

Abstract
Since the advent of organotransuranium chemistry six decades ago, structurally verified complexes remain restricted to π-bonded carbocycle and σ-bonded hydrocarbyl derivatives. Thus, transuranium-carbon multiple or dative bonds are yet to be reported. Here, utilizing diphosphoniomethanide precursors we report the synthesis and characterization of transuranium-carbene derivatives, namely, diphosphonio-alkylidene- and N-heterocyclic carbene–neptunium(III) complexes that exhibit polarized-covalent σ2π2 multiple and dative σ2 single transuranium-carbon bond interactions, respectively. The reaction of [NpIIII3(THF)4] with [Rb(BIPMTMSH)] (BIPMTMSH = {HC(PPh2NSiMe3)2}1–) affords [(BIPMTMSH)NpIII(I)2(THF)] (3Np) in situ, and subsequent treatment with the N-heterocyclic carbene {C(NMeCMe)2} (IMe4) allows isolation of [(BIPMTMSH)NpIII(I)2(IMe4)] (4Np). Separate treatment of in situ prepared 3Np with benzyl potassium in 1,2-dimethoxyethane (DME) affords [(BIPMTMS)NpIII(I)(DME)] (5Np, BIPMTMS = {C(PPh2NSiMe3)2}2–). Analogously, addition of benzyl potassium and IMe4 to 4Np gives [(BIPMTMS)NpIII(I)(IMe4)2] (6Np). The synthesis of 3Np–6Np was facilitated by adopting a scaled-down prechoreographed approach using cerium synthetic surrogates. The thorium(III) and uranium(III) analogues of these neptunium(III) complexes are currently unavailable, meaning that the synthesis of 4Np–6Np provides an example of experimental grounding of 5f- vs 5f- and 5f- vs 4f-element bonding and reactivity comparisons being led by nonaqueous transuranium chemistry rather than thorium and uranium congeners. Computational analysis suggests that these NpIII=C bonds are more covalent than UIII=C, CeIII=C, and PmIII=C congeners but comparable to analogous UIV=C bonds in terms of bond orders and total metal contributions to the M=C bonds. A preliminary assessment of NpIII=C reactivity has introduced multiple bond metathesis to transuranium chemistry, extending the range of known metallo-Wittig reactions to encompass actinide oxidation states III-VI.
Introduction
The growing wealth of structurally authenticated Th and U covalent multiple bond chemistry that has been realized in recent years has redrawn the known boundaries and molecular-level comprehension of these early members of the 5f-block actinide (An) series.1−4 In contrast, structurally authenticated examples of molecular non-dioxo(actinyl) transuranium-element multiple bonds are limited to a high-valent NpV bis(imido) complex,5 that is, an isolobal N-donor actinyl analogue, and one NpV terminal mono(oxo) complex.6 Low-valent transuranium-element multiple bonds remain restricted to spectroscopically detected [AnE]n+ (E = O, S; n = 0–2) species.7−13 Indeed, in contrast to the dominance of lanthanide (Ln) chemistry in the trivalent state, An-ligand (L) multiple bonding is generally found for AnIV-VI ions. Nonetheless, with more attention given to the pursuit of transuranium-ligand multiply bonded motifs it may be possible to access AnIII=L/AnIV=L and AnIII=L/LnIII=L comparisons that are currently not possible from the study of Th and U alone. The mixed-valent hexauranium ring complexes [{UIII(BIPMTMS)}3{UIV(BIPMTMS)}3(μ-I)3(μ-η6:η6-C6H5R)3] ((BIPMTMS)2– = {C(PPh2NSiMe3)2}2–, a bis(iminophosporano)methanediide; R = H, CH3), formally containing UIII=C bonds represent examples of UIII-ligand multiple bonding, but the presence of noninnocent arene bridges clouds assignments.14 Although organotransuranium chemistry has begun to mature over the past 5 years or so, this still sparsely populated area remains dominated by π-bonded ligands, such as the venerable cyclopentadienyl, arene, and cyclooctatetraenyl ligand sets,15−37 and only two σ-bonded hydrocarbyl Np complexes have been structurally validated.38,39
As fundamental species in organometallic chemistry,40 there is enduring interest in the chemistry of metal-carbene complexes; for example, Fischer carbenes, and of particular pertinence to this work polarized-covalent M=C double bonds, that is, alkylidenes, and dative M ← C bonds such as those from N-heterocyclic carbene (NHC) complexes.40−45 The first structurally characterized An-carbene complex was the phosphonio-alkylidene complex [UIV(CHPMe2Ph)(η5-C5H5)3] reported in 1981 (Figure 1, type I).46 Subsequently, a range of phosphonio- and diphosphonio-alkylidene complexes of U and Th have emerged (Figure 1, type II and III),41,47,48 and more recently, phosphino-silyl-alkylidene ({C(PPh2)(SiMe3)}2–),49−51 arsonium-alkylidene ({CHAsPh3}1–),52 and allenylidene ({CCCPh2}2–)53 derivatives have been reported (Figure 1, type IV-VI). The first An-NHC complexes (Figure 1, type VII), [UO2Cl2{C(NMesCX)2}2] (Mes = 2,4,6-Me3C6H2; X = H or Cl), were reported in 2001.54,55 More recently, the {C(NMeCMe)2} (IMe4) NHC has proven to be useful for supporting uranium(III)56 and (IV)57,58 and for providing comparison to mesoionic carbene derivatives (Figure 1, type VIII).59 To date, there are no transuranium-carbon multiple bonds for any transuranium oxidation state and no transuranium-NHC complexes.
Figure 1.
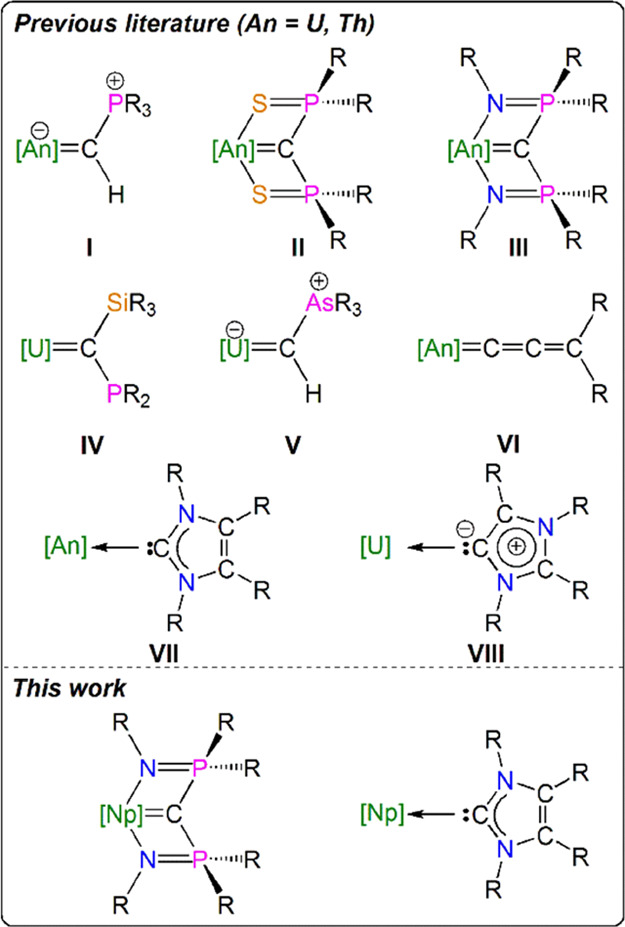
Key An=C and An ← C linkage types in early An-chemistry reported previously and in this work. Use of bracketed [An] (An = U or Th), [U], and [Np] is to acknowledge the various range of metal oxidation states and coligands that are omitted for clarity.
Here, we report the preparation of a diphosphoniomethanide-Np complex, which contains a polarized-covalent transuranium-carbon single σ2-bond. This methanide complex provides an entry-point to transuranium-carbene complexes, including two diphosphonio-alkylidene-NpIII and two NpIII-NHC derivatives that constitute transuranium-carbon polarized-covalent σ2π2 multiple bond and dative σ2 single bond interactions, respectively, Figure 1. The synthesis of these low-valent Np complexes produces clear-cut AnIII-ligand multiple bonding free of redox-active ancillary ligands and was facilitated by adopting a scaled-down prechoreographed approach using Ce as a synthetic surrogate. The analogous Th and U complexes remain experimentally unavailable, and so these Np complexes provide an instance where, instead of Th and U, it is low-valent transuranium chemistry that provides the precedent for experimentally benchmarking comparisons of homologous 5f and 4f electronic structure and bonding.
Results and Discussion
Synthetic Considerations
Previously, we found that the reaction of half an equivalent of [Li2{C(PPh2NSiMe3)2}]2 ([Li2BIPMTMS]2) with [UIVCl4(THF)3] straightforwardly and reliably afforded [(BIPMTMS)UIV(Cl)(μ-Cl)2Li(THF)2] or [{(BIPMTMS)UIV(Cl)(μ-Cl)(THF)}2] depending on the work-up conditions employed.60−62 In contrast, we find that the analogous reaction between [Li2BIPMTMS]2 and [NpIVCl4(DME)2], Scheme 1a, mostly results in intractable, dark product mixtures. However, a small crop of crystals of [(BIPMTMSH)NpIII(Cl)(μ-Cl)3NpIII{μ-(Cl)Li(DME)(OEt2)}(BIPMTMSH)] (1) was isolated on one occasion. Here, NpIV has been reduced to NpIII, and each (BIPMTMS)2– dianion has become protonated to its (BIPMTMSH)1– anion form ((BIPMTMSH)1– = {HC(PPh2NSiMe3)2}1–, a bis(iminophosphorano)methanide). We note that repeating the reaction under identical conditions, except using [UIVCl4(DME)2] instead of [NpIVCl4(DME)2], results in the isolation of [{(BIPMTMS)UIV}2(μ-Cl)6{Li(DME)}2] (2), analogously to our earlier reports.60−62 In this case, UIV ions are retained and no protonation of the (BIPMTMS)2– dianion occurs, Scheme 1a. These different observations for U and Np highlight the greater redox stability of UIV compared to NpIV,63 but the presence of occluded LiCl in [(BIPMTMS)UIV(Cl)(μ-Cl)2Li(THF)2] and 2 also suggested that using Li/Cl combinations could complicate reaction product outcomes. We therefore concluded that Li-reagents should be avoided and that a NpIII starting material could facilitate a rational route to access Np=C bonds without undesired redox chemistry.
Scheme 1. Synthesis of 1, 2, and 3M–6M (M = Ce, Np)a.

