

Summary
Hypomethylating agents (HMA) prolong survival and improve cytopenias in individuals with higher-risk myelodysplastic syndrome (MDS). Only 30-40% of patients, however, respond to HMAs, and responses may not occur for more than 6 months after HMA initiation. We developed a model to more rapidly assess HMA response by analyzing early changes in patients’ blood counts. Three institutions’ data were used to develop a model that assessed patients’ response to therapy 90 days after the initiation using serial blood counts. The model was developed with a training cohort of 424 patients from 2 institutions and validated on an independent cohort of 90 patients. The final model achieved an area under the receiver operating characteristic curve (AUROC) of 0.79 in the train/test group and 0.84 in the validation group. The model provides cohort-wide and individual-level explanations for model predictions, and model certainty can be interrogated to gauge the reliability of a given prediction.
Subject areas: Drugs, cancer, artificial intelligence
Graphical abstract
Highlights
-
•
We developed a model to more rapidly assess patients’ response to hypomethylating agents
-
•
The model’s predictions use exclusively routinely collected blood count data
-
•
The model confirmed prior findings and identified potential new prognostic factors
-
•
Model predictions are interpretable on both the individual and cohort-wide level
Drugs; Cancer; Artificial intelligence
Introduction
For patients with intermediate-to higher-risk myelodysplastic syndrome (MDS), the hypomethylating agents (HMA) azacitidine and decitabine represent the current standard of care. HMA improve patients’ cytopenias, prolong survival, and forestall acute myeloid leukemia (AML) transformation (Montalban-Bravo and Garcia-Manero, 2018; Stomper and Lübbert, 2019). In one accepted model of HMA mechanism of action, HMAs counteract aberrant methylation patterns and maturation signals, thereby tilting the balance toward normal myeloid maturation and away from malignant proliferation (Ogawa, 2020; Lindblad et al., 2017).
Owing to their mechanism of action, HMA response can be significantly delayed; typically, at least six 28-day cycles are administered before treatment is considered a failure (Montalban-Bravo and Garcia-Manero, 2018). This prolonged time to response, coupled with the myelosuppressive effects of HMAs, makes it difficult to evaluate responses and to predict which patients will ultimately benefit. For MDS patients with “high” and “very high” risk of leukemic transformation and death as assessed by the Revised International Prognostic Scoring System (IPSS-R), the median time between diagnosis and death is 1.6 and 0.8 years, respectively (Greenberg et al., 2012). Treatment with HMA in higher-risk individuals may preclude other lines of treatment entirely, thereby complicating trials of new agents.
No well-validated models to predict HMA response are in widespread use. Although some studies have identified patient subsets for whom non-response can be predicted based on genomic data, mutational data are not generally used to inform HMA treatment decisions in MDS, given the paucity of available therapies (Nazha et al., 2019; Stomper et al., 2019). Peripheral blood count changes, such as early increments in platelet counts and fetal hemoglobin, may predict HMA response and overall survival in patients with MDS and AML (Stomper et al., 2017, 2019; Huang et al., 2018; Itzykson et al., 2018) as sentinel events for the resumption of normal hematopoiesis. Indeed, in vivo evidence has directly related improved peripheral platelet counts with increased megakaryocyte differentiation (Stomper et al., 2019). These relationships between complete blood count (CBC) parameters and patient outcomes, however, have been assessed with conventional statistics, in isolated lineages, and with a limited number of timepoints.
We hypothesized that a machine learning approach examining serial complete blood cell count data from multiple lineages during the first 90 days of HMA therapy would yield accurate, interpretable response predictions, thereby providing a means for more promptly identifying patients to benefit from continued therapy.
Inclusion criteria, data collection, and definition of hypomethylating agent response
Patient data were collected from the Cleveland Clinic Taussig Cancer Center (CCF) (Cleveland, OH, USA), Moffitt Cancer Center (Tampa, FL, USA), and Sunnybrook Cancer Center (SHC) (Toronto, CA). All adult patients at participating institutions treated with HMA for MDS, chronic myelomonocytic leukemia (CMML), or myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes (MDS/MPN) between January 2004 and January 2019 were screened for inclusion. Patients were 18 or older, received at least four cycles of HMA treatment with no greater than 45 days in between cycles of treatment during the first four months of treatment (a maximum delay of one half-month), and had CBCs available at intervals of two weeks or less during the first 90 days of treatment with HMA and had been followed and evaluated at the treating institution for at least six months after the initiation of HMA therapy.
