telomere length, that dictates life expectancy [39]. In contrast, in some species, such as the wild Soay sheep, shorter average leukocyte telomeres are associated with an increased risk of early mortality [27, 40, 41]. In humans, females have been reported to possess longer blood cell telomere lengths than do males [42], and shorter average telomere lengths correlate with several human disorders [43, 44].
Other biological clocks have been established that reflect chronological age, such as the methylome clock, which is defined as age‐associated changes in the pattern and extent of methylated DNA [45, 46]. The telomere clock appears to more closely reflect a ‘replication clock’ rather than a chronological clock [46, 47, 48]. For example, only a weak correlation between telomere length and chronological age has been found in two independent studies [48, 49]. A relationship between the methylated status of 140 CpG islands and telomere length has also been described but might more closely reflect the population doubling time of the cell population [48].
2.1. Telomere erosion: a harbinger of senescence
Telomere attrition to a critical threshold length that is no longer able to sustain telomere integrity is one of several known inducers of cellular senescence [50]. This type of senescence, termed telomere‐induced or replicative senescence, can be triggered by the well‐known tumour suppressor p53 and its downstream effector p21, a cyclin‐dependent kinase inhibitor that represses the cell cycle (as discussed in an accompanying article in this special series [51, 52]). Mathematical models and studies of budding yeast have suggested that the shortest telomeres are responsible for senescence induction [53, 54]. In human cancer cells, the minimum length of a functional telomere is 12 bp [55], beyond which telomere‐telomere fusions are observed. Importantly, damage at a single telomere is enough to induce cell cycle arrest in budding yeast [56] and to induce cycles of chromosome breakage‐fusion‐bridge in cancer cells [57, 58, 59, 60] (Figs 1 and 2).
Fig. 1.
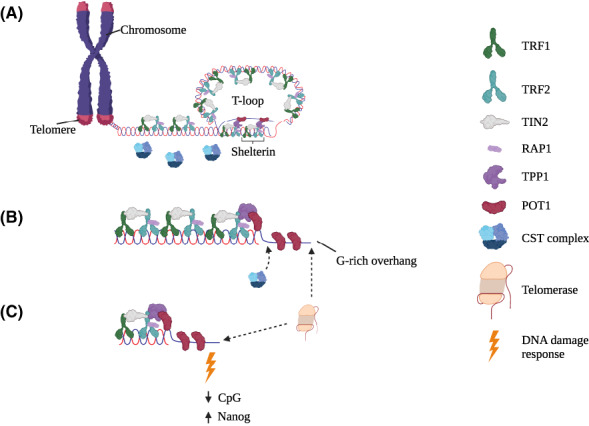
Overview of mammalian telomere structure. (A) Schematic representation of the chromosome end, or telomere in mammals. Telomeric DNA is composed of the hexanucleotide sequence TTAGGG with a single stranded G‐rich 3′ overhang that is looped back into the telomeric DNA to form a telomeric loop (T‐loop). The shelterin complex (composed of the proteins TRF1, TRF2, TIN2, RAP1, TPP1 and POT1) protects telomeres from being recognized as a DNA double‐stranded break. (B) During the S‐phase of the cell cycle, the T‐loop is opened and the G‐rich strand becomes accessible for extension by telomerase. After telomerase has extended the G‐rich strand, the CST complex (composed of the subunits CTC1, STN1, TEN1) together with the DNA replication machinery carries out fill‐in DNA synthesis of the complimentary C‐rich strand. (C) Critically short telomeres exhibit defects not only in telomere integrity (due to a DNA damage response) but can also perturb the epigenetic state (reducing CpG methylation and increasing Nanog expression). [Colour figure can be viewed at wileyonlinelibrary.com]
Fig. 2.
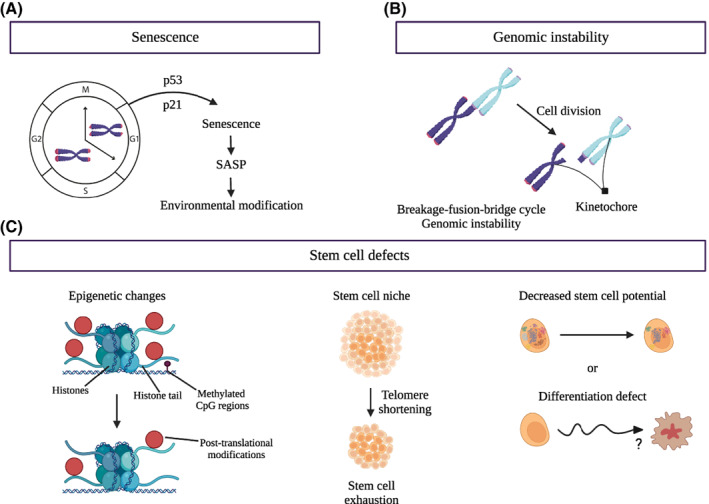
Consequences of telomere shortening. (A) Senescence is referred to as an irreversible exit from the cell cycle. This state can occur via a variety of mechanisms that include eroded telomeres. When telomeres reach a critically short threshold, cells undergo telomere‐induced (or replicative) senescence, which is triggered by p53 and its downstream effector p21, a cyclin‐dependent kinase inhibitor that arrests the cell cycle. Senescent cells can secrete factors that modify neighbouring tissues and cells and enhance aging, via a process termed the senescence‐associated secretory phenotype (SASP). (B) If a cell bypasses senescence and continues to divide, telomere fusions can occur, leading to a cycle of chromosome breakage‐fusion‐bridge formation and genetic instability. (C) Epigenetic modifications are also linked to telomere shortening. Such modifications can influence transcriptional activity and cell fate. In the left image, DNA wrapped around the histones (represented by the green spheres) may contain extensive post‐translational modifications (red sphere) on the histone tail, and the DNA itself can be methylated (small black sphere). Murine embryonic stem cells are particularly sensitive to epigenetic modifications driven by telomere shortening, which can reduce stem cell renewal potential (middle image). Telomere shortening can also affect the ability of stem cells to maintain a functional pluripotent state and/or differentiated state (right image). [Colour figure can be viewed at wileyonlinelibrary.com]
Telomere shortening is not the only telomeric damage that leads to senescence. The oxidation of telomeric DNA (to form 8‐oxoguanosine) induces senescence without telomere shortening, underlining the critical contribution of the DNA damage response in this process [61]. Moreover, cellular senescence can be induced by factors other than telomere erosion, including by persistent DNA damage across the genome, oncogene activation, loss of a tumour suppressor or other stressors, and thus critically short telomeres are not the sole hallmark of senescent cells [62]. Senescence is also triggered by two different pathways, one through p53, which is reversible, and the other through Rb, which is permanent [63, 64].
The presence of senescent cells in a tissue profoundly modifies tissue function. Senescent cells secret specific small molecules or exosomes (EV) that interact with and influence the nearby environment. This phenomenon is known more generally as the Senescence‐Associated Secretory Phenotype (SASP) and is a phenotype common to many senescent states, including that induced by eroded telomeres (please see accompanying review) [51, 52, 62, 65, 66] (Fig. 2). Among the molecules secreted by senescent cells exhibiting a SASP are pro‐inflammatory cytokines that promote the recruitment of immune cells to clear the growth‐arrested cells [67]. Senescence is thought to represent a normal process for somatic cells, as the presence of the SASP stimulates tissue regeneration by encouraging stem cell proliferation and differentiation [68, 69]. This rejuvenation process is important for ensuring tissue function, as it enables growth‐arrested somatic cells to be cleared away and replaced by newly differentiated cells.
However, the accumulation of senescent cells that arise via a telomere induced DDR or other stresses can also negatively affect nearby cells, promoting aging and tissue dysfunction. For instance, the Von Zglinicki lab has shown that the xenotransplantation of senescent cells into mouse muscle or skin induces senescence in cells near the transplantation site [70]. In addition, more recent studies have shown that the accumulation of senescent cells during aging can impact cells far from their location. EV and other microvesicles in circulation in the body carry small molecules, such as miRNA, that can affect the behavior of cells along their journey. With age, the composition of such vesicles changes. For example, microRNA‐183‐5p has been shown by Davis et al. [71] to increase in microvesicles with age, resulting in the suppression of bone marrow stromal stem cell proliferation and accompanied by senescence. These and other studies suggest that the in vivo removal or manipulation of senescent cells could have physiologically relevant benefits in ameliorating aging and age‐associated disease [72].
