

Abstract
The current COVID-19 pandemic caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) created a global health crisis. The ability of vaccines to protect immunocompromised individuals and from emerging new strains are major concerns. Hence antiviral drugs against SARS-CoV-2 are essential. The SARS-CoV-2 main protease Mpro is vital for replication and an important target for antivirals. Using CMap analysis and docking studies, withaferin A (wifA) and withanone (win), two natural products from the medicinal herb Withania somnifera (ashwagandha), were identified as promising candidates that can covalently inhibit the viral protease Mpro. Cell culture, enzymatic, LC-MS/MS, computational, and equilibrium dialysis based assays were performed. DFT calculations indicated that wifA and win can form stable adducts with thiols. The cytotoxicity of Mprowas significantly reduced by wifA and win. Both wifA and win were found to irreversibly inhibit 0.5 μM Mpro with IC50 values of 0.54 and 1.8 μM, respectively. LC-MS/MS analysis revealed covalent adduct formation with wifA at cysteines 145 and 300 of Mpro. The natural products wifA and win can irreversibly inhibit the SARS-CoV-2 main protease Mpro. Based on the work presented here we propose that both wifA and win have the potential to be safely used as preventative and therapeutic interventions for COVID-19.
The ongoing COVID-19 pandemic caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has created a global crisis of epic proportions since December 2019. (1) To date, more than 517 million people have been infected with SARS-CoV-2 and over 6 million have succumbed to it. Although the majority of patients infected with SARS-CoV-2 are either asymptomatic or mildly symptomatic, about 4% of individuals suffer from severe symptoms with dysregulated immune response, reduced lung function, and/or multiorgan failure. (2–4) SARS-CoV-2 has spread worldwide, with new strains emerging.
Coronaviruses are enveloped positive-strand RNA viruses classified into four genera, α-, β-, γ, and δ. (5,6) The β-coronavirus class comprises notable human pathogens including SARS-CoV, Middle East respiratory syndrome (MERS) coronavirus, and SARS-CoV-2. (7,8) SARS-CoV and SARS-CoV-2 are closely related, with 79.6% sequence similarity, and have the largest viral genome, ∼30 kb size. (9) The genome of SARS-CoV-2 is fairly complex, encoding a total of 29 proteins, of which 16 are nonstructural, 4 structural, and 8 accessory proteins. The structural proteins are spike protein (S), membrane protein (M), envelope protein (E), and nucleocapsid protein (N). (9,10) The viral RNA is translated into two long polypeptides, pp1a and pp1ab, which undergo extensive proteolytic cleavage to generate functional proteins critical for viral replication. (9,11) Two proteases in SARS-CoV-2 perform the proteolytic cleavage, a papain-like cysteine (Cys) protease (PLpro) and a chymotrypsin-like Cys protease (3CLpro, also known as Main Protease, Mpro). (12,13) Mpro plays a pivotal role in the viral life cycle. (6) Active Mpro exists as a dimer, with each monomer consisting of three domains and a noncanonical catalytic dyad consisting of His41 (histidine) and Cys145 in its active site. (6) A water molecule with at least three hydrogen-bonding interactions with surrounding residues in the active site substitutes for the third catalytic residue.
Because of immunocompromised individuals and emerging new strains of SARS-CoV-2 with enhanced virulence, infectivity, and morbidity (delta, omicron, and XE), there is a desperate need for antiviral drugs. (6) Although multiple vaccines have already reached the clinic, concerns regarding the time frame of their efficacies and their abilities to protect from evolving new strains remain. Moreover, there are numerous coronaviruses in nature that have the potential to mutate and jump species to infect humans. (14) Hence, a broad-spectrum antiviral that will be effective against coronaviruses at large is needed.
The ubiquitous coronavirus protein Mpro has no human homologue, thus making it a lucrative target for antiviral drug development. (11) Using various approaches including high-throughput and in silico screening, drug repurposing, and drug design, a plethora of potential reversible and irreversible inhibitors (including aldehydes, Michael acceptors (MAs), and epoxides) of Mpro have been identified. (9,11–13,15–18) Boceprevir, GC-376, and calpain inhibitors II and XII are a few examples of known Mpro inhibitors. (19,20) The Pfizer Mpro inhibitor nirmatrelvir is FDA approved and currently in the clinics. However, it is marketed as Paxlovid in combination with the 3A4 inhibitor ritonavir, which significantly limits the use of other drugs during its administration.
The health emergency resulting from the SARS-CoV-2 outbreak has prompted us to explore drug candidates that are time-tested for safety in humans. Natural products, medicinal extracts, and their active ingredients, which have been safely used for years, are rational substitutes. (21,22)Withania somnifera (also known as ashwagandha, Indian ginseng, or winter cherry) is a medicinal plant that has been safely used for thousands of years in India and its neighboring countries as remedies against various diseases. (23,24) WifA and win (Figure 1C) are two major components of W. somnifera. (25,26) Structurally, both wifA and win are C22 steroidal lactones with a withanolide-type A skeleton. Both compounds contain three electrophilic functional groups, two Michael acceptors (one α,β-unsaturated ketone and another methyl-substituted α,β-unsaturated lactone) and an epoxide functional group (Figure 1C). The active site Cys145 thiol of Mpro is a good nucleophile (at physiological pH ∼50% of the thiol exists in its deprotonated thiolate form) (16) that can react with both Michael acceptors and epoxides. (6,27) Therefore, we speculated that wifA and win may form a covalent bond with the active site Cys145 thiol of the viral protease Mpro and irreversibly inhibit it. Moreover, various studies indicated that both wifA and win can inhibit viral replication. (28–31) Clinical trials with ashwagandha are currently being pursued. Herein, using experimental approaches, we show that wifA and win, two major metabolites of W. somnifera, are covalent inhibitors of Mpro. Both wifA and win have the potential for immediate safe clinical use for treating COVID-19. (32)
Figure 1.
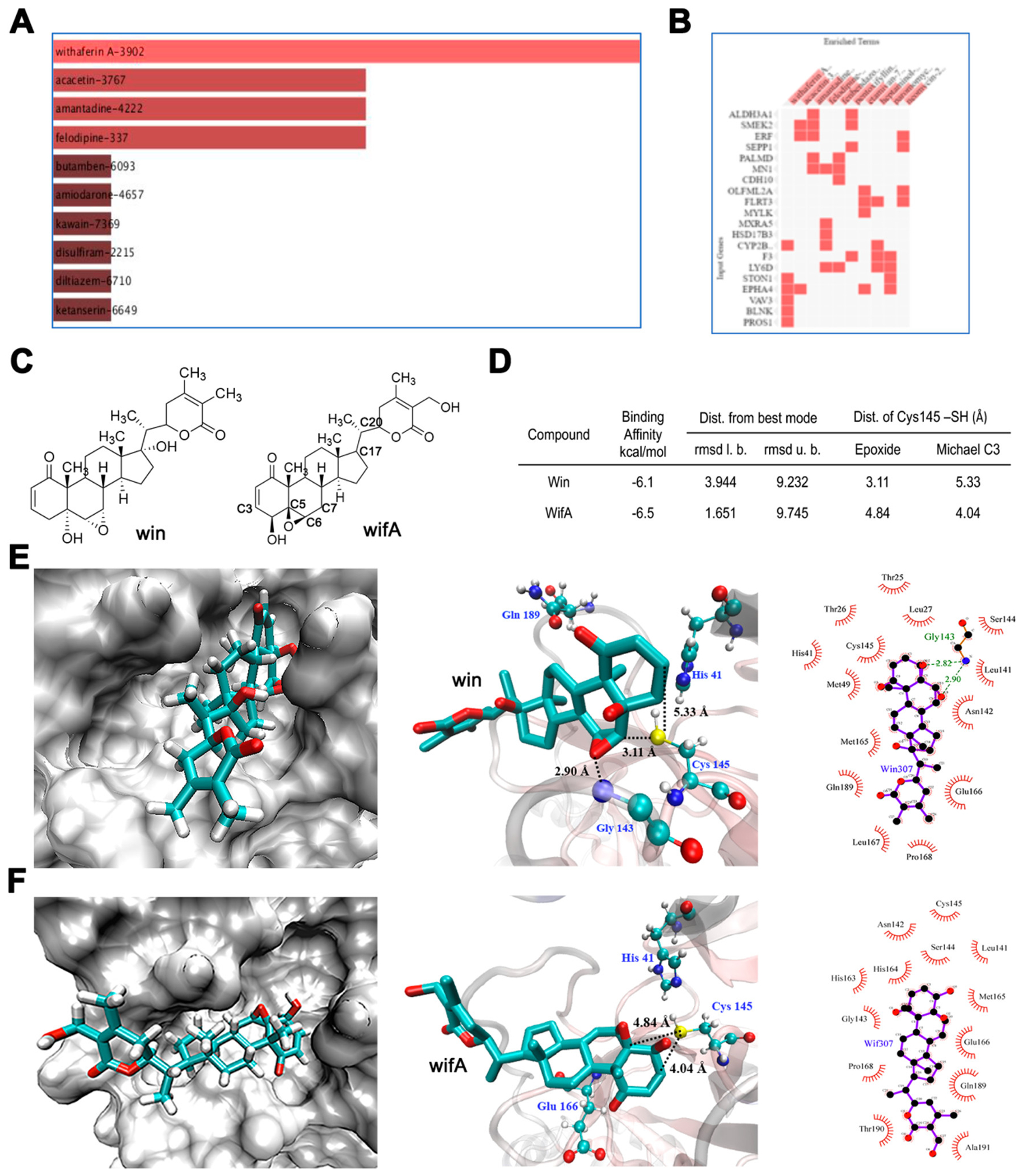
Identification of wifA and win as potential drug candidate against SARS-CoV-2. (A) Top drug candidates revealed by CMap analysis with genes uniquely upregulated in SARS-CoV-2-infected lung cells. (B) Genes associated with each drugs. In A and B all candidates shown are statistically significant at p < 0.05. (C) Structures of wifA and win. (D) Noncovalent binding of wifA and win at the active site of the SARS-CoV-2 cysteine protease Mpro using in silico docking studies. Binding parameters of wifA and win at the active site of Mpro. (E) Representative binding modes of win at the active site of Mpro in three different representations showing key distances and the interactions in Mpro-win. (F) Representative binding modes of wifA at the active site of Mpro in three different representations showing key distances and interactions in Mpro-wifA.