Response to therapy was determined using the 2006 International Working Group (IWG) response criteria for MDS (Cheson et al., 2006). Treatment was considered successful if patients had complete remission (CR) or hematologic improvement (HI). The time of response was defined as the first date that a patient met either of these endpoints.
Data processing
Serial CBC data pose a challenge for machine learning model development because of their irregularity and high variable number, an example of the “curse of dimensionality,” wherein the number of variables nears or exceeds the number of samples, impairing both model performance and generalizability. From the standpoint of examining serial CBC data over 90 days, using each day as a distinct data point would result in 90 unique variables for each component of a CBC included in the model. Thus, including several cell lineages in a model would entail hundreds or even thousands of variables, a prohibitively high level of dimensionality given the relatively low incidence of MDS and size of datasets available.
Data irregularity poses additional challenges. If data points are simply entered in order for each patient, the timeline fed into a machine learning model will be distorted. For example, without appropriately spacing data, two patients, one of whom had seven serial CBCs drawn over the course of a hospital stay, and one of whom had seven serial CBCs drawn over the course of 14 weeks, would appear to have been observed over similar time frames (i.e., timepoints would be entered as “one” through “seven,” without regard for the interval between each point). Additionally, the number of labs drawn varies between patients. Without appropriate pre-processing, a patient who has four labs drawn during a time period and a patient with eight labs drawn during that same period would have disparate numbers of variables. Thus, serial CBC data need to be processed in a manner that creates the same number of variables and at the same intervals, for each patient.
In order to make CBC data uniform between test, training, and validation cohorts, and to reduce dimensionality, laboratory values were downsampled and interpolated into regular time intervals. Downsampling from 1-day increments to 10 days increments was done to reduce the number of variables by an order of magnitude (i.e., reducing 90 data points per laboratory value to 9 data points). Given that the effects of HMA are seen over periods of weeks to months, we reasoned that this strategy would capture data in an appropriately granular manner.
Blood counts were placed on a timeline, with missing values (i.e., days when CBCs were not drawn for a patient) filled by linear interpolation (Figure S1). Using only the values for given time points might fail to capture the context for those values. For instance, a patient might have a platelet count of 80,000/mL on day 60 of treatment, but the significance of that value would differ vastly depending on whether their platelet count had been 15,000/mL or 250,000/mL at day zero. Thus, in addition to the downsampling strategy used for lab values, the change in laboratory values from the baseline (i.e., the value at day 0) was also considered as a separate set of variables in order to better capture the individual context of a given lab value (Figure S2). Both absolute laboratory values as well as change from baseline were used by the model. Transfusion requirements were not included in the model owing to inconsistent availability.
Model selection and study design
Multiple machine learning models were evaluated; random forests (RF) and gradient-boosted decision tree (GBDT) models XGBoost and LightGBM were selected. Although not explicitly designed for handling sequential data, tree-based methods performed best in preliminary analyses. Model performance was optimized using a hyperparameter grid search strategy. Recurrent neural network and convolutional neural network approaches were also employed, but discarded in light of poor performance in this setting.
Model development used a 5-fold train/test approach using data from CCF and Moffitt, with the SHC data held out as a validation cohort (Figure S3). The area under the receiver operating characteristic curve (AUROC) and area under the precision-recall curve (PR-AUC) were used to evaluate model performance. In the train/test group, 5 distinct models were developed, with model performance determined on the basis of the models’ pooled performance scores to reduce variability. Bootstrapping was used to generate 95% confidence intervals for model performance by repeatedly using sampling with replacement to make resampled versions (“bootstraps”) that allow for non-parametric estimation of confidence intervals. 200 bootstraps were used for each fold of cross-validation for a total of 1000 bootstraps; 95% confidence intervals were generated by taking the 2.5th and 97.fifth percentile values of the total bootstraps.
