

ABSTRACT
Neurodegenerative dementias are characterized by the abnormal accumulation of misfolded proteins. However, its diagnostic criteria are still based on the clinical phenotype. The development of biomarkers allowed in vivo detection of pathophysiological processes. This article aims to make a non-systematic review of the use of molecular neuroimaging as a biomarker. Molecular neuroimaging is based on the use of radiotracers for image acquisition. The radiotracer most used in PET is 18F-fluorodeoxyglucose (FDG), with which it is possible to study the regional brain glucose metabolism. The pattern of regional hypometabolism provides neuroanatomical information on the neurodegenerative process, which, in turn, has a good specificity for each type of proteinopathy. FDG is very useful in the differential diagnosis of neurodegenerative dementias through the regional pattern of involvement, including dementia with Lewy bodies and the spectrum of frontotemporal dementia. More recently, radiotracers with specific ligands to some of the pathological proteins have been developed. Pittsburgh compound B (PIB) labeled with 11C and the ligands that use 18F (florbetapir, florbetaben and flutemetamol) are the most used radiotracers for the detection of insoluble β-amyloid peptide in Alzheimer's disease (AD). A first generation of ligands for tau protein has been developed, but it has some affinity for other non-tau protein aggregates. A second generation has the advantage of having a higher affinity for hyperphosphorylated tau protein, including in primary tauopathies.
Keywords: Dementia, Positron-Emission Tomography, Amyloid, tau Proteins, Alzheimer Disease
RESUMO
As demências neurodegenerativas caracterizam-se pelo acúmulo anormal de proteínas mal dobradas. Entretanto, os seus critérios diagnósticos ainda se baseiam no fenótipo clínico. O desenvolvimento de biomarcadores permitiu a detecção in vivo do processo fisiopatológico. O objetivo deste artigo é fazer uma revisão não-sistemática sobre o papel da neuroimagem molecular como biomarcador. A neuroimagem molecular baseia-se no uso de radiotraçadores para aquisição da imagem. O mais usado no PET é o 18F-fluorodeoxiglicose (FDG), com o qual é possível estudar o metabolismo regional de glicose cerebral. O padrão de hipometabolismo regional fornece uma informação neuroanatômica do processo neurodegenerativo que, por sua vez, tem uma boa especificidade para cada tipo de proteinopatia. O PET-FDG é muito útil no diagnóstico diferencial das demências neurodegenerativas através do padrão de acometimento regional, incluindo a demência com corpos de Lewy e o espectro das demências frontotemporais. Mais recentemente, radiotraçadores com ligantes específicos a algumas das proteínas patológicas têm sido desenvolvidos. O composto B de Pittsburgh (PIB) com 11C e os ligantes dos que usam 18F (florbetapir, florbetaben e flutemetamol) são os radiotraçadores mais usados para a detecção de peptídeo β-amiloide insolúvel na doença de Alzheimer (DA). Uma primeira geração de ligantes para proteína tau foi desenvolvida, mas apresenta alguma afinidade a outros agregados proteicos não-tau. Uma segunda geração tem a vantagem de apresentar uma maior afinidade à proteína tau hiperfosforilada, incluindo nas taupatias primárias.
Palavras-chave: Demência, Tomografia por Emissão de Pósitrons, Amiloide, Proteínas tau, Doença de Alzheimer
INTRODUCTION
Throughout the 20th century, neurodegenerative dementias were initially described based on their phenotypes and post-mortem pathological studies. In these studies, in addition to the findings of neuronal loss and brain atrophy, typical alterations of each disease were described, such as the senile plaques and neurofibrillary tangles described by Alois Alzheimer 1 , the intracellular eosinophilic inclusions observed by Fritz Heinrich Lewy 2 and the intraneuronal argyrophilic inclusions reported by Arnold Pick 3 . Only in the last decades, with the development of immunohistochemical methods, it was discovered that these pathologies are due to the intra- or extracellular accumulation of misfolded proteins 4 . Therefore, neurodegenerative dementias could be classified by both clinical syndromes and underlying proteinopathies (Figure 1).
Figure 1. Summary of the molecular imaging findings in the different neurodegenerative diseases. First line: spectrum of proteinopathies causing neurodegenerative dementias and their respective clinical phenotypes. Pathological proteins: TDP-43, FUS, Tau, beta-amyloid, and alpha-synuclein. Second line: Patterns of regional glucose hypometabolism on brain FDG-PET that suggest or support each proteinopathy.;Third line: Patterns of amyloid PET with 11C-PIB in each condition. Fourth line: Patterns of uptake in dopamine transporter (DAT) SPECT or PET tracers or 18F-Fluordopa PET (SPECT images with 99mTc-TRODAT). Fifth line: Patterns of uptake in cardiothoracic 123I-mIBG scintigraphy, a tracer of the sympathetic innervation. The white arrow in the first image on the left, third line indicates hypometabolism in the left motor cortex in motor neuron disease (ALS/Mills Syndrome); yellow arrows in the last images on the right, third line: hypometabolism in both putamen and the cerebellum seen in MSA. Orange arrow in the last image on the right in the fifth line: absence of myocardial uptake of 123I-mIBG seen in synucleinopathies.
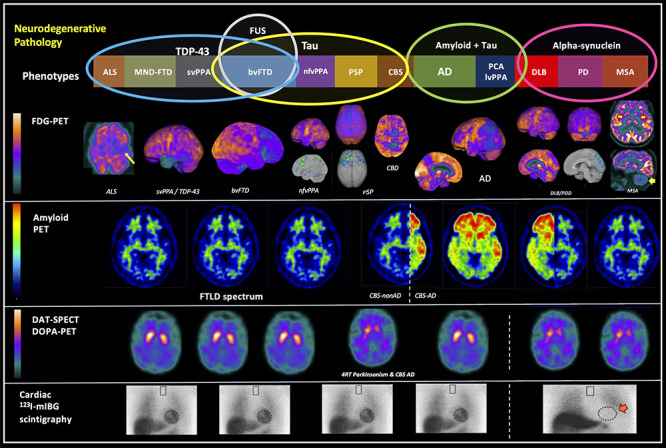
FTD: frontotemporal dementia; ALS: amyotrophic lateral sclerosis; MND: motor neuron disease; svPPA: Semantic variant primary progressive aphasia; nfvPPA: Nonfluent (or agrammatic) variant primary progressive aphasia; CBD: corticobasal degeneration; CBS: corticobasal syndrome; PSP: progressive supranuclear palsy; AD: Alzheimer's disease; lvPPA: Logopenic variant primary progressive aphasia; DLB: dementia with Lewy bodies; PD: Parkinson's disease; PDD: Parkinson's disease with dementia; MSA: Multiple Systems Atrophy; PET: Positron Emission Tomography; SPECT: single photon emission tomography; FDG: 18F-Fluorodeoxyglucose; PIB: Pittsburgh Compound-B labeled with carbon-11.
Despite advances in the pathological characterization of neurodegenerative dementias, its diagnostic criteria are still based on clinical phenotypes and not on proteinopathies 4 . However, this leads to some problems 4 , 5 . First, the accuracy of the clinical diagnosis of degenerative dementias is not adequate. Second, a single proteinopathy can lead to different phenotypes (for example, Alzheimer Disease (AD) can manifest as several variants: amnestic, aphasic or logopenic, visuospatial, dysexecutive and behavioral) 5 . The opposite also occurs: different proteinopathies can lead to the same phenotype, as is the case with the behavioral variant of frontotemporal dementia (vbFTD) 5 . Third, diagnostic criteria are not very sensitive to detect preclinical or prodromal stages. Finally, efforts in the development of disease-modifying treatments have been directed towards drugs that target these pathological proteins.
