

ABSTRACT
Introduction
Tecovirimat (TPOXX®; ST-246) was approved for the treatment of symptomatic smallpox by the USFDA in July of 2018 and has been stockpiled by the US government for use in a smallpox outbreak. While there has not been a reported case of smallpox since 1978 it is still considered a serious bioterrorism threat.
Areas covered
A brief history of smallpox from its proposed origins as a human disease through its eradication in the late 20th century is presented. The current smallpox threat and the current public health response plans are described. The discovery, and development of tecovirimat through NDA submission and subsequent approval for treatment of smallpox are discussed. Google Scholar and PubMed were searched over all available dates for relevant publications.
Expert opinion
Approval of tecovirimat to treat smallpox represents an important milestone in biosecurity preparedness. Incorporating tecovirimat into the CDC smallpox response plan, development of pediatric liquid and intravenous formulations, and approval for post-exposure prophylaxis would provide additional health security benefit.
Tecovirimat shows broad efficacy against orthopoxviruses in vitro and in vivo and could be developed for use against emerging orthopoxvirus diseases such as monkeypox, vaccination-associated adverse events, and side effects of vaccinia oncolytic virus therapy.
KEYWORDS: Smallpox, tecovirimat, ST-246, biodefense, health security, monkeypox, vaccinia, variola, Animal Rule
1.
1. Introduction
1.1. Smallpox history
The impact of smallpox on the course of human history cannot be overstated. Smallpox has been called ‘the most terrible of the ministers of death’ by English historian Thomas Macaulay [1], and ‘The Greatest Killer’ by Donald R. Hopkins [2]. Smallpox may have emerged as early as 10,000 BCE [3], and some of the earliest historical descriptions of a disease consistent with smallpox appear between 1350 and 1122 BCE [3,4]. Nearly all susceptible individuals exposed to variola virus (VARV), the causative agent of smallpox, will contract disease [5]. Historically, up to 30% of unvaccinated individuals succumbed to disease [5] which, up until the 18th century, may have killed as many as one person in ten [6]. Smallpox is considered to have been a factor in the fall of at least three empires including the Aztecs, the Incas, and the Romans [7]. In the 20th century alone, smallpox is estimated to have claimed 300 to 500 million lives [8].
The World Health Organization (WHO) announced the eradication of smallpox in 1980 [9] and immunization of the general population was discontinued [10]. The last reported case of naturally occurring smallpox occurred in Somalia in 1977 [11] and the last known case of smallpox was a laboratory acquired case in England in 1978 [12]. After official declaration of eradication, all known stocks of VARV were supposed to be destroyed or deposited in one of two WHO approved laboratories, the United States Centers for Disease Control and Prevention (USCDC) in Atlanta, Georgia (USA), or the Moscow Research Institute for Viral Preparations, USSR [13]. In 1994, VARV stocks being held in Moscow were moved to the State Research Center of Virology and Biotechnology (VECTOR) in Koltsovo, Russian Federation [14].
1.2. Current smallpox threats
Although naturally occurring smallpox no longer exists, there is concern that unregistered stocks of VARV may remain. In 2014 vials containing VARV were discovered in a cold storage room at the National Institutes of Health and other such discoveries may have occurred and not been reported [15]. According to a 2010 report published in the Washington Post [16], four nations, including Iraq, North Korea, and France may have retained undocumented stocks of VARV, and Russia may have undeclared stocks of VARV in addition to those reported to the WHO. In addition, recent developments in synthetic biology show that it is possible to recreate smallpox from commercially available materials using published genomic sequences and standard laboratory equipment [17].
Concern that VARV could be used as a biological weapon has increased interest in planning for a possible outbreak or intentional release [18]. VARV is classified as a Category A Bioterrorism Agent by the USCDC, defined as agents that are easily transmitted from person to person, result in high mortality rates, have potential for large public health impact, may cause social disruption and panic if released, and require specific public health preparedness actions [19]. The USCDC considers that deliberate release of VARV is possible [20], and VARV is considered a national security risk by the US Department of Homeland Security [21]. A single confirmed case of smallpox anywhere in the world would be a public health emergency [20] and it is suggested that VARV has the potential to be a weapon of mass destruction [18].
The threat of smallpox is exacerbated by the current immunological status of the general public. Population immunity to smallpox has declined since 1980 when routine vaccination of the general public was ceased [5,22], and the fraction of the population that is at least partly immunodeficient has increased since the conclusion of the eradication program. HIV infection, cancer therapies, and immunosuppression therapy for autoimmune disease and organ transplant contribute to increased prevalence of immunodeficiency in the population [18,22]. In addition, today’s population is more mobile than during the eradication era, making contact tracing and outbreak containment more difficult [22], and diagnosis of initial cases may be delayed due to the lack of health care personnel familiar with clinical smallpox [23]. These factors combined suggest that the public is more susceptible to a smallpox outbreak than ever [18,23].
1.3. The need for antiviral countermeasures and the smallpox response plan
The Project Bioshield Act was signed into law in 2004 as part of the US government’s strategy to defend against attacks using chemical, biological, radiological, and nuclear (CBRN) agents [24]. Project Bioshield provides funding for the advanced development and procurement of medical countermeasures to CBRN threats, allows for expedited processing of grants and contracts for the development of critical countermeasures by NIH, and gives the US Food and Drug Administration (USFDA) Emergency Use Authorization (EUA) authority to allow access to the best available medical countermeasures in emergency situations [25]. In 2006, the Biomedical Advanced Research and Development Authority (BARDA) was established under the U.S. Department of Health and Human Services to manage Project Bioshield and serve as the official interface between the biomedical industry and the US government [26].
In 1999, in consideration of the potentially devastating impact of a smallpox outbreak, the Committee on the Assessment of Future Scientific Needs for Variola Virus of the Institute of Medicine recommended development of medical countermeasures to meet this threat including the development of antiviral drugs and safer vaccines [22]. The USCDC issued guidance for smallpox response planning in 2001 [27,28], followed by periodic updates [29,30]. The most recent update, issued in 2015 [31], includes the addition of non-replicating vaccines (IMVAMUNE®, JYNNEOSTM) to the medical countermeasures available for use in a smallpox emergency.
The current strategy for response to a smallpox outbreak following confirmed diagnosis of human smallpox relies heavily on isolation and treatment of confirmed cases and focused vaccination of first and second degree contacts of confirmed and suspected cases (ring vaccination) with a live replicating vaccine (e.g. ACAM2000) [29]. Individuals with risk factors for receiving ACAM2000 but without known exposure to VARV could be given JYNNEOS [31]. Healthcare providers, first responders, laboratory workers, and others likely to come into contact with smallpox patients or clinical samples possibly containing VARV would also be vaccinated [29]. Large scale vaccination of all persons within a community, city, or larger region may be recommended if an outbreak is sufficiently large or dispersed [29]. At the present time, the official smallpox response plan does not incorporate use of antiviral drugs (e.g. TPOXX®, (tecovirimat)) in an outbreak.
Post-exposure vaccination with ACAM2000 provides some protection if administered within three days of exposure, diminishing rapidly afterward [32,33]. Prior to tecovirimat approval no USFDA approved antiviral drugs were available for treatment of smallpox. To date, approximately two million courses of the oral capsule formulation of tecovirimat, which received USFDA approval for the treatment of smallpox in 2018, have been procured by BARDA and delivered to the United States Strategic National Stockpile (SNS) for possible use in a smallpox emergency. It is likely that tecovirimat would be provided in all confirmed smallpox cases according to current full prescribing information on the label, but could also be provided ‘off-label’ to asymptomatic individuals suspected of exposure to VARV, concurrently with vaccination [34,35].
1.4. Discovery and early characterization of tecovirimat (ST-246)
1.4.1. Initial identification
The discovery and early characterization of tecovirimat (ST-246) are reviewed in detail in Jordan et al., 2010 [36] and Grosenbach et al., 2011 [37]. Using a high-throughput screening assay to evaluate 356,240 compounds from a structurally diverse chemical library 759 compounds were identified that inhibited vaccinia virus (VACV) or cowpox virus (CPXV)-induced cytopathic effect (CPE) >50% at a concentration of 5 μM in cultured cells [38,39]. Further examination of these initial hits identified a chemically related group of compounds that inhibited virus-induced CPE by 50% (EC50) at concentrations from 0.013 to 5 μM [39]. Structural analogs were generated from this group based on nascent structure-activity relationships to improve potency and metabolic stability and were tested in the cell-culture CPE assay against VACV and CPXV. Based on the observed low EC50 values in this assay and favorable metabolic stability, tecovirimat (Figure 1) was selected for further study [38].
Figure 1.
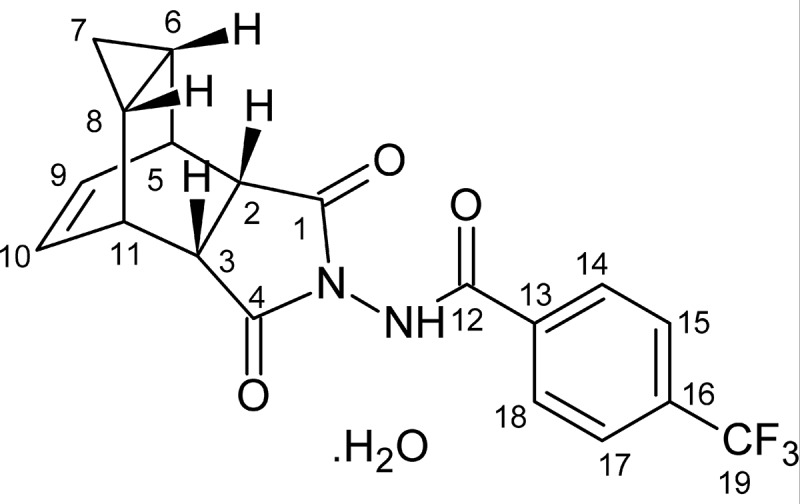
Molecular structure of tecovirimat (ST-246; 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop [f]isoindol-2(1 H)-yl)-benzamide)
1.4.2. Activity spectrum selectivity, cytotoxicity, and molecular target/mechanism of action
In CPE assays against a diverse panel of orthopoxviruses, including multiple strains of VARV, tecovirimat showed strong antiviral activity with an observed EC50 range from 0.01 to 0.07 μM [38,40,41] and was highly selective for orthopoxviruses as demonstrated by its inability to inhibit virus-induced CPE at concentrations up to 40 μM for multiple other classes of viruses tested (Table 1) [38,40]. Measurements of cytotoxicity in cell lines derived from mouse, rabbit, monkey, and human showed median 50% cytotoxic concentrations >50 μM in all cell lines tested [36,42].
