

Summary
Artemisia argyi, as famous as Artemisia annua, is a medicinal plant with huge economic value in the genus of Artemisia and has been widely used in the world for about 3000 years. However, a lack of the reference genome severely hinders the understanding of genetic basis for the active ingredient synthesis of A. argyi. Here, we firstly report a complex chromosome‐level genome assembly of A. argyi with a large size of 8.03 Gb, with features of high heterozygosity (2.36%), high repetitive sequences (73.59%) and a huge number of protein‐coding genes (279 294 in total). The assembly reveals at least three rounds of whole‐genome duplication (WGD) events, including a recent WGD event in the A. argyi genome, and a recent burst of transposable element, which may contribute to its large genome size. The genomic data and karyotype analyses confirmed that A. argyi is an allotetraploid with 34 chromosomes. Intragenome synteny analysis revealed that chromosomes fusion event occurred in the A. argyi genome, which elucidates the changes in basic chromosome numbers in Artemisia genus. Significant expansion of genes related to photosynthesis, DNA replication, stress responses and secondary metabolism were identified in A. argyi, explaining the extensive environmental adaptability and rapid growth characteristics. In addition, we analysed genes involved in the biosynthesis pathways of flavonoids and terpenoids, and found that extensive gene amplification and tandem duplication contributed to the high contents of metabolites in A. argyi. Overall, the reference genome assembly provides scientific support for evolutionary biology, functional genomics and breeding in A. argyi and other Artemisia species.
Keywords: Artemisia argyi genome, genome evolution, chromosome fusion, gene expansion, secondary metabolism
Artemisia argyi is a medicinal plant with huge economic value. Here, Miao et al construct a chromosome‐level genome of A. argyi, revealing the chromosome fusion event and extensive expansion of genes related to flavonoids and terpenoids synthesis.
Introduction
Artemisia is a large plant genus in the Asteraceae family that comprises approximately 500 species and subspecies (Bora and Sharma, 2011; Lee et al., 2006). These species are mainly distributed in the temperate northern hemisphere regions (Naß and Efferth, 2018) and most are widely used in various fields such as herb, food, cosmetics, spices, forage and ornamentals (Torrell et al., 2003). In 2015, the discovery of artemisinin, an antimalarial ingredient isolated from Artemisia annua, won the Nobel Prize in Physiology or Medicine, drawing global attention to study other species of the genus Artemisia (Efferth et al., 2015; Su and Miller, 2015).
Previous cytogenetic studies have contributed to the knowledge of the systematic and evolutionary relationship within the Artemisia species. Three basic chromosome numbers were reported in the genus based on numerous chromosome counts from approximately 373 taxa; x = 9 is the most common (85.6%), and x = 8 is less frequent (9.7%). Both basic chromosome numbers exhibited polyploid series, with known levels up to 16 x for x = 9 and hexaploid for x = 8. In addition, a chromosome number of 2 n = 34 occurs in a few species, such as Artemisia vulgaris, Artemisia rubipes and Artemisia argyi (Hoshi et al., 2003), suggesting that a third base number x = 17 may exist. The diversity of chromosome number and polyploidy level results in a 7.4‐fold variation in Artemisia genome size, from 4.11 Gb of A. dolosa (2 n = 2 x = 18) to 30.45 Gb of A. medioxima (2 n = 16 x = 144) (Pellicer et al., 2010). The chromosome numbers observed in Artemisia species suggest that, in addition to polyploidization, variation in basic chromosome numbers may also play an important role in the evolution of the genus. Chromosomal fusion and fission are considered the predominant causes for the evolution of basic chromosome numbers in animal and plant kingdoms. For example, the origin of human chromosome 2 was derived from head‐to‐head fusion of two ancestral ape chromosomes (Baldini et al., 1991). Chromosome fusion affects genetic diversity and environmental adaptation of Heliconius (Cicconardi et al., 2021). Large‐scale chromosomal fission/fusion events promote the speciation of the wild Morus notabilis (x = 6) and the cultivated Morus alba (x = 14) (Xuan et al., 2022). There are far more examples of chromosome fusion that could be discussed. In Artemisia, fluorechrome‐banded karyotypes of A. vulgaris provide some evidence that a centric (Robertsonian) chromosome fusion may also occur and cause the reduction of its basic chromosome number from x = 9 to x = 8 (Xirau and Siljak‐Yakovlev, 1997). However, despite a wide knowledge of cytological studies on Artemisia, the role of chromosome fusion in basic chromosome numbers variation of Artemisia has not been fully verified and highly valued.
Among the Artemisia genus, A. argyi (also called ‘Chinese mugwort’) is one of the well‐known species and is widely distributed in Asian countries, such as China, Korea and Japan (Mei et al., 2016). The dried leaves of A. argyi, known in Chinese as ‘Aiye’, have been used as TCM (traditional Chinese medicine) for about 3000 years (Lv et al., 2018; Song et al., 2019). A. argyi was first recorded in ‘Shi Jing’ (a famous China classical literature) near 1100 BC and was first recognized as medicine in ‘Wu Shi Er Bing Fang’ in the Han Dynasty (A.D. 220). In addition, the medical applications of A. argyi were also listed in many other classic clinical and medical literature such as ‘Huang Di Nei Jing’, ‘Ming Yi Bie Lu’, ‘Jin Gui Yao Lve’ and ‘Ben Cao Gang Mu’ (Li, 1957; Mawangdui Han Danasty Tomb bamboo books research group, 1979; Zhang, 1997). In the long‐term practice of traditional Chinese medicine, A. argyi is believed to have the properties of bitterness, warmth and pungency and has the effects of dispelling cold and dampness, warming menstruation, haemostasis, and preventing abortion (Chinese Pharmacopoeia Commission, 2020). Recent pharmacological studies have demonstrated that A. argyi also exhibits anti‐inflammatory (Yun et al., 2016; Zimmermann‐Klemd et al., 2020), anti‐allergic (Lv et al., 2018), antimicrobial (Pagning et al., 2016), antioxidant (Kim et al., 2015) and anticancer (Seo et al., 2003) activities. Clearly, A. argyi has a long history of application and is still widely used in clinical practice. In 2020, the annual value of the A. argyi market was over 40 billion RMB, making it the largest herbal medicine industry chain in China. In addition, acupuncture and moxibustion (world‐renowned medicinal products derived from A. argyi) were recognized as the World Intangible Cultural Heritage in 2010, and traditional Chinese medicine/therapy (including moxibustion) has been recommended by WHO (World Health Organization) in 2019. Therefore, acupuncture and moxibustion have successfully spread worldwide as a reliable alternative therapy for multiple diseases. As the major source of moxibustion, A. argyi is also becoming increasingly popular worldwide.
These amazing economic and medicinal values of A. argyi are due to the large number of secondary metabolites in its leaves, which include volatile oils, flavonoids, terpenoids, phenolic acids and other compounds (Song et al., 2019). More than 100 nature metabolites have been identified in A. argyi volatile oil, primarily comprising monoterpenes, sesquiterpenes and their derivatives (Guan et al., 2019). These metabolites contribute to the aromatic odours of A. argyi and pharmacological activities against asthma, eczema and cough (Du et al., 2021; Ge et al., 2016). A. argyi leaves are also rich in flavonoids, including flavonoids, flavonols, flavonols and chalcone (Lv et al., 2018). Among them, eupatilin, jaceosidin, apigenin, luteolin, quercetin, and naringin are representative components and have been proven to have biological activities for preventing oxidative damage, inflammation, allergies and tumours (Maleki et al., 2019; Nabavi et al., 2015; Serafini et al., 2010). Remarkably, eupatilin has been confirmed as the pharmacodynamic component of Stillen® (DA‐9601) which has been approved as a phytomedicine for gastritis in Korea. Thereby, it is necessary to investigate the biosynthesis pathways of these active secondary metabolites in A. argyi.
However, despite the huge economic and medical value, A. argyi's evolution and the molecular basis of the biosynthesis of the abundant active ingredients are rarely reported due to the lack of a high‐quality reference genome. Here, we constructed a chromosome‐level genome of A. argyi through an integrative approach combining PacBio sequencing (SMRT sequencing high fidelity, HiFi) and high‐throughput chromatin conformation capture (Hi‐C) technology. In addition, whole‐genome duplication (WGD) events and expansion and contraction of gene families in the A. argyi genome were also investigated through phylogenetic and comparative genomic analysis. Furthermore, pivotal candidate genes involved in the biosynthesis of terpenoids and flavonoids were also identified based on genomic and transcriptomic analyses. Briefly, as the first chromosome‐level genome in Artemisia, this reference genome will provide valuable resources for exploring the genetic and evolutionary biology of A. argyi and other Artemisia species.
