

Abstract
Aberrant activation of the PI3K/AKT pathway is considered in many malignant tumors and plays a crucial role in mediating malignancy progression, metastasis, and chemoresistance. Consequently, development of PI3K/AKT pathway targeted drugs is currently an attractive research field for tumor treatment. In this study, twenty-six flavonoid-based amide derivatives were synthesized and evaluated for their antiproliferation effects against seven cancer cell lines, including MDA-MB-231, MCF-7, HCC1937, A549, HepG2, GTL-16 and HeLa. Among them, compound 7t possessed the best specific cytotoxicity against triple negative breast cancer MDA-MB-231 cells with an IC50 value of 1.76 ± 0.91 μM and also presented inhibitory ability on clonal-formation, migration and invasion of MDA-MB-231 cells. Further cell-based mechanistic studies demonstrated that compound 7t caused cell cycle arrest of MDA-MB-231 cells at the G0/G1 phase and induced apoptosis. Meanwhile, the western blot assay revealed that compound 7t could down-regulate the expression of p-PI3K, p-AKT, and Bcl-2 and up-regulate the production of PTEN, Bax, and caspase-3. Molecular docking also showed a possible binding mode of 7t with PI3Kα. Together, compound 7t was eligible as a potential TNBC therapeutic candidate for further development.
Flavonoid-based amide 7t possesses excellent cytotoxicity against MDA-MB-231 cells and its antitumor activity is achieved by affecting the PI3K/AKT pathway with inducing apoptosis.
1. Introduction
Despite efforts over the last 20 years to find effective therapeutic medicines, cancer remains a global concern. According to the statistics of the World Health Organization in 2020,1 cancer was the top cause of death in 185 nations globally and more than 2.26 million cases were newly recorded. Among them, breast cancer has taken over lung cancer as the leading cause of female death. Despite the fact that conventional medications, combination therapies, and targeted treatments have significantly improved 5 year survival rates, patients with high-grade and metastatic malignancies still encounter a poor outlook.2 The molecular etiology of most cancers has not been completely elucidated and some types of cancer still lack efficient therapeutic drugs, resulting in a huge challenging and urgent demand to develop highly effective therapies.
Phosphatidylinositol 3-kinases (PI3Ks) are a family of lipid kinases associated with oncogene products which are one of the significant upstream components of the PI3K/AKT signal transduction pathway that has tremendous impact on tumor occurrence and development,3 whereas AKT is a downstream component of this pathway.4,5 In response to stimulation by growth factors such as VEGF, HGF, insulin or signal transduction complexes, RTKs and GPCRs on the cell membrane are activated, promoting the phosphorylation response of PI3K and leading to the initiation of the PI3K/AKT pathway.6,7 Generally, the activated PI3K catalyzes the phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3) with second messenger function.8–10 PIP3 then binds to the N-terminal PH domain of AKT and aggregates it to the plasma membrane, where AKT is activated by phosphorylating the Thr308 and Ser473 residues of PDK1 and PDK2, respectively.11 The activated AKT is subsequently detached from the cell membrane and released into the nucleus or free in the cytoplasm to further activate related downstream proteins that regulate cell growth, apoptosis, invasion, metastasis, and angiogenesis.12,13 Consequently, the PI3K/AKT pathway plays a crucial role in regulation of multiple physiological processes critical to mediate malignancy progression, metastasis, and chemoresistance.14–16 Hyper-activation of such a pathway is considered a common event in cancers including breast, non-small cell lung, and colorectal cancers.17–19 As multiple pathways contribute to the activation of the PI3K/AKT pathway, investigation of drugs targeting this system can provide new options for potent tumor therapies.20
As mentioned previously, development of a PI3K/AKT pathway targeted regulatory agent is currently an attractive research field in view of the significance for tumor treatment.21–23 Some typical PI3K/AKT inhibitors are depicted in Fig. 1. Galangin is a natural flavonoid with strong antiproliferation and apoptosis induction activities against many cancer cells, such as breast, melanoma, lung, and liver cancers through targeting the PI3K/AKT signal pathway.24–27 Wortmannin is an irreversible PI3K inhibitor isolated from fungal metabolites, which possesses obvious cytotoxicity and kinase inhibitory potency.28 However, its low bioavailability and high toxicity limit its clinical value. LY294002 is a reversible PI3K inhibitor originating from the natural flavonoid quercetin,29 which can decrease the formation of tumor tissue and autophagosomes by influencing the cell cycle and inducing cell apoptosis.30 But the clinical application is restricted due to its short half-life, poor lipid solubility, low bioavailability, and susceptibility to metabolic inactivation.31–33 BKM120 (Buparlisib) is a broad-spectrum PI3K inhibitor with high oral bioavailability, which can not only suppress the expression of NRF2 and enhance the sensitivity of Keap1 or NFE2L2 mutant squamous cell lung cancer, but also block VEGF induced neovascularization.34,35 Unfortunately, the phase III clinical research of BKM120 has been terminated because of its toxicity. Thus, exploration of low toxicity PI3K/AKT pathway inhibitors remains challenging.
Fig. 1. The structure of PI3K/AKT pathway targeted regulatory agents.

Our group make continuous efforts to explore new anticancer candidates36 and have developed a kind of ligustrazine–chalcone hybrid as an anti-triple negative breast cancer (TNBC) agent in the previous study.37 Thus, inspired by the efficacy of flavonoids having an outstanding anticancer activity via targeting the PI3K/AKT pathway38,39 and structural relevance between flavonoids and chalcone, we herein report the synthesis of flavonoid-based amide derivatives and evaluation of their anticancer ability. Analysis of the structural characteristics of the target compounds shows that a 4-fluorophenyl and diverse amides were introduced at the 7- and 3′-positions of the benzopyran-4-one mother nucleus respectively. Compared to galangin and LY294002, removal of excess hydroxyl groups at 5- and 7-positions could avoid inactivation of glycosylation metabolism, and introduction of the amide group and fluorine atom was expected to improve the anticancer capability. The anticancer evaluation involved cytological experiments and Western blot assay. The former was performed to assess the in vitro antiproliferative effect and related mechanism, while the latter was used to determine the influence of potential compounds on the expression of PI3K/AKT signal pathway related proteins.