Table 1.
Tecovirimat selectivity, and spectrum of antiviral activity
| Virus (Strain) | Family | Classification | EC50 | Reference |
|---|---|---|---|---|
| Vaccinia (NYCBH) | Orthopoxviridae | Double stranded DNA | 0.009 | [38] |
| Cowpox (Brighton Red) | Orthopoxviridae | Double stranded DNA | 0.050 | [38] |
| Cowpox cidofovir resistant (Brighton Red) | Orthopoxviridae | Double stranded DNA | 0.030 | [38] |
| Ectromelia | Orthopoxviridae | Double stranded DNA | 0.068 | [38] |
| Camelpox | Orthopoxviridae | Double stranded DNA | 0.012 | [38] |
| Monkeypox (Zaire ‘79) | Orthopoxviridae | Double stranded DNA | 0.014 | [38] |
| Variola (BUT) | Orthopoxviridae | Double stranded DNA | 0.016 | [38] |
| Variola (BSH) | Orthopoxviridae | Double stranded DNA | 0.046 | [38] |
| DQ441422_VARV_Bangladesh_1974_Solaiman | Orthopoxviridae | Double stranded DNA | 0.028 | [40] |
| DQ437588_VARV_Nepal_197 3 | Orthopoxviridae | Double stranded DNA | 0.021 | [40] |
| DQ441437_VARV_Sierra_Leone_1969 V68_258 | Orthopoxviridae | Double stranded DNA | 0.037 | [40] |
| DQ441419_VARV_Brazil_196 6 v66_39_Sao_Paulo | Orthopoxviridae | Double stranded DNA | 0.067 | [40] |
| DQ441430_VARV_Japan_195 1 Harper | Orthopoxviridae | Double stranded DNA | 0.026 | Unpublished |
| DQ441418_VARV_Botswana_ 1973 v73_225 | Orthopoxviridae | Double stranded DNA | 0.014 | Unpublished |
| DQ441417_VARV_Botswana_ 1972 v72_143 | Orthopoxviridae | Double stranded DNA | 0.011 | Unpublished |
| DQ441444_VARV_United_Kingdom_1946_Harvy | Orthopoxviridae | Double stranded DNA | 0.034 | Unpublished |
| DQ441416_VARV_Benin Dahomey_1968 v68_59 | Orthopoxviridae | Double stranded DNA | 0.015 | Unpublished |
| DQ437590_VARV_Somalia_1 977_V77-2479_Ali | Orthopoxviridae | Double stranded DNA | 0.028 | Unpublished |
| DQ441440_VARV_Sudan_1947 Juba | Orthopoxviridae | Double stranded DNA | 0.019 | [40] |
| X69198_VARV_major_India_1967 | Orthopoxviridae | Double stranded DNA | 0.005 | [41] |
| DQ437591_VARV_Sumatra_1 970_V70_222 | Orthopoxviridae | Double stranded DNA | 0.019 | Unpublished |
| DQ441442_VARV_Sumatra_1 970_V70_228 | Orthopoxviridae | Double stranded DNA | 0.020 | Unpublished |
| Herpes Simplex Virus Type-1 | Herpesviridae | Double stranded DNA | >40 | [38] |
| Cytomegalovirus | Herpesviridae | Double stranded DNA | >40 | [38] |
| Respiratory Syncytial Virus | Paramyxoviridae | Negative single strand RNA | >40 | [38] |
| Rotavirus | Reoviridae | Double stranded RNA | >40 | [38] |
| Rift Valley Fever Virus | Bunyaviridae | Negative single strand RNA | >40 | [38] |
| Tacaribe Virus | Arenaviridae | Ambisense RNA | >40 | [38] |
| Lymphocytic Choriomeningitis Virus | Arenaviridae | Ambisense RNA | >40 | [38] |
The VP37 protein of orthopoxviruses is critical for envelopment of intracellular mature virus with Golgi-derived membrane to form enveloped virus [38], which may then be released from the cell, and has been shown to have an important role in dissemination of virus from the site of infection and viral virulence in VACV [43,44]. Orthopoxviruses with impaired VP37 function resulting from deletion [45,46] or targeted point mutation [47,48] do not disseminate efficiently in cell culture and show a small plaque phenotype. Maturation of virus particles in orthopoxvirus VP37 knockouts does not proceed past the VP37 mediated envelopment step and these knockout strains are highly attenuated in vivo [46,49] and are unaffected by the presence of tecovirimat due to the absence of its target [50]. Tecovirimat targets the VP37 protein, which blocks the final steps in virus maturation and release from the infected cell, interfering with spread of virus in the host, as shown in Figure 2.
Figure 2.
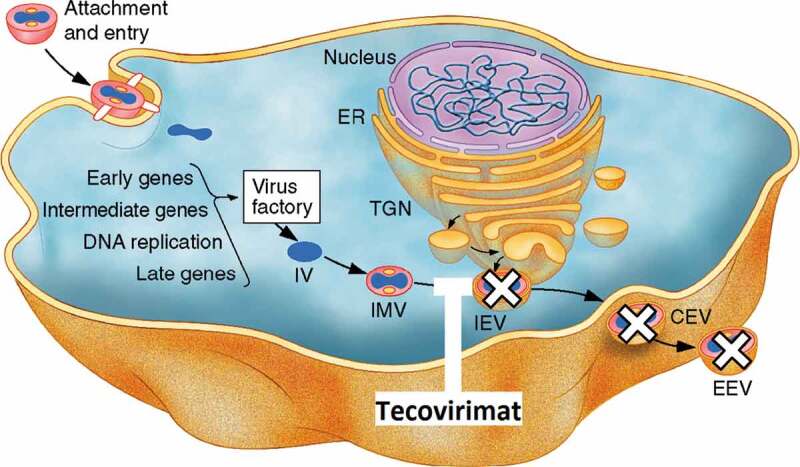
The molecular mechanism of action of tecovirimat. Image adapted from [51]. Following viral entry into a permissive cell, orthopoxviruses replicate in the cytoplasm. New viral particles are formed in regions called virus factories. These immature virus (IV) particles undergo membrane envelopment to form infectious intracellular mature virus (IMV). IMV may be further enveloped with a double membrane layer derived from early endosomes or trans-Golgi network (TGN) to form intracellular enveloped virus (IEV). These triple membrane-enveloped particles may then be transported to the cell surface where their outermost membrane fuses with the cytoplasmic membrane, releasing the virions as either cell-associated enveloped virus (CEV) which remain associated with the cell membrane, or extracellular enveloped virus (EEV), which disseminate from the site of infection [44]. The envelopment of IMV to form IEV requires the participation of several viral proteins including the VP37 protein, which is the target of tecovirimat. Inhibition of the VP37 protein prevents the formation of IEV from IMV
2. Methods
2.1. Literature search
PubMed and Google Scholar were searched for relevant references with no restrictions on publication dates using multiple combinations of applicable keywords for each topic discussed in the text.
2.2. Non-clinical animal efficacy studies
Four pivotal studies in non-human primates and two pivotal studies in rabbits were conducted. Studies included dose exploration to determine the minimum fully effective dose and infected-animal pharmacokinetic (PK) studies in both animal models, as well as studies of treatment delay and treatment duration in non-human primates (NHPs). Animal care and use protocols were approved by the institutional animal care and use committees of the institutions at which the studies were conducted (the Southern Research Institute, the Army Medical Institute of Infectious Diseases, and the Lovelace Respiratory Research Institute). Treatment of animals adhered to U.S. Government ‘Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Education’, the Guide for the Care and Use of Laboratory Animals [52], the Animal Welfare Act, and other applicable public laws and regulations.
In all studies in NHPs, cynomolgus macaques were challenged on day 0 with a lethal dose of monkeypox virus (MPXV) (Zaire 1979 strain) by the intravenous route with 5 × 107 plaque-forming units (PFU) per animal. In rabbit studies, New Zealand white rabbits were challenged on day 0 with a lethal dose of rabbitpox virus Utrecht (RPXV) by the intradermal route with 1000 PFU per animal.
Tecovirimat treatment was started four days after challenge or later, after clinical signs of disease (pock lesions, on body surfaces or oral mucosa, in non-human primates; fever and viremia in rabbits) were apparent in all study animals. Survival was the primary end point for all studies. Secondary pharmacodynamic endpoints in these studies included lesion formation (NHPs) and viral load (both animal models).
Pharmacokinetic and pharmacodynamic (PK/PD) models were developed for both animal models to establish the relationship between tecovirimat plasma exposures and survival following lethal challenge with orthopoxvirus. These PK/PD model-based analyses were used to compare the exposures in NHPs and rabbits treated at the most effective dose for each species to identify the more conservative animal model (i.e. the model requiring higher tecovirimat exposures for maximal benefit).
2.3. Clinical trials
PK and safety data from Phase 1 and Phase 2 dose-escalation and repeat-dose studies [53,54] were used to model the dosing in humans. The Phase 3 pivotal expanded safety trial was a multicenter, randomized, double-blind, placebo-controlled safety, tolerability, and pharmacokinetics trial involving healthy volunteers 18–79 years of age [55]. The tecovirimat dose selected for this trial targeted an exposure level that was several-fold in excess of that required for maximal efficacy in NHPs, and to keep the maximum plasma levels in human below maximum safety limit. Protocols used in the clinical studies were approved by a central institutional review board and all trial subjects provided written informed consent.