Results and Discussion
Identification of WifA as a Top Drug Candidate against SARS-CoV-2
The easy accessibility of publicly available large genomic and transcriptomic data sets from diseased samples has rendered computational drug repurposing fairly quick and adaptable. The Connectivity Map (CMap) project launched by the Broad Institute curates a comprehensive catalogue of gene expression signatures from cells exposed to genetic and pharmacological perturbagens. Such signatures can be used to predict previously unrecognized connections between drugs, genes, and diseases, serving as powerful computational tool for drug-repurposing studies. (33) We performed CMap analyses of differentially expressed genes (DEGs) from SARS-CoV-2- and SARS-CoV-infected human lung epithelial cells with Enrichr: interactive and collaborative HTML5 gene set enrichment analysis web tool. (34) These DEGs were originally derived by Jha et al. from the publicly available GSE147507 and GSE17400 data sets for SARS-CoV-2 and SASR-CoV, respectively. (35) We compared 76 and 383 upregulated genes out of 221 and 1315 unique SARS-CoV-2- and SARS-CoV-specific DEGs (Supplemental Tables S1 and S2) with DEGs downregulated in cancer cell lines treated with over 13 000 drugs. The analysis revealed two lists of drugs with wifA as the topmost candidate against SARS-CoV-2 and genistein against SARS-CoV, based on the cutoff of adjusted p value < 0.05 (Supplemental Tables S3 and S4; Figure 1A and B). None of the top candidates are overlapped, indicating that they have the potential for reversing the gene signatures unique to SARs-CoV-2- and CoV-infected lung cells (Supplemental Tables S3 and S4). A literature search further revealed wifA, the SARS-CoV-2-specific top hit, identified as a prospective drug candidate by two other groups who used comparable computation-based repurposing tools. (36,37) Six genes associated with wifA were absent in the SARS-CoV-specific upregulated gene list (Supplemental Tables S3, S4 and Figure 1B), indicating that wifA is a drug candidate for selective treatment of SARS-CoV-2 infection.
WifA and Win Bind to the Active Site of Mpro with the Electrophilic Functional Groups in Close Proximity to Cys145 Thiol
WifA also contains two reactive functional groups, a Michael acceptor and an epoxide, both of which can react with the active site thiolate. To evaluate the potential of wifA and its analog win to irreversibly inhibit Mpro through covalent adduct formation with the active site Cys145, we assessed the relative orientation of the electrophilic Michael acceptor and epoxide groups of wifA and win with respect to the Cys145 thiolate. Accordingly, docking studies were performed on Mpro with both wifA and win. We found multiple binding modes where wifA and win strongly bind to the active site of Mpro in a way that favors covalent adduct formation with Cys145 (Figure 1E and F, only the most proximally placed thiol to the electrophiles is represented). In the case of win, the thiol of Cys145 is within 3.11 Å of the epoxide group of win and 5.33 Å of the Michael acceptor. The binding orientation also favors backside attack on the epoxide carbon. Similarly, for wifA, the distance of the Cys145 thiol is 4.84 and 4.04 Å from the epoxide and the Michael acceptor groups, respectively. The binding affinities are also strong (≥6 kcal/mol). The results indicate the potential for both wifA and win to bind and form covalent adducts with the Cys145 thiol of Mpro.
WifA and Win Form Stable Adducts with Cysteine (Cys) and GSH
Although wifA is known to form adducts with Cys thiols in certain proteins, (38,39) binding orientation and pKa may have a significant role. Hence, to determine if the active site Cys145 of Mpro can react with wifA or win to form covalent adducts, we reacted wifA or win with Cys under physiological conditions. The logic is that if win or wifA forms covalent adducts with Cys thiol in aqueous solution, it will definitely react with Cys145 thiolate to form covalent adducts once bound in the right orientation at the active site of Mpro. Although Cys primarily exists in the thiol form under physiological conditions, 50% of the active site Cys145 of Mpro exist in the reactive thiolate form, which should facilitate adduct formation with electrophiles. (16) Accordingly, we treated win or wifA with Cys for 6 h in aqueous pH 7.4 buffer at 37 °C. Using LC-tandem MS, we observed Cys covalent adducts with wifA and win. Extracted ion chromatogram (EIC) analysis of the reaction of win with Cys showed two peaks eluting between 10.9 and 11.2 min and having m/z 592.2967 and 592.2972 ([M + H]+, calculated 592.2939) (Figures 2A and S1, Supporting Information). Collision-induced dissociation (CID) of the two peaks produced a similar fragmentation pattern. One interesting fragment observed is m/z384, which corresponds to the cleavage between C17 and C20 (Figure S2, Supporting Information). The m/z 384 fragment clearly indicates that Cys is covalently linked either to the epoxide or to the Michael acceptor functional group. In the case of wifA, reaction with Cys generated two peaks at 10.5 and 10.9 min having m/z 592.2987 and 592.2976 ([M + H]+, calculated 592.2939) (Figures 2A and S1, Supporting Information). Similar to win, a fragment corresponding to the cleavage between C17 and C20 with m/z 402 was observed (Figure S2, Supporting Information).
Figure 2.
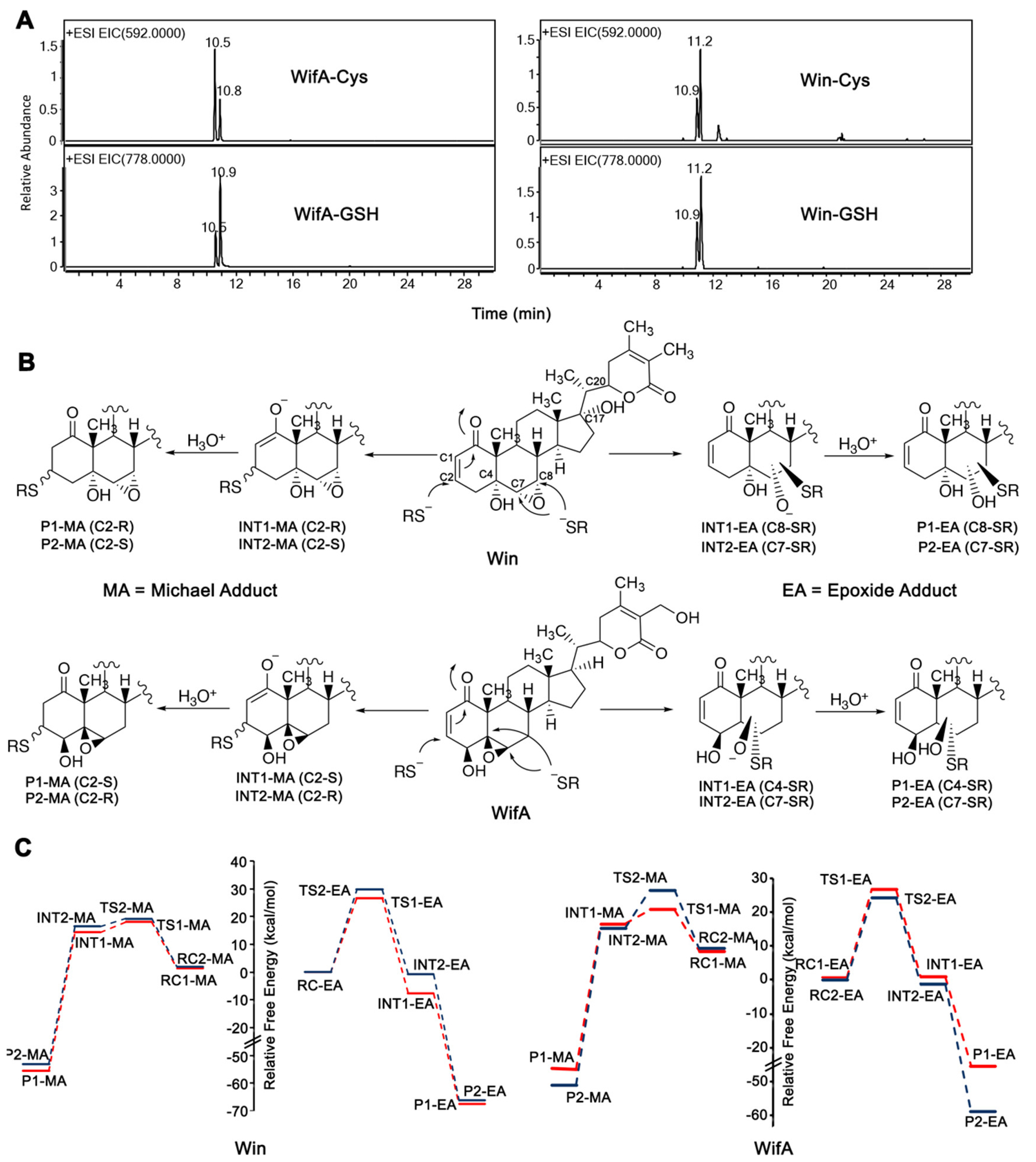
Proposed mechanism and calculated energy diagrams of the reaction of wifA and win with thiolate anion. (A) LC-MS chromatogram of Cys and GSH adducts of wifA and win. (B) Proposed reaction mechanism of wifA and win with the thiolate anion of Cys showing the formation of four possible covalent adducts. (C) Free energy profiles showing all four possible pathways for the reaction of wifA and win with the thiolate anion of Cys. Relative free energies were computed at the M06-2X/Def2-TZVP//M06-2X/6-31G* level of theory.