For all these reasons, in recent decades biomarkers have been developed to enable in vivo detection of the pathophysiological process, thus providing a better understanding of the natural history, an improvement in the sensitivity and specificity of the diagnostic criteria, and the development of disease-modifying treatments 6 - 9 . This article aims to conduct a non-systematic review of the literature on the use of molecular neuroimaging as a biomarker of the main neurodegenerative pathologies that lead to cognitive and/or behavioral decline (Alzheimer's disease, dementia with Lewy bodies and the frontotemporal dementia spectrum).
DEFINING BIOMARKERS AND THE ROLE OF MOLECULAR NEUROIMAGING IN THE DIAGNOSIS OF NEURODEGENERATIVE DEMENTIAS
Biomarkers can be defined as measures or indicators of physiological or pathological processes or responses to a therapeutic intervention 10 . An ideal biomarker should 11 : 1) reflect a fundamental aspect of pathophysiology; 2) indicate the real presence of the pathology and not merely an increased risk; 3) exhibit high sensitivity and specificity (80% or more); 4) be effective in predicting early or pre-clinical stages; 5) allow monitoring of disease severity or rate of progression; 6) indicate the effectiveness of the therapeutic intervention and 7) be non-invasive, economically feasible and available. Although this consensus was originally proposed in the context of AD, it can also be applied to biomarkers in other neurodegenerative diseases.
The biomarkers used in neurodegenerative diseases can be schematized according to the detection methods, including cerebrospinal fluid (CSF), serum and structural and molecular neuroimaging biomarkers 12 . Molecular neuroimaging methods have gained prominence in the study of biomarkers in neurodegenerative diseases for several reasons 13 . First, the molecular neuroimaging studies allow detection of pathological changes prior to morphological changes observed in structural neuroimaging exams, such as magnetic resonance imaging (MRI). Second, new radiotracers with affinity to bind to pathological proteins, like amyloid β peptide (Aβ) plaques and tau protein, have increasingly been developed. Moreover, neuroimaging allows the study of proteinopathy, its anatomical distribution, and neuronal networks.
Molecular neuroimaging methods are based on the use of radioisotopes (or radionuclides) for image acquisition. The basic principle is the administration of a radiotracer, a molecule with biological properties bound to the radionuclide and whose emission of the radioactive signal is detected by a scanner, forming a radiotracer 14 . There are two methods for acquiring molecular neuroimaging: positron emission tomography (PET) and single photon emission tomography (SPECT) 14 .
Currently the most used radioisotope in the SPECT neuroimaging study is technetium-99m (99mTc). Radiotracers marked with 99mTc are fat-soluble compounds that cross the blood-brain barrier and are distributed according to cerebral blood flow 14 . Therefore, most SPECT radiotracers assess brain perfusion. The advantage of SPECT is its low acquisition cost, longer half-life, greater simplicity in radiotracer synthesis and, consequently, greater availability. However, brain images acquired by SPECT have lower spatial resolution, which makes them less sensitive and specific than brain PET. Therefore, currently, the use of SPECT as a molecular neuroimaging biomarker is not recommended, except when PET is not available.
On the other hand, the most used radionuclides in PET are carbon-11 ( 11 C) and fluor-18 ( 18 F), which are more unstable isotopes with shorter half-lives. Therefore, PET requires radiopharmacy laboratories and production centers (cyclotrons) closer to the image acquisition center, which makes the method less available and more expensive. However, despite these disadvantages, PET images usually have a better accuracy as biomarkers of neurodegenerative diseases. Currently, semiquantitative analysis software has been incorporated into the routine analysis of PET scans, thus increasing the accuracy of the diagnostic method, and allowing better inter-examiner comparison 14 , 15 . Some of these software tools, such as 3D-SSP (Cortex ID Suite software, GE Healthcare or Scenium software, Siemens Healthineers), compare the examinations with a database of normal individuals, reporting the results as z-scores (or standard deviation) from the mean of normality.
The oldest PET method is the use of 18 F-fluorodeoxyglucose (FDG) as a radiotracer, with which it is possible to study the regional brain glucose metabolism (rBGM) 14 , 15 . rBGM, in turn, is an indirect measure of neuronal and glial synaptic activity 16 . Therefore, areas of neuronal and synaptic injury can be indirectly determined by regions of regional brain hypometabolism and, thus, FDG-PET plays a role as a biomarker of neurodegeneration 17 . The regional hypometabolism pattern provides neuroanatomical information on the neurodegenerative process which, in turn, has a good specificity for each type of neurodegenerative dementia (Table 1) 18 . Not surprisingly, in several groups of neurodegenerative diseases, the pattern of hypometabolism was included in the diagnostic criteria, as will be discussed later.
Table 1. Patterns of regional glucose hypometabolism on brain FDG-PET in the main groups of neurodegenerative dementias.
| Neurodegenerative dementia | Variants | Typical hypometabolism pattern |
|---|---|---|
| Alzheimer's disease | Amnestic (or limbic) | Temporoparietal association cortex, precuneus and posterior cingulate. In more advanced cases, there is an extension to the prefrontal cortex. |
| Logopenic variant of primary progressive aphasias | Temporoparietal association cortex, precuneus and posterior cingulate, asymmetrical, worse on the left | |
| Visual (or posterior cortical atrophy) | Temporoparietal association cortex, precuneus and posterior cingulate with occipital extension | |
| dysexecutive/ behavioral | Temporoparietal association cortex, precuneus and posterior cingulate with frontal extension | |
| Corticobasal syndrome | Temporoparietal association cortex, precuneus and asymmetric posterior cingulate, with involvement of the basal ganglia | |
| Lewy bodies dementias | Dementia with Lewy bodies | Temporoparietal and occipital association cortex, precuneus with posterior cingulate preservation (“cingulate island sign”) |
| Parkinson's disease dementia | Temporoparietal and occipital association cortex, with frontal extension, precuneus with preservation of the posterior cingulate. | |
| Frontotemporal dementias | Behavioral variant | Dorsolateral prefrontal cortex, orbitofrontal, anterior cingulate, anterior insula, and anterior temporal regions. It may have an asymmetry, worse on the right. |
| Non-fluent variant of primary progressive aphasias | Left inferior frontal region and left insula, with extension to the anterior cingulate and dorsolateral frontal region | |
| Semantic variant of primary progressive aphasias | Bilateral anterior temporal pole, worse on the left. | |
| Progressive supranuclear palsy | Dorsolateral, ventrolateral, and medial frontal cortex, including supplementary motor area and premotor cortex, caudate, thalamus, and midbrain | |
| 4R tauopathy corticobasal syndrome (corticobasal degeneration) | Dorsolateral frontoparietal cortex, including sensorimotor cortex, thalamus, and striatum, of asymmetrical pattern |
PET: Positron Emission Tomography; FDG: [18F] Fluorodeoxyglucose.
Despite its good accuracy, FDG-PET is still regarded as a biomarker of neurodegeneration. Therefore, the current frontiers of molecular neuroimaging techniques in cognitive decline have reached the detection of misfolded protein deposits 14 , 15 , 18 . These new methods were only possible after the development of radiotracers that bind specifically to these pathological proteins. In the following topics we will address some of these molecular neuroimaging modalities in the context of the main groups of neurodegenerative dementias.