A lead-in cohort of 40 subjects was randomly assigned in a 4:1 ratio, in either a fed or a fasted state, to receive 600 mg (three 200 mg capsules) of tecovirimat, or matching placebo, twice daily. After a blinded interim analysis review of safety and PK data demonstrated that sufficient tecovirimat plasma levels had been achieved, and USFDA review of the data, the trial was expanded to provide a large enough database for evaluation of product safety by randomly assigning an additional 412 participants at 11 sites to receive tecovirimat or placebo in the fed state only. Participants recorded all adverse events and concomitant medications were captured in subject diaries and during interviews with clinic study staff from the start of the trial period to completion of the trial at the Day 28 follow-up visit. Adverse events were also recorded during physical examinations and through laboratory evaluations on scheduled clinic days. A follow-up visit or telephone contact at Day 45 for the evaluation of adverse events was conducted only for participants in whom adverse events or serious adverse events were present on the Day 28 follow-up visit.
3. Animal studies
3.1. The animal efficacy rule
Smallpox is a strictly human disease [56] and no longer circulates in the population. Since cases of smallpox no longer occur it is not possible to evaluate efficacy of antiviral drugs in humans. Therefore, development of tecovirimat was conducted according to recommendations in the USFDA Animal Efficacy Rule (Animal Rule) [57]. In 2002 the USFDA established the ‘Animal Efficacy Rule’ to provide a pathway for development of drug (21 CFR 314.600 through 314.650) and biological (and 21 CFR 601.90 through 601.95) products, ‘when human efficacy studies are not ethical or feasible.’ [57,58].
The Animal Rule regulations state: USFDA may grant marketing approval for a new drug product for which safety has been established and for which the requirements of 21 CFR 314.600 are met based on adequate and well-controlled animal studies when the results of those animal studies establish that the drug product is reasonably likely to produce clinical benefit in humans. In assessing the sufficiency of animal data, the agency may take into account other data, including human data, available to the agency. USFDA will rely on the evidence from studies in animals to provide substantial evidence of the effectiveness of these products only when:
There is a reasonably well-understood pathophysiological mechanism of the toxicity of the substance and its prevention or substantial reduction by the product;
The effect is demonstrated in more than one animal species expected to react with a response predictive for humans, unless the effect is demonstrated in a single animal species that represents a sufficiently well-characterized animal model for predicting the response in humans;
The animal study endpoint is clearly related to the desired benefit in humans, generally the enhancement of survival or prevention of major morbidity; and
The data or information on the kinetics and pharmacodynamics of the product or other relevant data or information, in animals and humans, allows selection of an effective dose in humans [59].
Animal models of smallpox are especially challenging in that the host range of VARV is restricted to humans. VARV-induced lethal disease is difficult to achieve in non-human primates, requiring high viral challenge doses (108–109 PFU) and routes of infection that are not biologically relevant [60]. The disease resulting from intravenous VARV challenge poorly mimics the clinical characteristics of classic human smallpox at the higher challenge dose, more closely resembling the rare hemorrhagic presentation (reviewed in [61]). At the lower challenge dose (108 PFU), intravenous VARV challenge results in disease that resembles classic human smallpox following the asymptomatic incubation period, just after the prodromal stage of disease (see Figure 3 for timeline of human smallpox pathology), and is approximately 30% lethal [62]. The low mortality observed in this model, the requirement that VARV studies would have to be conducted at the USCDC, and the limited animal capacity in the USCDC ABSL-4 make the design and conduct of studies with sufficient statistical power to evaluate efficacy of antivirals against VARV in the intravenous challenge model of VARV in NHPs prohibitive.
Figure 3.
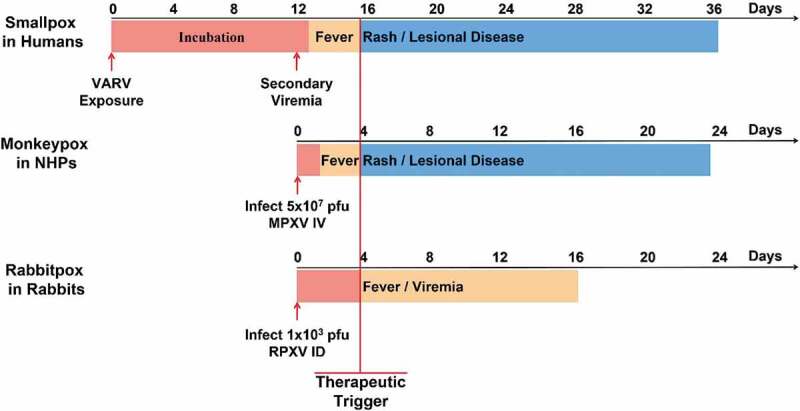
Comparison of the course of smallpox in humans with monkeypox in NHPs and rabbitpox in rabbits. In evaluating the non-human primate model for human smallpox for demonstrating the efficacy of antiviral therapeutics, a number of parameters have to be considered: First, it is important to determine the stage of disease progression during the course of human smallpox at which treatment would no longer be considered prophylactic, but therapeutic. The most distinctive and unambiguous identification of smallpox was the appearance of a synchronous, centrifugal rash that progressed to pustules beginning a few days after severe fever. The time course and associated pathology of monkeypoxvirus in the non-human primate versus human smallpox is nearly identical from the secondary viremia onward. In the intravenous challenge non-human primate model, lesions appear 3–4 days post-challenge and continue to increase in number and progress through stages typical of human smallpox until death. In this model a uniformly lethal challenge of 5 × 107 plaque-forming units was used. At this challenge dose all animals in our pivotal NHP efficacy studies showed signs of illness by 4 days following challenge at which time tecovirimat intervention is considered therapeutic. Recent literature also suggests that rabbitpox disease in rabbits closely mimics the stages of smallpox disease in humans [63]. After the initial infection, there is a symptom-free incubation period, followed by fever and the dissemination of virus in the blood and the establishment of a secondary systemic infection, followed by death. Following the USFDA recommendation at the 2011 advisory committee the intradermal challenge model was used for evaluation of tecovirimat efficacy. In this model a lethal viral challenge of 1,000 plaque-forming units of rabbitpox virus was chosen. The trigger in this model was different from the non-human primate model, since most rabbits die quickly, before developing lesions. The therapeutic trigger used in this model is fever, which is always observed prior to day 4. Therefore treatment started at day 4 in the rabbit models
Since an acceptable animal model of human smallpox using VARV has not been established it was necessary to develop animal models using surrogate pathogens that would approximate key properties of VARV pathogenesis in humans and to define model endpoints that were clearly related to the desired benefit in humans, such as enhancement of survival and mitigation of morbidity, in order to satisfy the animal rule requirements for development of tecovirimat as a smallpox therapeutic.
3.2. In vivo safety and efficacy
Over 50 animal studies of tecovirimat efficacy and safety have been conducted in support of the development of tecovirimat as a smallpox therapeutic. In pre-clinical animal safety pharmacology and toxicology studies in mice and NHPs no serious adverse effects were observed after 3 months of repeated dosing at tecovirimat plasma levels up to 23-fold over the recommended human dose in mice, and 2.5-fold over the recommended human dose in NHPs [36]. In dogs, seizure was observed following a single dose of 300 mg/kg [64], which is 4-fold higher than the highest observed human exposure, based on Cmax, at the recommended human dose. A follow up study in NHPs found no evidence of seizures following 12 days of high dose tecovirimat (300 mg/kg once per day), indicating that the dog is uniquely sensitive to this effect [36].
No effects were observed on reproductive function or early embryonic development in female mice at a dose of 100 mg/kg/day. In male mice receiving the same dose increased percent abnormal sperm and decreased sperm motility was observed, but it must be stipulated that the observed tecovirimat exposures at the dose given were 24-fold higher than the maximum exposure observed at the recommended human dose [64].
Tecovirimat showed protective efficacy against lethal challenge with every orthopoxvirus tested, in multiple animal models. Studies include ectromelia virus, CPXV, and VACV in mice [38,65,66,67,68], VARV and MPXV in NHPs [35,62,69,70,71,72], and MPXV in prairie dog [73] and golden ground squirrel models [74].
Pivotal in vivo studies of tecovirimat efficacy were conducted in NHPs challenged intravenously with a lethal dose of MPXV and rabbits challenged by the intradermal route with a lethal dose of RPXV. Tecovirimat treatment was initiated four days following challenge in both models. Based on the outcome of an Advisory Committee convened by the USFDA in December 2011 to specifically address the issue of animal models for smallpox, NHPs and rabbits were chosen as suitable primary models for clinical effects, mimicking certain aspects of human smallpox, following orthopoxvirus infection as shown in Figure 3. Specifically, the intravenous administration of MPXV (5 × 107 pfu) to NHPs and the intradermal administration of RPXV (1,000 pfu) to rabbits results in uniform mortality with clear indications of disease progression prior to death. Oral (gavage) administration of tecovirimat resulted in a reduction in the primary and secondary outcomes of orthopoxvirus infection in both species.
Primary studies in NHPs demonstrated that dose levels of ≥ 3 mg/kg/day for 14 days (initiated no later than post-infection Day 4) resulted in decreases in the incidences of mortality (> 95% survival compared to < 5% survival in NHPs treated with tecovirimat at dose levels of ≥ 3 mg/kg/day and <3 mg/kg/day, respectively). Treatment of NHPs with tecovirimat at dose levels of ≥ 10 mg/kg/day for 14 days (initiated no later than post-infection Day 4) did not result in further increases in survival but did result in lower circulating MPXV DNA levels, fewer pock lesions (verifying the findings of the in vitro studies that tecovirimat reduces viral dissemination), and fewer clinical signs of infection (unresponsiveness, dyspnea and fever, and lymphadenopathy) [37,55,72].