2. Results and discussion
2.1. Chemistry
The synthetic routine of target compounds 7a–7z is described in Scheme 1. Initially, the Suzuki–Miyaura coupling reaction was conducted between 1-(4′-bromo-2-hydroxyphenyl) ethan-1-one 1 and (4-fluorophenyl) boronic acid 2 to give intermediate 3. It was then reacted with 3-aminobenzaldehyde via aldol condensation in the presence of K2CO3 in MeOH to afford compound 4. Reduction of the nitro in intermediate 4 by SnCl2 produced the corresponding amino product 5 and it was subsequently cyclized under H2O2 and NaOH using methanol as a solvent to obtain intermediate 6. Finally, flavonoid compound 6 was condensed with a series of acyl chlorides in acetone and pyridine to provide the target compounds 7a–7z, which were characterized through nuclear magnetic resonance (NMR) and high-resolution mass spectrometry (HRMS).
Scheme 1. Synthetic route of target compounds. Reagents and conditions: (i) K2CO3, Pd(OAc)2, dioxane, 110 °C, 5 h; (ii) K2CO3, MeOH, 50 °C, reflux, 16 h; (iii) SnCl2, AcOH, H2O/MeOH, 90 °C, 12 h; (iv) 30%H2O2, NaOH, MeOH, 0–5 °C, rt, 10 h; (v) acetone, pyridine, rt, 3 h.

2.2. Antiproliferative assay in vitro
All synthesized compounds (7a–7z) were evaluated for their in vitro antiproliferation activities against seven human cancer cell lines including MDA-MB-231 (human breast cancer cell line), MCF-7 (human breast cancer cell line), HCC1937 (human breast cancer cell line), A549 (human non-small cell lung cancer cell line), HepG2 (human hepatocellular liver carcinoma cell line), GTL-16 (human gastric cancer cell line), and HeLa (human cervical carcinoma cell line) through the Cell Counting Kit-8 (CCK8) assay. LY294002 and 5-fluorouracil (5-Fu) were used as positive controls.
As depicted in Table 1, many compounds exhibited good cytotoxicity against these seven cell lines manifesting as IC50 values that were much lower than that of the well-known antitumor cytotoxic drug 5-Fu, while the other control flavonoid LY294002 showed a relatively less efficient antiproliferation potency. Notably, some compounds had excellent specific antiproliferative activities on breast cancer related cell lines such as MDA-MB-231, MCF-7, and HCC1937. For instance, compounds 7d, 7g, 7r, 7t, and 7u presented favorable antiproliferative effects against these three cell lines with IC50 values ranging from 1.76 μM to 9.05 μM, all of which were superior to the positive control 5-Fu.
Cytotoxicity of target compounds 7a–7z against MDA-MB-231, MCF-7, HCC1937, A549, HepG2 and GTL-16 cells in vitro.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R | IC50a (mean ± SD μmol L−1) | ||||||
| MDA-MB-231 | MCF-7 | HCC1937 | A549 | Hep G2 | GTL-16 | HeLa | ||
| 7a |
|
7.24 ± 0.47 | 7.57 ± 0.27 | 10.76 ± 0.43 | 4.41 ± 0.83 | 10.14 ± 0.91 | 17.44 ± 1.10 | 5.63 ± 0.48 |
| 7b |
|
16.02 ± 0.89 | 8.59 ± 0.78 | 14.23 ± 0.90 | 6.93 ± 0.55 | 9.28 ± 0.83 | 14.64 ± 1.39 | 5.35 ± 1.05 |
| 7c |
|
16.52 ± 1.03 | 6.25 ± 0.64 | 5.49 ± 0.63 | 8.70 ± 1.03 | 8.42 ± 0.71 | 21.07 ± 1.84 | 4.82 ± 1.69 |
| 7d |
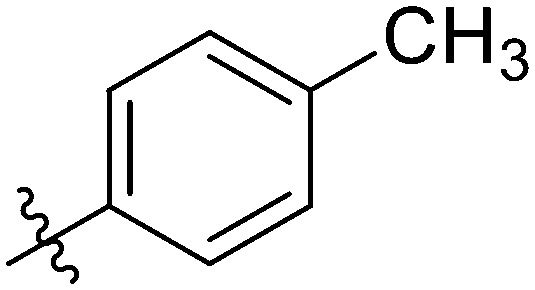
|
4.35 ± 0.94 | 2.40 ± 0.61 | 7.11 ± 0.94 | 6.91 ± 0.32 | 2.32 ± 0.57 | 12.92 ± 1.08 | 6.19 ± 1.08 |
| 7e |
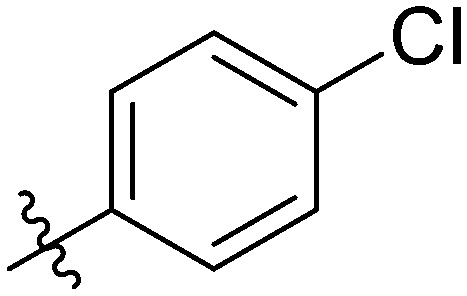
|
12.80 ± 0.98 | 4.73 ± 0.71 | 4.03 ± 0.73 | 10.82 ± 0.66 | 11.01 ± 1.25 | 9.14 ± 0.87 | 3.67 ± 1.26 |
| 7f |
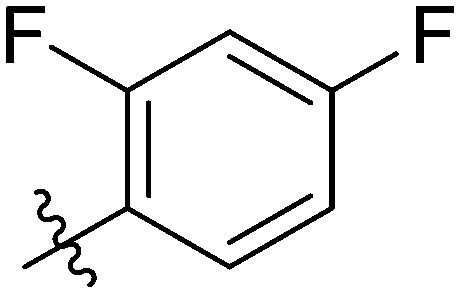
|
11.17 ± 0.63 | 13.45 ± 0.41 | 6.98 ± 0.38 | 20.46 ± 0.74 | 26.35 ± 0.82 | 9.64 ± 0.60 | 10.73 ± 1.70 |
| 7g |
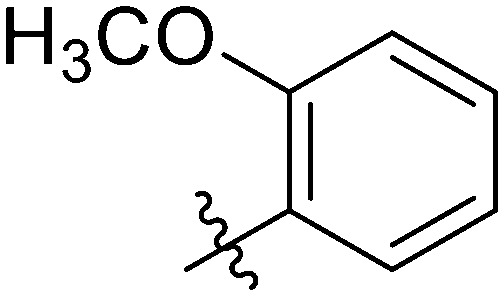
|
5.11 ± 0.95 | 2.10 ± 0.86 | 9.05 ± 1.10 | 11.66 ± 0.74 | 14.51 ± 1.47 | 6.37 ± 0.91 | 14.07 ± 0.38 |
| 7h |
|
4.91 ± 1.08 | 2.28 ± 0.62 | 9.90 ± 0.82 | 17.55 ± 0.99 | 19.39 ± 1.43 | 24.78 ± 1.29 | 11.86 ± 0.61 |
| 7i |
|
29.39 ± 0.49 | 32.62 ± 0.99 | 7.35 ± 0.81 | 19.37 ± 0.93 | 42.70 ± 0.83 | 33.01 ± 1.76 | 17.35 ± 1.11 |
| 7j |
|
24.93 ± 0.89 | 24.02 ± 0.69 | 8.63 ± 0.99 | 13.51 ± 0.98 | 13.89 ± 1.08 | 25.44 ± 0.86 | 9.74 ± 0.88 |
| 7k |
|
9.16 ± 0.97 | 15.27 ± 0.51 | 18.89 ± 0.76 | 10.80 ± 0.35 | 9.95 ± 0.96 | 12.46 ± 1.76 | 5.65 ± 1.00 |
| 7l |
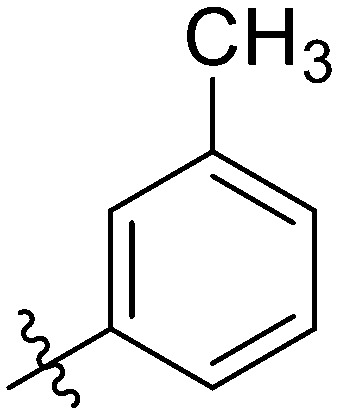
|
2.84 ± 0.20 | 7.85 ± 1.13 | 8.03 ± 1.12 | 16.45 ± 0.77 | 13.49 ± 0.86 | 10.89 ± 1.11 | 6.65 ± 1.00 |
| 7m |
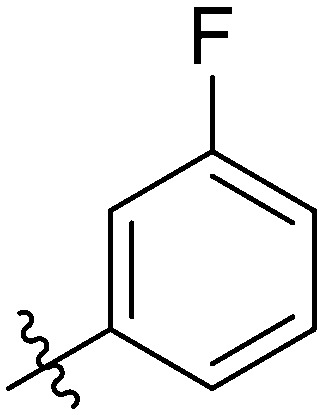
|
2.51 ± 0.93 | 5.77 ± 0.90 | 6.59 ± 0.61 | 15.39 ± 0.94 | 15.48 ± 1.06 | 15.82 ± 1.40 | 8.78 ± 1.15 |
| 7n |
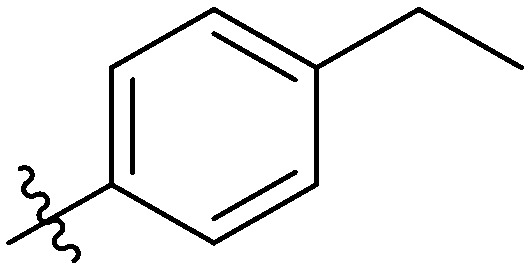
|
9.07 ± 0.51 | 22.77 ± 1.08 | 13.24 ± 0.34 | 18.47 ± 0.99 | 20.62 ± 1.04 | 24.56 ± 1.36 | 21.01 ± 1.55 |
| 7o |
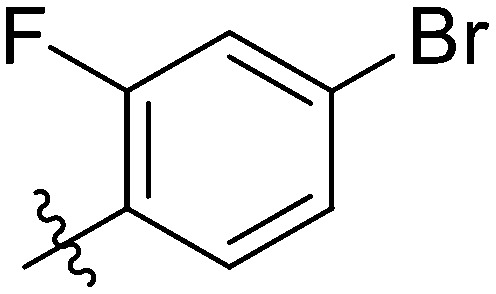
|
NT | NT | NT | 46.11 ± 1.06 | >100 | NT | NT |
| 7p |
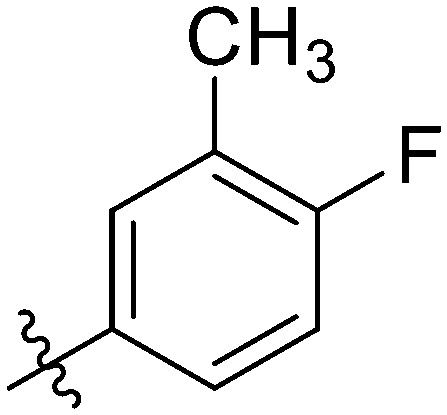
|
7.44 ± 0.37 | 7.71 ± 1.01 | 5.42 ± 0.78 | 11.45 ± 0.83 | 9.54 ± 0.76 | 12.10 ± 0.92 | 18.86 ± 1.53 |
| 7q |
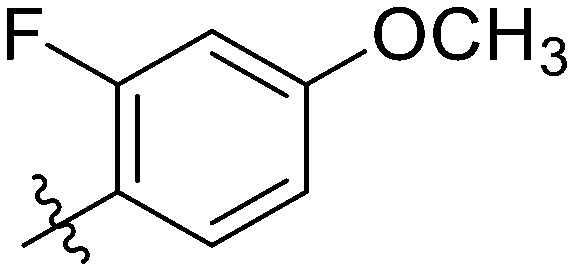
|
17.56 ± 0.84 | 20.03 ± 1.01 | 24.45 ± 0.64 | 39.95 ± 1.25 | 45.33 ± 1.14 | 15.30 ± 1.62 | 16.09 ± 1.05 |
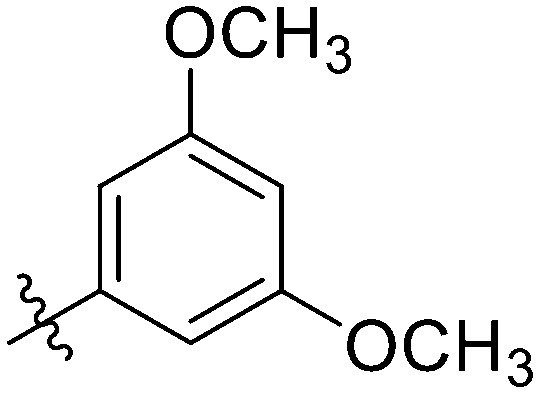
| 7r |
|
4.04 ± 0.31 | 3.47 ± 1.08 | 6.25 ± 0.47 | 2.39 ± 0.41 | 23.47 ± 0.64 | 19.58 ± 1.44 | 2.70 ± 1.03 |
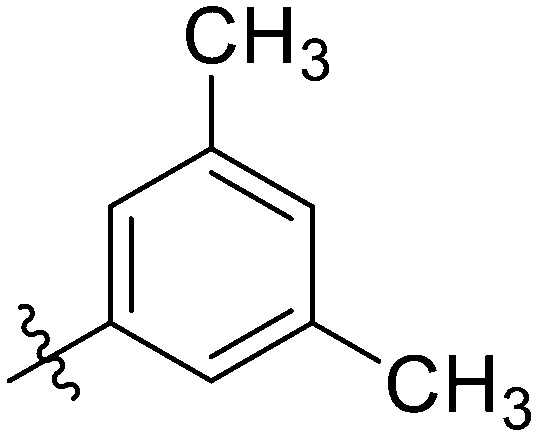
| 7s |
|
11.94 ± 1.00 | 20.43 ± 1.50 | 4.86 ± 0.88 | 3.74 ± 0.35 | 13.37 ± 1.31 | 13.51 ± 1.36 | 5.54 ± 1.10 |
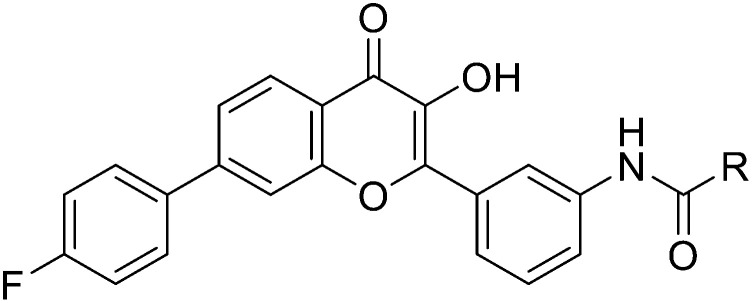
| 7t |
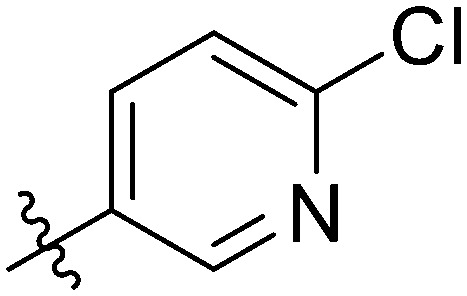
|
1.76 ± 0.91 | 2.89 ± 0.86 | 7.55 ± 0.95 | 3.58 ± 0.62 | 4.61 ± 0.49 | 7.52 ± 0.94 | 4.08 ± 1.19 |
| 7u |
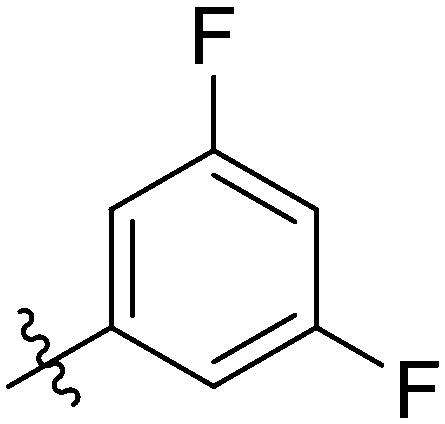
|
2.44 ± 0.76 | 2.49 ± 0.44 | 2.07 ± 1.06 | 7.62 ± 1.10 | 4.96 ± 0.93 | 7.41 ± 0.87 | 3.38 ± 0.66 |
| 7v |
|
NT | NT | NT | >100 | >100 | NT | NT |
| 7w |
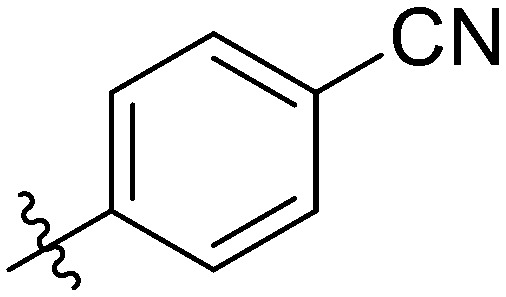
|
11.60 ± 0.92 | 2.66 ± 1.02 | 2.99 ± 0.56 | 2.36 ± 0.45 | 7.11 ± 0.99 | 31.81 ± 1.04 | 23.30 ± 1.40 |
| 7x |
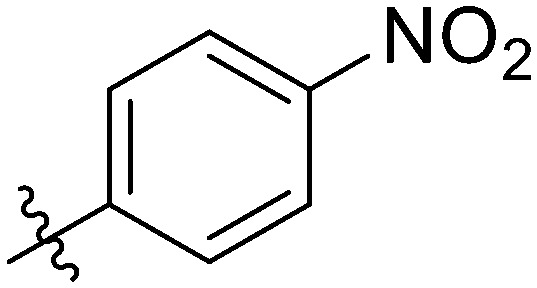
|
14.73 ± 0.83 | 3.97 ± 0.36 | 5.64 ± 0.41 | 2.44 ± 0.55 | 1.86 ± 0.35 | 18.22 ± 0.89 | 12.34 ± 0.69 |
| 7y |
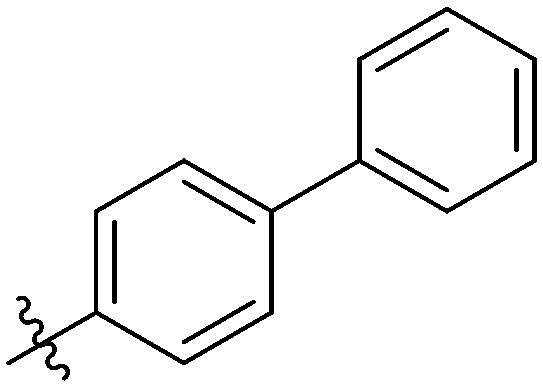
|
63.24 ± 1.02 | 38.49 ± 0.94 | 44.08 ± 1.15 | >100 | 88.48 ± 1.40 | NT | >100 |
| 7z |
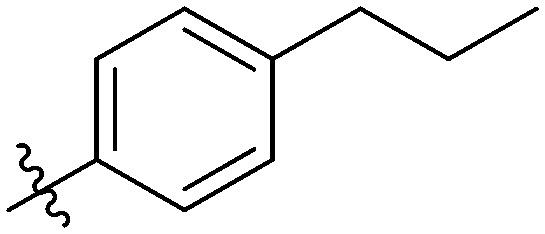
|
32.18 ± 1.13 | 36.77 ± 1.23 | 15.35 ± 1.32 | 47.62 ± 1.20 | 51.52 ± 1.11 | 20.44 ± 1.16 | 8.79 ± 1.23 |
| 5-Fub | 7.75 ± 0.82 | 4.42 ± 0.89 | 25.94 ± 1.12 | 2.65 ± 0.12 | 2.88 ± 0.88 | 0.84 ± 0.19 | 1.94 ± 0.23 | |
| LY294002b | 31.76 ± 0.71 | 39.62 ± 1.80 | 37.84 ± 1.26 | 34.13 ± 1.53 | 30.86 ± 1.74 | 24.38 ± 1.06 | 16.97 ± 1.48 | |
Antitumor activity was assayed by exposure to the substances for 72 h and is expressed as the concentration required to inhibit tumor cell proliferation by 50% (IC50); values were the means of three replicates ± standard deviation (SD).