As senescence is generally driven by cellular dysfunction, a logical, proposed role for senescence is to suppress cancer [73, 74]. Oncogene‐induced senescence is one such example of a suppressive mechanism (reviewed in [75]). While senescence and SASP induction is tumor‐suppressive in many contexts, it can also drive tumorigenesis (reviewed in [76]). For instance, cancer incidence increases with age, concomitant with a rise in tissue senescence [64]. Among the SASP secreted molecules, VEGF and tissue inhibitors of metalloproteinases (TIMP) promote cellular migration, including the potential of cancer cells to undergo metastasis [77, 78]. Other recent studies have shown that the presence of senescent cells, whether via perturbations in the DNA damage response or loss of telomere integrity, can modify tissue homeostasis during aging, which could also act to promote cancer [79, 80].
2.2. Telomeres and stem cells
Adult stem cells remain in the body throughout life, mainly in a protected environment called the stem cell niche, and can regenerate tissues via self‐renewal and differentiation. Thus, the sustained presence of functional stem cells is essential for tissue homeostasis, and the loss of the stem cell pool can lead to disease (Fig. 2).
Despite the ongoing expression of telomerase in certain stem cell and progenitor populations, telomere lengths, on average, are shorter in cells from older individuals than in cells from younger individuals. This telomere erosion can have varying effects on stem cells, from limiting their proliferation potential to inducing severe cell dysfunction [81, 82, 83]. Perturbations in stem cell function can impact the stem cell pool that is available to rejuvenate tissue, which in turn can contribute to age‐associated issues like stem cell depletion [84]. Similar age‐associated impacts on stem cells pools have been observed in diseases driven by telomere or telomerase insufficiencies. For example, some symptoms observed in dyskeratosis congenita or pulmonary fibrosis occur due to defects in the stem cell compartment and via the loss of cellular self‐renewal potential [85, 86, 87].
In some instances, these barriers to stem cell proliferation that develop with age can lead to the senescence of adult stem cells. For example, stem cell senescence has been described in bone marrow stromal stem cells and in mesenchymal stem cells [88]. Moreover, senescence impairs pluripotent cell reprogramming [89]. This limit on stem cell proliferation with age could have implications for stem cell therapy and its use of amplified stem cells. In addition, the presence of senescent cells in the stem cell niche could induce senescence in neighboring cells or could increase inflammation in the niche [71, 90, 91, 92].
In addition to limiting their capacity to proliferate, telomere attrition in stem cells can lead to the generation of dysfunctional differentiated cells (Fig. 2). For example, in aged hematopoietic stem cells, some progenitor cells differentiate inappropriately, therefore reducing the number of functional blood cells [93]. Other studies also suggest a differentiation bias during aging, i.e. the skewing of certain subpopulations in a manner that perturbs overall tissue function and resiliency [94].
Several unanswered questions remain as to the precise mechanisms by which telomeres influence stem cell behaviour. As mentioned earlier, the arrest of cell proliferation once a critical level of telomere attrition is reached is mainly instigated by DNA damage signaling. However, the signals that regulate differentiation are more complex and are tighly regulated by epigenetic mechanisms. For example, the chromatin of stem cells is known to be open and dynamic. However, during the process of differentiation, chromatin acquires repressive marks and a closed conformation, with the exception of particular lineage‐specific genetic regulatory elements, which become decorated with chromatin marks associated with either transcriptional activity or repression [95, 96]. Telomere erosion itself is also linked with marked alterations in the epigenetic state [94, 97, 98, 99]. For example, when telomere protection or telomere erosion are perturbed in mice, genome‐wide transcriptional and epigenetic alterations ensue [82, 100, 101, 102, 103, 104, 105, 106, 107, 108]. Furthermore, during senescence, or upon the distruption of telomere maintenance or Trf1 function, associated changes in the chromatin landscape occur that are linked to the dysregulation of the chromatin modifying complex, Polycomb Repressive Complex 2 (PRC2) and/or to changes in histone or DNA methylation status [81, 82, 109].
To elaborate on one such example, in mESCs with critically eroded telomeres (via prolonged propagation in the absence of Tert), cells fail to fully consolidate a differentiated state upon treatment with a differentiation‐inducing agent (all‐trans retinoic acid, ATRA). These cells are also prone to re‐acquire a transcriptional and chromatin landscape similar to that of undifferentiated cells [81, 82]. In this experimental context, the overexpression of the DNA methyltransferase Dnmt3b or the chemical/genetic modulation of PRC2 activity partially rescues the ability of Tert −/− mESCs to differentiate in response to the appropriate cues such as ATRA [81, 82]. Telomere erosion or perturbed telomere protection complexes are also associated with genome‐wide epigenetic alterations in budding yeast [110], which underscores the evolutionary conservation of this complex interrelationship.
A better understanding of the ways in which telomere erosion is linked to the genetic and epigenetic alterations that underlie the aging process has important implications for cancer, which leads us to the topics we discuss in the next section. For example, Ideker et al. [45] have shown that the methylome of older individuals shares similarities with the methylomes of patient‐derived cancers. Cancer stem cells share important characteristics with normal stem cells, such as unlimited proliferation, self renewal and differentiation capacity. Emerging evidence suggests that some cancer types, particularly glioblastoma and acute myeloid leukemia (AML), have reverted to a more primitive cell state via genetic and epigenetic alterations that exhibit striking similarities [111]. Thus, as seen in normal stem cells, the epigenetic alterations of cancer stem cells should not necessarily be considered in isolation from other cancer‐associated characteristics, such as short telomeres [112, 113].
3. Telomeres and telomerase in cancer
As mentioned above, the co‐existence of eroded telomeres and telomerase activity in cancer cells is similar to that observed during aging of stem cells. While telomerase is re‐activated in 85% of cancers [114], telomeres are relatively short in these cells [33, 115]. The upregulation of telomerase is a common occurrence in human cancers and occurs via multiple mechanisms [116]. In 10–15% of human cancers, telomerase is not reactivated, and cancer cells can survive without telomerase, usually via mechanisms that involve alternative lengthening of telomeres (ALT) (reviewed in [117]). These alternative mechanisms are more prevalent in certain types of tumours, such as in osteosarcomas [118], soft tissue sarcomas [119], glioblastoma [120] and neuroblastoma [121]. One of the most common mechanisms of telomerase upregulation in tumours is the presence of mutations at the TERT promoter. Indeed, mutations at the TERT promoter region are the most frequent, non‐coding mutation in cancer [122]. These mutations create binding motifs for the Erythroid Transformation Specific (ETS) transcription factors, which result in increased expression of TERT [123, 124, 125, 126]. Even a modest increase in TERT expression, which results in slightly more telomerase activity, is sufficient to confer a proliferative advantage. These TERT promoter mutations are thought to act as driver mutations because they confer a fitness advantage and thus may promote or drive cancer progression. Such mutations were first identified in two unrelated cohorts of melanoma patients [123, 124] and have subsequently been identified in subsets of glioblastoma, gliomas, thyroid cancer, hepatocellular carcinomas, bladder cancers and mantle cell lymphomas [127, 128, 129, 130].
3.1. TERT expression and telomere length: clinical indicators
Rare, somatic mutations within the TERT promoter region are also found in non‐malignant diseases, such as in idiopathic pulmonary fibrosis and aplastic anemia [131, 132]. In these instances, these mutations are often found in cis to the wild‐type TERT allele, and it is thought that that compensatory upregulation of the wild‐type allele may promote somatic gene rescue [131]. As in cancer, these mutations at the TERT promoter increase TERT transcription only marginally (by twofold or less), but nonetheless this increase is still sufficient to delay replicative senescence in human fibroblasts [123, 124, 125].
Epigenetic modifications can also increase TERT expression. For example, a hypermethylated region of the TERT promoter known as THOR (TERT Hypermethylated Oncological Region) was first detected in a group of pediatric brain tumours and later shown to drive the expression of the TERT locus in various tumour types, such as in brain tumours, and in bladder, breast, colon, lung and prostate cancers [133, 134, 135]. Although present in a wide range of tumour types, this hypermethylation pattern is rare in normal tissues [135], and is considered a potential tumour biomarker. As yet, the correlation between THOR hypermethylation and prognosis remains to be elucidated. In patients with bladder cancer and melanoma, THOR hypermethylation combined with TERT promoter mutations are associated with an increased risk of tumour progression and with a decreased progression‐free survival [136, 137]. However, THOR hypermethylation does not correlate with disease progression and/or survival in medulloblastoma, esophageal cancer or in meningioma, among other tumour types (reviewed in [138]).