In theory, the reaction of win or wifA with Cys should produce four distinct adducts, two isomers resulting from an attack on the two carbon atoms of the epoxide group and two diastereomers resulting from an attack on the alkene of the Michael acceptor from the re or si face (Figure 2B). The fact that we obtained two adducts for wifA and win (Figure 2A) suggests that there is selectivity, unless multiple adducts are coeluting. In the case of the reaction of win/wifA with Cys145 of Mpro, though, selectivity is expected and should depend on the binding orientation of the electrophiles (proximity of the Cys145 thiolate and the electrophile) and the thermodynamic/kinetic feasibility of the reactions.
Glutathione (GSH) is a tripeptide (l-γ-glutamyl-l-cysteinyl-glycine) containing a thiol (SH) group and serves as an important part of the cellular defense system against electrophiles and free radicals. (40) Conjugation with GSH, often catalyzed by GSH transferase, renders toxic molecules and reactive intermediates inert and hydrophilic for rapid elimination. (41) Because both wifA and win contain electrophilic groups, conjugation with GSH may result in their detoxication. Accordingly, we treated win or wifA with the tripeptide GSH for 6 h in aqueous pH 7.4 buffer at 37 °C. Consistent with the Cys assays, the reaction of GSH with wifA resulted in two peaks eluting at 10.5 and 10.9 min and having m/z 778.3634 and 778.3633 ([M + H]+, calculated 778.3579) (Figures 2A and S1, Supporting Information). Similarly, when win was used, two peaks between 10.9 and 11.2 min having m/z 778.3538 and 778.3603 ([M + H]+, calculated 778.3579) (Figures 2A and S1 Supporting Information) were observed. These results indicate that the cellular detoxication system involving GSH can form conjugates with both wifA and win. Although GSH adduct formation might lower the efficacy of both wifA and win, it should be mentioned here that the host protein involved in the biosynthesis of GSH, glutamate-cysteine ligase, is a proposed substrate of Mpro. (42) Hence, the attenuated levels of GSH in the infected cells would allow wifA and win to inhibit Mpro, while the normal level of GSH in noninfected cells would play a protective role against any toxic effect of wifA and win. Additionally, depletion of GSH in SARS-CoV-2-infected cells may also occur due to excessive ROS production due to acute inflammation. (43) A role of GSH in regulating virus replication and cytotoxicity is not unprecedented. (44)
Thermodynamic Energy Calculations for the Reactions of Cys Thiolate with Win/WifA
To gain insights on the thermodynamic stability and kinetic viability of the adduct formation, density functional theory (DFT) calculations of the reactions of wifA and win with Cys thiolate anion were performed. Based on the proposed mechanism of Mpro, (45) the calculations were performed using a deprotonated Cys thiolate. The thermodynamic energy calculations provide theoretical support for the feasibility of the covalent adduct formation between Cys thiolate and win/wifA and shed light onto the product selectivity out of the possible four.
Our calculations indicate considerable thermodynamic stability for all four adducts shown in Figure 2; however, reaction at the epoxide carbons results in the largest change in free energy (ΔG, measured relative to the most stable reaction complex) for both wifA and win. Thus, the most stable adducts are P1-EA and P2-EA for win with ΔG = −67.5 and −68.5 kcal/mol, respectively, and P2-EA for wifA with ΔG = −58.9 kcal/mol. Thus, the DFT results along with the experimental observations indicate the marked stability of the covalent adducts of wifA and win with Cys and, hence, the potential of wifA and win to be irreversible inhibitors of Mpro.
Inhibition of Mpro-Mediated Cytotoxicity by WifA and Win
To determine if wifA and win can inhibit Mpro, we performed a modified version of a cell-based assay recently developed by Resnick et al. (46) In this assay a mammalian expression construct (pCMV3-2019-nCoV-3CLpro) containing the SARS-CoV-2 Mpro cDNA was transfected into A549 (lung) and HepG2 (liver) cell lines. Because the expression vector confers hygromycin resistance, cells were kept under a constant selection pressure of hygromycin. Following transfection, cells were exposed to the drug or vehicle control. Viral proteases like Mpro result in significant toxicity (46) when transfected in mammalian cells (and refs 7-13 therein). Inhibition of Mpro, therefore, should reverse this cytotoxicity and can be measured by comparing cell proliferation. Consistent with our expectation, when HepG2 and A549 cell lines were transfected with the pCMV3-2019-nCoV-3CLpro construct, notable reduction in cell number was observed compared to the nontransfected control (Figure 3A). The addition of win or wifA (5 μM) to these transfected cells caused a significant increase in cell number compared to the DMSO control (Figure 3A). The effect was particularly pronounced in lung A549 cells. A similar increase in cell number was also observed for the known cysteine protease inhibitor E64, which was used as a positive control. Of the three drugs used here, wifA was found to be most efficient in mitigating the cytotoxic effect of Mpro. This observation is significant because both win and wifA (5 μM) are themselves toxic to HepG2 and A549 cells, resulting in attenuated cell proliferation compared to no-drug control (Figure S3, Supporting Information). Hence, the data presented here suggest that both wifA and win can reverse the cytotoxic effect of Mpro, possibly through inhibition of its proteolytic activity.
Figure 3.
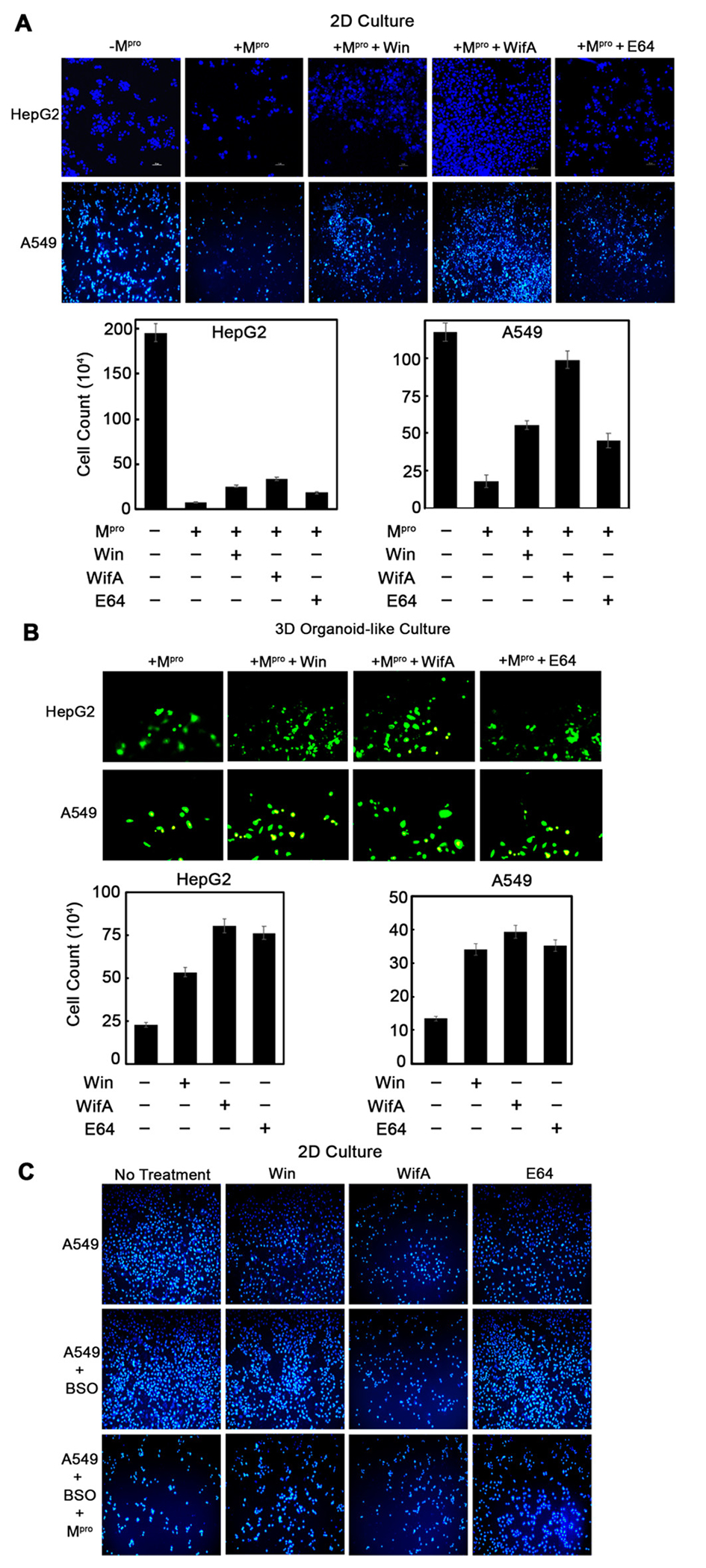
Cytotoxic effect of Mpro is mitigated by wifA and win. (A) 2-D assay showing inhibition of Mpro-mediated cytotoxicity in HepG2 and A549 cells treated with 5 μM win, wifA, or E64 (48 h treatment). Images were taken in either a fluorescent microscope (Leica Microsystems, Wetzlar, Germany) or a confocal microscope (Nikon Corporation, Tokyo, Japan) at 10–20× magnification. NucBlue Live ReadyProbes reagent (Hoechst 33342) stained the nucleus. The bottom panel is the quantification of the top panel microscopic images. (B) 3-D (organoid-like culture) assay confirming inhibition of Mpro-mediated cytotoxicity in HepG2 and A549 cell lines following exposure to win, wifA, and E64 (at above-mentioned conditions). Microscopic images of calcien AM-stained viable cells were taken at 10× for HepG2 and 20× for A549 magnification. The bottom panel indicates the quantitation of the top panel microscopic images. (C) Effect of buthionine sulfoximine (BSO) on cytotoxicity of Mpro-expressing A549 cells stained with NucBlue Live ReadyProbes reagent (Hoechst 33342). Images were taken at 10× magnification. The error bars represent standard error of three replicates.