Supporting studies in rabbits demonstrated that oral (gavage) administration of tecovirimat at dose levels of ≥ 20 mg/kg/day for 14 days (administered no later than post-infection Day 4) resulted in decreases in the incidences of mortality, blood RPXV levels, and clinical signs of infection (including dyspnea, fever, and respiratory distress). Survival for all dose levels (ranging from 20 to 120 mg/kg/day) was similar and exceeded 90% while all untreated rabbits succumbed to disease [55].
4. Determination of dose for human safety trials
The selection of a dose for product approval under the USFDA Animal Rule requires the use of animal efficacy data in order to establish the human dose in the absence of efficacy data in humans. In order to provide confidence that the human dose will be effective during a smallpox emergency, it is important to understand the PK factors that influence efficacy and, ideally, to provide a margin of drug exposure in humans above that which is efficacious in animals. The recommended human dose that meets the requirements for efficacy must also be safe at the effective dose [57,75].
The in vivo ability of tecovirimat to protect a host against a lethal poxvirus disease was demonstrated in many animal models and against many orthopoxviruses [36,37]. In order to bridge the effective animal dose to the proposed clinical human dose, population PK, PK/PD, and exposure-response analyses were performed with data from non-clinical studies in rabbits and NHPs [72,76]. Population PK analyses were conducted in these animal models to develop a robust structural PK/PD model for tecovirimat to determine critical PK parameters and plasma levels of tecovirimat associated with efficacy and to support clinical dose selection [72,76].
Appropriate human dosing was determined by triangulation of safety, PK, and efficacy data from two pivotal animal models, and human PK and safety data. Human dosing and target exposures are based on PK/PD data (shown in Table 2) from the NHP model [55] since, in the NHP model, higher plasma exposures are required for maximal efficacy in comparison to the rabbit model, making it the more rigorous standard. This makes sense as the high-dose intravenous challenge in the NHP model bypasses the early stages of natural infection and immediately establishes a systemic disease and widely disseminated viral infection typical of late stage smallpox in humans as opposed to the rabbit model in which a low intradermal dose results in local replication of the virus at the site of injection followed later by systemic dissemination of the virus.
Table 2.
Geometric mean of non-compartmental exposures in NHPs, rabbits, and humans
| Comparisons | Treatment Day | Cmax (ng/mL) | Cmin (ng/mL) | Cavg (ng/mL) | AUC0-24 (ng.h/mL) |
|---|---|---|---|---|---|
| NZ Rabbit 40 mg/kd, daily | 14 (Steady state) |
374 | 25 | 138 | 3,318 |
| NHP 10 mg/kg, daily | 1444 | 169 | 598 | 14,352 | |
| NHP to Rabbit Ratio | 3.9 | 6.8 | 4.3 | 4.3 | |
| Human 600 mg BID | 2209 | 690 | 1270 | 30,632 | |
| Human to NHP Ratio | 1.5 | 4.1 | 2.1 | 2.1 |
AUC0-24 = area under the concentration–time curve over 24 hours; Cavg = average (mean) concentration; Cmax = maximum concentration; Cmin = minimum concentration.
BID = twice daily; NHP = non-human primate; NZ Rabbit = New Zealand White Rabbit. Adapted from [55]
The endpoint for product evaluation is the prevention of mortality and, using this endpoint, a statistically significant reduction of mortality was observed at 3 mg/kg in the intravenous MPXV/NHP model [72]. However, at a dose of 10 mg/kg, a reduction in morbidity, as represented by clinical scores, lesion count, and viral DNA load, was observed [72], in addition to reducing mortality. In order to provide a greater margin of error, and more closely represent a preferred operational strategy where reduction in morbidity will additionally reduce transmission and patient suffering, the human dose is based on the higher dose of 10 mg/kg in the NHP model.
Exposures following 600 mg twice daily dosing in humans clearly exceed efficacious exposures in non-human primates for AUC0-24and Cmin [55] (Table 2) and keep Cmax in human below maximum safety limit (5575 ng/mL) determined in animal toxicology studies. Notably, for Cmin which is the most predictive PK parameter for efficacy [76], the proposed dose of tecovirimat provides exposures 4.1-fold higher than those demonstrated to be efficacious in the NHP model.
5. Clinical trials, demonstration of human safety profile
Safety and tolerability of tecovirimat in humans was assessed in 11 clinical studies: one Phase 3 pivotal study, three supportive multiple-dose studies, and seven supportive single-dose studies (Table 3) [53,54,55,77,78]. It was determined that absorption is enhanced when tecovirimat is administered with food, with plasma exposure (Cmax and AUC0-24) increasing by up to 45% at steady state [55,77]. Because of this, the expanded phase of the pivotal safety trial was conducted solely in fed subjects.
Table 3.
Summary of studies in the clinical development of tecovirimat
| ClinicalTrials.gov Identifier | Type of Study | Na | Objective(s) | Reference |
|---|---|---|---|---|
| NCT02474589 | Pivotal Phase 3, proposed clinical dose of oral tecovirimat 600 mg or placebo twice daily for 14 days | 419 | Safety, tolerability, and PK in fed and fasted healthy subjects | [55] |
| Registration not required | Phase 1 multiple-dose (600 mg tecovirimat twice daily for 15 days); DDI study | 77 | Safety, tolerability, and effect of repeated doses of tecovirimat on single-dose PK of probe substrates flurbiprofen, omeprazole, midazolam, repaglinide, and bupropion | Unpublished |
| NCT00431951 | Phase 1 multiple-dose (250, 400, or 800 mg tecovirimat or placebo once daily for 21 days) | 19 | Safety, tolerability, and PK in fed state | [54] |
| NCT00907803 | Phase 2 multiple-dose (400 mg or 600 mg tecovirimat or placebo once daily for 14 days) | 101 | Safety, tolerability, and PK in fed state | [53] |
| Registration not required | Phase 1 single-dose (500, 1000, or 2000 mg tecovirimat or placebo) | 37 | Safety, tolerability, and PK in fed and fasted state | [77] |
| NCT00728689 | Phase 1 single-dose, bioavailability of 2 forms (I and V) of tecovirimat (400 mg) | 11 | Safety, tolerability, and PK of 2 forms (I and V) of tecovirimat in fed state | [78] |
| Registration not required | Phase 1 single-dose (600 mg tecovirimat and 100 µCi of [14 C]-tecovirimat), mass balance | 6 | Safety, tolerability, mass balance, and routes of elimination of [14 C] | Unpublished |
| Registration not required | Phase 1 single supratherapeutic dose (1000 mg tecovirimat), effects of tecovirimat; thorough ECG study | 48 | ECG, safety, tolerability, and PK of single doses of tecovirimat 1000 mg, moxifloxacin 400 mg, and placebo in the fed state | Unpublished |
| Registration not required | Phase 1 single-dose (600 mg tecovirimat), effect of renal impairment | 37 | PK, safety, and tolerability in subjects with varying degrees of renal impairment including end-stage renal disease requiring HD; effect of HD on the removal of tecovirimat from the bloodstream | Unpublished |
| Registration not required | Phase 1 single-dose (600 mg tecovirimat), effect of hepatic impairment | 32 | PK, safety, and tolerability in subjects with varying degrees of hepatic impairment | Unpublished |
| Registration not required | Phase 1 single-dose (100, 200, or 600 mg tecovirimat), effect of mixing capsule contents with food or liquid | 47 | PK, safety, and tolerability after administration of a single dose as capsule contents mixed with a food or liquid compared to a single dose as intact capsules | Unpublished |
aN is the number of subjects completing the study.
KEY: DDI = drug–drug interaction; ECG = electrocardiogram; HD = hemodialysis; PK = pharmacokinetic
5.1. Pivotal clinical trial
In the Phase 3 pivotal study subjects who were administered tecovirimat at the proposed human dose of 600 mg twice daily for 14 days (N = 359) and subjects who were administered placebo (N = 90) had similar incidences of: treatment-emergent adverse events (TEAEs) (37.3% tecovirimat, 33.3% placebo), treatment-related TEAEs (19.8% tecovirimat, 16.7% placebo), and TEAEs leading to treatment discontinuation (1.7% tecovirimat, 2.2% placebo) during the study. A single pivotal study subject receiving tecovirimat had a fatal serious adverse event (SAE) of pulmonary embolism. The investigator determined that there was no causal association or relationship between the drug and the event [55].
5.1.1. Common adverse events
For both tecovirimat and placebo treated groups in the Phase 3 pivotal study, the most frequent TEAEs were headache (17.0% tecovirimat, 14.4% placebo) and nausea (5.6% each). The incidence and pattern of all TEAEs were generally similar between groups, with the difference in incidence being less than 3%.
5.2. Supportive multi-dose trials
In the supportive multiple-dose studies, the percentage of subjects with at least one TEAE ranged from 25.0–75.0%, with no clear dose- or duration-related trends across studies. Similarly, treatment-related TEAEs occurred in 0–62.5% of tecovirimat-treated subjects across studies and did not show clear dose- or duration-related trends across studies. Consistent with the pivotal study, TEAEs leading to treatment discontinuation were infrequent. No deaths or SAEs occurred during these studies.
5.2.1. Common adverse events
Consistent with the Phase 3 pivotal study, headache and nausea were among the most frequently observed TEAEs in subjects who were administered tecovirimat in the supportive multiple-dose studies. Across studies, the incidence of TEAEs of headache and nausea was higher in subjects receiving 21 versus 14 or 15 days of tecovirimat dosing. No other consistent patterns in the most common TEAEs were evident across the supportive multiple-dose studies.