MOLECULAR NEUROIMAGING IN ALZHEIMER'S DISEASE AND THE ATN CLASSIFICATION
AD is pathologically defined by the presence of senile plaques (extracellular deposit of Aβ) and neurofibrillary tangles (intraneuronal accumulation of hyperphosphorylated tau protein), in addition to signs of a neurodegenerative process (neuronal and synaptic loss, astrogliosis and microglial activation) 12 , 19 . The first diagnostic criteria were clinical, following the exclusion of others causes associated with cognitive decline 20 , 21 . However, changes in AD diagnostic criteria have been observed in recent decades as new knowledge of AD's pathophysiology and natural history have been better elucidated. Biomarkers played a vital role in these paradigm shifts.
In 2011, a working group from the National Institute on Aging and Alzheimer’s Association (NIA-AA) developed the current diagnostic criteria for AD 6 , 20 . These new criteria proposed the concept of stages of disease evolution, based on the “amyloid cascade” hypothesis 9 , 22 . According to this model, the disease follows a continuous course that starts from a preclinical phase (defined as the absence of cognitive decline and the presence of positive biomarkers for AD pathology), followed by a stage of mild cognitive impairment (MCI) and, finally, a stage of dementia due to AD 6 , 20 . For the first time, the preclinical stage, and the possibility of non-amnestic AD variants (posterior cortical atrophy, logopenic variant of primary progressive aphasias and dysexecutive/behavioral variant) were admitted.
In the 2011 criteria, the biomarkers are recommended only in selected cases: pre-senile onset dementias (< 65 years), rapidly progressive dementias, atypical dementias or for differential diagnoses with other neurodegenerative dementias. Later studies show that these 2011 clinical criteria, without the use of biomarkers, have a sensitivity of 70.9 to 87.3% and a low specificity of 44.3 to 70.8% when compared to pathological diagnosis 17 . Biomarkers have also been incorporated into the diagnosis of MCI due to AD 23 .
More recently, in 2018, the NIA-AA proposed new criteria for the biological definition of AD based on biomarkers according to an “ATN” system, especially targeting clinical trials, in which certainty of the pathological process involved is required (Table 2) 24 . According to the pathological process, AD biomarkers have been divided into biomarkers of amyloid pathology (A), tau pathology (T) and neurodegeneration (N), summarizing the ATN classification (Table 2) 7 , 24 , 25 .
Table 2. Alzheimer's disease biomarkers classified according to the pathological process (ATN system). Adapted from Jack et al. 28 .
| Pathology | Biomarker |
|---|---|
| Amyloid Pathology (A) | Decreased Aβ42 in CSF Positive amyloid PET |
| Tau pathology (T) | Increased hyperphosphorylated protein in CSF Positive tau PET |
| Neurodegeneration (N) | Increase total Tau protein in CSF Cortical atrophy on magnetic resonance imaging Regional glucose hypometabolism on FDG-PET |
CSF: cerebrospinal fluid; Aβ: beta-amyloid peptide; PET: Positron Emission Tomography; FDG: [18F] Fluorodeoxyglucose.
The purpose of these 2018 criteria is to apply them in clinical research, while the 2011 criteria would remain valid for use in clinical care(Table 3) 24 , 26 . According to these 2018 criteria, AD is defined by the positivity of biomarkers for amyloid (A+) and tau (T+), regardless of the presence or absence of neurodegeneration (N- or +). When only Aβ (A+) is present, but without evidence of tau (T-) pathology, the denomination should be “Alzheimer's pathological changes”. It therefore represents the initial stage of AD (amyloidosis without tauopathy) according to the “amyloid cascade” hypothesis. When there is positivity for tau protein (T+) and/or for neurodegeneration (N+), but negativity for amyloid pathology (A-), the diagnosis of Alzheimer`s pathology must be excluded, and it should be called SNAP (suspected non-Alzheimer’s pathophysiology). The term "clinical AD syndrome" was reserved for the situation in which the patient meets the clinical criteria for AD (amnestic or non-amnestic variants), but there is no information on amyloid or tau biomarkers.
Table 3. Diagnostic categories according to the ATN system (Amyloid-Tau-Neurodegeneration) within the continuum of the biological definition of Alzheimer's disease. Adapted from Jack et al. 28 .
| ATN profile | Category according to biomarker profile |
|---|---|
| A- T- N- | Normal Alzheimer disease D biomarkers |
| A+ T- N- | Alzheimer's continuum |
| A+ T+ N- | Alzheimer's disease (without neurodegeneration) |
| A+ T+ N+ | Alzheimer's disease (with neurodegeneration) |
| A+ T - N+ | Alzheimer's continuum + Non-Alzheimer Pathology |
| A- T+ N- | Suspected non-Alzheimer’s pathophysiology |
| A- T- N+ | Suspected non-Alzheimer’s pathophysiology |
| A- T+ N+ | Suspected non-Alzheimer’s pathophysiology |
In 2021, the International Working Group (IWG), in opposition to the 2018 NIA-AA criteria, criticized the excessive importance of the use of biomarkers for the diagnosis of AD 27 . They claim that several studies show an increased frequency of amyloidosis and neurodegeneration associated with aging even in cognitively normal individuals 8 , 28 , 29 , although the presence of amyloidosis in normal elderly is associated with higher rates of progression to cognitive decline 8 , 28 , 30 - 34 . Nevertheless, the IWG recommendation is that the diagnosis of AD should be restricted to people with A+T+ biomarkers and who have a cognitive syndrome compatible with AD 27 . This IWG definition, therefore, would fulfill the diagnosis of AD only in the MCI and dementia states with evidence of A+T+ biomarkers, which, on the one hand, increases specificity. On the other hand, the disease usually begins many years earlier and studies are continuously focusing on pre-clinical diagnosis and treatment, which are not possible when using the IWG recommendation.
Amyloid PET
Since the early 2000s, radiotracers for the detection of insoluble Aβ peptide deposited in neuritic plaques (amyloid PET) have been widely used in AD studies 15 , 18 . The first radiotracer developed was the Pittsburgh Compound-B labeled with carbon-11 (PIB), widely used in clinical research 13 , 18 . Sequentially several other Aβ peptide ligands were developed, all labeled with fluorine-18. These radiotracers (florbetapir, florbetaben and flutemetamol) are already cleared by the US regulatory agency for commercial clinical use 18 . All these radiotracers are clinically equivalent, but those linked to fluorine-18 have the advantage of a longer half-life of 110 minutes compared to 11 C, whose half-life is only 20 minutes 14 , 15 , 35 . In practice, due to carbon-11’s low half-life, 11 C-PIB needs to be synthesized in a cyclotron located in the same building of the imaging site 14 , 15 , 35 . But despite this disadvantage, studies with PIB have shown this to be a radioligand with a high accuracy in the detection and anatomical location of neuritic plaques.