Complex 3Np was not isolated. DME = 1,2-dimethoxyethane; Bn = benzyl; IMe4 = {C(NMeCMe)2}.
We identified [NpIIII3(THF)4] as a suitable starting material from which Np=C bonds could be prepared using BIPMTMS, given that a new, convenient Np0-metal-free route was recently reported to access this starting material.64 The combination of limited Np stocks (in comparison to Th, U, and Ln materials) coupled with the relatively high-specific radioactivity of 237Np, and its daughter isotopes, necessitated a small-scale use strategy (typically <30 mg Np) and preoptimized reaction and crystallization conditions based on surrogate trials. Thus, to manage the use of valuable Np we first choreographed scaled-down reactions using [CeIIII3(THF)4] as a synthetic surrogate for [NpIIII3(THF)4]. The synthesis of [(BIPMTMSH)CeIII(I)2(THF)] (3Ce) using [Rb(BIPMTMSH)] and [CeIIII3(THF)4] on multigram (>10 mmol) scales has been reported previously.65 To determine compatibility with our Np experimental protocols, we optimized the synthesis of 3Ce with those reagents at ∼0.04 mmol scale, Scheme 1b, and found that on this scale reactions can be performed rapidly and still yield crystalline 3Ce. During reaction optimizations, we noted that the THF in 3Ce is seemingly labile presenting opportunities for decomposition, which can present a major impediment on small scales. We therefore prepared the new derivative [(BIPMTMSH)CeIII(I)2(IMe4)] (4Ce, IMe4 = {C(NMeCMe)2}), Scheme 1b, because the IMe4 is a strong, kinetically inert donor and its use would pave the way to introducing NHC ligands to transuranium chemistry. With small-scale preparations of crystalline 3Ce and 4Ce in hand, we treated each with benzyl potassium in the presence of DME and IMe4,66,67 respectively, yielding crystalline samples of previously reported [(BIPMTMS)CeIII(I)(DME)] (5Ce) and the new derivative [(BIPMTMS)CeIII(I)(IMe4)2] (6Ce), Scheme 1b.
With the small-scale synthesis of 3Ce–6Ce accomplished, we attempted the synthesis of the NpIII analogues, Scheme 1b. At a small scale (∼0.03–0.04 mmol of Np), utilizing [Rb(BIPMTMSH)] and [NpIIII3(THF)4] we could not isolate [(BIPMTMSH)NpIII(I)2(THF)] (3 Np), possibly because of the THF-lability issue observed for 3Ce. However, adding IMe4 to 3Np prepared in situ afforded [(BIPMTMSH)NpIII(I)2(IMe4)] (4 Np) as ruby-red crystals in 16% yield. Likewise, the reaction between benzyl potassium and 3Np (prepared in situ) in DME afforded [(BIPMTMS)NpIII(I)(DME)] (5Np) as orange crystals in 37% isolated yield. Finally, treatment of 4Np with benzyl potassium and IMe4 afforded [(BIPMTMS)NpIII(I)(IMe4)2] (6Np) as red-purple crystals in 32% yield. Though the yields are low, which is attributed to the small scales and quite soluble nature of these complexes, they are reproducible.
Previously, it has been found that UIII disproportionates when paired with the (BIPMTMS)2– dianion, requiring arene buffers in inverse-sandwich-arene complexes to stabilize this combination via extensive U-arene δ-bonding interactions.14,61 We therefore revisited the synthesis of the U-analogues under these new preparative conditions using [UIIII3(THF)4], because these small-scale reactions are performed quickly. Although [(BIPMTMSH)UIII(I)2(THF)] (3U) can be made and isolated as per our previous report,14 all attempts to deprotonate and isolate the resulting product either result in disproportionation and/or decomposition or the formation of inverse-sandwich-arene complexes, highlighting the intrinsically less stable nature of UIII compared to NpIII.68 Nevertheless, the isolation of 4Np–6Np permits opportunities to make experimentally benchmarked AnIII vs AnIV and AnIII vs LnIII M=C (M = An, Ln) bonding comparisons that would otherwise remain lacking.69−72
Solid-State Structures
To define the metrical details of 4Np–6Np, their solid-state molecular structures were determined by single-crystal X-ray diffraction. For completeness, and as part of the choreographing scaled-down verification process, the structures of isomorphous (for each metal pair) 3Ce–6Ce were determined (see the Supporting Information for full details), noting that 3Ce and 5Ce were structural redeterminations, whereas those of 4Ce and 6Ce are reported for the first time. Before we discuss the Np–C interactions in detail, we note that the Np–N distances in 4Np–6Np are either statistically indistinguishable (by the 3σ-criterion) or are only marginally shorter than the corresponding Ce–N distances in 4Ce–6Ce, meaning that clear-cut conclusions cannot be drawn about any M–N bond length trends in these complexes. Where the M–I distances are concerned, the Np–I and Ce–I distances vary consistently as expected for the different coordination environments of 4Np–6Np and 4Ce–6Ce, but we note that for each isomorphous pair the Np–I distances are consistently shorter (∼0.02–0.05 Å) than the corresponding Ce–I distances.
The structure of 4Np, Figure 2, reveals a highly irregular six-coordinate Np ion, where the diphosphoniomethanide ligand adopts an “open-book” geometry.48 The two iodide ligands are approximately trans, though with quite an acute I-Np-I angle of 135.87(2)°, and the IMe4 NHC sits approximately trans to the methanide center, although again distorted far from the ideal (HC–Np–CNHC = 134.6(2)°). The Np–CHBIPM and the Np ← CNHC distances are 2.753(7) and 2.678(8) Å, respectively; we note that the former is ∼0.07 Å longer than the latter, despite their respective formal anionic and neutral charge states, likely reflecting the strongly donating nature of IMe4 and constraints of the (BIPMTMSH)1– anion chelate framework. The Np–CHBIPM distance in 4Np sits between the Np–C bond lengths of 2.574(4)–2.592(4) Å in [NpIII{C6H5C(H)NMe2}3]39 and 2.831(4) and 2.838(4) Å in 1, is slightly shorter than the Ce–CHBIPM distance of 2.806(9) Å in 3Ce, but is statistically indistinguishable (by the 3σ-criterion) from the Ce–CHBIPM distance of 2.768(6) Å in isostructural 4Ce; as expected, all are longer than the Np–C bonds (2.440(10) and 2.454(12) Å) in a previously reported NpIV silylamide double cyclometallate complex.38 The Np–CHBIPM distance in 4Np is ∼0.08 Å shorter than the U–CHBIPM distance of 2.827(3) Å in 3 U. However, we note that the M–CHBIPM distance in 3U is ∼0.02 Å longer than the corresponding distance in 3Ce, as expected from Shannon’s revised ionic radii (6-coordinate ions, Ce = 1.01; U = 1.03 Å)73 so it seems likely that the different M–CHBIPM distances in 4Np and 3U reflect the absence of the NHC ligand in the latter rather than an underlying Np vs U difference. There are no transuranium-NHC distances with which to compare the Np–CNHC distance in 4Np, but we note that the U ← CNHC distance in [U{N(SiMe3)2}3(IMe4)]56 is statistically indistinguishable at 2.672(5) Å. The Ce ← CNHC distance of 2.731(8) Å in 4Ce is significantly (∼0.06 Å) longer than the analogous distance in 4Np even though according to Shannon’s revised ionic radii, six-coordinate Ce and Np are both 1.01 Å.62
Figure 2.
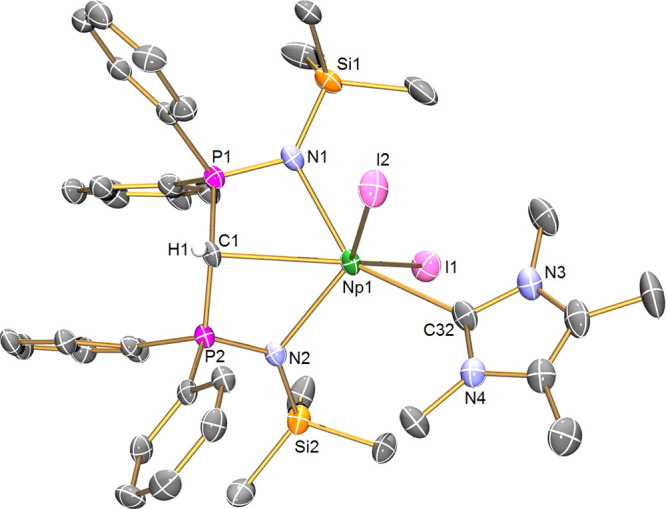
Solid-state molecular structure of complex 4Np at 100 K. Displacement ellipsoids are set at 50% probability and nonmethanide hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Np1-I1 3.0727(6), Np1-I2 3.1798(6), Np1-N1 2.423(6), Np1-N2 2.458(6), Np1-C1 2.753(7), Np1-C32 2.676(8), P1-N1 1.612(6), P1-C1 1.749(7), P2-N2 1.618(6), P2-C1 1.723(7), I1-Np1-I2 135.87(2), N1-Np1-I1 93.1(2), N1-Np1-I2 84.7(12), N1-Np1-N2 105.8(2), N1-Np1-C1 63.3(2), N1-Np1-C32 144.1(2), N2-Np1-I1 91.1(2), N2-Np1-I2 131.9(2), N2-Np1-C1 62.0(2), N2-Np1-C32 110.0(2), C1-Np1-I1 133.8(2), C1-Np1-I2 83.8(2), C32-Np1-I1 88.2(2), C32-Np1-I2 70.0(2), C32-Np1-C1 134.6(2), and P1-C1-P2 128.6(4).
