

Abstract
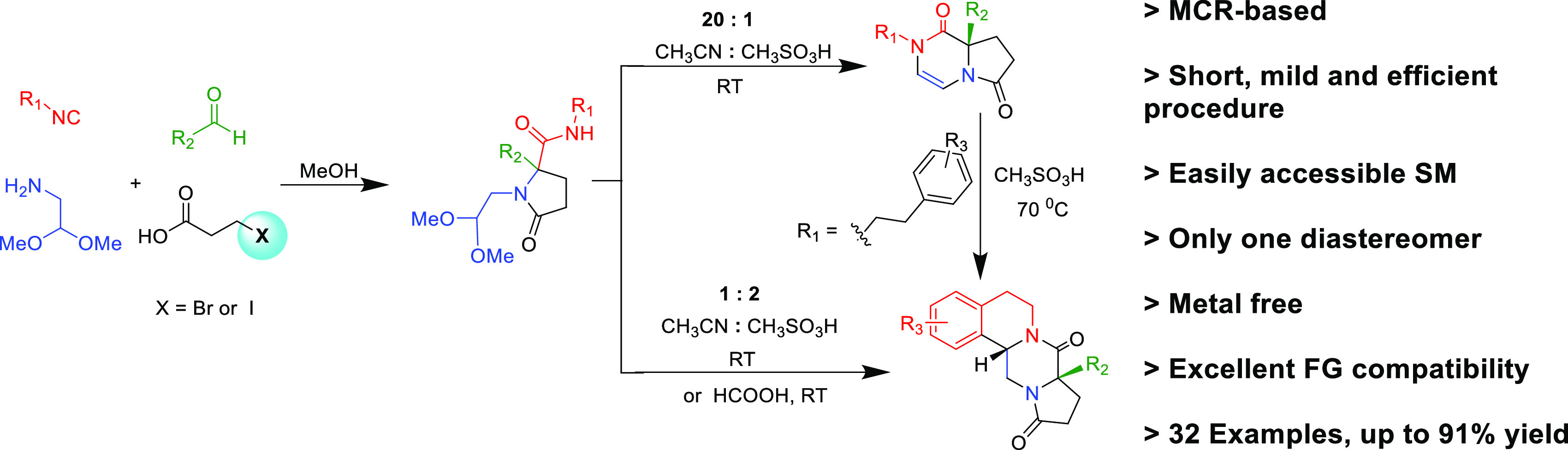
Discovering novel synthetic routes for rigid nitrogen-containing polyheterocycles using sustainable, atom-economical, and efficient (= short) synthetic pathways is of high interest in organic chemistry. Here, we describe an operationally simple and short synthesis of the privileged scaffold dihydropyrrolo[1,2-a]pyrazine-dione from readily accessible starting materials. The alkaloid-type polycyclic scaffold with potential bioactivity was achieved by a multicomponent reaction (MCR)-based protocol via a Ugi four-component reaction and Pictet–Spengler sequence under different conditions, yielding a diverse library of products.
Introduction
Heterocyclic compounds, particularly nitrogen(N)-containing heterocycles, have important roles in organic chemistry due to the application in almost all branches of organic chemistry including pharmaceutical research, functional materials, catalysis, and coordination chemistry.1 Among the N-containing heterocycles, polyheterocycles as constituents of diverse natural alkaloids and pharmaceutical agents have drawn much attention from organic and bioorganic chemists during the past decades.2 Some of them are an integral part of many biologically active molecules, and many currently marketed drugs hold polyheterocycles as their core structure, such as praziquantel (anthelmintic agent),3 tadalafil (male erectile dysfunction),4 or fumitremorgin C (BCRP specific inhibitor)5 (Figure 1A). Polyheterocycles due to their conformational stiffness often have very good receptor binding and membrane diffusion properties. Therefore, the quest for the novel synthetic routes for nitrogen (N)-containing polyheterocycles using sustainable, atom-economical, and efficient pathways is in high demand.6 Multicomponent reactions (MCR) are one of the most powerful tools to synthesize novel scaffold compounds for drug discovery in general and polyheterocycles specifically.7 The wide functional group compatibility of isocyanide-based MCRs allows for the introduction of orthogonal groups, which can be further cyclized.8 One such strategy is the Pictet–Spengler reaction of electron-rich β-arylethylamine undergoing condensation with an aldehyde or ketone followed by ring closure. The Pictet–Spengler reaction not only has been explored as a convenient method for the asymmetric synthesis of isoquinoline alkaloids9 but also was widely used for the synthesis of alkaloid-like polycyclic compounds by combining with MCR chemistry in recent years (Figure 1B).10
Figure 1.

(A) Polyheterocyclic drugs. (B) Some previous and (C) current work on Ugi/Pictet–Spengler sequences for the synthesis of polycyclic scaffolds (the colors indicate the origin of atoms from different starting material classes: red, isocyanide; green, oxo component; blue, amine; and black, acid component).
The union of the Ugi reaction with a Pictet–Spengler-type ring closure is very popular and has recently led to a sustainable and efficient access to 2-tetrazolo-substituted indoles,11 steroidal pyrazinoisoquinolines,12 isoindolinones,13 2-tetrazolylmethyl-2,3,4,9-tetrahydro-1H-β-carbolines,14 praziquantel and derivatives,15 polycycles,16 or chiral-pure synthetic alkaloids,17 among others, and is reviewed (Figure 1B).18
Results and Discussion
Inspired by Orru’s17 and our own work,19 we report here the highly stereoselective synthesis of 8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione species 6 by a Ugi/Pictet–Spengler sequence starting from isocyanides 1, aldehydes 2, 2,2-dimethoxyethan-1-amine 3, and 3-bromopropanoic acids 4 with excellent stereoselectivity in the final target 7. Moreover, we achieved the post-transformation of 6 to novel fused tricyclic systems 7 by treatment with methanesulfonic acid (Figure 1). To start, the Ugi-4CR of (2-isocyanoethyl)benzene 1a, picolinaldehyde 2a, 2,2-dimethoxyethan-1-amine 3a, and 3-bromopropanoic acid 4a in methanol at room temperature resulted in the expected Ugi-adduct 5a in an excellent yield of 98% after 15 h. With compound 5a in hand, we screened several cyclization conditions (Table 1). Noteworthy is that Dömling19 and Nadzan20 have reported the formation of 2-oxopiperazines by Ugi-N-acyliminium ion cyclization with good yield using HCOOH and TFA as acid components, respectively. To our surprise, TFA (entry 1), CH3COOH (entry 2), and 37% HCl(aq) solution (entry 5) in dioxane failed to give any 2-oxopiperazines product, while HCOOH afforded 6a in 26% yield (entry 3). The combination of CH3COOH and conc. H2SO4 was found to induce an improvement of this cyclization, which afforded 6a in 38% yield (entry 4). Disappointingly, methanesulfonic acid, which has been previously described as an appropriate acid for Ugi/Pictet–Spengler reactions, led to the formation of 65% 6a and 31% 7a when methanesulfonic acid in acetonitrile (v/v = 1:3) was used (entry 13). However, too much methanesulfonic acid turned out to be harmful for 6a formation, which only afforded 6a in 15% yield in two steps (entry 14). The reaction yield increased to 91% over two steps when the methanesulfonic acid ratio decreased and the time was increased to 12 h (entry 11). Interestingly, when the intermediate compound 5a was treated with a high concentration of methanesulfonic acid for 12 h, polycyclic compound 7a was formed with an excellent yield of 90% (entry 15). Unfortunately, the intermediate compound 5a with solventless methanesulfonic acid only formed polycyclic compound 7a at a good yield of 81%. To our delight, this reaction shows excellent stereoselectivity; only a single spatial configuration was formed in the products 6a and 7a.
Table 1. Optimization of Reaction Conditionsa.


Reaction conditions: 5a (0.2 mmol), 25 °C. The green color indicates the best conditions screened for 6a and 7a.
With these optimized conditions in hand, we went on to examine the substrate scope of the reaction. We first reacted aldehyde 2, carboxylic acid 3, and bifunctional amine 4 with different isocyanides 1 in methanol at room temperature for 15 h (Scheme 1). The Ugi 4-CR proceeded well, and the desired product 5 was isolated in good yield (Supporting Infromation). To our delight, the subsequent Pictet–Spengler reaction of 5 went smoothly, and the desired product 6 was isolated in good yield from 45 to 91%; both aromatic and aliphatic isocyanides work well in this reaction (Scheme 1).
Scheme 1. Synthesis of Heterocycles by Different Isocyanides and Aldehydes.

After successfully demonstrating that different isocyanides work well, we then focused on screening different aldehydes. Pyridine aldehyde was utilized in several cases and results in good yield, like picolinaldehyde (6a) and isonicotinaldehyde (6u) with 91 and 55% yields, respectively, while nicotinaldehyde did not work. Pyridine aldehyde with electron-donating and -withdrawing groups such as 5-Me,5-Br, 6-Cl, and 6-Br also reacted smoothly giving 68, 45, 71, and 40% yields, respectively. When quinoline-2-carbaldehyde was applied, it gave the corresponding products in a good yield of 72%. To our great surprise, utilizing aldehydes rather than pyridine-derived ones, neither compound 5 nor 6v was formed, and we could not observe the compounds 6w and 6x (Scheme 1).