Model interpretation: cohort-wide predictive performance
The machine learning interpretation software SHAP (Shapley Additive Explanations) (Lundberg and Lee, 2017) was employed in order to generate model explanations using a game theoretical approach (Figure S4). SHAP values were calculated to determine which variables carried the most weight in determining model predictions. Shapley values are calculated by taking a model and iteratively removing a single variable at a time to observe how the model’s prediction changes (Lundberg and Lee, 2017). If the removal of a variable decreases the likelihood of an event predicted by the model, that variable can in turn be inferred to have a positive influence on the likelihood of an event. Conversely, variables whose removal increases the model’s predicted likelihood of an event can be inferred to negatively influence the likelihood of an event (Figure S4). For example, if the default likelihood of an event is 0.6, a feature with a SHAP value of −0.1 decreases the overall likelihood from 0.6 to 0.5. For the ease of global model interpretation, raw SHAP values were converted to a percentage of overall SHAP values on a per-patient basis by taking the absolute value of the SHAP value for a given feature and dividing it by the sum of the absolute values of all SHAP values for that patient. The result describes what proportion of a model’s prediction a given variable is responsible for. Although SHAP values do not determine why a given variable impacts model predictions, they do describe which variables are important, and how important they are relative to one another. Predictions were generated by examining both pooled laboratory values (e.g., all WBC values over 90 days) and individual time points (e.g., the WBC values for the first ten days of treatment). SHAP values for laboratory values represent both the value of a given CBC parameter as well as its change from the parameter’s value at the start of treatment.
To better visualize model predictions, traditional statistics were used to interrogate model predictions. Model predictions were converted to percentiles of likelihood relative to the rest of the cohort (e.g., a patient predicted to have a 0% chance of response would be at the 0th percentile, whereas a predicted 100% chance of response would be at the 99th percentile). Percentile likelihood as described by the model was then compared to actual HMA response rates both by comparing event rates between discrete groups and by modeling event rates as a function of the predicted likelihood of response.
For the categorical approach, patients’ percentile likelihoods were divided into quintiles to mirror the current IPSS-R categorical approach to risk stratification, with response rates compared between adjacent quintiles by the chi-squared test. The continuous approach employed the methods used by Paik et al. (2004), which have subsequently been widely adopted in risk-stratifying breast cancer. Their approach estimates a continuous relationship between model predictions and response rates by using logistic regression to model the relationship between percentile likelihood of response and actual clinical outcomes (i.e., breast cancer recurrence or documented HMA response versus treatment failure). For all comparative statistics, a p value < 0.05 was considered significant.
Additional material
The file “public_code.zip,” related to the STAR Methods is attached in the supplemental material.
Results
Five hundred fourteen patients in total met eligibility criteria: 324 from CCF, 100 from Moffitt, and 90 from SHC. Patients’ mean age was 72 years (range: 40-94 years) with 30% female patients, demographic characteristics which are consistent with other cohorts. Patient risk strata varied between institutions; a greater proportion of patients from CCF (60%) were in the “Very Low,” “Low,” or “Medium” risk IPSS-R risk strata, compared to 41% from MCC and 15% from SHC (p < 0.001). In keeping with this, a smaller proportion (36.1%) of patients with CCF had the WHO-defined higher-risk subtypes MDS with excess blasts-1 (MDS-EB1) or MDS with excess blasts-2 (MDS-EB2) compared with the MCC (57%) and SHC (57%) cohorts (p < 0.001). Patients received similar numbers of cycles (mean number of cycles: 12.5 versus 12.9) of HMA between CCF and Moffitt, respectively (data were missing for SHC). Rates of progression to AML and mean follow-up time were similar between groups. Characteristics for the entire cohort as well as individual institutions are summarized in Table 1.
Table 1.