Results
Artemisia argyi genome sequencing, assembly and annotation
Qichun (Hubei Province) is the authentic production area of A. argyi in China. Based on preliminary resource evaluation, a highly volatile oil‐ and flavonoid‐producing A. argyi cultivar, ‘Xiang Ai’ from Qichun, was selected for de novo genome sequencing and assembly (Figure 1a). The somatic cells of A. argyi contained 34 chromosomes by the cytological observation method (Figure 1b) and the genome size was approximately 7.44 Gb by flow cytometry estimation (Figure S1). A 21‐mer analysis of genome survey sequencing shows that A. argyi is a tetraploid, with a monoploid genome size of ~1.96 Gb and a whole‐genome size of ~7.84 Gb (Figure S2). Compared to 12 other genome‐sequenced species in Asteraceae (Erigeron breviscapus, Helianthus annuus, Lactuca sativa, Artemisia annua, Cynara cardunculus, Conyza canadensis, Chrysanthemum nankingense, Chrysanthemum seticuspe, Carthamus tinctorius, Mikania micrantha, Tanacetum cinerariifolium, Taraxacum kok‐saghyz Rodin) (Badouin et al., 2017; Lin et al., 2018; Liu et al., 2020; Peng et al., 2014; Reyes‐Chin‐Wo et al., 2017; Scaglione et al., 2016; Shen et al., 2018; Song et al., 2018; Wu et al., 2021; Yamashiro et al., 2019; Yang et al., 2017), A. argyi features the largest and most complex genome. It was also estimated that the genome of A. argyi had a relatively high heterozygosity (2.36%) and a large proportion of repetitive sequences (75.86%) (Figure S2, Table S1), which increased the challenge of de novo assembly of this genome.
Figure 1.
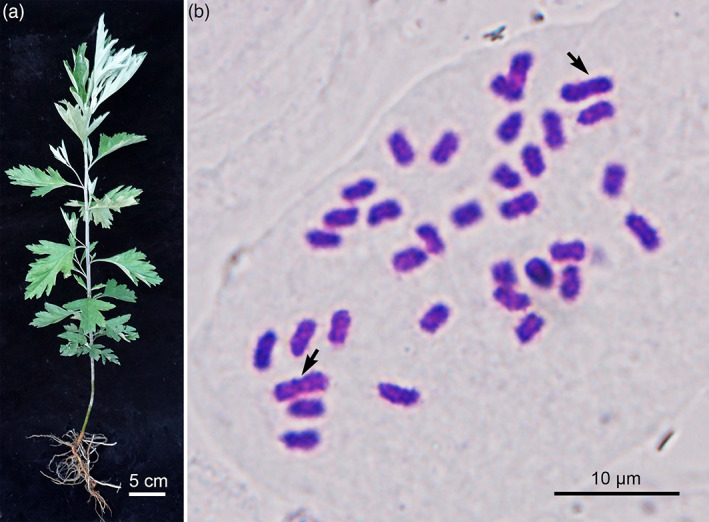
Plant morphology and somatic chromosome number of A. argyi. (a). A plant of A. argyi cultivar ‘Xiang Ai’. (b). The karyotype of A. argyi. [Colour figure can be viewed at wileyonlinelibrary.com]
To overcome the assembly challenging of the A. argyi genome caused by polyploidy, high heterozygosity and repetitive sequences, an integrated strategy was adopted by combining Illumina short paired‐end reads, PacBio long high‐fidelity (HiFi) reads, and Hi‐C sequencing (Figure S3). A total of 161.3 Gb of PacBio circular consensus sequencing (CCS) reads with an average length of 15 154 bp and 19‐fold whole‐genome coverage was obtained from 7 flow cells of the PacBio Sequel II platform (Table S2). These CCS reads were assembled into an initial genome with a total length of approximately 8.03 Gb, containing 10 274 contigs, with an N50 of 8.32 Mb and a longest contig of 43.52 Mb (Table S3) by Hifiasm assembly (Cheng et al., 2021). Subsequently, a total of 407 Gb clean Hi‐C paired‐end reads were used for scaffold extension and chromosome mounting (Table S4). With the assistance of Hi‐C sequence data, the assembled contigs were anchored to 34 super‐scaffolds, which covered 91.4% (7.34 Gb) of the size of the assembled genome (Tables S5 and S4). In summary, the chromosome‐level A. argyi genome assembly has a total size of approximately 8.03 Gb, containing 12 449 scaffolds, with a scaffold N50 size of 206.40 Mb and a contig N50 of 6.25 Mb (Table 1 and Figure 2a).
Table 1.
Major features of the Artemisia argyi genome assembly
| Assembly feature | Size/Number | |
|---|---|---|
| Hifi assembled | Hi‐C Anchored | |
| Assembly size (Mb) | 8029.08 | 8030.38 |
| GC % | 35.46 | 35.46 |
| Repeat (%) | 73.59 | 73.59 |
| Number of scaffolds | – | 12 449 |
| Scaffold N50 size (Mb) | – | 206.40 |
| Scaffold N90 size (Mb) | – | 180.30 |
| Longest scaffolds (Mb) | – | 342.73 |
| Number of contigs | 10 274 | 15 063 |
| Contig N50 size (Mb) | 8.32 | 6.25 |
| Contig N90 size (Mb) | 1.46 | 0.64 |
| Longest contig (Mb) | 43.52 | 40.67 |
Figure 2.
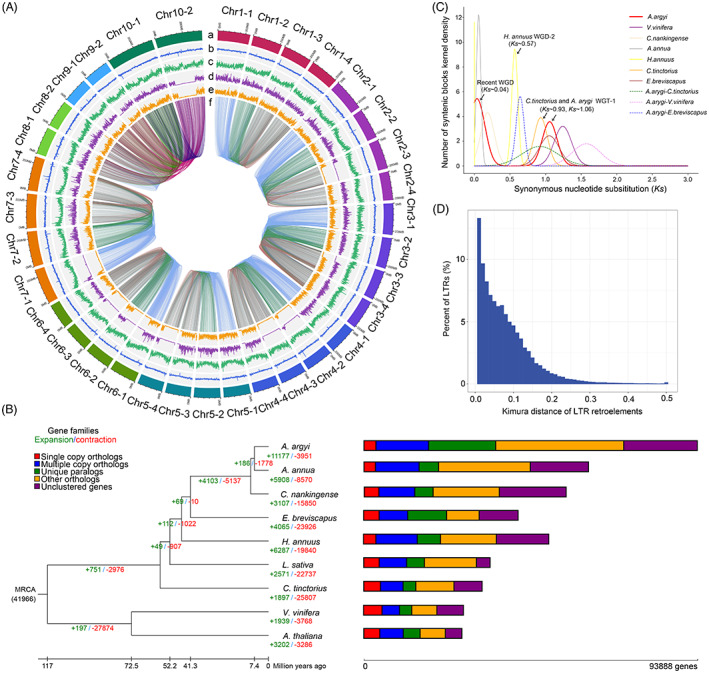
Assembly and genomic features of the A. argyi genome. (A). The circos diagram of A. argyi draft. (a) the genomic landscape of the 34 A. argyi pseudochromosomes. (b) the density of gene. (c) repeat coverage. (d) the density of SNP. (e) the density of Indel. f. synteny relationship between pseudo‐chromosomes. (B). Phylogenetic tree of seven species from the Asteraceae based on the information of single copy genes. And Arabidopsis and V. vinifera were used as the outgroup. The expanded gene families were marked with green and the contracted gene families were marked with red. (C). The Ks distributions of paralogous genes in A. argyi, A. annua, C. nankingense, H. annuus, E. breviscapus and C. tinctorius of the Asteraceae, and the eudicot species V. vinifera. (D). Kimula distance of LTR retroelements. [Colour figure can be viewed at wileyonlinelibrary.com]
To assess the completeness of the assembly of A. argyi genome, the Illumina short reads and PacBio isoform sequencing (Iso‐Seq) data were aligned to the assembled genome, resulting in high mapping rates of 99.89% and 99.70%, respectively (Table S6). Furthermore, benchmarking universal single‐copy orthologue (BUSCO) analysis (Simao et al., 2015) was also employed to assess the quality of the assembly. The obtained results showed that 95.5% (2221 out of 2236 BUSCOs) of the BUSCOs were completely present in the A. argyi genome (2192 of the 2236 BUSCO genes were complete) (Table S7). In short, these results demonstrated that the assembled A. argyi genome had high completeness.
We further applied a combination of ab initio and homology‐based approaches to identify the repetitive sequences. A total of 73.59% of the assembly was identified as repetitive sequences, including 1.37% DNA transposons, 1.29% interspersed nuclear elements (LINEs) and 39.08% long terminal repeats (LTRs) (Table S8). In addition, higher ratio of Gypsy than Copia elements was observed, each accounting for 24.47% and 13.80% of the genome, respectively, which is similar to the genomic characteristics of other Asteraceae species such as sunflower, stevia, and lettuce. Next, combined with RNA‐seq and full‐length transcriptome data generated from seven different tissues and organs (root, rhizome, stem and leaf A‐D) (Data S1), a total of 279 294 high‐quality protein‐coding genes were annotated for this tetraploid‐resolved genome. We attempted to separate the subgenomes by using K‐mers to examine the potential bias in genome characteristics. However, we failed to distinguish homologous chromosome pairs into distinct A and B subgenomes using the enrichment pattern of K‐mers. Therefore, we selected the longest chromosome from each pair of homologous chromosomes as one set of chromosomes of A. argyi, except for chromosome 10, because it was fused from chromosomes 8 and 9 (We address this further below). This monoploid genome contained 64 354 genes. We also counted the genes in contigs that were not anchored into chromosomes and 29 534 genes were obtained. The gene number ranks A. argyi as the most gene‐enriched species among the sequenced Asteraceae plants, and this gene number is about 2.5 times the average number of genes (36 795) reported for plant genomes (Ramírez‐Sánchez et al., 2016). The average lengths of gene and coding DNA sequence (CDS) were 3416 and 1256 bp, respectively, with an average of 5 exons and 4 introns per gene (Table S9). A total of 93.87% and 93.98% of these genes were functionally annotated in the Nr and TrEMBL databases, and 72.30% of the genes were classified by Gene Ontology (GO) terms, and 31.13% of the genes were annotated to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Table S10). In addition, 1033 miRNAs, 19 784 tRNAs, 310 rRNAs and 3606 SnRNAs were identified in the A. argyi genome (Table S11).
