

Abstract

Enantioselective [2 + 2] cycloaddition of C(1)-ammonium enolates generated catalytically using the isothiourea HyperBTM with N-alkyl isatins gives spirocyclic β-lactones. In situ ring opening with an amine nucleophile generates isolable highly enantioenriched products in up to 92:8 dr and in >99:1 er.
β-Lactones are versatile synthetic building blocks and significant components of many bioactive natural products.1,2 As a consequence, a range of enantioselective synthetic methods for their preparation has been developed, with both Lewis acid and Lewis base catalyzed approaches common.3 In terms of Lewis base catalysis using tertiary amines, the use of cinchona alkaloids and chiral DMAP derivatives has been extensively used to promote β-lactone formation through the generation of an intermediate C(1)-ammonium enolate.4 Although versatile, these methods typically rely on the generation of reactive monosubstituted ketenes (formed in situ from acyl chlorides) or isolable but sensitive disubstituted ketenes as starting materials.5 In an alternative approach, Romo introduced the NCAL (nucleophile-catalyzed aldol-lactonization) process to prepare β-lactones from keto-acids (Scheme 1a).6 Key to this protocol was the development of carboxylic acids as the C(1)-ammonium enolate precursor, with a modified Mukaiyama reagent used for in situ generation of a reactive ester. Addition of either a cinchona alkaloid or isothiourea catalyst was used to generate the desired C(1)-ammonium enolate, with subsequent intramolecular formal [2 + 2]-cycloaddition onto the pendant carbonyl giving highly enantioenriched β-lactones. Building on this work, we previously demonstrated the use of symmetric arylacetic anhydrides as alternative C(1)-ammonium enolate precursors.7 These anhydrides are generally readily prepared from the parent carboxylic acid, are easy to handle, and can be used in conjunction with isothiourea catalysts without requiring the excess base that is a recognized limitation of alternative protocols using carboxylic acids as starting materials. This approach was applied to the HyperBTM-catalyzed enantioselective intermolecular formation of β-lactones with perfluoroalkyl ketones and arylacetic anhydrides (Scheme 1b). Mechanistic studies using natural abundance 13C kinetic isotope effect experiments, together with computational analyses, indicated the operation of a concerted asynchronous [2 + 2] cycloaddition.8 To date, the isothiourea-catalyzed intermolecular [2 + 2] cycloaddition approach has not been demonstrated using cyclic ketones as substrates; in this manuscript the application of N-protected isatins as electrophiles to initially generate spirooxindole β-lactones is investigated.9 Attempted isolation of the β-lactones led to spontaneous decarboxylation, but postcatalysis addition of an amine nucleophile led to β-lactone ring opening to give isolable highly enantioenriched products (Scheme 1c). Mechanistic studies are consistent with in situ epimerization of the initially formed β-lactone, leading to a mixture of β-hydroxy amide diastereoisomers in highly enantioenriched form (up to 92:8 dr, >99:1 er for both diastereoisomers).
Scheme 1. Tertiary Amine-Catalyzed β-Lactone Syntheses: (a) Romo’s NCAL Intramolecular β-Lactone Synthesis; (b) Previous Work: β-Lactone Synthesis with Perfluoroalkyl Ketones; (c) This Work: β-Lactone Synthesis with Isatins Followed by Ring Opening.