A potential mechanistic explanation is given below. Next, we investigated the Pictet–Spengler cyclization of suitable 6 derived from (substituted) phenylethyl isocyanides to yield tetracyclic 7. High-concentration methanesulfonic acid was found to be good for monomethoxy phenylethyl or phenylethyl Ugi products, yielding 35–90% of 7 (Scheme 2). However, more electron-rich dimethoxyphenyl Ugi product 5 was too reactive; using methanesulfonic acid and formic acid was found superior for the cyclization to at of polyheterocycle 7h.
Scheme 2. Structures of Polyheterocycles,,,

The isocyanide, aldehyde, amine, and carboxylic acid components are depicted in red, green, blue, and black, respectively. The Ugi reaction conditions are as follows: 1 (1.0 mmol), 2 (1.0 mmol), 3 (1.0 mmol), 4 (1.0 mmol), MeOH (1.0 mL), 15 h, and 25 °C.
Reaction conditions: 5 (0.5 mmol), CH3SO3H (0.1 mL), CH3CN (2.0 mL), 25 °C, and 12 h.
Reaction conditions: 5 (0.5 mmol), CH3SO3H (1.0 mL), CH3CN (0.5 mL), 25 °C, 12 h. or HCOOH (2.0 mL), 25 °C, and 4 h.
Reaction conditions: CH3SO3H (2.0 mL), 70 °C, and 1 h.
The structures of 6a and 7a were confirmed by X-ray crystallography (Figure 2). Noteworthy is the pyridyl residue exit orthogonal to the bicyclic and tricyclic flat ring systems. The pyridyl ring of 7a forms a short T-shaped stacking interaction with the phenyl group of a neighboring molecule, thus forming a dimer in the crystal. Next, the scalability of this method was investigated (Scheme 3). A four-component reaction of (2-isocyanoethyl)benzene, picolinaldehyde, 2,2-dimethoxyethan-1-amine, and 3-bromopropanoic acid was conducted on a 8 mmol scale, which upon further treatment with methanesulfonic acid conditions produced 6a and polyheterocyclic product 7a in 90% (2.4 g) and 89% (2.3 g) yields, respectively.
Figure 2.
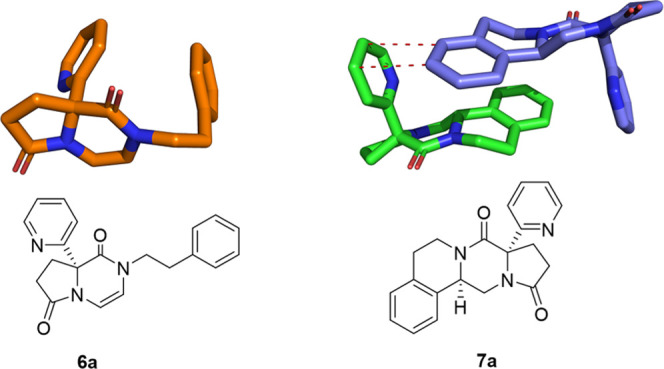
X-ray structures of the selected products CCDC 2089898 (6a) and CCDC 2089897 (7a) shown as sticks rendered using PyMol. Short intermolecular contacts (3.7 Å) in 7a are shown as red dotted lines.
Scheme 3. Further Synthetic Applications.

To further underscore the usefulness of the herein-described 8a-(pyridin-2-yl)-8,8a-dihydropyrrolo [1,2-a]pyrazine-1,6(2H,7H)-diones, we performed several late-stage functionalizations. In compound 6a, we reduced the double bond with Pd/C to give the product 8a in a quantitative yield (Scheme 3). In another application, the bromo group of 6t was coupled with 1,4-benzodioxane-6-boronic acid to give the derivative 8b (Scheme 3). C–C coupling reactions such as the Suzuki reaction are highly preferred “privileged” reactions in drug discovery.21
The findings of the high substrate specificity of the overall sequence, working with pyridine and pyrimidine derivatives, is unusual and deserves some mechanistic discussion. A hypothesized mechanism of this tandem reaction is shown in Scheme 4.
Scheme 4. Proposed Reaction Mechanism.

The Ugi α-adduct intermediate A forms under standard conditions, and a pyridine side chain-induced resonance structure B is conceivable. Supported by the vicinal pyridine, the Ugi product (C) formed through the Mumm rearrangement. The Ugi product (C) with a double bond cyclizes through an intramolecular substitution to yield the stable compound 5a (derivative 5x crystal, see the Supporting Infromation). The Ugi-cyclized product 5a when treated under strongly acidic conditions further cyclizes as previously described by Nadzan20 and Dömling16 toward the target product 6a and 7a, respectively. The exclusive formation of the syn-diastereomers 7a can be explained by the less-hindered reaction of the phenylethyl moiety anti to the pyridine. The mechanism involving a nucleophilic substitution differentiates the synthesis from previous ones and explains why only 2- and 4-pyridyl but not 3-pyridyl carbaldehyde or benzaldehyde used in the Ugi reaction can undergo further cyclization. Using MeOD and CD3CN as the solvent to perform certain control experiments, all the NMR data show that nothing changes.
Conclusions
In summary, we have developed a straightforward method to assemble rigid nitrogen (N)-containing polyheterocycles. The Ugi postcyclization strategy is probably the most powerful tool in MCR chemistry to create structural diversity and large compound numbers while keeping the number of synthetic steps low. It has already attracted much attention from medicinal chemists in the field of drug discovery. Our new Ugi/Pictet–Spengler strategy is an expedited and convergent access to skeletally diverse compounds. Significantly, N-heterocycles and the novel fused tricyclic compound can now be constructed in just two steps with this method. We have also shown scalability of our method to gram-scale and several “late-stage” modifications. The process uses readily available starting materials and is simple to operate, provides a diverse compound library, and will therefore become a synthetical useful addition to the method arsenal in organic synthesis and medicinal chemistry.
Experimental Section
General Remarks
All chemicals (aldehydes 2, 2,2-dimethoxyethan-1-amine 3, 3-bromopropanoic acid 4, and amines) were purchased from commercial suppliers and used without any purification unless otherwise noted. All isocyanides 1 were prepared according to the procedures reported from our lab, and all data are given in the Supporting Infromation.11,16,22 Nuclear magnetic resonance spectra were recorded on a Brucker 500 MHz. Chemical shifts for 1H NMR were reported as δ values, and coupling constants were reported in hertz (Hz). The following abbreviations were used for spin multiplicity: s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, quin = quintet, dd = double of doublets, ddd = double of doublet of doublets, and m = multiplet. Chemical shifts for 13C NMR were reported in ppm relative to the solvent peak. Thin-layer chromatography was performed on silica gel plates (0.20 mm thickness, particle size 25 μm). Flash chromatography was performed using RediSep Rf normal-phase silica flash columns (Silica Gel 60 Å, 230–400 mesh). High-resolution mass spectra were recorded using a LTQOrbitrap-XL (Thermo) at a resolution of 60000@m/z400. Melting points were obtained on a melting point apparatus and were uncorrected. Yields given refer to chromatographically purified and spectroscopically pure compounds unless otherwise stated.
General Experimental Procedure and Characterization Procedure A
General procedure for heterocycle 6: A solution of isocyanide 1 (1 mmol, 1.0 equiv), aldehyde 2 (1 mmol, 1.0 equiv), amine 3 (1 mmol, 1.0 equiv), and acid 4 (1 mmol, 1.0 equiv) in methanol (1 mL) was stirred at room temperature for 15 h. The solvents were removed under vacuum. Then, the crude Ugi-adduct 5 was dissolved in 4 mL of acetonitrile, and 0.2 mL of methanesulfonic acid was added. The resulting mixture was stirred at room temperature for 12 h. The reaction was diluted with dichloromethane and quenched with saturated sodium bicarbonate solution at 0–5 °C. The resulting solution was extracted with dichloromethane (10 mL × 3). The solvents were removed under vacuum, and the crude product was purified by flash column chromatography to give pure product 6.
Procedure B
General procedure for polyheterocycle 7: A solution of isocyanide 1 (1 mmol, 1.0 equiv), aldehyde 2 (1 mmol, 1.0 equiv), amine 3 (1 mmol, 1.0 equiv), and acid 4 (1 mmol, 1.0 equiv) in methanol (1 mL) was stirred at room temperature for 15 h. The solvents were removed under vacuum. Then, the crude Ugi-adduct 5 was dissolved in 1 mL of acetonitrile, and 2 mL of methanesulfonic acid was added. The resulting mixture was stirred at 25 °C for 12 h. The reaction was diluted with dichloromethane and quenched with saturated sodium bicarbonate solution at 0–5 °C. The resulting solution was extracted with dichloromethane (10 mL × 3). The solvents were removed under vacuum, and the crude product was purified by flash column chromatography to give pure product 7.
