

Abstract
Methylmalonic acidemia (MMA) is a severe and potentially lethal autosomal recessive inborn error of metabolism most frequently caused by mutations in the methylmalonyl-CoA mutase (MMUT) gene. Proof-of-concept adeno-associated virus (AAV) gene therapy studies using mouse models of MMA have demonstrated promise for this therapeutic approach but translation to the clinic could be limited by preexisting capsid immunity and vector potency. Here we explore the efficacy of a novel clade E capsid, 44.9, as a serotype for systemic AAV gene therapy for MMA. An anti-AAV44.9 neutralizing antibody (NAb) survey in adult volunteers (n = 19) and a large cohort of MMA patients (n = 48) revealed a seroprevalence rate of ∼26% and 13%, respectively. The efficacy of AAV44.9 gene delivery was examined in two murine models of MMA, representing neonatal lethal and juvenile phenotypes of MMA. Systemic delivery of the AAV44.9-Mmut vector prevented lethality and lowered disease-related metabolites in MMA mice. Tissue biodistribution and transgene expression studies in treated MMA mice showed that AAV44.9 was efficient at transducing the liver and heart. In summary, we establish that AAV44.9 exhibits a low prevalence of preexisting NAb in humans, is highly efficacious in the treatment of clinically severe MMA mouse models and is therefore a promising vector for clinical translation.
Keywords: AAV 44.9, neutralizing antibodies, MMA, methylmalonic acidemia, systemic gene therapy, liver directed gene therapy
Graphical abstract
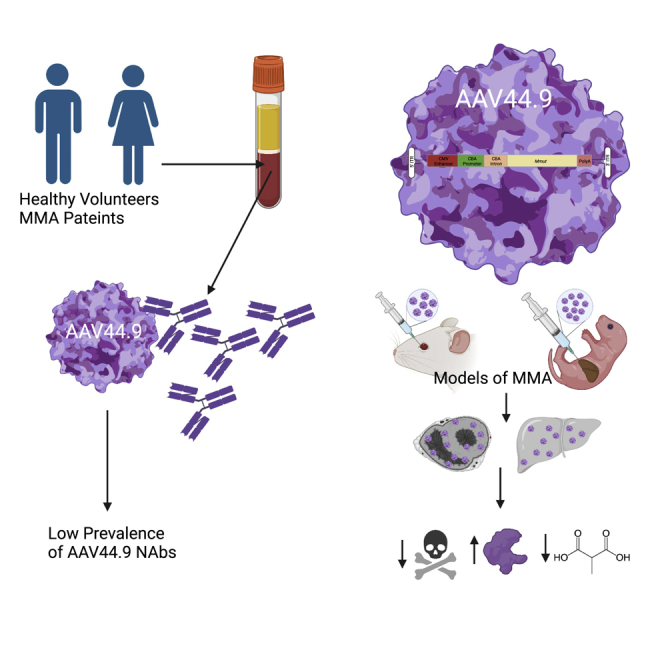
The novel capsid AAV44.9 was assessed for use in human gene therapy. In patients with methylmalonic acidemia (MMA) and adult blood donors, the seroprevalence of anti-capsid neutralizing antibodies was low, and an AAV44.9 therapeutic vector was potent in MMA mouse models. Our studies support broad clinical translational of AAV44.9 vectors.
Introduction
Methylmalonic acidemia (MMA) is a severe and heterogeneous metabolic disorder most commonly caused by mutations in the methylmalonyl-CoA mutase (MMUT) gene.1 The methylmalonyl-CoA mutase enzyme (MMUT) is responsible for the conversion of methylmalonyl-CoA to succinyl-CoA in the mitochondrial matrix, an important step in the catabolism of branched chain amino acids, odd chain fatty acids, and cholesterol. Decreased MMUT activity results in elevations of the disease-related biomarkers, methylmalonic acid and methylcitrate. Historically, MMA caused by MMUT deficiency was divided into two subgroups, mut0 and mut−, based on the amount of MMUT enzymatic activity measured in fibroblasts.2 Patients with no MMUT activity are categorized as mut0 and have a poor medical prognosis, whereas patients with some residual MMUT activity are categorized as mut− and have a milder disease presentation.3,4 MMA manifests as a multisystemic disease, with recurrent metabolic decompensations as a nearly universal feature of the clinical phenotype.5 Because dietary protein restriction and carnitine supplementation are not effective at controlling disease progression, liver transplantation to increase hepatic MMUT enzymatic activity has been offered as a surgical treatment for MMA, especially those who are more severely affected and suffer from recurrent metabolic instability.6, 7, 8, 9, 10, 11, 12 However, limited organ availability, the potential complications arising during or after surgery, the requirement for life-long immunosuppression, and uncertain long-term clinical sequalae post-transplantation have motivated the pursuit of alternative treatment approaches for patients with MMA.13
Adenoviral, lentiviral, mRNA, and systemic adeno-associated virus (AAV) gene delivery using both gene-addition and gene-editing approaches have all demonstrated efficacy in murine models of MMA.14, 15, 16, 17, 18, 19, 20, 21 As a result of promising pre-clinical studies, two clinical trials to treat patients with MMA have been enabled, one uses an AAV-mediated nuclease-free gene-editing approach with an AAV LK03 vector, and the other relies upon lipid nanoparticles to deliver MMUT mRNA to the liver.22,23 Conventional AAV gene addition experiments have demonstrated impressive efficacy in the most severe MMA mouse models, especially with serotype 8 and 9 AAV vectors, and the observation that many MMA patients lack preexisting NAb against AAV2, AAV8, and AAV9 capsids supports clinical translation.15,17,24
In an effort to further expand the spectrum of gene therapies that might be useful in the treatment of MMA and related disorders, we have explored the use of the novel capsid AAV44.9,25,26 originally isolated as a contaminant in a laboratory stock of simian adenovirus SV15 rhesus monkey kidney cell culture. Phylogenetically, AAV44.9 has the most homology to AAVrh.8 and falls between clades E and D. Clade E capsids are reported to interact with galactose for cellular entry but a receptor that interacts with clade D capsids has not been identified.27 AAV44.9 vectors have shown enhanced transduction and spread in subretinal injections in primate models as well as enhanced acinar cell transduction in the salivary gland when pseudoserotyped for gene delivery applications. However, the broader tissue tropisms following systemic delivery of AAV44.9 vectors have not been explored, despite the demonstrated efficacy of related AAVrh.8 vectors in the treatment of metabolic disorders and recent translation to humans with Tay-Sachs disease.28, 29, 30
After testing for AAV44.9 NAb in a large population of MMA patients and a group of healthy volunteers, we prepared AAV44.9 vectors, explored vector trophism with reporter cassettes, and then treated murine models of MMA, representing neonatal lethal (Mmut−/−) and juvenile phenotypes (Mmut−/− MCK-Mmut+) with an AAV44.9 vector that used a ubiquitous promoter to express a murine Mmut cDNA.31,32 Systemic delivery of the therapeutic AAV44.9 vector rescued Mmut−/− mice from lethality and lowered disease-related metabolites in Mmut−/− MCK-Mmut+ mice. Tissue biodistribution and transgene expression studies in treated MMA mice showed that AAV44.9 was efficient at transducing the liver and heart and penetrated the CNS. Our patient and murine studies support the pursuit of AAV44.9 vectors in the treatment of MMA, and by extension, other related disorders of intermediary metabolism.
Results
Prevalence of preexisting antibodies against AAV44.9 capsid in healthy volunteers and MMA patients
A major limitation to the application of systemic gene therapy using AAV is the presence of NAb against the respective vector serotypes, which can not only interfere with transduction, but also potentially initiate severe untoward immune reactions. Little is known about the seroprevalence of NAb to AAV44.9 in the general population or specific disease cohorts. To first approximate the general population, AAV2 and AAV44.9 NAb titers were measured in serum collected from healthy adult blood donors (n = 19, mean age 47 ± 11 years). The seroprevalence at a titer of 1:16 of AAV44.9 NAb was 27% compared with AAV2 NAb, which were reactive in 37% of the same individuals at much higher titers (Figure 1A). In addition, only one of the seven or 14% of the healthy volunteers were co-seropositive for both AAV2 and AAV44.9 NAb (individual data not presented). Hence, the pattern of seroreactivity to AAV44.9 was lower in frequency, with lower titer NAb, and showed distinct reactivity compared with AAV2 in the same group, but these differences were not statistically significant (one-way ANOVA).