Another distinguishing feature of adult cancers is their relatively short average telomere lengths, which have been linked to disease outcome. For example, patients with a range of cancers (including chronic lymphocytic leukemia, breast cancer, non–small cell lung cancer, myelodysplastic syndromes and multiple myeloma), whose leukocytes have short telomeres, have an unfavourable prognosis relative to those whose telomere lengths are within the normal range (reviewed in [139]). One might thus assume that short telomeres would always be detrimental. However in the case of esophageal cancer [140], prostate cancer [141], clear renal cell carcinoma [142], melanoma [143] and hepatocellular carcinoma [144], longer telomeres are associated with a poorer prognosis, possibly related to difficulties in replicating long telomeric tracts. As Rivera and colleagues showed, long telomeres might also be detrimental to cell fitness [145]. Using human embryonic stem cells and human induced pluripotent stem cells, the group showed that long telomeres led to the formation of C‐circles (the C‐rich single‐stranded telomeric DNA complimentary to the G‐rich strand; also a common biomarker of the ALT pathway [146]). Excessively long telomeres can lead to increased sensitivity to replication stress [145], increased sensitivity to DNA damage [147] and telomere fragility [145, 148]. Altogether, these studies suggest that there exists an optimal telomere range for cancer cell fitness, that was referred to as the telomere ‘Goldilocks Zone’ [149].
3.2. Therapeutic approaches to telomerase inhibition
The effects of telomerase inhibition have been widely studied in different cell types. Telomerase inhibition leads to telomere shortening, followed by a reduction in cancer cell growth and/or apoptosis [150, 151, 152]. Even in proliferating tumour cells, genome rearrangements such as chromothripsis (numerous clustered chromosomal rearrangements that arise in a single event) and hypermutational regions (kataegis) appear to be driven, in part, by telomere instability [153, 154, 155, 156].
Unfortunately, an FDA‐approved telomerase inhibitor is not yet available. Currently, numerous approaches are being pursued that target telomerase or telomere integrity in preclinical and clinical settings, including telomerase inhibitors, the targetting of telomere replication, vaccines, and other immunological approaches (reviewed in ref. [157]). The only direct telomerase inhibitor used in clinical trials so far, Imetelstat, is a modified 13‐mer oligonucleotide that shares complementarity to the human TR (telomerase RNA component) template region [158, 159]. Imetelstat competes with the telomeric DNA substrate, leading to telomerase inhibition and consequently to telomere shortening [159]. Currently, Imetelstat is being used in phase II/III clinical trials in myelodysplastic syndromes and refractory myelofibrosis. In a phase II study in patients with intermediate/high‐risk myelofibrosis, treatment with Imetelstat exhibited a better response in patients bearing the JAK2‐V617F mutation [160]. However, the molecular mechanism behind this response is unclear, and there was no significant reduction in telomere length in the patients who exhibited a partial or complete response [160].
Another inhibitor that is not clinically approved but is widely used in preclinical models is the small molecule BIBR1532, a synthetic and non‐competitive inhibitor that shares some similarities to non‐nucleosidic inhibitors of HIV1 reverse transcriptase [161, 162]. The mechanism‐of‐action of this small molecule occurs via the non‐competitive inhibition of the catalytic site [162]. BIBR1532 binds to the C‐terminal region of TERT, in a hydrophobic motif named FVYL, that is close to the region important for the interaction of TERT with telomerase RNA (TR) [163]. BIBR1532 showed promising effects in preclinical models of solid and hematological malignancies [164, 165, 166, 167, 168]. Other telomerase inhibitors have also been found to induce telomere‐induced cell death in cell‐based models, including the nucleoside analogues azidothymidine (AZT) [169, 170] and 6‐thio‐dG [171]. Several compounds that specifically destabilize telomeric DNA have also been identified that induce apoptosis in human cancer lines, such as pyridostatin, telomestatin, BRACO‐19 and RHPS4 [172, 173, 174, 175] (Fig. 3). Also, peptide vaccines against TERT are under active investigation. One of these vaccines, called GV1001, is already in phase III clinical trials (see refs [157, 176] for a more detailed information on immunotherapies against telomerase).
Fig. 3.
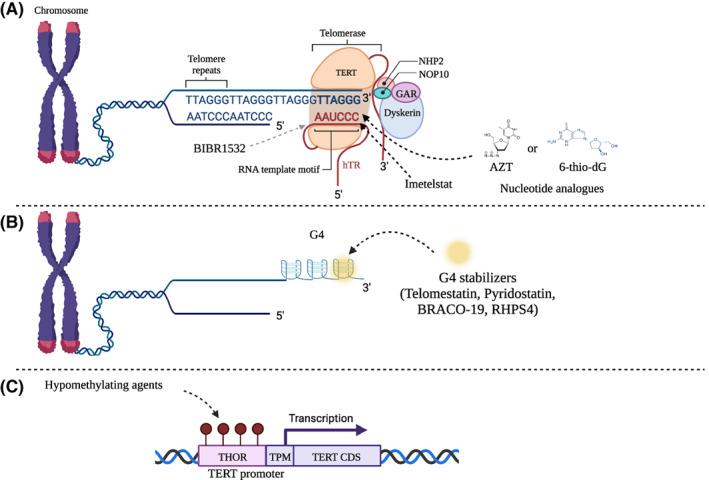
Current and emerging telomere‐targeting treatments. (A) Therapeutic strategies that target telomerase use compounds (such as Imetelstat and BIBR1532) that can directly inhibit the catalytic core of the telomerase enzyme, or they may also affect telomere DNA replication (such as the nucleotide analogue AZT or the nucleoside analogue 6‐thio‐dG). The telomeric DNA sequence is indicated (TTAGGG repeats in the 5′ to 3′ direction). Telomerase is a multi‐subunit complex that contains TERT (orange), the telomerase RNA (hTR), and associated subunits (GAR, NOP10, NHP2, and dyskerin). For simplicity only 1 telomeric repeat of the hTR template motif is shown; the full template contains 1.5 telomeric repeats (CAAUCCCAAUC) that is reverse‐transcribed in an iterative process. (B) More recently, compounds that affect telomere structure, such as G‐quadraplex (G4) stabilizers, have been investigated, as they induce immediate cell death upon telomere uncapping and do not exhibit a therapeutic lag, as is the case with telomere attrition. The telomeric G‐quadruplexes are located at the 3′ end of the G‐rich strand. The yellow circle represents the G4‐stabilizers as indicated. (C) As the expression of TERT is regulated by DNA methylation status (through the THOR region), chromatin‐modifying agents might also have therapeutic benefit in the treatment of telomerase‐positive tumours. THOR stands for TERT Hypermethylated Oncological Region. Red spheres indicate methylated CpG dinucleotides at the THOR region that may be a potential target site for the use of hypomethylating agents. TPM indicates the promoter region where the TERT promoter mutations are commonly found, upstream of the coding sequence (TERT CDS). [Colour figure can be viewed at wileyonlinelibrary.com]
3.3. Therapeutic challenges to modulation of telomerase activity
As telomere erosion is one of the hallmarks of aging and is associated with other deleterious age‐associated phenotypes [177], one strategy being considered is telomerase reactivation. The re‐expression of telomerase reverse transcriptase in normal human cells is sufficient to avert senescence and leads to the capacity to divide indefinitely, called immortalization [178, 179]. Immortalized cells can maintain a relatively stable karyotype even after prolonged culture [180, 181, 182]. However, reactivation of telomerase can be detrimental in other contexts. For example, in Tert null mice bred for successive generations until telomeres reached a critically eroded state, Tert reintroduction led to a marked increase in tumorigenesis [183]. Thus, while the reactivation of telomerase rescues phenotypes associated with aging in certain cell‐based and animal models [87, 184, 185, 186], it could have deleterious consequences if used as a therapy in older individuals who have acquired somatic mutations in blood or other tissues or in those with genetic predispositions that affect genome integrity (e.g. familial cancer syndromes).
Conversely, what are the prospects for telomerase inhibition as a viable treatment for cancer? The crystal structure of TERT shows that the enzyme has a catalytic core that is similar to HIV and should, in principle, be druggable [187, 188, 189, 190, 191]. Aside from the usual issues of bioavailability and toxicity, there are at least two challenges that arise with the clinical use of a telomerase inhibitor. The first challenge is the possibility of drug resistance. So far, no study has established whether genetic alterations in telomerase would lead to resistance to Imetelstat (currently in phase II/III clinical trials) or BIBR1532 (used in preclinical studies) [168, 192, 193, 194]. It is possible that amino acid alterations in TERT or nucleotide substitutions in hTR could promote drug resistance; however, many of these substitutions would also be likely to reduce enzyme activity [195, 196]. For example, in TERT, mutations at the FVYL pocket (the predicted binding site of BIBR1532) reduce telomerase activity [163]. In terms of disease, there are known alterations in TERT that are generally loss‐of‐function mutations and that are found in patients with telomere biology disorders, such as dyskeratosis congenita and pulmonary fibrosis [197, 198, 199]. It is worth mentioning that BIBR1532 is an allosteric inhibitor [163] and, thus, we cannot rule out the possibility that mutations outside the FVYL pocket might affect sensitivity to BIBR1532 inhibition. If resistant mutations are identified, presumably the inhibitors could be refined to act against these new variants, in the same manner as there are now later‐generation EGFR or BRAF inhibitors that specifically target their respective protein variants that led to the acquired resistant phenotype [200, 201, 202].