Cohort demographics
| Missing | Total | CCF | Moffitt | SHC | p value | |
|---|---|---|---|---|---|---|
| Number | 514 | 324 | 100 | 90 | ||
| Female | 0 | 154 (30) | 101 (31) | 28 (28) | 25 (28) | 0.723 |
| Age, mean (SD) | 0 | 71.8 (9) | 71.6 (10) | 69.6 (8) | 74.8 (9) | 0.001 |
| Days of follow-up, mean (SD) | 752.8 (722) | 776.1 (685) | 714.3 (595) | 712.5 (948) | ||
| De novo, n (%) | 91 | 336 (79) | 260 (81) | 76 (76) | Missing | 0.406 |
| Number of cycles, mean (SD) | 92 | 12.6 (12) | 12.5 (12) | 12.9 (12) | Missing | |
| Azacitidine (%) | 0 | 428 (83) | 256 (79) | 90 (90) | 84 (92) | <0.001 |
| Decitabine (%) | 0 | 86 (17) | 68 (21) | 10 (10) | 6 (8) | |
| Prior lenalidomide (%) | 90 | 29 (7) | 20 (8) | 9 (10) | Missing | 0.208 |
| Other prior treatment (%) | 90 | 12 (3) | 8 (3) | 4 (4) | Missing | 0.419 |
| % Marrow Blasts, mean (SD) | 4 | 3.3 (6) | 0.1 (0.1) | 7.6 (6) | 10.1 (7.7) | <0.001 |
| Progression to AML (%) | 0 | 170 (33) | 111 (34) | 31 (31) | 28 (31) | 0.757 |
| IPSS-R cytogenetic score | 17 | 0.023 | ||||
| 0 | 12 (2) | 9 (3) | 1 (1) | 2 (2) | ||
| 1 | 260 (51) | 183 (56) | 51 (51) | 26 (29) | ||
| 2 | 77 (15) | 49 (15) | 12 (12) | 16 (18) | ||
| 3 | 148 (29) | 83 (26) | 35 (35) | 30 (33) | ||
| Missing | 17 (3) | 0 (0) | 1 (1) | 16 (18) | ||
| IPSS-R category | 17 | <0.001 | ||||
| Very Low | 21 (4) | 19 (6) | 1 (1) | 1 (1) | ||
| Low | 124 (24) | 101 (31) | 21 (21) | 2 (2) | ||
| Intermediate | 105 (20) | 74 (23) | 19 (19) | 12 (13) | ||
| High | 110 (21) | 64 (20) | 21 (21) | 25 (28) | ||
| Very High | 137 (27) | 66 (20) | 37 (37) | 34 (38) | ||
| Missing | 17 (3) | 0 (0) | 1 (1) | 16 (18) | ||
| Sub-type | 0 | <0.001 | ||||
| CMML | 59 (12) | 47 (15) | 3 (3) | 9 (10) | ||
| MDS-5q | 7 (1) | 5 (2) | 0 (0) | 2 (2) | ||
| MDS-NOS | 71 (14) | 56 (17) | 4 (4) | 11 (12) | ||
| MDS-U | 12 (2) | 10 (3) | 2 (2) | 0 | ||
| RARS | 36 (7) | 22 (7) | 12 (12) | 2 (2) | ||
| MDS-EB1 | 91 (18) | 49 (15) | 23 (23) | 19 (21) | ||
| MDS-EB2 | 133 (26) | 68 (21) | 34 (34) | 32 (36) | ||
| RCMD | 100 (20) | 63 (19) | 22 (22) | 15 (17) | ||
| RCUD | 5 (1) | 4 (1) | 0 | 1 (1) |
Continuous variables are compared between cohorts using an ANOVA test, and categorical variables are compared using the chi-squared test.
Model performance
Model performance was evaluated for RF, XGBoost, and LightGBM. Demographic data and results of next-generation sequencing panels were included in preliminary models; however, they did not improve model performance and were removed thereafter. When predicting HMA response after 90 days of treatment, the RF model achieved an AUROC of 0.78 [0.60-0.89] and PR-AUC of 0.77 [0.61-0.89] in the train/test cohort, with an AUROC of 0.84 [0.76-0.92] and PR-AUC of 0.84 [0.71-0.93] in the validation cohort. XGBoost achieved an AUROC of 0.79 [0.67-0.90] and PR-AUC of 0.78 [0.6211-0.90] in the train/test cohort, with an AUROC of 0.82 [0.74-0.91] and PR-AUC and of 0.84 [0.72-0.92] in the validation cohort. LightGBM achieved an AUROC of 0.81 [0.66-0.91] and PR-AUC of 0.79 [0.64-0.92] in the train/test cohort, with an AUROC of 0.83 [0.74-0.91] and PR-AUC of 0.83 [0.70-0.92] in the validation cohort. XGBoost was selected from these three models for further analysis on the basis of achieving the best-combined performance in the training and validation cohorts.
When feature importance was assessed by calculating Shapely values for both laboratory values and change in laboratory values from baseline (Figure 1), the most influential parameter was hemoglobin, followed by platelets, red cell distribution width (RDW), and white blood cell (WBC) count in both the train/test and validation cohorts. The magnitude of importance was similar between the train/test and validation cohorts. When the importance of laboratory values was examined along individual time points (Figure 2), hemoglobin, platelets, and absolute neutrophil count (ANC) carried more weight during days 60-90 of treatment. RDW was most influential during days 30-60 of treatment. The influences of absolute laboratory values and change from baseline were also considered separately. In this case, the change in hemoglobin and platelets was most influential at the end of the treatment period, and the change in RDW was most influential during days 31-60 (Figure S5). When absolute laboratory values were considered separately, hemoglobin values were most influential during the last 30 days of treatment; conversely, WBC, platelet, monocyte, ANC, and RDW absolute values were more influential during the first half of treatment (Figure S5).