Figure 1.

Neutralizing antibodies (NAb) against AAV capsids in healthy volunteers and MMA patients
(A) Seroreactivity in healthy volunteers against AAV2 and AAV44.9 at NAb titers ≥1:16. (B) Seroreactivity in MMA patients with MMUT deficiency against AAV9 and AAV44.9 at NAb titers ≥1:16. NA, not available; NT, not transplanted; T, transplanted. p < 0.05 one-way ANOVA with post hoc Tukey test compared with AAV NAb in healthy volunteers.
We next studied a large and diverse cohort of MMA patients with MMUT deficiency (n = 48). Table 1 presents the age, sex, the enzymatic subtype (mut0 or mut− if known) of each patient, and the respective AAV9 and 44.9 NAb titers measured. The seroprevalence at a titer of 1:16 in the entire MMA patient group was 15% for AAV9 and 13% for AAV44.9 NAb (Figure 1B). In the subgroup of MMA patients that were not transplanted (n = 38), the seroprevalence was slightly lower than that of the entire MMA cohort at 11% for AAV9 NAb and 8% for AAV44.9 NAb. The seroprevalence rates for AAV44.9 and AAV9 NAb in the non-transplanted subgroup of mut0 MMA patients (n = 26) was low, at 4% for both AAV capsids (Figure 1B). Of note, the mut0 MMA patients who were not transplanted (n = 26) had a lower seroprevalence for AAV44.9 and AAV9 NAb than the transplanted mut0 MMA subgroup (n = 11), the mut− MMA (not transplanted) subgroup (n = 5) and the MMA subgroup without transplantation or a mut enzymatic subtyping (n = 6). A total of six of the seven Nab-positive MMA patients or 86% were positive for both AAV9 and AAV44.9 NAb. In contrast to the healthy volunteers, the MMA patients had a lower seroprevalence of AAV44.9 NAb (13%) compared with the healthy volunteers (27%), and further, the most severe, non-transplanted mut0 subgroup was largely seronegative, with NAb titers 1:16 when reactive. The differences observed in seropositivity between different MMA patient subgroups and AAV capsids was not statistically significant (one-way ANOVA). However, the difference between seropositivity of AAV2 in healthy volunteers and the seropositivity of AAV9 or AAV44.9 in the mut0 MMA subgroup was significant (p < 0.05, one-way ANOVA).
Table 1.
MMA patient demographics and NAb status
| Subject | Age | Sex | Subtype | Variant 1 | Variant 2 | Transplant status | AAV9 Ab titer |
AAV44.9 Ab titer |
|---|---|---|---|---|---|---|---|---|
| 1 | 3.4 | M | NA | c.372_375dup p.(Lys124_Asp125insGlu) | c.842T>C p.(Leu281Ser) |
not transplanted | Negative | Negative |
| 2 | 3.5 | F | mut0 | c.682C>T (p.Arg228Ter) | c.1287C>G p.(Tyr429Ter) |
not transplanted | Negative | Negative |
| 3 | 3.8 | F | NA | c.850G>T p.(Gly284Ter) |
c.1043G>T p.(Arg348Ile) |
not transplanted | Negative | Negative |
| 4 | 4.2 | F | mut0 | c.682C>T (p.Arg228Ter) | c.1106G>A p.(Arg369His) | LT | Negative | Negative |
| 5 | 4.2 | M | mut0 | c.1207C>T p.(Arg403Ter) |
Unknown | not transplanted | Negative | Negative |
| 6 | 5.4 | M | mut0 | c.1106G>A p.(Arg369His) | c.1630_1631delGGinsTA p.(Gly544∗) | not transplanted | Negative | Negative |
| 7 | 5.7 | M | mut0 | c.322C>T p.(Arg108Cys) |
c.682C>T (p.Arg228Ter) | not transplanted | Negative | Negative |
| 8 | 5.9 | M | mut– | c.91C>T p.(Arg31Ter) |
c.2150G>T p.(Gly717Val) | not transplanted | Negative | Negative |
| 9 | 5.9 | M | NA | c.88C>T p.(Gln30Ter) |
c.299A>G p.(Tyr100Cys) |
not transplanted | Negative | Negative |
| 10 | 6.1 | F | mut0 | c.2179C>T p.(Arg727Ter) |
c.2179C>T p.(Arg727Ter) |
LT | Negative | Negative |
| 11 | 6.4 | F | mut0 | c.682C>T p.(Arg228Ter) |
c.1108A>C p.(Thr370Pro) |
not transplanted | Negative | Negative |
| 12 | 6.8 | F | mut0 | c.607G>A p.(Gly203Arg) |
c.607G>A p.(Gly203Arg) |
not transplanted | Negative | Negative |
| 13 | 7.8 | M | NA | c.1106G>A p.(Arg369His) | c.1324G>C p.(Ala442Pro) |
not transplanted | Negative | Negative |
| 14 | 8.7 | M | mut0 | c.671_678dup p.(Val227Asnfs∗16) | c.1022dup p.(Asn341Lysfs∗20) | not transplanted | Negative | Negative |
| 15 | 9.3 | M | mut– | c.1181T>A p.(Leu394Ter) | c.1924G>C p.(Gly642Arg) | not transplanted | Negative | Negative |
| 16 | 9.5 | F | NA | c.643G>T p.(Gly215Cys) |
c.2080C>T p.(Arg694Trp) | not transplanted | Negative | Negative |
| 17 | 9.8 | M | mut0 | c.349G>T p.(Glu117Ter) |
c.1038_1040del p.(Leu347del) | LKT | Negative | Negative |
| 18 | 9.8 | M | mut0 | c.52C>T p.(Gln18Ter) |
c.1038_1040del p.(Leu347del) | not transplanted | Negative | Negative |
| 19 | 10.3 | M | NA | c.1106G>A p.(Arg369His) | c.1324G>C p.(Ala442Pro) |
not transplanted | 1:32 | 1:128 |
| 20 | 10.8 | F | mut0 | c.1924G>C p.(Gly642Arg) | c.1924G>C p.(Gly642Arg) | not transplanted | Negative | Negative |
| 21 | 11.7 | F | mut0 | c.878A>C p.(Gln293Pro) |
c.878A>C p.(Gln293Pro) |
LKT | Negative | Negative |
| 22 | 11.8 | M | mut0 | c.1560G>C p.(Lys520Asn) | c.1560G>C p.(Lys520Asn) | not transplanted | Negative | Negative |
| 23 | 11.8 | F | mut– | c.281G>T p.(Gly94Val) |
c.2150G>T p.(Gly717Val) | not transplanted | Negative | Negative |
| 24 | 11.8 | M | mut0 | c.146_147ins279 p.? |
c.372_374dup p.(Lys124_Asp125insGlu) | not transplanted | Negative | Negative |
| 25 | 11.9 | F | mut0 | c.927G>A p.(Trp309Ter) |
c.983T>C p.(Leu328Pro) |
not transplanted | Negative | Negative |
| 26 | 13.0 | M | mut0 | c.2179C>T p.(Arg727Ter) |
c.2179C>T p.(Arg727Ter) |
LKT | Negative | Negative |
| 27 | 13.1 | M | mut0 | c.682C>T p.(Arg228Ter) |
c.1332+1del p.? |
not transplanted | Negative | Negative |
| 28 | 13.2 | M | mut0 | c.655A>T p.(Asn219Tyr) |
c.2159_2160delAT p.(Asn720Serfs∗17) | not transplanted | Negative | Negative |
| 29 | 13.3 | F | mut0 | c.607G>A p.(Gly203Arg) |
c.682C>T (p.Arg228Ter) | not transplanted | Negative | Negative |
| 30 | 13.6 | F | mut0 | c.1106G>A p.(Arg369His) | c.1196_1197del p.(Val399GlufsTer24) | not transplanted | Negative | Negative |
| 31 | 13.8 | M | mut0 | c.753 + 2T>A p.? |
c.1741C>T p.(Arg581Ter) |
not transplanted | Negative | Negative |
| 32 | 15.2 | F | mut0 | c.91C>T p.(Arg31Ter) |
c.2053_2055dup p.(Leu685dup) | not transplanted | Negative | Negative |