Initial studies began with the reaction of (2R,3S)-HyperBTM 1 (5 mol %) with phenylacetic anhydride 2 and N-benzyl isatin 3, in CH2Cl2. Although β-lactone 5 could be observed by 1H NMR spectroscopic analysis of the crude reaction product, attempted chromatographic purification resulted in the isolation of alkene 4 in 58% yield [66:34 (E)/(Z)], consistent with decarboxylation of β-lactone 5.10 As an alternative, the crude reaction mixture was treated in situ with excess benzylamine (3.0 equiv) to give the isolable β-hydroxyamide derivative 6 (76:24 dr).11 Chromatographic purification gave the separable diastereoisomers in 81% combined yield and >99:1 er for each diastereomer (Scheme 2a). In situ formation of a mixed anhydride, using phenylacetic acid and pivaloyl chloride, and subsequent HyperBTM-catalyzed cycloaddition followed by ring opening led to slightly decreased levels of diastereoselectivity (69:31 dr, >99:1 er) and product yield (57%). Further attempted optimization through variation in catalyst, solvent, and reaction temperature gave no significant improvement in either product dr or yield (see Supporting Information (SI) for full information) with consistent high enantioselectivity observed. Intrigued by the observation that both product diastereoisomers were highly enantioenriched (>99:1 er), further investigations probed if the stereochemical outcome (76:24 dr, >99:1 er) was intrinsic to the catalyzed process, or alternatively a consequence of in situ epimerization of the β-lactone 5 or β-hydroxyamide product 6. Control experiments showed that retreatment of a single diastereoisomer of β-hydroxyamide 6 (>95:5 dr, 99:1 er) to the reaction conditions, or with excess iPr2NEt, led to no change in dr or er. In situ reaction monitoring at room temperature using 1H NMR spectroscopy allowed the concentration and dr of β-lactone 5 to be quantified over the reaction course (Scheme 2b). After 10 min, a single diastereoisomer of β-lactone 5 (60% conversion of isatin 3) was observed, with the dr gradually reducing with time to 85:15 dr after 2 h (∼80% conversion). Extending the reaction time to 16 h gave the β-lactone 5 in 70:30 dr and reduced yield (70%) due to the observation of (E)/(Z) alkenes 4 from decarboxylation in the reaction mixture. These results are consistent with an initial highly stereoselective catalytic process giving β-lactone 5 in high diastereo- and enantioselectivity, with in situ epimerization giving a mixture of diastereoisomers.12 To further probe this process, variation of the reaction time before addition of benzylamine, plus the use of alternative amine nucleophiles for derivatization, was investigated (Scheme 2c). At 0 °C, addition of benzylamine after a 30-min reaction time gave product 6 in an improved 95:5 dr and >99:1 er but with reduced yield (36%) compared to the standard 3 h reaction time (81%, 76:24 dr, >99:1 er). The use of morpholine and pyrrolidine gave the corresponding products 7 and 8 respectively in uniformly excellent enantioselectivity (>99:1 er), but varying diastereoselectivity (85:15 and 68:32 dr respectively). The variation in dr presumably reflects competition between the basicity (promoting epimerization alongside iPr2NEt) and nucleophilicity (promoting ring opening) of these amines and their relative reaction rates. Consistent with these observations, X-ray crystal structure analysis allowed the relative and absolute configuration of the product minor diastereoisomer 9 to be unambiguously determined (Scheme 2d).13 The observed (S)-configuration at C(2) is opposite to that expected based upon the established selectivity of (2R,3S)-HyperBTM in C(1)-ammonium enolate reactions,14 consistent with epimerization of β-lactone 5.
Scheme 2. Optimization and β-Lactone Epimerization.

Yield of isolated products. Reported er of major diastereoisomer (er always >99:1 for minor diastereoisomer). 1H NMR of the crude reaction product was used to determine dr. (b) 30 min reaction time before addition of amine; (c) 3 h reaction time before addition of amine.
Following these observations, the scope and limitations of this process were examined through variation of the isatin, anhydride, and ring-opening nucleophile reaction components (Figure 1). Alternative N-substituents within the isatin were tolerated, with N-methyl, N-allyl protected isatins giving the corresponding products 10 and 11 in good yields and consistently high enantioselectivity (>99:1 er). Interestingly, using N-Boc protected isatin led to product 12 in >99:1 er as a result of ring opening of the β-lactone and the oxindole presumably facilitated by the N-Boc substituent.15