A three-dimensional (3-D) system provides a cellular environment that is a better representation of the in vivo situation than growing cells in two-dimensional (2-D) monolayers. (47) Although animal models are widely used, there are many variables in their physiology, genetic makeup, and level of stress that prevent meaningful extrapolation of in vivo animal data to clinical settings. The agreement between data obtained in preclinical animal models and clinical trials is far from satisfactory, with an average rate of similarity barely reaching 8%. (48) For understanding the pharmacology and toxicology of drugs and small molecules, 3-D culture systems are gaining acceptance as an effective alternative to animal models. Accordingly, we repeated the above 2-D assays in 3-D organoid-like cultures of HepG2 and A549 cells. We observed a similar trend, providing additional evidence that wifA and win can mitigate the cytotoxic effect of Mpro, with wifA being more effective than win (Figure 3B).
Both wifA and win contain reactive electrophilic groups that can efficiently form adducts with GSH (Figure 2A), which may result in their detoxication. Therefore, we repeated the above experiment with A549 cells in the presence of buthionine sulfoximine (BSO), which reduces the intracellular GSH levels by inhibiting glutamate-cysteine ligase. (49) Reduction in GSH levels will decrease the GSH-mediated conjugation of win/wifA and, in turn, increase their half-life. Thus, the addition of BSO should in principle increase the win/wifA-mediated inhibition of Mpro, resulting in a further increase in proliferation/number of Mpro-expressing A549 cells. Under the conditions used here, BSO alone has a negligible effect on cell proliferation (Figure 3C). Mpro-transfected A549 cells showed a significant reduction in cell number. Addition of win or wifA resulted in a slight increase in the number of Mpro-expressing A549 cells compared to a no-drug control (Figure 3C). In the case of E64 a significant increase in cell number compared to a no-drug control was observed. Contrary to our expectation, addition of BSO resulted in a similar/reduced protection of Mpro-expressing A549 cells by win/wifA when compared to no BSO treatment. Possible explanations may be that (1) both wifA and win are cytotoxic to A549 cells under conditions of reduced GSH levels, (2) the GSH level is already low in Mpro-expressing cells (as proposed in the literature (42)), and (3) the GSH adducts of wifA and win can also inhibit Mpro. To test our third hypothesis, we performed docking studies with the win-/wifA-GSH adducts (described in a later section, Figure S6, Supporting Information).
WifA and Win Covalently Bind to Mpro
To determine if wifA and win bind to Mpro, we performed an equilibrium dialysis experiment. In this experiment a buffered solution of Mpro was dialyzed against a similar buffered solution of win or wifA for 16 h. When a drug binds to the target protein, its concentration in the dialysate (solution outside the dialysis chamber) decreases compared to the no-protein control. Consistent with our expectation, when Mpro was present in the dialyzer (dialysis chamber) an ∼18% and ∼14% decrease in the concentration of wifA and win in the dialysate was observed, respectively (Figure 4A). Although, apparently the change may seem small, it is significant if we consider the stoichiometry of drug in the dialysis solution (volume is 10 times more). We then analyzed the solution in the dialyzer. After denaturing the protein, the concentration of the drug inside the dialyzer was compared with that of the dialysate solution. The premise here is that, if the drug is noncovalently bound to the protein, denaturation should lead to its release into the solution, resulting in a significant increase in its concentration inside the bag. On the contrary, if covalently bound, the drug concentration should remain similar to that of the dialysate. We found that the win/wifA concentration remained similar after denaturation of the protein, indicating covalent binding to the protein (Figure 4A).
Figure 4.
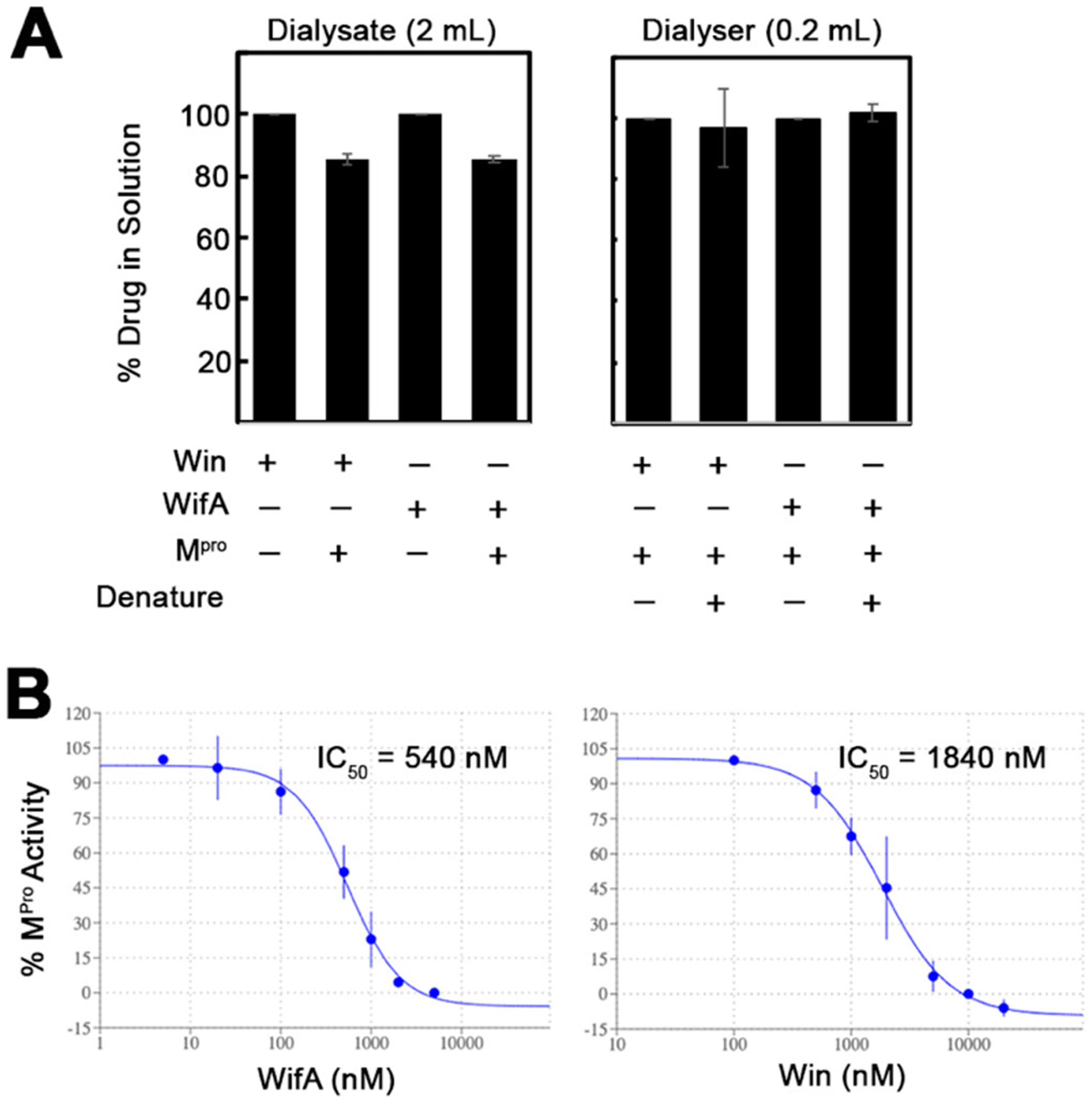
WifA and win directly bind and inhibit the protease activity of Mpro. (A) Equilibrium dialysis experiment to demonstrate the direct covalent interaction between Mpro and win or wifA. Mpro (30 μg, 200 μL) was dialyzed against a buffered solution of win or wifA (2 μM, 2 mL) for 16 h. The concentrations of drug in the dialysate were quantified by LC-MS (wifA) or LC-MSMS (win). In the case of the solution in the dialyzer, the proteins were denatured by heating, followed by LC-MS (wifA) or LC-MS/MS (win) quantitation of the drugs. (B) FRET-based kinetic assay demonstrating the percent inhibition of protease activity of the recombinant Mpro (0.5 μM) following preincubation with an increasing concentration of wifA or win. The error bars represent the standard deviation of three replicates.