| 33 | 15.6 | F | mut0 | c.281G>T p.(Gly94Val) |
c.1108A>C p.(Thr370Pro) |
not transplanted | Negative | Negative |
| 34 | 16.9 | F | mut0 | c.2078del p.(Gly693AspfsTer12) | c.2078del p.(Gly693AspfsTer12) | not transplanted | 1:16 | 1:16 |
| 35 | 17.1 | M | mut0 | c.1106G>A p.(Arg369His) | c.1782_1786del p.(Ser594Argfs∗11) | LT | Negative | Negative |
| 36 | 17.6 | F | mut0 | c.29dupT p.(Leu10PhefsTer39) | c.1658del p.(Val553GlyfsTer17) | not transplanted | Negative | Negative |
| 37 | 18.1 | F | mut0 | c.323G>A p.(Arg108His) |
c.1867G>C p.(Gly623Arg) | not transplanted | Negative | Negative |
| 38 | 18.6 | M | mut0 | c.1105C>T p.(Arg369Cys) | c.1207C>T p.(Arg403Ter) |
LKT | Negative | Negative |
| 39 | 18.9 | F | mut0 | c.643G>T p.(Gly215Cys) |
c.643G>T p.(Gly215Cys) |
not transplanted | Negative | Negative |
| 40 | 23.7 | M | mut0 | c.1207C>T p.(Arg403Ter) |
c.1360G>A p.(Gly454Arg) | not transplanted | Negative | Negative |
| 41 | 26.2 | M | mut0 | c.1741C>T p.(Arg581Ter) |
c.1741C>T p.(Arg581Ter) |
KT | 1:64 | 1:256 |
| 42 | 26.7 | M | mut– | c.1760A>C p.(Tyr587Ser) |
c.2150G>T p.(Gly717Val) | not transplanted | 1:16 | 1:128 |
| 43 | 26.9 | F | mut0 | c.1106G>A p.(Arg369His) | c.1106G>A p.(Arg369His) | LKT | 1:16 | 1:64 |
| 44 | 27.1 | M | mut0 | c.655A>T p.(Asn219Tyr) |
c.1048C>T p.(His350Tyr) |
KT | Negative | Negative |
| 45 | 28.1 | F | mut0 | c.322C>T p.(Arg108Cys) |
c.623_624del p.(Val208AlafsTer2) | not transplanted | Negative | Negative |
| 46 | 28.5 | F | mut– | c.2150G>T p.(Gly717Val) | c.2150G>T p.(Gly717Val) | not transplanted | 1:32 | 1:256 |
| 47 | 33.7 | M | mut0 | c.753 + 2T>A p.? |
c.1560 + 1G>T p.? |
not transplanted | Negative | Negative |
| 48 | 38.6 | F | mut0 | c.826G>T p.(Glu276Ter) |
c.1106G>A p.(Arg369His) | LKT | 1:64 | Negative |
NA (not available) indicates individuals who did not have fibroblast 13C propionate incorporation studies and mut0 or mut– designation could not be determined based on genotype. KT, kidney transplant; LKT, liver kidney transplant; LT, liver transplant.
In addition, we compared the AAV9 NAb results from nine of the same plasma samples here with those reported previously and noted a 100% concordance between the seronegative samples between the two studies, which use slightly different NAb assays.24 Furthermore, the single seropositive subject positive for AAV9 NAbs previously reported had an NAb titer of 1:80 and when re-assayed in the present study, it was 1:64.
AAV44.9 reporter studies in wild-type and ROSAmT/mG (Tomato) mice
Tissue tropism following systemic delivery of AAV44.9 to mice was evaluated using two different reporter-based approaches. First, wild-type young adult Balb/c mice were treated with AAV44.9 carrying a firefly luciferase reporter gene expressed under the control of the CMV promoter at a dose of 1 × 1012 vg/mouse via tail vein injection. The mice (n = 3) were then imaged 8 weeks after the injection for luciferase expression by measuring luminescence in live mice, which demonstrated a strong signal coming from the area of the heart and the liver (Figure 2A), with inferior expression in other tissues (Figure S1). The reporter signal was therefore additionally quantitated in the hearts and livers after necropsy of three mice (Figures 2B and 2C).
Figure 2.

AAV44.9 vectors efficiently transduce liver and heart in mice following systemic delivery
(A) A representative image of a wild-type young adult Balb/c mouse treated by tail vein injection with 1 × 1012 vg/mouse AAV44.9 expressing a firefly luciferase reporter gene. Luciferase expression was assessed in live mice 2 months later using the IVIS Lumina platform to quantify luminescence, which is depicted with a color bar scale in the inset of the panel. (B) Luminescence of three dissected and isolated livers and hearts, respectively, are displayed, each image is complemented by a color bar scale. (C) Luciferase transduction quantification from organs in (B). Luminescence is expressed as Average Radiance n = 3. ∗p < 0.05. (D and E) Transduction efficiency and specificity of AAV44.9 Cre-recombinase in ROSAmT/mG (Tomato) transgenic mice as assessed by fluorescence expression of GFP. Mice received 1 × 1012 vg/mouse by tail vein administration. One month after treatment, the liver (D) and heart (E) were collected, and analyzed by confocal microscopy. Panels, from left to right, show labeled cells with DAPI (blue), mTomato (red), mGFP (green), and finally mTomato/mGFP (merged), in their respective channels. Near complete conversion to green fluorescence is appreciated in both tissues following Cre expression. The untreated control organs maintain the membrane-localized Tomato fluorescence.
To further confirm transduction and expression of the AAV transgene in vivo, ROSAmT/mG (Tomato) transgenic mice were used to determine the expression of Cre after AAV44.9 exposure. These mice harbor a cell membrane-targeted, two-color fluorescent Cre-reporter allele. Prior to Cre recombination, cell membrane-localized tdTomato (mT) fluorescence expression is widespread in cells/tissues. Cre-recombinase results in membrane-localized GFP (mGFP) fluorescence expression replacing the red fluorescence. Tissues were harvested 4 weeks after treatment and imaged for red and green fluorescence. Systemic delivery of AAV44.9 CMV-Cre resulted in close to a 100% red to green fluorescence transition induced by Cre-recombinase expression in liver and heart (Figures 2D and 2E). In summary, these results demonstrate that AAV44.9 has strong tropism for the liver and heart in mice.
Treatment of murine models of MMA with a therapeutic AAV44.9 vector
A previously validated vector transgene was used to test the efficacy of AAV44.9 gene delivery in two well-established murine models of MMA that replicate the severe neonatal lethal and juvenile phenotypes seen in MMA patients.15,31,32 This transgene used a cytomegalovirus (CMV) enhancer and chicken beta-actin (CBA) promoter to ubiquitously express a murine Mmut cDNA. This AAV vector, CBA-Mmut, was packaged into an AAV44.9 capsid (AAV44.9-CBA-Mmut).