Comparative genomic analysis of A. argyi
To gain insights into the evolution of the A. argyi genome, a comparative genomic analysis was performed using Arabidopsis thaliana, Vitis vinifera and six other Asteraceae species (A. annua, C. nankingense, E. breviscapus, H. annus, L. sativa and C. tinctorius). According to the sequence homology among these nine species, 93 888 protein‐coding genes (including 64 354 genes in the monoploid genome of A. argyi and 29 534 genes in scattered contigs) comprised 3384 single‐copy orthologues, 14 817 multiple‐copy orthologues, 18 902 unique paralogues, 36 066 other paralogues and 20 719 unclustered genes (Figure 2b and Table S12). These genes were clustered into 27 915 gene families; among them, 5321 (19.06%) gene families contained 18 902 unique genes in A. argyi (Table S13). GO enrichment analysis showed that the biological functions of these specific gene families were enriched in RNA‐directed DNA polymerase activity (GO: 0003964), DNA polymerase activity (GO: 0034061), nucleotidyltransferase activity (GO: 0016779) and zinc ion binding (GO: 0008270) (Figure S5 and Data S2). Meanwhile, KEGG enrichment results showed that these genes were mainly enriched in pathways involved in phagosome (ko04145), protein processing in endoplasmic reticulum (ko04141), mismatch repair (ko03430) and terpenoid backbone biosynthesis (ko00900) (Figure S6 and Data S3). Furthermore, gene family evolution analysis showed that 40.04% (11 177/27915) of the gene families were expanded and 14.15% (3951/27 915) of the gene families were contracted in the A. argyi genome (Figure 2b). Compared to the other six species in the Asteraceae family, whose contracted genes were larger than the expanded genes, the number of expanded gene families in A. argyi was approximately three times that of the contracted gene families. GO enrichment analysis indicated that the functions of the expanded genes were significantly related in terms of binding, catalytic activity, photosynthetic electron transport in photosystem II and oxidoreductase activity (Figure S7 and Data S4). KEGG analysis revealed that expanded genes were enriched in photosynthesis, DNA replication, homologous recombination and several secondary metabolic pathways (Figure S8 and Data S5). In particular, several markedly expanded genes were identified including PsbA genes (encoded photosystem II P680 reaction center protein D1) in PSII, replication factor A1 (RPA1) participating in homologous recombination and DNA replication, and heat shock protein genes (HSPs, HSP40, HSP70, HSP73) and terpene synthase genes (TPSs) (Table S14). It is well known that these genes are related to plant growth and stress responses. Given the ability of A. argyi to adapt to a wide variety of habitat conditions, these largely expanded genes may contribute to its successful expansion across the landscape and rapid growth.
Subsequently, we constructed a time‐calibrated phylogenetic tree by using a concatenated sequence alignment of 944 single‐copy orthologues shared by these nine species. These results verified the close evolutionary relationship between A. argyi and A. annua, and the divergence time of A. argyi and A. annua was approximately 7.4 million years ago (Mya). The most recent common ancestor (MRCA) of A. argyi and A. annua diverged from the MRCA of C. nankingense ~9.3 Mya, which together diverged from the MRCA of E. breviscapus ~41.3 Mya and further from the MRCA of L. sativa ~52.2 Mya (Figure 2b).
Whole‐genome duplication (WGD) is considered the main factor driving genome evolution and expansion (Yan et al., 2021). In A. argyi, the WGD events were examined by distributions of synonymous substitutions (Ks) within genes in syntenic blocks compared with six other species (A. annua, C. nankingense, H. annuus, E. breviscapus, C. tinctorius and V. vinifera). The distribution of Ks for the paralogous genes of the A. argyi genome showed two prominent peaks at ~0.04 and ~1.06, indicating that A. argyi has experienced two rounds of WGD (recent WGD and WGT‐1) events. We further estimated that the most recent WGD event of A. argyi occurred at ~2.2 Mya, which was a species‐specific duplication event and did not occur in the genomes of E. breviscapus, H. annuus and C. tinctorius, but occurred in A. annua and C. nankingense. The WGT‐1 event in A. argyi was a conserved whole‐genome triplication event shared with E. breviscapus, C. tinctorius and other Asterid‐II plants (Badouin et al., 2017), occurring at approximately 62.9 Mya. Moreover, the Ks dot plot of retained paralogues in A. argyi genome also supported the occurrence of the WGD events (Figure 3a). Based on the Ks value (~0.6) of orthologous peaks for A. argyi and E. breviscapus, we predicted that their divergence time was ~38.2 Mya, which was close to the phylogenetic results. The relative age (Kimura distance) computed for LTR retroelements also indicates a recent increasing transposon activity (Figure 2d). The most recent WGD event and the recent outbreak of LTRs in A. argyi may be one of the most important reasons for its large genome size.
Figure 3.
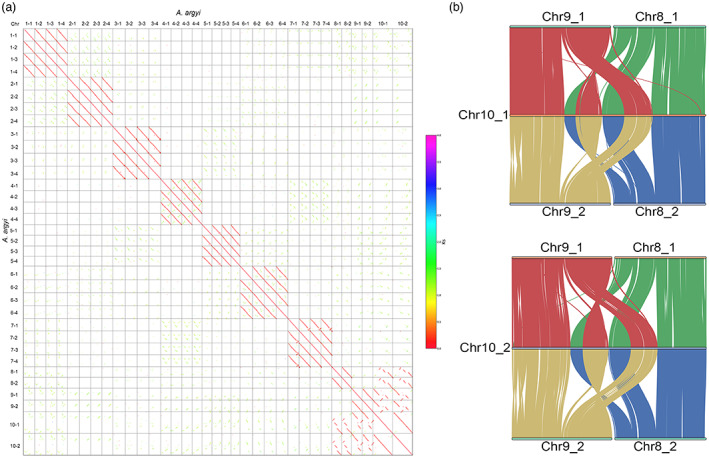
Chromosomal collinearity patterns and chromatin structures between chromosome 10 and chromosomes 8 and 9. (a). Syntenic dot plot between all chromosomes of A. argyi. (b). Syntenic blocks between chromosomes 8, 9 and 10. [Colour figure can be viewed at wileyonlinelibrary.com]
Chromosome fusion in the A. argyi genome
Synteny analysis showed that the 34 pseudochromosomes of A. argyi comprised 10 homologous groups, of which seven groups each had four sets of monoploid chromosomes, and three groups had two chromosomes in each (Figure S9). Moreover, each of the four chromosomes in the 1–7 chromosome groups can be divided into two subgroups according to the gene synteny analysis, which indicates that A. argyi is an allotetraploid (Figure S10), and this result was also consistent with that of survey analysis. Importantly, the length of chromosome 10 was almost the sum of the lengths of chromosomes 8 and 9 (Figure 3a and Table S5), and intragenome synteny analysis showed that chromosomes 8 and 9 shared close syntenic regions with chromosome 10 (Figure 3b). In addition, 11 780 (92.05%, total 12 797) genes on chromosome 10 were homologous with the genes on chromosomes 8 and 9 based on the BLAST results. Together, these data allow us to postulate that the ancestral 8 and 9‐like chromosomes were fused into the chromosome 10 in A. argyi, and chromosome 10 appears as the end‐to‐end fusion of ancestral 8 and 9‐like chromosomes, accompanied by at least one inversion and two intrachromosomal translocation events (Figure 3b).
By comparing the number of homologous genes on chromosome 10 with those on chromosomes 8 and 9, we found that 1129 genes were missing and that 1017 genes were newly formed on chromosome 10. These genes were mainly concentrated in the biological process category, including heme transport, iron coordination entity transport (lost genes), snRNA binding and oxidative phosphorylation (novel genes) (Data S6). In addition, by comparing the expression levels of homologous genes on chromosome 10 with those on chromosomes 8 and 9, we found that 411 genes were upregulated and 404 genes were downregulated on chromosome 10. GO enrichment analysis showed that the biological functions of these upregulated genes were significantly enriched in response to external biotic stimulus and cellular response to salicylic acid stimulus, and the downregulated genes were enriched in the methylerythritol 4‐phosphate pathway (Data S7).