Inhibition of the Protease Activity of Mpro by WifA and Win
To determine if wifA and win can inhibit Mpro, we performed a fluorescence resonance energy transfer (FRET)-based assay. (15)Purified Mpro (0.5 μM) was preincubated for 60 min with various concentrations of either of the drugs before adding the substrate DABCYL-KTSAVLQSGFRKME-EDANS, with DABCYL being the quencher and EDANS being the fluorophore. The nonfluorescent substrate becomes fluorescent upon cleavage by Mpro between Q and S, and the change in the fluorescence intensity was measured over time to follow the proteolytic activity of Mpro (Figure 4B). The IC50 for the inhibition of Mpro, calculated from the initial rate of proteolysis, was found to be 0.54 and 1.8 μM for wifA and win, respectively (Figure 4B). We then performed the same assay with a fixed concentration of wifA but varying time of incubation with Mpro (Figure S5, Supporting Information). When 0.5 μM Mpro was incubated with 5 μM wifA, complete inhibition was observed within 5 min. However, when 0.25 μM Mpro was incubated with 0.5 μM wifA, ∼80% inhibition of enzyme activity was observed after 30 min of incubation. Consistent with the cell-based experiments, the enzyme inhibition assay confirms that both wifA and win can efficiently inhibit Mpro.
Because wifA and win have been shown to react with thiols, dithiothreitol (DTT) or GSH reducing agents were not used in the reaction buffer. Nonspecific oxidation of the Cys thiols in Mpro due to the absence of DTT or GSH resulting in reduction of proteolytic activity is a concern in our assay. Hence, we also repeated the above-mentioned FRET-based assay with wifA in the presence of 5 mM GSH. The IC50 for the inhibition of Mpro in the presence of 5 mM GSH was found to be 0.63 μM (Figure S5, Supporting Information). This slight increase is expected considering that GSH can form covalent adducts with wifA.
WifA and Win Form Covalent Adducts with Mpro
To further confirm the covalent binding of wifA and win to the active site of Mpro, we treated Mpro with wifA, digested with chymotrypsin, and identified wifA-adducted chymotryptic peptides using LC-tandem MS. Because win undergoes significant fragmentation (multiple loss of water molecules) in the source, we decided to perform the LC-tandem MS assay with wifA only. Under the conditions used here, wifA does not show much fragmentation in the source and the protonated molecular ion is observed as the major peak. The extracted ion chromatogram of m/z 1297.6, which corresponds to the wifA-adducted NGSC145GSVGF peptide, resulted in a peak eluting at 10.9 min and having a mass of 1297.5983 (M + H)+ (calculated 1297.6023). Further analysis of the data also revealed adduct formation on the peptide DVVRQC300SGVTF (Figure 5). Of the 12 Cys residues in Mpro, five─at positions 22, 85, 145, 156, and 300─are solvent-exposed. Hence the alkylation of Cys145 and -300 of Mpro by wifA is not unusual. Covalent adduct formation at reactive Cys residues of proteins by wifA is not unprecedented. It has been shown to form adducts at Cys303 of α-tubulin, at Cys239 and Cys303 of β-tubulin, at Cys238 of vimentin, and at Cys294 of glial fibrillary acidic protein (GFAP). (38,39,50,51) While Cys145 is involved in catalysis, Cys300 has a role in the dimerization of Mpro, which is necessary for its catalytic activity. (52)
Figure 5.
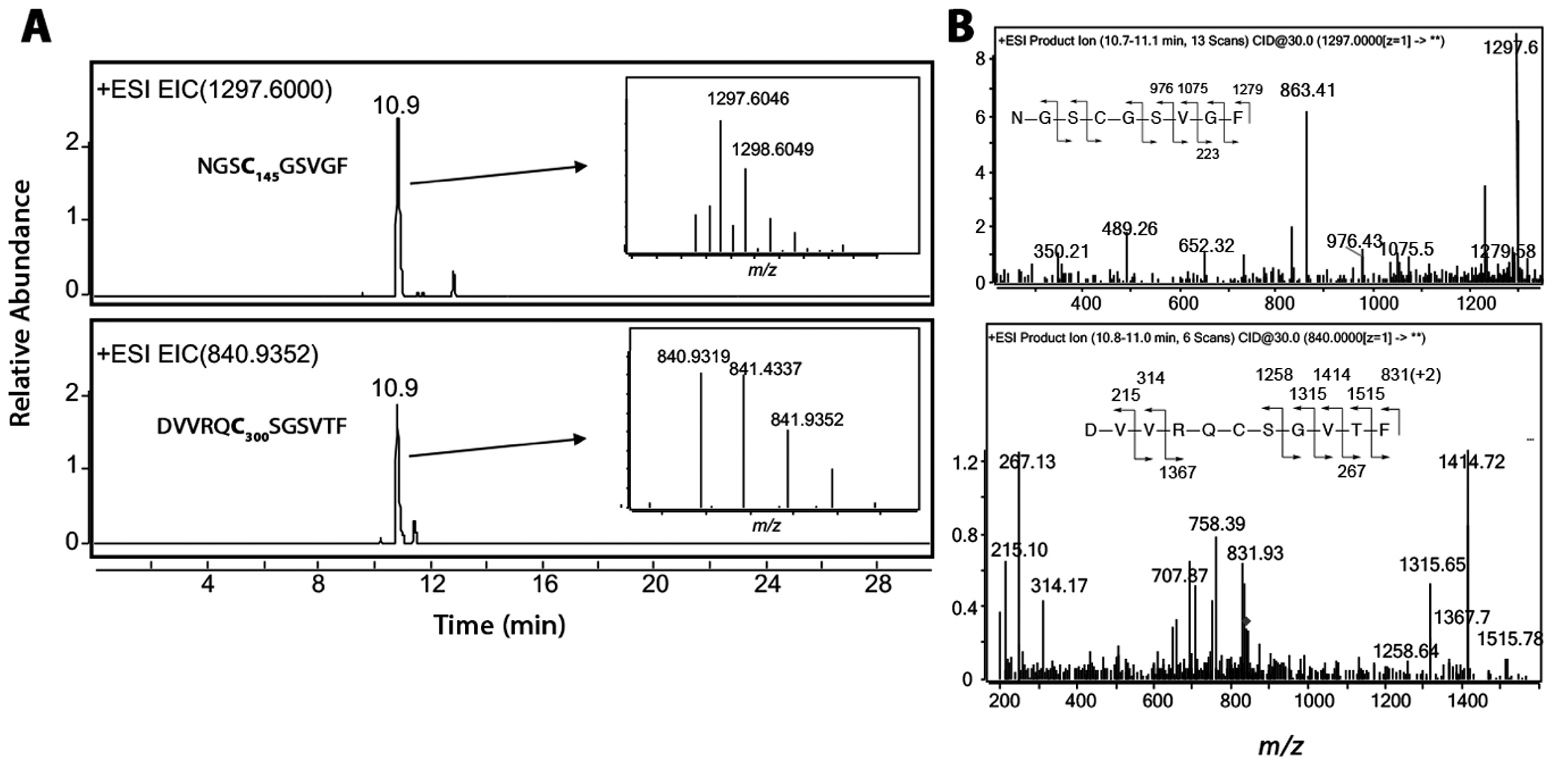
LC-HRMS and LC-MSMS detection and identification of wifA adducted chymotryptic peptides of Mpro. (A) Extracted ion chromatogram showing peaks corresponding to two different Cys-containing peptides of Mpro covalently adducted to wifA. The HRMS spectra are shown in the insets. (B) LC-MS/MS spectrum and fragmentation analysis of the two adducted peptides.