Mice with the neonatal lethal form of MMA (Mmut−/−) harbor a deletion of a critical exon in Mmut and replicate the severe early-onset phenotype observed in humans.32 These mice do not produce Mmut mRNA or MMUT protein, have elevated levels of plasma methylmalonic acid, and perish in the first 72 h after birth. The ability of an AAV vector to rescue lethal phenotype of the Mmut−/− mice is a quick and definitive way to initially screen the potency of an AAV capsid to treat MMA. Mmut−/− mice were therefore treated with a dose of 2 × 1011 vg per pup at birth using a direct intrahepatic injection route and survival was compared with untreated Mmut−/− mice. A significant increase in survival (p < 0.005, Log rank (Mantel-Cox) test) was observed in the AAV44.9 treated Mmut−/− mice (n = 8) versus the untreated Mmut−/− mice (n = 10) (Figure 3A).
Figure 3.

Survival of Mmut−/− mice, vector biodistribution, and MMUT protein expression after treatment with AAV44.9-CBA-Mmut
Mmut−/− mice received an intrahepatic injection of 2 × 1011 vg of AAV44.9-CBA-Mmut per pup at birth, and were sacrificed on day of life 60. (A) AAV44.9-CBA-Mmut treated Mmut−/− mice had a significant increase in survival in comparison with untreated Mmut−/− mice (∗∗∗p < 0.005, Log rank (Mantel-Cox) test). (B) Vector biodistribution of AAV44.9-CBA-Mmut in Mmut−/− treated mice at day 60, n = 4 for all tissues studied (∗∗p < 0.01, two-way ANOVA). (C) MMUT hepatic protein expression in AAV44.9-CBA-Mmut treated Mmut−/− mice (n = 3) determined by western blot analysis at day 60. Beta-actin served as a loading control. (D) Quantification of hepatic MMUT protein expression in (C) normalized to beta-actin. NS, not significant (one-way ANOVA with Kruskal-Wallis test); WT, wild type. Error bars = SEM.
Three of the AAV44.9 treated Mmut−/− mice were culled on day 60 and their tissues were harvested to evaluated vector biodistribution and MMUT expression (Figures 3B and 3C). The vector copy (vc) number, determined by RT-PCR, was highest in the liver (1 vc/diploid genome) and heart (8.5 vc/diploid genome) and was much less abundant (<0.4 vc/diploid genome) in the brain, pancreas, spleen, kidney, and skeletal muscle (Figure 3B). The vector copy number in the heart was significantly higher than all tissues tested (p < 0.01, two-way ANOVA). MMUT expression was restored to the levels was observed in a control wild-type (Mmut+/− genotype) mouse, while MMUT protein was undetectable in an untreated Mmut−/− mouse (Figures 3C and 3D). This initial study demonstrated proof-of-concept rescue of a lethal murine phenotype, a promising vector biodistribution, and robust hepatic MMUT protein expression, which inspired application of AAV44.9 in a viable model of MMA to study the effects of treatment on other disease-related phenotypes that are associated with MMA.
We next studied a hypomorphic murine model of MMA that was created by transgenesis, which restores methylmalonyl-CoA mutase enzymatic activity only in the cardiac and skeletal muscle in Mmut−/− mice.31 Mmut−/−MCK-Mmut+ mice recapitulate the phenotype of juvenile MMA, and usually survive until weaning, making it possible to compare changes such as weight and metabolites in age-matched AAV44.9-CBA-Mmut treated and untreated MMA mice, which is not feasible due to the very early lethality seen in Mmut−/− mice. Of note, historical values of methylmalonic acid levels in the blood of Mmut−/−MCK-Mmut+ mice are massively elevated, in the 2 millimolar range, and because these animals have substantial lethality after weaning and beyond, age-matched untreated cohorts were not bled for concurrent comparison with the AAV44.9-CBA-Mmut-treated cohort.
Mmut−/−MCK-Mmut+ received a retro-orbital dose of 7 × 1012 vg/kg of AAV44.9-CBA-Mmut at 1 month of life, near the time of weaning. The levels of the disease-related metabolites methylmalonic acid and the propionyl-CoA-derived metabolite, 2-methylcitrate, were significantly lower (p < 0.0004 and p < 0.04, respectively; one-way ANOVA with Kruskal-Wallis test) 10 days after treatment in Mmut−/−MCK-Mmut+ mice relative to their levels prior to treatment (Figures 4A and 4B). AAV44.9-CBA-Mmut gene therapy effect was also durable, and treated Mmut−/−MCK-Mmut+ mice maintained lower metabolite levels at 5 months post-treatment (Figure 4B).
Figure 4.

Improvement in disease-related metabolites and growth in MMA mice after AAV44.9-CBA-Mmut treatment
Mmut−/−MCK-Mmut+ received 7 × 1012 vg/kg of AAV44.9-CBA-Mmut at 1 month of life by retro-orbital delivery. (A) Methylmalonic acid levels were measured in the plasma and compared with pretreatment levels. (∗∗p < 0.004, one-way ANOVA with Kruskal-Wallis test). (B) Methylcitrate levels were measured in the plasma and compared with pretreatment levels. (∗p < 0.04, one-way ANOVA with Kruskal-Wallis test). (C) Growth in mice before and after AAV44.9-CBA-Mmut treatment in female Mmut−/−MCK-Mmut+ relative to untreated Mmut−/−MCK-Mmut+ and wild-type (WT) mice. (D) Growth in mice before and after AAV44.9-CBA-Mmut treatment in male Mmut−/−MCK-Mmut+ relative to untreated Mmut−/−MCK-Mmut+ and WT mice (∗p < 0.05, ∗∗p < 0.005, two-way ANOVA). Error bars = SEM.
The mean weight of the AAV44.9-treated Mmut−/− MCK-Mmut+ female mice was not significantly different (two-way ANOVA) from the mean weight of wild-type control female mice at 2 weeks and 5 months post-treatment (Figure 4C). The mean weight of the AAV44.9-treated Mmut−/− MCK-Mmut+ male mice was significantly different from the mean weight of wild-type male controls at the pretreatment time point (two-way ANOVA); however, 5 months after AAV44.9 treatment, the mean weights between the Mmut−/− MCK-Mmut+ male mice was not significantly different (two-way ANOVA) from the mean weight of wild-type male controls (Figure 4D).
Discussion
AAV-mediated gene delivery has been successfully used to treat a growing number of genetic diseases, but the presence of preexisting neutralizing antibodies against the AAV vector capsid can prevent the achievement of a therapeutic benefit in patients, especially in liver-targeted systemic gene therapy applications.33, 34, 35 Therefore, choosing a capsid that has a low prevalence of preexisting NAb in the target patient population remains a critical consideration. For this reason, the presence of preexisting AAV capsid NAb to a recently discovered capsid, AAV44.9, for the treatment of MMA in both healthy volunteers and a group of MMA patients was conducted prior to testing of AAV44.9 gene delivery in two murine models of MMA.
The seroprevalence of AAV44.9 was first compared with AAV2 in healthy blood donors. AAV2 NAbs were examined due to the high prevalence of seropositivity that is well documented after natural infections.33,36,37 While others have reported lower limits of NAb detection at 1:2 or 1:5 dilutions, our assay relies upon measuring a secreted luciferase delivered by an AAV reporter, and produces a background signal with heat-inactivated, control sera at a 1:16 dilution, which we therefore consider as the upper limit of detection by our platform. Variation in NAb titer measurements between labs is well documented, and may be related to the use of different cell lines, AAV reporters, and NAb cutoffs when reporting seroprevalence, which, in turn, allows for only very broad and general comparisons between different AAV capsid NAb studies.38,39 A further complicating variable is that some groups report titers of AAV capsid immunoglobin G (IgG) to determine the presence of capsid binding antibodies by ELISA.40,41 Hence, we interpret our results relative to the standards we have validated with our platform to determine whether a sample is seropositive.