Figure 1.
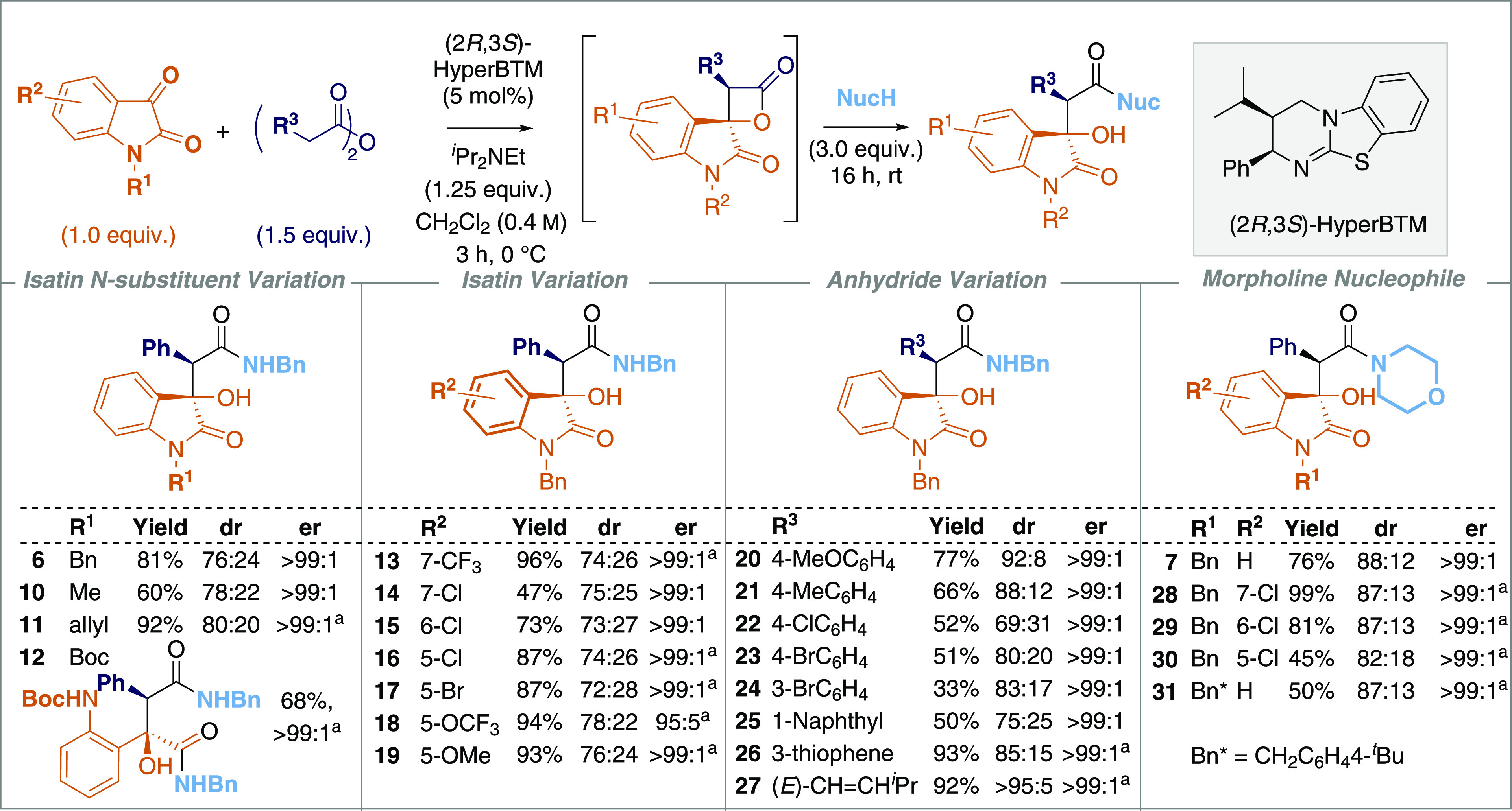
Scope of the reaction. Combined yield of isolated diastereoisomers. Reported er of major diastereoisomer. Reaction performed on 0.40 mmol scale under air atmosphere. 1H NMR of the crude reaction product used to determine dr. a(2S,3R)-HyperBTM used, product has opposite absolute configuration to that shown.
Variation of the isatin component was expanded to incorporate 5-, 6-, and 7-substituted isatins, as well as variation of the N-substituent. Substitution at the 5-, 6-, and 7-position was consistently tolerated, with good product yields observed for both electron-withdrawing and electron-donating substituents, giving products 13–19 with excellent enantioselectivity (>99:1 er) and consistent diastereoselectivity (from 72:28 to 78:22 dr). Unfortunately, 4-substitution of the isatin was not tolerated, with 4-Cl isatin giving <10% conversion to products. The poor conversion in this case is ascribed to the spatial proximity to the reaction center, disfavoring nucleophilic addition.
A selection of anhydrides was next synthesized from the parent carboxylic acids and tested in this cycloaddition/ring-opening process. Notably, incorporating the 4-MeOC6H4-substituent within the anhydride gave product 20 in 77% yield with 92:8 dr and >99:1 er. The higher dr (compared to 6) presumably reflects the electron-donating nature of the aryl substituent that reduces the acidity of the C(3)–H within the β-lactone intermediate. This effect was also observed for the 4-MeC6H4-substrate 21 (88:12 dr, >99:1 er). Halogen-containing phenylacetic anhydrides were tolerated, giving the corresponding products 22–24 in variable diastereoselectivity (from 69:31 to 83:17 dr) but with excellent enantiocontrol. A 1-naphthyl-derivative 25 was prepared in 50% yield and >99:1 er, while extension to a 3-thiophene derivative 26 was also tolerated (85:15 dr, >99:1 er). While the use of simple alkyl anhydride derivatives did not generate any product (see SI for further information), the use of an (E)-alkenyl substituent was tolerated, giving 27 in excellent yield, dr, and er (91%, >95:5 dr, >99:1 er) that was amenable to scale-up to 10 mmol scale, giving >4.1 g of product. Finally, five examples using morpholine as the derivatizing agent were demonstrated (7, 28–31). Generally, good yields and higher diastereocontrol were observed than in the corresponding benzylamides, with excellent enantiocontrol maintained (>99:1 er). Reduced yield was observed for the 5-chloro 30 and N-(para-tert-butylbenzyl) 31 derivatives, with the low solubility of 30 complicating the purification process.
Based upon these observations, alongside previous work in this area, a catalytic cycle is proposed (Figure 2). Initial addition of (2R,3S)-HyperBTM 1 to the phenylacetic anhydride 2 results in the formation of acyl ammonium ion pair 32. Deprotonation at C(2)- gives the corresponding (Z)-ammonium enolate 33,16 with a stabilizing 1,5-O···S chalcogen bonding interaction (nO to σ*S–C)17−19 providing a conformational bias and ensuring coplanarity between the 1,5-O- and S-atoms. The observed product configuration is consistent with that observed in the related [2 + 2]-cycloaddition of C(1)-ammonium enolates and trifluoromethylketones,8 so by analogy a similar concerted asynchronous [2 + 2]-cycloaddition pathway via transition state assembly 34 to give 35 is proposed. Subsequent catalyst release generates the β-lactone 5 in high diastereo- and enantioselectivity. In situ epimerization of the lactone at C(3)- leads to a mixture of β-lactone diastereoisomers, with the subsequent addition of an amine nucleophile promoting ring opening to give the isolable β-hydroxyamide products.
Figure 2.

Proposed catalytic cycle for the intermolecular [2 + 2] cycloaddition to form β-lactones.