Figure 1.

Global feature importance, determined by Shapley values
Bar plots shown depict the relative importance of different laboratory values for predicting HMA response in the model, with bar length corresponding to the relative importance of a given feature.
Figure 2.

Feature importance by time point
The heatmaps shown depict different laboratory values in rows, with individual columns corresponding to groups of ten days. Tile color corresponds to relative feature importance as calculated by Shapley values.
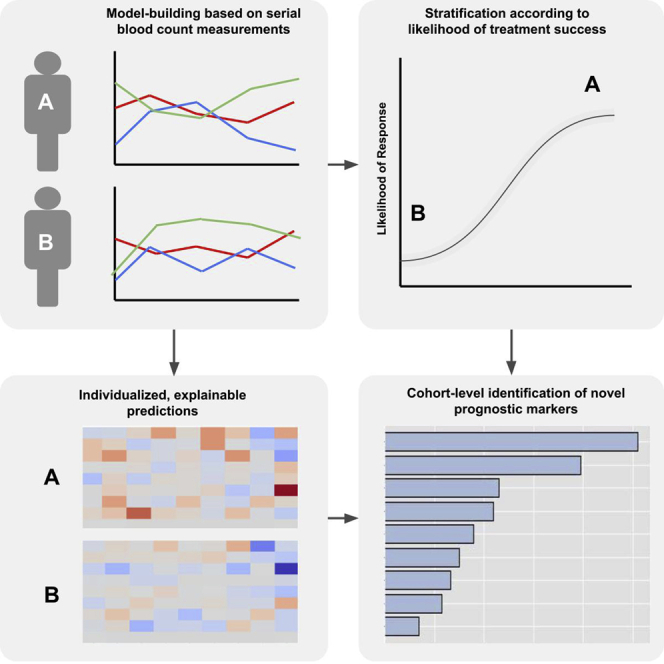
Shapley values also provide predictions about individual patients’ response to HMA. Figure 3 shows two examples of personalized predictions: a patient predicted not to respond (“low” likelihood; second quintile of likelihood) and a patient with a high (fourth quintile) likelihood of response.
Figure 3.

Personalized predictions of HMA response
The pairs of graphs shown depict patients’ hemoglobin, platelet, and ANC values in line graphs during the first 90 days of treatment, and the factors contributing to model predictions in the corresponding heatmaps. Heatmap tiles in blue represent factors making response less likely; red tiles represent factors favoring a response.
Logistic regression was used to estimate the relationship between response rates and the percentile of the predicted response generated by the model (i.e., how likely a given patient was to respond compared to the rest of the cohort). A 0.69 pseudo Rˆ2 value (p = 0.000) was observed between predicted percentile and actual events for the train/test cohort; the pseudo-Rˆ2 value for the validation cohort was 0.26 (p = 6.9 × 10−37, Figure 4). When the percentile of response likelihood was split into quintiles (labeled “very low,” “low,” “intermediate,” “high,” and “very high” likelihoods of response) significant differences (p < 0.05 for all comparisons) were observed between all adjacent quintiles in both the train/test and validation cohorts (Figure 5). Assessment of the relationship between response likelihood, both via estimating by logistic regression and by dividing into different risk strata, demonstrated that the model effectively discriminates between patients with different likelihoods of responding to HMA.
Figure 4.

Estimated HMA response rates (logistic regression) versus percentile of predicted response likelihood
Individual patients are arranged along the x axis in order of predicted likelihood of response from those predicted least likely to respond to those predicted most likely to respond. Response rates as estimated by logistic regression are shown on the y axis.
Figure 5.

HMA response rates by quintile likelihood of response
Bar graphs depict the proportion of patients achieving a response when patients are sorted according to model predictions of response likelihood. p-values between adjacent quintiles are obtained via the chi-squared test.
Discussion
We describe a novel approach to more rapidly assess HMA response in patients with MDS based on routine serial monitoring of patient CBC values during the first 90 days of therapy. The cohorts used to generate the model, taken from three different academic hospitals, are similar to those previously described in clinical trials with regard to patient age, biological sex, and response rates. Cohorts were similar in terms of mean follow-up time and length of treatment. Although the proportion of disease risk (per the IPSS-R) and disease subtypes varied significantly between cohorts, IPSS-R has not been shown to be predictive of response, and similar rates of AML transformation were observed between groups (Stomper et al., 2019).