Genes involved in flavonoid biosynthesis
Eupatilin, jaceosidin, hispidulin, schaftoside, isoschaftoside and vitexicarpin are representative bioactive flavonoids in A. argyi that contribute to versatile pharmacological effects such as anti‐inflammatory, antioxidation and anti‐tumor effects (Maleki et al., 2019; Nabavi et al., 2015; Serafini et al., 2010). Ultra‐performance liquid chromatography (HPLC) was used to quantify these flavonoids in seven different tissues (roots, rhizome, stem, and four different developmental stages of leaves A–D) of ‘Xiang Ai’. The obtained results showed that these bioactive flavonoids were more abundant in leaves than in other tissues. Among them, the content of eupatilin was the highest, and it increased with the growth stage of leaves (Figure S11). However, the genes that participated in the biosynthesis of these flavonoids such as hispidulin, jaceosidin and eupatilin in A. argyi remain largely unknown. Based on extensive investigations of the flavonoid biosynthesis pathway in other plants (Saito et al., 2013), we proposed the possible biosynthesis routes of these compounds in A. argyi (Figure 4a,c).
Figure 4.
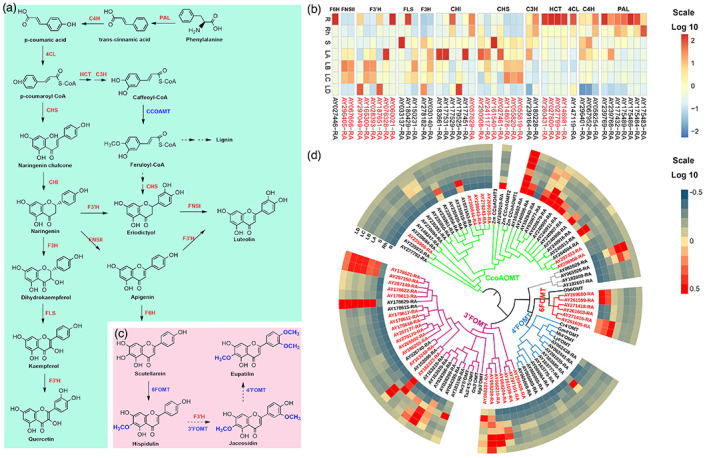
The identification and expression profiles of genes related to the biosynthesis of flavonoids in A. argyi. (a). The proposed flavonoid biosynthesis pathway in A. argyi. Red fonts represented the abbreviations of enzymes participating in the catalytic steps. And the full names of relative enzymes were shown in Data S9. (b). The expression patterns of candidate genes involved in flavonoids biosynthesis pathway in different tissues. The expanded genes were marked in red. R, root; Rh, rhizome; S, stem; LA, leaf buds, 0 day; LB, young leaves 15 days; LC, mature leaves 30 days; LD, old leaves 45 days. (c). Proposed biosynthesis pathways for hispidulin, jaceosidin and eupatilin. (d). Expression profile and phylogenetic tree of all members of the flavonoid O‐methyltransferase (FOMT) gene family in A. argyi. The genes in red were expanded genes. [Colour figure can be viewed at wileyonlinelibrary.com]
In total, 44 candidate genes encoding 12 key enzymes in flavonoid biosynthesis pathway were identified by homologue searching and functional annotation. Of note, nearly half of the candidates especially were expanded genes and the number of phenylalanine ammonia‐lyase (PAL), 4‐hydroxylase (C4H), hydroxycinnamoyl transferase (HCT), chalcone synthase (CHS), flavanone hydroxylase (F3′H, F3H) homologues in A. argyi was dramatically increased relative to that in Arabidopsis (Table S15). We mapped these genes to A. argyi genome and found that the PAL exhibited tandem repeats on chromosomes 5 (Data S8). Subsequently, a transcriptomic analysis was performed using samples from roots and leaf organs (Figure S12) to identify differentially expressed genes (DEGs) between different tissues and different developmental stages of leaves. Based on their expression patterns, almost all candidate genes were expressed in seven selected tissues, but the expression levels of the first four genes in this pathway, especially the expression levels of HCT genes in root samples, were higher than those in leaf samples, while the expression levels of the downstream genes, especially the CHS genes, were higher in leaves than in roots (Figure 4b). HCT is a key enzyme in lignin synthesis (Baucher et al., 2003), while CHS is the first rate‐limiting enzyme in plant flavonoid synthesis (Dixon and Paiva, 1995). Therefore, the expression patterns of the HCT and CHS genes were crucial for the regulation of lignin and flavonoid synthesis in A. argyi, which probably facilitates its rapid adaptation to heterogeneous environments. Furthermore, most of the DEGs involved in flavonoid biosynthesis were upregulated in the leaf tissues, and this correlates well with the fact that flavonoids, for example, hispidulin, jaceosidin and eupatilin, are mainly enriched in A. argyi leaves.
Flavonoid O‐methyltransferase (FOMT) is a key enzyme for the postmodification of flavonoid compounds. Studies have confirmed that O‐methylated flavonoids have stronger antioxidant, anti‐inflammatory, and anti‐cancer functions (Zhao et al., 2019). In consideration of the active ingredients, including hispidulin, jaceosidin, eupatilin and vitexicarp, in A. argyi that were all O‐methylated flavonoids, we further investigated the whole‐genome FOMT genes in A. argyi using the conserved domain and the reported FOMT genes as queries. A total of 83 FOMT were identified. Phylogenetic analysis showed that these FOMTs were clustered into five main subclades based on their catalytic sites including 31 3′FOMT, six 6FOMT, 10 4′FOMT, 32 caffeoyl‐CoA O‐methyltransferase (CCoAOMT) and four unclassified FOMTs (Figure 4d). We found that almost all types of FOMTs were significantly expanded in the A. argyi genome and exhibited a pattern of tandem duplication (Data S9). Most of the CCoAOMTs were primarily expressed in roots, while the FOMTs catalysing methylation at the 3′‐OH, 4′‐OH and 6‐OH groups were mainly expressed in leaves (Figure 4d). Hispidulin has a methoxy on its C6 position, indicating that its enzymatic synthesis requires 6FOMT to mediate C6‐methoxylation. Based on the chemical structure and a previous study (Zhang et al., 2016), scutellarein may be a precursor for hispidulin biosynthesis. Jaceosidin is a di‐methoxyflavone with one methyl group on the C6 position of the A ring and another on the C3′ position of the B ring, suggesting that its synthesis requires an additional 3′FOMT by comparison with hispidulin. Eupatilin is a trimethoxyflavone that has an extra methyl group at the C4 ‘position of the B ring compared to jaceosidin, suggesting that there may be a 4′FOMT catalysing the conversion of jaceosidin to eupatilin (Figure 4c). In summary, the identification of these FOMT candidates based on genomic and transcriptome analyses will accelerate the enzymatic synthesis pathway of hispidulin, jaceosidin, and eupatilin.
Genes involved in terpenoid biosynthesis
Volatile oil is a significant pharmacodynamic component in the leaves of A. argyi due to a rich content of terpenoids. The volatile oil from A. argyi leaves contains abundant monoterpenoids and sesquiterpenoids (Figure 5a). Although terpenoids are diverse and have various structures, they all come from the common precursors isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). IPP and DMAPP are mainly produced through the mevalonate pathway (MVA) pathway in the cytoplasm and the methylerythritol phosphate (MEP) pathway in the plastid (Sapir‐Mir et al., 2008; Vranova et al., 2013). Candidate genes participating in MVA and MEP pathways were screened by using the methods of homoloy searching and functional annotation. The obtained results indicated that a total of 66 genes encoding 14 gene families were involved in these two pathways in A. argyi (Figure 5b). These genes were widely distributed on A. argyi chromosomes, especially on chromosomes 7 (Data S10). The key genes identified in the MEP pathway were greatly expanded compared with the genes identified in the MVA pathway. RNA‐seq analysis demonstrated that genes related to the MEP pathway were more specifically expressed in leaves than those in the MVA pathway (Figure 5c), indicating that the terpenoids in A. argyi leaves mainly come from the MEP pathway.
Figure 5.