Procedure C
General procedure for polyheterocycle 7h: A solution of isocyanide 1 (1 mmol, 1.0 equiv), aldehyde 2 (1 mmol, 1.0 equiv), amine 3 (1 mmol, 1.0 equiv), and acid 4 (1 mmol, 1.0 equiv) in methanol (1 mL) was stirred at room temperature for 15 h. The solvents were removed under vacuum. Then, the crude Ugi-adduct 5 was dissolved in 3.0 mL of HCOOH. The resulting mixture was stirred at room temperature for 4 h. The solvents were removed under vacuum; then, the crude product was diluted with dichloromethane and quenched with saturated sodium bicarbonate solution at 0–5 °C. The resulting solution was extracted with dichloromethane (10 mL × 3). The solvents were removed under vacuum, and the crude product was purified by flash column chromatography to give pure product 7h.
Procedure D
General procedure for heterocycle 6 transformation to polyheterocycle 7: A solution of heterocycle 6 (1 mmol, 1.0 equiv) in 3.0 mL of methanesulfonic acid was stirred at 70 °C for 1 h. The reaction was diluted with dichloromethane and quenched with saturated sodium bicarbonate solution at 0–5 °C. The resulting solution was extracted with dichloromethane (10 mL × 3). The solvents were removed under vacuum, and the crude product was purified by flash column chromatography to give pure product 7.
Gram-Scale synthesis of 6a and 7a
A solution of isocyanide 1a (8 mmol, 1.0 equiv), aldehyde 2a (8.0 mmol, 1.0 equiv), amine 3a (8.0 mmol, 1.0 equiv), and acid 4a (8.0 mmol, 1.0 equiv) in methanol (8 mL) was stirred at room temperature for 15 h. The solvents were removed under vacuum. Then, the crude Ugi-adduct 5a was dissolved in 20 mL of acetonitrile, and 1.0 mL of methanesulfonic acid was added. The resulting mixture was stirred at room temperature for 12 h. The reaction was diluted with dichloromethane and quenched with saturated sodium bicarbonate solution at 0–5 °C. The resulting solution was extracted with dichloromethane (80 mL × 3). The solvents were removed under vacuum, and the crude product was purified by flash column chromatography to give pure product 6a (2.4 g, 90%). Then, a solution of heterocycle 6a (2.4 g, 7.2 mmol, 1.0 equiv) in 10 mL of methanesulfonic acid was stirred at 70 °C for 1 h. The reaction mixture was diluted with dichloromethane and quenched with saturated sodium bicarbonate solution at 0–5 °C. The resulting solution was extracted with dichloromethane (100 mL × 3). The solvents were removed under vacuum, and the crude product was purified by flash column chromatography to give pure product 7a (2.3 g, 96%).
Procedure E
Synthesis of product 8a: Compound 6a (100 mg, 0.3 mmol, 1.0 equiv) was dissolved in 5.0 mL of THF, Pd/C (6.4 mg, 0.06 mmol, 0.2 equiv) was added under a N2 atmosphere; the reaction mixture was stirred under a H2 atmosphere for 12 h. The reaction mixture was filtered, and the filtrate was removed under vacuum and purified by flash column chromatography to give pure product 8a (0.1 g, 98%).
Procedure F
Synthesis of product 8b: A solution of compound 6t (99 mg, 0.24 mmol, 1.0 equiv), 1,4-benzodioxane-6-boronic acid (52.0 mg, 0.29 mmol, 1.2 equiv), Pd(PPh3)4 (14 mg, 0.012 mmol, 0.05 equiv), and K2CO3 (100 mg, 0.72 mmol, 3.0 equiv) in DMF/H2O (8/2 mL) was stirred at 80 °C under a N2 atmosphere for 16 h. After cooling, 10 mL of water was added to dilute the reaction mixture. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine and dried over anhydrous Na2SO4. After removal of the EtOAc, the residue was purified by column chromatography (silica gel, methanol/dichloromethane = 5:95) to afford the product 8b (0.1 g, 91%).
(R)-2-Phenethyl-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6a)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6a (303 mg, 91%) as a yellow solid; 1H NMR (500 MHz, chloroform-d) δ 8.62 (ddd, J = 4.7, 1.9, 1.1 Hz, 1H), 7.67 (td, J = 7.8, 1.8 Hz, 1H), 7.26 (ddd, J = 7.6, 4.8, 1.3 Hz, 1H), 7.23–7.17 (m, 4H), 7.08–7.04 (m, 2H), 6.49 (d, J = 5.7 Hz, 1H), 5.49 (d, J = 5.7 Hz, 1H), 3.90 (ddd, J = 14.0, 7.9, 6.3 Hz, 1H), 3.74 (dt, J = 13.4, 7.4 Hz, 1H), 2.93–2.82 (m, 2H), 2.76 (ddd, J = 12.5, 11.0, 9.5 Hz, 1H), 2.63 (ddd, J = 17.3, 11.0, 8.7 Hz, 1H), 2.55–2.43 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.5, 165.2, 157.6, 149.8, 137.9, 137.0, 128.9, 128.5, 126.6, 123.2, 119.7, 116.8, 106.7, 68.8, 48.3, 34.6, 30.7, 29.7. HRMS (ESI) m/z calculated for C20H20N3O2 [M + H]+: 334.1530; found [M + H]+: 334.1531.
(R)-2-(4-Chlorophenyl)-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6b)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6b (245 mg, 72%) as a brown solid. 1H NMR (500 MHz, chloroform-d) δ 8.67 (d, J = 4.7 Hz, 1H), 7.80–7.72 (m, 1H), 7.45–7.36 (m, 3H), 7.32 (dd, J = 7.5, 4.8 Hz, 1H), 7.28–7.17 (m, 2H), 6.67 (d, J = 5.5 Hz, 1H), 5.87 (d, J = 5.7 Hz, 1H), 2.89 (dt, J = 13.2, 10.4 Hz, 1H), 2.71 (ddd, J = 17.3, 11.0, 8.9 Hz, 1H), 2.58 (dd, J = 17.4, 9.9 Hz, 1H), 2.49 (dd, J = 13.3, 8.9 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.3, 165.0, 157.7, 150.0, 138.1, 137.3, 133.4, 129.4, 127.2, 123.5, 119.4, 117.2, 107.5, 69.2, 30.1, 29.6. HRMS (ESI) m/z calculated for C18H15ClN3O2 [M + H]+: 340.0821; found [M + H]+: 340.0827.
(R)-2-(3-Fluorobenzyl)-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6c)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6c (276 mg, 82%) as a brown solid. 1H NMR (500 MHz, chloroform-d) δ 8.63 (d, J = 5.2 Hz, 1H), 7.70 (t, J = 7.8 Hz, 1H), 7.26 (dd, J = 13.2, 8.3 Hz, 3H), 6.96 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 9.8 Hz, 1H), 6.57 (d, J = 5.7 Hz, 1H), 5.64 (d, J = 5.7 Hz, 1H), 4.92 (d, J = 15.1 Hz, 1H), 4.63 (d, J = 15.1 Hz, 1H), 2.92–2.80 (m, 1H), 2.72–2.59 (m, 1H), 2.59–2.48 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.4, 165.5, 164.0, 162.0, 157.6, 149.8, 138.6, 138.5, 137.0, 130.3, 130.2, 123.4, 123.3, 123.3, 119.6, 115.9, 114.9, 114.8, 114.6, 107.4, 68.9, 49.0, 30.4, 29.6. HRMS (ESI) m/z calculated for C19H17FN3O2 [M + H]+: 338.1235; found [M + H]+: 338.1238.
(R)-2-(4-Methoxybenzyl)-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6d)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6d (227 mg, 65%) as a brown solid. 1H NMR (500 MHz, chloroform-d) δ 8.61 (d, J = 5.5 Hz, 1H), 7.66 (td, J = 7.8, 1.9 Hz, 1H), 7.25 (dd, J = 7.6, 4.2 Hz, 2H), 7.13 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 6.53 (d, J = 5.7 Hz, 1H), 5.65 (d, J = 5.7 Hz, 1H), 4.82 (d, J = 14.7 Hz, 1H), 4.61 (d, J = 14.7 Hz, 1H), 3.80 (s, 3H), 2.88–2.77 (m, 1H), 2.75–2.65 (m, 1H), 2.56–2.47 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.5, 165.4, 159.3, 157.7, 149.8, 136.9, 129.4, 128.1, 123.3, 119.6, 116.0, 114.1, 107.1, 68.8, 55.3, 49.1, 30.5, 29.7. HRMS (ESI) m/z calculated for C20H20N3O3 [M + H]+: 350.1452; found [M + H]+: 350.1457.