Studies comparing amyloid PET with post-mortem pathological diagnosis show a sensitivity of 96% and a specificity of 100% in cases of dementia due to AD 18 . Sensitivity is not 100% because all radiopharmaceuticals have high affinity for insoluble fibrillar amyloid in neuritic plaques, with good anatomopathological correlation, but they are insensitive to soluble Aβ oligomers 18 , 36 - 38 . Also, amyloid PET may be less sensitive in earlier stages of the disease 36 , mostly because of the dichotomized nature of its interpretation: scans are usually classified as positive or negative, which most commonly include individuals with mild deposition as negative. Therefore, changes in CSF Aβ may precede changes in amyloid PET. Amyloid PET is usually classified as "positive" if there is a loss of gray and white matter differentiation in at least two of the following six areas: frontal, temporal, lateral parietal, precuneus, anterior cingulate, and posterior cingulate cortex(Figure 1) 39 , 40 . On the other hand, the amyloid PET image is classified as "negative" when there is a clear contrast between gray and white and no significant uptake in the cortex. In the quantitative analysis of amyloid PET, it was agreed to determine the standard uptake values ratio (SUVr) of cortical areas normalized for gray matter of the cerebellum, as this is a region that does not present significant Aβ deposits in AD 39 , 40 . Interestingly, amyloid radiotracers have high affinity for the myelin sheath in the absence of Aβ deposits, leading to their use in the investigation of demyelinating diseases such as multiple sclerosis.
The pattern of radiotracer uptake for amyloid in the cortex follows the pattern commonly seen in post-mortem studies, with involvement of the frontal regions, followed by the precuneus and posterior cingulate and finally areas of temporoparietal association, medial temporal region, primary cortical areas. and striatum 36 . Furthermore, as was already known, the distribution of amyloid pathology is not distinguished between AD variants, unlike tau pathology, whose neuroanatomical distribution is reflected in the clinical phenotype 41 .
It is important to note that a positive amyloid PET alone does not fulfill the criteria for the diagnosis of AD in any of the existent criteria 6 , 20 , 24 , 26 , as cognitively normal individuals, with MCI, or even with other forms of dementia (particularly dementia with Lewy bodies) may have positive tests for the presence of amyloid 6 . While between 70-90% of patients clinically diagnosed with AD test positive for amyloid, about 30-40% of cognitively normal older adults over 80 years also test positive for amyloid 6 . Rates of positive amyloid PET in cognitively normal older adults vary among studies, some reporting high rates of 20% at around 65 years and 60% at 85 years 14 . A PET-based study of our group found 18% of amyloid positivity in controls (71.19 ± 6.1 years old), and 76% in clinically-defined AD, and as low as 37% of positivity in amnestic MCI (mean ages of 73.7 ± 7.3 and 73.0 ± 5.8, respectively) 42 .
These data indicate that the process of extracellular Aβ peptide deposition is a common process in brain aging. Therefore, diagnostic criteria based on biomarkers indicate that the diagnosis of AD should show positivity for both amyloid and tau biomarkers (A+T+). On the other hand, studies show that cognitively normal older adults with positive amyloid PET have a higher chance of evolving to AD in the next 10 to 20 years, indicating that part of these subjects may be in a prodromal or preclinical stage of AD 43 , 44 . Indeed, in older adults below 80 years of age who have cognitive decline, a negative amyloid PET excludes the diagnosis of AD. Table 4 summarizes the advantages and dis-advantages of amyloid PET.
Table 4. Advantages and disadvantages of amyloid PET.
| Advantage | Disadvantages |
|---|---|
| Negative amyloid PET excludes AD; Very useful for differentiating pre-senile onset dementias; Allows you to differentiate from primary tauopathies and TDP-43pathies; Positive amyloid PET has high predictive power for conversion to AD. | Above 80 years of age, 30 to 40% of normal older adults have a positive amyloid PET; Not very useful in differentiating from alpha-synucleinopathies; High cost and little availability; Not yet incorporated into AD clinical criteria. |
PET: Positron Emission Tomography; AD: Alzheimer disease; TDP-43: transactive response DNA binding protein of 43 kDa.
Tau PET
Tau protein plays a role in the proteins that give support and stability to the microtubules that are found in axons. Hyperphosphorylation of the tau protein leads to formation of insoluble filaments, which are deposited as intracellular inclusions, ultimately leading to cell death 12 . Tau exists in six isoforms distributed in two groups in equal proportions: three isoforms have three repeats (tau 3R), and the other three have four repeats (tau 4R) of the sequence of amino acids that bind to microtubules 15 , 45 . AD is characterized by a tauopathy with the presence of two subtypes of tau (3R and 4R). However, according to the amyloid cascade hypothesis, AD is considered a tauopathy secondary to amyloid pathology 22 . Other 3R and 4R tauopathies are chronic traumatic encephalopathy, primary age-related tauopathy (PART) and some cases of FTD by a mutation in the MAPT gene 45 , 46 .
On the other hand, primary tauopathies are divided between the 3R and 4R. 3R-tauopathies are found in some cases of FTD and can aggregate in characteristic intracytoplasmic inclusions called Pick bodies 45 , 46 . The group of 4R-tauopathies includes corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), FTD with parkinsonism linked to chromosome 17 and most cases of the nonfluent/agrammatic variant of primary progressive aphasia (nfvPPA) 45 , 46 .
Studies of tau biomarkers have shown a recent increase in interest for a few reasons: failure of clinical trials of anti-amyloid therapies, a more significant correlation between tauopathy and AD progression (unlike amyloid pathology), recent studies suggesting pathways of tauopathy progression independent of amyloid pathology and the diagnosis of primary tauopathies 46 - 49 .
Since the 2010s, several ligands for tau protein detection (tau PET) have been developed and validated from post-mortem comparative studies 46 , 49 - 51 . 18 F-FDDNP was the first radiotracer developed, however, it had low specificity, binding to neurofibrillary tangles and amyloid aggregates, which caused it to be discontinued 45 . Other first-generation radiopharmaceuticals have been developed: 11 C-PBB3, 18 F-flortaucipir (previously 18 F-AV1451 or 18 F-T807), 18 F-THK5317, and 18 F-THK5351 (45,46). In 2020, 18 F-flortaucipir was the first FDA-approved for clinical and commercial use 52 . These radiotracers bind to regions typically affected regions in AD patients, such as the lateral temporal, lateral and medial parietal (precuneus) and posterior cingulate 45 , 46 , 49 , 51 . Studies with flortaucipir in different AD phenotypes show that the distribution of the tau protein follows the anatomical distribution associated with each cognitive manifestation of the variants 45 , 46 , 49 , 51 .
Although these radiotracers have a higher affinity for tau protein compared to 18 F-FDDNP, they still have some affinity for other protein aggregates, such as TDP-43, Aβ, and alpha-synuclein. For example, studies show that PET with 18 F-flortaucipir ( 18 F-AV1451) has regional uptake in the semantic variant of primary progressive aphasias, whose pathology is most commonly TDP-43 45 , 46 . Furthermore, these radiotracers have low specificity in distinguishing the type of tauopathy (3R vs. 4R) and the level of maturity of the tau deposit (pre-tangle, mature neurofibrillary tangle, and phantom tangle), as well as the type of cell affected (neurons versus glia). It is known, for example, that in primary tauopathies, especially in PSP and CBD, glial cells also show deposits of hyperphosphorylated tau 45 , 46 . The low affinity for other non-3R/4R tauopathies limited the use of first-generation tracers in the clinic.