5.3. Drug-drug interactions
Tecovirimat is a weak inducer of cytochrome P450 (CYP)3A and a weak inhibitor of CYP2C8 and CYP2C19 [64]. However, the effects are not expected to be clinically relevant for most substrates of those enzymes based on the magnitude of interactions and the duration of treatment of tecovirimat. In diabetic patients on repaglinide therapy tecovirimat may cause increased levels of repaglinide which could result in hypoglycemia. In patients receiving midazolam, administration of tecovirimat may reduce levels of midazolam requiring monitoring of midazolam effect, dose adjustment, or alternative sedatives.
5.4. Human safety summary
At the recommended adult human dose of 600 mg orally twice daily for 14 days (N = 359), the incidence of TEAEs was 37.7%, which was similar to the incidence of TEAEs among subjects receiving placebo (33.3%); N = 90) Most TEAEs were mild or moderate. The most common TEAEs were headache and nausea, which occurred in 17.0% and 5.6% of subjects receiving tecovirimat, respectively, in the pivotal study. There were no drug-related SAEs or pregnancies in clinical trials, and the incidence of TEAEs leading to treatment discontinuation was low (1.7% in the pivotal study).
Based on the data available, tecovirimat dosing at 600 mg orally twice daily with a meal for 14 days demonstrates an adequate safety profile in humans. Headache was the only AE occurring with higher frequency in subjects administered tecovirimat relative to those who received placebo. Most reported AEs were mild, all resolved without sequelae, and withdrawals/discontinuations were few.
The non-clinical safety and efficacy data, and the clinical safety data demonstrate that the tecovirimat dose of 600 mg orally twice daily for 14 days is safe and provides exposures in excess of those that were effective in the animal models. The proposed dose of tecovirimat 600 mg twice daily for 14 days has met the essential standard of the Animal Rule in that it is ‘reasonably likely to produce clinical benefit in humans’ in the event of a smallpox emergency.
6. Conclusion
6.1. Brief discussion and summary of tecovirimat development
Tecovirimat (ST-246) was first reported as a promising antiviral candidate to treat orthopoxvirus infection in 2005 [38]. In vitro studies confirmed that tecovirimat inhibited replication of multiple orthopoxviruses in cell culture, including multiple isolates of VARV, the causative agent of smallpox. Tecovirimat demonstrated oral bioavailability [38] and broad-spectrum protective efficacy in multiple lethal animal models of orthopoxvirus disease [35,38,62,65,66,67,68,69,70,71,72,74] including all orthopoxviruses known to be human pathogens.
It was shown in the lethal aerosol challenge MPXV model in cynomolgus macaques that a dose of 10 mg/kg of tecovirimat twice daily for 14 days provided significant survival benefit when treatment was initiated from 1 and 7 days following challenge [70]. In the lethal intravenous challenge model of MPXV in cynomolgus macaques the treatment regimen described above provides significant survival benefit when initiated up to 5 days following challenge [55]. Data from these studies suggests that tecovirimat will provide therapeutic benefit in human smallpox cases if treatment is initiated at the first signs of lesional disease, and may also have therapeutic benefit should treatment be delayed for several days following appearance of lesions. In animal models earlier initiation of treatment correlated with increased survival benefit and reduction in signs of illness. In studies where treatment was initiated one day following challenge animals showed negligible signs of illness, suggesting that tecovirimat has potential for post-exposure prophylaxis.
Non-clinical pivotal efficacy studies conducted in the NHP/MPXV and NZW rabbit/RPXV models defined the minimum efficacious doses and plasma exposures required for protection from disease and established the NHP model as the more conservative model for selection of an appropriate human dose. Pharmacodynamic analysis of plasma exposure and efficacy data from NHP studies was used to define minimal efficacy exposure for humans [76]. Based on this, a human dose of tecovirimat 600 mg twice daily was predicted to provide tecovirimat plasma exposure several fold in excess of that necessary for maximal efficacy in animal studies [55].
The pivotal Phase 3 human safety study was conducted using the established human dose regimen of 600 mg tecovirimat twice daily for 14 days taken with food. Results of this study showed that this regimen was well tolerated. Adverse events were generally mild, incidence was similar between placebo and treated groups, and no tecovirimat-associated safety issues were identified in study subjects [55].
7. Expert opinion
7.1. Update the smallpox response program
At the time of this writing, the current US Civilian Smallpox Preparedness and Response Program does not include guidelines for the use of tecovirimat in a smallpox outbreak. Computer simulations of smallpox outbreak scenarios clearly indicate that the availability of antiviral drugs, such as tecovirimat, improve the ability to control a smallpox outbreak [79]. Modeling of antiviral treatment of patients with confirmed diagnosis of smallpox shows a clear improvement in time to resolution of an outbreak and in total case load, and these parameters are further improved if antivirals are used for prophylaxis [79]. Official guidance from public health authorities for the use of antiviral countermeasures in a smallpox emergency is desirable.
7.2. Tecovirimat label expansion
7.2.1. Emerging orthopoxvirus pathogens and vaccination complications
Tecovirimat was granted approval by the USFDA in 2018 explicitly for use as a therapeutic intervention in positively diagnosed cases of smallpox. While tecovirimat shows good protective efficacy in animal models of all orthopoxviruses known to infect humans, it is not currently approved for any indications other than diagnosed smallpox.
Due to cessation of routine vaccination of the general public following the declaration of smallpox eradication in 1980, immunity to smallpox and other diseases caused by orthopoxvirus human pathogens has declined in the global population. At the same time, increased incidence of monkeypox [80,81], cowpox [82], buffalopox [83,84], and vaccinia [85] have been observed in many parts of the world. These emergent orthopoxviruses present a growing public health concern in many parts of the world [86]. Although outbreaks of these diseases are generally infrequent and modest in size there is concern that these diseases could become more prevalent [87,88]. Therefore, there is interest in development of treatments for these infections, especially monkeypox, which has been showing a significant increase in outbreaks in West and Central Africa [81,89,90], as well as imported cases in the US [91], Europe [92], Middle East [93], and Asia [94].
Certain specific populations such as laboratory workers involved in orthopoxvirus research, members of the military being deployed overseas, and civilian smallpox response team volunteers are routinely vaccinated with ACAM2000 and are at risk for vaccination related adverse events [95,96,97]. These groups would benefit from the availability of an approved treatment for adverse events related to ACAM2000 vaccination.
Tecovirimat has been used under e-IND/Compassionate use authorization in several cases of orthopoxvirus illness in the US and Europe [98,99,100,101]. Outcomes of these cases were generally encouraging although the efficacy of tecovirimat could not be specifically determined because of concomitant administration of other antiviral interventions, such as cidofovir, and/or vaccinia immune globulin intravenous (VIGIV). Also, in each of these cases there were preexisting risk factors for VACV complications, such as dermatological disorders, or immunodeficiencies. It should be noted that tecovirimat efficacy may be reduced in immunodeficient patients [64] making determination of protective effects of treatment challenging. Depending on the severity of a patients immunodeficiency extended treatment beyond the label recommendation of 14 days may be necessary to allow sufficient time for the immune system to control a VACV infection. In one instance, an active duty member of the military received an ACAM2000 vaccination prior to an overseas deployment and was subsequently diagnosed with leukemia. The patient was treated with VIGIV and tecovirimat concurrently with initiation of chemotherapy. It was necessary to continue tecovirimat therapy for 62 days to control the VACV infection and the patient went on to fully recover [102]. In a similar case, another member of the military received ACAM2000 vaccination and was later diagnosed with leukemia. In this case chemotherapy was initiated prior to the administration of antiviral therapies including VIGIV, brincidofovir, and tecovirimat oral and topical formulations [103]. Tecovirimat was continued for 73 days and ultimately the progressive VACV infection was resolved.
All together, these factors strongly support the pursuit of efforts, including the conduct of human clinical efficacy trials, to expand the label indications for tecovirimat to include emerging orthopoxvirus infections and complications of smallpox vaccination.
7.2.2. Liquid suspension pediatric formulation
Tecovirimat, supplied in 200 mg capsules, is currently approved for the treatment of smallpox disease in adults and pediatric patients weighing at least 13 kg. Pediatric patients weighing less than 13 kg would require less than 1 capsule (200 mg) of tecovirimat, making accurate dosing difficult. In a smallpox outbreak emergency the pediatric patient population would benefit from the availability of a liquid oral tecovirimat formulation to ensure accuracy of dosing.
To address this unmet need, as of this writing development of an oral liquid suspension formulation of tecovirimat for pediatric patients weighing less than 13 kg who are not currently covered by the approved label indication for tecovirimat is ongoing.
7.2.3. Intravenous formulation
In a smallpox outbreak patients may present who are critically ill and may be unable to take tecovirimat due to severity of illness or other reasons and who would benefit from antiviral therapeutic intervention. These patients could be treated with intravenous tecovirimat until they were sufficiently recovered to complete their regimen with tecovirimat, or until they had completed a full 14-day course of treatment.
To address this unmet need a tecovirimat injection formulation for intravenous infusion would be beneficial. Development of such a formulation is currently in progress.
7.2.4. Post-exposure prophylaxis
While the tecovirimat label indication approved by the USFDA is restricted to therapeutic application in cases of diagnosed smallpox there is abundant pre-clinical evidence in multiple animal orthopoxvirus disease models [38,65,70,74] that tecovirimat administered shortly after lethal orthopoxvirus exposures but prior to onset of clear signs of disease is highly protective against mortality and dramatically reduces morbidity. Model simulations of smallpox outbreaks suggest that therapeutic intervention with a smallpox antiviral in confirmed cases of disease, in addition to the vaccination program described in the smallpox response plan, improves ability to control an outbreak [79]. Furthermore, prophylactic intervention in exposed asymptomatic individuals additionally improves outbreak control by reducing virus shedding and disease transmission.