(R)-2-(Benzo[d][1,3]dioxol-5-ylmethyl)-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6e)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6e (301 mg, 83%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.60 (d, J = 4.7 Hz, 1H), 7.67 (td, J = 7.8, 1.8 Hz, 1H), 7.28–7.23 (m, 2H), 6.71 (d, J = 7.9 Hz, 1H), 6.69–6.62 (m, 2H), 6.52 (d, J = 5.7 Hz, 1H), 5.94–5.90 (m, 2H), 5.64 (d, J = 5.7 Hz, 1H), 4.77 (d, J = 14.7 Hz, 1H), 4.56 (d, J = 14.7 Hz, 1H), 2.82 (ddd, J = 12.8, 11.0, 9.5 Hz, 1H), 2.65 (ddd, J = 17.5, 11.0, 9.0 Hz, 1H), 2.56–2.45 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.4, 165.4, 157.7, 149.7, 148.0, 147.4, 137.0, 129.8, 123.4, 121.5, 119.6, 115.9, 108.4, 108.3, 107.1, 101.2, 68.8, 49.3, 30.5, 29.7. HRMS (ESI) m/z calculated for C20H18N3O4 [M + H]+: 364.1248; found [M + H]+: 364.1249.
(R)-2-Benzyl-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6f)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6f (246 mg, 77%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.62 (dt, J = 5.2, 1.6 Hz, 1H), 7.67 (td, J = 7.7, 1.9 Hz, 1H), 7.38–7.23 (m, 5H), 7.17 (dd, J = 7.5, 2.1 Hz, 2H), 6.54 (d, J = 5.7 Hz, 1H), 5.66 (d, J = 5.7 Hz, 1H), 4.89 (d, J = 14.8 Hz, 1H), 4.69 (d, J = 14.8 Hz, 1H), 2.90–2.77 (m, 1H), 2.67 (ddd, J = 17.7, 11.0, 8.9 Hz, 1H), 2.58–2.48 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.5, 165.4, 157.63, 149.8, 137.0, 136.0, 128.8, 127.9, 127.9, 123.4, 119.6, 116.1, 107.2, 68.9, 49.5, 30.6, 29.7. HRMS (ESI) m/z calculated for C19H18N3O2 [M + H]+: 320.1357; found [M + H]+: 320.1359.
(R)-8a-(Pyridin-2-yl)-2-(p-tolyl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6g)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6g (182 mg, 57%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.66 (dt, J = 4.7, 1.4 Hz, 1H), 7.75 (td, J = 7.7, 1.8 Hz, 1H), 7.42 (dt, J = 7.9, 1.2 Hz, 1H), 7.32–7.29 (m, 1H), 7.23 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.5 Hz, 2H), 6.63 (d, J = 5.5 Hz, 1H), 5.88 (d, J = 5.7 Hz, 1H), 2.88 (ddd, J = 13.1, 11.0, 9.7 Hz, 1H), 2.72 (ddd, J = 17.3, 11.0, 8.9 Hz, 1H), 2.61–2.43 (m, 2H), 2.38 (s, 3H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.4, 165.1, 157.9, 149.9, 137.7, 137.1, 137.1, 129.9, 125.7, 123.4, 119.5, 117.9, 107.0, 69.2, 30.3, 29.7, 21.1. HRMS (ESI) m/z calculated for C19H18N3O2 [M + H]+: 320.1355; found [M + H]+: 320.1358.
Methyl (R)-3-(1,6-Dioxo-8a-(pyridin-2-yl)-6,7,8,8a-tetrahydropyrrolo[1,2-a]pyrazin-2(1H)-yl)propanoate (6h)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6h (142 mg, 45%) as yellow oil. 1H NMR (500 MHz, chloroform-d) δ 8.84–8.24 (m, 1H), 7.68 (td, J = 7.8, 1.8 Hz, 1H), 7.28–7.20 (m, 2H), 6.50 (s, 1H), 5.77 (d, J = 5.7 Hz, 1H), 3.88–3.83 (m, 2H), 3.62 (s, 3H), 2.85–2.69 (m, 2H), 2.71–2.57 (m, 2H), 2.59–2.42 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.4, 171.7, 165.3, 157.7, 149.7, 137.0, 123.3, 119.5, 117.3, 106.7, 68.7, 51.8, 43.3, 32.8, 30.2, 29.6. HRMS (ESI) m/z calculated for C16H18N3O4 [M + H]+: 316.1252; found [M + H]+: 316.1256.
Methyl (R)-2-(1,6-Dioxo-8a-(pyridin-2-yl)-6,7,8,8a-tetrahydropyrrolo[1,2-a]pyrazin-2(1H)-yl)acetate (6i)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6i (181 mg, 60%) as yellow oil. 1H NMR (500 MHz, chloroform-d) δ 8.60 (ddd, J = 4.9, 1.9, 1.0 Hz, 1H), 7.70 (td, J = 7.7, 1.8 Hz, 1H), 7.38 (dt, J = 8.0, 1.1 Hz, 1H), 7.25 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 6.57 (d, J = 5.5 Hz, 1H), 5.64 (d, J = 5.7 Hz, 1H), 4.40 (d, J = 17.3 Hz, 1H), 4.24 (d, J = 17.2 Hz, 1H), 3.75 (s, 3H), 2.81–2.67 (m, 2H), 2.57–2.47 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.5, 168.2, 165.9, 157.2, 149.8, 136.9, 123.4, 120.0, 116.4, 107.5, 68.7, 52.5, 47.5, 30.6, 29.8. HRMS (ESI) m/z calculated for C15H16N3O4 [M + H]+: 302.1126; found [M + H]+: 302.1129.
(R)-2-Phenyl-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6j)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6j (171 mg, 56%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.67 (dt, J = 4.7, 1.3 Hz, 1H), 7.76 (d, J = 1.9 Hz, 1H), 7.44 (td, J = 7.6, 2.2 Hz, 3H), 7.38–7.27 (m, 4H), 6.65 (d, J = 5.7 Hz, 1H), 5.91 (d, J = 5.7 Hz, 1H), 2.95–2.84 (m, 1H), 2.73 (ddd, J = 17.3, 11.0, 8.9 Hz, 1H), 2.58 (ddd, J = 17.3, 9.8, 1.7 Hz, 1H), 2.50 (dd, J = 13.2, 7.3 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.4, 165.0, 157.8, 149.9, 139.6, 137.2, 129.3, 127.7, 125.9, 123.4, 119.5, 117.7, 107.2, 69.2, 30.2, 29.7. HRMS (ESI) m/z calculated for C18H16N3O2 [M + H]+: 306.1265; found [M + H]+: 306.1267.
(R)-2-(2-Methoxyphenethyl)-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6k)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6k (276 mg, 76%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.62–8.60 (m, 1H), 7.66 (td, J = 7.7, 1.8 Hz, 1H), 7.27–7.17 (m, 3H), 6.91 (dd, J = 7.3, 1.8 Hz, 1H), 6.83 (d, J = 7.1 Hz, 1H), 6.77 (td, J = 7.4, 1.1 Hz, 1H), 6.46 (d, J = 5.7 Hz, 1H), 5.52 (d, J = 5.7 Hz, 1H), 3.85 (t, J = 6.4 Hz, 1H), 3.82 (s, 3H), 3.82–3.78 (m, 1H), 3.01–2.94 (m, 1H), 2.84–2.77 (m, 1H), 2.76–2.70 (m, 1H), 2.68–2.58 (m, 1H), 2.53–2.44 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.5, 165.3, 157.7, 157.6, 149.7, 136.9, 130.7, 128.0, 126.2, 123.1, 120.4, 119.7, 117.0, 110.2, 106.5, 68.8, 55.3, 46.6, 30.8, 29.7, 29.6. HRMS (ESI) m/z calculated for C21H22N3O3 [M + H]+: 364.1624; found [M + H]+: 364.1627.
(R)-2-(4-Fluorophenethyl)-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6l)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6l (250 mg, 71%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.64–8.59 (m, 1H), 7.66 (td, J = 7.8, 1.8 Hz, 1H), 7.26 (dd, J = 7.6, 4.7 Hz, 1H), 7.18 (d, J = 7.9 Hz, 1H), 7.04–6.93 (m, 2H), 6.92–6.86 (m, 2H), 6.51 (d, J = 5.5 Hz, 1H), 5.49 (d, J = 5.7 Hz, 1H), 3.90 (ddd, J = 13.9, 7.7, 6.4 Hz, 1H), 3.75–3.64 (m, 1H), 2.90–2.79 (m, 2H), 2.78–2.70 (m, 1H), 2.60 (ddd, J = 17.7, 11.0, 8.9 Hz, 1H), 2.53–2.45 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.4, 165.2, 162.7, 160.7, 155.6, 149.8, 136.9, 133.5, 130.3, 123.2, 119.6, 116.4, 115.4, 115.2, 106.9, 68.8, 48.1, 33.7, 30.8, 29.6. HRMS (ESI) m/z calculated for C20H19FN3O2 [M + H]+: 352.1471; found [M + H]+: 352.1473.