As has been repeatedly noted by others, we documented a relatively high seroprevalence of modest to high-titer AAV2 NAb in 37% of the healthy blood donors, which is lower than the reactivity against AAV44.9 NAb at 26%, most of whom had NAb detected at the lower limit of the assay. An unexpected observation was the very low co-seropositivity between AAV2 and AAV44.9-positive individuals, suggesting that exposure to a common serotype and/or shared antigenic epitopes between the related capsids does not likely drive the serological profile of AAV44.9 at the population level. The importance of this finding is that it illustrates the potential for the wider applications of AAV44.9 vectors in adult populations where exposure to AAV2 is associated with co-seropositivity to other serotypes, such as AAV8, and thus might severely restrict eligibility for an AAV gene therapy. We then extended our study to a cohort of largely pediatric patients (Table 1) with the rare metabolic disorder, MMA, a group we have previously examined for NAb against AAV2, AAV8, and AAV9.24 Specifically, we wondered whether a study of the seroprevalence of AAV44.9 could expand the scope of capsids that might be candidates for use in systemic AAV gene therapy to treat the MMA patients, especially since current vectors use either LK03 or propose AAV8, and with certainty, some children will be excluded from receiving potentially life-saving therapies because of preexisting anti-capsid immunity. We therefore measured AAV44.9, and for comparison, AAV9, NAb. The seroprevalence (≥1:16) of AAV44.9 and AAV9 NAb in the entire MMA cohort was 13% and 15%, respectively. Previous studies of a healthy human population (n = 85) reported a seroprevalence (≥1:14) for AAV9 of 37% and in our previous study, the MMA patient cohort (n = 42) had a seroprevalence (≥1:20) for AAV9 of 24%.24,42 The relatively consistent findings between the current and published studies of neutralizing antibodies to AAV9, given the aforementioned differences between platforms and laboratories, demonstrates that the methods used in the current study are indeed robust.
While the ability of anti-AAV capsid antibodies to impede transduction and limit therapeutic efficacy is well established, the NAb or IgG titer at which anti-capsid antibodies interferes with gene delivery in vivo remains uncertain. A hemophilia B clinical study with AAV2 reported that a patient with an AAV2 NAb titer of >1:17 failed to respond, while a hemophilia B clinical study with an AAV5 vector reported that a patient with a NAb titer of 1:340 and IgG titer of 1:256 expressed factor IX (FIX) after treatment.33,43 Currently, there are no established AAV NAb or IgG cutoff titers for clinical trials, and the specific parameters and assays are established by the sponsor of the clinical trial and approved by the Food and Drug Administration (FDA) for each indication. The highly effective and FDA-approved AAV9 systemic gene therapy for spinal muscle atrophy type 1 requires a <1:50 AAV9 IgG titer, determined using an ELISA, as a cutoff for enrollment.44 Based on our assay performance, we selected <1:16 as a lower hypothetical limit for systemic AAV44.9 gene delivery to treat MMA. At this titer threshold, approximately 89% of the MMA population without transplantation, and 95% of those in the mut0 MMA subgroup, could be candidates to receive an AAV44.9 vector.
Phylogenetically, AAV44.9 is most like AAVrh.8, and efficiently transduces multiple cell types, including salivary gland cells, liver cells, photoreceptors, and different types of neurons.25 AAVrh.8 is also reported to be neurotropic and can cross the blood brain barrier.45,46 A structural study of AAVrh.8 in comparison with serotypes AAV2, AAV8, and AAV9, which share 84%, 91%, and 87% VP sequence identity with AAVrh.8, identified differences in the surface loops known to mediate receptor binding, transduction efficiency, and antigenicity.47 Biochemical assays showed that AAVrh.8 is unable to bind heparin like AAV2,47 suggesting that AAV44.9 transduction is perhaps mediated by a distinct receptor and may help explain why AAV2 and AAV44.9 have low NAb co-seropositivity in the adult blood donors we studied. Further investigations will be required to understand if cell surface glycans play any role in determining AAV44.9 transduction activity.
Studies in wild-type mice demonstrated the hepatic and cardiac trophism by AAV44.9 following systemic delivery of luciferase and Cre reporters and enabled a proof-of-concept extension in an animal model of a severe metabolic disease. The AAV44.9 vector studied here showed similar efficacy in rescuing neonatal lethal MMA mice when compared with similarly configured AAV8 and AAV9 vectors,15,17 and afforded broad transduction, including the CNS. In addition, treatment of a milder transgenic murine model of MMA with AAV44.9 demonstrated a reduction in disease-related metabolites and improved growth, as has been reported for the treatment of this model with AAV8 and AAV-Anc80.31,48
Our studies provide additional evidence to support continued efforts to examine the seroprevalence of AAV capsids, in control and target populations. Preexisting AAV capsid NAb in humans can vary greatly, with unexpected patterns, as best exemplified by the finding here that there was almost no co-seropositivity between AAV44.9 and AAV2 NAb in healthy adult blood donors. The pattern seen in the MMA cohort was also notable in that patients in the most severe subset were nearly seronegative, in total, against AAV44.9. The pronounced efficacy of correction seen in MMA mouse models further enables AAV44.9 for possible translation of systemic gene delivery to humans, and especially as a viable alternative to AAV8 or AAV9 where hepatic and/or cardiac transduction of the vector is desired.
Materials and methods
Patients and samples
Blood specimens from healthy donors were obtained from NIH Blood Bank, and sample collection was approved by the NIH Institutional Review Board (IRB) under the protocol “Collection and Distribution of Blood Components From Healthy Donors for In Vitro Research Use” (clinicaltrials.gov identifier: NCT00001846). The MMA patients were evaluated at the NIH Clinical Center under the protocol “Clinical and Basic Investigations of Methylmalonic Acidemia and Related Disorders” (clinicaltrials.gov identifier: NCT00078078). The study was approved by the NIH IRB, and the research adhered to the tenets of the Declaration of Helsinki. Informed consent from patients and/or guardians was obtained. This protocol evaluates clinical and genetic features of patients with MMA and allows for research specimen collection. Blood samples were drawn in Na heparin tubes and centrifuged at 1000–2000 × g at 4°C for 10 min. Plasma was removed and aliquoted in 1-mL volumes, and samples were stored at −80°C until use for this study. Plasma samples were thawed on wet ice. From the thawed samples, one aliquot was removed for AAV9 and AAV44.9 NAb analysis.
AAV neutralizing antibody assay
This method is an adaptation of a previously published method, which used an enhanced GFP reporter.49 Before performing the assay, each AAV containing CAG-Gaussia luciferase reporter gene was first titrated in COS cells as follows: 1.5 × 104 cells were seeded in 96-well plates the day before. Cells were then incubated for 2 h with a serial dilution of each AAV. After extensive wash, cells were incubated for 48 h. The dilution of virus that gave, in a Gaussia luciferase assay, a luminescence of ∼1 × 105 Relative Light Units (RLU) was chose to perform the neutralizing antibody assay.
Each patient serum sample was diluted 1:16 followed by six further 1:2 dilutions, then was incubated with AAV2, 9, or 44.9 Gaussia luciferase reporters for 2 hours in COS cells in quadruplicate. Incubation was followed by three washes of medium. Finally, cells were placed in DMEM containing 10% fetal calf serum for 48 h, at which time the supernatant medium was assayed for luciferase assay. AAV was considered neutralizing at the serum dilution at which RLU values were less than 50% of the control.
Gaussia luciferase assay
Ten microliters of supernatant medium from each well was mixed, in 96-well White Microlite 1+ plates (Thermo Fisher) with 200 μL of buffer containing the Gaussia coelenterazine substrate (GLuc Glow Assay Kit #D0720, NanoLight Technology) and immediately quantified for luminescence by the multi wells reader luminometer Victor X2 (PerkinElmer). Gaussia reading was performed using the software PerkinElmer 2030 Manager with protocol settings standard for Luminescence.