The model demonstrated the ability to reliably predict HMA response after three months in the train/test cohort and retained its ability to do so when tested on an independent validation cohort, suggesting good external validity. Assessing the usefulness of a model based on its AUROC is highly contextual. However, in the setting of trying to predict future events, we believe these models would be of benefit for clinicians, especially in the case of predictions where the model had a high level of certainty about the likelihood of response. Despite concordance between the train/test and validation cohorts’ performance as measured by AUROC and PR-AUROC, there was a marked discrepancy in the cohorts’ pseudo Rˆ2 values when a continuous risk-stratification approach was used. We interpret this as evidence that while the model performs well in the aggregate, it is less reliable for stratifying the likelihood of response between patients with similar risks of treatment failure (e.g., it may reliably identify high- and low-risk patients, but may have trouble discriminating between two high-risk patients’ likelihood of treatment failure).
Unsurprisingly, hemoglobin and platelets were important predictors of response, although we should highlight that the only data used were those preceding patients’ having met clinical endpoints for response. The predictive value of absolute platelet count early in treatment is concordant with previous findings (Stomper et al., 2017, 2019). Beyond this, the model identified several other predictive factors such as RDW and monocyte count which are not typically considered when assessing HMA response. Incorporating these factors into the model resulted in more robust predictions than could be made by examining three CBC parameters alone. These changes might represent sentinel events in the resumption of normal hematopoiesis and an early indicator of which patients are likely to benefit from continuing therapy.
A reliable, personalized model for HMA response has several potential uses. Although our model does not assist in the upfront decision about whether to initiate HMA therapy, it does provide a means for making more prompt, data-driven decisions about treatment duration. Similar precedents exist for other hematologic malignancies; in Hodgkin lymphoma, where high cure rates need to be balanced against lifelong toxicity, response-adapted therapy with fluorodeoxyglucose-positron emission tomography (FDG-PET) imaging has emerged as a means for balancing toxicity and efficacy (Broadfoot and Johnson, 2017; Barrington et al., 2016). Conversely, in multiple myeloma, recent work has focused on selecting patients for treatment intensification in those at risk for treatment failure (Jackson et al., 2019). In this case, an electronic health record-integrated model could aid decision-making about HMA treatment via automated queries of laboratory data in a manner similar to decision aids such as statin support tools.
In addition to clinician-level decision-making, a reliable means for promptly appraising HMA response has implications for clinical trial design. HMAs are widely used for higher-risk MDS and for ethical and regulatory reasons, trials of new therapies are typically conducted either alongside HMA therapy or after HMA failure. This increases the likelihood of unacceptable toxicities or of poorer performance status and more advanced disease in the setting of HMA failure. Identifying likely non-responders to HMA therapy early would allow patients to enroll in trials earlier and impact the clinical trial design of HMA backbone combination therapies; trials in patients with better performance status and less advanced disease would be more likely to identify potentially beneficial novel agents.
Limitations of the study
Certain factors of our model require further study. Because of the data requirements of our model, all of the patients used to train and validate come from MDS referral centers rather than the community setting. The exclusion of such patients might have skewed our training data toward MDS patients with more aggressive disease, greater numbers of comorbidities, or other adverse prognostic indicators. Conversely, the inclusion of patients in clinical trials might select fitter patients. In either case, an important future focus will be validating the model in other practice settings. Time from diagnosis to treatment initiation, which has been associated with lower response rates, was also not factored into this model (Komrokji et al., 2021). Additionally, because transfusion records were not available, the model does not directly account for transfusion requirements, although it may do so implicitly based on platelet and hemoglobin values. Models that explicitly consider transfusion requirements may be able to more accurately predict HMA success or failure. Factors such as changing transfusion requirements may also be more apparent to clinicians than to statistical models. Finally, while the number of cycles was provided and all patients’ inclusion was contingent on having met inclusion criteria for both minimum number of cycles received and lack of treatment interruptions, the precise timing of HMA administration was not available in our dataset. Thus, the effect of treatment timing could not be assessed with the data available.