The structure of 5Np, Figure 3, reveals a distorted octahedral Np ion, where the diphosphonio-alkylidene C-center is trans to a DME oxygen donor atom, the iodide is trans to the other DME oxygen donor atom, and the nitrogen donors can be considered to be trans to one another. Notably, the iodide is thus oriented cis with respect to the diphosphonio-alkylidene C-atom. The carbene adopts a T-shaped geometry, with a P-C-P angle of 170.4(5)° and a sum of angles at the CBIPM center of 359.9°, which in principle orients it favorably to engage in a double-bonding interaction with Np. The Np–CBIPM distance is found to be 2.425(7) Å. There is no transuranium precedent to compare the Np=CBIPM distance in 5Np to; however, we note that the Ce=CBIPM distance in isostructural 5Ce (2.477(2) Å) is longer by 0.052(2) Å. The Np=CBIPM distance in 5Np fits nicely into the trend established by previous U=CBIPM complexes, with, for example, [UVI(BIPMTMS)(Cl)2(O)], UVI=C = 2.184(3) Å; [UV(BIPMTMS)(Cl)2(I)], UV=C = 2.268(10) Å; [{UIV(BIPMTMS)(μ-Cl)(Cl)(THF)}2], UIV=C = 2.322(4) Å; [{UIII(BIPMTMS)}3{UIV(BIPMTMS)}3(μ-I)3(μ-η6:η6-C7H8)3], UIII=C = 2.413(8) and 2.47(2) Å, UIV=C = 2.398(7) and 2.30(3) Å.60,61 The P–C distances in 5Np (av. 1.640 Å) are contracted (∼0.1 Å) compared to the P–C distances in 4Np (av. 1.736 Å), reflecting the increased charge at the central carbon atom of (BIPMTMS)2– in 5Np instead of (BIPMTMSH)1– in 4Np. This can be rationalized by invoking dipolar electrostatic shortening rather than hyperconjugation or delocalization effects.74
Figure 3.
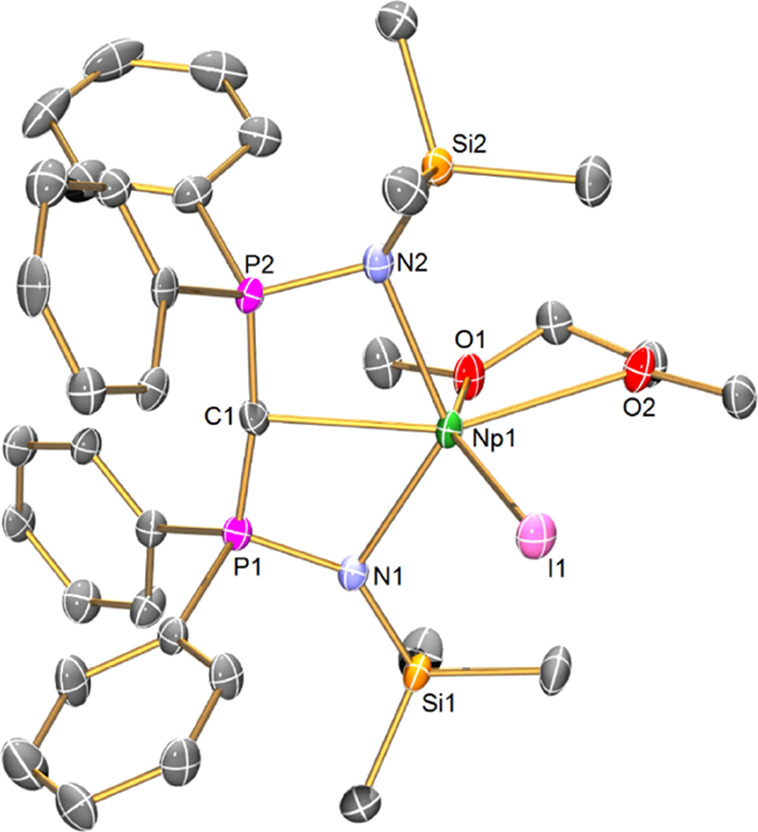
Solid-state molecular structure of complex 5Np at 100 K. Displacement ellipsoids are set at 50% probability and hydrogen atoms and lattice solvent are omitted for clarity. Selected bond lengths [Å] and angles [°]: Np1-I1 3.1065(5), Np1-O1 2.524(5), Np1-O2 2.636(5), Np1-N1 2.431(6), Np1-N2 2.414(6), Np1-C1 2.425(7), P1-N1 1.602(6), P1-C1 1.627(7), P2-N2 1.631(6), P2-C1 1.652(7), O1-Np1-I1 153.9(2), O1-Np1-O2 61.8(2), O2-Np1-I1 95.3(2), N1-Np1-I1 103.6(2), N1-Np1-O1 81.3(2), N1-Np1-O2 122.4(2), N2-Np1-I1 95.2(2), N2-Np1-O1 101.5(2), N2-Np1-O2 102.2(2), N2-Np1-N1 128.8(2), N2-Np1-C1 65.2(2), C1-Np1-I1 109.3(2), C1-Np1-O1 95.9(2), C1-Np1-O2 152.9(2), C1-Np1-N1 63.6(2), and P1-C1-P2 170.4(5).
The structure of 6Np, Figure 4, reveals an irregular six-coordinate Np ion, where the diphosphonio-alkylidene adopts an open book geometry,48 the two IMe4 NHCs are cis with respect to each other residing on the more open face presented by the (BIPMTMS)2– ligand, and the iodide resides on the opposite, more closed face. The P-C-P angle is 136.5(3)° and the sum of angles at the CBIPM center is 322.5°. This pyramidalization of the CBIPM in principle would be expected to make it a poorer donor center than the one in 5Np, which is consistent with the Np=CBIPM distance of 2.490(6) Å in 6Np, which is ∼0.07 Å longer than the corresponding Np=CBIPM distance in 5Np; the pyramidalization of the CBIPM center may reflect steric congestion and also that with so many strong donors the Np ion in 6Np may be quite electron-rich, which is consistent with the optical data (vide infra). Consistent with these observations, the P–C distances in 6Np (av. 1.673 Å) are slightly (0.03 Å) longer than the P–C distances in 5Np. Nevertheless, the Np=CBIPM distance in 6Np is slightly shorter than the corresponding Ce=CBIPM distance of 2.519(2) Å in isostructural 6Ce. The two Np ← CNHC distances are 2.677(5) and 2.751(6) Å – clearly longer than the formal Np=CBIPM distance, and one is statistically indistinguishable to the analogous Np ← CNHC distance in 4Np. One Np ← CNHC distance is indistinguishable from the U ← CNHC distance in [U{N(SiMe3)2}3(IMe4)], but the other is ∼0.07 Å longer, likely reflecting steric congestion at the Np ion. This pattern is also found in isostructural 6Ce with Ce ← CNHC distances of 2.737(3) and 2.806(2) Å, revealing that the Np ← CNHC distances in 6Np are consistently ∼0.06 Å shorter than the Ce ← CNHC distances in 6Ce despite the identical Shannon ionic radii of Np and Ce.
Figure 4.
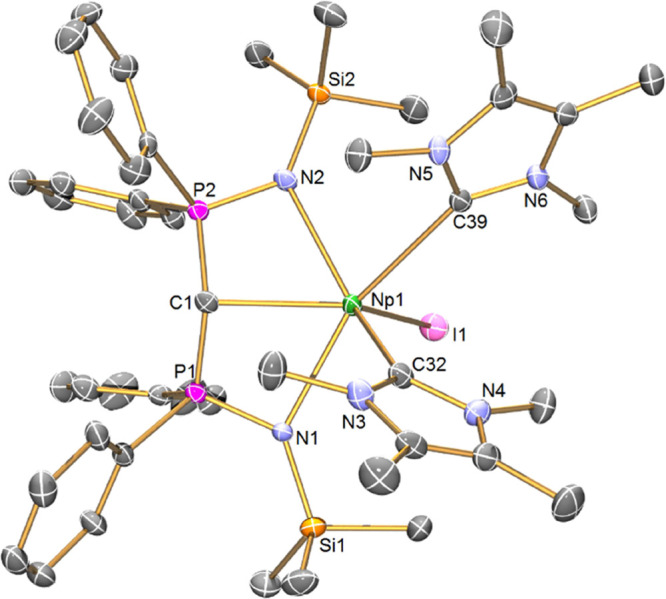
Solid-state molecular structure of complex 6Np at 120 K. Displacement ellipsoids are set at 50% probability and hydrogen atoms and lattice solvent are omitted for clarity. Selected bond lengths [Å] and angles [°]: Np1-I1 3.1571(4), Np1-N1 2.485(4), Np1-N2 2.492(5), Np1-C1 2.490(6), Np1-C32 2.677(5), Np1-C39 2.751(6), P1-N1 1.620(5), P1-C1 1.675(6), P2-N2 1.614(5), P2-C1 1.671(5), N1-Np1-I1 91.8(2), N1-Np1-N2 120.9(2), N1-Np1-C1 64.0(2), N1-Np1-C32 81.4(2), N1-Np1-C39 154.2(2), N2-Np1-I1 98.1(2), N2-Np1-C32 132.7(2), N2-Np1-C39 81.0(2), C1-Np1-I1 127.5(2), C1-Np1-N2 64.1(2), C1-Np1-C32 98.7(2), C1-Np1-C39 123.6(2), C32-Np1-I1 124.3(2), C32-Np1-C39 73.2(2), C39-Np1-I1 98.7(2), and P1-C1-P2 136.5(3).
Overall, the solid-state molecular structures for 4Np–6Np reveal that there are no substantial differences between NpIII and UIII where these Np–CHBIPM, Np=CBIPM, and Np ← CNHC distances are concerned – at least across the small range of comparable molecules. However, a clear trend emerges where Np exhibits consistently shorter Np=CBIPM and Np ← CNHC distances (∼0.03 and ∼0.06 Å, respectively) compared to isostructural Ce=CBIPM and Ce ← CNHC distances. The Np–CHBIPM and Ce–CHBIPM distances do not exhibit any statistically significant differences. While (BIPMTMS)2– enjoys considerable conformational flexibility, allowing facile variation in its donor strength to metals, and hence M=C distances, (BIPMTMSH)1– is more rigid. Thus, the similarity in Np–CHBIPM and Ce–CHBIPM bond distances likely reflects the constraints of this chelate.