The second challenge is the time delay required for a telomerase inhibitor to elicit cell death or arrest, that is, the fact that several cell divisions are needed before telomere erosion triggers a DNA damage response. This is known as the lag period, and evidence suggests that initial telomere length can affect the time required to elicit cell death in response to telomerase inhibition [150, 151, 152]. In other words, tumours with short telomeres would require fewer cell doublings to trigger growth arrest or cell death. By contrast, tumours possessing longer telomeres would require a prolonged treatment period before the telomeres reach a critical threshold. In one study, the removal of telomerase from engineered human tumor cells with long telomeres did not significantly impact cell proliferation or tumour formation for several months in culture, until telomeres eventually reached critically shortened lengths [203]. This study underscores the issues surrounding the lag phase, and further raises a concern that longer periods of treatment with an inhibitor might facilitate the development of drug resistance.
The third challenge in the use of telomerase inhibitors is the checkpoint status of the cancer cells. For example, cells with critically short telomeres activate a DNA damage response and, potentially, trigger cell death (e.g. via activation of the Rb or p53 checkpoint pathway) [204, 205]. Conversely, if a tumor lacks the p53 checkpoint, the cells can continue to proliferate at the expense of genomic instability [205]. Hence, the use of telomerase inhibitors in tumors p53‐deficient could, in principle, increase the mutational burden of the tumors.
One approach to address these issues, which is now commonly employed in cancer treatment, is to combine telomerase inhibitors with inhibitors that target a gene or network whose function is critical for proliferation in the absence of telomerase. This approach of targetting synthetic‐sick‐lethal (SSL) interactions might shorten the lag period required to elicit tumour cell death upon telomere erosion and forestall the emergence of drug resistance. This approach has been used successfully in the development of poly‐ADP‐ribose polymerase (PARP) inhibitors, which are specifically lethal to tumour cells with BRCA1/2 mutations [206, 207]. Indeed, the factors to which cells with eroded telomeres are sensitized overlap with many of the other hallmarks of aging. One example of a newly identified target that is SSL with telomerase loss is the previously unannotated gene C16orf72/TAPR1 [208]. C16orf72/TAPR1 acts to taper p53 activation in response to eroded telomeres, and its loss was also identified as SSL in the presence of oxidative damage or DNA damage, and in a genome‐wide screen for deletions that sensitize cells to ATR inhibition [209, 210, 211].
Telomerase inhibitors might also be SSL with epigenetic regulators. As an example, murine stem cells with eroded telomeres are unable to stably differentiate. Inhibitors of the enzyme that deposits methyl groups on H3K27, PRC2, further exacerbate this unstable differentiation state, and inhibitors that repress the de‐methylation of H3K27me3 partially rescue the unstable differentiation phenotype [82]. Thus, one might imagine that treatments that target the epigenetic vulnerabilities of cancer, many of which are now in clinical trial, could be effective in combination with telomerase inhibitors. The only exception would be tumours that do not rely on telomerase but instead use ALT, in which case telomere replication fidelity or stability could be targeted rather than telomerase itself.
4. Conclusions and perspectives
Evolutionary studies in multiple organisms, including mammals, have established that telomerase and telomere‐associated factors are under positive selective pressure [212, 213, 214, 215, 216, 217]. Scientists examining this question posit that such selective pressures could arise as a result of adaptive mechanisms [217, 218]. This is also a relevant question with respect to age‐associated diseases, including cancer, whose incidence increases later in life after the reproductive period. Thus, scientists have pondered whether short telomeres later in life are the result of antagonistic pleitropy (i.e. a trait under selective pressure to be beneficial earlier in life, that later in life that becomes deleterious) [219], or of other adaptive measures independent of early‐life selection. Interesting models have been proposed to account for how mammals have evolved to cope with counterselective pressures (e.g. long versus short telomeres) in relation to aging and cancer [73]. In this post‐genomics era, there will now be an opportunity to put some of these theories to test at the molecular level, and to determine how the modulation of these selective pressures during cell/tissue aging and cancer can be exploited therapeutically to ameliorate disease.
We are at a watershed in our understanding of the molecular underpinnings of aging and cancer. The ongoing challenge is to understand how to best apply these new insights in the diagnosis, treatment, and management of cancer and other age‐associated diseases. Indeed, age is the largest single risk factor for cancer (cruk.org/cancerstats). Therefore, interventions that ameliorate age‐associated disease must be considered carefully in the context of cancer incidence and prognosis. These questions are at the nexus of the challenges and opportunities that lied ahead for those pursuing the mechanisms of telomere integrity, aging and cancer.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
GB, MC and LH wrote and edited the manuscript. GB, with input from MC, generated the figures, which were edited by LH.
Acknowledgements
We thank Jane Alfred and anonymous reviewers for their constructive input on this review. Funding to LH is provided by the Canadian Institutes of Health Research and GB was supported by a scholarship from the FRQ‐S. Figures were generated using BioRender (www.biorender.com).
Gustavo Borges and Mélanie Criqui contributed equally to this article
References
- 1. Varela E, Munoz‐Lorente MA, Tejera AM, Ortega S, Blasco MA. Generation of mice with longer and better preserved telomeres in the absence of genetic manipulations. Nat Commun. 2016;7:11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calado RT, Dumitriu B. Telomere dynamics in mice and humans. Semin Hematol. 2013;50:165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meyne J, Ratliff RL, Moyzis RK. Conservation of the human telomere sequence (TTAGGG)(n) among vertebrates. Proc Natl Acad Sci USA. 1989;86:7049–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peska V, Garcia S. Origin, diversity, and evolution of telomere sequences in plants. Front Plant Sci. 2020;11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wellinger RJ, Zakian VA. Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. Genetics. 2012;191:1073–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wicky C, Villeneuve AM, Lauper N, Codourey L, Tobler H, Müller F. Telomeric repeats (TTAGGC)n are sufficient for chromosome capping function in Caenorhabditis elegans . Proc Natl Acad Sci USA. 1996;93:8983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–14. [DOI] [PubMed] [Google Scholar]
- 8. Stansel RM, de Lange T, Griffith JD. T‐loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang. EMBO J. 2001;20:5532–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Timashev LA, De Lange T. Characterization of t‐loop formation by TRF2. Nucleus. 2020;11:164–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tomaska L, Nosek J, Kar A, Willcox S, Griffith JD. A new view of the T‐loop junction: implications for self‐primed telomere extension, expansion of disease‐related nucleotide repeat blocks, and telomere evolution. Front Genet. 2019;10:792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–10. [DOI] [PubMed] [Google Scholar]
- 12. Liu D, O'Connor MS, Qin J, Songyang Z. Telosome, a mammalian telomere‐associated complex formed by multiple telomeric proteins. J Biol Chem. 2004;279:51338–42. [DOI] [PubMed] [Google Scholar]
- 13. Gao H, Cervantes RB, Mandell EK, Otero JH, Lundblad V. RPA‐like proteins mediate yeast telomere function. Nat Struct Mol Biol. 2007;14:208–14. [DOI] [PubMed] [Google Scholar]
- 14. Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, et al. RPA‐like mammalian Ctc1‐Stn1‐Ten1 complex binds to single‐stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell. 2009;36:193–206. [DOI] [PubMed] [Google Scholar]
- 15. Surovtseva YV, Churikov D, Boltz KA, Song X, Lamb JC, Warrington R, et al. Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol Cell. 2009;36:207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen LY, Redon S, Lingner J. The human CST complex is a terminator of telomerase activity. Nature. 2012;488:540–4. [DOI] [PubMed] [Google Scholar]
- 17. Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, et al. 53BP1‐RIF1‐shieldin counteracts DSB resection through CST‐ and Polalpha‐dependent fill‐in. Nature. 2018;560:112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cicconi A, Chang S. Shelterin and the replisome: at the intersection of telomere repair and replication. Curr Opin Genet Dev. 2020;60:77–84. [DOI] [PubMed] [Google Scholar]