Safety Profiles of WifA and Win and the Potential of Win/WifA-GSH Adducts to Inhibit Mpro
Based on the data presented here it can be concluded that both wifA and win bind and form covalent adducts with reactive Cys thiols of Mpro. Although this property renders wifA and win as candidates for irreversible inhibition of Mpro, a concern exists regarding potential toxicity due to off-target inhibition and adduct formation. W. somnifera like many (but not all) herbal supplements is considered generally safe for consumption without considerable side effects. No reports of serious adverse events or hepatoxicity resulting from W. somnifera extracts are available in clinical trials. (53) However, a few cases of liver toxicity in the USA and Iceland resulting from commercially available W. somnifera products have been reported recently. (54) The symptoms usually resolve with discontinued use, and fatalities or chronic injuries are yet to be reported. One possible explanation for this safety profile irrespective of being a bis-electrophile is GSH-mediated detoxication of wifA and win. We have shown that both wifA and win can form adducts with GSH under physiological conditions in the absence of glutathione-S-transferase (GST). The GSH adducts of wifA and win (Figure S6, Supporting Information) should, in principle, have lower efficacy as potential irreversible inhibitors of Mpro. However, the Mpro-cytotoxicity assays with BSO indicated otherwise (Figure 3C). Considering the flexible active site of Mpro, we used docking studies to examine if the GSH adducts of wifA and win bind to the active site of Mpro. Docking studies suggested that the GSH adducts of wifA and win may bind to the active site of Mpro with better affinity compared to the drug alone (Figure S6, Supporting Information).
Although exciting, one question still remains to be addressed, i.e., if GSH adducts of wifA and win will behave as covalent inhibitors of Mpro. Both wifA and win have two accessible electrophilic centers. While one forms a covalent linkage with GSH, the other is still available for the reaction with Cys thiolate of Mpro. Docking studies with win-GSH-MA and wifA-GSH-MA adducts showed various binding modes with strong binding affinities. This proposed property of wifA and win, where the apparent detoxicated GSH adducts have an enhanced potential to bind to Mpro, not only strengthens their candidature as Mpro inhibitors but also explains the safety associated with wifA, win, and their plant extracts in normal cells.
Conclusion
Using various in silico and biochemical studies we have shown here that the bis-electrophiles wifA and win from the medicinal plant W. somnifera are good and safe inhibitors of Mpro. Docking studies revealed that wifA and win have the potential to bind to Mpro. Both wifA and win can form thermodynamically stable adducts with Cys and GSH under physiological conditions. 2-D and 3-D cell culture assays and fluorescence-based kinetic assays showed that wifA and win can inhibit Mpro with IC50 values of 0.54 and 1.8 μM for wifA and win, respectively. Equilibrium dialysis experiments and LC-HRMS/MS analysis indicated that wifA and win covalently bind to Mpro. Finally, docking studies revealed that the GSH adducts of wifA and win also have the potential to bind Mproand form covalent adducts. Taken together, we have shown here that two major metabolites of the widely used medicinal plant W. somnifera can form covalent adducts with Mpro and inhibit SARS-CoV-2 replication. We strongly believe that wifA and win along with W. somnifera extracts have the potential for both prevention and treatment of COVID-19.
Experimental Section
Reagents and Cell Lines
WifA and win were purchased from Sigma-Aldrich (now Merck, St. Louis, MO, USA) and were of >98% purity. Unless otherwise mentioned all other reagents were of the highest purity available (>95%) and obtained from Sigma-Aldrich (now Merck, St. Louis, MO, USA); ethanol, DMSO, NaCl, imidazole, ethylenediamine tetraacetic acid (EDTA), Tris-Cl, glycine, and Luria broth were obtained from HiMedia (Mumbai, India); acetonitrile was from JT Baker (Center Valley, PA, USA).
Cell line HepG2 was acquired from National Center for Cell Science (Pune, Maharashtra, India), and A549 was a generous gift from Dr. Naga Suresh Veerapu, Shiv Nadar University, India. All cell culture reagents and fluorescent dyes were purchased from HiMedia and Thermo Fisher Scientific (Waltham, MA, USA).
No unexpected or unusually high safety hazards were encountered.
Connectivity Map Analysis
The CMap project launched by the Broad Institute consists of a comprehensive catalogue of gene expression signatures from cells with genetic alterations or treated with pharmacological agents, which are useful for predicting previously unrecognized connections between drugs, genes, and diseases. To identify drugs/pharmacological agents that might be useful for treating Covid-19 infection, first we identified the differentially expressed genes specific for SASRs-CoV-2-infected lung cells from previously published papers and publicly available databases (GSE147507). As a control, we also took the DEGs specific for SARS-CoV-infected lung cells (GSE17400). Next we compared the upregulated DEGs from both data sets against the DEGs from drug-treated cells deposited in the CMap portal. This was done by the freely available software Enrichr. The analysis generated drugs/pharmacological agents that can potentially reverse the upregulated DEG signature generated from each virus-infected lung cell. We selected the drugs from the top of the lists that have an adjusted p value of <0.05. CMap also provided the list of genes that are tentatively responsible for sensitizing the infected cells to a candidate drug.
Docking Studies
The crystal structure of SARS-CoV-2 main protease (Mpro) in complex with an inhibitor N3-I (PDB id: 6LU7) was downloaded from the RCSB protein data bank and processed before molecular docking studies were performed. (15) At first, we deleted the ligated inhibitor N3-I and crystal waters to make space for the substrates (proposed inhibitors) to bind at the active site. Then, we added the missing hydrogen atoms using Chimera version 1.13.1 software. (55) We performed the molecular docking simulations for 10 different substrates. Initially we performed docking studies for wifA and win. Later, similar docking simulations were carried out for four possible win-glutathione (win-GSH) adducts and for four possible withaferin-A-glutathione (wifA-GSH) adducts. The binding modes of the substrate-bound Mpro were generated by docking the substrates into the binding site using AutoDock Vina software. (56) For all docking studies, a grid box of suitable dimensions centered at the same coordinates of the previously deleted crystallized ligand (x: −8.58, y: 10.85, z: 68.72) was generated. We used an exhaustiveness setting of 7, and the maximum energy difference among the different binding modes was kept at 3 kcal/mol for all docking calculations. The representative binding modes of the substrates docked at the active site of Mpro were chosen based on the proximity of the electrophilic centers (epoxide or the Michael acceptor) of the substrates with a Cys145 residue. The contact analysis and the interaction between the residues in the binding pocket and the corresponding ligand were made with the Visual Molecular Dynamics (VMD) software. (57) The 2-D protein–ligand interaction plot was generated by LigPlot+ version v.1.4.5 software. (58)
Density Functional Theory Calculations
The mechanistic studies of the covalent adduct formation between cysteine and win/wifA were performed using DFT. (59) All the structures were optimized at the M06-2X/6-31G* level (B1) of theory using the Gaussian 09 package. (60) The optimizations were carried out in an implicit solvent using the CPCM solvent model, with parameters corresponding to water, which is the solvent used in the experimental studies. (61,62) The nature of stationary points was characterized by frequency calculations at the B1 level of theory. The single-point energy evaluations in water solvent were done on the optimized geometries at the M06-2X/Def2-TZVP level (B2) of theory. (63) The zero-point energy and thermal correction to Gibbs free energy obtained from the B1 frequency analysis were added to the B2 single-point energies. The relative free energies were evaluated at 298.15 K.
Formation of WifA- or Win-Thiol Adducts
To form a win/wifA-thiol adduct, win/wifA (100 μM) was incubated with GSH (5 mM) or cysteine (5 mM) in 100 mM potassium phosphate (pH 7.5) at 37 °C for 6 h. All reagents were procured from Sigma-Aldrich, MO, USA.
LC-MS and LC-MSMS Analysis
An Agilent 6540 UHD Accurate-Mass Q-TOF LCMS system (Agilent Technologies, Santa Clara, CA, USA) with an Agilent UHPLC system was used for LC-tandem MS analysis. Phenomenex (Torrance, CA, USA) Kinetex Polar C18 columns (column A: 5 μm, 2.1 mm × 10 mm; column B; 2.6 μm, 2.1 mm × 100 mm) were used for chromatography.
Various compounds and adducts were separated using solvent A (containing 0.1% HCO2H and water v/v) and solvent B (containing 0.1% HCO2H and CH3CN, v/v) following a gradient program with a flow rate of 200-300 μL min-1: 0-5 min, 97% A (v/v); 5-20 min, linear gradient to 100% B; 20-24 min, hold at 100% B (v/v); 24-25 min, linear gradient to 98% A (v/v); 25-30 min, hold at 97% A (v/v). The temperature of the column was maintained at 30 °C, and samples (20 μL) were infused with an autosampler.
ESI conditions were as follows: gas temperature 325 °C, drying gas flow rate 8 L/min, nebulizer 35 psi, sheath gas temperature 300 °C, sheath gas flow rate 10 L/min, capillary voltage 2500 V, nozzle voltage 1000 V, capillary current 0.054 μA, chamber current 4.23 μA, fragmenter voltage 80 V, skimmer voltage 70 V. MS/MS was performed in targeted mode using a normalized collision energy of 35%. MS-grade solvents were purchased from Sigma-Aldrich.