Plasmid vector and rAAV production
Production and purification of recombinant AAV9 and AAV44.9 were obtained using a three- (AAV9) or four-plasmid (AAV44.9) procedure, as previously described.26 Human embryonic kidney 293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA); 293T cells were grown at 37°C under a 5% CO2 humidified atmosphere in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U of penicillin/mL, and 0.1 mg of streptomycin/mL. Briefly, 293T cells were transfected by calcium phosphate with three or four plasmids as follows: an adenovirus helper plasmid (pAd12) containing VA RNA and coding the E2 and E4 proteins; one AAV9 or AAV2 helper plasmid containing rep and cap or two AAV helper plasmids containing the AAV rep and the AAV44.9 capsid genes; and a vector plasmid including the AAV2 inverted terminal repeats flanking the appropriate reporter gene expression cassette. The mixture, to transfect five 15-cm-diameter plates, contained 32 μg DNA of each plasmid, except pAd12, 72 μg. The cells were harvested 48 h post-transfection and a crude viral lysate was obtained after one freeze-thaw cycle. The lysate was treated with 0.5% deoxycholic acid (DOC) and 100 U/mL DNase (Benzonase, Sigma) for 30 min at 37°C. The vector particles present in the clarified lysate (obtained by further low-speed centrifugation) were further purified by CsCl (Sigma) gradient ultracentrifugation, and the vector titer was determined by quantitative real-time PCR (Applied Biosystems, Foster City, CA, USA). The vector doses were dialyzed against 0.9% NaCl using Slide-A-Lyzer 10K cassettes (Thermo Fisher). Vector was concentrated using a centrifugal filter unit (Amicon Ultra 0.5mL 100k).
AAVr particles were quantified by qPCR. One microliter of diluted recombinant preparation was added to a PCR containing 1× SYBR Green Master Mix (ABI) and 0.25 pmol/μL forward and reverse primers. Amplification was detected using an ABI 7700 sequence detector (ABI). Specific primers for Gaussia and CMV were used: HGLuc. forward 5′-CACGCCCAAGATGAAGAAGT -3′, HGLuc. reverse 5′-GAACCCAGGAATCTCAGGAATG-3′, CMV forward 5′-CATCTACGTATTAGTCATCGCTATTACCAT-3′, CMV reverse 5′-TGGAAATCCCCGTGAGTCA-3′. Following denaturation at 96°C for 10 min, cycling conditions were 96°C for 15 s, 60°C for 1 min for 40 cycles. The viral DNA in each sample was quantified by comparing the fluorescence profiles with a set of DNA standards.
The University of Pennsylvania Vector Core provided the expression vector, p-AAV2-CI-CB7-RBG. This vector contains transcriptional control elements from the cytomegalovirus enhancer/chicken β-actin promoter, cloning sites for the insertion of a cDNA, and the rabbit β-globin polyA signal. Terminal repeats from AAV serotype 2 flank the expression cassette. The murine Mmut cDNA was cloned into p-AAV2-CI-CB7-RBG.
AAV44.9-CBA-Mmut titer was determined by quantitative real-time PCR analysis with TaqMan gene expression assays murine methylmalonyl-CoA mutase (Mm00485312_m1; Applied Biosystems, Foster City, CA).
Animal studies and models
Animal work was approved by and performed in accordance with the guidelines for animal care at NIH and the Guide for the Care and Use of Laboratory Animals. All mice were housed in micro-isolators, maintained under a 12-h light/12-h dark cycle. Mice were maintained on a standard mouse chow (PicoLab Mouse Diet 20; LabDiet, St. Louis, MO) and water, which was available ad libitum. Both MMA mouse models were created at NIH on a mixed C57BL/6 × 129SV/Ev × FvBN genetic background and were previously described; Mmut−/− and Mmut−/−;MCK-Mmut+.31,32 Tomato floxed-GFP mice, B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,−EGFP)Luo/J, and BALB/c mice were obtained from The Jackson Laboratory.
Live body imaging of luciferase expression
Mice were anesthetized in an isoflurane vaporizing chamber and then injected with 200 μL of luciferase substrate d-luciferin potassium salt (GoldBio) dissolved in 40 mg/mL PBS intraperitoneally. Mice were then transferred to a Lumina in vivo Imaging System (IVIS) dark chamber equipped with an anesthetic vaporizer and imaged. Luciferase activity was quantified and expressed as Average Radiance (p/s/cm2/sr) using Living Image software, version 2.60.1 (Caliper Life Sciences).
Confocal imaging microscopy
Livers and hearts of mTomato/mGFP mice, treated with AAV44.9 CMV-Cre or untreated, were fixed in 4% formaldehyde overnight at 4°C and then frozen in dry ice and stored at −80°C. Cryosections were cut, 30 μm, using a Leica CM3050S cryostat, after which samples were mounted in Fluoromount G with DAPI on glass slides and covered with a #1.5 coverslip. Confocal images were acquired using a FluoView 1000 (Olympus).
Images were processed using the ImageJ software.
Vector genome copy number
Genome copy (GC) number was measured by quantitative real-time PCR analysis with TaqMan gene expression assays (mouse GAPD 4352932E and murine methylmalonyl-CoA mutase [Mm00485312_m1]; Applied Biosystems, Foster City, CA). Samples were analyzed in an Applied Biosystems 7500 fast real-time PCR system, in accordance with the manufacturer’s protocols. A standard curve was prepared, using serial dilutions of a plasmid carrying the murine Mmut cDNA. Genomic DNA was extracted from murine liver samples and 100 ng of DNA was used to determine the GC number of rAAV at 60 days of life.
Western blot analysis
Samples were prepared and analyzed as previously described.21 Previously snap frozen livers were homogenized in 1 mL of T-PER Tissue Protein Extraction Reagent (Thermo Scientific, cat.# 78,510) with a Wheaton 15-mL Tenbroeck tissue grinder (Wheaton, fr. #357426). The lysate was centrifuged with a Thermo Sorvall Legend Micro 17 Centrifuge at 5,000 rpm for 10 min and supernatant was used for gel electrophoresis. Protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Scientific, cat.# 23,225) according to the manufacture’s protocol. A total of 50 μg of total protein was separated by gel electrophoresis using XCell SureLock Mini-Cell Electrophoresis System (Thermo Scientific, cat.# EI0001) on a mini 4% to 12% NuPAGE Bis-Tris gel with NuPAGE MES SDS running buffer according to the manufacture’s protocol. Protein transfer was done using an iBlot 2 Dry Blotting System (Invitrogen, cat.# IB21001) and Invitrogen iBlot 2 Transfer Stacks, nitrocellulose, mini (Invitrogen, cat.# IB23002). Intercept (TBS) blocking buffer (LI-COR, 927-60001) was used for blocking and antibody hybridization. The following antibodies were used for the detection of MMUT (Abcam, ab134956) and β-Actin (Proteintech, 66009-1-Ig) at a dilution of 1:2,000 in conjunction with the following secondary antibodies at a dilution of 1:20,000 (LI-COR, 925-32211 and LI-COR, 926-68072). Blots were imaged using LI-COR’s Odyssey DLx imaging system and LI-COR Acquisition Software.
Metabolic measurements
Blood samples were collected using retro-orbital sinus plexus sampling with sterile heparinized glass capillary tubes. Samples were immediately centrifuged and plasma was collected, diluted in water, and frozen at −80°C until measurements were performed. Plasma methylmalonic acid and 2-methylcitrate levels were quantified by gas chromatography-mass spectrometry with stable isotopic internal calibration as previously described.50,51
Statistical analyses
Prism 9 version 9.2.0 by GraphPad was used to analyze all data. Results are expressed as mean ± SEM. Values of p < 0.05 were considered statistically significant. Depending on the experimental design, a one-way ANOVA, two-way ANOVA, or log rank (Mantel-Cox) test were used to test for statistical significance and the test used is specified in the results section and figure legends.
Acknowledgments
The expert assistance from Darwin Romero with animal husbandry and care, and other members of the NHGRI laboratory and animal support program, and Yeap Ng, Roberto Weigert and William Swain for imaging assistance. R.J.C., S.M, B.T.H., J.L.S., I.M., and C.P.V. were supported by the Intramural Research Program of the NHGRI through 1ZIAHG200318-16. G.D, T.K, and J.A.C. were supported by the Intramural Research Program of the NIDCR through Z01 DE00695.