In summary, a procedure for the generation of highly enantioenriched β-hydroxyamides (>99:1 er) has been developed. This protocol involves the in situ preparation of β-lactones from isatins and 2-arylacetic anhydrides using the isothiourea HyperBTM 1 to promote a concerted asynchronous [2 + 2]-cycloaddition, followed by in situ ring opening with a nucleophile. Mechanistic studies suggest a base-promoted epimerization leads to a reduction in the diastereoselectivity of the initially formed β-lactone product, giving rise to a diastereoisomeric mixture of isolable products each in high er (>99:1 er). Further work from this laboratory is focused upon alternative application of isothioureas and other Lewis bases in enantioselective catalysis.
Acknowledgments
The research leading to these results has received funding from the EPSRC (EP/S019359/1, C.M.Y.; EP/T023643/1, J.D.; K.K.) as well as Syngenta and the EPSRC Centre for Doctoral Training in Critical Resource Catalysis [CRITICAT, EP/L016419/1 (W.C.H.)].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c02170.
Accession Codes
CCDC 2153991 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
§ Y.A. and K.K. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews, see:; a Mukherjee S.; Biju A. Recent advances in the organocatalytic enantioselective synthesis of functionalized β-lactones. Chem.—Asian J. 2018, 13, 2333. 10.1002/asia.201800902. [DOI] [PubMed] [Google Scholar]; b Romo D.; Tennyson R. L.; Wang Y. β-Lactones as intermediates for natural product total synthesis and new transformations. Heterocycles 2004, 64, 605–658. 10.3987/REV-04-SR(P)3. [DOI] [Google Scholar]
- For selected examples see;; a Pommier A.; Pons J.-M. The synthesis of natural 2-oxetanones. Synthesis 1995, 1995, 729–744. 10.1055/s-1995-4011. [DOI] [Google Scholar]; b Lowe C.; Vederas J. C. Naturally occurring β-lactones: Occurrence, syntheses and properties. A review. Org. Prep. Proced. Int. 1995, 27, 305–346. 10.1080/00304949509458466. [DOI] [Google Scholar]; c Yang H. W.; Romo D. Methods for the synthesis of optically active β-lactones. Tetrahedron 1999, 55, 6403–6434. 10.1016/S0040-4020(99)00185-4. [DOI] [Google Scholar]; d Böttcher T.; Sieber S. A. β-Lactams and β-lactones as activity-based probes in chemical biology. Med. Chem. Comm. 2012, 3, 408–417. 10.1039/c2md00275b. [DOI] [Google Scholar]
- For reviews see:; a Schneider C. Catalytic, enantioselective syntheses of β-lactones-versatile synthetic building blocks in organic chemistry. Angew. Chem., Int. Ed. 2002, 41, 744–746. 10.1002/1521-3773(20020301)41:5<744::AID-ANIE744>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; b Douglas J.; Morrill L. C.; Richmond E.; Smith A. D. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki N., Ed.; Wiley: Hoboken NJ, 2014; ch. 3, pp 89–114. [Google Scholar]; c Van K. N.; Morrill L. C.; Smith A. D.; Romo D. In Lewis Base Catalysis in Organic Synthesis; Vedejs E., Denmark S. E., Eds.; Wiley-VCH: Weinheim, 2016; Vol. 2, ch. 13, pp 527–653. [Google Scholar]
- a Wynberg H.; Staring E. G. J. Asymmetric synthesis of (S)- and (R)-malic acid from ketene and chloral. J. Am. Chem. Soc. 1982, 104, 166–168. 10.1021/ja00365a030. [DOI] [Google Scholar]; b Wynberg H.; Staring E. G. J. Catalytic asymmetric synthesis of chiral 4-substituted oxetanones. J. Org. Chem. 1985, 50, 1977–1979. 10.1021/jo00211a039. [DOI] [Google Scholar]; c Wynberg H.; Staring E. G. J. The absolute configuration of 4-(trichloromethyl)oxetan-2-one; a case of double anchimeric assistance with inversion. J. Chem. Soc., Chem. Commun. 1984, 1181–1182. 10.1039/c39840001181. [DOI] [Google Scholar]; d Calter M. A. J. Org. Chem. 1996, 61, 8006–8007. 10.1021/jo961721c. [DOI] [PubMed] [Google Scholar]; e Tennyson R.; Romo D. Use of In Situ Generated Ketene in the Wynberg β-Lactone Synthesis: New Transformations of the Dichlorinated β-Lactone Products. J. Org. Chem. 2000, 65, 7248–7252. 