The second generation of radiotracers was more recently developed. Some examples are: 18F-MK-6240, 18F-RO-948, 18F-PI-2620, 18F-GTP1, 18F-PM-PBB3, 18F-JNJ311 and 18F-JNJ-067 45 , 46 . These second-generation radiotracers have the advantage of having a higher affinity for hyperphosphorylated tau protein and less binding to non-tau targets compared to first-generation radiotracers 53 - 55 . Second-generation radiotracers have shown that tau PET has a good accuracy in predicting the trajectory of cognitive decline in cognitively normal subjects or those with MCI, superior to amyloid PET 50 , 56 .
Two of these tracers ( 18 F-MK-6240 and 18 F-PI-2620) are in an advanced stage of testing in clinical settings and showed promising results in 4R tauopathies, such as CBD and PSP. The main advantage of this generation of tau PET is its use in primary tauopathies. PSP has the advantage of being exclusively a tauopathy (unlike CBD or even FTD which can be phenotypes of different proteinopathies). Some first-generation radiotracers show good affinities for tau deposits in other non-AD tauopathies, and the uptake pattern may indicate whether it is a primary or secondary AD tauopathy 45 , 49 . These radiopharmaceuticals also have an affinity for monoamine oxidase B (MAO-B), which is abundantly present in base nuclei and may lead to false positives 45 , 49 . 18 F-flortaucipir, the most used first-generation tracer, cannot reliably differentiate the types of non-AD tauopathies (PSP, CBD or FTD) and may not detect the early stages of Braak (I to III) 57 , 58 . 18 F-RO-948 was also shown to have good accuracy in differentiating AD from non-AD tauopathies in a comparative post-mortem study 55 .
If second-generation tau PET tracers continuously prove their value, they have the potential to replace amyloid and FDG-PET in some scenarios in clinical practice (e.g., in the different types of tauopathies), providing “one-stop-shop” studies of pathology and disease staging at the same time.
FDG-PET in Alzheimer's disease
Despite advances in PET ligands specific for pathological protein deposits, FDG-PET remains the main molecular neuroimaging method available in clinical practice. Studies show that a pattern of regional hypometabolism in areas of association temporoparietal, medial temporal, precuneus and posterior cingulate have a high specificity for AD ranging from 86 to 98%, compared to pathological diagnosis (Figure 1) 17 , 35 . Its accuracy in the evaluation of AD and FTD variants is comparable to amyloid PET, with the advantage of providing insights on disease stage 17 . Furthermore, FDG PET findings precede the structural changes seen on MRI, another biomarker of neurodegeneration. Patients with MCI who present this “AD pattern” have a 75 to 100% accuracy in predicting conversion to dementia 35 Interestingly, the posterior cingulate has been shown to be the region whose involvement best predicts MCI conversion to dementia due to AD 35 .
FDG-PET is also very useful in the differential diagnosis between AD and other neurodegenerative dementia, by showing specific regional patterns of hypometabolism in each condition (Table 1). This good specificity even helps in the differential diagnosis between AD variants and other groups of neurodegenerative dementias. For example, anterior cingulate hypometabolism makes it possible to accurately differentiate a bvFTD from a dysexecutive/behavioral variant of AD 41 , 59 . In CBS, temporoparietal and posterior cingulate hypometabolism suggest an AD pathology and predicts a positive amyloid PET with 88.5% of accuracy and 100% of positive predictive value, whereas asymmetric hypometabolism of the dorsolateral frontoparietal cortex, including sensorimotor cortex, thalamus, and striatum, suggests non-AD CBS (generally, 4R tauopathy) 60 .
Comparative studies of tau PET and FDG-PET imaging indicate an overlap between areas of tau protein accumulation and hypometabolism. This overlap, on the other hand, does not occur between amyloid PET and FDG PET. This has two consequences: first, that tauopathy correlates more directly with neuronal injury and synaptic dysfunction than does amyloid pathology, and second, that FDG PET may be a better substitute for tau PET than amyloid PET in places where this test is unavailable 42 by showing simultaneously specific patterns of neurodegeneration and helping to stage the disease. This use of FDG-PET as a proxy for tau pathology was previously foreseen 26 .
Additionally, the combination of amyloid and FDG-PET provided incredibly high sensitivity and specificity of 97% and 98%, if both are congruent, in a post-mortem study 17 . This was replicated in a PET study using the rationale of the “ATN” staging, where an FDG-PET with an AD-pattern predicted amyloid positivity in 93% of cases (27 of 29 FDG-positive individuals), and provided alternative diagnostic hypotheses (e.g. frontal hypometabolism suggestive of FTD) in amyloid-negative individuals 42 . However, it is essential to emphasize that according to the amyloid cascade hypothesis, the changes in tau PET would precede those observed in FDG-PET, a less specific biomarker of neurodegeneration.
MOLECULAR NEUROIMAGING IN DEMENTIA WITH LEWY BODIES
Dementia with Lewy bodies (DLB), like Parkinson's disease (PD) and multiple system atrophy (MSA), is part of a group of neurodegenerative diseases characterized by the presence of alpha-synuclein neuronal inclusions 61 . DLB is described as the second cause of neurodegenerative dementia after the age of 65. The current diagnostic criteria for DLB were defined by a consensus in 2017 that included, among other points, biomarkers 62 . These biomarkers were classified as indicative, whose specificity is high enough to define the probability of DLB, and supportive biomarkers, with lower sensitivity, but with good accuracy to allow clinical suspicion. The indicative biomarkers are polysomnography, myocardial sympathetic innervation scintigraphy, and PET/SPECT with radiotracer for dopamine transporter (DAT). Supportive biomarkers included FDG-PET, MRI and electroencephalogram. None of these biomarkers define the presence of alpha-synuclein in vivo, but somehow reflect the repercussions of the pathophysiological process.
PET or SPECT using dopamine tracers aim to assess the integrity of the nigrostriatal dopaminergic pathways through the study of dopamine transporter, a presynaptic protein with a role in dopamine uptake 63 . The most used radiotracer is 123-I-ioflupane (DATSCAN), but some countries use other tracers like TRODAT-1 (a tropane derivative labeled with 99mTc), whose images are acquired with SPECT, a more spread technology than PET. In DLB and PD, there is a low uptake in basal ganglia, demonstrating the loss of dopaminergic neurons from the nigrostriatal pathways(Figure 1). The specificity and sensitivity to distinguish from AD is 90% and 78%, respectively, which would justify it as an indicative biomarker 62 . It is also very useful for differentiating neurodegenerative parkinsonism from secondary ones (such as medication), and from essential tremors. However, it would not have good specificity to distinguish from other dementia with parkinsonism (eg, PSP and CBD), which makes it questionable as an indicative biomarker in a patient exclusively with parkinsonism and dementia, without other main symptoms 64 .
Myocardial scintigraphy with iodine-123-labeled meta-iodo-benzyl-guanidine (123I-mIBG) is another biomarker with good sensitivity (69%) and high specificity (87%, reaching 94% in mild cases) to differentiate DLB from AD 62 . mIBG is a molecule captured by pre-synaptic noradrenergic neurons and is therefore a biomarker of postganglionic sympathetic innervation. In patients with DLB, there is a denervation of sympathetic fibers in the myocardium and, consequently, there is a low cardiac uptake of 123I-mIBG on scintigraphy. Care should be taken in diabetic patients, autonomic neuropathies, heart disease and users of tricyclics antidepressants, as there may be false-positive low uptake in these individuals.