- 19. Sfeir A, De Lange T. Removal of shelterin reveals the telomere end‐protection problem. Science. 2012;336:593–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shubin CB, Mayangsari R, Swett AD, Greider CW. Rif1 regulates telomere length through conserved HEAT repeats. Nucleic Acids Res. 2021;49:3967–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shubin CB, Greider CW. The role of rif1 in telomere length regulation is separable from its role in origin firing. Elife. 2020;9:1–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greider CW, Harley B, H. & Futcher, A. B. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60. [DOI] [PubMed] [Google Scholar]
- 23. Sholes SL, Karimian K, Gershman A, Kelly TJ, Timp W, Greider CW. Chromosome specific telomere lengths and the minimal functional telomere revealed by nanopore sequencing. Genome Res. 2021;32:616–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972;239:197–201. [DOI] [PubMed] [Google Scholar]
- 25. Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973;41:181–90. [DOI] [PubMed] [Google Scholar]
- 26. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. [DOI] [PubMed] [Google Scholar]
- 27. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60. [DOI] [PubMed] [Google Scholar]
- 28. Daniali L, Benetos A, Susser E, Kark JD, Labat C, Kimura M, et al. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat Commun. 2013;4:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Szostak JW. Telomerase: the beginning of the ends. Nature. 1989;337:303–4. [DOI] [PubMed] [Google Scholar]
- 30. Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–9. [DOI] [PubMed] [Google Scholar]
- 31. Shippen‐Lentz D, Blackburn EH. Functional evidence for an RNA template in telomerase. Science. 1990;247:546–52. [DOI] [PubMed] [Google Scholar]
- 32. Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB, Lansdorp PM. Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc Natl Acad Sci USA. 1994;91:9857–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zimmermann S, Martens UM. Telomeres, senescence, and hematopoietic stem cells. Cell Tissue Res. 2008;331:79–90. [DOI] [PubMed] [Google Scholar]
- 34. López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gorbunova V, Seluanov A. Coevolution of telomerase activity and body mass in mammals: from mice to beavers. Mech Ageing Dev. 2009;130:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lorenzini A, Johnson FB, Oliver A, Tresini M, Smith JS, Hdeib M, et al. Significant correlation of species longevity with DNA double strand break recognition but not with telomere length. Mech Ageing Dev. 2009;130:784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blasco MA. Mice with bad ends: mouse models for the study of telomeres and telomerase in cancer and aging. EMBO J. 2005;24:1095–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Munoz‐Lorente MA, Cano‐Martin AC, Blasco MA. Mice with hyper‐long telomeres show less metabolic aging and longer lifespans. Nat Commun. 2019;10:4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Buddhachat K, Brown JL, Kaewkool M, Poommouang A, Kaewmong P, Kittiwattanawong K, et al. Life expectancy in marine mammals is unrelated to telomere length but is associated with body size. Front Genet. 2021;12:737860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Froy H, Bird EJ, Wilbourn RV, Fairlie J, Underwood SL, Salvo‐Chirnside E, et al. No evidence for parental age effects on offspring leukocyte telomere length in free‐living Soay sheep. Sci Rep. 2017;7:9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Froy H, Underwood SL, Dorrens J, Seeker LA, Watt K, Wilbourn RV, et al. Heritable variation in telomere length predicts mortality in Soay sheep. Proc Natl Acad Sci USA. 2021;118:e2020563118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gardner M, Bann D, Wiley L, Cooper R, Hardy R, Nitsch D, et al. Gender and telomere length: systematic review and meta‐analysis. Exp Gerontol. 2014;51:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Epel ES, Lin J, Wilhelm FH, Wolkowitz OM, Cawthon R, Adler NE, et al. Cell aging in relation to stress arousal and cardiovascular disease risk factors. Psychoneuroendocrinology. 2006;31:277–87. [DOI] [PubMed] [Google Scholar]
- 44. Epel ES, Merkin SS, Cawthon R, Blackburn EH, Adler NE, Pletcher MJ, et al. The rate of leukocyte telomere shortening predicts mortality from cardiovascular disease in elderly men. Aging (Albany NY). 2008;1:81–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome‐wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA. 1992;89:10114–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu AT, Seeboth A, Tsai PC, Sun D, Quach A, Reiner AP, et al. DNA methylation‐based estimator of telomere length. Aging. 2019;11:5895–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marioni RE, Harris SE, Shah S, McRae AF, von Zglinicki T, Martin‐Ruiz C, et al. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int J Epidemiol. 2016;45:424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. 2019;179:813–27. [DOI] [PubMed] [Google Scholar]
- 51. Wang C, Hao X, Zhang R. Targeting cellular senescence to combat cancer and ageing. Mol Oncol. 2022;16(18):3319–32. 10.1002/1878-0261.13266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Takasugi M, Yoshida Y, Ohtani N. Cellular senescence and the tumour microenvironment. Mol Oncol. 2022;16(18):3333–51. 10.1002/1878-0261.13268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abdallah P, Luciano P, Runge KW, Lisby M, Géli V, Gilson E, et al. A two‐step model for senescence triggered by a single critically short telomere. Nat Cell Biol. 2009;11:988–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eugène S, Bourgeron T, Xu Z. Effects of initial telomere length distribution on senescence onset and heterogeneity. J Theor Biol. 2017;413:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Capper R, Britt‐Compton B, Tankimanova M, Rowson J, Letsolo B, Man S, et al. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21:2495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sandell LL, Zakian VA. Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell. 1993;75:729–39. [DOI] [PubMed] [Google Scholar]
- 57. Sabatier L, Ricoul M, Pottier G, Murnane JP. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol Cancer Res. 2005;3:139–50. [DOI] [PubMed] [Google Scholar]
- 58. Hemann MT, Hackett J, IJpma A, Greider CW. Telomere length, telomere‐binding proteins, and DNA damage signaling. Cold Spring Harb Symp Quant Biol. 2000;65:275–9. [DOI] [PubMed] [Google Scholar]
- 59. Meznikova M, Erdmann N, Allsopp R, Harrington LA. Telomerase reverse transcriptase‐dependent telomere equilibration mitigates tissue dysfunction in mTert heterozygotes. Dis Model Mech. 2009;2:620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goldman F, Bouarich R, Kulkarni S, Freeman S, Du HY, Harrington L, et al. The effect of TERC haploinsufficiency on the inheritance of telomere length. Proc Natl Acad Sci USA. 2005;102:17119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barnes RP, de Rosa M, Thosar SA, Detwiler AC. Telomeric 8‐oxoguanine drives rapid premature senescence in the absence of telomere shortening. bioRxiv 2021; 10.1101/2021.05.05.442662 [PREPRINT] [DOI] [PMC free article] [PubMed]
- 62. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. 2021;9:645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–22. [DOI] [PubMed] [Google Scholar]
- 64. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Malaquin N, Tu V, Rodier F. Assessing functional roles of the senescence‐associated secretory phenotype (SASP). Methods Mol Biol. 2019;1896:45–55. [DOI] [PubMed] [Google Scholar]
- 67. Lujambio A. To clear, or not to clear (senescent cells)? That is the question. Inside the Cell. 2016;1:87–95. [Google Scholar]
- 68. Da Silva‐Álvarez S, Guerra‐Varela J, Sobrido‐Cameán D, Quelle A, Barreiro‐Iglesias A, Sánchez L, et al. Cell senescence contributes to tissue regeneration in zebrafish. Aging Cell. 2020;19:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence‐associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31:172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. da Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell. 2019;18:e12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Davis C, Dukes A, Drewry M, Helwa I, Johnson MH, Isales CM, et al. MicroRNA‐183‐5p increases with age in bone‐derived extracellular vesicles, suppresses bone marrow stromal (stem) cell proliferation, and induces stem cell senescence. Tissue Eng Part A. 2017;23:1231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gasek NS, Kuchel GA, Kirkland JL, Xu M. Strategies for targeting senescent cells in human disease. Nat Aging. 2021;1:870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Risques RA, Promislow DEL. All's well that ends well: why large species have short telomeres. Philos Trans R Soc Lond B Biol Sci. 2018;373:20160448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell. 2007;11:461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Paramos‐de‐Carvalho D, Jacinto A, Saude L. The right time for senescence. Elife. 2021;10:e72449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cuollo L, Antonangeli F, Santoni A, Soriani A. The senescence‐associated secretory phenotype (SASP) in the challenging future of cancer therapy and age‐related diseases. Biology (Basel). 2020;9:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–74. [DOI] [PubMed] [Google Scholar]