10.1021/jo001010l. [DOI] [PubMed] [Google Scholar]; f Calter M. A.; Orr R. K.; Song W. Org. Lett. 2003, 5, 4745–4748. 10.1021/ol0359517. [DOI] [PubMed] [Google Scholar]; g Zhu C.; Shen X.; Nelson S. G. J. Am. Chem. Soc. 2004, 126, 5352–5353. 10.1021/ja0492900. [DOI] [PubMed] [Google Scholar]; h Calter M. A.; Tretyak O. A.; Flaschenriem C. Org. Lett. 2005, 7, 1809–1812. 10.1021/ol050411q. [DOI] [PubMed] [Google Scholar]; i Green M. E.; Rech J. C.; Floreancig P. E. Angew. Chem., Int. Ed. 2008, 47, 7317–7320. 10.1002/anie.200802548. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Jiang X.; Fu C.; Ma S. Chem.—Eur. J. 2008, 14, 9656–9664. 10.1002/chem.200801363. [DOI] [PubMed] [Google Scholar]; k Chandra B.; Fu D.; Nelson S. G. Angew. Chem., Int. Ed. 2010, 49, 2591–2594. 10.1002/anie.200906245. [DOI] [PubMed] [Google Scholar]; l Vargo T. R.; Hale J. S.; Nelson S. G. Angew. Chem., Int. Ed. 2010, 49, 8678–8681. 10.1002/anie.201004925. [DOI] [PubMed] [Google Scholar]; m Wan S.; Wu F.; Rech J. C.; Green M. E.; Balachandran R.; Horne W. S.; Day B. W.; Floreancig P. E. J. Am. Chem. Soc. 2011, 133, 16668–16679. 10.1021/ja207331m. [DOI] [PubMed] [Google Scholar]
- a Wilson J. E.; Fu G. C. Asymmetric synthesis of highly substituted β-lactones by nucleophile-catalyzed [2 + 2] cycloadditions of disubstituted ketenes with aldehydes. Angew. Chem., Int. Ed. 2004, 43, 6358–6360. 10.1002/anie.200460698. [DOI] [PubMed] [Google Scholar]; b Zhu C.; Shen X.; Nelson S. G. Cinchona alkaloid-lewis acid catalyst systems for enantioselective ketene–aldehyde cycloadditions. J. Am. Chem. Soc. 2004, 126, 5352–5353. 10.1021/ja0492900. [DOI] [PubMed] [Google Scholar]; For a selected NHC-catalyzed process, see:; c He L.; Lv H.; Zhang Y.; Ye S. Formal cycloaddition of disubstituted ketenes with 2-oxoaldehydes catalyzed by chiral N-heterocyclic carbenes. J. Org. Chem. 2008, 73, 8101–8103. 10.1021/jo801494f. [DOI] [PubMed] [Google Scholar]; d Douglas J.; Taylor J. E.; Churchill G.; Slawin A. M. Z.; Smith A. D. NHC-Promoted asymmetric β-lactone formation from arylalkylketenes and electron-deficient benzaldehydes or pyridinecarboxaldehydes. J. Org. Chem. 2013, 78, 3925–3938. 10.1021/jo4003079. [DOI] [PubMed] [Google Scholar]; e Davies A. T.; Slawin A. M. Z.; Smith A. D. Enantioselective NHC-Catalyzed Redox [2 + 2] Cycloadditions with Perfluoroketones; A Route to Fluorinated Oxetanes. Chem.—Eur. J. 2015, 21, 18944–18948. 10.1002/chem.201504256. [DOI] [PubMed] [Google Scholar]
- a Cortez G. S.; Tennyson R. L.; Romo D. Intramolecular, nucleophile-catalyzed aldol-lactonization (NCAL) reactions: catalytic, asymmetric synthesis of bicyclic β-lactones. J. Am. Chem. Soc. 2001, 123, 7945–7946. 10.1021/ja016134+. [DOI] [PubMed] [Google Scholar]; b Oh S. H.; Cortez G. S.; Romo D. Asymmetric Synthesis of Bicyclic β-Lactones via the Intramolecular, Nucleophile-Catalyzed Aldol Lactonization: Improved Efficiency and Expanded Scope. J. Org. Chem. 2005, 70, 2835–2838. 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]; c Henry-Riyad H.; Lee C.; Purohit V. C.; Romo D. Bicyclic- and Tricyclic-β-lactones via Organonucleophile-Promoted Bis-Cyclizations of Keto Acids: Enantioselective Synthesis of (+)-Dihydroplakevulin. Org. Lett. 2006, 8, 4363–4366. 10.1021/ol061816t. [DOI] [PubMed] [Google Scholar]; d Ma G.; Nguyen H.; Romo D. Concise Total Synthesis of (±)-Salinosporamide A, (±)-Cinnabaramide A, and Derivatives via a Bis-cyclization Process: Implications for a Biosynthetic Pathway?. Org. Lett. 2007, 9, 2143–2146. 10.1021/ol070616u. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Purohit V. C.; Matla A. S.; Romo D. Concise Synthesis of Spirocyclic, Bridged γ-Butyrolactones via Stereospecific, Dyotropic Rearrangements of β-Lactones Involving 1,2-Acyl and δ-Lactone Migrations. J. Am. Chem. Soc. 2008, 130, 10478–10479. 10.1021/ja803579z. [DOI] [PubMed] [Google Scholar]; f Leverett C. A.; Purohit V. C.; Romo D. Enantioselective, Organocatalyzed, Intramolecular Aldol Lactonizations with Keto Acids Leading to Bi- and Tricyclic β-Lactones and Topology-Morphing Transformations. Angew. Chem., Int. Ed. 2010, 49, 9479–9483. 