FDG-PET is considered a supportive biomarker of DLB. As in AD, the pattern is usually of temporoparietal hypometabolism. However, unlike in AD, there may be also a characteristic occipital hypometabolism in DLB, with a sensitivity of 70% and specificity of 74% (Figure 1) when present 62 . In addition, there is a relative preservation of the posterior cingulate (which is classically affected in AD), called the “posterior cingulate island sign” 65 . Amyloid PET, in turn, is of little use in distinguishing between AD and DLB, as there is a variable frequency of deposition of amyloid pathology in patients with DLB.
MOLECULAR NEUROIMAGING IN FRONTOTEMPORAL DEMENTIAS
Frontotemporal dementias (or frontotemporal lobar degeneration) correspond to a group of neurodegenerative diseases where there is a selective involvement of the frontal and temporal lobes 66 . From a clinical phenotype point of view, they comprise two large groups: the behavioral variant (vbFTD) and primary progressive aphasias (PPA). Among the PPAs, in turn, there are three variants: the agrammatic or non-fluent (nfvPPA) and the semantic (svPPA). The logopenic variant of PPA is not part of the FTD syndrome, because of the common underlying AD pathology. More recently, some authors propose a third group of FTD: the motor variants, which would include atypical parkinsonism (PSP and CBD) or motor neuron disease (FTD with amyotrophic lateral sclerosis) 67 . bvFTD corresponds to the second cause of degenerative dementia in the pre-senile age group, after AD.
Just as there is phenotypic heterogeneity, FTD is characterized by pathological variability (Figure 1). Three proteins are associated with FTD: tau, TDP-43 (transactive response DNA binding protein of 43 kDa) and FUS (fused in sarcoma protein). To date, there is no specific radioligand for the last two proteins. In the case of tau protein, studies focus mainly on the use of tau PET in AD, but some studies have been published in samples of patients with FTD, PSP and CBD. As previously described, PSP has been studied as a model of primary tauopathy because, unlike the other phenotypes, PSP is almost exclusively caused by deposits of 4R-tau protein 68 .
Some PET studies highlight the importance of tau protein deposition in FTD. In a study with 18 F-flortaucipir in patients with various FTD phenotypes, including genetic forms of FTD (MAPT and C9orf72 mutations), there was an increase in uptake in the left inferior frontal gyrus compared to the right in cases of nfvPPA. Half of the cases of bvFTD had increased uptake in the frontotemporal region, being more intense in MAPT mutation carriers. Furthermore, uptake of 18 F-flortaucipir was observed in cases of svPPA and mutation of C9orf72 mutation, whose proteinopathy is usually TDP-43 69 . In another study, using the radiopharmaceutical 18 F-MK-6240 in patients with genetic FTD, a mild uptake was noted in cases of symptomatic MAPT carriers. Only one case of a patient with a non-tau mutation (in this case, C9orf72 mutation) showed minimal tracer uptake, suggesting that 18 F-MK-6240 may be a potential marker for primary tauopathies 70 .
However, FDG-PET is still the most-used molecular neuroimaging method in FTD. The presence of a clinical syndrome with typical pattern of hypometabolism makes the diagnosis of FTD or PPA likely, according to diagnostic criteria 71 , 72 . In bvFTD, the pattern of hypometabolism is the involvement of the orbitofrontal region, dorsolateral prefrontal cortex, ventromedial prefrontal cortex, anterior cingulate, temporal poles, and basal ganglia, often asymmetrically (Figure 1). In patients whose underlying pathology is a tauopathy (particularly in Pick's disease), anterior frontotemporal atrophy may be very pronounced compared to the posterior temporoparietal cortex ("knife-edge" pattern). Patients with MAPT mutations present a relatively symmetrical pattern of hypometabolism of the orbitofrontal, dorsolateral prefrontal, and especially the anterior temporal lobes. In individuals carrying the GRN mutations, hypometabolism is asymmetric, with extension to the parietal lobe 73 . In patients with FUS pathology, there is a characteristic involvement of the caudate nuclei and ventral frontal cortex 73 , 74 .
In PPAs, hypometabolism reflects the pattern of pathology involvement within the language neural network. In nfvPPA, the left inferior frontal cortex is affected, commonly extending to the anterior insula. The hypometabolism of the temporal poles, more to the left, with extension to the lateral temporal cortex, is the hallmark of the svPPA pattern. lvPPA is characterized by left temporoparietal hypometabolism, a typical pattern of AD 72 .
WHEN AND HOW SHOULD MOLECULAR NEUROIMAGING TESTS BE REQUESTED IN COGNITIVE DECLINE IN CLINICAL PRACTICE?
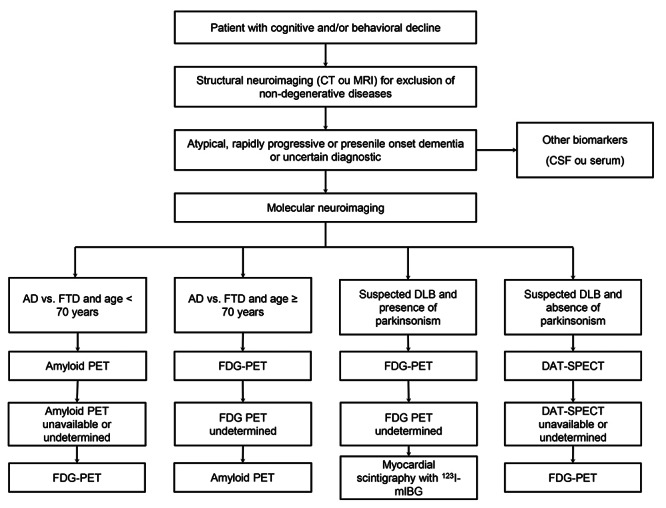
In clinical practice, molecular neuroimaging should be requested carefully, as it is a high-cost and low-availability exam. But it is very useful in situations where the diagnosis of cognitive decline is uncertain, especially when the clinical manifestations are atypical, early-onset or rapidly progressive dementias. It is important to emphasize that before requesting a molecular neuroimaging, the recommendation is always to perform a structural neuroimaging, in order to exclude structural pathologies. CSF biomarkers (in the future, plasma biomarkers) are alternatives to molecular neuroimaging. However, CSF biomarkers have also high-cost, in addition to presenting some measurement problems, especially for the beta-amyloid peptide (pre-analytical errors, lack of interlaboratory reliability) which can lead to false-results. FDG-PET is the most available molecular neuroimaging test. It is important to emphasize that FDG-PET should ideally be performed in clinics whose evaluation is not only visual, but also a semi-quantitative analysis is performed to increase the accuracy of the method. Amyloid PET is restricted to a few specialized centers and, so far, tau PET is not available in our country. Regardless of availability, FDG has a good accuracy for the differential diagnosis between neurodegenerative dementias. We propose a flowchart in the request for molecular neuroimaging in the diagnostic investigation of cognitive and/or behavioral decline (Figure 2).
Figure 2. Flowchart showing a proposal in the sequence of the request of molecular neuroimaging in the diagnostic investigation of cognitive and/or behavioral decline in the clinical practice. Abbreviations: FTD: frontotemporal dementia; AD: Alzheimer's disease; DLB: dementia with Lewy bodies.; PET: Positron Emission Tomography; SPECT: single photon emission tomography; FDG: 18F-Fluorodeoxyglucose; DAT: dopamine transporter; 123I-mIBG: iodine-123-labeled meta-iodo-benzyl-guanidine; CSF: cerebrospinal fluid; MRI: magnetic resonance imaging; CT: computed tomography.