Author contributions
R.J.C. and G.D. prepared and edited the manuscript, and designed and supervised the studies; J.L.S, I.M., and C.P.V. characterized patients, analyzed data, and edited the manuscript; B.T.H. performed studies, analyzed data, and edited the manuscript; S.M. characterized patients; T.K. performed studies; J.A.C. and C.P.V. supervised studies, edited the manuscript, and secured funding.
Declaration of interests
R.J.C., G.D., J.A.C., and C.P.V. are inventors on a patent application filed by NIH on their behalf on the use of AAV44.9 as a gene therapy vector to treat MMA.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.09.001.
Supplemental information
Data availability
The materials presented here are available for distribution after the execution of an MTA with the NIH.
References
- 1.Manoli I., Sloan J.L., Venditti C.P. In: Gene Reviews((R)) Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W., Mirzaa G.M., Amemiya A., editors. 1993. Isolated methylmalonic acidemia. [Google Scholar]
- 2.Willard H.F., Rosenberg L.E. Inherited methylmalonyl CoA mutase apoenzyme deficiency in human fibroblasts: evidence for allelic heterogeneity, genetic compounds, and codominant expression. J. Clin. Invest. 1980;65:690–698. doi: 10.1172/JCI109715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsui S.M., Mahoney M.J., Rosenberg L.E. The natural history of the inherited methylmalonic acidemias. N. Engl. J. Med. 1983;308:857–861. doi: 10.1056/NEJM198304143081501. [DOI] [PubMed] [Google Scholar]
- 4.Hörster F., Baumgartner M.R., Viardot C., Suormala T., Burgard P., Fowler B., Hoffmann G.F., Garbade S.F., Kölker S., Baumgartner E.R. Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut-cblA, cblB) Pediatr. Res. 2007;62:225–230. doi: 10.1203/PDR.0b013e3180a0325f. [DOI] [PubMed] [Google Scholar]
- 5.Oberholzer V.G., Levin B., Burgess E.A., Young W.F. Methylmalonic aciduria. An inborn error of metabolism leading to chronic metabolic acidosis. Arch. Dis. Child. 1967;42:492–504. doi: 10.1136/adc.42.225.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van 't Hoff W.G., Dixon M., Taylor J., Mistry P., Rolles K., Rees L., Leonard J.V. Combined liver-kidney transplantation in methylmalonic acidemia. J. Pediatr. 1998;132:1043–1044. doi: 10.1016/s0022-3476(98)70407-x. [DOI] [PubMed] [Google Scholar]
- 7.Sloan J.L., Manoli I., Venditti C.P. Liver or combined liver-kidney transplantation for patients with isolated methylmalonic acidemia: who and when? J. Pediatr. 2015;166:1346–1350. doi: 10.1016/j.jpeds.2015.03.026. [DOI] [PubMed] [Google Scholar]
- 8.Niemi A.K., Kim I.K., Krueger C.E., Cowan T.M., Baugh N., Farrell R., Bonham C.A., Concepcion W., Esquivel C.O., Enns G.M. Treatment of methylmalonic acidemia by liver or combined liver-kidney transplantation. J. Pediatr. 2015;166:1455–1461.e1. doi: 10.1016/j.jpeds.2015.01.051. [DOI] [PubMed] [Google Scholar]
- 9.Fraser J.L., Venditti C.P. Methylmalonic and propionic acidemias: clinical management update. Curr. Opin. Pediatr. 2016;28:682–693. doi: 10.1097/MOP.0000000000000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manoli I., Myles J.G., Sloan J.L., Shchelochkov O.A., Venditti C.P. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonic acidemias. Genet. Med. 2016;18:386–395. doi: 10.1038/gim.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillai N.R., Stroup B.M., Poliner A., Rossetti L., Rawls B., Shayota B.J., Soler-Alfonso C., Tunuguntala H.P., Goss J., Craigen W., et al. Liver transplantation in propionic and methylmalonic acidemia: a single center study with literature review. Mol. Genet. Metab. 2019;128:431–443. doi: 10.1016/j.ymgme.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu T.H., Chien Y.H., Lin H.Y., Liao H.C., Ho H.J., Lai C.J., Chiang C.C., Lin N.C., Yang C.F., Hwu W.L., et al. Methylmalonic acidemia/propionic acidemia - the biochemical presentation and comparing the outcome between liver transplantation versus non-liver transplantation groups. Orphanet J. Rare Dis. 2019;14:73. doi: 10.1186/s13023-019-1045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandler R.J., Venditti C.P. Gene therapy for methylmalonic acidemia: past, present, and future. Hum. Gene Ther. 2019;30:1236–1244. doi: 10.1089/hum.2019.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandler R.J., Venditti C.P. Adenovirus-mediated gene delivery rescues a neonatal lethal murine model of mut(0) methylmalonic acidemia. Hum. Gene Ther. 2008;19:53–60. doi: 10.1089/hum.2007.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandler R.J., Venditti C.P. Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy. Mol. Ther. 2010;18:11–16. doi: 10.1038/mt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandler R.J., Venditti C.P. Pre-clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl-CoA mutase in a murine model of methylmalonic acidemia (MMA) Mol. Genet. Metab. 2012;107:617–619. doi: 10.1016/j.ymgme.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sénac J.S., Chandler R.J., Sysol J.R., Li L., Venditti C.P. Gene therapy in a murine model of methylmalonic acidemia using rAAV9-mediated gene delivery. Gene Ther. 2012;19:385–391. doi: 10.1038/gt.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong E.S.Y., McIntyre C., Peters H.L., Ranieri E., Anson D.S., Fletcher J.M. Correction of methylmalonic aciduria in vivo using a codon-optimized lentiviral vector. Hum. Gene Ther. 2014;25:529–538. doi: 10.1089/hum.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An D., Schneller J.L., Frassetto A., Liang S., Zhu X., Park J.S., Theisen M., Hong S.J., Zhou J., Rajendran R., et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017;21:3548–3558. doi: 10.1016/j.celrep.2017.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneller J.L., Lee C.M., Venturoni L.E., Chandler R.J., Li A., Myung S., Cradick T.J., Hurley A.E., Lagor W.R., Bao G., Venditti C.P. In vivo genome editing at the albumin locus to treat methylmalonic acidemia. Mol. Ther. Methods Clin. Dev. 2021;23:619–632. doi: 10.1016/j.omtm.2021.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandler R.J., Venturoni L.E., Liao J., Hubbard B.T., Schneller J.L., Hoffmann V., Gordo S., Zang S., Ko C.W., Chau N., et al. Promoterless, nuclease-free genome editing confers a growth advantage for corrected hepatocytes in mice with methylmalonic acidemia. Hepatology. 2021;73:2223–2237. doi: 10.1002/hep.31570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gene Therapy with hLB-001 in Pediatric Patients with Severe Methylmalonic Acidemia (SUNRISE). NCT04581785, ClinicalTrials.gov. October 9, 2020 ed. National Library of Medicine.
- 23.A Study to Assess Safety, Pharmacokinetics, and Pharmacodynamics of mRNA-3705 in Participants with Isolated Methylmalonic Acidemia. NCT04899310, Clinicaltrials.gov. December 21, 2021 ed. National Library of Medicine.