Used as a positive control.
The most potent antiproliferation activity against the TNBC cell line MDA-MB-231 was obtained from compound 7t whose R group at the 3′-position of the amide fragment was 6-chloro-3-pyridyl with an IC50 value of 1.76 ± 0.91 μM, which was nearly four times the potency of 5-Fu (IC50 value of 7.75 ± 0.82 μM). Compounds 7u and 7m whose R group was 3,5-difluorophenyl and 3-fluorophenyl, respectively, showed the second and third highest antiproliferative capabilities on MDA-MB-231 cells with IC50 values of 2.49 ± 0.44 μM and 2.51 ± 0.93 μM, respectively. These results indicated that, for the cytotoxicity on MDA-MB-231 cells, a pyridyl substituent on the R group was preferred over a phenyl, and a fluorine-substituted phenyl moiety was better than other phenyl substituents. Besides its excellent cytotoxicity on MDA-MB-231, compound 7u also displayed favorable antiproliferative capacity against MCF-7 and HCC1937 cells with IC50 values of 2.49 ± 0.44 μM and 2.07 ± 1.06 μM, respectively. These results suggested that compound 7u was not only the optimum compound with a comprehensive inhibitory effect on these breast cancer cells, but also the strongest cytotoxic agent against the TNBC cell line HCC1937 which exceeded the effectiveness of 5-Fu by ten-fold (IC50: 25.94 ± 1.12 μM). Compound 7w whose R group was 4-cyanophenyl exhibited the second powerful antiproliferation capability on HCC1937 (IC50: 2.99 ± 0.56 μM) and gratifying proliferation inhibition activity on MCF-7, but a relatively reduced cytotoxicity against MDA-MB-231.
In addition, further investigations indicated that the 3,5 positions on the benzene ring in the R group being doubly substituted by fluorine was more favorable to enhance potent and broad-spectrum cytotoxicity against breast cancer cells compared to the mono-fluorophenyl or other mono-substituted phenyl substituted compounds. These conclusions were consistent with observations from compound 7g bearing 2-methoxyphenyl in the R group. Compared to compounds 7t and 7u, compound 7g only showed forceful proliferative inhibition on MCF-7 with an IC50 value of 2.10 ± 0.86 μM, while obvious decreased antiproliferation effects on the other two TNBC cells, MDA-MB-231 and HCC1937, were afforded.
It was worth noting that different selectivities for wild-type versus mutant TNBC cells were obtained from compounds 7t and 7w. The antiproliferation on the wild TNBC cell line MDA-MB-231 of compound 7t was superior to that of the mutant TNBC cell line HCC1937 (IC50: 1.76 vs. 7.55 μM) and the selectivity ratio between the mutants and wild types was 1 : 4.29. In contrast, compound 7w possessed preferred cytotoxicity on the mutant type than the wild type (IC50: 11.60 vs. 2.99 μM) with a selectivity ratio of 3.88 : 1. Such observations were of great significance for the development of TNBC therapeutic drugs for different stages and types.
To our delight, some of the synthesized compounds also showed comparable cytotoxicity against non-small cell lung cancer A549 and hepatocellular carcinoma HepG2 cells to the positive control 5-Fu. For example, compound 7x with an electron-withdrawing substituent 4-nitrophenyl on the R group possessed the most antiproliferative potency on HepG2 among all target compounds and its IC50 value was 1.86 ± 0.35 μM which was superior to that of 5-Fu (IC50 = 2.88 ± 0.88 μM). Moreover, a comparable cytotoxicity on A549 to that of 5-Fu was obtained from compound 7x (IC50: 2.44 ± 0.55 μM vs. 2.65 ± 0.12 μM). In addition to 7x, compounds 7r and 7w also had almost equivalent antiproliferation activities against A549 cells to 5-Fu, whereas a slightly better proliferative inhibition effect on HepG2 cells was afforded by compound 7d with an IC50 value of 2.32 ± 0.57 μM.
A preliminary analysis of the structural characteristics affecting the specific antiproliferative ability against A549 and HepG2 cells revealed that substituents at the 4-position on the benzene ring in the R group exhibited a significant influence. The nitro group was the best substituent and methyl or cyano moieties also achieved satisfactory outcomes. Meanwhile substituents with hydrogen or chain alkyl such as ethyl gave poor activities. Compared to 2- or 3-positions, substituents on the 4-position were preferred. Disubstituted derivatives of the benzene ring in the R group showed less efficient antiproliferation activities than 4-position substituted compounds. Moreover, replacement of phenyl with pyridyl in the R group displayed comparable cytotoxicity but pyrimidinyl substitution resulted in complete inactivation.
Nevertheless, the target compounds exhibited less efficient cytotoxicity on human cervical carcinoma HeLa cells. The IC50 value of the best efficiency compound could be as low as 2.70 ± 1.03 μM, but was still slightly less effective than 5-Fu (IC50 = 1.94 ± 0.23 μM). Unfortunately, the antiproliferative capability of the synthesized compounds against human gastric cancer GTL-16 cells was limited and all IC50 values were much worse that of than 5-Fu. The most active compound only gave an IC50 value of 6.37 μM, which was nearly eight-fold higher than that of 5-Fu (IC50: 0.84 μM).
Taken together, due to the remarkable specific cytotoxicity of compound 7t, combined with extremely limited clinical treatment options and drugs for TNBC, we selected it for further investigation in order to provide a basis for exploration of new candidates with a high therapeutic index.
2.3. Colony-forming assay of compound 7t
To further explore the proliferation inhibitory effect on MDA-MB-231 cells of compound 7t, a plate colony-forming assay was performed to determine its influence on the number and size of colony-forming cells and analyze the changes of colony-forming ability of MDA-MB-231cells.
As described in Fig. 2, the number and size of MDA-MB-231 cells were inhibited dramatically by treatment with compound 7t with the concentration increasing (0.25IC50, 0.5IC50, IC50 and 2IC50) and time prolonging (24 h, 48 h and 72 h). It was indicated that compound 7t had colony-formation inhibitory abilities on MDA-MB-231 cells in both a dose- and time-dependent manner.
Fig. 2. (A and C) Effect of compound 7t on colony-formation inhibition of MDA-MB-231 cells. (B and D) Numbers of colonies counted by image, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 compared to the control.

2.4. Hoechst staining and flow cytometry assays of compound 7t
To confirm whether the antiproliferation effect of compound 7t on MDA-MB-231 cells was associated with induction of apoptosis, Hoechst staining and flow cytometry assays were carried out. According to the Hoechst staining image in Fig. 3A, with the increase in concentration of compound 7t (0.5IC50, IC50, 2IC50), a growing number of morphological changes from MDA-MB-231 cells occurred obviously such as chromatin concentration and margination, half-moon, loop and ellipse shaped fragment formation, and emergence of apoptotic bodies; it further showed that the floating cells with fluorescent spots grew dramatically. Thus, it was suggested that compound 7t could significantly induce apoptosis of MDA-MB-231 cells in a concentration-dependent manner as summarized in Fig. 3B.
Fig. 3. The effect of compound 7t on MDA-MB-231 cell apoptosis and the cell cycle. MDA-MB-231 cells were treated with the indicated concentrations of 7t for 48 h, and apoptotic cells were detected by Hoechst 33342 staining and flow cytometry, respectively. All data represent the mean ± SD, n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 compared to the control. (A) The effect of compound 7t on apoptosis of MDA-MB-231 cells by Hoechst staining. (B) Statistical apoptosis cell proportion of MDA-MB-231 with different dose concentrations of compound 7t. (C) The effect of compound 7t on the cell cycle in MDA-MB-231 cells obtained through the flow cytometry assay. (D) The flow cytometry data analysis as a means to measure the percentages of G0/G1, S, and G2/M. (E) Effect of compound 7t on apoptosis of MDA-MB-231 cells measured through the Annexin V-FITC/PI assay. (F) The flow cytometry data analysis as a means to obtain the percentages of survival cells, early and late apoptotic cells.

On the basis of the cell cycle assay depicted in Fig. 3C and D, the proportion of the G0/G1 phase of MDA-MB-231 cells elevated obviously with increasing concentration (0.5IC50, IC50 and 2IC50) in each dose group administrated with compound 7t after 48 h compared with the blank group. On the contrary, a dramatic decrease in the proportion of the S-phase combined with a slight reduction in the proportion of the G2/M-phase of MDA-MB-231 cells was observed, indicating that compound 7t inhibited the growth cycle transition of MDA-MB-231 cells and blocked the cell growth cycle at the G0/G1 phase.
Furthermore, the apoptosis graphic of MDA-MB-231 cells was afforded by flow cytometry utilizing the Annexin V-FITC/PI double staining method. In the light of the statistical cell proportion illustrated in Fig. 3E and F, compound 7t strongly induced the apoptosis of MDA-MB-231 cells and the overall apoptosis rate raised gradually with increasing concentration (0.5IC50, IC50, 1.25IC50, 2IC50 and 4IC50). None of the evident early apoptotic cells were observed under the concentrations of 0.5IC50 while the proportions of early and late apoptotic cells gradually rose when the concentrations were greater than IC50 (the early/late apoptosis ratios were 46.96%/3.81% and 68.96%/14.4% for concentrations of IC50 and 2IC50, respectively). Such observations demonstrated that compound 7t induced a concentration-dependent apoptosis induction on MDA-MB-231 cells.
2.5. Transwell simulated cell invasion and wound-healing assays of compound 7t
Owing to the invasion of cancer cells which is a process that is inextricably linked to cell metastasis, a Transwell simulated cell invasion assay was applied to examine the effect of compound 7t on the invasive ability of MDA-MB-231 cells at different concentrations. As shown in Fig. 4A and B, the number of cells crossing the microporous membrane decreased gradually at the concentrations of 0.5IC50, IC50, and 2IC50, indicating that compound 7t displayed significant potency to suppress the invasion capability of MDA-MB-231 cells in a concentration-dependent manner.
Fig. 4. The effect of compound 7t on MDA-MB-231 cell invasion and migration. All data represent the mean ± SD, n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 compared to the control. (A) The effect of compound 7t on Transwell cell invasion assays. (B) Statistical number of invaded MDA-MB-231 cells with treatment with compound 7t at different dose concentrations. (C) The wound healing assay for compound 7t on MDA-MB-231 cells. (D) Statistics of scratch mobility from MDA-MB-231 cells with compound 7t at different dose concentrations.

As the metastasis of cancer cells is related to their migration ability, a wound healing assay was conducted to probe the inhibitory effect of compound 7t on the migration of MDA-MB-231 cells at different concentrations and times. The corresponding results could be determined by measurement of the scratch distance and statistics of scratch mobility. As demonstrated in Fig. 4C and D, the scratch healing speed slowed down and the movement migration ability was affected via administration of compound 7t at the concentration of 0.5IC50 after 12 h compared with the blank group. Subsequently, the relative migration rate of MDA-MB-231 cells abated obviously with increasing dose concentrations and the relative migration rates were 64.13%, 42.32%, 35.41% and 23.11% at concentrations of 0, 0.5IC50, IC50, and 2IC50, respectively after 48 h, which meant that the motility of MDA-MB-231 cells was significantly inhibited by compound 7t at high lever concentrations. All these outcomes suggested that compound 7t suppressed the migration capacity of MDA-MB-231 cells in a concentration-dependent manner.