(R)-2-(3-Methoxyphenyl)-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6m)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6m (354 mg, 90%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.66 (d, J = 6.6 Hz, 1H), 7.75 (td, J = 7.7, 1.8 Hz, 1H), 7.41 (d, J = 7.9 Hz, 1H), 7.35–7.31 (m, 1H), 7.31–7.29 (m, 1H), 6.89 (t, J = 2.2 Hz, 1H), 6.87 (t, J = 2.0 Hz, 1H), 6.85 (t, J = 2.2 Hz, 1H), 6.64 (d, J = 5.7 Hz, 1H), 5.90 (d, J = 5.5 Hz, 1H), 3.83 (s, 3H), 2.89 (ddd, J = 13.2, 11.0, 9.7 Hz, 1H), 2.72 (ddd, J = 17.2, 10.9, 8.8 Hz, 1H), 2.58 (ddd, J = 17.2, 9.8, 1.6 Hz, 1H), 2.49 (ddd, J = 13.2, 9.0, 1.7 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.4, 165.0, 160.2, 157.8, 149.9, 140.7, 137.2, 130.0, 123.5, 119.5, 118.1, 117.7, 113.4, 111.9, 107.1, 69.2, 55.5, 30.2, 29.7. HRMS (ESI) m/z calculated for C19H18N3O3 [M + H]+: 336.1325; found [M + H]+: 336.1328.
(R)-2-Cyclohexyl-8a-(pyridin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6n)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6n (159 mg, 51%) as yellow oil. 1H NMR (500 MHz, chloroform-d) δ 8.61–8.57 (m, 1H), 7.67 (td, J = 7.7, 1.8 Hz, 1H), 7.28 (d, J = 6.8 Hz, 1H), 7.24 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 6.54 (d, J = 5.8 Hz, 1H), 5.77 (d, J = 5.9 Hz, 1H), 4.52 (tt, J = 11.8, 3.8 Hz, 1H), 2.84–2.75 (m, 1H), 2.74–2.65 (m, 1H), 2.62–2.43 (m, 2H), 1.83 (d, J = 9.1 Hz, 2H), 1.69 (d, J = 6.4 Hz, 2H), 1.51–1.25 (m, 5H), 1.16–1.07 (m, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.6, 165.1, 158.0, 149.7, 136.9, 123.20, 119.5, 112.6, 106.9, 68.8, 52.9, 31.6, 30.6, 30.6, 29.8, 25.7, 25.6, 25.3. HRMS (ESI) m/z calculated for C18H22N3O2 [M + H]+: 312.1634; found [M + H]+: 312.1638.
(R)-3-(1,6-Dioxo-8a-(pyridin-2-yl)-6,7,8,8a-tetrahydropyrrolo[1,2-a]pyrazin-2(1H)-yl)propanenitrile (6o)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6o (150 mg, 53%) as brown oil. 1H NMR (500 MHz, chloroform-d) δ 8.59 (dd, J = 5.4, 1.9 Hz, 1H), 7.71 (td, J = 7.8, 1.8 Hz, 1H), 7.28–7.23 (m, 2H), 6.58 (d, J = 5.7 Hz, 1H), 5.76 (d, J = 5.5 Hz, 1H), 3.92–3.77 (m, 2H), 2.85–2.74 (m, 2H), 2.69–2.60 (m, 2H), 2.57–2.44 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.2, 165.5, 157.4, 149.8, 137.2, 123.5, 119.4, 117.1, 116.3, 107.6, 68.7, 43.3, 30.0, 29.5, 16.9. HRMS (ESI) m/z calculated for C15H15N4O2 [M + H]+: 283.1145; found [M + H]+: 283.1149.
(R)-2-Phenethyl-8a-(quinolin-2-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6p)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6p (276 mg, 72%) as a brown solid. 1H NMR (500 MHz, chloroform-d) δ 8.16 (d, J = 8.7 Hz, 1H), 8.09 (d, J = 8.5 Hz, 1H), 7.85 (dd, J = 11.2, 8.2 Hz, 1H), 7.73 (ddd, J = 8.5, 6.9, 1.6 Hz, 1H), 7.65–7.50 (m, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.21–7.11 (m, 3H), 7.06 (dd, J = 7.8, 1.8 Hz, 2H), 6.58 (d, J = 5.5 Hz, 1H), 5.48 (d, J = 5.5 Hz, 1H), 3.91 (dt, J = 13.7, 7.1 Hz, 1H), 3.75 (dt, J = 13.4, 7.5 Hz, 1H), 2.89 (t, J = 7.5 Hz, 2H), 2.86–2.77 (m, 1H), 2.71–2.61 (m, 1H), 2.57–2.50 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.5, 165.3, 157.1, 147.3, 137.9, 137.5, 130.0, 129.9, 128.8, 128.5, 127.6, 127.4, 127.0, 126.6, 117.4, 116.4, 107.4, 69.4, 48.3, 34.6, 30.6, 29.7. HRMS (ESI) m/z calculated for C24H22N3O2 [M + H]+: 384.1632; found [M + H]+: 384.1636.
(R)-8a-(5-Methylpyridin-2-yl)-2-phenethyl-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6q)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6q (257 mg, 68%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.44 (d, J = 2.8 Hz, 1H), 7.46 (dd, J = 8.0, 2.4 Hz, 1H), 7.28–7.16 (m, 3H), 7.12–7.03 (m, 3H), 6.48 (d, J = 5.7 Hz, 1H), 5.49 (d, J = 5.7 Hz, 1H), 3.91–3.82 (m, 1H), 3.81–3.69 (m, 1H), 2.93–2.81 (m, 2H), 2.79–2.69 (m, 1H), 2.67–2.56 (m, 1H), 2.54–2.44 (m, 2H), 2.35 (s, 3H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.3, 164.8, 158.6, 151.5, 139.6, 137.7, 128.9, 128.6, 126.7, 124.2, 118.3, 116.5, 107.0, 68.3, 48.0, 34.5, 30.7, 29.7. HRMS (ESI) m/z calculated for C21H22N3O2 [M + H]+: 348.1615; found [M + H]+: 348.1637.
(R)-8a-(6-Chloropyridin-2-yl)-2-phenethyl-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6r)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6r (314 mg, 71%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 7.59 (t, J = 7.8 Hz, 1H), 7.28–7.25 (m, 2H), 7.25–7.20 (m, 2H), 7.15–7.09 (m, 2H), 7.07 (d, J = 7.6 Hz, 1H), 6.48 (d, J = 5.7 Hz, 1H), 5.56 (d, J = 5.7 Hz, 1H), 3.98–3.89 (m, 1H), 3.76 (dt, J = 13.9, 7.3 Hz, 1H), 2.90 (td, J = 7.3, 3.9 Hz, 2H), 2.78–2.66 (m, 2H), 2.56–2.41 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.5, 165.3, 154.6, 150.3, 137.9, 137.4, 132.9, 128.9, 128.5, 126.6, 119.1, 116.7, 106.7, 68.6, 48.3, 34.6, 30.7, 29.7, 18.1. HRMS (ESI) m/z calculated for C20H19ClN3O2 [M + H]+: 368.1162; found [M + H]+: 368.1165.
(R)-8a-(6-Bromopyridin-2-yl)-2-phenethyl-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6s)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6s (165 mg, 40%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 7.50–7.42 (m, 2H), 7.27–7.21 (m, 3H), 7.14–7.08 (m, 3H), 6.48 (d, J = 5.7 Hz, 1H), 5.56 (d, J = 5.7 Hz, 1H), 3.94 (dt, J = 14.3, 7.3 Hz, 1H), 3.76 (dt, J = 13.6, 7.2 Hz, 1H), 2.91 (td, J = 7.3, 2.3 Hz, 2H), 2.77–2.67 (m, 2H), 2.54–2.44 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.3, 164.8, 159.0, 142.1, 139.2, 137.7, 128.9, 128.6, 128.0, 126.7, 118.7, 116.4, 107.1, 68.3, 46.4, 34.1, 30.7, 29.7. HRMS (ESI) m/z calculated for C20H19BrN3O2 [M + H]+: 412.0653; found [M + H]+: 412.0656.
(R)-8a-(5-Bromopyridin-2-yl)-2-phenethyl-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6t)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6t (186 mg, 45%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.64 (d, J = 2.4 Hz, 1H), 7.76 (dd, J = 8.5, 2.4 Hz, 1H), 7.24 (dd, J = 5.0, 2.2 Hz, 3H), 7.10–7.06 (m, 3H), 6.49 (d, J = 5.5 Hz, 1H), 5.54 (d, J = 5.7 Hz, 1H), 3.96–3.89 (m, 1H), 3.74 (dt, J = 13.7, 7.2 Hz, 1H), 2.94–2.82 (m, 2H), 2.78–2.70 (m, 1H), 2.59 (ddd, J = 17.3, 11.0, 8.5 Hz, 1H), 2.53–2.42 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 173.3, 164.8, 156.1, 150.8, 139.6, 137.6, 128.8, 128.6, 126.7, 121.1, 120.5, 116.5, 106.8, 68.4, 48.0, 34.5, 30.7, 29.5. HRMS (ESI) m/z calculated for C20H19BrN3O2 [M + H]+: 412.0655; found [M + H]+: 412.0658.