- 24.Harrington E.A., Sloan J.L., Manoli I., Chandler R.J., Schneider M., McGuire P.J., Calcedo R., Wilson J.M., Venditti C.P. Neutralizing antibodies against adeno-associated viral capsids in patients with mut methylmalonic acidemia. Hum. Gene Ther. 2016;27:345–353. doi: 10.1089/hum.2015.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boye S.L., Choudhury S., Crosson S., Di Pasquale G., Afione S., Mellen R., Makal V., Calabro K.R., Fajardo D., Peterson J., et al. Novel AAV44.9-based vectors display exceptional characteristics for retinal gene therapy. Mol. Ther. 2020;28:1464–1478. doi: 10.1016/j.ymthe.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Pasquale G., Perez Riveros P., Tora M., Sheikh T., Son A., Teos L., Grewe B., Swaim W.D., Afione S., Zheng C., et al. Transduction of salivary gland acinar cells with a novel AAV vector 44.9. Mol. Ther. Methods Clin. Dev. 2020;19:459–466. doi: 10.1016/j.omtm.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr. Opin. Virol. 2016;21:75–80. doi: 10.1016/j.coviro.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCurdy V.J., Johnson A.K., Gray-Edwards H.L., Randle A.N., Bradbury A.M., Morrison N.E., Hwang M., Baker H.J., Cox N.R., Sena-Esteves M., Martin D.R. Therapeutic benefit after intracranial gene therapy delivered during the symptomatic stage in a feline model of Sandhoff disease. Gene Ther. 2021;28:142–154. doi: 10.1038/s41434-020-00190-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gray-Edwards H.L., Randle A.N., Maitland S.A., Benatti H.R., Hubbard S.M., Canning P.F., Vogel M.B., Brunson B.L., Hwang M., Ellis L.E., et al. Adeno-associated virus gene therapy in a sheep model of tay-sachs disease. Hum. Gene Ther. 2018;29:312–326. doi: 10.1089/hum.2017.163. [DOI] [PubMed] [Google Scholar]
- 30.Flotte T.R., Cataltepe O., Puri A., Batista A.R., Moser R., McKenna-Yasek D., Douthwright C., Gernoux G., Blackwood M., Mueller C., et al. AAV gene therapy for Tay-Sachs disease. Nat. Med. 2022;28:251–259. doi: 10.1038/s41591-021-01664-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manoli I., Sysol J.R., Epping M.W., Li L., Wang C., Sloan J.L., Pass A., Gagné J., Ktena Y.P., Li L., et al. FGF21 underlies a hormetic response to metabolic stress in methylmalonic acidemia. JCI Insight. 2018;3:124351. doi: 10.1172/jci.insight.124351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandler R.J., Sloan J., Fu H., Tsai M., Stabler S., Allen R., Kaestner K.H., Kazazian H.H., Venditti C.P. Metabolic phenotype of methylmalonic acidemia in mice and humans: the role of skeletal muscle. BMC Med. Genet. 2007;8:64. doi: 10.1186/1471-2350-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B.P., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 34.Jiang H., Couto L.B., Patarroyo-White S., Liu T., Nagy D., Vargas J.A., Zhou S., Scallan C.D., Sommer J., Vijay S., et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood. 2006;108:3321–3328. doi: 10.1182/blood-2006-04-017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scallan C.D., Jiang H., Liu T., Patarroyo-White S., Sommer J.M., Zhou S., Couto L.B., Pierce G.F. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood. 2006;107:1810–1817. doi: 10.1182/blood-2005-08-3229. [DOI] [PubMed] [Google Scholar]
- 36.Calcedo R., Morizono H., Wang L., McCarter R., He J., Jones D., Batshaw M.L., Wilson J.M. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin. Vaccine Immunol. 2011;18:1586–1588. doi: 10.1128/CVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li C., Narkbunnam N., Samulski R.J., Asokan A., Hu G., Jacobson L.J., Manco-Johnson M.J., Monahan P.E., Joint Outcome Study Investigators Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2012;19:288–294. doi: 10.1038/gt.2011.90. [DOI] [PubMed] [Google Scholar]
- 38.Meliani A., Leborgne C., Triffault S., Jeanson-Leh L., Veron P., Mingozzi F. Determination of anti-adeno-associated virus vector neutralizing antibody titer with an in vitro reporter system. Hum. Gene Ther. Methods. 2015;26:45–53. doi: 10.1089/hgtb.2015.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber T. Anti-AAV antibodies in AAV gene therapy: current challenges and possible solutions. Front. Immunol. 2021;12:658399. doi: 10.3389/fimmu.2021.658399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Day J.W., Finkel R.S., Mercuri E., Swoboda K.J., Menier M., van Olden R., Tauscher-Wisniewski S., Mendell J.R. Adeno-associated virus serotype 9 antibodies in patients screened for treatment with onasemnogene abeparvovec. Mol. Ther. Methods Clin. Dev. 2021;21:76–82. doi: 10.1016/j.omtm.2021.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy S.L., Li H., Mingozzi F., Sabatino D.E., Hui D.J., Edmonson S.A., High K.A. Diverse IgG subclass responses to adeno-associated virus infection and vector administration. J. Med. Virol. 2009;81:65–74. doi: 10.1002/jmv.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mimuro J., Mizukami H., Shima M., Matsushita T., Taki M., Muto S., Higasa S., Sakai M., Ohmori T., Madoiwa S., et al. The prevalence of neutralizing antibodies against adeno-associated virus capsids is reduced in young Japanese individuals. J. Med. Virol. 2014;86:1990–1997. doi: 10.1002/jmv.23818. [DOI] [PubMed] [Google Scholar]
- 43.Majowicz A., Nijmeijer B., Lampen M.H., Spronck L., de Haan M., Petry H., van Deventer S.J., Meyer C., Tangelder M., Ferreira V. Therapeutic hFIX activity achieved after single AAV5-hFIX treatment in hemophilia B patients and NHPs with pre-existing anti-AAV5 NABs. Mol. Ther. Methods Clin. Dev. 2019;14:27–36. doi: 10.1016/j.omtm.2019.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Zaidy S.A., Mendell J.R. From clinical trials to clinical practice: practical considerations for gene replacement therapy in SMA type 1. Pediatr. Neurol. 2019;100:3–11. doi: 10.1016/j.pediatrneurol.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 45.Yang B., Li S., Wang H., Guo Y., Gessler D.J., Cao C., Su Q., Kramer J., Zhong L., Ahmed S.S., et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. 2014;22:1299–1309. doi: 10.1038/mt.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giove T.J., Sena-Esteves M., Eldred W.D. Transduction of the inner mouse retina using AAVrh8 and AAVrh10 via intravitreal injection. Exp. Eye Res. 2010;91:652–659. doi: 10.1016/j.exer.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halder S., Van Vliet K., Smith J.K., Duong T.T.P., McKenna R., Wilson J.M., Agbandje-McKenna M. Structure of neurotropic adeno-associated virus AAVrh.8. J. Struct. Biol. 2015;192:21–36. doi: 10.1016/j.jsb.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ilyinskii P.O., Michaud A.M., Rizzo G.L., Roy C.J., Leung S.S., Elkins S.L., Capela T., Chowdhury A., Li L., Chandler R.J., et al. ImmTOR nanoparticles enhance AAV transgene expression after initial and repeat dosing in a mouse model of methylmalonic acidemia. Mol. Ther. Methods Clin. Dev. 2021;22:279–292. doi: 10.1016/j.omtm.2021.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corden A., Handelman B., Yin H., Cotrim A., Alevizos I., Chiorini J.A. Neutralizing antibodies against adeno-associated viruses in Sjogren's patients: implications for gene therapy. Gene Ther. 2017;24:241–244. doi: 10.1038/gt.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marcell P.D., Stabler S.P., Podell E.R., Allen R.H. Quantitation of methylmalonic acid and other dicarboxylic acids in normal serum and urine using capillary gas chromatography-mass spectrometry. Anal. Biochem. 1985;150:58–66. doi: 10.1016/0003-2697(85)90440-3. [DOI] [PubMed] [Google Scholar]
- 51.Allen R.H., Stabler S.P., Savage D.G., Lindenbaum J. Elevation of 2-methylcitric acid I and II levels in serum, urine, and cerebrospinal fluid of patients with cobalamin deficiency. Metabolism. 1993;42:978–988. doi: 10.1016/0026-0495(93)90010-l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The materials presented here are available for distribution after the execution of an MTA with the NIH.