10.1002/anie.201004671. [DOI] [PubMed] [Google Scholar]; g Morris K. A.; Arendt K. M.; Oh S. H.; Romo D. Double Diastereoselective, Nucleophile-Catalyzed Aldol Lactonizations (NCAL) Leading to β-Lactone Fused Carbocycles and Extensions to β-Lactone Fused Tetrahydrofurans. Org. Lett. 2010, 12, 3764–3767. 10.1021/ol101388h. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Leverett C. A.; Purohit V. C.; Johnson A. G.; Davis R. L.; Tantillo D. J.; Romo D. Dyotropic Rearrangements of Fused Tricyclic β-Lactones: Application to the Synthesis of (−)-Curcumanolide A and (−)-Curcumalactone. J. Am. Chem. Soc. 2012, 134, 13348–13356. 10.1021/ja303414a. [DOI] [PubMed] [Google Scholar]; i Kong W.; Romo D. Diastereo- and enantioselective synthesis of bi- and tricyclic N-heterocycle-fused β-lactones. J. Org. Chem. 2017, 82, 13161–13170. 10.1021/acs.joc.7b02235. [DOI] [PubMed] [Google Scholar]
- Morrill L. C.; Ledingham L. A.; Couturier J.-P.; Bickel J.; Harper A. D.; Fallan C.; Smith A. D. 2-Arylacetic anhydrides as ammonium enolate precursors. Org. Biomol. Chem. 2014, 12, 624–636. 10.1039/C3OB41869C. [DOI] [PubMed] [Google Scholar]
- a Barrios Antunez D.; Greenhalgh M. D.; Brueckner A. C.; Walden D. M.; Elías-Rodríguez P.; Roberts P.; Young B.; West T. W.; Slawin A. M. Z.; Cheong P. H-Y.; Smith A. D. Catalytic enantioselective synthesis of perfluoroalkyl-substituted β-lactones via a concerted asynchronous [2 + 2] cycloaddition: a synthetic and computational study. Chem. Sci. 2019, 10, 6162–6173. 10.1039/C9SC00390H. [DOI] [PMC free article] [PubMed] [Google Scholar]; For alternative [2 + 2] cycloadditions with isothioureas, see:; b Smith S. R.; Douglas J.; Prevet H.; Shapland P.; Slawin A. M. Z.; Smith A. D. Isothiourea-catalyzed asymmetric synthesis of β-lactams and β-amino esters from arylacetic acid derivatives and N-sulfonyl aldimines. J. Org. Chem. 2014, 79, 1626–1639. 10.1021/jo402590m. [DOI] [PubMed] [Google Scholar]; c Morrill L. C.; Smith S. M.; Slawin A. M. Z.; Smith A. D. Isothiourea-Mediated Asymmetric Functionalization of 3-Alkenoic Acids. J. Org. Chem. 2014, 79, 1640–1655. 10.1021/jo402591v. [DOI] [PubMed] [Google Scholar]
- For a related [2 + 2]-cycloaddition to generate spirocyclic β-lactams using isothioureas, see:Jin J.-H.; Zhao J.; Yang W.-L.; Deng W.-P. Asymmetric Synthesis of Spirooxindole β-lactams via Isothiourea-catalyzed Mannich/lactamization Reaction of Aryl Acetic Acids with Isatin-derived Ketimines. Adv. Synth. Catal. 2019, 361, 1592–1596. 10.1002/adsc.201801621. [DOI] [Google Scholar]
- For selected studies concerning the decarboxylation of β-lactones, see:; a Noyce D. S.; Banitt E. H. The Stereochemistry of the Decarboxylation of β-Lactones to Form Olefins. J. Org. Chem. 1966, 31, 4043–4047. 10.1021/jo01350a037. [DOI] [Google Scholar]; b Mulzer J.; Zippel M.; Brüntrup G. Thermal Decarboxylation of β-Lactones: Steric Hindrance of Mesomerism as Indication of a Zwitterionic Intermediate. Angew. Chem., Int. Ed. 1980, 19, 465–466. 10.1002/anie.198004651. [DOI] [Google Scholar]; c Ocampo R.; Dolbier W. R.; Bartberger M. D.; Paredes R. A. Kinetic Study of the Thermal Decarboxylation of α,α-Difluoro β-Lactones. J. Org. Chem. 1997, 62, 109–114. 10.1021/jo961648q. [DOI] [PubMed] [Google Scholar]; d Ocampo R.; Dolbier W. R.; Zuluaga F. Synthesis of α-Fluoro-β-lactones and Their Thermal Conversion to 1-Fluoroalkenes. Collect. Czech. Chem. Commun. 2002, 67, 1325–1334. 10.1135/cccc20021325. [DOI] [Google Scholar]; e Ramachandran P. V.; Otoo B. Facile synthesis of 1-trifluoromethylalkenes via the decarboxylation of α-trifluoromethyl-β-lactones. Chem. Commun. 2015, 51, 12388–12390. 10.1039/C5CC03230J. [DOI] [PubMed] [Google Scholar]
- Attempted ring opening of the β-lactone 5 with MeOH led to a complex product distribution so this was not followed further.