Spectroscopic Analysis
The 1H nuclear magnetic resonance (NMR) spectra of 4Np–6Np are consistent with their NpIII 5f4 formulations, exhibiting paramagnetically shifted resonances in spectral windows up to 67 ppm. In particular, the CHBIPM resonance for 4Np is found at −54.6 ppm. The 31P NMR spectra are also characteristically paramagnetically shifted, and it is notable that the 31P chemical shift for 4Np (−488 ppm) shifts significantly when converted to 5Np and 6Np (−789 and −740 ppm), which for the latter are similar chemical shifts to [BIPMTMSUIV(X)n] complexes (X = alkyl, amide, imido; n = 2, 2, 1, respectively) which are 5f2 congeners that typically span the range −605 to −905 ppm.75−77
The UV–vis–NIR spectra of 4Np–6Np, Figure 5, are consistent with those of their NpIII formulations.6,38,64,71,78−80 In particular, broad pairs of absorptions, presumed to be Laporte allowed f-d transitions, are found in the 16,000–26,000 cm–1 region (ε = ∼1500 M–1 cm–1). As the ligand fields change from 4Np to 5Np to 6Np the pairs of bands shift and can be ordered energetically as 5Np > 4Np > 6Np. The most electron-rich Np would, simplistically, be expected to have the smallest f-d energy gap, and indeed, this is consistent with the ligand field at 6Np and that the pair of f-d absorptions for 6Np are lowest in energy of the series. While comparisons using 5Np are complicated by it being the sole complex with DME in the coordination sphere of Np, 4Np and 6Np are more closely related, with BIPM, iodide, and NHC ligands common to both and here the f-d energy ordering of 5Np > 6Np is clear, reflecting the presence of the strongly donating diphosphonio-alkylidene in the latter. The NIR regions of the UV–vis–NIR spectra of 4Np–6Np, Figure 5 inset, exhibit multiple weak absorptions assigned as Laporte forbidden f-f transitions whose overall patterns are characteristic of NpIII.6,38,64,71,78−80 The absorption bands at ∼10,000, ∼11,500, and ∼12,500 cm–1 can be assigned to the 5I7, 5F3, and 5G3/5I8/5S2 transitions of NpIII,81 and we note that the intensities and fwhm values for 5Np (ε = ∼90 M–1 cm–1, fwhm = 530, 1000, 1108, av. 879 cm–1) and 6Np (ε = ∼75 M–1 cm–1, fwhm = 426, 909, 1095, av. 810 cm–1) are slightly larger than those for 4Np (ε = ∼50 M–1 cm–1, fwhm = 418, 842, 1045, av. 768 cm–1). Though the changes are modest, this may reflect the presence of the strong diphosphonio-alkylidene donors in the former pair compared to the diphosphoniomethanide in the latter, which in turn would invoke Np 5f-orbital contributions to the bonding of these complexes, which is indeed supported by density functional theory (DFT) calculations (vide infra).
Figure 5.

Comparison of solution UV–vis–NIR spectra of 4Np (black line, 0.49 mM), 5Np (blue line, 0.51 mM), and 6Np (red line, 0.58 mM), all in toluene shown between 7000 and 35,000 cm–1 (1429–286 nm) at ambient temperature. Inset: Expanded view of ∼8000–16,000 cm–1 region.
Quantum Chemical Calculations
To probe the nature of the Np–C interactions in 4Np–6Np, we performed DFT calculations. Because 4Np–6Np are experimentally authenticated, we calculated the experimentally inaccessible, and hence hypothetical, 4U–6U complexes, using structurally validated 4Np–6Np to give confidence in the DFT results and provide 5f3 UIII vs 5f4 NpIII comparisons. Given the ionic radii size match of NpIII and CeIII, and with 4Ce–6Ce structurally authenticated, we also performed DFT calculations on 4Ce–6Ce to provide an isostructural LnIII vs AnIII comparison. Noting that CeIII is a good size match to NpIII, but with different f-electron counts of 4f1 and 5f4, respectively, we also performed DFT calculations on hypothetical 4Pm–6Pm because PmIII is 4f4 and isoelectronic to 5f4 NpIII. Confidence in the hypothetical Pm models is afforded by the structurally confirmed 4Ce–6Ce. Given the good agreement between the gas-phase geometry optimized and solid-state metrical data, where comparisons are available, the DFT data can be considered to represent a reliable qualitative model of the electronic structures (Table 1) and hence a representative picture to probe bonding differences suggested by the solid-state metrical data. Note that there is little structural variance in the computed M–N bonding (Δmax/av. +0.04/+0.02 Å between Np and Ce structures), in line with the experimental solid-state metrical data, hence we focus discussion on the M–C bond interactions.
Table 1. Selected Computed Properties for 4M–6M (M = Np, U, Ce, Pm).
| bond and
indices |
chargesd |
spin densitiese |
NBO M-C
σ-bond component (%)f |
NBO M-C π-bond component (%)f |
QTAIMh |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpda | bondb | BIc | M | C | M | C | Mg | Cg | M s/p/d/f | M | C | M s/p/d/f | ρ | ε |
| 4Np | Np–CHBIPM | 0.59 | 1.51 | –1.64 | 4.21 | –0.01 | 9 | 91 | 9/0/45/46 | 0.04 | 0.05 | |||
| Np ← CNHC | 0.83 | –0.43 | –0.02 | 0 | 100 | 0.05 | 0.01 | |||||||
| 5Np | Np=CBIPM | 1.40 | 1.54 | –1.96 | 4.36 | –0.07 | 17 | 83 | 4/1/32/63 | 14 | 86 | 0/0/38/62 | 0.08 | 0.21 |
| 6Np | Np=CBIPM | 1.20 | 1.51 | –1.64 | 4.22 | –0.05 | 15 | 85 | 9/1/39/51 | 10 | 90 | 0/1/43/56 | 0.08 | 0.18 |
| Np ← CNHC | 0.65 | –0.44 | –0.03 | 0 | 100 | 0.04 | 0.03 | |||||||
| Np ← CNHC | 0.69 | –0.46 | –0.03 | 0 | 100 | 0.05 | 0.03 | |||||||
| 4U | U–CHBIPM | 0.58 | 1.58 | –1.65 | 3.09 | –0.01 | 9 | 91 | 8/0/47/45 | 0.04 | 0.05 | |||
| U ← CNHC | 0.82 | –0.45 | –0.02 | 0 | 100 | 0.05 | 0.01 | |||||||
| 5U | U=CBIPM | 1.28 | 1.57 | –2.00 | 3.28 | –0.04 | 14 | 86 | 4/1/42/53 | 13 | 87 | 0/0/40/60 | 0.08 | 0.20 |
| 6U | U=CBIPM | 1.17 | 1.62 | –1.67 | 3.08 | –0.04 | 14 | 86 | 10/1/46/43 | 10 | 90 | 0/1/50/49 | 0.08 | 0.17 |
| U ← CNHC | 0.77 | –0.50 | –0.03 | 0 | 100 | 0.05 | 0.03 | |||||||
| U ← CNHC | 0.81 | –0.48 | –0.03 | 0 | 100 | 0.05 | 0.03 | |||||||
| 4Ce | Ce–CHBIPM | 0.46 | 1.20 | –1.54 | 1.04 | –0.01 | 0 | 100 | 0.04 | 0.05 | ||||
| Ce ← CNHC | 0.60 | –0.31 | –0.01 | 0 | 100 | 0.04 | 0.01 | |||||||
| 5Ce | Ce=CBIPM | 1.05 | 1.32 | –1.82 | 1.07 | –0.01 | 10 | 90 | 1/1/61/37 | 8 | 92 | 0/0/65/35 | 0.07 | 0.22 |
| 6Ce | Ce=CBIPM | 0.96 | 1.29 | –1.53 | 1.01 | –0.01 | 9 | 91 | 7/1/65/27 | 7 | 93 | 2/1/60/37 | 0.07 | 0.19 |
| Ce ← CNHC | 0.52 | –0.38 | –0.01 | 0 | 100 | 0.04 | 0.03 | |||||||
| Ce ← CNHC | 0.58 | –0.36 | –0.01 | 0 | 100 | 0.04 | 0.03 | |||||||
| 4Pm | Pm–CHBIPM | 0.31 | 1.06 | –1.47 | 4.38 | –0.04 | 10 | 90 | 5/0/32/63 | 0.04 | 0.05 | |||
| Pm ← CNHC | 0.39 | –0.26 | –0.05 | 0 | 100 | 0.04 | 0.05 | |||||||
| 5Pm | Pm=CBIPM | 0.94 | 1.26 | –1.76 | 4.40 | –0.02 | 18 | 82 | 1/0/24/75 | 19 | 81 | 0/0/20/80 | 0.07 | 0.16 |
| 6Pm | Pm=CBIPM | 0.76 | 1.11 | –1.48 | 4.39 | –0.02 | 15 | 85 | 5/1/31/63 | 14 | 86 | 1/0/24/75 | 0.06 | 0.13 |
| Pm ← CNHC | 0.28 | –0.31 | –0.02 | 0 | 100 | 0.03 | 0.05 | |||||||
| Pm ← CNHC | 0.33 | –0.29 | –0.02 | 0 | 100 | 0.04 | 0.02 | |||||||
All compounds geometry optimized without symmetry constraints at the BP86 TZP/ZORA (all-electron) level.
M–C bond: M–CHBIPM = methanide of (BIPMTMSH)1–; M ← CNHC = IMe4 NHC carbene; M=CBIPM = diphosphonio-alkylidene of (BIPMTMS)2–.
Nalewajski–Mrozek bond indices.
MDCq charges.
MDCm spin densities.
Natural bond orbital (NBO) analysis.
Values of 0% for the total M contribution to the M–C bond mean that the M contribution is below the cut-off threshold of NBO (5%).
Quantum Theory of Atoms in Molecules (QTAIM) bond critical point topological electron density (ρ) and ellipticity (ε) analysis.
As expected, the Np–CHBIPM bond order in 4Np (0.59) reveals a polarized linkage, but conversion of that linkage to Np=CBIPM gives bond indices for 5Np and 6Np (1.40 and 1.20) that are at least double that of 4Np, reflecting their formal polarized-covalent single- and double-bond natures, respectively. The dative Np ← CNHC bond orders are broadly similar to the covalent Np–CHBIPM bond index, reflecting the strong donor nature of IMe4, but this metric is noticeably larger for the Np ← CNHC bond in 4Np (0.83) than 6Np (av. 0.67) reflecting the weaker donor strength of (BIPMTMSH)1– compared to (BIPMTMS)2–. The computed charges and spin densities are overall consistent with NpIII ions (av. 1.52, 4.26, respectively) and charge donation from the ligands to Np centers. Highly electrostatic Np–CHBIPM and Np ← CNHC interactions are returned by NBO analyses, but polarized-covalent Np=CBIPM twofold bonding interactions with Np contributions of 10–17% are confirmed, as exemplified by the Np=CBIPM bond of 5Np, Figure 6 and Table 1 (and see the Supporting Information). QTAIM analysis of 4Np–6Np confirms the anticipated polarized-covalent nature of the Np=CBIPM bonds in 5Np and 6Np, again with more covalent Np=CBIPM than Np ← CNHC bonds. The bond critical point ellipticity values reveal cylindrical single bonds for the Np–CHBIPM and Np ← CNHC interactions (ε values close to zero) and asymmetric double-bond interactions for the Np=CBIPM linkages (ε values that deviate substantially from zero, for example, benzene and ethene have ε values of 0.23 and 0.45, respectively).82
Figure 6.