- 78. Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–77. [DOI] [PubMed] [Google Scholar]
- 79. Ghadaouia S, Olivier MA, Martinez A, Kientega T, Qin J, Lambert‐Lanteigne P, et al. Homologous recombination‐mediated irreversible genome damage underlies telomere‐induced senescence. Nucleic Acids Res. 2021;49:11690–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fleury H, Malaquin N, Tu V, Gilbert S, Martinez A, Olivier MA, et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat Commun. 2019;10:2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pucci F, Gardano L, Harrington L. Short telomeres in ESCs lead to unstable differentiation. Cell Stem Cell. 2013;12:479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Criqui M, Qamra A, Chu TW, Sharma M, Tsao J, Henry D, et al. Telomere dysfunction cooperates with epigenetic alterations to impair murine embryonic stem cell fate commitment. Elife. 2020;9:e47333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Martinez P, Blasco MA. Telomere‐driven diseases and telomere‐targeting therapies. J Cell Biol. 2017;216:875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, et al. Longevity, stress response, and cancer in aging telomerase‐deficient mice. Cell. 1999;96:701–12. [DOI] [PubMed] [Google Scholar]
- 85. Armanios M. Telomeres and age‐related disease: how telomere biology informs clinical paradigms. J Clin Invest. 2013;123:996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Batista LFZ, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, et al. Telomere shortening and loss of self‐renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011;474:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase‐deficient mice. Nature. 2011;469:102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Li Y, Wu Q, Wang Y, Li L, Bu H, Bao J. Senescence of mesenchymal stem cells (Review). Int J Mol Med. 2017;39:775–82. [DOI] [PubMed] [Google Scholar]
- 89. Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cárdenes N, Álvarez D, Sellarés J, Peng Y, Corey C, Wecht S, et al. Senescence of bone marrow‐derived mesenchymal stem cells from patients with idiopathic pulmonary fibrosis. Stem Cell Res Ther. 2018;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kornienko JS, Smirnova IS, Pugovkina NA, Ivanova JS, Shilina MA, Grinchuk TM, et al. High doses of synthetic antioxidants induce premature senescence in cultivated mesenchymal stem cells. Sci Rep. 2019;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu J, Ding Y, Liu Z, Liang X. Senescence in mesenchymal stem cells: functional alterations, molecular mechanisms, and rejuvenation strategies. Front Cell Dev Biol. 2020;8:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Udroiu I, Sgura A. Growing and aging of hematopoietic stem cells. World J Stem Cells. 2021;13:594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Brunet A, Rando TA. Interaction between epigenetic and metabolism in aging stem cells. Curr Opin Cell Biol. 2017;45:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Klein DC, Hainer SJ. Chromatin regulation and dynamics in stem cells. Curr Top Dev Biol. 2020;138:1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Martello G, Smith A. The nature of embryonic stem cells. Annu Rev Cell Dev Biol. 2014;30:647–75. [DOI] [PubMed] [Google Scholar]
- 97. Adelman ER, Huang HT, Roisman A, Olsson A, Colaprico A, Qin T, et al. Aging human hematopoietic stem cells manifest profound epigenetic reprogramming of enhancers that may predispose to leukemia. Cancer Discov. 2019;9:1080–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kane AE, Sinclair DA. Epigenetic changes during aging and their reprogramming potential. Crit Rev Biochem Mol Biol. 2019;54:61–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rando TA, Chang HY. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell. 2012;148:46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Platt JM, Ryvkin P, Wanat JJ, Donahue G, Ricketts MD, Barrett SP, et al. Rap1 relocalization contributes to the chromatin‐mediated gene expression profile and pace of cell senescence. Genes Dev. 2013;27:1406–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sen P, Lan Y, Li CY, Sidoli S, Donahue G, Dou Z, et al. Histone acetyltransferase p300 induces de novo super‐enhancers to drive cellular senescence. Mol Cell. 2019;73:684–698 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Barry RM, Sacco O, Mameri A, Stojaspal M, Kartsonis W, Shah P, et al. Rap1 regulates TIP60 function during fate transition between two‐cell‐like and pluripotent states. Genes Dev. 2022;36:313–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yeung F, Ramirez CM, Mateos‐Gomez PA, Pinzaru A, Ceccarini G, Kabir S, et al. Nontelomeric role for Rap1 in regulating metabolism and protecting against obesity. Cell Rep. 2013;3:1847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ferrara‐Romeo I, Martinez P, Blasco MA. Mice lacking RAP1 show early onset and higher rates of DEN‐induced hepatocellular carcinomas in female mice. PLoS One. 2018;13:e0204909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Martinez P, Gomez‐Lopez G, Garcia F, Mercken E, Mitchell S, Flores JM, et al. RAP1 protects from obesity through its extratelomeric role regulating gene expression. Cell Rep. 2013;3:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Martinez P, Gomez‐Lopez G, Pisano DG, Flores JM, Blasco MA. A genetic interaction between RAP1 and telomerase reveals an unanticipated role for RAP1 in telomere maintenance. Aging Cell. 2016;15:1113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Martinez P, Thanasoula M, Carlos AR, Gomez‐Lopez G, Tejera AM, Schoeftner S, et al. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol. 2010;12:768–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Marion RM, Montero JJ, Lopez de Silanes I, Grana‐Castro O, Martinez P, Schoeftner S, et al. TERRA regulate the transcriptional landscape of pluripotent cells through TRF1‐dependent recruitment of PRC2. Elife. 2019;8:e44656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ito T, Teo YV, Evans SA, Neretti N, Sedivy JM. Regulation of cellular senescence by polycomb chromatin modifiers through distinct DNA damage‐ and histone methylation‐dependent pathways. Cell Rep. 2018;22:3480–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Song S, Johnson FB. Epigenetic mechanisms impacting aging: a focus on histone levels and telomeres. Genes (Basel). 2018;9:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Goethe E, Carter BZ, Rao G, Pemmaraju N. Glioblastoma and acute myeloid leukemia: malignancies with striking similarities. J Neurooncol. 2018;136:223–31. [DOI] [PubMed] [Google Scholar]
- 112. Wainwright EN, Scaffidi P. Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer. 2017;3:372–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shukla S, Meeran SM. Epigenetics of cancer stem cells: pathways and therapeutics. Biochim Biophys Acta. 2014;1840:3494–502. [DOI] [PubMed] [Google Scholar]
- 114. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–5. [DOI] [PubMed] [Google Scholar]
- 115. Telomeres Mendelian Randomization Collaboration , Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, et al. Association between telomere length and risk of cancer and non‐neoplastic diseases: a Mendelian randomization study. JAMA Oncol. 2017;3:636–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Barthel FP, Wei W, Tang M, Martinez‐Ledesma E, Hu X, Amin SB, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49:349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Claude E, Decottignies A. Telomere maintenance mechanisms in cancer: telomerase, ALT or lack thereof. Curr Opin Genet Dev. 2020;60:1–8. [DOI] [PubMed] [Google Scholar]
- 118. Bryan TM, Englezou A, Dalla‐Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor‐derived cell lines. Nat Med. 1997;3:1271–4. [DOI] [PubMed] [Google Scholar]
- 119. Johnson JE, Varkonyi RJ, Schwalm J, Cragle R, Klein‐Szanto A, Patchefsky A, et al. Multiple mechanisms of telomere maintenance exist in liposarcomas. Clin Cancer Res. 2005;11:5347–55. [DOI] [PubMed] [Google Scholar]
- 120. Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 2005;11:217–25. [PubMed] [Google Scholar]
- 121. Onitake Y, Hiyama E, Kamei N, Yamaoka H, Sueda T, Hiyama K. Telomere biology in neuroblastoma: telomere binding proteins and alternative strengthening of telomeres. J Pediatr Surg. 2009;44:2258–66. [DOI] [PubMed] [Google Scholar]
- 122. Mularoni L, Sabarinathan R, Deu‐Pons J, Gonzalez‐Perez A, Lopez‐Bigas N. OncodriveFML: a general framework to identify coding and non‐coding regions with cancer driver mutations. Genome Biol. 2016;17:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61. [DOI] [PubMed] [Google Scholar]