A post-exposure prophylaxis indication may require a longer period of tecovirimat treatment than that the approved human treatment regimen to account for possible variability in disease progression between the animal models and human smallpox, thus necessitating human safety studies of longer duration than those conducted for the approval of tecovirimat for smallpox treatment. And on account of the likelihood that smallpox vaccines could be used concurrently with tecovirimat in an outbreak it would be beneficial to conduct tecovirimat/vaccine interaction studies to determine whether tecovirimat interferes with establishment of vaccination-induced immunity.
To address this potentially beneficial application of tecovirimat a program has been established to facilitate expansion of the tecovirimat label to include an indication for post-exposure prophylaxis.
7.3. Resistance
Naturally occurring tecovirimat resistant orthopoxviruses have not been observed to date, although tecovirimat resistance may develop under drug selection. In tecovirimat isolates of CPXV, VACV, and camelpox virus isolated under drug selection amino acid substitutions and insertions in the VP37 protein were noted [38,50]. In one EUA case in a patient with progressive vaccinia, resistant VACV was isolated late in disease following extended systemic tecovirimat treatment at plasma levels lower than the targeted therapeutic human dose, and concurrent topical tecovirimat application [99].
The emergence of tecovirimat resistant virus is an important concern. However, the circumstances under which the emergence of tecovirimat resistant viruses has been observed, extended suboptimal dosing in an immunocompromised patient [99], and drug selection over multiple passages in cell culture over a period of months [38], differ substantially from the conditions likely to be prevalent in a smallpox outbreak and may not be predictive of the likelihood of tecovirimat resistance emergence in that situation. One approach to mitigate the risk of emergent resistance to tecovirimat in an outbreak is combination therapy using a second antiviral with a different mechanism of action than tecovirimat. As such it is important that research into other smallpox antiviral drugs continues, directed toward eventually obtaining approval of a second drug to treat smallpox.
7.4. Oncolytic virus therapy
Oncolytic virotherapy is a promising approach to treatment of several currently difficult to manage cancers [104,105,106,107]. Oncolytic orthopoxviruses, such as VACV, have shown potential therapeutic benefit in pre-clinical animal models of multiple types of cancers [106]. In the case of VACV there is a delicate balance between delivering a sufficient dose to have a meaningful clinical effect on cancerous cells and avoiding a dose that may result in unacceptable side effects. Since higher doses of oncolytic VACV are more likely to be effective, therapeutic benefit could be improved if there was a method to increase the dose of VACV that could be safely delivered. Also, while oncolytic virus vectors generally appear to be safe adverse events could emerge if these new therapies were used in larger populations.
Tecovirimat is protective against VACV in lethal animal models of infection, inhibits formation of vaccination site lesions in animal models of vaccination, and has potential to support the development and use of oncolytic VACV immunotherapies. Tecovirimat could be used as an adjunct to VACV oncolytic virotherapy in a few ways. Antiviral intervention could be a safety net to rescue patients should severe adverse events result from oncolytic virotherapy, or as a planned component of a specific oncolytic regimen where a high dose of VACV is followed by tecovirimat after a specified interval to prevent or reduce adverse effects. The availability of a potent antiviral drug against VACV provides additional assurance to patients receiving these promising investigational therapies, their physicians, and regulators.
We have recently entered into a collaboration with Turnstone Biologics to provide tecovirimat in connection with Turnstone’s proprietary SKV vaccinia oncolytic immunotherapy platform [108]. The collaboration will provide Turnstone with access to tecovirimat oral antiviral capsules for use if required in future clinical programs.
7.5. 5 year outlook on smallpox and emerging poxvirus countermeasures
Over the next several years we expect that the civilian smallpox response plan will be revised and updated to explicitly include tecovirimat in the list of available countermeasures to be used in case of a smallpox bioterrorist attack or other smallpox outbreak. Programs being conducted to expand the tecovirimat label to include intravenous and liquid oral formulations are expected to culminate in New Drug Application (NDA) submissions to the USFDA for approval of these formulations for smallpox treatment. Regulatory submissions in support of expansion of the tecovirimat indications to include post-exposure prophylaxis, administration to individuals known or suspected to have been exposed to VARV in the course of a smallpox outbreak, but prior to the onset of clinical symptoms, are expected. In addition, the global threat of emerging orthopoxvirus disease outbreaks, such as monkeypox, buffalopox, cowpox, and vaccinia, suggests that tecovirimat label expansion to include these indications would be beneficial, and human clinical efficacy trials for these indications are recommended. With the potential for expansion of tecovirimat label indications along with the global population growth new models of smallpox outbreak scenarios should be developed and simulations based on these models used to inform government decisions on the inventory levels of tecovirimat, and other anti-smallpox countermeasures, that would need to be maintained in the US Strategic National Stockpile to provide an adequate level of preparedness for a hypothetical smallpox outbreak.
Finally, research efforts to develop a second smallpox antiviral drug with a different mechanism of action than tecovirimat are expected to be ongoing and should continue.
Article highlights
Smallpox has a potential 30% fatality rate and was responsible for approximately 300 million deaths worldwide in the 20th century alone. Up until the 18th century, as many as one in 10 people may have died as a result of smallpox
Smallpox was officially declared eradicated by the World Health Organization in 1980, but is still considered a serious public health threat for multiple reasons and the need for medical countermeasures against this threat is well established.
Tecovirimat was approved for the treatment of positively diagnosed smallpox under the USFDA Animal Efficacy Rule which provides a mechanism to obtain approval of new countermeasures for indications where human efficacy trials cannot be conducted.
In animal studies tecovirimat was shown to be highly efficacious against every known human orthopoxvirus pathogen in multiple animal models. In animal studies and human clinical safety trials tecovirimat was shown to be safe and well tolerated at the recommended human dose, or its equivalent.
The molecular target of tecovirimat, the orthopoxvirus protein VP37, which has no identified human homologs, is necessary for membrane envelopment of intracellular mature virus particles to form enveloped virus which are then released from the cell. Enveloped virus is implicated in both cell-to-cell spread and long range dissemination of virus within the host.
Future investigation into expanded indications for therapeutic use of tecovirimat for non-variola orthopoxvirus pathogens and other potential applications, such as a component of oncolytic vaccinia virus therapy, are proposed.
Declaration of interest
All authors are employees of SIGA Technologies, Inc. and may hold stock or equity interest in the company. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Review disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Macaulay TB, Trevelyan HMM. The history of England from the accession of James II. Cambridge University Press; New York, NY; 2011
- 2.Hopkins DR. The greatest killer: smallpox in history, with a new introduction. 2002. [Google Scholar]
- 3.Hopkins DR, Lythcott GI. Princes and peasants: smallpox in history. University of Chicago Press; Chicago, IL. 1983 [Google Scholar]
- 4.Behbehani AM. The smallpox story: life and death of an old disease.. Microbiol Rev. 1983;47(4):455–509. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Comprehensive overview of the history of smallpox and the eradication program.
- 5.Henderson DA, Inglesby TV, Bartlett JG, et al. Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA. 1999;281(22):2127–2137. [DOI] [PubMed] [Google Scholar]
- 6.Melamed S, Israely T, Paran N. Challenges and achievements in prevention and treatment of smallpox. Vaccines (Basel). 2018;6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barquet N, Domingo P. Smallpox: the triumph over the most terrible of the ministers of death. Ann Intern Med. 1997;127(8 Pt 1):635–642. [DOI] [PubMed] [Google Scholar]
- 8.Thèves C, Biagini P, Crubézy E. The rediscovery of smallpox. Clin Microbiol Infect. 2014;20(3):210–218. [DOI] [PubMed] [Google Scholar]
- 9.Henderson DA. Smallpox eradication. 433021. 1980;95(5):422–426. [PMC free article] [PubMed] [Google Scholar]
- 10.Breman JG, Arita I. The confirmation and maintenance of smallpox eradication. N Engl J Med. 1980;303(22):1263–1273. [DOI] [PubMed] [Google Scholar]
- 11.Deria A, Jezek Z, Markvart K, et al. The world’s last endemic case of smallpox: surveillance and containment measures. Bull World Health Organ. 1980;58(2):279–283. [PMC free article] [PubMed] [Google Scholar]
- 12.Barclay WR. The conquest of smallpox. JAMA. 1978;240(18):1991–1992. [DOI] [PubMed] [Google Scholar]
- 13.Fenner F, Henderson DA, Arita I, et al. Smallpox and its eradication. World Health Organization; Geneva, Switzerland; 1988.•• This is the definitive resource for detailed information about smallpox including but not limited to history, virology, clinical aspects of disease, and eradication [Google Scholar]
- 14.Leitenberg M, Zilinskas RA, Kuhn JH. The Soviet biological weapons program: a history. Harvard University Press; Cambridge, MA. 2012 [Google Scholar]
- 15.Cohen J. Alarm over biosafety blunders. Science. 2014;345(6194):247–248. [DOI] [PubMed] [Google Scholar]
- 16.Gellman B Four nations thought to possess smallpox. The Washington Post, 2002;5, 4–5. [Google Scholar]
- 17.Noyce RS, Lederman S, Evans DH. Construction of an infectious horsepox virus vaccine from chemically synthesized DNA fragments. PLoS One. 2018;13(1):e0188453. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes de novo synthesis of an extinct orthopoxvirus (horsepox virus) using publically available information, and commercially available equipment and supplies. Impacts the significance of the variola stock retention discussion since it shows that variola could be resurrected without great difficulty.