2.6. Western blot assay in MDA-MB-231 cells of compound 7t
With the aim of making a further effort on the mechanism exploration of the antiproliferation potency against MDA-MB-231 cells of compound 7t, a Western blot assay was employed to discuss its influence on the PI3K/AKT signaling pathway namely the relevant protein expression of PI3K, p-PI3K, AKT, p-AKT, and PTEN, and apoptosis related protein expression, such as caspase-3, Bcl-2, and Bax. As illustrated in Fig. 5, by treatment with compound 7t on MDA-MB-231 cells at concentrations of 1 μM, 5 μM, and 10 μM for 48 h, respectively, the production of phosphorylated PI3K and phosphorylated AKT was inhibited in a concentration-dependent manner. The expression of tumor suppressor gene PTEN was up-regulated. Besides, the downstream anti-apoptotic protein Bcl-2 was down-regulated and the pro-apoptotic protein Bax as well as the apoptotic execution protein caspase-3 was up-regulated, and the apoptosis of MDA-MB-231 cells was thereby induced. Consequently, compound 7t possessed excellent antiproliferation performance via inhibition of the PI3K/AKT signaling pathway and induction of apoptosis as it could not only suppress the phosphorylation of PI3K and AKT to inactivate downstream targets, but also regulate cell death through endogenous pathways.
Fig. 5. Western blot assay of MDA-MB-231 treated with compound 7t. (A) Western blot analysis of proteins involved in the PI3K/AKT signaling pathway. (B) Regulation statistics on the expression of PI3K/AKT signaling pathway-related proteins in MDA-MB-231 cells. (C) Western blot analysis of apoptosis-associated proteins. (D) Regulation statistics on the expression of apoptosis-related proteins in MDA-MB-231 cells.

2.7. Modeling of compound 7t on PI3Kα
A molecular docking study was carried out to predict the possible binding mode between compound 7t and PI3Kα (PDB: 4tv3), which could be responsible for the potent antiproliferation activity. The molecular simulation protocol was initially validated by re-docking of the co-crystalized ligand into the binding sites of PI3Kα and the reliability was verified by a high similarity value between the re-docked and original ligand poses from the Sybyl-X 2.1 software. As shown in Fig. 6, compound 7t can perfectly be docked into the active site of 4tv3 as evidenced by satisfactory scoring functions such as total-score and c-score. The critical hydrogen bond of carbonyl and hydroxyl groups with Val851 was formed similar to LY294002. The carbonyl group in the amide fragment formed another key hydrogen bond with residue Arg770. In addition, the two phenyl groups of the flavonol scaffold formed π–π stacking interaction with Tyr836 and Trp780, respectively. The benzene ring on the 2-position with guanidyl of the Arg770 residue produced an important π–cation interaction. Furthermore, the chlorine atom on the pyridine ring formed a halogen bond with residue Asn853. All these interactions may contribute to its significant antiproliferative potency.
Fig. 6. Binding mode of compound 7t in the active site of PI3Kα obtained from molecular docking. 7t was represented in stick colored with green carbon, red oxygen and blue nitrogen respectively, while the surrounding key residues were depicted in stick with brown carbon, red oxygen and blue nitrogen. Hydrogen and halogen bonds were shown in dashed lines colored yellow and purple respectively. π–π stacking and π–cation interactions were described in dotted lines colored cyan and green respectively.

3. Conclusions
In summary, a series of novel flavonoid-based amide compounds were synthesized and evaluated for their in vitro anticancer bioactivities against seven tumor cell lines. Among them, compounds 7t and 7u presented significant specific antiproliferation activities on breast cancer cell lines. Compound 7u was the strongest cytotoxic agent against mutant type TNBC cell HCC1937 with an IC50 value of 2.07 ± 1.06 μM which was exceeded the positive control 5-Fu by ten-fold. Compound 7t possessed the most antiproliferative ability against wild type TNBC cell MDA-MB-231 with an IC50 value of 1.76 ± 0.91 μM, nearly four times the potency of 5-Fu. In addition, compound 7t presented inhibitory ability on clonal-formation, migration and invasion of MDA-MB-231 cells. Further cell-based mechanistic studies indicated that compound 7t caused cell cycle arrest of MDA-MB-231 cells in a dose dependent manner at the G0/G1 phase and induced apoptosis. Moreover, the western blot assay demonstrated that compound 7t could down-regulate the expression of p-PI3K, p-AKT, and Bcl-2 and up-regulate the expression of PTEN, Bax, and caspase-3 so as to inhibit the PI3K/AKT pathway from achieving its significant antiproliferative effects. Molecular docking simulation also showed a possible binding mode of compound 7t with PI3Kα. Overall, the obtained compounds 7t and 7u were eligible as potential regulators of the PI3K/AKT pathway to be further developed and provided a basis for TNBC curative candidates.
4. Experimental
4.1. General information
The 1H NMR and 13C NMR spectra were recorded using TMS as the internal standard on a Bruker Advance III 400 spectrometer (Bruker, Switzerland) at 400 MHz and 101 MHz in either CDCl3 or DMSO-D6. Chemical shifts (δ) were referenced to TMS as an internal standard (1H NMR, 13C NMR). Coupling constants are given in Hz. High-resolution mass (HRMS) spectra were obtained with a Thermo-DFS mass spectrometer (ThermoFisher Scientific, USA). Reaction progress was monitored using analytical thin layer chromatography (TLC) on precoated silica gel GF254 (Qingdao Haiyang Chemical Plant, Qingdao, China) plates, and the spots were detected under UV light (254 nm). Flash column chromatography was performed with silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. All other chemical materials were obtained from commercial suppliers and were used without further purification.
All cancer cell lines MDA-MB-231, MCF-7, HCC1937, A549, HepG2, GTL-16, and Hela were purchased from iCell Bioscience Inc, Shanghai. Biological materials including plasma, fetal bovine serum (FBS), DMEM medium and pancreatin were purchased from Shanghai Beyotime Biotechnology Co., Ltd. Cell Counting Kit-8, annexin V-APC/PI apoptosis analysis kits, and flow cytometry kits for apoptosis were purchased from Jiangsu KeyGEN BioTECH Co., Ltd. Penicillin–streptomycin liquid, Hoechst 33258 staining solution and phosphate buffer saline (PBS) were purchased from Hangzhou Genom Biotechnology Co., Ltd.
4.2. Synthesis of compound 3
4-Bromo-2-hydroxyacetophenone (4.0 g, 18.6 mmol), p-fluorophenylboronic acid (3.9 g, 27.9 mmol), palladium acetate (0.42 g, 1.86 mmol), and potassium carbonate (7.7 g, 55.8 mmol) were added sequentially to a 100 mL flask and charged with nitrogen. 1,4-Dioxane (20 ml) and water (4 ml) were slowly injected into the flask via a syringe, and the reaction mixture was stirred at 110 °C with reflux for 5 h. After completion monitored by thin-layer chromatography (TLC), the mixture was acidified to pH 6–7 with an aqueous solution of acetic acid. The mixture solution was then filtered and the filtrate was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and the solvent was evaporated under vacuum. The obtained crude product was purified by flash chromatography on silica gel (Vethyl acetate/Vpetroleum ether = 1/40) to give 2.623 g of product as a white solid. Yield 65%. 1H NMR (400 MHz, DMSO-d6) δ 12.13 (s, 1H), 7.94 (d, J = 8.3 Hz, 1H), 7.80–7.74 (m, 2H), 7.33–7.27 (m, 2H), 7.24 (dd, J = 8.2, 1.9 Hz, 1H), 7.21 (d, J = 1.7 Hz, 1H), 2.65 (s, 3H);13C NMR (101 MHz, DMSO-d6) δ 204.68, 164.34, 161.85, 147.05, 135.43, 132.64, 129.73, 129.65, 119.58, 118.06, 116.52, 116.30, 115.59, 27.94; HR-ESI-MS calcd for C14H11FO2 [M + H]+ 231.0743; found 231.0813.
4.3. Synthesis of compound 4
Compound 3 (2.0 g, 8.7 mmol) and 3-nitrobenzaldehyde (1.3 g, 8.7 mmol) were dissolved in methanol (20 mL) and KOH (2.4 g, 43.5 mmol) was added subsequently. The reaction mixture was then stirred at 50 °C for 16 h. After completion monitored by TLC, the reaction mixture was acidified to pH 6–7 with an aqueous solution of acetic acid to allow the yellow solid to precipitate. The solid was collected by filtration and the obtained crude product was purified by flash chromatography on silica gel (Vethyl acetate/Vpetroleum ether = 1/15) to give 1.16 g of product as a yellow solid. Yield 60%. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (t, J = 1.9 Hz, 1H), 8.33 (d, J = 8.5 Hz, 1H), 8.28 (d, J = 7.8 Hz, 1H), 8.23–8.20 (m, 1H), 8.20–8.17 (m, 1H), 7.87 (d, J = 15.6 Hz, 1H), 7.78–7.74 (m, 2H), 7.70–7.66 (m, 1H), 7.26 (d, J = 8.9 Hz, 2H), 7.23 (d, J = 1.6 Hz, 1H), 7.20 (d, J = 1.8 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 193.4, 163.0, 161.97, 148.9, 147.4, 142.6, 136.9, 135.9, 135.3, 132.4, 130.9, 129.9, 129.8, 125.5, 125.0, 123.8, 120.0, 118.1, 116.6, 116.4, 115.8; HR-ESI-MS calcd for C21H14FNO4 [M + H]+ 364.0907; found 364.0966.
4.4. Synthesis of compound 5
To a solution of compound 4 (2.0 g, 5.5 mmol) in methanol (20 mL) were added SnCl2 (4.2 g, 22.0 mmol) and acetic acid (5 mL) sequentially. The suspension solution was reacted at 90 °C with reflux overnight. After completion monitored by TLC, the reaction mixture was filtered and the filtrate was then diluted with water and extracted with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. The crude product was obtained through removing the solvent under vacuum and further purified by flash chromatography on silica gel (Vethyl acetate/Vpetroleum ether = 1/4) to give 1.43 g of product as a yellow solid. Yield 80%. 1H NMR (400 MHz, DMSO-d6) δ 8.23 (d, J = 8.3 Hz, 1H), 7.85 (d, J = 15.5 Hz, 1H), 7.80–7.76 (m, 2H), 7.67 (d, J = 15.4 Hz, 1H), 7.30 (s, 1H), 7.10 (t, J = 7.7 Hz, 1H), 7.05 (dt, J = 7.5, 1.1 Hz, 1H), 6.98 (t, J = 1.9 Hz, 1H), 6.67 (ddd, J = 7.8, 2.2, 1.2 Hz, 1H), 5.21 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 193.5, 164.4, 162.9, 161.9, 149.7, 147.0, 146.5, 135.5, 135.4, 131.9, 130.0, 129.8, 129.7, 121.1, 120.1, 118.1, 117.5, 116.6, 116.3, 115.8, 114.6; HR-ESI-MS calcd for C21H16FNO2 [M + H]+ 334.1165; found 334.1233.
4.5. Synthesis of compound 6
To a mixture of compound 5 (1.0 g, 3.0 mmol) and NaOH (1.2 g, 30.0 mmol) in methanol/water mixed solvents (Vmethanol/Vwater = 4/1) was slowly added 30% hydrogen peroxide dropwise (6 mL) at 0–4 °C. The reaction mixture was then stirred at the same temperature for 16 h. After completion monitored by TLC, the mixture was acidified to pH 6–7 with an aqueous solution of hydrochloric acid to allow the yellow solid to precipitate. The solid was collected by filtration and the obtained crude product was purified by flash chromatography on silica gel (Vethyl acetate/Vpetroleum ether = 1/3) to give 0.58 g of product as a yellow solid. Yield 70%. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (d, J = 8.4 Hz, 1H), 8.13 (d, J = 1.5 Hz, 1H), 8.07–8.03 (m, 2H), 7.91 (dd, J = 8.4, 1.6 Hz, 1H), 7.65 (t, J = 1.9 Hz, 1H), 7.59–7.56 (m, 1H), 7.54–7.49 (m, 2H), 7.36 (t, J = 7.9 Hz, 1H), 6.87 (ddd, J = 7.8, 2.2, 0.8 Hz, 1H), 5.46 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.3, 164.4, 161.9, 155.4, 149.2, 146.8, 144.5, 139.8, 135.2, 132.4, 129.9, 129.4, 126.0, 123.6, 120.6, 116.7, 116.4, 116.2, 116.1, 116.0, 113.4; HR-ESI-MS calcd for C21H14FNO3 [M + H]+ 348.0958; found 348.1026.
4.6. General procedures for the preparation of 7a–7z
To a solution of compound 6 (0.5 g, 1.5 mmol) and pyridine (0.25 mL) in acetone (20 mL) were added the corresponding benzoyl chloride analogs (2.6 mmol) dropwise in an ice bath. The reaction mixture was then warm to room temperature and stirred for another 12 h. After completion monitored by TLC, the reaction solution was diluted with distilled water and extracted with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. Removing the solvent under vacuum afforded the crude product which was purified by flash chromatography on silica gel (Vethyl acetate/Vpetroleum ether = 1 : 5) to give the corresponding target compounds 7a–7z.