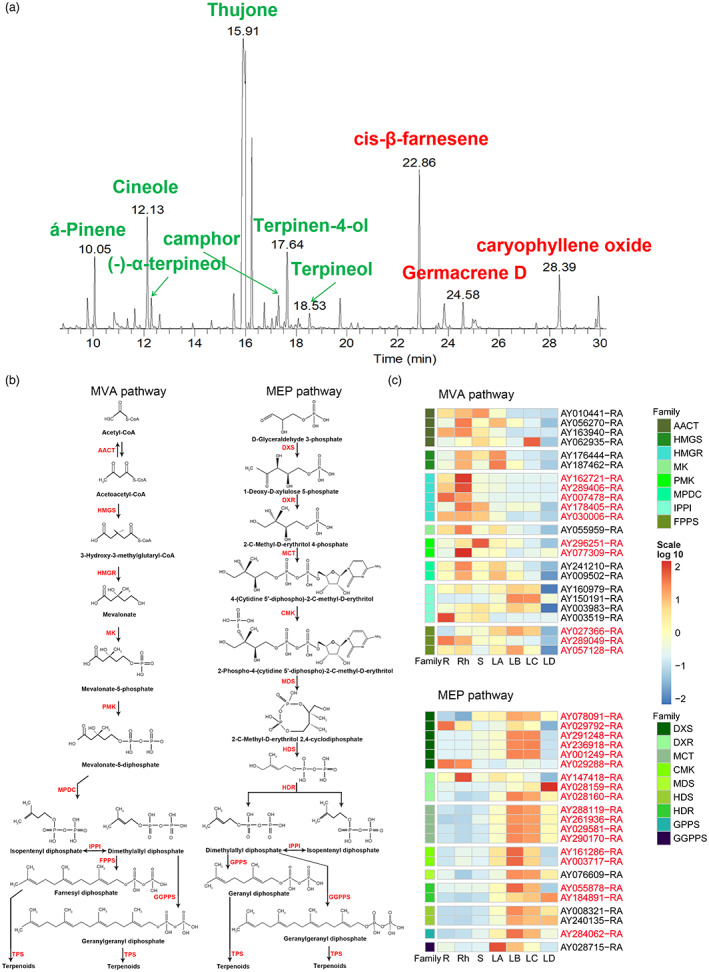
Volatiles compounds in A. argyi leaves and their biosynthesis pathways. (a). Gas chromatogram of volatile oils from A. argyi leaves. Monoterpenes and sesquiterpenes were marked green and red, respectively. (b). The proposed MVA and MEP pathways in A. argyi. Red fonts indicated the abbreviations of enzymes participating in these two pathways. And the full names of these enzymes were listed in Data S10. (c). The expression patterns of candidate genes involved in MVA and MEP pathways in different tissues. The expanded genes were marked in red. R, root; Rh, rhizome; S, stem; LA, leaf buds, 0 day; LB, young leaves 15 days; LC, mature leaves 30 days; LD, old leaves 45 days. [Colour figure can be viewed at wileyonlinelibrary.com]
Terpene synthase (TPS) family genes are responsible for the biosynthesis and structural diversity of terpenoids (Chen et al., 2011). We found that TPS genes were extremely expanded in the A. argyi genome. In total, we identified 135 TPS genes in the A. argyi genome (Data S11). According to phylogenetic analyses, these TPSs were grouped into five subfamilies, including TPS‐a, TPS‐b, TPS‐c, TPS‐d and TPS‐e/f (Figure 6a). More than 80% of TPSs belonged to TPS‐a and TPS‐b subfamilies, which are mainly involved in the biosynthesis of monoterpene, sesquiterpene, and diterpene, indicating the remarkable expansion of these two TPS subfamilies. The expression patterns of TPSs in different tissues were analysed, most of which had relatively high expression levels in leaves compared to other tissues (Figure S13). These results were consistent with the abundant terpenoids in the leaves of A. argyi. Chromosome localization showed that the TPS genes were not uniformly distributed throughout the chromosomes (Figure S14). For example, there were 12 TPS‐a genes clustered on chromosome 6 and 13 TPS‐b genes clustered on chromosome 3, with a pattern of tandem duplication (Figure 6b,c). Expression analysis indicated that most of the TPS genes in the cluster were not actively expressed, except for AY184877‐RA (TPS‐a), AY184880‐RA (TPS‐a) and AY075453 (TPS‐b), which were significantly expressed in leaf tissues (Figure 6b,c). This finding demonstrates that some duplicated TPSs may have undergone neofunctionalization.
Figure 6.
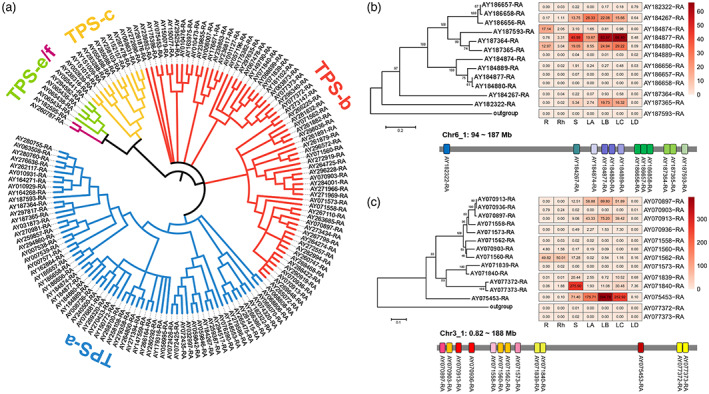
Expansion of terpene synthase‐encoding genes and their gene clusters on chromosomes in the Artemisia argyi genome. (a). Phylogeny of TPSs identified in A. argyi genome. (b). Phylogeny, expression profiles and chromosomal position of TPS‐a clade gene cluster on Chr6_1. The outgroup gene of the phylogenetic tree was TPS‐e subfamily gene with gene ID AY065441‐RA. (c). Phylogeny, expression profiles and chromosomal position of TPS‐b clade gene cluster on Chr3_1. The outgroup gene of the phylogenetic tree was TPS‐e subfamily gene with gene ID AY294582‐RA. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
The high‐quality A. argyi genome sequence in this study represents the first species in the genus Artemisia for which chromosome‐level assembly has been constructed. This well‐annotated genome will be the foundation for evolutionary and molecular biological studies of this economically and medicinally important plant. However, the high heterozygosity (2.36%), large proportion of repetitive sequences (75.86%) and polyploidy (allotetraploid) present significant challenges for the genome assembly of A. argyi. In this study, an integrated strategy combing PacBio long HiFi reads, Hi‐C sequencing and Illumina short reads greatly facilitated the assembly of the complex polyploidy A. argyi genome. Compared with the other 14 species in Asteraceae whose genomes have been released, A. argyi features the largest genome with a size of 8.03 Gb. The scaffold N50 (206.4 Mb) of the A. argyi genome was the longest among them, and the contig N50 (6.25 Mb) is only shorter than that of safflower (Wu et al., 2021). In brief, the A. argyi genome is the fifth chromosomal‐level genome in Asteraceae, and the first chromosome‐level genome in Artemisia.
WGD events and TE amplification are the main causes of large genomes (Bennetzen, 2002; Van de Peer et al., 2009). Here, the analysis of Ks distribution indicated that two rounds of WGD events occurred in the A. argyi genome (Figure 2c). The most recent WGD (~2.2 Mya) is an A. argyi and its closely related species (A. annua and C. nankingense) specific event, which is distinct from the WGD‐2 event in the sunflower genome (Badouin et al., 2017). Combined with a WGT‐γ event occurring in all eudicots and a basal WGT‐1 event occurring in Asteraceae, we conclude that A. argyi underwent at least three rounds of WGDs. The additional WGD events may also contribute to gene family expansion. In comparison with other sequenced Asteraceae species, we found that the number of expanded gene families in A. argyi was approximately three times that of the contracted gene families (Figure 2b), which accounts for a prominently large number of genes in the A. argyi genome. In particular, PsbA genes in photosystem II, RPA1 in DNA replication, HSPs and TPS were markedly expanded, all of which participate in plant growth and development and stress responses. Moreover, the transposable elements occupied 73.59% of the A. argyi genome, and the LTR/Gypsy subfamilies (24.47%) were the most abundant. The distribution of Ks also hints that LTR retrotransposons explosion occurred recently in the A. argyi genome (Figure 2d). Together, the additional WGD event and the recent proliferation of LTRs in A. argyi may be responsible for the expansion of the genome and may play vital roles in A. argyi adaptations to challenging environments.
Variation in basic chromosome numbers is not a rare phenomenon in some genera in the Asteraceae family, such as Melampodium whose basic chromosome numbers are x = 11, 9 and 14 (McCann et al., 2016). Artemisia is reported to have at least two basic chromosome numbers, with x = 9 being the common chromosome number and x = 8 being less frequent (Pellicer et al., 2010). The somatic chromosome numbers of A. argyi also exhibited considerable variation, including 2n = 18, 34 and 36, through chromosome count and karyotype analysis. The number of basic chromosomes of A. argyi is nominally x = 9 (Pellicer et al., 2010). Chromosomal fusion/fission and polyploidy are the main causes of chromosome number evolution (Mitros et al., 2020; Xuan et al., 2022). Our chromosomal‐scale genome of A. argyi provides reliable evidence for the chromosome number variation of this species. Synteny analysis showed that the 34 chromosomes of A. argyi comprise 10 homologous groups, of which seven groups each have four sets of monoploid chromosomes, and three groups have two chromosomes in each, and chromosomes 8 and 9 shared closely syntenic regions and high rates of homologous genes with chromosome 10 (Figure 3a,b). We proposed that the ancestral 8‐ and 9‐like chromosomes were fused into chromosome 10 in A. argyi. This single fusion explains the odd base chromosome number of A. argyi. Moreover, the existence of x = 8 base chromosomes suggests that the fusion may occur before allohybridization. Polyploidy and variation in basic chromosome numbers in Artemisia may facilitate the species differentiation and survival in extremely arid habitats.