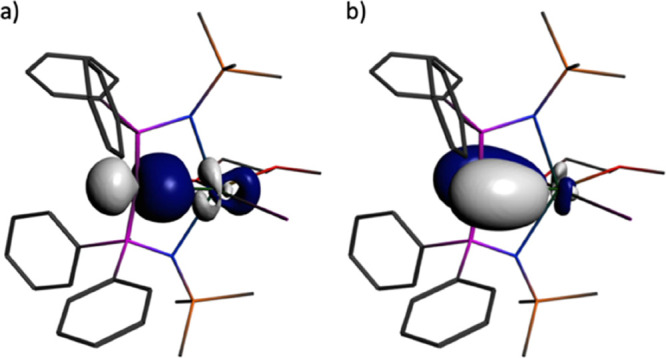
NBO representations of the Np=CBIPM σ- and π-bond interaction in 5Np. (a) Np=CBIPM σ-bond. (b) Np=CBIPM π-bond. Hydrogen atoms are omitted for clarity.
Previous work on UIV=CBIPM complexes, using the same level of theory for the calculations on 5Np and 6Np (BP86, all-electron ZORA TZP), found total UIV contributions averaging ∼15–18%, with bond orders of ∼1.4. Those data are remarkably similar to those found for NpIII5Np and 6Np. To account for this, two competing effects merit consideration. For equivalent oxidation states, Np has a greater effective nuclear charge than U, so the radial distribution of the 5f- and 6d-orbitals will be smaller for Np than U. However, as the oxidation state is decreased, the 5f- and 6d-orbitals will in principle expand. Therefore, we tentatively suggest when considering NpIII in relation to UIII, and then UIV, that 5f- and 6d-orbital contraction from increasing Zeff between equivalent oxidation states may be offset when moving to U with an oxidation state one unit higher (at least in the context of these An=CBIPM bonding interactions). The result would be that, due to net equalization of these competing effects, NpIII in the An=CBIPM ligand field is roughly equivalent to UIV with a result that total Np contributions to the NpIII=CBIPM bonds in 5Np and 6Np appears to be similar to UIV=CBIPM congeners, noting that the 5f orbital character dominates overall. However, the NpIII=CBIPM σ- and π-bonds of 5Np and 6Np (av. 58% 5f character) exhibit a considerably less 5f character than UIV=CBIPM complexes (∼70–90% 5f character), which is counterbalanced for Np by increased 7s and 6d orbital participation for the σ-bonds and mainly 6d orbital participation for the π-bonds. An additional difference is the consistently significant 7s contributions (4–9%) from Np to the σ-components of the NpIII=CBIPM bonds which has not been found for UIV=CBIPM bonds.
The above analysis provides a baseline from which Np3+-containing 4Np–6Np is compared to hypothetical U3+-containing 4U–6U congeners with confidence. The M–CHBIPM bond orders for 4Np and 4U are very similar (∼0.59), with 5Np having a higher bond order (1.40) than 5U (1.28), but the opposite is found for 6U (1.17) vs 6Np (1.20). The NBO data reveal that the U ← CNHC bonds are largely electrostatic and invariant, which is the same as the Np ← CNHC bonds. The UIII=CBIPM bonds are similar to the NpIII=CBIPM bonds but slightly more polarized in terms of total metal contributions (9–14 vs 9–17%, respectively). While the π-bonds are little changed from NpIII to UIII, still having dominant 5f character (but reduced compared to UIV=CBIPM complexes), the σ-bonds for U have approximately equal 6d vs 5f orbital contributions but, as was found for Np, significant 7s contributions (4–10%) are revealed. Thus, it would seem that AnIII=CBIPM bonds for both An = U and Np consistently exhibit significant (∼4–10%) 7s contributions that are not present in UIV=CBIPM complexes. We note that the QTAIM data for 4U–6U are quite similar to those of 4Np–6Np and confirm the presence of UIII=CBIPM double-bond interactions. Previously, we extrapolated bond orders and U% contributions to the UIII=CBIPM bonds in the mixed-valent [{UIII(BIPMTMS)}3{UIV(BIPMTMS)}3(μ-I)3(μ-η6:η6-C7H8)3] molecule of ∼1.2 and 13%, respectively.14 These data certainly fit well into the trends of U over oxidation states of III-VI (UIII, ∼13; UIV, ∼18; UV, ∼26; UVI, ∼28%) and the data in Table 1. However, the presence of arene bridges and the large number of basis functions for [{UIII(BIPMTMS)}3{UIV(BIPMTMS)}3(μ-I)3(μ-η6:η6-C7H8)3] meant that these were extrapolations at best. It is thus notable that those values compare very well to the computed values for 4U–6U (9, 14, and 14% respectively), which has been enabled with confidence by being benchmarked against 4Np–6Np, demonstrating the value of accessing transuranium targets when U congeners are experimentally unavailable.
Next, we turn our attention to comparing the data for 4Np–6Np to 4Ce–6Ce because both are experimentally validated series of complexes containing central metal ions with identical Shannon ionic radii. We noted in the analysis of structural data above that the M–CHBIPM bond distances are largely the same for 4Np vs 4Ce, but the M=CBIPM and M ← CNHC bonds tended to be shorter in 5Np and 6Np vs 5Ce and 6Ce. In line with those data, the computed bond metrics largely follow the same pattern, resulting in lower bond orders on a like-for-like basis for Ce (Ce–CBIPM 0.46, Ce=CBIPM av. 1.00, Ce ← CNHC av. 0.57) vs Np (Np–CBIPM 0.59, Np=CBIPM av. 1.30, Np ← CNHC av. 0.72). This pattern translates through to the NBO analysis, where for each system the Ce contributions (7–10%) to given polarized-covalent bonds are about 60% of the corresponding Np values (9–17%). The Ce=CBIPM σ- and π-bonding components are dominated by 5d character (av. 63%), with an approximate 2:1 ratio of 5d:4f character. Also, analogously to Np, 6s character is found (1–7%) for the Ce=CBIPM σ-bonds, which is significant but slightly lower than the corresponding Np 7s contributions to the Np=CBIPM bonds (4–9%).
Noting the clear differences between the computed electronic structures of 5f44Np–6Np and 4f14Ce–6Ce, we finally compare isoelectronic 5f44Np–6Np vs 4f44Pm–6Pm, where the experimentally anchored calculations on 4Ce–6Ce provide confidence in extending the models and computational methods to 4Pm–6Pm. Notably, the computed Pm–C distances are, like-for-like, always slightly longer for 4Pm–6Pm vs 4Np–6Np, but, as anticipated, the Pm–C distances are shorter than the corresponding Ce–C distances because of the increased effective nuclear charge and lanthanide contraction. However, while the Np–C bond orders (0.59–1.40) are like-for-like larger than the Pm–C bond orders (0.28–0.94), the latter are also consistently lower than the corresponding Ce–C bond orders (0.46–1.05). Inspection of the NBO data of 4Pm–6Pm reveals two notable points. First, the Pm% contributions to the Pm–CHBIPM (10%) and Pm=CBIPM (14–19%) bonds of 4Pm–6Pm are similar to the Np% of the Np–CHBIPM (9%) and Np=CBIPM (10–17%) bonds of 4Np–6Np and are thus significantly larger than the corresponding Ce data (7–10%). Second, whereas the Ce bonding is dominated by 5d character (av. 63%), for 4Pm–6Pm the bonding is dominated by 4f character (av. 71%), more than the 5f character of the Np congeners (av. 56%), along with 6s contributions (1–5%). The high Pm% contributions to the bonding but low bond orders at first sight may seem contradictory, but we suggest that this is an example of 4f mixing due to a good energy match with the CBIPM orbitals but poor spatial overlap.83,84 Though the differences are small, this would appear to be the case based on the QTAIM data, where the Pm values are consistently smaller than the Np values.
The computational results can be summarized as follows: (i) decreased f orbital contributions to bonding with MIII ions (relative to MIV ions) can be compensated for by s and d contributions; (ii) the NpIII=CBIPM systems are overall comparable to UIV=CBIPM analogues; (iii) Np is the most covalent (by total metal% contribution to the bonding) of Np, U, Ce, and Pm for these MIII=CBIPM complexes; (iv) the bonding of the Ce complexes is dominated by 5d character; (v) the bonding of the Pm complexes is dominated by 4f character, but here the covalency is likely due to good energy matching and not spatial overlap; (vi) the M–CNHC bonding is consistently highly electrostatic for all complexes; and (vii) by isolating and characterizing NpIII=CBIPM complexes it has been possible to complete benchmarking of U contributions in U=CBIPM bonding over U oxidation states of III-VI.
Preliminary Reactivity Assessment
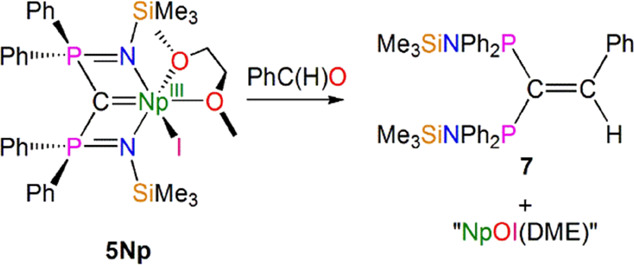
A preliminary reactivity study of 5Np reveals metallo-Wittig reactivity, as anticipated for a species with a formal Np=C double-bond interaction.47,62,85 In particular, benzaldehyde reacts with 5Np to afford the alkene product PhC(H)=C(PPh2NSiMe3)2 (7), Scheme 2, as evidenced by 1H and 31P NMR spectroscopic data.62 As far as we are aware, the reaction of 5Np with benzaldehyde constitutes the first multiple bond metathesis reaction in transuranium chemistry, and indeed AnIII-chemistry more broadly given the dearth of AnIII-ligand multiple bonding. The metallo-Wittig reactivity of 5Np is complementary to the metallo-Wittig reactivity already established for U-analogues in oxidation states IV-VI,47,62,85 showing now that An=C double bonds can execute multiple bond metathesis over the full range of commonly accessible An oxidation states (III-VI). The reactivity of 5Np also forges a link to the metallo-Wittig reactivity of LnIII=C bonds, providing a bridge between trivalent M=C bond metathesis chemistry of Ln and An ions.
Scheme 2. Reaction of 5Np with Benzaldehyde to Produce the Alkene 7.