It should be noted that our model was developed specifically in patients receiving 28-day cycles of parenteral HMA, the standard of care for the last two decades. It is unknown how our model would translate to other dosing schedules, routes of delivery, or combination therapies. Specifically, orally bioavailable HMA are being actively studied in MDS and AML (Garcia-Manero et al., 2020). The pharmacokinetics of oral HMA may differ from parenteral, and the convenience of oral therapy might lead to alternative dosing schedules or even continuous therapy. There is also increasing interest in using venetoclax in the treatment of MDS and AML (Garcia, 2020). No patients in our study received oral HMA or venetoclax, so it is unknown how well a model developed for parenteral HMA monotherapy would translate. The timing and trajectory of CBC changes in either of these scenarios might necessitate the development of alternative models.
Finally, it should be noted that while next-generation sequencing (NGS) data were available for a small number of patients in our cohorts, sub-analyses examining the utility of NGS data in predicting HMA responsiveness did not add predictive value to our model. Therefore, NGS data were excluded in favor of having a larger sample with which to develop our model. CBC values likely serve as a proxy for underlying disease biology, and as such it is appealing to enrich existing models with genomic data, which are more proximal to underlying drivers of MDS. Sample size (given the low incidence of most mutations, only a handful of which are represented in a sample of hundreds of patients) is a limiting factor in integrating such data, and larger, multi-institutional datasets are likely needed, as prior work has, indeed, demonstrated predictive utility for genomic data (Stomper et al., 2017).
Conclusion
We developed a machine learning model to accurately assess HMA response in patients with MDS. By sampling serial CBC information every 1-2 weeks during the first 90 days of treatment and considering both absolute laboratory values and changes in those values compared to baseline, the model makes accurate, interpretable predictions about which patients are likely to benefit from continued HMA administration, and which ones should consider the cessation of therapy or an investigational agent. Future work should focus on the refinement and validation of this approach in community oncology settings, and the integration of next-generation sequencing data in order to develop models that more directly reflect disease biology.
STAR★Methods
Key resources table
| REAGENT or RESOURCES | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | This paper | https://doi.org/10.17632/5jx94ndznh.1 |
| Software and algorithms | This paper | https://doi.org/10.17632/5jx94ndznh.1 |
Resource availability
Lead contact
Inquiries should be directed to the lead author, Aziz Nazha, at azizn38@yahoo.com.
Acknowledgments
We would like to acknowledge the contributions of the patients who provided the data used to conduct this project. There are no funding sources or grants related to this project.
Author contributions
Conceptualization, A.N and NR; Methodology, A.N and NR; Software, A.N and NR; Formal Analysis, A.N and NR; Investigation, A.N and NR; Resources, D.S., R.B., A.B., A.D., S.M., R.K., J.M., M.S., A.N., Y.G., M.H., B.J.; Data Curation, N.R., S.M., N.A., J.S., Y.R., A.P., A.M., M.S., T.K., Y.G., M.H., B.J.; Writing – Original Draft, N.R.; Writing – Review and Editing, A.N., N.R., R.B., A.B., A.D., S.M., R.K., N.A., M.S.; Visualization, N.R.; Supervision, A.N.; Project Administration, A.N.
Declaration of interests
The authors of this article declare no competing conflicts of interest related to its contents and affirm that its contents are not submitted for review in any other journals.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104931.
Supplemental information
5
Data and code availability
No new physical resources were used for this product. Publically available code and de-identified data are listed in the key resources table. Code is also attached as a.zip file. Other inquiries are welcome via the lead author.