(R)-2-Phenethyl-8a-(pyridin-4-yl)-8,8a-dihydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (6u)
Synthesis according to procedure A in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 6u (183 mg, 55%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.62 (d, J = 6.3 Hz, 2H), 7.19 (ddd, J = 13.1, 5.0, 2.1 Hz, 5H), 6.99–6.91 (m, 2H), 6.52 (d, J = 5.7 Hz, 1H), 5.48 (d, J = 5.7 Hz, 1H), 3.88 (dt, J = 13.6, 6.8 Hz, 1H), 3.69–3.61 (m, 1H), 2.89–2.73 (m, 3H), 2.53–2.34 (m, 3H). 13C{1H} NMR (126 MHz, chloroform-d) δ 172.9, 164.7, 150.4, 147.3, 137.4, 128.7, 128.6, 126.7, 120.0, 117.1, 105.8, 67.0, 48.3, 34.4, 32.5, 29.0. HRMS (ESI) m/z calculated for C20H20N3O2 [M + H]+: 334.1526; found [M + H]+: 334.1528.
(8aR,13aS)-8a-(Pyridin-2-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (7a)
Synthesis according to procedure B in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 7a (300 mg, 90%) as a white solid. 1H NMR (500 MHz, chloroform-d) δ 8.65 (ddd, J = 4.7, 1.7, 1.1 Hz, 1H), 7.78 (td, J = 7.7, 1.9 Hz, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.32–7.29 (m, 1H), 7.24–7.16 (m, 3H), 7.04 (dd, J = 6.9, 2.2 Hz, 1H), 4.66 (ddd, J = 12.6, 4.7, 3.2 Hz, 1H), 4.42 (dd, J = 11.0, 5.5 Hz, 1H), 3.93 (dd, J = 12.6, 5.5 Hz, 1H), 3.60–3.54 (m, 1H), 3.09–3.02 (m, 2H), 2.93 (ddd, J = 16.1, 11.3, 4.7 Hz, 1H), 2.82 (dt, J = 15.8, 3.4 Hz, 1H), 2.61–2.53 (m, 1H), 2.42 (ddd, J = 17.0, 9.9, 3.5 Hz, 1H), 2.26 (ddd, J = 13.1, 9.3, 3.5 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 175.1, 170.0, 159.3, 149.8, 137.5, 134.8, 132.5, 128.9, 127.6, 126.9, 125.6, 123.2, 119.6, 70.9, 52.1, 46.6, 39.2, 31.3, 29.5, 28.9. HRMS (ESI) m/z calculated for C20H20N3O2 [M + H]+: 334.1543; found [M + H]+: 334.1548.
(8aR,13aS)-2-Methoxy-8a-(pyridin-2-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (7b)
Synthesis according to procedure B in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 7b (200 mg, 55%) as a white solid. 1H NMR (500 MHz, chloroform-d) δ 8.65 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 7.78 (td, J = 7.7, 1.8 Hz, 1H), 7.52 (dt, J = 7.9, 1.0 Hz, 1H), 7.30 (ddd, J = 7.4, 4.8, 1.1 Hz, 1H), 7.08 (dd, J = 8.4, 4.3 Hz, 1H), 6.78 (dd, J = 8.4, 2.7 Hz, 1H), 6.55 (d, J = 2.7 Hz, 1H), 4.64 (ddd, J = 12.8, 4.7, 3.3 Hz, 1H), 4.38 (dd, J = 11.0, 5.4 Hz, 1H), 3.92 (dd, J = 12.6, 5.5 Hz, 1H), 3.77 (s, 3H), 3.60–3.54 (m, 1H), 3.09–2.98 (m, 2H), 2.92–2.80 (m, 1H), 2.75 (dt, J = 15.4, 3.5 Hz, 1H), 2.62–2.53 (m, 1H), 2.42 (ddd, J = 17.0, 9.9, 3.4 Hz, 1H), 2.26 (ddd, J = 13.2, 9.3, 3.4 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 175.1, 169.9, 159.3, 158.5, 149.8, 137.5, 133.4, 129.9, 126.8, 123.2, 119.6, 113.7, 110.7, 70.9, 55.4, 52.2, 46.6, 39.5, 31.4, 29.5, 28.0.. HRMS (ESI) m/z calculated for C21H22N3O3 [M + H]+: 364.1631; found [M + H]+: 364.1634.
(8aR,13aS)-4-Methoxy-8a-(pyridin-2-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (7c)
Synthesis according to procedure B in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 7c (164 mg, 45%) as a white solid. 1H NMR (500 MHz, chloroform-d) δ 8.67–8.62 (m, 1H), 7.78 (td, J = 7.8, 1.9 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.33–7.28 (m, 1H), 7.17 (t, J = 8.0 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 6.64 (d, J = 7.7 Hz, 1H), 4.71 (ddd, J = 12.9, 5.1, 2.8 Hz, 1H), 4.42 (dd, J = 10.8, 5.4 Hz, 1H), 3.90 (dd, J = 12.5, 5.0 Hz, 1H), 3.84 (s, 3H), 3.59–3.51 (m, 1H), 3.10–2.99 (m, 2H), 2.99–2.88 (m, 1H), 2.69–2.49 (m, 2H), 2.43 (dd, J = 9.9, 3.3 Hz, 1H), 2.26 (ddd, J = 12.8, 9.3, 3.4 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 175.0, 169.9, 159.3, 156.7, 149.8, 137.5, 133.8, 127.4, 123.7, 123.2, 119.7, 117.5, 108.8, 70.8, 55.5, 52.0, 46.5, 38.8, 31.3, 29.5, 22.0. HRMS (ESI) m/z calculated for C21H22N3O3 [M + H]+: 364.1642; found [M + H]+: 364.1645.
(8aR,13aS)-8a-(6-Methylpyridin-2-yl) 5,6,9,10, 13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (7d)
Synthesis according to procedure B in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 7d (167 mg, 48%) as a white solid. 1H NMR (500 MHz, chloroform-d) δ 7.64 (t, J = 7.7 Hz, 1H), 7.29 (s, 1H), 7.24–7.20 (m, 2H), 7.20–7.17 (m, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.06–7.02 (m, 1H), 4.65 (ddd, J = 12.6, 4.7, 3.2 Hz, 1H), 4.48 (dd, J = 11.0, 5.4 Hz, 1H), 3.94 (dd, J = 12.5, 5.4 Hz, 1H), 3.60–3.52 (m, 1H), 3.09–2.99 (m, 2H), 2.98–2.90 (m, 1H), 2.82 (dt, J = 15.8, 3.4 Hz, 1H), 2.61 (d, J = 7.7 Hz, 1H), 2.58 (s, 3H), 2.40 (ddd, J = 16.9, 9.9, 3.3 Hz, 1H), 2.27 (ddd, J = 12.9, 9.3, 3.2 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 175.0, 170.2, 158.8, 158.4, 137.5, 134.8, 132.7, 128.9, 127.5, 126.9, 125.6, 122.6, 116.5, 70.9, 52.1, 46.52, 39.2, 31.3, 29.6, 28.9, 24.8. HRMS (ESI) m/z calculated for C21H22N3O2 [M + H]+: 348.1635; found [M + H]+: 348.1638.
(8aR,13aS)-8a-(Quinolin-2-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (7e)
Synthesis according to procedure B in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 7e (180 mg, 47%) as a white solid. 1H NMR (500 MHz, chloroform-d) δ 8.26 (d, J = 8.7 Hz, 1H), 8.15 (d, J = 7.6 Hz, 1H), 7.87 (d, J = 9.8 Hz, 1H), 7.83–7.73 (m, 1H), 7.66 (d, J = 8.5 Hz, 1H), 7.60 (t, J = 7.5 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 7.24–7.18 (m, 2H), 7.04–6.98 (m, 1H), 4.67 (dt, J = 12.5, 4.0 Hz, 1H), 4.51 (dd, J = 11.0, 5.4 Hz, 1H), 4.12 (dd, J = 12.5, 5.5 Hz, 1H), 3.65–3.57 (m, 2H), 3.15–3.03 (m, 1H), 3.00–2.91 (m, 1H), 2.89–2.83 (m, 2H), 2.66–2.52 (m, 1H), 2.50–2.32 (m, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 175.0, 170.0, 161.1, 158.7, 147.4, 138.5, 138.0, 134.7, 132.5, 130.1, 130.0, 128.9, 128.8, 128.7, 127.5, 127.1, 126.9, 126.7, 125.6, 117.4, 71.6, 52.1, 46.9, 39.3, 35.5, 31.0, 28.9. HRMS (ESI) m/z calculated for C24H22N3O2 [M + H]+: 384.1613; found [M + H]+: 384.1616.