Conclusions
To conclude, we have reported the synthesis and characterization of transuranium-carbon polarized-covalent σ2π2 multiple and dative σ2 single bond interactions. The new diphosphoniomethanide-, diphosphonio-alkylidene-, and N-heterocyclic carbene-neptunium(III) derivatives reported here include unambiguous examples of trivalent An-ligand multiple bonds, and their synthesis was facilitated by adopting a scaled-down prechoreographed approach using CeIII synthetic surrogates. The elucidation of periodic trends across the 5f series, and comparisons to the 4f elements, is often grounded in the ability to isolate and characterize homologous series of molecules and driven by the establishment of Th and U chemistry first and subsequently followed by transuranium congeners. Consequently, the work reported here highlights an instance where nonaqueous low-valent transuranium chemistry provides the bonding motif precedent enabling comparison of M=C bonding to LnIII congeners and early An=C bonding in other An oxidation states. A preliminary assessment of reactivity has introduced multiple bond metathesis to transuranium chemistry, together with prior examples of AnIV-VI reactivity now extending the range of An metallo-Wittig reactions to encompass oxidation states III-VI overall and providing an AnIII comparison to LnIII congeners.
Acknowledgments
We gratefully acknowledge funding and support from the UK EPSRC (EP/P001386/1 and EP/M027015/1), EU ERC (GoG612724), U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, Heavy Element Chemistry Program at Los Alamos National Laboratory (LANL) (A.J.G., J.M., B.L.S.; contract DE-AC52-06NA25396) for experimental Np chemistry, LANL Laboratory Directed Research and Development program for a Distinguished J. R. Oppenheimer Postdoctoral Fellowship (C.A.P.G.; LANL-LDRD 20180703PRD1), and The University of Manchester including computational resources and associated support services of the Computational Shared Facility. S.T.L. thanks the Alexander von Humboldt Foundation for a Friedrich Wilhelm Bessel Research Award. We thank the anonymous referees for their constructive and helpful comments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c02152.
Experimental and computational details and X-ray crystallographic, spectroscopic, magnetic, and quantum chemical calculations (PDF)
Accession Codes
CCDC 2125323–2125331 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033. All other data are available from the authors on reasonable request.
The authors declare no competing financial interest.
Supplementary Material
References
- Hayton T. W. Metal-ligand multiple bonding in uranium: structure and reactivity. Dalton Trans. 2010, 39, 1145–1158. 10.1039/B909238B. [DOI] [PubMed] [Google Scholar]
- Liddle S. T. The Renaissance of Non-Aqueous Uranium Chemistry. Angew. Chem., Int. Ed. 2015, 54, 8604–8641. 10.1002/anie.201412168. [DOI] [PubMed] [Google Scholar]
- Hartline D. R.; Meyer K. From Chemical Curiosities and Trophy Molecules to Uranium-Based Catalysis: Developments for Uranium Catalysis as a New Facet in Molecular Uranium Chemistry. JACS Au 2021, 1, 698–709. 10.1021/jacsau.1c00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayton T. W. Recent developments in actinide-ligand multiple bonding. Chem. Commun. 2013, 49, 2956–2973. 10.1039/C3CC39053E. [DOI] [PubMed] [Google Scholar]
- Brown J. L.; Batista E. R.; Boncella J. M.; Gaunt A. J.; Reilly S. D.; Scott B. L.; Tomson N. C. A Linear trans-Bis(imido) Neptunium(V) Actinyl Analog: NpV(NDipp)2(tBu2bipy)2Cl (Dipp = 2,6-iPr2C6H3). J. Am. Chem. Soc. 2015, 137, 9583–9586. 10.1021/jacs.5b06667. [DOI] [PubMed] [Google Scholar]
- Dutkiewicz M. S.; Goodwin C. A. P.; Perfetti M.; Gaunt A. J.; Griveau J.-C.; Colineau E.; Kovács A.; Wooles A. J.; Caciuffo R.; Walter O.; Liddle S. T. A Terminal Neptunium(V)-Mono(Oxo) Complex. Nat. Chem. 2022, 14, 342–349. 10.1038/s41557-021-00858-0. [DOI] [PubMed] [Google Scholar]
- Pereira C. C.; Marsden C. J.; Marçalo J.; Gibson J. K. Actinide sulfides in the gas phase: experimental and theoretical studies of the thermochemistry of AnS (An = Ac, Th, Pa, U, Np, Pu, Am and Cm). Phys. Chem. Chem. Phys. 2011, 13, 12940–12958. 10.1039/C1CP20996E. [DOI] [PubMed] [Google Scholar]
- Infante I.; Kovacs A.; La Macchia G.; Shahi A. R.; Gibson J. K.; Gagliardi L. Ionization energies for the actinide mono- and dioxides series, from Th to Cm: theory versus experiment. J. Phys. Chem. A 2010, 114, 6007–6015. 10.1021/jp1016328. [DOI] [PubMed] [Google Scholar]
- Marçalo J.; Gibson J. K. Gas-phase energetics of actinide oxides: an assessment of neutral and cationic monoxides and dioxides from thorium to curium. J. Phys. Chem. A 2009, 113, 12599–12606. 10.1021/jp904862a. [DOI] [PubMed] [Google Scholar]
- Gibson J. K.; Haire R. G.; Marçalo J.; Santos M.; Pires de Matos A.; Mrozik M. K.; Pitzer R. M.; Bursten B. E. Gas-Phase Reactions of Hydrocarbons with An+ and AnO+ (An = Th, Pa, U, Np, Pu, Am, Cm): The Active Role of 5f Electrons in Organoprotactinium Chemistry. Organometallics 2007, 26, 3947–3956. 10.1021/om700329h. [DOI] [Google Scholar]
- Gibson J. K. Actinide Gas-Phase Chemistry: Reactions of An+ and AnO+ [An = Th, U, Np, Pu, Am] with Nitriles and Butylamine. Inorg. Chem. 1999, 38, 165–173. 10.1021/ic980908e. [DOI] [Google Scholar]
- Gibson J. K. Gas-Phase Transuranium Organometallic Chemistry: Reactions of Np+, Pu+, NpO+, and PuO+ with Alkenes. J. Am. Chem. Soc. 1998, 120, 2633–2640. 10.1021/ja973182e. [DOI] [Google Scholar]
- Kovács A.; Konings R. J. M.; Gibson J. K.; Infante I.; Gagliardi L. Quantum chemical calculations and experimental investigations of molecular actinide oxides. Chem. Rev. 2015, 115, 1725–1759. 10.1021/cr500426s. [DOI] [PubMed] [Google Scholar]
- Wooles A. J.; Mills D. P.; Tuna F.; McInnes E. J. L.; Law G. T. W.; Fuller A. J.; Kremer F.; Ridgway M.; Lewis W.; Gagliardi L.; Vlaisavljevich B.; Liddle S. T. Uranium(III)-carbon multiple bonding supported by arene δ-bonding in mixed-valence hexauranium nanometre-scale rings. Nat. Commun. 2018, 9, 2097. 10.1038/s41467-018-04560-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long B. N.; Beltrán-Leiva M. J.; Celis-Barros C.; Sperling J. M.; Poe T. N.; Baumbach R. E.; Windorff C. J.; Albrecht-Schönzart T. E. Cyclopentadienyl coordination induces unexpected ionic Am-N bonding in an americium bipyridyl complex. Nat. Commun. 2022, 13, 201. 10.1038/s41467-021-27821-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick B. D.; Sattelberger A. P.; Avens L. R.. Transuranium Organometallics Elements: The Next Generation. In Transuranium Elements: A Half Century, Morss L. R.; Fuger J., Eds.; American Chemical Society: Washington DC, 1992; 239–247. [Google Scholar]
- Marks T. J.; Fischer R. D., Eds. Organometallics of the f-Elements: Proceedings of the NATO Advanced Study Institute, Sogesta, Urbino, Italy, Sept, 1978; Springer: Dordrecht, 1978. [Google Scholar]
- De Ridder D. J. A.; Rebizant J.; Apostolidis C.; Kanellakopulos B.; Dornberger E. Bis(cyclooctatetraenyl)neptunium(IV). Acta Crystallogr. C 1996, 52, 597–600. 10.1107/S0108270195013047. [DOI] [Google Scholar]
- Magnani N.; Apostolidis C.; Morgenstern A.; Colineau E.; Griveau J. C.; Bolvin H.; Walter O.; Caciuffo R. Magnetic memory effect in a transuranic mononuclear complex. Angew. Chem., Int. Ed. 2011, 50, 1696–1698. 10.1002/anie.201006619. [DOI] [PubMed] [Google Scholar]
- Walter O. Actinide Organometallic Complexes with π-Ligands. Chem. – Eur. J. 2019, 25, 2927–2934. 10.1002/chem.201803413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold P. L.; Dutkiewicz M. S.; Walter O. Organometallic Neptunium Chemistry. Chem. Rev. 2017, 117, 11460–11475. 10.1021/acs.chemrev.7b00192. [DOI] [PubMed] [Google Scholar]
- Goodwin C. A. P.; Su J.; Stevens L. M.; White F. D.; Anderson N. H.; Auxier J. D. II; Albrecht-Schönzart T. E.; Batista E. R.; Briscoe S. F.; Cross J. N.; Evans W. J.; Gaiser A. N.; Gaunt A. J.; James M. R.; Janicke M. T.; Jenkins T. F.; Jones Z. R.; Kozimor S. A.; Scott B. L.; Sperling J. M.; Wedal J. C.; Windorff C. J.; Yang P.; Ziller J. W. Isolation and Characterization of a Californium Metallocene. Nature 2021, 599, 421–424. 10.1038/s41586-021-04027-8. [DOI] [PubMed] [Google Scholar]