- For other examples where this phenomenon has been observed using isothioureas, see:; a Reference (8).; b Ji D.-S.; Liang H.; Yang K.-X.; Feng Z.-T.; Luo Y.-C.; Xu G.-Q.; Gu Y.; Xu P.-F. Solvent directed chemically divergent synthesis of β-lactams and α-amino acid derivatives with chiral isothiourea. Chem. Sci. 2022, 13, 1801–1807. 10.1039/D1SC06127E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCDC 2153991 contains crystallographic data for the minor (2S,3R)-diastereoisomer 9.
- For representative examples, see:; a Morrill L. C.; Lebl T.; Slawin A. M. Z.; Smith A. D. Catalytic asymmetric α-amination of carboxylic acids using isothioureas. Chem. Sci. 2012, 3, 2088–2093. 10.1039/c2sc20171b. [DOI] [Google Scholar]; b Morrill L. C.; Douglas J.; Lebl T.; Slawin A. M. Z.; Fox D. J.; Smith A. D. Isothiourea-mediated asymmetric Michael-lactonisation of trifluoromethylenones: a synthetic and mechanistic study. Chem. Sci. 2013, 4, 4146–4155. 10.1039/c3sc51791h. [DOI] [Google Scholar]; c Smith S. R.; Leckie S. M.; Holmes R.; Douglas J.; Fallan C.; Shapland P.; Pryde D.; Slawin A. M. Z.; Smith A. D. α-Ketophosphonates as Ester Surrogates: Isothiourea-Catalyzed Asymmetric Diester and Lactone Synthesis. Org. Lett. 2014, 16, 2506–2509. 10.1021/ol500873s. [DOI] [PubMed] [Google Scholar]; d Zhang S.; Taylor J. E.; Slawin A. M. Z.; Smith A. D. Isothiourea-Catalyzed Enantioselective Functionalization of 2-Pyrrolyl Acetic Acid: Two-Step Synthesis of Stereodefined Dihydroindolizinones. Org. Lett. 2018, 20, 5482–5485. 10.1021/acs.orglett.8b02423. [DOI] [PubMed] [Google Scholar]; e Zhang S.; Greenhalgh M. D.; Slawin A. M. Z.; Smith A. D. Tandem sequential catalytic enantioselective synthesis of highly-functionalised tetrahydroindolizine derivatives. Chem. Sci. 2020, 11, 3885–3892. 10.1039/D0SC00432D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples, see:; a Franke A. Synthesis and reactions of (o-acylamino)phenylglyoxylic amides. Liebigs. Annalen der Chemie 1982, 1982, 794–804. 10.1002/jlac.198219820420. [DOI] [Google Scholar]; b Ibrahim M. F.; Al-Karewi; Asma A. A.; Khattab S. N.; Hamed E. A.; El-Faham A. Aminolysis of isatin and N-acetyl isatin in acetonitrile and mixed acetonitrile water systems. Asian. J. Org. Chem. 2014, 26, 8029–8038. 10.14233/ajchem.2014.16968. [DOI] [Google Scholar]
- Wang C.; Li S.-J.; Zhang M.; Wei D.; Ding L. Origin of stereoselectivity in an isothiourea catalyzed Michael addition reaction of aryl ester with vinyl sulfone. New. J. Chem. 2020, 44, 17906–17911. 10.1039/D0NJ03540H. [DOI] [Google Scholar]
- For discussions of S···O interactions in isothiourea catalysis:; a Birman V. B.; Li X.; Han Z. Nonaromatic Amidine Derivatives as Acylation Catalysts. Org. Lett. 2007, 9, 37–40. 10.1021/ol0623419. [DOI] [PubMed] [Google Scholar]; b Liu P.; Yang X.; Birman V. B.; Houk K. N. Origin of Enantioselectivity in Benzotetramisole-Catalyzed Dynamic Kinetic Resolution of Azlactones. Org. Lett. 2012, 14, 3288–3291. 10.1021/ol301243f. [DOI] [PubMed] [Google Scholar]; c Abbasov M. E.; Hudson B. M.; Tantillo D. J.; Romo D. Acylammonium Salts as Dienophiles in Diels–Alder/Lactonization Organocascades. J. Am. Chem. Soc. 2014, 136, 4492–4495. 10.1021/ja501005g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Robinson E. R. T.; Walden D. M.; Fallan C.; Greenhalgh M. D.; Cheong P. H.-Y.; Smith A. D. Non-bonding 1,5-S···O interactions govern chemo- and enantioselectivity in isothiourea-catalyzed annulations of benzazoles. Chem. Sci. 2016, 7, 6919–6927. 10.1039/C6SC00940A. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Greenhalgh M. D.; Smith S. M.; Walden D. M.; Taylor J. E.; Brice Z.; Robinson E. R. T.; Fallan C.; Cordes D. B.; Slawin A. M. Z.; Richardson H. C.; Grove M. A.; Cheong P. H.-Y.; Smith A. D. A C=O···Isothiouronium Interaction Dictates Enantiodiscrimination in Acylative Kinetic Resolutions of Tertiary Heterocyclic Alcohols. Angew. Chem., Int. Ed. 2018, 57, 3200–3206. 