- 124. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer‐associated TERT promoter mutations abrogate telomerase silencing. Elife. 2015;4:e07918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The chromatin accessibility landscape of primary human cancers. Science. 2018;362:eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK, et al. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer. 2013;20:603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self‐renewal. Proc Natl Acad Sci USA. 2013;110:6021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac‐Sage P, Laurent C, et al. High frequency of telomerase reverse‐transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Panero J, Alves‐Paiva RM, Roisman A, Santana‐Lemos BA, Falcao RP, Oliveira G, et al. Acquired TERT promoter mutations stimulate TERT transcription in mantle cell lymphoma. Am J Hematol. 2016;91:481–5. [DOI] [PubMed] [Google Scholar]
- 131. Maryoung L, Yue Y, Young A, Newton CA, Barba C, van Oers NS, et al. Somatic mutations in telomerase promoter counterbalance germline loss‐of‐function mutations. J Clin Invest. 2017;127:982–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Gutierrez‐Rodrigues F, Donaires FS, Pinto A, Vicente A, Dillon LW, Cle DV, et al. Pathogenic TERT promoter variants in telomere diseases. Genet Med. 2019;21:1594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Castelo‐Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol. 2013;14:534–42. [DOI] [PubMed] [Google Scholar]
- 134. Lee DD, Leao R, Komosa M, Gallo M, Zhang CH, Lipman T, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest. 2019;129:1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lee DD, Komosa M, Sudhaman S, Leao R, Zhang CH, Apolonio JD, et al. Dual role of allele‐specific DNA hypermethylation within the TERT promoter in cancer. J Clin Invest. 2021;131:e146915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Leao R, Lee D, Figueiredo A, Hermanns T, Wild P, Komosa M, et al. Combined genetic and epigenetic alterations of the TERT promoter affect clinical and biological behavior of bladder cancer. Int J Cancer. 2019;144:1676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Seynnaeve B, Lee S, Borah S, Park Y, Pappo A, Kirkwood JM, et al. Genetic and epigenetic alterations of TERT are associated with inferior outcome in adolescent and young adult patients with melanoma. Sci Rep. 2017;7:45704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Lee DD, Komosa M, Nunes NM, Tabori U. DNA methylation of the TERT promoter and its impact on human cancer. Curr Opin Genet Dev. 2020;60:17–24. [DOI] [PubMed] [Google Scholar]
- 139. Cleal K, Norris K, Baird D. Telomere length dynamics and the evolution of cancer genome architecture. Int J Mol Sci. 2018;19:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lv Y, Zhang Y, Li X, Ren X, Wang M, Tian S, et al. Long telomere length predicts poor clinical outcome in esophageal cancer patients. Pathol Res Pract. 2017;213:113–8. [DOI] [PubMed] [Google Scholar]
- 141. Svenson U, Roos G, Wikstrom P. Long leukocyte telomere length in prostate cancer patients at diagnosis is associated with poor metastasis‐free and cancer‐specific survival. Tumour Biol. 2017;39:1010428317692236. [DOI] [PubMed] [Google Scholar]
- 142. Svenson U, Ljungberg B, Roos G. Telomere length in peripheral blood predicts survival in clear cell renal cell carcinoma. Cancer Res. 2009;69:2896–901. [DOI] [PubMed] [Google Scholar]
- 143. Llorca‐Cardenosa MJ, Pena‐Chilet M, Mayor M, Gomez‐Fernandez C, Casado B, Martin‐Gonzalez M, et al. Long telomere length and a TERT‐CLPTM1 locus polymorphism association with melanoma risk. Eur J Cancer. 2014;50:3168–77. [DOI] [PubMed] [Google Scholar]
- 144. Liu HQ, An JZ, Liu J, Yang YF, Zhang HX, Zhao BY, et al. Leukocyte telomere length predicts overall survival in hepatocellular carcinoma treated with transarterial chemoembolization. Carcinogenesis. 2012;33:1040–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rivera T, Haggblom C, Cosconati S, Karlseder J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat Struct Mol Biol. 2017;24:30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Henson JD, Reddel RR. Assaying and investigating alternative lengthening of telomeres activity in human cells and cancers. FEBS Lett. 2010;584:3800–11. [DOI] [PubMed] [Google Scholar]
- 147. Fairlie J, Harrington L. Enforced telomere elongation increases the sensitivity of human tumour cells to ionizing radiation. DNA Repair (Amst). 2015;25:54–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Harrington L, Pucci F. In medio stat virtus: unanticipated consequences of telomere dysequilibrium. Philos Trans R Soc Lond B Biol Sci. 2018;373:20160444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhang X, Mar V, Zhou W, Harrington L, Robinson MO. Telomere shortening and apoptosis in telomerase‐inhibited human tumor cells. Genes Dev. 1999;13:2388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Herbert B, Pitts AE, Baker SI, Hamilton SE, Wright WE, Shay JW, et al. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc Natl Acad Sci USA. 1999;96:14276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, et al. Inhibition of telomerase limits the growth of human cancer cells. Nat Med. 1999;5:1164–70. [DOI] [PubMed] [Google Scholar]
- 153. Cleal K, Jones RE, Grimstead JW, Hendrickson EA, Baird DM. Chromothripsis during telomere crisis is independent of NHEJ, and consistent with a replicative origin. Genome Res. 2019;29:737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Cortes‐Ciriano I, Lee JJ, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole‐genome sequencing. Nat Genet. 2020;52:331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Maciejowski J, Chatzipli A, Dananberg A, Chu K, Toufektchan E, Klimczak LJ, et al. APOBEC3‐dependent kataegis and TREX1‐driven chromothripsis during telomere crisis. Nat Genet. 2020;52:884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Guterres AN, Villanueva J. Targeting telomerase for cancer therapy. Oncogene. 2020;39:5811–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Herbert BS, Pongracz K, Shay JW, Gryaznov SM. Oligonucleotide N3′‐‐>P5′ phosphoramidates as efficient telomerase inhibitors. Oncogene. 2002;21:638–42. [DOI] [PubMed] [Google Scholar]
- 159. Asai A, Oshima Y, Yamamoto Y, Uochi TA, Kusaka H, Akinaga S, et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003;63:3931–9. [PubMed] [Google Scholar]
- 160. Tefferi A, Lasho TL, Begna KH, Patnaik MM, Zblewski DL, Finke CM, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373:908–19. [DOI] [PubMed] [Google Scholar]
- 161. Damm K, Hemmann U, Garin‐Chesa P, Hauel N, Kauffmann I, Priepke H, et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001;20:6958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Pascolo E, Wenz C, Lingner J, Hauel N, Priepke H, Kauffmann I, et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non‐nucleosidic drug candidate. J Biol Chem. 2002;277:15566–72. [DOI] [PubMed] [Google Scholar]
- 163. Bryan C, Rice C, Hoffman H, Harkisheimer M, Sweeney M, Skordalakes E. Structural basis of telomerase inhibition by the highly specific BIBR1532. Structure. 2015;23:1934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ward RJ, Autexier C. Pharmacological telomerase inhibition can sensitize drug‐resistant and drug‐sensitive cells to chemotherapeutic treatment. Mol Pharmacol. 2005;68:779–86. [DOI] [PubMed] [Google Scholar]
- 165. El Daly H, Martens UM. Telomerase inhibition and telomere targeting in hematopoietic cancer cell lines with small non‐nucleosidic synthetic compounds (BIBR1532). Methods Mol Biol. 2007;405:47–60. [DOI] [PubMed] [Google Scholar]
- 166. Roth A, Durig J, Himmelreich H, Bug S, Siebert R, Duhrsen U, et al. Short telomeres and high telomerase activity in T‐cell prolymphocytic leukemia. Leukemia. 2007;21:2456–62. [DOI] [PubMed] [Google Scholar]
- 167. Parsch D, Brassat U, Brummendorf TH, Fellenberg J. Consequences of telomerase inhibition by BIBR1532 on proliferation and chemosensitivity of chondrosarcoma cell lines. Cancer Invest. 2008;26:590–6. [DOI] [PubMed] [Google Scholar]
- 168. Liu W, Yin Y, Wang J, Shi B, Zhang L, Qian D, et al. Kras mutations increase telomerase activity and targeting telomerase is a promising therapeutic strategy for Kras‐mutant NSCLC. Oncotarget. 2017;8:179–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Strahl C, Blackburn EH. The effects of nucleoside analogs on telomerase and telomeres in Tetrahymena. Nucleic Acids Res. 1994;22:893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Strahl C, Blackburn EH. Effects of reverse transcriptase inhibitors on telomere length and telomerase activity in two immortalized human cell lines. Mol Cell Biol. 1996;16:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Mender I, Gryaznov S, Dikmen ZG, Wright WE, Shay JW. Induction of telomere dysfunction mediated by the telomerase substrate precursor 6‐thio‐2'‐deoxyguanosine. Cancer Discov. 2015;5:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Muller S, Sanders DA, Di Antonio M, Matsis S, Riou JF, Rodriguez R, et al. Pyridostatin analogues promote telomere dysfunction and long‐term growth inhibition in human cancer cells. Org Biomol Chem. 2012;10:6537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Tauchi T, Shin‐ya K, Sashida G, Sumi M, Okabe S, Ohyashiki JH, et al. Telomerase inhibition with a novel G‐quadruplex‐interactive agent, telomestatin: in vitro and in vivo studies in acute leukemia. Oncogene. 2006;25:5719–25. [DOI] [PubMed] [Google Scholar]