References
- Barrington S., Kirkwood A., Franceschetto A., Fulham M., Roberts T., Almquist H., Brun E., Hjorthaug K., Viney Z., Pike L., et al. PET-CT for staging and early response: results from the Response-Adapted Therapy in Advanced Hodgkin Lymphoma study. Blood. 2016;127:1531–1538. doi: 10.1182/blood-2015-11-679407. [DOI] [PubMed] [Google Scholar]
- Broadfoot J., Johnson P. Response-adapted therapy in Hodgkin lymphoma. Hematologiacal Oncol. 2017;35:33–36. doi: 10.1002/hon.2398. [DOI] [PubMed] [Google Scholar]
- Cheson B., Bennett J., Kantajian H., Pinto A., Schiffer C., Nimer S., Pinto A., Beran M., de Witte T., Stone R., et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419–425. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- Garcia J.S. Prospects for venetoclax in myelodysplastic syndromes. Hematol. Oncol.. Clin North Am. 2020;34:441–448. doi: 10.1016/j.hoc.2019.10.005. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G., Griffiths E.A., Steensma D.P., Roboz G.J., Wells R., McCloskey J., Odenike O., DeZern A., Yee K., Busque L., et al. Oral cedazuridine/decitabine for MDS and CMML: a phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood. 2020;136:674–683. doi: 10.1182/blood.2019004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg P.L., Tuechler H., Schanz J., Sanz G., Garcia-Manero G., Solé F., Bennet J., Bowen D., Fenaux P., Dreyfus F., et al. Revised international prognostic scoring System for myelodysplastic syndromes. Blood. 2012;120:2454–2465. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Zhao H., Hong M., Zhu H., Zhu Y., Lian Y., Li S., Li J., Qian S. Early recovery of the platelet count after decitabine-based induction chemotherapy is a prognostic marker of superior response in elderly patients with newly diagnosed acute myeloid leukaemia. BMC Cancer. 2018;18 doi: 10.1186/s12885-018-5160-5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6299938/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzykson R., Crouch S., Travaglino E., Smith A., Symeonidis A., Hellström-Lindberg E., Sanz G., Cermak J., Stauder R., Elena C., et al. Early platelet count kinetics has prognostic value in lower-risk myelodysplastic syndromes. Blood Adv. 2018;2:2079–2089. doi: 10.1182/bloodadvances.2018020495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson G.H., Davies F.E., Pawlyn C., Cairns D.A., Striha A., Collett C., Waterhouse A., Jones J., Kishore B., Garg M., et al. Response-adapted intensification with cyclophosphamide, bortezomib, and dexamethasone versus no intensification in patients with newly diagnosed multiple myeloma (Myeloma XI): a multicentre, open-label, randomised, phase 3 trial. Lancet Haematol. 2019;6:e616–e629. doi: 10.1016/S2352-3026(19)30167-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komrokji R., Al Ali N., Padron E., Lancet J., Nazha A., Steensma D., DeZern A., Roboz G., Garcia-Manero G., Sekeres M., Sallman D. What is the optimal time to initiate hypomethylating agents (HMAs) in higher risk myelodysplastic syndromes (MDSs)? Leuk. Lymphoma. 2021;62:2762–2767. doi: 10.1080/10428194.2021.1938028. [DOI] [PubMed] [Google Scholar]
- Lindblad K.E., Goswami M., Hourigan C.S., Oetjen K.A. Immunological effects of hypomethylating agents. Expert Rev. Hematol. 2017;10:745–752. doi: 10.1080/17474086.2017.1346470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg S.M., Lee S.-I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 2017;30:4765–4774. [Google Scholar]
- Montalban-Bravo G., Garcia-Manero G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018;93:129–147. doi: 10.1002/ajh.24930. [DOI] [PubMed] [Google Scholar]
- Nazha A., Sekeres M.A., Bejar R., Rauh M.J., Othus M., Komrokji R.S., Barnard J., Hilton C., Kerr C., Steensma D., et al. Genomic biomarkers to predict resistance to hypomethylating agents in patients with myelodysplastic syndromes using artificial intelligence. JCO Precision Oncology. 2019:1–11. doi: 10.1200/PO.19.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S. Genetic basis of myelodysplastic syndromes. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2020;96:107–121. doi: 10.2183/pjab.96.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik S., Skak S., Tang G., Kim C., Baker J., Cronin M., Baehner F., Walker M., Watson D., Park T., et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004 doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- Stomper J., Lübbert M. Can we predict responsiveness to hypomethylating agents in AML? Semin. Hematol. 2019;56:118–124. doi: 10.1053/j.seminhematol.2019.02.001. [DOI] [PubMed] [Google Scholar]
- Stomper J., Ihorst G., Suciu S., Sander P.N., Becker H., Wijermans P.W., Bisse Fetal hemoglobin (HbF) induction during initial decitabine (DAC) treatment of elderly high-risk MDS and AML patients: a potential dynamic biomarker for outcome. Blood. 2017;130:4261. doi: 10.3324/haematol.2017.187278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stomper J., May A.M., Joeckel T.E., Bronsert P., Werner M., Lübbert M. Decitabine-induced early platelet response, a predictor of favorable outcome during hypomethylating treatment of MDS, is associated with in vivo megakaryocytic differentiation. Blood. 2019;13:4265. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
5
Data Availability Statement
No new physical resources were used for this product. Publically available code and de-identified data are listed in the key resources table. Code is also attached as a.zip file. Other inquiries are welcome via the lead author.