4.6.1. 2-Fluoro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)benzamide (7a)
Light yellow solid. Yield 56%. Mp 224.6–226.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 9.70 (s, 1H), 8.57 (s, 1H), 8.13 (d, J = 8.4 Hz, 1H), 7.97–7.93 (m, 2H), 7.88 (d, J = 2.0 Hz, 1H), 7.87 (d, J = 3.5 Hz, 1H), 7.85 (d, J = 5.4 Hz, 1H), 7.74 (dd, J = 8.4, 1.7 Hz, 1H), 7.67 (td, J = 7.6, 1.7 Hz, 1H), 7.60–7.54 (m, 1H), 7.54–7.50 (m, 1H), 7.37–7.33 (m, 1H), 7.33 (t, J = 2.3 Hz, 1H), 7.31 (d, J = 2.4 Hz, 1H), 7.31–7.28 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 162.7, 159.4, 155.4, 145.6, 144.8, 140.0, 139.6, 135.2, 133.1, 132.3, 130.5, 130.0, 129.9, 129.5, 127.1, 126.1, 125.5, 125.4, 125.1, 123.8, 121.9, 121.9, 120.7, 119.7, 116.7, 116.5, 116.4; HR-ESI-MS calcd for C28H17F2NO4 [M + H]+ 470.1126; found 470.1196.
4.6.2. 4-Fluoro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)benzamide (7b)
Light yellow solid. Yield 58%. Mp 260.8–261.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.62 (d, J = 20.0 Hz, 1H), 9.72 (s, 1H), 8.64 (dt, J = 9.4, 1.8 Hz, 1H), 8.26 (dd, J = 8.8, 5.5 Hz, 1H), 8.19–8.12 (m, 2H), 8.12–8.05 (m, 2H), 7.94 (d, J = 3.7 Hz, 1H), 7.93 (s, 1H), 7.92–7.89 (m, 1H), 7.55 (td, J = 8.0, 3.9 Hz, 1H), 7.42–7.38 (m, 2H), 7.37 (s, 1H), 7.37–7.32 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 171.3, 165.1, 162.9, 156.6, 156.1, 155.5, 145.8, 144.7, 140.2, 139.9, 139.7, 133.8, 132.2, 131.1, 131.0, 130.0, 129.9, 122.1, 120.7, 116.7, 116.5, 116.5, 116.0, 116.0, 115.8, 115.8, 100.0; HR-ESI-MS calcd for C28H17F2NO4 [M + H]+ 470.1126; found 470.1181.
4.6.3. N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-4-methoxy-benzamide (7c)
Light yellow solid. Yield 65%. Mp 278.8–279.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 9.62 (s, 1H), 8.57 (s, 1H), 8.10 (d, J = 8.3 Hz, 1H), 7.97 (s, 1H), 7.95 (s, 1H), 7.91 (d, J = 7.3 Hz, 1H), 7.87 (s, 1H), 7.85 (s, 1H), 7.83 (s, 1H), 7.72 (d, J = 8.1 Hz, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.29 (t, J = 8.6 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 3.77 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 165.6, 162.5, 155.5, 145.9, 144.7, 143.4, 140.0, 139.9, 135.2, 132.1, 130.3, 130.0, 129.9, 129.2, 127.2, 126.1, 123.8, 123.6, 122.8, 120.7, 120.4, 116.70, 116.5, 116.3, 114.1, 56.0; HR-ESI-MS calcd for C29H20FNO5 [M + H]+ 482.1326; found 482.1382.
4.6.4. N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-4-methylbenzamide (7d)
Light yellow solid. Yield 60%. Mp 274.5–275.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 9.69 (s, 1H), 8.64 (s, 1H), 8.16 (d, J = 8.4 Hz, 1H), 8.00 (s, 1H), 7.98 (d, J = 8.2 Hz, 1H), 7.94 (s, 1H), 7.92 (s, 1H), 7.91–7.86 (m, 2H), 7.80–7.75 (m, 1H), 7.53 (t, J = 8.0 Hz, 1H), 7.37 (s, 1H), 7.35 (s, 2H), 7.33 (s, 1H), 2.38 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 166.1, 161.9, 155.5, 145.8, 144.7, 142.3, 139.9, 139.9, 135.1, 135.1, 132.3, 132.1, 130.0, 129.9, 129.5, 129.2, 128.3, 126.1, 123.7, 122.8, 120.7, 120.4, 116.7, 116.5, 116.2, 21.6; HR-ESI-MS calcd for C29H20FNO4 [M + H]+ 466.1376; found 466.1440.
4.6.5. 4-Chloro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl) phenyl)benzamide (7e)
Light yellow solid. Yield 70%. Mp 238.3–239.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 9.58 (s, 1H), 8.47 (s, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 1.8 Hz, 1H), 7.87 (s, 1H), 7.86 (s, 1H), 7.85 (d, J = 5.0 Hz, 1H), 7.77 (s, 1H), 7.76 (s, 1H), 7.74 (s, 1H), 7.64 (dd, J = 8.4, 1.5 Hz, 1H), 7.49–7.46 (m, 2H), 7.41 (t, J = 8.0 Hz, 1H), 7.21 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.3, 165.2, 162.0, 155.5, 145.7, 144.8, 139.9, 139.6, 137.1, 135.1, 133.9, 132.2, 130.2, 130.2, 130.0, 129.9, 129.4, 129.0, 126.1, 124.0, 123.8, 122.8, 120.7, 120.5, 116.7, 116.5, 116.2; HR-ESI-MS calcd for C28H17ClFNO4 [M + H]+ 486.0830; found 486.0888.
4.6.6. 2,4-Difluoro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)benzamide (7f)
Light yellow solid. Yield 69%. Mp 244.4–245.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.73 (d, J = 17.7 Hz, 1H), 9.73 (s, 1H), 8.58 (s, 1H), 8.15 (dd, J = 8.4, 1.9 Hz, 1H), 7.99 (s, 1H), 7.98–7.96 (m, 1H), 7.92–7.90 (m, 1H), 7.89 (s, 1H), 7.88 (s, 1H), 7.82–7.78 (m, 1H), 7.78–7.75 (m, 1H), 7.55 (t, J = 8.1 Hz, 1H), 7.46–7.40 (m, 1H), 7.35 (t, J = 8.8 Hz, 2H), 7.23 (td, J = 8.9, 2.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 162.7, 162.0, 155.5, 145.6, 144.8, 140.0, 139.5, 135.2, 135.1, 133.6, 132.3, 130.0, 129.9, 129.5, 126.9, 126.1, 123.9, 123.8, 121.9, 120.7, 119.8, 116.7, 116.5, 116.2, 112.5, 112.2, 105.2; HR-ESI-MS calcd for C28H16F3NO4 [M + H]+ 488.1031; found 488.1104.
4.6.7. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-2-methoxybenzamide (7g)
Light yellow solid. Yield 69%. Mp 218.9–219.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 9.68 (s, 1H), 8.62 (s, 1H), 8.16 (d, J = 8.4 Hz, 1H), 7.99 (d, J = 1.5 Hz, 1H), 7.96 (dt, J = 8.0, 1.3 Hz, 1H), 7.92–7.90 (m, 1H), 7.89 (d, J = 3.4 Hz, 1H), 7.89–7.86 (m, 1H), 7.78 (dd, J = 8.4, 1.7 Hz, 1H), 7.64 (dd, J = 7.5, 1.7 Hz, 1H), 7.54 (d, J = 8.2 Hz, 1H), 7.38–7.33 (m, 2H), 7.21–7.16 (m, 1H), 7.07 (td, J = 7.5, 0.9 Hz, 1H), 3.91 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 165.4, 164.4, 161.9, 157.0, 155.4, 145.8, 144.7, 139.9, 139.8, 135.1, 132.6, 132.3, 130.1, 130.0, 129.9, 129.3, 126.1, 125.5, 123.8, 122.0, 121.0, 120.7, 119.7, 116.7, 116.5, 116.3, 112.5, 56.4; HR-ESI-MS calcd for C29H20FNO5 [M + H]+ 482.1326; found 488.1379.
4.6.8. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-2-methylbenzamide (7h)
Light yellow solid. Yield 61%. Mp 235.3–237.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 9.68 (s, 1H), 8.63 (s, 1H), 8.16 (d, J = 8.4 Hz, 1H), 7.98 (d, J = 1.7 Hz, 1H), 7.97–7.94 (m, 1H), 7.92 (d, J = 5.9 Hz, 1H), 7.91 (d, J = 3.2 Hz, 1H), 7.90–7.87 (m, 1H), 7.78 (dd, J = 8.4, 1.7 Hz, 1H), 7.56–7.48 (m, 2H), 7.42–7.37 (m, 1H), 7.36 (d, J = 7.0 Hz, 1H), 7.35–7.33 (m, 1H), 7.31 (d, J = 7.4 Hz, 2H), 2.40 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 168.6, 161.9, 155.5, 145.8, 144.7, 140.0, 139.9, 137.6, 135.8, 135.1, 132.2, 131.1, 130.2, 130.0, 129.9, 129.4, 127.8, 126.2, 126.1, 123.8, 123.6, 121.8, 120.7, 119.6, 116.7, 116.5, 116.2, 19.9; HR-ESI-MS calcd for C29H20FNO4 [M + H]+ 466.1376; found 466.1435.
4.6.9. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-4-(trifluoromethyl)benzamide (7i)
Light yellow solid. Yield 67%. Mp 272.3–273.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 9.73 (s, 1H), 8.70 (s, 1H), 8.25 (d, J = 8.2 Hz, 2H), 8.17 (d, J = 8.4 Hz, 1H), 8.02 (dd, J = 7.7, 1.2 Hz, 2H), 8.00–7.96 (m, 1H), 7.94 (d, J = 7.2 Hz, 1H), 7.92 (s, 1H), 7.91 (s, 1H), 7.90–7.87 (m, 1H), 7.78 (dd, J = 8.4, 1.7 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.41–7.32 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 165.1, 161.9, 155.5, 145.7, 144.7, 139.9, 139.5, 135.1, 135.1, 132.2, 130.0, 129.3, 126.1, 125.9, 125.9, 125.8, 124.0, 123.7, 123.1, 122.8, 120.7, 120.5, 116.7, 116.5, 116.2, 31.2; HR-ESI-MS calcd for C29H17F4NO4 [M + H]+ 520.1094; found 520.1152.
4.6.10. 4-Bromo-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)benzamide (7j)
Light yellow solid. Yield 69%. Mp 268.2–268.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.55 (d, J = 8.9 Hz, 1H), 9.73 (s, 1H), 8.62 (s, 1H), 8.17 (d, J = 8.4 Hz, 1H), 8.04–7.98 (m, 2H), 7.98–7.95 (m, 2H), 7.95–7.93 (m, 1H), 7.92 (d, J = 3.2 Hz, 1H), 7.90 (d, J = 5.3 Hz, 1H), 7.81–7.78 (m, 1H), 7.78 (s, 1H), 7.77 (d, J = 1.9 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.36 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 165.2, 162.0, 155.5, 145.7, 144.7, 139.9, 139.6, 135.2, 134.3, 132.2, 132.0, 130.4, 130.0, 129.9, 129.3, 126.1, 126.0, 123.9, 123.7, 122.7, 120.7, 120.5, 116.7, 116.5, 116.2; HR-ESI-MS calcd for C28H17BrFNO4 [M + H]+ 530.0325; found 530.0381. [M + 2 + H]+ 532.0325; found 532.0359.
4.6.11. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-3-methoxybenzamide (7k)
Light yellow solid. Yield 71%. Mp 243.6–244.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 9.70 (s, 1H), 8.70 (t, J = 1.9 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 8.01 (ddd, J = 8.0, 1.7, 1.0 Hz, 1H), 7.97 (ddd, J = 8.2, 2.1, 1.0 Hz, 1H), 7.94–7.90 (m, 2H), 7.79 (dd, J = 8.4, 1.7 Hz, 1H), 7.64 (ddd, J = 7.7, 1.5, 0.9 Hz, 1H), 7.60 (dd, J = 2.5, 1.6 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 7.39–7.34 (m, 2H), 7.18 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 3.87 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 165.9, 162.0, 159.7, 155.5, 145.8, 144.7, 139.9, 139.8, 136.6, 135.2, 135.1, 132.2, 130.1, 130.0, 129.9, 126.1, 123.8, 123.8, 122.9, 120.7, 120.6, 118.1, 116.7, 116.5, 116.3, 113.5, 55.9; HR-ESI-MS calcd for C29H20FNO5 [M + H]+ 482.1326; found 482.1385.