Flavonoids and terpenoids are the main medicinal active ingredients in A. argyi. Based on the high‐quality genome, we proposed backbone biosynthetic pathways of flavonoids and terpenoids and identified the key gene families in both pathways. In plants, the biosynthesis of flavone O‐methyl derivatives is catalysed by FOMTs, which transfer the methyl group of S‐adenosyl‐L‐methionine (SAM) to the hydroxyl group of flavonoids. Thus, FOMTs play significant roles in the biosynthesis of O‐methylated flavones in A. argyi. In this study, a relatively complete biosynthetic pathway for the three O‐methylated flavones, hispidulin, jaceosidin, and eupatilin, was conjected based on synergistic analysis of genome sequencing, transcriptomics and metabolomics. A large number of FOMT genes were identified in the A. argyi genome, and the FOMTs catalysing methylation at the 3′‐OH, 4′‐OH and 6‐OH groups are important candidates for the synthesis of hispidulin, jaceosidin and eupatilin (Figure 4). For terpenoid synthesis, The high and special expression of genes in the MEP pathway in A. argyi genome demonstrates that this pathway is a main source of terpenoid precursors. A. argyi leaves contain more than 100 kinds of terpenoids (mainly rich in monoterpenes and sesquiterpenes), and the diversity of terpenoids is mainly determined by TPS family genes. Whole‐genome‐wide identification of TPS family genes shows that the extensive expansion of TPS‐a and TPS‐b subfamily genes (Figure 6) in the A. argyi genome may be one explanation for the accumulation of diverse monoterpenes and sesquiterpenes. We also found remarkable tandem duplications in the FOMT and TPS family genes, which may contribute to the high contents of flavonoids and volatile oil in A. argyi.
In conclusion, our study of a high‐quality A. argyi genome provides valuable information for the finite genomic resources of complicated polyploids in Artemisia and offers a comprehensive insights into the mechanism of economically and medicinally important traits and environmental adaptation.
Methods
Plant materials
The cultivated mugwort ‘Xiang Ai’ was used for the construction of the reference genome. The young leaves of 90‐day‐old ‘Xiang Ai’ plants were sampled to extract high‐quality genomic DNA for genome sequencing and Hi‐C analysis. Roots®, rhizomes (Rh), stems (S), leaf buds (0 day, LA), young leaves (15 days, LB), mature leaves (30 days, LC) and old leaves (45 days, LD) of ‘Xiang Ai’ were collected for RNA‐seq analysis.
Flow cytometry
The nuclear DNA content of ‘Xiang Ai’ was measured by flow cytometry according to the method described previously (Dolezel et al., 2007). Briefly, leaves from ‘Xiang Ai’ and Chrysanthemum nankingense plants were finely chopped with a razor blade in 2 mL Galbra’th's buffer, respectively. After the suspension was filtered through a 48 μm nylon membrane, 200 μL of PI (50 μg/mL) and 100 μg/mL RnaseA were added immediately. Following 30 min of incubation on ice, the samples were detected by flow cytometry (BD FACSCalibur, BD Biosciences, Wuhan, China).
Estimation of genome size
Illumina‐seq generated approximately 215 Gb of clean reads, which were subjected to K‐mer analysis to estimate the genome size of ‘Xiang Ai’. The 21‐mer frequency distribution was shown in Figure S2.
Whole‐genome sequencing
For PacBio SMRT sequencing, high‐quality DNA from ‘Xiang Ai’ was first sheared and concentrated to construct 15‐Kb DNA sequencing libraries and subsequently run on a PacBio Sequel II platform according to the manufactuer's instruction with seven cells.
For genome survey sequencing, 5150‐bp paired‐end (PE) libraries were constructed for sequencing on an Illumina NovaSeq 6000 platform and ~215 Gb of raw sequencing data were obtained.
For Hi‐C sequencing, two Hi‐C libraries digested with MboI restriction enzyme were sequenced on BGI MGISEQ‐2000 to generate ~422 Gb of valid data from 150 PE reads.
Genome assembly and evaluation
Briefly, we performed de novo assembly using HiCanu v 2.2 (Koren et al., 2017) and Hifiasm (version 0.13‐r308) (Cheng et al., 2021) and the result showed that the Hifiasm assembly (N50 = 8.32 Mb with 10 274 contigs) was better than the HiCanu assembly (4.69 Mb with 48 227 contigs). Then, the draft genome from the Hifiasm assembly was further assembled into scaffolds with Hi‐C data using the 3D‐DNA pipeline (version 180922) (van Berkum et al., 2010). These scaffolds were roughly split by Juicebox (version 1.11.08) (Robinson et al., 2018) and another round of scaffolding by 3D‐DNA. Genome assembly completeness was assessed by BUSCOs (Simao et al., 2015) and transcriptome data.
Genome annotation
Transposable elements of A. argyi assembly were identified by homology‐ and de novo‐based methods. Repbase (version 2017‐01‐27) was used to build the homology repeat library (Jurka et al., 2005). Then, RepeatModeler v2.0.1 (Jurka et al., 2005) was used to construct the de novo repeat library. Finally, RepeatMasker v4.1.0 (Tempel, 2012) was used to annotate the repetitive elements in the Repbase and the de novo repeat library.
Protein coding genes of A. argyi genome were annotated by MAKER according to three complementary methods: de novo prediction, homology‐based prediction and transcriptome‐based prediction (Campbell et al., 2014). First of all, Isoseq3 was employed to identify full‐length high‐quality transcripts from PacBio Sequel II (Singh et al., 2016). HISAT2 (version 2.1.0; Pertea et al., 2016) and Cufflinks (version 2.2.1) (Trapnell et al., 2012) were used to predict the genes by using the RNA‐seq data and the assembled genome. In the second round, Augustus v 3.2.1 (Keller et al., 2011), GeneMark‐ES Suite v 4.61_lic (Lomsadze et al., 2005) and SNAP (version 2013‐02‐16) (Johnson et al., 2008) tools were used for gene model training for de novo prediction. Finally, precise gene annotation was performed by using non‐redundant proteins from Artemisia annua and Helianthus annuus based on the homology‐based approach. Complete BUSCO hits were used to evaluate the gene annotation results of A. argyi genome (Simao et al., 2015). Following the gene annotation, BLAST analyses against several functional databases (NR, EggNOG, SwissProt and TrEMBL) were performed on the predicted protein‐coding genes to identify homologous proteins in other species using diamond (version v2.0.4.142; Buchfink et al., 2015). In addition, possible GO terms were obtained using the SwissProt database and Trembl database ID mapping. Gene pathways were accomplished with KEGG analysis by using KOBAS v 3.0 (Xie et al., 2011).
tRNAs were annotated by tRNAscan‐SE (version 1.3.1) (Lowe and Eddy, 1997), rRNAs were annotated by BLASTN (version 2.10.1) (Gardner et al., 2009). miRNAs and snRNAs were annotated by searching the Rfam database (version 14.5) using BLASTN and INFERNAL (version 1.1.2) (Nawrocki et al., 2009).
Sequence alignment and variation analysis
Burrows–Wheeler Aligner (BWA, version 0.7.17) software was used to map all clean reads to the assembled genome (Li and Durbin, 2009) with the methods described by He et al. (2021). In brief, SAMtools (version 1.9) were employed to convert the mapping results into the BAM format (Li et al., 2009). Picard package (Version 1.96) was used for the filtration of duplicated reads. Genome Analysis Toolkit (GATK, version 3.8‐0‐ge9d806836) was used to realign reads around Indels (McKenna et al., 2010). Variations were detected with both SAMtools mpileup and GATK HaplotypeCaller packages, and only concordance results were retained. Raw variations were filtered by GATK VariantFiltration packages with the following parameters ‘‐‐filter‐name FilterQual ‐‐filter‐expression “QUAL < 60.0” ‐‐filter‐name FilterQD ‐‐filter‐expression “QD < 20.0” ‐‐filter‐name FilterFS ‐‐filter‐expression “FS > 13.0” ‐‐filter‐name FilterMQ ‐‐filter‐expression “MQ < 30.0” ‐‐filter‐name FilterMQRankSum ‐‐filter‐expression “MQRankSum < ‐1.65” ‐‐filter‐name FilterReadPosRankSum ‐‐filter‐expression “ReadPosRankSum < ‐1.65” ‐‐cluster‐window‐size 10 ‐‐cluster‐size 2’. SNPs within 10 bp with an indel were removed. Finally, SnpEff software was used to annotate the identified SNPs and Indels (Cingolani et al., 2012).
Gene family, phylogenomic analysis and WGD identification
Gene families of A. argyi genome and Arabidopsis thaliana, Vitis vinifera and six other Asteraceae species were identified by OrthoMCL (version 2.0.9) with default parameters (Li et al., 2003). RaxML (version 8.2.12) were used to construct the phylogenetic tree by using the single‐copy orthologues of the nine species (Stamatakis, 2014). The divergence times of A. thaliana and A. argyi from TimeTree (http://timetree.org/) were used for calibration and the divergence times of phylogenetic tree were estimated by r8s (version 1.81) (Sanderson, 2003). The expanded and contracted gene families were calculated by CAFE (version 4.2) in each lineage (De Bie et al., 2006). WGD events in the A. argyi genome were searched according to the WGDi pipeline (Sun et al., 2021).
RNA sequencing
Total RNA of different tissues was extracted using TRIzol reagent. An mRNA sequencing library of seven different tissues was constructed on an Illumina Novaseq 6000 platform by 150 bp PE sequencing. For full‐length transcriptome sequencing, a mixed RNA library from different tissues was prepared according to the PacBio ISO‐Seq experimental workflow and subsequently run on a PacBio Sequel II platform. For RNA‐seq analysis, 2 μg RNA from each sample was sequenced on the Illumina platform. Three replications were performed for each sample.