(8aR,13aS)-8a-(6-Bromopyridin-2-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a] isoquinoline-8,11(8aH)-dione (7f)
Synthesis according to procedure B in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 7f (350 mg, 85%) as a brown solid. 1H NMR (500 MHz, chloroform-d) δ 7.62 (t, J = 7.8 Hz, 1H), 7.50 (dd, J = 7.7, 5.0 Hz, 2H), 7.25 (dd, J = 5.8, 3.4 Hz, 2H), 7.19 (dd, J = 5.4, 3.5 Hz, 1H), 7.13 (dd, J = 5.5, 3.6 Hz, 1H), 4.63 (ddd, J = 12.6, 4.8, 3.3 Hz, 1H), 4.53 (dd, J = 10.4, 5.5 Hz, 1H), 3.97 (dd, J = 12.7, 5.4 Hz, 1H), 3.65 (dd, J = 12.7, 10.0 Hz, 1H), 3.06 (ddd, J = 12.6, 11.2, 3.5 Hz, 1H), 3.01–2.91 (m, 2H), 2.89–2.79 (m, 1H), 2.67–2.54 (m, 1H), 2.40 (ddd, J = 16.9, 9.8, 3.0 Hz, 1H), 2.30 (ddd, J = 13.4, 9.2, 3.0 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 174.7, 169.4, 160.4, 142.2, 139.7, 134.7, 132.5, 128.9, 127.9, 127.7, 127.1, 125.5, 118.8, 70.3, 52.5, 45.8, 39.6, 31.6, 29.4, 28.8. HRMS (ESI) m/z calculated for C20H19BrN3O2 [M + H]+: 412.0625; found [M + H]+: 412.0629.
(8aR,13aS)-8a-(Pyridin-4-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (7g)
Synthesis according to procedure B in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 7g (237 mg, 71%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.72 (d, J = 6.3 Hz, 2H), 7.45–7.39 (m, 2H), 7.27–7.21 (m, 2H), 7.20 (dd, J = 6.5, 2.5 Hz, 1H), 7.07–7.01 (m, 1H), 4.70–4.62 (m, 1H), 4.45 (dd, J = 11.3, 5.2 Hz, 1H), 3.90 (dd, J = 12.8, 5.3 Hz, 1H), 3.59 (t, J = 12.1 Hz, 1H), 3.13–2.99 (m, 2H), 2.98–2.90 (m, 1H), 2.83 (dt, J = 15.8, 3.2 Hz, 1H), 2.58–2.47 (m, 1H), 2.43 (ddd, J = 17.2, 9.9, 3.6 Hz, 1H), 2.17 (ddd, J = 13.2, 9.1, 3.7 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 175.2, 169.1, 151.0, 149.4, 134.7, 131.7, 129.1, 127.8, 127.1, 125.6, 119.4, 68.7, 51.9, 46.9, 39.3, 32.6, 29.0, 28.8. HRMS (ESI) m/z calculated for C20H20N3O2 [M + H]+: 334.1532; found [M + H]+: 334.1535.
(8aR,13aS)-2,3-Dimethoxy-8a-(pyridin-2-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (7h)
Synthesis according to procedure C in the 1 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 7h (299 mg, 76%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 8.58–8.53 (m, 1H), 7.79 (td, J = 7.8, 1.9 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.32 (dd, J = 7.6, 4.7 Hz, 1H), 6.80–6.76 (m, 1H), 6.76–6.71 (m, 1H), 5.00 (s, 1H), 4.23 (dd, J = 14.0, 6.7 Hz, 1H), 3.97–3.89 (m, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.63 (dt, J = 13.4, 7.2 Hz, 1H), 3.32 (dd, J = 13.4, 3.6 Hz, 1H), 2.88 (t, J = 7.5 Hz, 2H), 2.70 (ddd, J = 13.1, 8.4, 2.7 Hz, 1H), 2.66–2.45 (m, 2H), 2.38 (ddd, J = 16.6, 9.1, 2.8 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 174.1, 170.3, 158.0, 149.1, 149.0, 147.7, 138.0, 131.2, 123.7, 121.4, 120.9, 112.0, 111.3, 78.7, 69.4, 55.94, 55.9, 46.5, 44.0, 33.9, 31.6, 29.9. HRMS (ESI) m/z calculated for C22H24N3O4 [M + H]+: 394.1733; found [M + H]+: 394.1738.
(8aR,13aS)-8a-(Pyrimidin-4-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (7i)
Synthesis according to procedure C in the 1 mmol scale and purification by column chromatography (silica gel, dichloromethane: methanol= 1:20) afforded 7i (117 mg, 35%) as a light-yellow solid. 1H NMR (500 MHz, chloroform-d) δ 9.26 (s, 1H), 8.83 (s, 1H), 7.60 (s, 1H), 7.26–7.14 (m, 3H), 7.08 (d, J = 6.3 Hz, 1H), 4.62 (dt, J = 12.6, 4.1 Hz, 1H), 4.43 (dd, J = 10.4, 5.2 Hz, 1H), 3.90 (dd, J = 12.8, 5.2 Hz, 1H), 3.68–3.60 (m, 1H), 3.04 (td, J = 11.9, 3.6 Hz, 1H), 3.00–2.88 (m, 2H), 2.82 (d, J = 15.8 Hz, 1H), 2.53 (dt, J = 18.6, 9.4 Hz, 1H), 2.40 (ddd, J = 17.0, 9.9, 3.1 Hz, 1H), 2.25 (td, J = 9.5, 4.6 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 174.7, 168.6, 167.9, 159.0, 158.1, 134.7, 132.1, 129.0, 127.8, 127.1, 125.4, 117.4, 70.0, 52.6, 45.7, 39.7, 31.4, 29.2, 28.7. HRMS (ESI) m/z calculated for C19H19N4O2 [M + H]+: 335.1432; found [M + H]+: 335.1437.
(R)-2-Phenethyl-8a-(pyridin-2-yl)tetrahydropyrrolo[1,2-a]pyrazine-1,6(2H,7H)-dione (8a)
Synthesis according to procedure E in the 0.3 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 8a (100 mg, 98%) as a white solid. 1H NMR (500 MHz, chloroform-d) δ 8.59 (ddd, J = 4.9, 1.9, 1.0 Hz, 1H), 7.69 (td, J = 7.7, 1.8 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.27–7.20 (m, 4H), 7.17–7.13 (m, 2H), 3.95–3.82 (m, 2H), 3.53 (dt, J = 13.6, 7.2 Hz, 1H), 3.30–3.18 (m, 2H), 3.02–2.92 (m, 1H), 2.90 (t, J = 6.5 Hz, 2H), 2.76 (dt, J = 13.1, 9.3 Hz, 1H), 2.64–2.53 (m, 1H), 2.46–2.32 (m, 2H). 13C{1H} NMR (126 MHz, chloroform-d) δ 174.4, 169.6, 158.9, 149.5, 138.4, 137.1, 128.9, 128.6, 126.6, 123.0, 120.4, 70.1, 49.2, 45.9, 36.5, 33.7, 31.2, 30.0. HRMS (ESI) m/z calculated for C20H22N3O2 [M + H]+: 336.1624; found [M + H]+: 336.1627.
(8aR,13aR)-8a-(5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)pyridin-2-yl)-5,6,9,10,13,13a-hexahydro-8H-pyrrolo[1′,2′:4,5]pyrazino[2,1-a]isoquinoline-8,11(8aH)-dione (8b)
Synthesis according to procedure F in the 0.24 mmol scale and purification by column chromatography (silica gel, petroleum ether/ethyl acetate = 1:3) afforded 8b (100 mg, 91%) as a white solid. 1H NMR (500 MHz, chloroform-d) δ 8.81 (dd, J = 2.4, 0.8 Hz, 1H), 7.89 (dd, J = 8.2, 2.4 Hz, 1H), 7.72–7.67 (m, 1H), 7.57 (td, J = 7.2, 1.5 Hz, 1H), 7.54 (d, J = 8.2 Hz, 1H), 7.51–7.46 (m, 1H), 7.21 (d, J = 13.7 Hz, 1H), 7.13 (d, J = 2.2 Hz, 1H), 7.08 (td, J = 8.7, 2.2 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.68 (dt, J = 12.6, 3.9 Hz, 1H), 4.51 (dd, J = 11.0, 5.4 Hz, 1H), 4.33 (s, 4H), 3.97 (dd, J = 12.6, 5.4 Hz, 1H), 3.63–3.55 (m, 1H), 3.07 (dt, J = 13.2, 9.5 Hz, 2H), 3.00–2.89 (m, 1H), 2.83 (dt, J = 15.8, 3.5 Hz, 1H), 2.70–2.54 (m, 1H), 2.44 (ddd, J = 16.9, 9.9, 3.4 Hz, 1H), 2.31 (ddd, J = 12.9, 9.3, 3.4 Hz, 1H). 13C{1H} NMR (126 MHz, chloroform-d) δ 175.1, 170.0, 157.4, 147.8, 144.1, 144.1, 135.4, 134.8, 132.2, 132.0, 130.4, 128.9, 128.6, 128.5, 127.6, 127.0, 125.6, 120.2, 119.5, 118.1, 115.9, 70.7, 64.4, 52.1, 46.6, 39.3, 31.4, 29.5, 28.9. HRMS (ESI) m/z calculated for C28H26N3O4 [M + H]+: 468.1815; found [M + H]+: 468.1818.