Cell Culture Assay
HepG2 cells were cultured in DMEM with 10% FBS and 1% pen–strep and were grown in 5% CO2 at 37 °C. A total of 300 000 cells of HepG2 or A549 were seeded in a 35 mm dish. After 16 h of seeding 3 μg of pCMV3-2019-nCoV-3CLpro plasmid (Sino Biological, Beijing, China) was transfected using Lipofectamine 2000 (Invitrogen, MA, USA) and kept in serum-free DMEM for 8 h. After 8 h of transfection, media was changed to complete media and cells were allowed to grow for another 12 h. The cells were subsequently trypsinised and counted by a Neubauer hemocytometer (Sigma-Aldrich). A total of 1 × 104 of these cells were again plated in 48-well plates with hygromycin B and either win, wifA, or E64. In another plate nontransfected cells were subjected to the same condition for use as a control. After 48 h of drug treatment, 1 drop of NucBlue Live Cell Stain Readyprobes Reagent (Invitrogen) stain was added, and the plate was incubated for 30 min at 37 °C and 5% CO2. After 30 min of incubation microscopic images were taken (Leica DFC450, Leica Microsystems, Wetzlar, Germany). Following imaging, cells were counted using a Neubauer hemocytometer.
For assays with BSO, pCMV3-2019-nCoV-3CLpro-transfected A549 cells were pretreated with 500 μM BSO. Twenty hours posttransfection, either 5 μM win, wifA, or E64 was added along with 1 mM BSO (Sigma-Aldrich). In another plate nontransfected cells were subjected to the same condition for use as a control. The used drug concentrations are as follows: win, 5 μM; wifA, 5 μM; E64, 5 μM; hygromycin B, 200 μg. Drugs and reagents were procured from Sigma-Aldrich.
For the 3-D assay, 3000 cells/μL were taken from both pCMV3-2019-nCoV-3CLpro-transfected and nontransfected A549 and HepG2 cells. A 1:1 mixture of growth factor-reduced Matrigel (Corning Incorporated, NY, USA) and cell suspensions were made, and media with respective drugs were added at the same time. After the formation of organoids, images were taken (Leica DFC450). The organoids were then remelted on ice and centrifuged to get the cell pellet. The pellet of the organoids was then trypsinized and counted using a Neubauer hemocytometer.
Recombinant SARS-CoV-2 Mpro Expression and Purification
The pETM33_Nsp5_Mpro construct for bacterial expression of SARS-CoV-2 Mpro was procured from Addgene, MA, USA. This was deposited to Addgene by Dr. Ylva Ivarsson, Uppsala University, Sweden. The expression plasmid (pETM33_NSP5_Mpro), encoding the SARS-CoV-2 Mpro cDNA, was transformed into E. coli (Rosetta) cells (gifted by Dr. Rajan Vyas, Shiv Nadar University) following standard protocols. A single transformed colony was picked and cultured in Luria broth medium containing 50 μg/mL kanamycin at 37 °C. When the optical density at 600 nm of the culture reached 0.6, cells were induced by adding 0.5 mM IPTG and grown for another 6 h at 30 °C. Cells were pelleted by centrifugation at 6000g for 15 min and stored at −70 for further use. For protein purification, the cell pellet was resuspended in lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM imidazole) and lysed by sonication for 20 min (5 s on, 20 s off, 40% amplitude) on ice followed by centrifuged at 22500g for 30 min. The supernatant was passed through a 5 μM syringe filter before transferring into a tube containing HIS-Select nickel affinity gel (Merck) and kept on a revolving mixer at 4 °C for 3 h. The mixture was centrifuged and the pellet was washed three times with 3 bed volumes of resuspension buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 20 mM imidazole). Because the Mpro protein was expressed as a GST fusion protein with a His tag linked to GST, Mpro was eluted from the beads by digesting the bound fusion protein with HRV3C protease. Beads were resuspended in buffer containing 50 mM Tris buffer, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, and 3 μL of HRV3C protease (GeneScript, 250 IU) and kept overnight at 4 °C. The beads with bound GST were removed by centrifugation, and the supernatant containing Mpro was transferred to an Amicon filter (10 kDa cutoff, Merck, Darmstadt, Germany) for buffer exchange and to concentrate. The purity and identity of the protein were checked by PAGE and Western blot analysis, respectively.
Western Blot
The purified protein was denatured using 6× denaturation buffer at 95 °C for 10 min and then resolved on a 12% SDS-polyacrylamide gel by electrophoresis. Resolved proteins were transferred onto a nitrocellulose membrane and then blocked by incubating in 5% skimmed milk to minimize nonspecific binding of antibodies. The blocked blot was submerged with primary antibodies for Mpro (Mpro, main protease antibody, rabbit, polyclonal, BPS Bioscience, San Diego, CA, USA) overnight at 4 °C and was washed three times (10 min each) with 1× Tris buffer saline with 0.1% Tween-20 (TBST) followed by incubation with HRP-conjugated secondary antibody (Sigma-Aldrich) for Mpro. Unbound antibodies were removed by washing with 1× TBST buffer (3 × 10 min each), and signal was recorded using SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA, USA). Images were taken in ImageQuant LAS 500 (GE Healthcare, Chicago, IL, USA).
Equilibrium Dialysis
To determine the binding of wifA and win to Mpro, equilibrium dialysis was performed. A solution containing 50 mM potassium phosphate buffer (pH 7.5), 2 μM win or wifA, and Mpro (30 μg) was dialyzed against the same solution without the protein at room temperature for 16 h. The dialysis membrane had a cutoff of 3.5 kDa (Thermo Fisher Scientific, MA, USA). Following dialysis, the relative amount of win or wifA in the dialysate was measured by LC-MS and compared against the no-protein control.
The solution inside the dialyzer was transferred into a tube, and an equal volume of cold acetonitrile was added to denature the proteins. The solution was vortexed and centrifuged, and the supernatant was subjected to LC-MS analysis to measure the relative concentration of win or wifA.
Enzyme Kinetic Assay
The activity of Mpro and its inhibition by wifA and win were measured by a continuous kinetic assay. The nonfluorescent Mprosubstrate DABCYL-KTSAVLQSGFRKME-EDANS (BPS Bioscience, San Diego, CA, USA) was used. Upon proteolytic cleavage by Mpro, the substrate becomes fluorescent, having wavelengths of 336 and 455 nm for excitation and emission, respectively. The assay was performed by preincubating Mpro (0.5 μM) with win or wifA (5 μM) in Tris buffer (50 mM, pH 7.4) containing 2 μM EDTA for 5-120 min at 37 °C. For certain reactions 5 mM GSH was used. Reactions were initiated by adding the substrate (20 μM). The activity of the enzyme was followed by monitoring the fluorescence intensity at 37 °C with a multimode plate reader (BioTek, Gen5, BioTek US, VT, USA). IC50 was calculated using the online tool provided by ATT Bioquest (Quest Graph IC50 Calculator, AAT Bioquest, Inc., https://www.aatbio.com/tools/ic50-calculator.
LC-MS and LC-MS/MS Detection of the WifA-Mpro Adduct
In a typical assay, 30 μg of Mpro was incubated with 30 μM wifA in 500 μL of 50 mM Tris-HCl pH 7.5 buffer for 12 h at 37 °C. Following incubation, 2 volumes of cold acetonitrile was added and centrifuged. The supernatant was discarded, and the pellet was washed twice with chloroform and once with acetonitrile. After air drying the pellet, it was resuspended in 10 mM CaCl2 and 100 mM Tris-HCl and digested with chymotrypsin (5 μg) overnight at 37 °C. The reaction was stopped by adding 5% formic acid (v/v) and loaded into a prepacked C18 column. The column was washed with water, and finally peptides were eluted using acetonitrile. The acetonitrile eluent was dried in a Speedvac, resuspended in 50% acetonitrile, and analyzed by LC-MS and LC-MS/MS.
Statistical Analysis
Each bar graph is the mean of two to three replicates along with the calculated standard deviation.
Supplementary Material
Acknowledgments
This work was supported in part by the DBT, Ramalingaswami Re-entry fellowships [BT/RLF/RE-ENTRY/18/2013 to G.C. and BT/RLF/RE-ENTRY/35/2012 to A.C.] and Shiv Nadar University. The authors thank Shazia Siddiqui for help in collecting data for Figure S2. D.M. acknowledges the support provided by PARAM Shakti under the National Supercomputing Mission (DST/NSM/R&D_HPC_Applications/2021/08), Government of India.
Footnotes
Supporting Information
- CMap, docking, LC-tandem MS, protein gel, kinetic assay, and microscopic data (PDF)
- Upregulated gene lists that are unique to SARS-CoV-2-infected lung cells (XLSX)
- Upregulated gene lists that are unique to SARS-CoV-infected lung cells (XLSX)
- CMap analysis-predicted list of drugs which can reverse the upregulated gene signature specific for SARS-CoV-2 (XLSX)
- CMap analysis-predicted list of drugs which can reverse the upregulated gene signature specific for SARS-CoV (XLSX)
The authors declare no competing financial interest.
Contributor Information
Shayantani Chakraborty, Department of Life Science, Shiv Nadar University, Greater Noida, UP 201314, India.
Dibyendu Mallick, Department of Chemistry, Presidency University, Kolkata 700073, India.
Mausumi Goswami, Department of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore, TN 632014, India.
F. Peter Guengerich, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37235, United States.
Anindita Chakrabarty, Department of Life Science, Shiv Nadar University, Greater Noida, UP 201314, India.
Goutam Chowdhury, Independent Researcher, Greater Noida, UP 201314, India.