4.6.12. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-3-methylbenzamide (7l)
Yellow solid. Yield 63%. Mp 239.5–240.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 9.67 (s, 1H), 8.62 (t, J = 1.9 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.02 (s, 1H), 8.01–8.00 (m, 1H), 7.98 (s, 1H), 7.95 (dt, J = 7.9, 1.0 Hz, 1H), 7.91 (dd, J = 7.4, 2.0 Hz, 1H), 7.88 (dd, J = 8.8, 5.4 Hz, 2H), 7.81 (s, 1H), 7.75 (dd, J = 8.4, 1.7 Hz, 1H), 7.51 (t, J = 8.0 Hz, 1H), 7.37 (d, J = 1.0 Hz, 1H), 7.32 (t, J = 8.8 Hz, 2H), 2.37 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.25, 166.34, 161.95, 155.49, 146.15, 145.82, 144.73, 142.71, 139.91, 138.22, 135.22, 135.13, 132.78, 132.17, 130.02, 129.94, 129.25, 128.82, 128.79, 127.63, 126.12, 125.48, 123.74, 120.71, 120.44, 116.70, 116.49, 116.27, 21.51; HR-ESI-MS calcd for C29H20FNO4 [M + H]+ 466.1376; found 466.1435.
4.6.13. 3-Fluoro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)benzamide (7m)
Light yellow solid. Yield 64%. Mp 242.2–243.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 9.72 (s, 1H), 8.65 (t, J = 1.7 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.05–8.00 (m, 2H), 7.97–7.94 (m, 1H), 7.93 (d, J = 5.5 Hz, 1H), 7.91 (s, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.87–7.83 (m, 1H), 7.79 (dd, J = 8.4, 1.3 Hz, 1H), 7.65–7.59 (m, 1H), 7.57 (t, J = 7.8 Hz, 1H), 7.51–7.44 (m, 1H), 7.37 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 162.4, 155.5, 146.2, 145.7, 144.7, 142.7, 139.9, 139.6, 137.5, 135.2, 132.2, 131.1, 130.0, 129.9, 129.3, 127.6, 127.6, 126.1, 124.6, 124.0, 122.8, 120.7, 120.6, 116.7, 116.5, 115.3, 115.1; HR-ESI-MS calcd for C28H17F2NO4 [M + H]+ 470.1126; found 470.1185.
4.6.14. 4-Ethyl-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)benzamide (7n)
Light yellow solid. Yield 73%. Mp 257.6–258.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 9.69 (s, 1H), 8.66 (t, J = 1.9 Hz, 1H), 8.17 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 1.6 Hz, 1H), 8.00–7.97 (m, 1H), 7.97 (s, 1H), 7.97–7.95 (m, 1H), 7.94 (dt, J = 2.2, 1.0 Hz, 1H), 7.93–7.88 (m, 2H), 7.78 (dd, J = 8.4, 1.6 Hz, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.38 (s, 1H), 7.37 (s, 1H), 7.36 (d, J = 3.3 Hz, 1H), 7.33 (d, J = 2.0 Hz, 1H), 2.69 (q, J = 7.6 Hz, 2H), 1.21 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 166.1, 161.9, 155.5, 148.4, 145.8, 144.7, 140.0, 139.9, 135.2, 135.1, 132.7, 132.2, 130.0, 129.9, 129.2, 128.4, 128.3, 126.1, 123.7, 122.7, 120.7, 120.4, 116.7, 116.5, 116.2, 28.6, 15.9; HR-ESI-MS calcd for C30H22FNO4 [M + H]+ 480.1533; found 480.1595.
4.6.15. 4-Bromo-2-fluoro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phe-nyl)benzamide (7o)
Yellow solid. Yield 61%. Mp 243.8–244.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 9.72 (s, 1H), 8.61–8.46 (m, 1H), 8.16 (dd, J = 8.4, 2.5 Hz, 1H), 8.02–7.99 (m, 1H), 7.99–7.97 (m, 1H), 7.93–7.91 (m, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.89 (d, J = 2.2 Hz, 1H), 7.80–7.73 (m, 2H), 7.70–7.65 (m, 1H), 7.58 (d, J = 1.9 Hz, 1H), 7.57–7.56 (m, 1H), 7.38–7.33 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 171.0, 162.7, 162.0, 145.6, 144.8, 140.0, 139.4, 135.1, 132.4, 132.1, 130.1, 130.0, 129.9, 129.5, 128.3, 126.1, 124.8, 123.9, 123.8, 121.9, 120.7, 120.2, 120.0, 119.8, 116.7, 116.5, 116.2; HR-ESI-MS calcd for C28H16BrF2NO4 [M + H]+ 548.0231; found 548.0297; [M + 2 + H]+ 550.0244; found 550.0275.
4.6.16. 4-fluoro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-3-meth-yl benzamide (7p)
Light yellow solid. Yield 66%. Mp 219.9–221.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 9.70 (s, 1H), 8.61 (s, 1H), 8.16 (dd, J = 8.3, 4.9 Hz, 1H), 8.00 (s, 1H), 7.96 (d, J = 1.9 Hz, 1H), 7.92 (s, 1H), 7.91 (s, 1H), 7.89 (s, 1H), 7.89 (s, 1H), 7.78 (d, J = 7.5 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.36 (t, J = 8.7 Hz, 2H), 7.33–7.27 (m, 1H), 2.32 (d, J = 1.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 178.0, 171.8, 170.0, 166.7, 160.2, 158.7, 150.5, 149.5, 144.8, 144.7, 144.6, 139.9, 137.0, 136.12136.1, 134.8, 134.7, 134.0, 129.8, 129.6, 128.5, 125.5, 121.5, 121.2, 120.4, 120.2, 19.5; HR-ESI-MS calcd for C29H19F2 NO4 [M + H]+ 484.1282; found 484.1350.
4.6.17. 2-Fluoro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-4-meth-oxybenzamide (7q)
Light yellow solid. Yield 67%. Mp 235.2–237.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 9.69 (s, 1H), 8.58 (t, J = 1.8 Hz, 1H), 8.16 (dd, J = 8.4, 1.8 Hz, 1H), 7.98 (d, J = 1.8 Hz, 1H), 7.98–7.94 (m, 1H), 7.93–7.90 (m, 1H), 7.89 (d, J = 1.6 Hz, 1H), 7.88 (d, J = 2.2 Hz, 1H), 7.77 (dd, J = 8.4, 1.7 Hz, 1H), 7.68 (t, J = 8.6 Hz, 1H), 7.53 (t, J = 8.0 Hz, 1H), 7.37–7.32 (m, 2H), 6.96 (dd, J = 12.5, 2.4 Hz, 1H), 6.90 (dd, J = 8.6, 2.5 Hz, 1H), 3.84 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 163.2, 162.5, 160.9, 155.5, 145.7, 144.7, 139.9, 139.7, 135.2, 132.3, 131.8, 131.8, 130.0, 129.9, 129.4, 126.1, 123.7, 120.7, 119.8, 117.2, 117.1, 116.7, 116.5, 116.2, 111.1, 102.5, 102.3, 56.5; HR-ESI-MS calcd for C29H19F2NO5 [M + H]+ 500.1231; found 500.1302.
4.6.18. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-3,5-dimethoxy-benzamide (7r)
Light yellow solid. Yield 72%. Mp 222.1–224.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 9.69 (s, 1H), 8.67–8.63 (m, 1H), 8.16 (dd, J = 8.4, 3.9 Hz, 1H), 8.01 (d, J = 1.6 Hz, 1H), 8.00–7.97 (m, 1H), 7.92 (d, J = 3.5 Hz, 1H), 7.91 (s, 1H), 7.90–7.88 (m, 1H), 7.78 (dd, J = 8.4, 1.7 Hz, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.38–7.32 (m, 2H), 7.18 (d, J = 2.3 Hz, 2H), 6.71 (t, J = 2.2 Hz, 1H), 3.82 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 178.0, 170.5, 166.7, 165.7, 160.2, 150.6, 149.5, 144.6, 144.5, 142.0, 139.9, 136.9, 134.8, 134.7, 134.0, 130.9, 128.7, 128.6, 127.7, 125.5, 121.4, 121.2, 121.0, 111.0, 108.8, 60.8; HR-ESI-MS calcd for C30H22FNO6 [M + H]+ 512.1431; found 512.1502.
4.6.19. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-3,5-dimethylbenzamide (7s)
Light yellow solid. Yield 69%. Mp 272.3–273.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 9.69 (s, 1H), 8.64 (t, J = 1.8 Hz, 1H), 8.17 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 1.5 Hz, 1H), 8.00–7.93 (m, 2H), 7.93–7.90 (m, 1H), 7.89 (dd, J = 5.2, 3.0 Hz, 1H), 7.78 (dd, J = 8.4, 1.6 Hz, 1H), 7.63 (s, 2H), 7.54 (t, J = 8.0 Hz, 1H), 7.36 (t, J = 8.8 Hz, 2H), 7.22 (s, 1H), 2.36 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 173.20, 166.41, 161.90, 155.43, 145.79, 144.69, 139.89, 139.83, 138.03, 138.03, 135.19, 135.12, 135.08, 133.42, 132.11, 129.96, 129.87, 129.18, 126.06, 125.94, 125.94, 123.72, 122.61, 120.65, 120.33, 116.64, 116.42, 116.17, 21.35, 21.35; HR-ESI-MS calcd for C30H24FNO4 [M + H]+ 480.1533; found 480.1604.
4.6.20. 6-Chloro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)nicotinamide (7t)
Yellow solid. Yield 75%. Mp 248.5–249.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 9.73 (s, 1H), 9.03 (d, J = 2.3 Hz, 1H), 8.67 (s, 1H), 8.46 (dd, J = 8.3, 2.5 Hz, 1H), 8.17 (d, J = 8.3 Hz, 1H), 8.03 (s, 1H), 8.01 (s, 1H), 7.96 (d, J = 8.2 Hz, 1H), 7.91 (dd, J = 8.4, 5.4 Hz, 2H), 7.78 (d, J = 8.1 Hz, 1H), 7.72 (d, J = 8.3 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.36 (t, J = 8.7 Hz, 2H);13C NMR (101 MHz, DMSO-d6) δ 173.2, 163.6, 161.9, 155.4, 153.3, 150.0, 145.5, 144.7, 139.9, 139.7, 139.3, 135.1, 132.2, 130.2, 129.9, 129.8, 129.3, 126.0, 124.6, 124.0, 123.7, 122.6, 120.6, 120.4, 116.6, 116.4, 116.2; HR-ESI-MS calcd for C27H16ClFN2O4 [M + H]+ 487.0783; found 487.0853.
4.6.21. 3,5-Difluoro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)benzamide (7u)
Light yellow solid. Yield 73%. Mp 246.4–247.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 9.71 (s, 1H), 8.65 (t, J = 1.6 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 8.02 (s, 1H), 8.00 (s, 1H), 7.96–7.92 (m, 1H), 7.90 (dd, J = 8.6, 5.4 Hz, 2H), 7.79 (d, J = 2.0 Hz, 1H), 7.77 (s, 1H), 7.76 (s, 1H), 7.58–7.54 (m, 1H), 7.51 (dt, J = 9.1, 2.3 Hz, 1H), 7.35 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 164.0, 163.5, 161.5, 155.4, 145.6, 144.7, 139.9, 139.3, 138.6, 135.1, 132.2, 130.0, 129.9, 129.3, 127.1, 126.0, 124.1, 123.7, 122.8, 120.6, 120.6, 116.6, 116.4, 116.2, 111.9, 111.6. HR-ESI-MS calcd for C28H16F3NO4 [M + H]+ 488.1031; found 488.1104.
4.6.22. 2,4-Dichloro-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)py-rimidine-5-carboxamide (7v)
Light yellow solid. Yield 72%. Mp 243.8–245.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H), 9.79 (s, 1H), 9.13 (s, 1H), 8.56 (t, J = 1.8 Hz, 1H), 8.16 (d, J = 8.4 Hz, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.99 (d, J = 1.5 Hz, 1H), 7.94–7.92 (m, 1H), 7.91–7.90 (m, 1H), 7.90–7.87 (m, 1H), 7.78 (dd, J = 8.4, 1.6 Hz, 1H), 7.60 (t, J = 8.1 Hz, 1H), 7.36 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 163.6, 161.9, 155.4, 153.3, 150.0, 145.5, 144.7, 139.9, 139.7, 139.3, 135.1, 132.2, 130.2, 129.9, 129.8, 129.3, 126.0, 124.6, 123.7, 122.7, 120.6, 120.4, 116.6, 116.4, 116.2. HR-ESI-MS calcd for C26H14Cl2FN3O4 [M + H]+522.3134; found 522.4187.