Acknowledgments
This research has been supported (to A.D.) by the National Institutes of Health (NIH) (2R01GM097082-05), the European Lead Factory (IMI) under Grant Agreement No. 115489, and the Qatar National Research Foundation (NPRP6-065-3- 012). The authors acknowledge Marcel de Vries (University of Groningen) for his help with HRMS analysis. B.Z. acknowledges the China Scholarship Council for support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c00244.
NMR spectra and crystal structure determinations (PDF)
Accession Codes
CCDC 2089897–2089898 and 2141575 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
CCDC 2089898 (6a) and CCDC 2089897 (7a) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif or by emailing data_request@ccdc.cam.ac.uk or by contacting the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Giraudo A.; Krall J.; Bavo F.; Nielsen B.; Kongstad K. T.; Rolando B.; Blasio R. D.; Gloriam D. E.; Löffler R.; Thiesen L.; Harpsøe K.; Frydenvang K.; Boschi D.; Wellendorph P.; Lolli M. L.; Jensen A. A.; Frølund B. Five-Membered N-Heterocyclic Scaffolds as Novel Amino Bioisosteres at γ-Aminobutyric Acid (GABA) Type A Receptors and GABA Transporters. J. Med. Chem. 2019, 62, 5797–5809. 10.1021/acs.jmedchem.9b00026. [DOI] [PubMed] [Google Scholar]; b Wu X.-F.; Neumann H.; Beller M. Synthesis of Heterocycles via Palladium-Catalyzed Carbonylations. Chem. Rev. 2013, 113, 1–35. 10.1021/cr300100s. [DOI] [PubMed] [Google Scholar]
- Ibarra I. A.; Islas-Jácome A.; González-Zamora E. Synthesis of polyheterocycles via multicomponent reactions. Org. Biomol. Chem. 2018, 16, 1402–1418. 10.1039/C7OB02305G. [DOI] [PubMed] [Google Scholar]
- Angelucci F.; Basso A.; Bellelli A.; Brunori M.; Mattoccia L. P.; Valle C. The anti-schistosomal drug praziquantel is an adenosine antagonist. Parasitology 2007, 134, 1215–1221. 10.1017/S0031182007002600. [DOI] [PubMed] [Google Scholar]
- Coward R. M.; Carson C. C. Tadalafi l in the treatment of erectile dysfunction. Ther. Clin. Risk Manage. 2008, 4, 1315–1329. 10.2147/TCRM.S3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.; Liu J.; Bi L.; Zhao M.; Wang C.; Baudy-Floc’h M.; Ju J.; Peng S. Toward Breast Cancer Resistance Protein (BCRP) Inhibitors: Design, Synthesis of A Series of New Simplified Fumitremorgin C Analogues. Tetrahedron 2007, 63, 5510–5528. 10.1016/j.tet.2007.04.045. [DOI] [Google Scholar]
- Zarganes-Tzitzikas T.; Zarganes-Tzitzikas T.; Chandgude A. L. Multicomponent Reactions, Union of MCRs and Beyond. Chem. Rec. 2015, 15, 981–996. 10.1002/tcr.201500201. [DOI] [PubMed] [Google Scholar]
- a Dömling A. Recent Developments in Isocyanide Based Multicomponent Reactions in Applied Chemistry. Chem. Rev. 2006, 106, 17–89. 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; b Dömling A.; Ugi I. Multicomponent reactions with isocyanides. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. [DOI] [PubMed] [Google Scholar]; c Dömling A.; Wang W.; Wang K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmanschap G.; Ruijter E.; Orru R. V. A. Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–598. 10.3762/bjoc.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heravi M.; Zadsirjan V.; Malmir M. Application of the Asymmetric Pictet–Spengler Reaction in the Total Synthesis of Natural Products and Relevant Biologically Active Compounds. Molecules 2018, 23, 943–991. 10.3390/molecules23040943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Znabet A.; Zonneveld J.; Janssen E.; De Kanter F. J. J.; Helliwell M.; Turner N. J.; Ruijter E.; Orru R. V. A. Asymmetric synthesis of synthetic alkaloids by a tandem biocatalysis/Ugi/Pictet–Spengler-type cyclization sequence. Chem. Commun. 2010, 46, 7706–7708. 10.1039/c0cc02938f. [DOI] [PubMed] [Google Scholar]
- Lei X. F.; Lampiri P.; Patil P.; Angeli G.; Neochoritis C. G.; Dömling A. A multicomponent tetrazolo indole synthesis. Chem. Commun. 2021, 57, 6652–6655. 10.1039/D1CC02384E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso F.; Galilea A.; Mañez P. A.; Acebedo S. L.; Cabrera G. M.; Otero M.; Barquero A. A.; Ramírez J. A. Beyond Pseudo-natural Products: Sequential Ugi/Pictet- Spengler Reactions Leading to Steroidal Pyrazinoisoquinolines That Trigger Caspase-Independent Death in HepG2 Cells. ChemMedChem 2021, 16, 1945–1955. 10.1002/cmdc.202100052. [DOI] [PubMed] [Google Scholar]
- Kurva M.; Kerim M. D.; Gàmez-Montaño R.; Kaim L El. Straightforward access to complex isoindolinones from the Ugi reaction of o-nitrobenzoic acid derivatives. Org. Biomol. Chem. 2019, 17, 9655–9659. 10.1039/C9OB02065A. [DOI] [PubMed] [Google Scholar]
- Cárdenas-Galindo L. E.; Islas-Jácome A.; Alvarez-Rodríguez N. V.; Kaim L. E.; Gámez-Montaño R. paper Synthesis of 2-Tetrazolylmethyl-2,3,4,9-tetrahydro-1H-β-carbolines by a One-Pot Ugi-Azide/Pictet–Spengler Process. Synthesis 2014, 46, 49–56. 10.1055/s-0033-1340051. [DOI] [Google Scholar]
- Dömling A.; Khoury K. Praziquantel and Schistosomiasis. ChemMedChem 2010, 5, 1420–1434. 10.1002/cmdc.201000202. [DOI] [PubMed] [Google Scholar]
- Wang W.; Ollio S.; Herdtweck E.; Dömling A. Polycyclic Compounds by Ugi-Pictet-Spengler Sequence. J. Org. Chem. 2011, 76, 637–644. 10.1021/jo102058s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Znabet A.; Zonneveld J.; Janssen E.; De Kanter F. J. J.; Helliwell M.; Turner N. J.; Ruijter E.; Orru R. V. A. Asymmetric synthesis of synthetic alkaloids by a tandem biocatalysis/Ugi/Pictet–Spengler-type cyclization sequence. Chem. Commun. 2010, 46, 7706–7708. 10.1039/c0cc02938f. [DOI] [PubMed] [Google Scholar]
- a Calcaterra A.; Mangiardi L.; Monache G. D.; Quaglio D.; Balducci Si.; Berardozzi S.; Iazzetti A.; Franzini R.; Botta B.; Ghirga F. The Pictet-Spengler Reaction Updates Its Habits. Molecules 2020, 25, 414–496. 10.3390/molecules25020414. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ingallina C.; D’Acquarica I.; Monache G. D.; Ghirga F.; Quaglio D.; Ghirga P.; Berardozzi S.; Markovic V.; Botta B. The Pictet-Spengler Reaction Still on Stage. Curr. Pharm. Des. 2016, 22, 1808–1850. 10.2174/1381612822666151231100247. [DOI] [PubMed] [Google Scholar]; c Dömling A.; Huang Y. Piperazine Scaffolds via Isocyanide-Based Multicomponent Reactions. Synthesis 2010, 2010, 2859–2883. 10.1055/s-0030-1257906. [DOI] [Google Scholar]
- Wang Y.; Patil P.; Kurpiewska K.; Kalinowska-Tluscik J.; Dömling A. Diverse Isoquinoline Scaffolds by Ugi/Pomeranz–Fritsch and Ugi/Schlittler–Müller Reactions. Org. Lett. 2019, 21, 3533–3537. 10.1021/acs.orglett.9b00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. F.; Chen M.; Arrhenius T.; Nadzan A. A convenient solution and solid-phase synthesis of Δ5-2-oxopiperazines via N-acyliminium ions cyclization. Tetrahedron Lett. 2002, 43, 6293–6295. 10.1016/S0040-4039(02)01403-X. [DOI] [Google Scholar]
- Buskes M. J.; Blanco M. Impact of Cross-Coupling Reactions in Drug Discovery and Development. Molecules 2020, 25, 3493–3515. 10.3390/molecules25153493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E.; Kurpiewska K.; Kalinowska-Tluscik J.; Dömling A. Cleavable β-Cyanoethyl Isocyanide in the Ugi Tetrazole Reaction. Org. Lett. 2016, 18, 4762–4765. 10.1021/acs.orglett.6b01826. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.