Gene identification in flavonoid and terpenoid biosynthesis pathways
The protein sequences of the enzymes (PAL, C4H, 4CL, HCT, C3H, CHS, CHI, F3H, and FLS) in flavonoid biosynthesis pathways of A. thaliana were obtained from the TAIR database, and those for F3′H and FNSII in fleabanes, FNSI in parsley and celery, and F6H in soybean were obtained from previous studies (He et al., 2021) and the NCBI database. These sequences were blasted against the A. argyi protein sequences using BLASTP (E‐value < 1e−5).
Functional proteins involved in the MEP and MVA pathways of terpenoid backbone biosynthesis in A. thaliana were obtained from the TAIR database. Homologues of these proteins in the genome of A. argyi were investigated by BLASTP with an E‐value cutoff of 1e−5. Fragments per kb of exon model per million mapped fragments (FPKM) not <10 were selected for heatmap analysis.
Identification of terpene synthase genes (TPSs) and flavonoid O‐methytransferase (FOMTs)
The protein sequences of TPSs were identified by screening with functional motifs PF01397 and PF03936. A total of 146 TPS genes were predicted in the ‘Xiang Ai’ genome.There are 11 genes were removed manually, since it was not possible to calculate genetic distance by MEGA6.
We searched the candidate FOMT genes by the combination of conserved domain (PF01596) and homologue‐based BLAST, and repetitive sequences were removed. A total of 83 FOMT genes were predicted in the A. argyi genome. 11 selected FOMT proteins downloaded from NCBI based on high gene homology and genome annotation were subjected to phylogenetic analysis.
Phylogenetic reconstruction of TPSs and FOMTs
The neighbour‐joining trees were constructed using the MEGA6 software (Tamura et al., 2013). Heatmap analysis based on RNA‐seq data was performed by the pheatmap package in R language (version 1.0.12).
HPLC analysis of flavonoids
Agilent 1260 Infinity HPLC system (Agilent Technologies) was used for HPLC analyses. The flavonoids were separated by ZORBAXRRHD Eclipse Plus 95A C18 column (2.1 × 100 mm, 1.8 μm, Agilent) and detected at 330 nm by UV. The mobile phases were acetonitrile (A) and 0.1% phosphoric acid (B) with the flow rate of 0.4 mL/min. The separation gradient conditions were as follows: 0–0.5 min, 2–5% A; 0.5–7 min, 5–25% A; 7–11 min, 25–330% A; 11–14 min, 30–33% A; 14–19.5 min, 33–45% A; 19.5–20.5 min, 45–85% A; 20.5–27 min, 85–98% A; 27–28 min, 98–2% A.
Conflicts of interest
The authors declare no competing interests.
Author contributions
L.Q.H., D.H.L., G.G. N. and Y.H.M., conceived and managed the project. D.D.L., T.T.Z., H.Z.D, J.C. and S.N.P prepared the samples for sequencing. Z.H.L. and G.G. N. contributed to the assembly and annotation. Y.H.M., D.D.L. and T.T.Z. analysed data and drafted the manuscript. Z.P.X. and H.Z.D revised the manuscript.
Supporting information
Figure S1 Estimate of the genome size of A. argyi (Xiang ai, 4‐6) by flow cytometry.
Figure S2 Estimate of the genome size and complexity of A. argyi by K‐mers method.
Figure S3 The flowchart of processing pipeline used to assemble the A. argyi genome.
Figure S4 The Hi‐C map of A. argyi genome assembly.
Figure S5 Enriched GO terms for gene families specific to A. argyi.
Figure S6 KEGG enrichment of A. argyi specific genes.
Figure S7 GO enrichment of A. argyi expanded genes.
Figure S8 KEGG enrichment of A. argyi expanded genes.
Figure S9 Syntenic relationships based on pairs of collinear genes of A. argyi.
Figure S10 Clustering of counts of 13‐mers.
Figure S11 The contents of representative flavonoids in seven different tissues of A. argyi. R, root; Rh, rhizome; S, stem; LA, leaf buds, 0 d; LB, young leaves 15 d; LC, mature leaves 30 d; LD, old leaves 45 d.
Figure S12 Expression correlation heat map of pairwise samples.
Figure S13 Expression profiles of TPSs in A. argyi.
Figure S14 Chromosome distribution of TPS family genes in the A. argyi genome.
Table S1 K‐mer analysis of the A. argyi genome using K‐mer = 21.
Table S2 Summary of PacBio sequencing.
Table S3 Statistics of pre‐assembly of the A. argyi genome by using Hifiasm and Hicanu.
Table S4 Summary of Hi‐C sequencing.
Table S5 Summary of chromosome level assembly based on Hi‐C data.
Table S6 Statistics of Illumina and isoform sequencing clean reads mapping rate of A. argyi genome assembly.
Table S7 The assessment of A. argyi genome and annotation completeness with BUSCO.
Table S8 Summary of repeats and transposable elements in the A. argyi genome assembly.
Table S9 Statistics of genes annotated in the A. argyi genome.
Table S10 Functional annotation of predicted protein‐coding genes in the A. argyi genome.
Table S11 Statistics of noncoding RNA genes in the A. argyi genome.
Table S12 Gene family categories.
Table S13 Comparisons of genes and gene families.
Table S14 Significantly expanded genes in the A. argyi genome.
Table 15 Copy numbers of gene families involved in flavonoid biosynthesis among plant species.
Data S1 Summary of RNA sequencing.
Data S2 Enriched GO terms for gene families specific to A. arygi.
Data S3 Enriched KEGG terms for gene families specific to A. arygi.
Data S4 Enriched GO terms for expanded genes in A. arygi.
Data S5 Enriched KEGG terms for expanded genes in A. arygi.
Data S6 Enriched GO terms for lost and novel genes in chromosome 10 by comparision with chromosme 8 and 9.
Data S7 Enriched GO terms for up‐ and down‐regulated genes in chromosome 10 by comparision with chromosme 8 and 9.
Data S8 Location of the flavonoids biosynthesis genes in the A. argyi genome.
Data S9 Genome wide identification, classification of flavonoid o‐methyltransferase (FOMT) gene family in A. argyi genome.
Data S10 Location of the already‐known terpenoids metabolism genes in the A. argyi genome.
Data S11 Genome wide identification, classification of terperne synthase (TPS) gene family in A. argyi genome.
Acknowledgements
We appreciate the funding support from the key project at the central government level: The ability establishment of sustainable use for valuable Chinese medicine resources (2060302), National key research and development program (No: 2017YFC1700704), the young qihuang scholars of the national administration of TCM, the special fund for the construction of modern agricultural industrial technology system (CARS‐21, CRAS‐23), Hubei Province outstanding young and middle‐aged scientific and technological innovation team ‐ Traditional Chinese medicine ecological agriculture (T2021008), innovation team and talents cultivation programme of national administration of traditional Chinese medicine. (No: ZYYCXTD‐D‐202005), and the Nature Science Foundation of Hubei Province (2021CFB225). We appreciate the support for computational work from BioSmartSeek company.
Contributor Information
Guogui Ning, Email: ggning@mail.hzau.edu.cn.
Dahui Liu, Email: liudahui@hbtcm.edu.cn.
Luqi Huang, Email: huangluqi01@126.com.
Data availability statement
The whole assembled genome data have been submitted in the National Genomics Data Center under the accession number PRJNA804646. The raw data of genome sequencing and RNA sequencing have been deposited at Sequence Read Archive (SRA) under the accession PRJNA804653.