4.6.23. 4-Cyano-N-(3-(7-(4-fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl) phenyl)benzamide (7w)
Yellow solid. Yield 69%. Mp 261.6–262.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 9.71 (s, 1H), 8.68 (s, 1H), 8.20 (d, J = 8.3 Hz, 2H), 8.16 (d, J = 8.4 Hz, 1H), 8.04 (s, 1H), 8.02 (s, 1H), 7.97 (q, J = 2.4, 1.6 Hz, 2H), 7.91 (dd, J = 8.7, 5.4 Hz, 2H), 7.79 (dd, J = 8.4, 1.4 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.36 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 164.8, 155.4, 145.6, 144.7, 143.6, 139.9, 139.5, 139.2, 136.9, 133.2, 132.9, 132.2, 130.4, 130.1, 129.9, 129.8, 129.6, 129.3, 129.2, 128.2, 126.1, 120.7, 120.6, 116.7, 116.5, 114.4; HR-ESI-MS calcd for C29H17FN2O4 [M + H]+ 477.1172; found 477.3286.
4.6.24. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl) -4-nitrobenzamide (7x)
Light yellow solid. Yield 70%. Mp 262.3–263.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.96 (s, 1H), 9.71 (s, 1H), 8.68 (t, J = 1.7 Hz, 1H), 8.36 (d, J = 8.9 Hz, 2H), 8.28 (d, J = 8.9 Hz, 2H), 8.16 (d, J = 8.4 Hz, 1H), 8.02 (d, J = 9.1 Hz, 2H), 8.00–7.95 (m, 1H), 7.90 (dd, J = 8.8, 5.4 Hz, 2H), 7.77 (dd, J = 8.4, 1.5 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.35 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 164.51, 161.90, 155.42, 149.67, 145.57, 144.67, 140.80, 139.90, 139.40, 135.11, 132.21, 129.94, 129.90, 129.90, 129.85, 129.28, 126.04, 124.12, 123.97, 123.97, 123.69, 122.75, 120.64, 120.54, 116.63, 116.42, 116.21; HR-ESI-MS calcd for C28H17FN2O6 [M + H]+ 497.1076; found 497.1976.
4.6.25. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-[1,1′-biphenyl]-4-carboxamide (7y)
Yellow solid. Yield 63%. Mp 254.8–256.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 9.71 (s, 1H), 8.72 (t, J = 1.7 Hz, 1H), 8.19 (d, J = 8.5 Hz, 1H), 8.18 (s, 1H), 8.15 (s, 1H), 8.04 (d, J = 1.4 Hz, 1H), 8.01 (dt, J = 8.2, 2.1 Hz, 2H), 7.96–7.91 (m, 2H), 7.86 (d, J = 8.4 Hz, 2H), 7.83–7.78 (m, 2H), 7.77 (s, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.52 (t, J = 7.6 Hz, 2H), 7.44 (dd, J = 8.3, 6.3 Hz, 1H), 7.38 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 165.8, 161.9, 155.5, 145.8, 144.7, 143.7, 139.9, 139.8, 139.6, 135.1, 135.1, 133.9, 132.1, 130.0, 129.9, 129.6, 129.2, 129.0, 128.7, 127.4, 127.0, 126.1, 123.8, 122.7, 120.7, 120.4, 116.7, 116.4, 116.2; HR-ESI-MS calcd for C34H22FNO4 [M + H]+ 528.1533; found 528.1605.
4.6.26. N-(3-(7-(4-Fluorophenyl)-3-hydroxy-4-oxo-4H-chromen-2-yl)phenyl)-4-propylbenzamide (7z)
Light yellow solid. Yield 64%. Mp 245.3–246.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 9.69 (s, 1H), 8.69 (t, J = 1.9 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 8.01–7.98 (m, 2H), 7.98–7.95 (m, 2H), 7.95–7.90 (m, 2H), 7.80 (dd, J = 8.4, 1.7 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.39 (d, J = 2.1 Hz, 1H), 7.37 (d, J = 1.9 Hz, 1H), 7.36 (s, 1H), 7.35 (d, J = 1.3 Hz, 1H), 2.68–2.61 (m, 2H), 1.64 (h, J = 7.3 Hz, 2H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.2, 166.1, 161.9, 155.4, 146.7, 145.8, 144.7, 140.0, 139.8, 135.1, 132.6, 132.1, 130.0, 129.9, 129.2, 128.8, 128.35, 126.1, 123.7, 122.7, 120.7, 120.1, 116.6, 116.4, 116.2, 37.5, 24.37, 14.1; HR-ESI-MS calcd for C31H24FNO4 [M + H]+ 494.1689; found 494.1760.
4.7. In vitro cytotoxicity against seven different cancer cell lines
The human cancer cell lines HCC1937, A549, HeLa, and GTL-16 were cultured in RPMI medium 1640 with 10% FBS, 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin, whereas MDA-MB-231, MCF-7, and Hep G2 were cultured in high sugar DMEM medium with 10% FBS, 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin. All the cells were incubated in an incubator at 37 °C with 5% CO2, and all the experiments were performed while the cells were in the logarithmic growth phase. Cells were inoculated in 96-well plates at a density of 4 × 103–6 × 103 cells per well and incubated for 24 h. After the cells were fully plastered, 100 μL of serum-free medium with various concentrations of compounds was added and incubated at 37 °C for 72 h. A cell viability assay (CCK8 assay) was performed after compound administration. LY294002 and 5-fluorouracil were selected as positive controls, and both blank and zeroed groups were set up. A 10% CCK8 solution was added to each well in a final volume of 110 μL and incubated at 37 °C for 2 h. The absorbance (optical density, OD) at 450 nm and 630 nm was measured on a multifunctional microplate analyzer (Mithras2 LB943, Berthold, Germany). The concentration of a compound that inhibited cell growth by 50% (IC50) was processed by SPSS 19.0 statistical software. The inhibition rate was calculated by the formula [1 − (experimental group − background group)/(blank group − zero-adjusted group)] × 100%. Five replicate wells were set up for each experimental group, and the results of three independent experiments were expressed as mean ± SD.
4.8. Colony-formation assay
The MDA-MB-231 cells were inoculated at 800 cells per well in 12-well plates with 3 replicate wells per group and incubated in an incubator at 37 °C with 5% CO2 for 5–7 days. When 30–50 cell clusters were formed, the drug was added and incubation was continued for 24 h, 48 h, and 72 h. The old medium was removed, the cells were washed twice with pre-cooled PBS, and fixed with 4% paraformaldehyde for 30 minutes. The fixative was removed, and a 0.5% crystal violet staining solution was added. The cells were then gently washed with water and air-dried. The clones were photographed using a gel imager and counted by ImageJ software.
4.9. Hoechst staining assay
The MDA-MB-231 cells were inoculated in 6-well plates at 2 × 105 –3 × 105 per well and incubated overnight in an incubator at 37 °C with 5% CO2. After the cells were plastered, the drug was added and incubated for 48 h. The old solution was removed, the cells were washed twice with PBS, and methanol was added to fix the cells for 30 minutes. The cells were washed twice with PBS to remove the methanol, 500 μL of Hoechst 33258 staining solution was added to each well and the cells were stained for 30 minutes at room temperature with protection from light. Cells were assessed morphologically using a fluorescence microscope.
4.10. Cell cycle assay
The MDA-MB-231 cells were treated with different concentrations of compound 7t for 48 h, digested with trypsin, and collected from the walled cells. After centrifugation, the supernatant was discarded, the cells were washed 2–3 times with PBS, and collected so that each sample contained approximately 1 × 106 cells per mL. 500 μL of pre-cooled 70% ethanol was added and fixed overnight at 4 °C. A centrifuge was used to remove the ethanol, the cells were resuspended with PBS, and the cell suspension was filtered through a 200-mesh nylon membrane. A CytoFLEX flow cytometer (Beckman Coulter, USA) was used to measure the cell cycle, and the excitation wavelength of red fluorescence was measured at 488 nm.
4.11. Annexin V-FITC/PI double staining assay for detection of apoptosis
The MDA-MB-231 cells were treated with different concentrations of compound 7t for 48 h, digested with trypsin, and collected from the walled cells. After centrifugation, the supernatant was discarded, the cells were washed 2–3 times with PBS, and collected so that each sample contained approximately 5 × 105 cells per mL. The cells were resuspended with 500 μL of binding buffer, 5 μL of Annexin V-FITC and 5 μL of PI were added, and the reaction was carried out at room temperature and protected from light for 15 minutes. Then the cells were detected by a CytoFLEX flow cytometer (Beckman Coulter, USA) with an excitation wavelength of Ex = 488 nm and an emission wavelength of Em = 530 nm.
4.12. Wound healing migration assay
The MDA-MB-231 cells were inoculated and cultured in 6-well plates at 4 × 105 cells per well overnight. After the cells were adhered to the wall, they were scribed with a sterilized 200 μL gun tip, washed with PBS solution to remove the scribed cells, and incubated with the normal medium or compound 7t. Photographs were taken with a phase contrast microscope at time points of 0, 12 h, 24 h, and 48 h of incubation.
4.13. Transwell invasion assays
The MDA-MB-231 cells in the logarithmic growth phase were counted by trypsin digestion with a counting plate, and single cell suspensions were prepared by resuspension in serum-free medium at a density of 5 × 105 cells per well. A total of 600 μL of complete medium was added to the lower chamber of the Transwell chamber. A cell suspension of 200 μL per well was uniformly added dropwise to the upper chamber, along with different concentrations of compound 7t, and placed in an incubator at 37 °C for 48 h. The upper chamber medium was removed and allowed to soak in methanol for 2 h for fixation. Non-invasive cells in the upper chamber were removed with a sterile swab containing PBS, then inverted and air-dried. Cells in the small chamber were stained with 500 μL of 0.5% crystalline violet for 30 minutes, then washed with PBS and air-dried, photographed with a microscope, and counted using ImageJ software.
4.14. Western blotting analysis
The MDA-MB-231 cells were incubated in 6-well plates with different concentrations of compound 7t at 37 °C for 12 h, trypsin digested, washed with PBS, and centrifuged to obtain cell samples. A protein lysis solution (10× RIPA + 1× PMSF) was added at 0 °C and the cells were fully lysed for 30 min. The samples were placed at 14 000 × g and centrifuged at 4 °C for 10 minutes, and the supernatant was the extracted protein. The protein concentration was determined using the BCA protein assay kit. Protein samples (40 μg) were processed by SDS-PAGE gel electrophoresis and then transferred to PVDF membranes. The PVDF membrane strips containing the target proteins were blocked with 5% skim milk powder and then incubated with the primary antibody at 4 °C overnight. The membrane strips were removed and washed three times with TBST. The strips are incubated with the secondary antibody for 2 h at room temperature. The strips were removed and washed three times with TBST for five minutes each time. The developing solution (1 : 1 ratio of liquid A to liquid B, which can be used directly) was evenly dropped onto the protein side of the strips and incubated for one minute at room temperature. After incubation, the strips were transferred to a gel imager for exposure imaging.