- Karraker D. G.; Stone J. A. Bis(cyclooctatetraenyl)neptunium(III) and -plutonium(III) compounds. J. Am. Chem. Soc. 1974, 96, 6885–6888. 10.1021/ja00829a012. [DOI] [Google Scholar]
- Karraker D. G.; Stone J. A.; Jones E. R.; Edelstein N. Bis(cyclooctatetraenyl)neptunium(IV) and bis(cyclooctatetraenyl)plutonium(IV). J. Am. Chem. Soc. 1970, 92, 4841–4845. 10.1021/ja00719a014. [DOI] [Google Scholar]
- Windorff C. J.; Sperling J. M.; Albrecht-Schönzart T. E.; Bai Z.; Evans W. J.; Gaiser A. N.; Gaunt A. J.; Goodwin C. A. P.; Hobart D. E.; Huffman Z. K.; Huh D. N.; Klamm B. E.; Poe T. N.; Warzecha E. A Single Small-Scale Plutonium Redox Reaction System Yields Three Crystallographically-Characterizable Organoplutonium Complexes. Inorg. Chem. 2020, 59, 13301–13314. 10.1021/acs.inorgchem.0c01671. [DOI] [PubMed] [Google Scholar]
- Apostolidis C.; Walter O.; Vogt J.; Liebing P.; Maron L.; Edelmann F. T. A Structurally Characterized Organometallic Plutonium(IV) Complex. Angew. Chem., Int. Ed. 2017, 56, 5066–5070. 10.1002/anie.201701858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J.; Windorff C. J.; Batista E. R.; Evans W. J.; Gaunt A. J.; Janicke M. T.; Kozimor S. A.; Scott B. L.; Woen D. H.; Yang P. Identification of the Formal +2 Oxidation State of Neptunium: Synthesis and Structural Characterization of {NpII[C5H3(SiMe3)2]3}1−. J. Am. Chem. Soc. 2018, 140, 7425–7428. 10.1021/jacs.8b03907. [DOI] [PubMed] [Google Scholar]
- Windorff C. J.; Chen G. P.; Cross J. N.; Evans W. J.; Furche F.; Gaunt A. J.; Janicke M. T.; Kozimor S. A.; Scott B. L. Identification of the Formal +2 Oxidation State of Plutonium: Synthesis and Characterization of {PuII[C5H3(SiMe3)2]3}−. J. Am. Chem. Soc. 2017, 139, 3970–3973. 10.1021/jacs.7b00706. [DOI] [PubMed] [Google Scholar]
- Apostolidis C.; Dutkiewicz M. S.; Kovács A.; Walter O. Solid-State Structure of Tris-Cyclopentadienide Uranium(III) and Plutonium(III). Chem. – Eur. J. 2018, 24, 2841–2844. 10.1002/chem.201704845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutkiewicz M. S.; Farnaby J. H.; Apostolidis C.; Colineau E.; Walter O.; Magnani N.; Gardiner M. G.; Love J. B.; Kaltsoyannis N.; Caciuffo R.; Arnold P. L. Organometallic neptunium(III) complexes. Nat. Chem. 2016, 8, 797–802. 10.1038/nchem.2520. [DOI] [PubMed] [Google Scholar]
- Laubereau P. G. The formation of dicyclopentadienylberkeliumchloride. Inorg. Nucl. Chem. Lett. 1970, 6, 611–616. 10.1016/0020-1650(70)80057-5. [DOI] [Google Scholar]
- Bagnall K. W.; Payne G. F.; Alcock N. W.; Flanders D. J.; Brown D. Actinide structural studies. Part 8. Some new oxygen-donor complexes of trichloro(cyclopentadienyl)neptunium(IV); the crystal structure of trichloro(η5-cyclopentadienyl)bis(methyldiphenylphosphine oxide)neptunium(IV). J. Chem. Soc., Dalton Trans. 1986, 783–787. 10.1039/DT9860000783. [DOI] [Google Scholar]
- Bagnall K. W.; Plews M. J.; Brown D.; Fischer R. D.; Klähne E.; Landgraf G. W.; Sienel G. R. Anionic tris(cyclopentadienyl)actinide(IV) complexes. J. Chem. Soc., Dalton Trans. 1982, 1999–2007. 10.1039/DT9820001999. [DOI] [Google Scholar]
- Bagnall K. W.; Plews M. J.; Brown D. Tris(cyclopentadienyl)plutonium(IV) chloride and thiocyanate, (η5-C5H5)3PuCl and (η5-C5H5)3Pu(NCS). J. Organomet. Chem. 1982, 224, 263–266. 10.1016/S0022-328X(00)85838-6. [DOI] [Google Scholar]
- Bagnall K. W.; Payne G. F.; Brown D. Phosphine oxide complexes of cyclopentadienyl neptunium(IV) and plutonium(IV) N-thiocyanates. J. Less Common Met. 1986, 116, 333–339. 10.1016/0022-5088(86)90666-1. [DOI] [Google Scholar]
- Karraker D. G.; Stone J. A. Covalency of neptunium(IV) tris(cyclopentadienyl) compounds from Mössbauer spectra. Inorg. Chem. 1979, 18, 2205–2207. 10.1021/ic50198a031. [DOI] [Google Scholar]
- Karraker D. G.Reaction of Plutonium Metal with Diiodoethane. In Plutonium Chemistry; American Chemical Society: 1983; 216, 41–48. [Google Scholar]
- Staun S. L.; Stevens L. M.; Smiles D. E.; Goodwin C. A. P.; Billow B. S.; Scott B. L.; Wu G.; Tondreau A. M.; Gaunt A. J.; Hayton T. W. Expanding the Nonaqueous Chemistry of Neptunium: Synthesis and Structural Characterization of [Np(NR2)3Cl], [Np(NR2)3Cl]−, and [Np{N(R)(SiMe2CH2)}2(NR2)]− (R = SiMe3). Inorg. Chem. 2021, 60, 2740–2748. 10.1021/acs.inorgchem.0c03616. [DOI] [PubMed] [Google Scholar]
- Myers A. J.; Tarlton M. L.; Kelley S. P.; Lukens W. W.; Walensky J. R. Synthesis and Utility of Neptunium(III) Hydrocarbyl Complex. Angew. Chem., Int. Ed. 2019, 58, 14891–14895. 10.1002/anie.201906324. [DOI] [PubMed] [Google Scholar]
- de Frémont P.; Marion N.; Nolan S. P. Carbenes: Synthesis, properties, and organometallic chemistry. Coord. Chem. Rev. 2009, 253, 862–892. 10.1016/j.ccr.2008.05.018. [DOI] [Google Scholar]
- Ephritikhine M. Uranium carbene compounds. C. R. Chim. 2013, 16, 391–405. 10.1016/j.crci.2012.12.001. [DOI] [Google Scholar]
- Nelson D. J.; Nolan S. P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. 10.1039/C3CS60146C. [DOI] [PubMed] [Google Scholar]
- Herrmann W. A.; Köcher C. N-Heterocyclic Carbenes. Angew. Chem., Int. Ed. 1997, 36, 2162–2187. 10.1002/anie.199721621. [DOI] [Google Scholar]
- Bourissou D.; Guerret O.; Gabbaï F. P.; Bertrand G. Stable Carbenes. Chem. Rev. 2000, 100, 39–92. 10.1021/cr940472u. [DOI] [PubMed] [Google Scholar]
- Hopkinson M. N.; Richter C.; Schedler M.; Glorius F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- Cramer R. E.; Maynard R. B.; Paw J. C.; Gilje J. W. A uranium-carbon multiple bond. Crystal and molecular structure of (η5-C5H5)3UCHP(CH3)2(C6H5). J. Am. Chem. Soc. 1981, 103, 3589–3590. 10.1021/ja00402a065. [DOI] [Google Scholar]
- Gregson M.; Wooles A. J.; Cooper O. J.; Liddle S. T. Covalent Uranium Carbene Chemistry. Comments Inorg. Chem. 2015, 35, 262–294. 10.1080/02603594.2015.1020154. [DOI] [Google Scholar]
- Liddle S. T.; Mills D. P.; Wooles A. J. Early metal bis(phosphorus-stabilised)carbene chemistry. Chem. Soc. Rev. 2011, 40, 2164–2176. 10.1039/C0CS00135J. [DOI] [PubMed] [Google Scholar]
- Lu E.; Wooles A. J.; Gregson M.; Cobb P. J.; Liddle S. T. A Very Short Uranium(IV)-Rhodium(I) Bond with Net Double-Dative Bonding Character. Angew. Chem., Int. Ed. 2018, 57, 6587–6591. 10.1002/anie.201803493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu E.; Boronski J. T.; Gregson M.; Wooles A. J.; Liddle S. T. Silyl-Phosphino-Carbene Complexes of Uranium(IV). Angew. Chem., Int. Ed. 2018, 57, 5506–5511. 10.1002/anie.201802080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu E.; Atkinson B. E.; Wooles A. J.; Boronski J. T.; Doyle L. R.; Tuna F.; Cryer J. D.; Cobb P. J.; Vitorica-Yrezabal I. J.; Whitehead G. F. S.; Kaltsoyannis N.; Liddle S. T. Back-bonding between an electron-poor, high-oxidation-state metal and poor π-acceptor ligand in a uranium(V)-dinitrogen complex. Nat. Chem. 2019, 11, 806–811. 10.1038/s41557-019-0306-x. [DOI] [PubMed] [Google Scholar]
- Seed J. A.; Sharpe H. R.; Futcher H. J.; Wooles A. J.; Liddle S. T. Nature of the Arsonium-Ylide Ph3As=CH2 and a Uranium(IV) Arsonium-Carbene Complex. Angew. Chem., Int. Ed. 2020, 59, 15870–15874. 10.1002/anie.202004983. [DOI] [PubMed] [Google Scholar]
- Kent G. T.; Yu X.; Wu G.; Autschbach J.; Hayton T. W. Synthesis and electronic structure analysis of the actinide allenylidenes, [{(NR2)3}An(CCCPh2)]− (An = U, Th; R = SiMe3). Chem. Sci. 2021, 12, 14383–14388. 10.1039/D1SC04666G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold P. L.; Casely I. J. F-block N-heterocyclic carbene complexes. Chem. Rev. 2009, 109, 3599–3611. 10.1021/cr8005203. [DOI] [PubMed] [Google Scholar]
- Oldham Jr W. J.; Oldham S. M.; Smith W. H.; Costa D. A.; Scott B. L.; Abney K. D., Synthesis and structure of N-heterocyclic carbene complexes of uranyl dichloride. Chem. Commun. 2001, 1348–1349. 10.1039/B102649F. [DOI] [Google Scholar]
- Nakai H.; Hu X.; Zakharov L. N.; Rheingold A. L.; Meyer K. Synthesis and characterization of N-heterocyclic carbene complexes of uranium(III). Inorg. Chem. 2004, 43, 855–857. 10.1021/ic035142j. [DOI] [PubMed] [Google Scholar]
- Evans W. J.; Kozimor S. A.; Ziller J. W. Bis(pentamethylcyclopentadienyl) U(III) oxide and U(IV) oxide carbene complexes. Polyhedron 2004, 23, 2689–2694. 10.1016/j.poly.2004.08.007. [DOI] [Google Scholar]