- 18.MacIntyre CR, Costantino V, Chen X, et al. Influence of population immunosuppression and past vaccination on smallpox reemergence. Emerg Infect Dis. 2018;24(4):646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention Atlanta G . Bioterrorism agents/diseases. Atlanta (GA): Centers for Disease Control and Prevention. [cited 2020 Jun 28]. Available from: https://emergency.cdc.gov/agent/agentlist-category.asp [Google Scholar]
- 20.Centers for Disease Control and Prevention Atlanta G . Smallpox: bioterrorism.Atlanta (GA): Centers for Disease Control and Prevention. [cited 2020 Jun 28]. Available from: https://www.cdc.gov/smallpox/bioterrorism/public/index.html [Google Scholar]
- 21.Smallpox . U.S. Department of Health & Human Services; 2004 [cited 2020 Jun 28]. Available from: https://medicalcountermeasures.gov/barda/cbrn/smallpox.aspx [Google Scholar]
- 22.Carpenter C, Arvin A, Beasley R, et al. Assessment of future scientific needs for live variola virus. Washington, DC: National Academy Press; 1999. [PubMed] [Google Scholar]
- 23.Sato H. Countermeasures and vaccination against terrorism using smallpox: pre-event and post-event smallpox vaccination and its contraindications. Environ Health Prev Med. 2011;16(5):281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dudley G, McFee RB. Preparedness for biological terrorism in the United States: project BioShield and beyond. J Am Osteopathic Assoc. 2005;105(9):417. [PubMed] [Google Scholar]
- 25.Services UDoHaH . Project bioshield overview. US Department of Health and Human Services; 2020 [cited 2020 Jun 28]. Available from: https://emergency.cdc.gov/agent/agentlist-category.asp [Google Scholar]
- 26.Kraft M, Marks E. US government counterterrorism: a guide to who does what. CRC Press; Boca Raton, FL. 2011. doi: 10.1201/b11296 [DOI] [Google Scholar]
- 27.Strikas RA, Neff LJ, Rotz L, et al. US civilian smallpox preparedness and response program, 2003. Clinl Infect Dis. 2008;46(Supplement_3):S157–S167. [DOI] [PubMed] [Google Scholar]
- 28.Rotz LD, Dotson DA, Damon IK, et al. Vaccinia (smallpox) vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2001. MMWR Recomm Rep. 2001;50(Rr–10):1–25; quiz CE21-27. [PubMed] [Google Scholar]
- 29.Prevention CfDCa . CDC guidance for post-event smallpox planning. Atlanta: Centers for Disease Control and Prevention; 2002. (Ed.^(Eds). [Google Scholar]
- 30.Wharton M, Strikas RA, Harpaz R, et al. Recommendations for using smallpox vaccine in a pre-event vaccination program. Supplemental recommendations of the Advisory Committee on Immunization Practices (ACIP) and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep. 2003;52(Rr–7):1–16. [PubMed] [Google Scholar]
- 31.Petersen BW, Damon IK, Pertowski CA, et al. Clinical guidance for smallpox vaccine use in a postevent vaccination program. MMWR Recomm Rep. 2015;64(Rr–02):1–26. [PubMed] [Google Scholar]
- 32.Keckler MS, Reynolds MG, Damon IK, et al. The effects of post-exposure smallpox vaccination on clinical disease presentation: addressing the data gaps between historical epidemiology and modern surrogate model data. Vaccine. 2013;31(45):5192–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Discussion of therapeutic efficacy and limitations of post-exposure smallpox vaccination. Establishes role of antivirals to to address unmet health security needs.
- 33.Massoudi MS, Barker L, Schwartz B. Effectiveness of postexposure vaccination for the prevention of smallpox: results of a delphi analysis. J Infect Dis. 2003;188(7):973–976. [DOI] [PubMed] [Google Scholar]; • Discussion of therapeutic efficacy and limitations of post-exposure smallpox vaccination. Establishes role of antivirals to to address unmet health security needs.
- 34.Merchlinsky M, Albright A, Olson V, et al. The development and approval of tecoviromat (TPOXX((R))), the first antiviral against smallpox. Antiviral Res. 2019;168:168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russo AT, Berhanu A, Bigger CB, et al. Co-administration of tecovirimat and ACAM2000 in non-human primates: effect of tecovirimat treatment on ACAM2000 immunogenicity and efficacy versus lethal monkeypox virus challenge. Vaccine. 2020;38(3):644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jordan R, Leeds JM, Tyavanagimatt S, et al. Development of ST-246(R) for treatment of poxvirus infections. Viruses. 2010;2(11):2409–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grosenbach DW, Jordan R, Hruby DE. Development of the small-molecule antiviral ST-246 as a smallpox therapeutic. Future Virol. 2011;6(5):653–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang G, Pevear DC, Davies MH, et al. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J Virol. 2005;79(20):13139–13149. [DOI] [PMC free article] [PubMed] [Google Scholar]; • First published description of tecovirimat (ST-246) as an orthopoxvirus inhibitor in vitro and in vivo.
- 39.Bailey TR, Rippin SR, Opsitnick E, et al. N-(3,3a,4,4a,5,5a,6,6a-Octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2-(1H)-yl)carboxamides: identification of novel orthopoxvirus egress inhibitors. J Med Chem. 2007;50(7):1442–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith SK, Olson VA, Karem KL, et al. In vitro efficacy of ST246 against smallpox and monkeypox. Antimicrob Agents Chemother. 2009;53(3):1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kabanov AS, Sergeev AA, Shishkina LN, et al. [A comparative study of the antiviral activity of chemical compounds concerning the orthopoxviruses experiments in vivo]. Vopr Virusol. 2013;58(4):39–43. [PubMed] [Google Scholar]
- 42.Duraffour S, Snoeck R, de Vos R, et al. Activity of the anti-orthopoxvirus compound ST-246 against vaccinia, cowpox and camelpox viruses in cell monolayers and organotypic raft cultures. Antivir Ther. 2007;12(8):1205–1216. [PubMed] [Google Scholar]
- 43.Payne LG. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J Gen Virol. 1980;50(1):89–100. [DOI] [PubMed] [Google Scholar]
- 44.Smith GL, Vanderplasschen A, Law M. The formation and function of extracellular enveloped vaccinia virus. J Gen Virol. 2002;83(Pt 12):2915–2931. [DOI] [PubMed] [Google Scholar]; • Detailed discussion of orthopoxvirus maturation and envelopment. Describes the role of enveloped viruses in dissemination from the site of infection and the significnce of dissemination with regard to orthopoxvirus virulence. Supports the validity in targeting envelopment as an antiviral strategy.
- 45.Blasco R, Moss B. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J Virol. 1991;65(11):5910–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roscoe F, Xu RH, Sigal LJ. Characterization of ectromelia virus deficient in EVM036, the homolog of vaccinia virus F13L, and its application for rapid generation of recombinant viruses. J Virol. 2012;86(24):13501–13507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roper RL, Moss B. Envelope formation is blocked by mutation of a sequence related to the HKD phospholipid metabolism motif in the vaccinia virus F13L protein. J Virol. 1999;73(2):1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sung TC, Roper RL, Zhang Y, et al. Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. Embo J. 1997;16(15):4519–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vliegen I, Yang G, Hruby D, et al. Deletion of the vaccinia virus F13L gene results in a highly attenuated virus that mounts a protective immune response against subsequent vaccinia virus challenge. Antiviral Res. 2012;93(1):160–166. [DOI] [PubMed] [Google Scholar]
- 50.Duraffour S, Lorenzo MM, Zoller G, et al. ST-246 is a key antiviral to inhibit the viral F13L phospholipase, one of the essential proteins for orthopoxvirus wrapping. J Antimicrob Chemother. 2015;70(5):1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hruby DE, Byrd CM. Less is more: poxvirus proteolysis. Microbe. 2006;1(2):70–75. [Google Scholar]
- 52.Council, NR. Guide for the care and use of laboratory animals. National Academies Press; Washington, DC. 2011 [PubMed]
- 53.Chinsangaram J, Honeychurch KM, Tyavanagimatt SR, et al. Safety and pharmacokinetics of the anti-orthopoxvirus compound ST-246 following a single daily oral dose for 14 days in human volunteers. Antimicrob Agents Chemother. 2012;56(9):4900–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jordan R, Chinsangaram J, Bolken TC, et al. Safety and pharmacokinetics of the antiorthopoxvirus compound ST-246 following repeat oral dosing in healthy adult subjects. Antimicrob Agents Chemother. 2010;54(6):2560–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grosenbach DW, Honeychurch K, Rose EA, et al. Oral tecovirimat for the treatment of smallpox. N Engl J Med. 2018;379(1):44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Comprehensive review of TPOXX development including detailed discussion of clinical safety and navigation of the Animal Rule.
- 56.Shchelkunov SN. Orthopoxvirus genes that mediate disease virulence and host tropism. Adv Virol. 2012;524743:2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.USFDA . Product development under the Animal Rule, guidance for industry. Rockville (MD): United States Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research/Center for Biologics Evaluation and Research; 2015 [cited 2020 Jun 28]. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM399217.pdf [Google Scholar]; •• Final guidance from FDA on drug development under the Animal Rule. Provides a framework for future drug development.