4.15. Molecular modeling
Surflex-dock in Sybyl 2.1 was used for molecular docking. The structures of small molecules were built in the Sybyl package with standard bond lengths and angles and minimized using the Powell method. The termination of the gradient, force field of Tripos and charge of Gasteiger–Hückel were applied for the minimization process. The AMBER FF99 force field was applied for the protein after extracting all the non-polar water and addition of hydrogen atoms. The standard docking procedures were performed with parameters set in default. The reliability of the docking strategy was verified by re-docking the original ligand into the crystal structure. After docking, residues within 5 Å of the inhibitors were identified and the conformations that appeared the most frequently and possessed the lowest binding free energy were selected for further analysis. Binding modes and interactions were analyzed in Pymol.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support from High-level University Construction Fund of Guangdong Province (grant number: 06-410-2107203, 06-410-2107252, 06-410-2107284); Medical Scientific Research Foundation of Guangdong Province of China (grant number: A2022006); National College Student Innovation and Entrepreneurship Training Program (grant number: 202110570033).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00148a
References
- Sung H. Ferlay J. Siegel R. L. Laversanne M. Soerjomataram I. Jemal A. Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca-Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Miller K. D. Nogueira L. Mariotto A. B. Rowland J. H. Yabroff K. R. Alfano C. M. Jemal A. Kramer J. L. Siegel R. L. Cancer treatment and survivorship statistics, 2019. Ca-Cancer J. Clin. 2019;69:363–385. doi: 10.3322/caac.21565. doi: 10.3322/caac.21565. [DOI] [PubMed] [Google Scholar]
- Fukui Y. Saltiel A. Hanafusa H. Phosphatidylinositol-3 kinase is activated in v-src, v-yes, and v-fps transformed chicken embryo fibroblasts. Oncogene. 1991;6:407–411. doi: 10.1016/0027-5107(91)90045-P. [DOI] [PubMed] [Google Scholar]
- Manning B. D. Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M. Bode A. M. Dong Z. Lee M. AKT as a therapeutic target for cancer. Cancer Res. 2019;79:1019–1031. doi: 10.1158/0008-5472.CAN-18-2738. doi: 10.1158/0008-5472.CAN-18-2738. [DOI] [PubMed] [Google Scholar]
- Bilanges B. Posor Y. Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019;20:515–534. doi: 10.1038/s41580-019-0129-z. doi: 10.1038/s41580-019-0129-z. [DOI] [PubMed] [Google Scholar]
- Ellis H. Ma C. X. PI3K inhibitors in breast cancer therapy. Curr. Oncol. Rep. 2019;21:110. doi: 10.1007/s11912-019-0846-7. doi: 10.1007/s11912-019-0846-7. [DOI] [PubMed] [Google Scholar]
- Burke J. E. Williams R. L. Synergy in activating class I PI3Ks. Trends Biochem. Sci. 2015;40:88–100. doi: 10.1016/j.tibs.2014.12.003. doi: 10.1016/j.tibs.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Zhang L. Li Y. Wang Q. Chen Z. Li X. Wu Z. Hu C. Liao D. Zhang W. Chen Z.-S. The PI3K subunits, P110α and P110β are potential targets for overcoming P-gp and BCRP-mediated MDR in cancer. Mol. Cancer. 2020;19:10. doi: 10.1186/s12943-019-1112-1. doi: 10.1186/s12943-019-1112-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D. A. Chiu H. Hopkins B. D. Bagrodia S. Cantley L. C. Abraham R. T. The PI3K pathway in human disease. Cell. 2017;170:605–635. doi: 10.1016/j.cell.2017.07.029. doi: 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J. Yao D. Liu W. Wang N. Lv H. Zhang G. Ji M. Xu L. He N. Shi B. Hou P. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer. 2012;12:50–61. doi: 10.1186/1471-2407-12-50. doi: 10.1186/1471-2407-12-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. A. Jain V. K. Rizwanullah M. Ahmad J. Jain K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: a review on drug discovery and future. Drug Discovery Today. 2019;24:2181–2191. doi: 10.1016/j.drudis.2019.09.001. doi: 10.1016/j.drudis.2019.09.001. [DOI] [PubMed] [Google Scholar]
- Lee Y. R. Chen M. Pandolfi P. P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 2018;19:547–562. doi: 10.1038/s41580-018-0015-0. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- Martorana F. Motta G. Pavone G. Motta L. Stella S. Vitale S. R. Manzella L. Vigneri P. AKT inhibitors: new weapons in the fight against breast cancer? Front. Pharmacol. 2021;12:662232. doi: 10.3389/fphar.2021.662232. doi: 10.3389/fphar.2021.662232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. Han X. Ou D. Liu T. Li Z. Jiang G. Liu J. Zhang J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020;104:575–587. doi: 10.1007/s00253-019-10257-8. doi: 10.1007/s00253-019-10257-8. [DOI] [PubMed] [Google Scholar]
- Robbins H. L. Hague A. The PI3K/Akt pathway in tumors of endocrine tissues. Front. Endocrinol. 2015;6:188. doi: 10.3389/fendo.2015.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe L. M. Yuzugullu H. Zhao J. J. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy B. Lu Y. Teng K. Y. Nuovo G. Li X. Shapiro C. L. Majumder S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012;72:5048–5059. doi: 10.1158/0008-5472.can-12-1248. doi: 10.1158/0008-5472.can-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. Zhang H. Xu C. Zhao X. Clinicopathological research and expression of PTEN/PI3K/Akt signaling pathway in non-small cell lung cancer. Zhongguo Feiai Zazhi. 2009;12:889–892. doi: 10.3779/j.issn.1009-3419.2009.08.11. [DOI] [PubMed] [Google Scholar]
- Zhu J. Hou T. Mao X. Discovery of selective phosphatidylinositol 3-kinase inhibitors to treat hematological malignancies. Drug Discovery Today. 2015;20:988–994. doi: 10.1016/j.drudis.2015.03.009. doi: 10.1016/j.drudis.2015.03.009. [DOI] [PubMed] [Google Scholar]
- Yadav R. R. Guru S. K. Joshi P. Mahajan G. Mintoo M. J. Kumar V. Bharate S. S. Mondhe D. M. Vishwakarma R. A. Bhushan S. Bharate S. B. 6-Aryl substituted 4-(4-cyanomethyl) phenylamino quinazolines as a new class of isoformselective PI3K-alpha inhibitors. Eur. J. Med. Chem. 2016;122:731–743. doi: 10.1016/j.ejmech.2016.07.006. doi: 10.1016/j.ejmech.2016.07.006. [DOI] [PubMed] [Google Scholar]
- Zhu W. Chen C. Sun C. Xu S. Wu C. Lei F. Xia H. Tu Q. Zheng P. Design, synthesis and docking studies of novel thienopyrimidine derivatives bearing chromone moiety as mTOR/PI3Kalpha inhibitors. Eur. J. Med. Chem. 2015;93:64–73. doi: 10.1016/j.ejmech.2015.01.061. doi: 10.1016/j.ejmech.2015.01.061. [DOI] [PubMed] [Google Scholar]
- Shao T. Wang J. Chen J. G. Wang X. M. Li H. Li Y. P. Li Y. Yang G. D. Mei Q. B. Zhang S. Q. Discovery of 2-methoxy-3-phenylsulfonamino-5-(quinazolin-6-yl or quinolin-6-yl)benzamides as novel PI3K inhibitors and anticancer agents by bioisostere. Eur. J. Med. Chem. 2014;75:96–105. doi: 10.1016/j.ejmech.2014.01.053. doi: 10.1016/j.ejmech.2014.01.053. [DOI] [PubMed] [Google Scholar]
- Zhu L. Luo Q. Bi J. Ding J. Ge S. Chen F. Galangin inhibits growth of human head and neck squamous carcinoma cells in vitro and in vivo. Chem.-Biol. Interact. 2014;224:149–156. doi: 10.1016/j.cbi.2014.10.027. doi: 10.1016/j.cbi.2014.10.027. [DOI] [PubMed] [Google Scholar]
- Abramson V. G. Troxel A. B. Feldman M. Mies C. Wang Y. Sherman L. McNally S. Diehl A. Demichele A. Cyclin D1 b in human breast carcinoma and co expression with cyclin D1 a is associated with poor outcome. Anticancer Res. 2010;30:1279–1285. [PMC free article] [PubMed] [Google Scholar]
- Cao J. Wang H. Chen F. Fang J. Xu A. Xi W. Zhang S. Wu G. Wang Z. Galangin inhibits cell invasion by supressing the epithelial-mesenchymal transition and inducing apoptosis in renal cell carainoma. Mol. Med. Rep. 2016;13:4238–4244. doi: 10.3892/mmr.2016.5042. doi: 10.3892/mmr.2016.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W. Zhang H. Yuan C. Galangin enhanced antitumor effects of apatinib in gastric cancer SGC-7901 cells via PI3K/Akt and p38-MAPK signaling pathway. Tianjin Yiyao. 2019;47:1020–1025. doi: 10.11958/20190272. [DOI] [Google Scholar]
- Walker E. H. Pacold M. E. Perisic O. Stephens L. Hawkins P. T. Wymann M. P. Williams R. L. Structural Determinants of Phosphoinositide 3-Kinase Inhibition by Wortmannin, LY294002, Quercetin, Myricetin and Staurosporine. Mol. Cell. 2000;6:909–919. doi: 10.1016/S1097-2765(05)00089-4. doi: 10.1016/S1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- Ikezoe T. Nishioka C. Bandobashi K. Yang Y. Kuwayama Y. Adachi Y. Takeuchi T. Koeffler H. P. Taguchi H. Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk. Res. 2007;31:673–682. doi: 10.1016/j.leukres.2006.08.001. doi: 10.1016/j.leukres.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Abdallah M. E. El-Readi M. Z. Althubiti M. A. Almaimani R. A. Ismail A. M. Idris S. Refaat B. Almalki W. H. Babakr A. T. Mukhtar M. H. Abdalla A. N. Idris O. F. Tamoxifen and the PI3K Inhibitor: LY294002 Synergistically Induce Apoptosis and Cell Cycle Arrest in Breast Cancer MCF-7 Cells. Molecules. 2020;25:3355. doi: 10.3390/molecules25153355. doi: 10.3390/molecules25153355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z. Pinson J. A. Mountford S. J. Orive S. Schoenwaelder S. M. Shackleford D. Powell A. Nelson E. M. Hamilton J. R. Jackson S. P. Jennings I. G. Thompson P. E. Discovery and antiplatelet activity of a selective PI3K beta inhibitor (MIPS-9922) Eur. J. Med. Chem. 2016;122:339–351. doi: 10.1016/j.ejmech.2016.06.010. doi: 10.1016/j.ejmech.2016.06.010. [DOI] [PubMed] [Google Scholar]
- Heffron T. P. Wei B. Olivero A. Staben S. T. Tsui V. Do S. Dotson J. Folkes A. J. Goldsmith P. Goldsmith R. Gunzner J. Lesnick J. Lewis C. Mathieu S. Nonomiya J. Shuttleworth S. Sutherlin D. P. Wan N. C. Wang S. Wiesmann C. Zhu B. Y. Rational design of phosphoinositide 3-kinase alpha inhibitors that exhibit selectivity over the phosphoinositide 3-kinase beta isoform. J. Med. Chem. 2011;54:7815–7833. doi: 10.1021/jm2007084. doi: 10.1021/jm2007084. [DOI] [PubMed] [Google Scholar]
- Chaussade C. Rewcastle G. W. Kendall J. D. Denny W. A. Cho K. Grønning L. M. Chong M. L. Anagnostou S. H. Jackson S. P. Daniele N. Shepherd P. R. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem. J. 2007;404:449–458. doi: 10.1042/BJ20070003. doi: 10.1042/BJ20070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abazeed M. Adams D. Hurov K. Tamayo P. Creighton C. Sonkin D. Giacomelli A. Schreiber S. Hammerman P. Meyerson M. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. 2013;73:6289–6298. doi: 10.1016/j.ijrobp.2013.06.356. doi: 10.1016/j.ijrobp.2013.06.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massacesi C. Di Tomaso E. Urban P. Germa C. Quadt C. Trandafifir L. Aimone P. Fretault N. Dharan B. Tavorath R. Hirawat S. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. OncoTargets Ther. 2016;9:203–210. doi: 10.2147/OTT.S89967. doi: 10.2147/OTT.S89967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G. Ma Y. Liang X. Xie G. Luo Y. Zha D. Wang S. Yu L. Zheng X. Wu W. Zhang C. Design, synthesis and antitumor evaluation of novel 5-methylpyrazolo[1,5-a]pyrimidine derivatives as potential c-Met inhibitors. Bioorg. Chem. 2020;104:104356. doi: 10.1016/j.bioorg.2020.104356. doi: 10.1016/j.bioorg.2020.104356. [DOI] [PubMed] [Google Scholar]
- Luo Y. Wu W. Zha D. Zhou W. Wang C. Huang J. Chen S. Yu L. Li Y. Huang Q. Zhang J. Zhang C. Synthesis and biological evaluation of novel ligustrazine-chalcone derivatives as potential anti-triple negative breast cancer agents. Bioorg. Med. Chem. Lett. 2021;47:128230. doi: 10.1016/j.bmcl.2021.128230. doi: 10.1016/j.bmcl.2021.128230. [DOI] [PubMed] [Google Scholar]
- Manikanta M. Vikas S. Pramila C. Suvarna V. A critical review on anticancer mechanisms of natural flavonoid puerarin. Anti-Cancer Agents Med. Chem. 2020;20:678–686. doi: 10.2174/1871520620666200227091811. doi: 10.2174/1871520620666200227091811. [DOI] [PubMed] [Google Scholar]
- Li D. Y. Du G. Gong X. P. Guo J. Zhang J. Chen C. Xue Y. Zhu H. Zhang Y. Hyperattenins L and M, two new polyprenylated acylphloro -glucinols with adamantyl and homoadamantyl core structures from Hypericum attenuatum. Fitoterapia. 2018;125:130–134. doi: 10.1016/j.fitote.2017.12.020. doi: 10.1016/j.fitote.2017.12.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.