References
- (1).Pascarella G; Strumia A; Piliego C; Bruno F; Del Buono R; Costa F; Scarlata S; Agrò FE J. Intern Med 2020, 288 (2), 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gulati A; Pomeranz C; Qamar Z; Thomas S; Frisch D; George G; Summer R; DeSimone J; Sundaram B Am. J. Med. Sci 2020, 360 (1), 5–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lake MA Clin Med. (Lond) 2020, 20 (2), 124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Oran DP; Topol EJ Ann. Int. Med 2020, 173 (5), 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ludwig S; Zarbock A Anesth Analg 2020, 131 (1), 93–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pillaiyar T; Manickam M; Namasivayam V; Hayashi Y; Jung SH J. Med. Chem 2016, 59 (14), 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Forster P; Forster L; Renfrew C; Forster M Proc. Natl. Acad. Sci. U. S. A 2020, 117 (17), 9241–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bzówka M; Mitusińska K; Raczyńska A; Samol A; Tuszyński JA; Góra A. Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. Int. J. Mol. Sci 2020, 21 (9), 3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li Q; Kang C Progress in Developing Inhibitors of SARS-CoV-2 3C-Like Protease. Microorganism 2020, 8 (8), 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ahsan W; Alhazmi HA; Patel KS; Mangla B; Al Bratty M; Javed S; Najmi A; Sultan MH; Makeen HA; Khalid A; et al. Front Public Health 2020, 8, 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dai W; Zhang B; Jiang XM; Su H; Li J; Zhao Y; Xie X; Jin Z; Peng J; Liu F; et al. Science 2020, 368 (6497), 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vuong W; Khan MB; Fischer C; Arutyunova E; Lamer T;Shields J; Saffran HA; McKay RT; van Belkum MJ; Joyce MA; et al. Nat. Commun 2020, 11 (1), 4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dömling A; Gao L Chem. 2020, 6 (6), 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Haake C; Cook S; Pusterla N; Murphy B Coronavirus Infections in Companion Animals: Virology, Epidemiology, Clinical and Pathologic Features. Viruse 2020, 12 (9), 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; et al. Nature 2020, 582 (7811), 289–293. [DOI] [PubMed] [Google Scholar]
- (16).Kneller DW; Phillips G; O’Neill HM; Tan K; Joachimiak A; Coates L; Kovalevsky A Room-temperature X-ray crystallography reveals the oxidation and reactivity of cysteine residues in SARS-CoV-2 3CL M(pro): insights into enzyme mechanism and drug design. IUCrJ. 2020, 7, 1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Asgian JL; James KE; Li ZZ; Carter W; Barrett AJ; Mikolajczyk J; Salvesen GS; Powers JC J. Med. Chem 2002, 45 (23), 4958–4960. [DOI] [PubMed] [Google Scholar]
- (18).Jackson PA; Widen JC; Harki DA; Brummond KMJ Med. Chem 2017, 60 (3), 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Fu L; Ye F; Feng Y; Yu F; Wang Q; Wu Y; Zhao C; Sun H; Huang B; Niu P; et al. Nat. Commun 2020, 11 (1), 4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; et al. Cell Res. 2020, 30 (8), 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Firenzuoli F; Antonelli M; Donelli D; Gensini GF; Maggini VJ Altern Complement Med. 2020, 26 (10), 851–853. [DOI] [PubMed] [Google Scholar]
- (22).Fuzimoto AD; Isidoro CJ Tradit Complement Med. 2020, 10 (4), 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mandlik Ingawale DS; Namdeo AG J. Diet Suppl 2020, 1–44. [DOI] [PubMed] [Google Scholar]
- (24).Mishra LC; Singh BB; Dagenais S Altern Med. Rev 2000, 5 (4), 334–346. [PubMed] [Google Scholar]
- (25).Siddiqui S; Ahmed N; Goswami M; Chakrabarty A; Chowdhury G Curr. Res. Toxicol 2021, 2, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Priyandoko D; Ishii T; Kaul SC; Wadhwa R PLoS One 2011, 6 (5), No. e19552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chowdhury G; Cho SH; Pegg AE; Guengerich FP Angew. Chem., Int. Ed. Engl 2013, 52 (49), 12879–12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Patil VS; Hupparage VB; Malgi AP; Deshpande SH; Patil SA; Mallapur SP Chinese Herbal Medicines 2021, 13, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Straughn AR; Kakar SS J. Ovarian Res 2020, 13 (1), 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kim C-H Anti-SARS-CoV-2 Natural Products as Potentially Therapeutic Agents. Frontiers in Pharmacology 2021, 12, Article 590509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kumar V; Dhanjal JK; Bhargava P; Kaul A; Wang J; Zhang H; Kaul SC; Wadhwa R; Sundar DJ Biomol. Struct. Dyn 2020, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dhanjal JK; Kumar V; Garg S; Subramani C; Agarwal S; Wang J; Zhang H; Kaul A; Kalra RS; Kaul SC; et al. Int. J. Biol. Macromol 2021, 184, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Jarada TN; Rokne JG; Alhajj RJ Cheminform 2020, 12 (1), 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chen EY; Tan CM; Kou Y; Duan Q; Wang Z; Meirelles GV; Clark NR; Ma’ayan A BMC Bioinformatics 2013, 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jha PK; Vijay A; Halu A; Uchida S; Aikawa M Front Cardiovasc Med. 2021, 7, 623012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Islam T; Rahman MR; Aydin B; Beklen H; Arga KY; Shahjaman M Eur. J. Pharmacol 2020, 887, 173594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pushparaj PN; Abdulkareem AA; Naseer MI Front Pharmacol 2021, 12, 688227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bargagna-Mohan P; Hamza A; Kim YE; Khuan Abby Ho Y; Mor-Vaknin N; Wendschlag N; Liu J; Evans RM; Markovitz DM; Zhan CG; et al. Chem. Biol 2007, 14 (6), 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bargagna-Mohan P; Paranthan RR; Hamza A; Dimova N; Trucchi B; Srinivasan C; Elliott GI; Zhan CG; Lau DL; Zhu H; et al. J. Biol. Chem 2010, 285 (10), 7657–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rahman I; MacNee W Eur. Respir. J 2000, 16 (3), 534–554. [DOI] [PubMed] [Google Scholar]
- (41).Hayes JD; Flanagan JU; Jowsey IR Annu. Rev. Pharmacol Toxicol 2005, 45, 51–88. [DOI] [PubMed] [Google Scholar]
- (42).Taylor EW; Radding W Front Nutr 2020, 7, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Khanfar A; Al Qaroot B Eur. Rev. Med. Pharmacol. Sci 2020, 24 (23), 12500–12509. [DOI] [PubMed] [Google Scholar]
- (44).Fraternale A; Brundu S; Magnani M Biol. Chem 2017, 398 (2), 261−275. Journal of Natural Products pubs.acs.org/jnp Article 10.1021/acs.jnatprod.2c00521 [DOI] [PubMed] [Google Scholar]; J. Nat. Prod 2022, 85, 2340–2350 2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ś widerek K; Moliner V. Chem. Sci 2020, 11 (39), 10626–10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Resnick SJ; Iketani S; Hong SJ; Zask A; Liu H; Kim S; Melore S; Lin FY; Nair MS; Huang Y; et al. J. Virol 2021, 95 (14), No. e0237420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Jensen G; Morrill C; Huang Y Acta Pharm. Sin B 2018, 8 (5), 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Hartung T Altex 2013, 30 (3), 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Griffith OW J. Biol. Chem 1982, 257 (22), 13704–13712. [PubMed] [Google Scholar]
- (50).Antony ML; Lee J; Hahm ER; Kim SH; Marcus AI; Kumari V; Ji X; Yang Z; Vowell CL; Wipf P; et al. J. Biol. Chem 2014, 289 (3), 1852–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yang J; Yan W; Li Y; Niu L; Ye H; Chen L Mol. Pharmacol 2019, 96 (6), 711–719. [DOI] [PubMed] [Google Scholar]
- (52).Davis DA; Bulut H; Shrestha P; Yaparla A; Jaeger HK; Hattori SI; Wingfield PT; Mieyal JJ; Mitsuya H; Yarchoan R mBio 2021, 12 (4), No. e0209421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Ashwagandha. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases, 2012. [Google Scholar]
- (54).Philips CA; Ahamed R; Rajesh S; George T; Mohanan M; Augustine P World J. Hepatol 2020, 12 (9), 574–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE J. Comput. Chem 2004, 25 (13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- (56).Trott O; Olson AJ J. Comput. Chem 2010, 31 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Humphrey W; Dalke A; Schulten KJ Mol. Graph 1996, 14 (1), 33–38 , 27−38. [DOI] [PubMed] [Google Scholar]
- (58).Laskowski RA; Swindells MB J. Chem. Inf Model 2011, 51 (10), 2778–2786. [DOI] [PubMed] [Google Scholar]
- (59).Zhao Y; Truhlar DG Theoretical chemistry accounts 2008, 120, 215–241. [Google Scholar]
- (60).Gaussian 09; Gaussian Inc.: Wallingford, CT, 2009. [Google Scholar]
- (61).Barone V; Cossi MJ Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- (62).Cossi M; Rega N; Scalmani G; Barone VJ Comput. Chem 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
- (63).Weigend F; Ahlrichs R Phys. Chem. Chem. Phys 2005, 7 (18), 3297–3305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.