- 58.Snoy PJ. Establishing efficacy of human products using animals: theUS food and drug administration’s “Animal Rule”. Vet Pathol. 2010;47(5):774–778. [DOI] [PubMed] [Google Scholar]
- 59.New drug and biological drug products; evidence needed to demonstrate effectiveness of new drugs when human efficacy studies are not ethical or feasible . Final rule. Fed Regist. 2002;67(105):37988–37998. [PubMed] [Google Scholar]
- 60.Jahrling PB, Hensley LE, Martinez MJ, et al. Exploring the potential of variola virus infection of cynomolgus macaques as a model for human smallpox. Proc Natl Acad Sci U S A. 2004;101(42):15196–15200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chapman JL, Nichols DK, Martinez MJ, et al. Animal models of orthopoxvirus infection. Vet Pathol. 2010;47(5):852–870. [DOI] [PubMed] [Google Scholar]
- 62.Huggins J, Goff A, Hensley L, et al. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob Agents Chemother. 2009;53(6):2620–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perry MR, Warren R, Merchlinsky M, et al. Rabbitpox in New Zealand white rabbits: a therapeutic model for evaluation of poxvirus medical countermeasures under the FDA animal rule. Front Cell Infect Microbiol. 2018;8(356). DOI: 10.3389/fcimb.2018.00356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.TPOXX® (tecovirimat) [package insert]. Corvallis, OR: SIGA Technologies, Inc; 2018. [Google Scholar]
- 65.Quenelle DC, Buller RM, Parker S, et al. Efficacy of delayed treatment with ST-246 given orally against systemic orthopoxvirus infections in mice. Antimicrob Agents Chemother. 2007;51(2):689–695. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berhanu A, King DS, Mosier S, et al. ST-246 inhibits in vivo poxvirus dissemination, virus shedding, and systemic disease manifestation. Antimicrob Agents Chemother. 2009;53(12):4999–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grosenbach DW, Berhanu A, King DS, et al. Efficacy of ST-246 versus lethal poxvirus challenge in immunodeficient mice. Proc Natl Acad Sci U S A. 2010;107(2):838–843. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parker S, Chen NG, Foster S, et al. Evaluation of disease and viral biomarkers as triggers for therapeutic intervention in respiratory mousepox - an animal model of smallpox. Antiviral Res. 2012;94(1):44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berhanu A, Prigge JT, Silvera PM, et al. Treatment with the smallpox antiviral tecovirimat (ST-246) alone or in combination with ACAM2000 vaccination is effective as a postsymptomatic therapy for monkeypox virus infection. Antimicrob Agents Chemother. 2015;59(7):4296–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Russo AT, Grosenbach DW, Brasel TL, et al. Effects of treatment delay on efficacy of tecovirimat following lethal aerosol monkeypox virus challenge in cynomolgus macaques. J Infect Dis. 2018;218(9):1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mucker EM, Goff AJ, Shamblin JD, et al. Efficacy of tecovirimat (ST-246) in nonhuman primates infected with variola virus (Smallpox). Antimicrob Agents Chemother. 2013;57(12):6246–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jordan R, Goff A, Frimm A, et al. ST-246 antiviral efficacy in a nonhuman primate monkeypox model: determination of the minimal effective dose and human dose justification. Antimicrob Agents Chemother. 2009;53(5):1817–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith SK, Self J, Weiss S, et al. Effective antiviral treatment of systemic orthopoxvirus disease: ST-246 treatment of prairie dogs infected with monkeypox virus. J Virol. 2011;85(17):9176–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sbrana E, Jordan R, Hruby DE, et al. Efficacy of the antipoxvirus compound ST-246 for treatment of severe orthopoxvirus infection. Am J Trop Med Hyg. 2007;76(4):768–773. [PubMed] [Google Scholar]
- 75.Aebersold P. FDA experience with medical countermeasures under the Animal Rule. Adv Prev Med. 2012;507571:2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leeds JM, Fenneteau F, Gosselin NH, et al. Pharmacokinetic and pharmacodynamic modeling to determine the dose of ST-246 to protect against smallpox in humans. Antimicrob Agents Chemother. 2013;57(3):1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jordan R, Tien D, Bolken TC, et al. Single-dose safety and pharmacokinetics of ST-246, a novel orthopoxvirus egress inhibitor. Antimicrob Agents Chemother. 2008;52(5):1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chinsangaram J, Honeychurch KM, Tyavanagimatt SR, et al. Pharmacokinetic comparison of a single oral dose of polymorph form i versus form V capsules of the antiorthopoxvirus compound ST-246 in human volunteers. Antimicrob Agents Chemother. 2012;56(7):3582–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finin P, Kosaraju A, Rose E, et al. The role of vaccination, antiorthopoxvirus drug, and social cooperativity in a mathematical model of smallpox control. Biosecur Bioterror. 2013;11(1):59–72. [DOI] [PubMed] [Google Scholar]; • Discussion of smallpox outbreak models and the impact of vaccination, antiviral drugs, and social cooperativity on outcome. Approach presented could be applied to other viral outbreak scenarios.
- 80.Beer EM, Rao VB. A systematic review of the epidemiology of human monkeypox outbreaks and implications for outbreak strategy. PLoS Negl Trop Dis. 2019;13(10):e0007791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petersen E, Abubakar I, Ihekweazu C, et al. Monkeypox - Enhancing public health preparedness for an emerging lethal human zoonotic epidemic threat in the wake of the smallpox post-eradication era. Int J Infect Dis. 2019;78:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vorou RM, Papavassiliou VG, Pierroutsakos IN. Cowpox virus infection: an emerging health threat. Curr Opin Infect Dis. 2008;21(2):153–156. [DOI] [PubMed] [Google Scholar]
- 83.Bhanuprakash V, Venkatesan G, Balamurugan V, et al. Zoonotic infections of buffalopox in India. Zoonoses Public Health. 2010;57(7–8):e149–155. [DOI] [PubMed] [Google Scholar]
- 84.Singh RK, Balamurugan V, Bhanuprakash V, et al. Emergence and reemergence of vaccinia-like viruses: global scenario and perspectives. Indian J Virol. 2012;23(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peres MG, Bacchiega TS, Appolinario CM, et al. Vaccinia virus in blood samples of humans, domestic and wild mammals in Brazil. Viruses. 2018;10(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moussatché N, Damaso CR, McFadden G. When good vaccines go wild: feral Orthopoxvirus in developing countries and beyond. J Infect Dev Ctries. 2008;2(3):156–173. [DOI] [PubMed] [Google Scholar]
- 87.Lewis-Jones S. Zoonotic poxvirus infections in humans. Curr Opin Infect Dis. 2004;17(2):81–89. [DOI] [PubMed] [Google Scholar]
- 88.Shchelkunov SN. An increasing danger of zoonotic orthopoxvirus infections. PLoS Pathog. 2013;9(12):e1003756–e1003756. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Discussion of emerging orthopoxvirus diseases that could be treated with TPOXX.
- 89.Reynolds MG, Doty JB, McCollum AM, et al. Monkeypox re-emergence in Africa: a call to expand the concept and practice of One Health. Expert Rev Anti Infect Ther. 2019;17(2):129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Durski KN, McCollum AM, Nakazawa Y, et al. Emergence of Monkeypox—West and Central Africa, 1970–2017. Morbidity Mortality Weekly Rep. 2018;67(10):306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kile JC, Fleischauer AT, Beard B, et al. Transmission of monkeypox among persons exposed to infected prairie dogs in Indiana in 2003. Arch Pediatr Adolesc Med. 2005;159(11):1022–1025. [DOI] [PubMed] [Google Scholar]
- 92.Vaughan A, Aarons E, Astbury J, et al. Two cases of monkeypox imported to the United Kingdom, September 2018. Euro Surveill. 2018;23(38):1800509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Erez N, Achdout H, Milrot E, et al. Diagnosis of imported Monkeypox, Israel, 2018. Emerg Infect Dis. 2019;25(5):980–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ng OT, Lee V, Marimuthu K, et al. A case of imported Monkeypox in Singapore. Lancet Infect Dis. 2019;19(11):1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beachkofsky TM, Carrizales SC, Bidinger JJ, et al. Adverse events following smallpox vaccination with ACAM2000 in a military population. Arch Dermatol. 2010;146(6):656–661. [DOI] [PubMed] [Google Scholar]
- 96.McNeil MM, Cano M, E RM, et al. Ischemic cardiac events and other adverse events following ACAM2000((R)) smallpox vaccine in the vaccine adverse event reporting system. Vaccine. 2014;32(37):4758–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Petersen BW, Harms TJ, Reynolds MG, et al. Use of vaccinia virus smallpox vaccine in laboratory and health care personnel at risk for occupational exposure to orthopoxviruses - recommendations of the advisory committee on immunization practices (ACIP), 2015. MMWR Morb Mortal Wkly Rep. 2016;65(10):257–262. [DOI] [PubMed] [Google Scholar]
- 98.Vora S, Damon I, Fulginiti V, et al. Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin Infect Dis. 2008;46(10):1555–1561. [DOI] [PubMed] [Google Scholar]
- 99.Lederman ER, Davidson W, Groff HL, et al. Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J Infect Dis. 2012;206(9):1372–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Whitehouse ER, Rao AK, Yu YC, et al. Novel treatment of a vaccinia virus infection from an occupational needlestick - San Diego, California, 2019. MMWR Morb Mortal Wkly Rep. 2019;68(42):943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Centers for Disease C, Prevention . Human vaccinia infection after contact with a raccoon rabies vaccine bait - Pennsylvania, 2009. MMWR Morb Mortal Wkly Rep. 2009;58(43):1204–1207. [PubMed] [Google Scholar]
- 102.Lindholm DA, Fisher RD, Montgomery JR, et al. Preemptive tecovirimat use in an active duty member presenting with acute myeloid leukemia after smallpox vaccination. Clin Infect Dis. 2019;69:2205–2207. [DOI] [PubMed] [Google Scholar]
- 103.Progressive vaccinia in a military smallpox vaccinee - United States, 2009. MMWR Morb Mortal Wkly Rep. 2009;58(19):532–536. [PubMed] [Google Scholar]
- 104.Guo ZS, Lu B, Guo Z, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer. 2019;7(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Al Yaghchi C, Zhang Z, Alusi G, et al. Vaccinia virus, a promising new therapeutic agent for pancreatic cancer. Immunotherapy. 2015;7(12):1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Torres-Domínguez LE, McFadden G. Poxvirus oncolytic virotherapy. Expert Opin Biol Ther. 2019;19(6):561–573. [DOI] [PubMed] [Google Scholar]
- 107.Pelin A, Boulton S, Tamming LA, et al. Engineering vaccinia virus as an immunotherapeutic battleship to overcome tumor heterogeneity. Expert Opin Biol Ther. 2020; (9):1–15 [DOI] [PubMed]
- 108.Pelin A, Huh M, Tang M, et al. Abstract PR19: utilizing novel oncolytic vaccinia virus for selective expression of immunotherapeutic payloads in metastatic tumors. Cancer Immunol Res. 2020;8(4 Supplement):PR19–PR19. [Google Scholar]