References
- Ryan NS, Rossor MN, Fox NC. Alzheimer’s disease in the 100 years since Alzheimer’s death. Brain. 2015;138(12):3816–3821. doi: 10.1093/brain/awv316. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9(1):13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- Kertesz A, Kalvach P. Arnold pick and German neuropsychiatry in Prague. Arch Neurol. 1996;53(9) 1:935–938. doi: 10.1001/archneur.1996.00550090147021. [DOI] [PubMed] [Google Scholar]
- Allegri RF. Moving from neurodegenerative dementias, to cognitive proteinopathies, replacing “where” by “what”…. Dement Neuropsychol. 2020;14(3):237–242. doi: 10.1590/1980-57642020dn14-030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espay AJ, Vizcarra JA, Marsili L, Lang AE, Simon DK, Merola A, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology. 2019;92(7):329–337. doi: 10.1212/wnl.0000000000006926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson O, Batrla R, Brix B, Carrillo MC, Corradini V, Edelmayer RM, et al. The Alzheimer’s Association international guidelines for handling of cerebrospinal fluid for routine clinical measurements of amyloid β and tau. Alzheimers Dement. 2021;17(9):1575–1582. doi: 10.1002/alz.12316. [DOI] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72(4):578–586. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–128. doi: 10.1016/s1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califf RM. Biomarker definitions and their applications. Exp Biol Med. 2018;243(3):213–221. doi: 10.1177/1535370217750088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE. Biological Markers of Alzheimer’ s Disease. Neurobiol Aging. 1998;19(2):145–147. doi: 10.1016/s0197-4580(98)00013-x. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Prim. 2021;7(1):33. doi: 10.1038/s41572-021-00269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Doré V, Burnham SC, Masters CL, Rowe CC. Imaging Tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol. 2018;14(4):225–236. doi: 10.1038/nrneurol.2018.9. [DOI] [PubMed] [Google Scholar]
- Laforce R, Jr, Soucy JP, Sellami L, Dallaire-Théroux C, Brunet F, Bergeron D, et al. Molecular imaging in dementia: Past, present, and future. Alzheimer’s Dement. 2018;14(11):1522–1552. doi: 10.1016/j.jalz.2018.06.2855. [DOI] [PubMed] [Google Scholar]
- Perani D, Iaccarino L, Lammertsma AA, Windhorst AD, Edison P, Boellaard R, et al. A new perspective for advanced positron emission tomography-based molecular imaging in neurodegenerative proteinopathies. Alzheimers Dement. 2019;15(8):1081–1103. doi: 10.1016/j.jalz.2019.02.004. [DOI] [PubMed] [Google Scholar]
- Zimmer ER, Parent MJ, Souza DG, Leuzy A, Lecrux C, Kim HI, et al. FDG PET signal is driven by astroglial glutamate transport. Nat Neurosci. 2017;20(3):393–395. doi: 10.1038/nn.4492. 18F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesman-Segev OH, La Joie R, Iaccarino L, Lobach I, Rosen HJ, Seo SW, et al. Diagnostic accuracy of Amyloid versus 18F-Fluorodeoxyglucose Positron Emission Tomography in Autopsy-Confirmed Dementia. Ann Neurol. 2021;89(2):389–401. doi: 10.1002/ana.25968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chételat G, Arbizu J, Barthel H, Garibotto V, Law I, Morbelli S, et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 2020;19(11):951–962. doi: 10.1016/S1474-4422(20)30314-8. [DOI] [PubMed] [Google Scholar]
- Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397(10284):1577–1590. doi: 10.1016/S0140-6736(20)32205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, Cummings J, Blennow K, Gao P, Jack CR, Jr, Vergallo A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol. 2021;17(9):580–589. doi: 10.1038/s41582-021-00520-w. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Haeberlein SB, Carrillo MC, Hendrix JA, Kerchner G, Margolin R, et al. The National Institute on Aging and the Alzheimer’s Association Research Framework for Alzheimer’s disease: perspectives from the research roundtable. Alzheimers Dement. 2018;14(4):563–575. doi: 10.1016/j.jalz.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Villain N, Frisoni GB, Rabinovici GD, Sabbagh M, Cappa S, et al. Clinical diagnosis of Alzheimer’s disease: recommendations of the International Working Group. Lancet Neurol. 2021;20(6):P484–P496. doi: 10.1016/s1474-4422(21)00066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Wiste HJ, Weigand SD, Rocca WA, Knopman DS, Mielke MM, et al. Age-specific population frequencies of cerebral β-amyloidosis and neurodegeneration among people with normal cognitive function aged 50-89 years: a cross-sectional study. Lancet Neurol. 2014;13(10):997–1005. doi: 10.1016/s1474-4422(14)70194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JB, Zetterberg H, Van Harten AC, Glodzik L, Martinez-Lage P, Bocchio-Chiavetto L, et al. Alzheimer’s disease cerebrospinal fluid biomarker in cognitively normal subjects. Brain. 2015;138(9):2701–2715. doi: 10.1093/brain/awv199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JB, Bjerke M, Chen K, Rozycki M, Jack CR, Jr, Weiner MW, et al. Memory, executive, and multidomain subtle cognitive impairment: Clinical and biomarker findings. Neurology. 2015;85(2):144–153. doi: 10.1212/wnl.0000000000001738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitz BE, Weissfeld LA, Lopez OL, Kuller LH, Saxton J, Singhabahu DM, et al. Cognitive trajectories associated with β-amyloid deposition in the oldest-old without dementia. Neurology. 2013;80(15):1378–1384. doi: 10.1212/wnl.0b013e31828c2fc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villain N, Chételat G, Grassiot B, Bourgeat P, Jones G, Ellis KA, et al. Regional dynamics of amyloid-β deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: a voxelwise PiB-PET longitudinal study. Brain. 2012;135(7):2126–2139. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by ¿11CÁPiB. Neurology. 2010;74(10):807–815. doi: 10.1212/wnl.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré V, Villemagne VL, Bourgeat P, Fripp J, Acosta O, Chetélat G, et al. Cross-sectional and longitudinal analysis of the relationship between aβ deposition, cortical thickness, and memory in cognitively unimpaired individuals and in alzheimer disease. JAMA Neurol. 2013;70(7):903–911. doi: 10.1001/jamaneurol.2013.1062. [DOI] [PubMed] [Google Scholar]
- Schilling LP Zimmer ER, Shin M Leuzy A, Pascoal TA Bene, et al. Imaging Alzheimer’s disease pathophysiology with PET. Dement Neuropsychol. 2016;10(2):79–90. doi: 10.1590/s1980-5764-2016dn1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray ME, Lowe VJ, Graff-Radford NR, Liesinger AM, Cannon A, Przybelski SA, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain. 2015;138(5):1370–1381. doi: 10.1093/brain/awv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri O, Sabbagh MN, Seibyl J, Barthel H, Akatsu H, Ouchi Y, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: Phase 3 study. Alzheimer’s Dement. 2015;11(8):964–974. doi: 10.1016/j.jalz.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Gatsonis C, Apgar C, Chaudhary K, Gareen I, Hanna L, et al. Association of amyloid positron emission tomography with subsequent change in clinical management among medicare beneficiaries with mild cognitive impairment or dementia. JAMA. 2019;321(13):1286–1294. doi: 10.1001/jama.2019.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa C, Ishii K, Hyodo T, Sakaguchi K, Usami K, Shimamoto K, et al. Investigation of 11C-PiB equivocal PET findings. Ann Nucl Med. 2015;29(2):164–169. doi: 10.1007/s12149-014-0924-8. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD, Jagust WJ, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11(1):1–15. doi: 10.1016/j.jalz.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford J, Yong KXX, Apostolova LG, Bouwman FH, Carrillo M, Dickerson BC, et al. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021;20(3):222–234. doi: 10.1016/S1474-4422(20)30440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho AM, Busatto GF, Porto FHG, Faria DP, Ono CR, Garcez AT, et al. Correction to: Brain PET amyloid and neurodegeneration biomarkers in the context of the 2018 NIA-AA research framework: an individual approach exploring clinical-biomarker mismatches and sociodemographic parameters. Eur J Nucl Med Mol Imaging. 2020;47(11):2715–2716. doi: 10.1007/s00259-020-04933-5. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Therneau TM, Wiste WHJ, Weigand SD, Knopman DS, Lowe VJ, et al. Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study. Lancet Neurol. 2016;15(1):P56–P64. doi: 10.1016/S1474-4422(15)00323-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):P357–P367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Wang YT, Edison P. Tau Imaging in Neurodegenerative Diseases Using Positron Emission Tomography. Curr Neurol Neurosci Rep. 2019;19(7):45. doi: 10.1007/s11910-019-0962-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Chiotis K, Lemoine L, Gillberg PG, Almkvist O, Rodriguez-Vieitez E, et al. Tau PET imaging in neurodegenerative tauopathies -still a challenge. Mol Psychiatry. 2019;24(8):1112–1134. doi: 10.1038/s41380-018-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of Tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21(1):21–35. doi: 10.1038/s41583-019-0240-3. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Altomare D, Thal DR, Ribaldi F, van der Kant R, Ossenkoppele R, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci. 2022;23(1):53–66. doi: 10.1038/s41583-021-00533-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Smith R, Cullen NC, Strandberg O, Vogel JW, Binette AP, et al. Biomarker-based prediction of longitudinal Tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2022;79(2):149–158. doi: 10.1001/jamaneurol.2021.4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Smith R, Mattsson-Carlgren N, Groot C, Leuzy A, Strandberg O, et al. Accuracy of Tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head-to-head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78(8):961–971. doi: 10.1001/jamaneurol.2021.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Pontecorvo MJ, Devous MD, Lu M, Arora AK, Truocchio SP, et al. Positron emission tomography imaging with ¿18FÁflortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 2020;77(7):829–839. doi: 10.1001/jamaneurol.2020.0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattay VS, Fotenos AF, Ganley CJ, Marzella L. Brain Tau imaging: food and drug administration approval of 18F-flortaucipir injection. J Nucl Med. 2020;61(10):1411–1412. doi: 10.2967/jnumed.120.252254. [DOI] [PubMed] [Google Scholar]
- Mormino EC, Toueg TN, Azevedo C, Castillo JB, Guo W, Nadiadwala A, et al. Tau PET imaging with 18F-PI-2620 in aging and neurodegenerative diseases. Eur J Nucl Med Mol Imaging. 2021;48(7):2233–2244. doi: 10.1007/s00259-020-04923-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarte M-L, Nordberg A, Lemoine L. Characterization of MK6240, a Tau PET tracer, in autopsy brain tissue from Alzheimer’s disease cases. Eur J Nucl Med Mol Imaging. 2021;48(4):1093–1102. doi: 10.1007/s00259-020-05035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Smith R, Ossenkoppele R, Santillo A, Borroni E, Klein G, et al. Diagnostic performance of RO948 F 18 Tau positron emission tomography in the differentiation of Alzheimer disease from other neurodegenerative disorders. JAMA Neurol. 2020;77(8):955–965. doi: 10.1001/jamaneurol.2020.0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Smith R, Cullen NC, Strandberg O, Vogel JW, Binette AP, et al. Biomarker-Based Prediction of Longitudinal Tau Positron Emission Tomography in Alzheimer Disease. JAMA Neurol. 2022;79(2):149–158. doi: 10.1001/jamaneurol.2021.4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani-Meigooni DN, Iaccarino L, La Joie R, Baker S, Bourakova V, Boxer AL, et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain. 2020;143(11):3477–3494. doi: 10.1093/brain/awaa276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel M, Barthel H, van Eimeren T, Marek K, Beyer L, Song M, et al. Assessment of 18F-PI-2620 as a biomarker in progressive supranuclear palsy. JAMA Neurol. 2020;77(11):1408–1419. doi: 10.1001/jamaneurol.2020.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Pijnenburg YAL, Perry DC, Cohn-Sheehy BI, Scheltens NME, Vogel JW, et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain. 2015;138(9):2732–2749. doi: 10.1093/brain/awv191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmera JB, Coutinho AM, Aranha MR, Studart-Neto A, Carneiro CG, de Almeida IJ, et al. FDG-PET patterns predict amyloid deposition and clinical profile in corticobasal syndrome. Mov Disord. 2021;36(3):651–661. doi: 10.1002/mds.28373. [DOI] [PubMed] [Google Scholar]
- Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet. 2015;386(10004):1683–1697. doi: 10.1016/S0140-6736(15)00462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies. Neurology. 2017;89(1):88–100. doi: 10.1212/WNL.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoessl AJ, Lehericy S, Strafella AP. Imaging insights into basal ganglia function, Parkinson’s disease, and dystonia. Lancet. 2014;384(9942):532–544. doi: 10.1016/S0140-6736(14)60041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmera JB, Brucki S, Nitrini R. Reader response: diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2018;90(6):299–300. doi: 10.1212/WNL.0000000000004917. [DOI] [PubMed] [Google Scholar]
- Graff-Radford J, Murray ME, Lowe VJ, Boeve BF, Ferman TJ, Przybelski SA, et al. Dementia with Lewy bodies: basis of cingulate island sign. Neurology. 2014;83(9):801–809. doi: 10.1212/WNL.0000000000000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):P1672–P1682. doi: 10.1016/S0140-6736(15)00461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A, et al. Structure-based classification of tauopathies. Nature. 2021;598(7880):359–363. doi: 10.1038/s41586-021-03911-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli M, Strafella AP. New advances in Tau imaging in parkinsonism. Int Rev Psychiatry. 2017;29(6):628–635. doi: 10.1080/09540261.2017.1396446. [DOI] [PubMed] [Google Scholar]
- Tsai RM, Bejanin A, Lesman-Segev O, Lajoie R, Visani A, Bourakova V, et al. 18F-flortaucipir (AV-1451) Tau PET in frontotemporal dementia syndromes. Alzheimers Res Ther. 2019;11(1):13. doi: 10.1186/s13195-019-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JP, Bezgin G, Savard M, Pascoal TA, Finger E, Laforce R, et al. 18F-MK-6240 Tau-PET in genetic frontotemporal dementia. Brain. 2022;145(5):1763–1772. doi: 10.1093/brain/awab392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Semin Neurol. 2007;27(1):48–57. doi: 10.1055/s-2006-956755. [DOI] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeter LH, Kaat LD, Rohrer JD, van Swieten JC. Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol. 2017;13(7):406–419. doi: 10.1038/nrneurol.2017.75. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, Dejesus-Hernandez M, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, Tau, progranulin and sporadics. Brain. 2012;135(3):794–806. doi: 10.1093/brain/aws001. [DOI] [PMC free article] [PubMed] [Google Scholar]