- 174. Burger AM, Dai F, Schultes CM, Reszka AP, Moore MJ, Double JA, et al. The G‐quadruplex‐interactive molecule BRACO‐19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res. 2005;65:1489–96. [DOI] [PubMed] [Google Scholar]
- 175. Salvati E, Leonetti C, Rizzo A, Scarsella M, Mottolese M, Galati R, et al. Telomere damage induced by the G‐quadruplex ligand RHPS4 has an antitumor effect. J Clin Invest. 2007;117:3236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zanetti M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat Rev Clin Oncol. 2017;14:115–28. [DOI] [PubMed] [Google Scholar]
- 177. Chakravarti D, LaBella KA, DePinho RA. Telomeres: history, health, and hallmarks of aging. Cell. 2021;184:306–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998;8:279–82. [DOI] [PubMed] [Google Scholar]
- 179. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life‐span by introduction of telomerase into normal human cells. Science. 1998;279:349–52. [DOI] [PubMed] [Google Scholar]
- 180. Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A, et al. Human endothelial cell life extension by telomerase expression. J Biol Chem. 1999;274:26141–8. [DOI] [PubMed] [Google Scholar]
- 181. Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, et al. Absence of cancer‐associated changes in human fibroblasts immortalized with telomerase. Nat Genet. 1999;21:115–8. [DOI] [PubMed] [Google Scholar]
- 182. Vaziri H, Squire JA, Pandita TK, Bradley G, Kuba RM, Zhang H, et al. Analysis of genomic integrity and p53‐dependent G1 checkpoint in telomerase‐induced extended‐life‐span human fibroblasts. Mol Cell Biol. 1999;19:2373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost‐Alimova M, Protopopov A, et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell. 2012;148:896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Le Saux CJ, Davy P, Brampton C, Ahuja SS, Fauce S, Shivshankar P, et al. A novel telomerase activator suppresses lung damage in a murine model of idiopathic pulmonary fibrosis. PLoS One. 2013;8:e58423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Gourronc FA, Robertson mM, Herrig AK, Lansdorp PM, Goldman FD, Klingelhutz AJ. Proliferative defects in dyskeratosis congenita skin keratinocytes are corrected by expression of the telomerase reverse transcriptase, TERT, or by activation of endogenous telomerase through expression of papillomavirus E6/E7 or the telomerase RNA component, TERC. Exp Dermatol. 2010;19:279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Westin ER, Chavez E, Lee KM, Gourronc FA, Riley S, Lansdorp PM, et al. Telomere restoration and extension of proliferative lifespan in dyskeratosis congenita fibroblasts. Aging Cell. 2007;6:383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem. 1999;274:17395–8. [DOI] [PubMed] [Google Scholar]
- 188. Sauerwald A, Sandin S, Cristofari G, Scheres SH, Lingner J, Rhodes D. Structure of active dimeric human telomerase. Nat Struct Mol Biol. 2013;20:454–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Ghanim GE, Fountain AJ, van Roon AM, Rangan R, Das R, Collins K, et al. Structure of human telomerase holoenzyme with bound telomeric DNA. Nature. 2021;593:449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Nguyen THD, Tam J, Wu RA, Greber BJ, Toso D, Nogales E, et al. Cryo‐EM structure of substrate‐bound human telomerase holoenzyme. Nature. 2018;557:190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Liu B, He Y, Wang Y, Song H, Zhou ZH, Feigon J. Structure of active human telomerase with telomere shelterin protein TPP1. Nature. 2022;604:578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Mascarenhas J, Harrison CN, Kiladjian JJ, Komrokji RS, Koschmieder S, Vannucchi AM, et al. Imetelstat in intermediate‐2 or high‐risk myelofibrosis refractory to JAK inhibitor: IMpactMF phase III study design. Future Oncol. 2022;18:2393–402. [DOI] [PubMed] [Google Scholar]
- 193. Mascarenhas J, Komrokji RS, Palandri F, Martino B, Niederwieser D, Reiter A, et al. Randomized, single‐blind, multicenter phase II study of two doses of imetelstat in relapsed or refractory myelofibrosis. J Clin Oncol. 2021;39:2881–92. [DOI] [PubMed] [Google Scholar]
- 194. Dogan F, Ozates NP, Bagca BG, Abbaszadeh Z, Sogutlu F, Gasimli R, et al. Investigation of the effect of telomerase inhibitor BIBR1532 on breast cancer and breast cancer stem cells. J Cell Biochem. 2018. 10.1002/jcb.27089 [Online ahead of print]. [DOI] [PubMed] [Google Scholar]
- 195. Marusic L, Anton M, Tidy A, Wang P, Villeponteau B, Bacchetti S. Reprogramming of telomerase by expression of mutant telomerase RNA template in human cells leads to altered telomeres that correlate with reduced cell viability. Mol Cell Biol. 1997;17:6394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Guiducci C, Cerone MA, Bacchetti S. Expression of mutant telomerase in immortal telomerase‐negative human cells results in cell cycle deregulation, nuclear and chromosomal abnormalities and rapid loss of viability. Oncogene. 2001;20:714–25. [DOI] [PubMed] [Google Scholar]
- 197. Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107:2680–5. [DOI] [PubMed] [Google Scholar]
- 198. Marrone A, Sokhal P, Walne A, Beswick R, Kirwan M, Killick S, et al. Functional characterization of novel telomerase RNA (TERC) mutations in patients with diverse clinical and pathological presentations. Haematologica. 2007;92:1013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352:1413–24. [DOI] [PubMed] [Google Scholar]
- 200. Amelia T, Kartasasmita RE, Ohwada T, Tjahjono DH. Structural insight and development of EGFR tyrosine kinase inhibitors. Molecules. 2022;27:819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Passaro A, Janne PA, Mok T, Peters S. Overcoming therapy resistance in EGFR‐mutant lung cancer. Nat Cancer. 2021;2:377–91. [DOI] [PubMed] [Google Scholar]
- 202. Cotto‐Rios XM, Agianian B, Gitego N, Zacharioudakis E, Giricz O, Wu Y, et al. Inhibitors of BRAF dimers using an allosteric site. Nat Commun. 2020;11:4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Taboski MA, Sealey DC, Dorrens J, Tayade C, Betts DH, Harrington L. Long telomeres bypass the requirement for telomere maintenance in human tumorigenesis. Cell Rep. 2012;1:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53‐ and ATM‐dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–5. [DOI] [PubMed] [Google Scholar]
- 205. Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–38. [DOI] [PubMed] [Google Scholar]
- 206. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434:913–7. [DOI] [PubMed] [Google Scholar]
- 207. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. [DOI] [PubMed] [Google Scholar]
- 208. Benslimane Y, Sanchez‐Osuna M, Coulombe‐Huntington J, Bertomeu T, Henry D, Huard C, et al. A novel p53 regulator, C16ORF72/TAPR1, buffers against telomerase inhibition. Aging Cell. 2021;20:e13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Hustedt N, Alvarez‐Quilon A, McEwan A, Yuan JY, Cho T, Koob L, et al. A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol. 2019;9:190156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Amici DR, Ansel DJ, Metz KA, Smith RS, Phoumyvong CM, Gayatri S, et al. C16orf72/HAPSTR1 is a molecular rheostat in an integrated network of stress response pathways. Proc Natl Acad Sci USA. 2022;119:e2111262119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Lü Y, Cho T, Mukherjee S, Malik A, Gonzalez‐Foutel NS, Oh R, et al. (2022) Genome‐wide CRISPR screens identify novel regulators of wild‐type and mutant p53 stability. bioRxiv 10.1101/2022.03.13.483372 [PREPRINT] [DOI] [PMC free article] [PubMed]
- 212. Lustig AJ. Hypothesis: paralog formation from progenitor proteins and paralog mutagenesis spur the rapid evolution of telomere binding proteins. Front Genet. 2016;7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Beilstein MA, Renfrew KB, Song X, Shakirov EV, Zanis MJ, Shippen DE. Evolution of the telomere‐associated protein POT1a in Arabidopsis thaliana is characterized by positive selection to reinforce protein‐protein interaction. Mol Biol Evol. 2015;32:1329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Wells TB, Zhang G, Harley Z, Vaziri H. Genetic hypervariability in two distinct deuterostome telomerase reverse transcriptase genes and their early embryonic functions. Mol Biol Cell. 2009;20:464–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Millner RW, Pepper JR. Cardiomyoplasty. BMJ. 1991;302:1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Pontremoli C, Forni D, Cagliani R, Pozzoli U, Clerici M, Sironi M. Evolutionary rates of mammalian telomere‐stability genes correlate with karyotype features and female germline expression. Nucleic Acids Res. 2018;46:7153–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Morgan CC, Mc Cartney AM, Donoghue MT, Loughran NB, Spillane C, Teeling EC, et al. Molecular adaptation of telomere associated genes in mammals. BMC Evol Biol. 2013;13:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Young AJ. The role of telomeres in the mechanisms and evolution of life‐history trade‐offs and ageing. Philos Trans R Soc Lond B Biol Sci. 2018;373:20160452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Wick G, Berger P, Jansen‐Durr P, Grubeck‐Loebenstein B. A Darwinian‐evolutionary concept of age‐related diseases. Exp Gerontol. 2003;38:13–25. [DOI] [PubMed] [Google Scholar]