- Gardner B. M.; McMaster J.; Liddle S. T. Synthesis and structure of a bis-N-heterocyclic carbene complex of uranium tetrachloride exhibiting short Cl···Ccarbene contacts. Dalton Trans. 2009, 6924–6926. 10.1039/B906000F. [DOI] [PubMed] [Google Scholar]
- Seed J. A.; Gregson M.; Tuna F.; Chilton N. F.; Wooles A. J.; McInnes E. J. L.; Liddle S. T. Rare-Earth- and Uranium-Mesoionic Carbenes: A New Class of f-Block Carbene Complex Derived from an N-Heterocyclic Olefin. Angew. Chem., Int. Ed. 2017, 56, 11534–11538. 10.1002/anie.201706546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O. J.; Mills D. P.; McMaster J.; Moro F.; Davies E. S.; Lewis W.; Blake A. J.; Liddle S. T. Uranium-carbon multiple bonding: facile access to the pentavalent uranium carbene [U{C(PPh2NSiMe3)2}(Cl)2(I)] and comparison of UV=C and UIV=C bonds. Angew. Chem., Int. Ed. 2011, 50, 2383–2386. 10.1002/anie.201007675. [DOI] [PubMed] [Google Scholar]
- Mills D. P.; Moro F.; McMaster J.; van Slageren J.; Lewis W.; Blake A. J.; Liddle S. T. A delocalized arene-bridged diuranium single-molecule magnet. Nat. Chem. 2011, 3, 454–460. 10.1038/nchem.1028. [DOI] [PubMed] [Google Scholar]
- Mills D. P.; Cooper O. J.; Tuna F.; McInnes E. J.; Davies E. S.; McMaster J.; Moro F.; Lewis W.; Blake A. J.; Liddle S. T. Synthesis of a uranium(VI)-carbene: reductive formation of uranyl(V)-methanides, oxidative preparation of a [R2C=U=O]2+ analogue of the [O=U=O]2+ uranyl ion (R = Ph2PNSiMe3), and comparison of the nature of UIV=C, UV=C, and UVI=C double bonds. J. Am. Chem. Soc. 2012, 134, 10047–10054. 10.1021/ja301333f. [DOI] [PubMed] [Google Scholar]
- Morss L. R.; Edelstein N. M.; Fuger J. (Eds), The Chemistry of the Actinide and Transactinide Elements; Springer: Dordrecht, 2011. [Google Scholar]
- Goodwin C. A. P.; Janicke M. T.; Scott B. L.; Gaunt A. J. [AnI3(THF)4] (An = Np, Pu) preparation bypassing An0 metal precursors: access to Np3+/Pu3+ nonaqueous and organometallic complexes. J. Am. Chem. Soc. 2021, 143, 20680–20696. 10.1021/jacs.1c07967. [DOI] [PubMed] [Google Scholar]
- Gregson M.; Lu E.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. A cerium(IV)-carbon multiple bond. Angew. Chem., Int. Ed. 2013, 52, 13016–13019. 10.1002/anie.201306984. [DOI] [PubMed] [Google Scholar]
- Talavera G.; Peña J.; Alcarazo M. Dihalo(imidazolium)sulfuranes: A Versatile Platform for the Synthesis of New Electrophilic Group-Transfer Reagents. J. Am. Chem. Soc. 2015, 137, 8704–8707. 10.1021/jacs.5b05287. [DOI] [PubMed] [Google Scholar]
- Ansell M. B.; Roberts D. E.; Cloke F. G.; Navarro O.; Spencer J. Synthesis of an [(NHC)2Pd(SiMe3)2] Complex and Catalytic cis-Bis(silyl)ations of Alkynes with Unactivated Disilanes. Angew. Chem., Int. Ed. 2015, 54, 5578–5582. 10.1002/anie.201501764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara S.; Yoshida Z.; Aoyagi H.; Maeda K.; Shirai O.; Kitatsuji Y.; Yoshida Y. A Critical Evaluation of the Redox Properties of Uranium, Neptunium and Plutonium Ions in Acidic Aqueous Solutions. Pure Appl. Chem. 1999, 71, 1771–1807. 10.1351/pac199971091771. [DOI] [Google Scholar]
- Jones M. B.; Gaunt A. J.; Gordon J. C.; Kaltsoyannis N.; Neu M. P.; Scott B. L. Uncovering f-element bonding differences and electronic structure in a series of 1 : 3 and 1 : 4 complexes with a diselenophosphinate ligand. Chem. Sci. 2013, 4, 1189–1203. 10.1039/C2SC21806B. [DOI] [Google Scholar]
- Macor J. A.; Brown J. L.; Cross J. N.; Daly S. R.; Gaunt A. J.; Girolami G. S.; Janicke M. T.; Kozimor S. A.; Neu M. P.; Olson A. C.; Reilly S. D.; Scott B. L. Coordination chemistry of 2,2′-biphenylenedithiophosphinate and diphenyldithiophosphinate with U, Np, and Pu. Dalton Trans. 2015, 44, 18923–18936. 10.1039/C5DT02976G. [DOI] [PubMed] [Google Scholar]
- Su J.; Cheisson T.; McSkimming A.; Goodwin C. A. P.; DiMucci I. M.; Albrecht-Schönzart T.; Scott B. L.; Batista E. R.; Gaunt A. J.; Kozimor S. A.; Yang P.; Schelter E. J. Complexation and redox chemistry of neptunium, plutonium and americium with a hydroxylaminato ligand. Chem. Sci. 2021, 12, 13343–13359. 10.1039/D1SC03905A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R. E.; Carter T. J.; Autillo M.; Stegman S. Thiocyanate complexes of the lanthanides, Am and Cm. Chem. Commun. 2020, 56, 2622–2625. 10.1039/C9CC07612C. [DOI] [PubMed] [Google Scholar]
- Shannon R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. 10.1107/s0567739476001551. [DOI] [Google Scholar]
- Orzechowski L.; Jansen G.; Harder S. Synthesis, Structure, and Reactivity of a Stabilized Calcium Carbene: R2CCa. J. Am. Chem. Soc. 2006, 128, 14676–14684. 10.1021/ja065000z. [DOI] [PubMed] [Google Scholar]
- Lu E.; Cooper O. J.; Tuna F.; Wooles A. J.; Kaltsoyannis N.; Liddle S. T. Uranium-Carbene-Imido Metalla-Allenes: Ancillary-Ligand-Controlled cis-/trans-Isomerisation and Assessment of trans Influence in the R2C=UIV=NR′ Unit (R=Ph2PNSiMe3; R′=CPh3). Chem. – Eur. J. 2016, 22, 11559–11563. 10.1002/chem.201602690. [DOI] [PubMed] [Google Scholar]
- Lu E.; Tuna F.; Lewis W.; Kaltsoyannis N.; Liddle S. T. Uranium Metalla-Allenes with Carbene Imido R2C=UIV=NR′ Units (R=Ph2PNSiMe3; R′=CPh3): Alkali-Metal-Mediated Push-Pull Effects with an Amido Auxiliary. Chem. – Eur. J. 2016, 22, 11554–11558. 10.1002/chem.201602603. [DOI] [PubMed] [Google Scholar]
- Gregson M.; Lu E.; Mills D. P.; Tuna F.; McInnes E. J.; Hennig C.; Scheinost A. C.; McMaster J.; Lewis W.; Blake A. J.; Kerridge A.; Liddle S. T. The inverse-trans-influence in tetravalent lanthanide and actinide bis(carbene) complexes. Nat. Commun. 2017, 8, 14137. 10.1038/ncomms14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin C. A. P.; Ciccone S. R.; Bekoe S.; Majumdar S.; Scott B. L.; Ziller J. W.; Gaunt A. J.; Furche F.; Evans W. J. 2.2.2-Cryptand complexes of neptunium(III) and plutonium(III). Chem. Commun. 2022, 58, 997–1000. 10.1039/d1cc05904a. [DOI] [PubMed] [Google Scholar]
- Carnall W. T. A systematic analysis of the spectra of trivalent actinide chlorides in D3h site symmetry. J. Chem. Phys. 1992, 96, 8713–8726. 10.1063/1.462278. [DOI] [Google Scholar]
- Apostolidis C.; Schimmelpfennig B.; Magnani N.; Lindqvist-Reis P.; Walter O.; Sykora R.; Morgenstern A.; Colineau E.; Caciuffo R.; Klenze R.; Haire R. G.; Rebizant J.; Bruchertseifer F.; Fanghänel T. [An(H2O)9](CF3SO3)3 (An=U-Cm, Cf): Exploring Their Stability, Structural Chemistry, and Magnetic Behavior by Experiment and Theory. Angew. Chem., Int. Ed. 2010, 49, 6343–6347. 10.1002/anie.201001077. [DOI] [PubMed] [Google Scholar]
- Carnall W. T.; Crosswhite H.; Crosswhite H. M.; Hessler J. P.; Edelstein N.; Conway J. G.; Shalimoff G. V.; Sarup R. Energy level analysis of Np3+:LaCl3 and Np3+:LaBr3. J. Chem. Phys. 1980, 72, 5089–5102. 10.1063/1.439797. [DOI] [Google Scholar]
- Bader R. F. W.; Slee T. S.; Cremer D.; Kraka E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061–5068. 10.1021/ja00353a035. [DOI] [Google Scholar]
- Kirker I.; Kaltsoyannis N. Does covalency really increase across the 5f series? A comparison of molecular orbital, natural population, spin and electron density analyses of AnCp3 (An = Th-Cm; Cp = η5-C5H5). Dalton Trans. 2011, 40, 124–131. 10.1039/C0DT01018A. [DOI] [PubMed] [Google Scholar]
- Su J.; Batista E. R.; Boland K. S.; Bone S. E.; Bradley J. A.; Cary S. K.; Clark D. L.; Conradson S. D.; Ditter A. S.; Kaltsoyannis N.; Keith J. M.; Kerridge A.; Kozimor S. A.; Löble M. W.; Martin R. L.; Minasian S. G.; Mocko V.; La Pierre H. S.; Seidler G. T.; Shuh D. K.; Wilkerson M. P.; Wolfsberg L. E.; Yang P. Energy-Degeneracy-Driven Covalency in Actinide Bonding. J. Am. Chem. Soc. 2018, 140, 17977–17984. 10.1021/jacs.8b09436. [DOI] [PubMed] [Google Scholar]
- Cooper O. J.; Mills D. P.; Lewis W.; Blake A. J.; Liddle S. T. Reactivity of the uranium(IV) carbene complex [U(BIPMTMS)(Cl)(μ-Cl)2Li(THF)2] (BIPMTMS = {C(PPh2NSiMe3)2}) towards carbonyl and heteroallene substrates: metallo-Wittig, adduct formation, C-F bond activation, and [2 + 2]-cycloaddition reactions. Dalton Trans. 2014, 43, 14275–14283. 10.1039/C4DT00909F. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.