10.1002/anie.201712456. [DOI] [PubMed] [Google Scholar]; f Young C. M.; Elmi A.; Pascoe D. J.; Morris R. K.; McLaughlin C.; Woods A. M.; Frost A. B.; de la Houpliere A.; Ling K. B.; Smith T. K.; Slawin A. M. Z.; Willoughby P. H.; Cockroft S. L.; Smith A. D. The Importance of 1,5-Oxygen···Chalcogen Interactions in Enantioselective Isochalcogenourea Catalysis. Angew. Chem., Int. Ed. 2020, 59, 3705–3710. 10.1002/anie.201914421. [DOI] [PubMed] [Google Scholar]; For use of S···O interactions in asymmetric synthesis:; g Nagao Y.; Miyamoto S.; Miyamoto M.; Takeshige H.; Hayashi K.; Sano S.; Shiro M.; Yamaguchi K.; Sei Y. Highly Stereoselective Asymmetric Pummerer Reactions That Incorporate Intermolecular and Intramolecular Nonbonded S···O Interactions. J. Am. Chem. Soc. 2006, 128, 9722–9729. 10.1021/ja056649r. [DOI] [PubMed] [Google Scholar]; For examples of S···O interactions in medicinal chemistry:; h Beno B. R.; Yeung K.-S.; Bartberger M. D.; Pennington L. D.; Meanwell N. A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]; For a discussion on the origin of chalcogen-bonding interactions:; i Pascoe D. J.; Ling K. B.; Cockroft S. L. The Origin of Chalcogen-Bonding Interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. 10.1021/jacs.7b08511. [DOI] [PubMed] [Google Scholar]; For an excellent short overview, see:; j Breugst M.; Koenig J. J. σ-Hole Interactions in Catalysis. Eur. J. Org. Chem. 2020, 2020, 5473–5487. 10.1002/ejoc.202000660. [DOI] [Google Scholar]
- For a review of 1,5-chalcogen interactions in organoselenium chemistry, see:; a Mukherjee A. J.; Zade S. S.; Singh H. B.; Sunoj R. B. Organoselenium Chemistry: Role of Intramolecular Interactions. Chem. Rev. 2010, 110, 4357–4416. 10.1021/cr900352j. [DOI] [PubMed] [Google Scholar]; For selected examples in catalysis, see:; b Fujita K. M.; Iwaoka M.; Tomoda S. Synthesis of Diaryl Diselenides Having Chiral Pyrrolidine Rings with C2 Symmetry. Their Application to the Asymmetric Methoxyselenenylation of trans-β-Methylstyrenes. Chem. Lett. 1994, 23, 923–926. 10.1246/cl.1994.923. [DOI] [Google Scholar]; c Fujita K.; Murata K.; Iwaoka M.; Tomoda S. Asymmetric intramolecular selenoetherification and selenolactonization using an optically active diaryl diselenide derived from D-mannitol. J. Chem. Soc., Chem. Commun. 1995, 1641–1642. 10.1039/c39950001641. [DOI] [Google Scholar]; d Fujita K.; Murata K.; Iwaoka M.; Tomoda S. Asymmetric methoxyselenenylation of olefins using an optically active diaryl diselenide derived from d-mannitol. Tetrahedron Lett. 1995, 36, 5219–5222. 10.1016/0040-4039(95)00976-J. [DOI] [Google Scholar]; e Wirth T. Asymmetric Reaction of Arylalkenes with Diselenides. Angew. Chem., Int. Ed. 1995, 34, 1726–1728. 10.1002/anie.199517261. [DOI] [Google Scholar]
- For an early theoretical investigation, see:; a Bleiholder C.; Gleiter R.; Werz D. B.; Köppel H. Theoretical Investigations on Heteronuclear Chalcogen–Chalcogen Interactions: On the Nature of Weak Bonds between Chalcogen Centers. Inorg. Chem. 2007, 46, 2249–2260. 10.1021/ic062110y. [DOI] [PubMed] [Google Scholar]; For a recent perspective, see:; b Kolb S.; Oliver G. A.; Werz D. B. Chemistry Evolves, Terms Evolve, but Phenomena Do Not Evolve: From Chalcogen–Chalcogen Interactions to Chalcogen Bonding. Angew. Chem., Int. Ed. 2020, 59, 22306–22310. 10.1002/anie.202007314. [DOI] [PubMed] [Google Scholar]; For examples of chalcogen bonding catalysis, see:; c Benz S.; López-Andarias J.; Mareda J.; Sakai N.; Matile S. Catalysis with Chalcogen Bonds. Angew. Chem., Int. Ed. 2017, 56, 812–815. 10.1002/anie.201611019. [DOI] [PubMed] [Google Scholar]; d Wonner P.; Vogel L.; Düser M.; Gomes L.; Kniep F.; Mallick B.; Werz D. B.; Huber S. M. Angew. Chem, Int. Ed. 2017, 56, 12009–12012. 10.1002/anie.201704816. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wang W.; Zhu H.; Liu S.; Zhao Z.; Zhang L.; Hao J.; Wang Y. Chalcogen–Chalcogen Bonding Catalysis Enables Assembly of Discrete Molecules. J. Am. Chem. Soc. 2019, 141, 9175–9179. 10.1021/jacs.9b03806. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.