References
- Badouin, H. , Gouzy, J. , Grassa, C.J. , Murat, F. , Staton, S.E. , Cottret, L. , Lelandais‐Briere, C. et al. (2017) The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature, 546, 148–152. [DOI] [PubMed] [Google Scholar]
- Baucher, M. , Halpin, C. , Petit‐Conil, M. and Boerjan, W. (2003) Lignin: genetic engineering and impact on pulping. Crit. Rev. Biochem. Mol. Biol. 38, 305–350. [DOI] [PubMed] [Google Scholar]
- Bennetzen, J.L. (2002) Mechanisms and rates of genome expansion and contraction in flowering plants. Genetica, 115, 29e36. [DOI] [PubMed] [Google Scholar]
- Bora, K.S. and Sharma, A. (2011) The genus Artemisia: a comprehensive review. Pharm. Biol. 49, 101–109. [DOI] [PubMed] [Google Scholar]
- Buchfink, B. , Xie, C. and Huson, D.H. (2015) Fast and sensitive protein alignment using DIAMOND. Nat. Methods, 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Campbell, M.S. , Holt, C. , Moore, B. and Yandell, M. (2014) Genome annotation and curation using MAKER and MAKER‐P. Curr. Protoc. Bioinformatics, 48, 4 11 11‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F. , Tholl, D. , Bohlmann, J. and Pichersky, E. (2011) The family of terpene synthases in plants: a mid‐size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. [DOI] [PubMed] [Google Scholar]
- Cheng, H. , Concepcion, G.T. , Feng, X. , Zhang, H. and Li, H. (2021) Haplotype‐resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods, 18, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicconardi, F. , Lewis, J.J. , Martin, S.H. , Reed, R.D. , Danko, C.G. and Montgomery, S.H. (2021) Chromosome fusion affects genetic diversity and evolutionary turnover of functional loci but consistently depends on chromosome size. Mol. Biol. Evol. 38, 4449–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wangle, L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S.J. et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinese Pharmacopoeia Commission (2020) Chinese Pharmacopoeia. Beijing: China Medica Science Press. [Google Scholar]
- De Bie, T. , Cristianini, N. , Demuth, J.P. and Hahn, M.W. (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics, 22, 1269–1271. [DOI] [PubMed] [Google Scholar]
- Dixon, R.A. and Paiva, N.L. (1995) Stress‐induced phenylpropanoid metabolism. Plant Cell, 7, 1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolezel, J. , Greilhuber, J. and Suda, J. (2007) Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2, 2233–2244. [DOI] [PubMed] [Google Scholar]
- Du, J. , Gao, R. and Zhao, J. (2021) The effect of volatile oil from Chinese Mugwort leaf on human Demodecid mites in vitro. Acta Parasitol. 66, 615–622. [DOI] [PubMed] [Google Scholar]
- Efferth, T. , Zacchino, S. , Georgiev, M.I. , Liu, L. , Wagner, H. and Panossian, A. (2015) Nobel prize for artemisinin brings phytotherapy into the spotlight. Phytomedicine, 22, A1–A3. [DOI] [PubMed] [Google Scholar]
- Gardner, P.P. , Daub, J. , Tate, J.G. , Nawrocki, E.P. , Kolbe, D.L. , Lindgreen, S. , Wilkinson, A.C. et al. (2009) Rfam: updates to the RNA families database. Nucleic Acids Res. 37, D136–D140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, Y.B. , Wang, Z.G. , Xiong, Y. , Huang, X.J. , Mei, Z.N. and Hong, Z.G. (2016) Anti‐inflammatory and blood stasis activities of essential oil extracted from Artemisia argyi leaf in animals. J. Nat. Med. 70, 531–538. [DOI] [PubMed] [Google Scholar]
- Mawangdui Han Danasty Tomb bamboo books research group (1979) Recipes for Fifty‐Two Ailments. Beijing: Cultural Relics Publishing House. [Google Scholar]
- Guan, X. , Ge, D. , Li, S. , Huang, K. , Liu, J. and Li, F. (2019) Chemical composition and antimicrobial activities of Artemisia argyi Levl. et Vant essential oils extracted by simultaneous distillation‐extraction, subcritical extraction and hydrodistillation. Molecules, 24, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, S. , Dong, X. , Zhang, G. , Fan, W. , Duan, S. , Shi, H. , Li, D. et al. (2021) High quality genome of Erigeron breviscapus provides a reference for herbal plants in Asteraceae. Mol. Ecol. Resour. 21, 153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi, Y. , Kondo, K. , Korobkov, A.A. , Tatarenko, I.V. , Kulikov, P.V. , Verkholat, V.P. , et al. (2003) Cytological study in the genus Artemisia L. (Asteraceae) from Russia. Chromosome. Sci. 7, 83–89. [Google Scholar]
- IJdo, J.W. , Baldini, A. , Ward, D.C. , Reeders, S.T. and Wells, R.A. (1991) Origin of human chromosome 2: an ancestral telomere‐telomere fusion. Proc. Natl. Acad. Sci. USA, 88, 9051–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, A.D. , Handsaker, R.E. , Pulit, S.L. , Nizzari, M.M. , O'Donnell, C.J. and de Bakker, P.I. (2008) SNAP: a web‐based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics, 24, 2938–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J. , Kapitonov, V.V. , Pavlicek, A. , Klonowski, P. , Kohany, O. and Walichiewicz, J. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Keller, O. , Kollmar, M. , Stanke, M. and Waack, S. (2011) A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics, 27, 757–763. [DOI] [PubMed] [Google Scholar]
- Kim, J.K. , Shin, E.C. , Lim, H.J. , Choi, S.J. , Kim, C.R. , Suh, S.H. , Kim, C.J. et al. (2015) Characterization of nutritional composition, antioxidative capacity, and sensory attributes of seomae mugwort, a native Korean variety of Artemisia argyi H. Lev. & Vaniot. J. Anal. Methods Chem. 2015, 916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B.P. , Berlin, K. , Miller, J.R. , Bergman, N.H. and Phillippy, A.M. (2017) Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M.Y. , Doh, E.J. , Park, C.H. , Kim, Y.H. , Kim, E.S. , Ko, B.S. and Oh, S.E. (2006) Development of SCAR marker for discrimination of Artemisia princeps and A. argyi from other Artemisia herbs. Bio. Pharm. Bull. 29, 629–633. [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with burrows‐wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J., Jr. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. (1957) Compendium of Materia Medica. Beijing: People's Medical Publishing House. [Google Scholar]
- Lin, T. , Xu, X. , Ruan, J. , Liu, S.Z. , Wu, S.G. , Shao, X.J. , Wang, X.B. et al. (2018) Genome analysis of Taraxacum kok‐saghyz Rodin provides new insights into rubber biosynthesis. Natl. Sci. Rev. 5, 78–87. [Google Scholar]
- Liu, B. , Yan, J. , Li, W. , Yin, L. , Li, P. , Yu, H. , Xing, L. et al. (2020) Mikania micrantha genome provides insights into the molecular mechanism of rapid growth. Nat. Commun. 11, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomsadze, A. , Ter‐Hovhannisyan, V. , Chernoff, Y.O. and Borodovsky, M. (2005) Gene identification in novel eukaryotic genomes by self‐training algorithm. Nucleic Acids Res. 33, 6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, T.M. and Eddy, S.R. (1997) tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, J.L. , Li, Z.Z. and Zhang, L.B. (2018) Two new flavonoids from Artemisia argyi with their anticoagulation activities. Nat. Prod. Res. 32, 632–639. [DOI] [PubMed] [Google Scholar]
- Maleki, S.J. , Crespo, J.F. and Cabanillas, B. (2019) Anti‐inflammatory effects of flavonoids. Food. Chem. 299, 125124. [DOI] [PubMed] [Google Scholar]
- McCann, J. , Schneeweiss, G.M. , Stuessy, T.F. , Villaseñor, J.L. and Weiss‐Schneeweiss, H. (2016) The impact of reconstruction methods, phylogenetic uncertainty and branch lengths on inference of chromosome number evolution in American daisies (Melampodium, Asteraceae). PLoS ONE, 11, e0162299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei, Q. , Chen, X. , Xiang, L. , Liu, Y. , Su, Y. , Gao, Y. , Dai, W. et al. (2016) DNA barcode for identifying folium Artemisiae argyi from counterfeits. Bio. Pharm. Bull. 39, 1531–1537. [DOI] [PubMed] [Google Scholar]
- Mitros, T. , Session, A.M. , James, B.T. , Wu, G.A. , Belaffif, M.B. , Clark, L.V. , Shu, S.Q. et al. (2020) Genome biology of the paleotetraploid perennial biomass crop miscanthus. Nat. Commun. 11, 5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naß, J. and Efferth, T. (2018) The activity of Artemisia spp. and their constituents against Trypanosomiasis. Phytomedicine, 47, 184–191. [DOI] [PubMed] [Google Scholar]
- Nabavi, S.F. , Braidy, N. , Gortzi, O. , Sobarzo‐Sanchez, E. , Daglia, M. , Skalicka‐Woźniak, K. and Nabavi, S.M. (2015) Luteolin as an anti‐inflammatory and neuroprotective agent: a brief review. Brain Res. Bull. 119, 1–11. [DOI] [PubMed] [Google Scholar]
- Nawrocki, E.P. , Kolbe, D.L. and Eddy, S.R. (2009) Infernal 1.0: inference of RNA alignments. Bioinformatics, 25, 1335–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagning, A.L.N. , Tamokouc, J.D. , Lateef, M. , Tapondjou, L.A. , Kuiate, J.R. , Ngnokam, D. and Ali, M.S. (2016) New triterpene and new flavone glucoside from Rhynchospora corymbosa (Cyperaceae) with their antimicrobial, tyrosinase and butyrylcholinesterase inhibitory activities. Phytochem. Lett. 16, 121–128. [Google Scholar]
- Pellicer, J. , Garcia, S. , Canela, M.A. , Garnatje, T. , Korobkov, A.A. , Twibell, J.D. and Valles, J. (2010) Genome size dynamics in Artemisia L. (Asteraceae): following the track of polyploidy. Plant Biol. 12, 820–830. [DOI] [PubMed] [Google Scholar]
- Peng, Y. , Lai, Z. , Lane, T. , Nageswara‐Rao, M. , Okada, M. , Jasieniuk, M. , O'Geen, H. et al. (2014) De novo genome assembly of the economically important weed horseweed using integrated data from multiple sequencing platforms. Plant Physiol. 166, 1241–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M. , Kim, D. , Pertea, G.M. , Leek, J.T. and Salzberg, S.L. (2016) Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and ballgown. Nat. Protoc. 11, 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez‐Sánchez, O. , Pérez‐Rodríguez, P. , Delaye, L. and Tiessen, A. (2016) Plant proteins are smaller because they are encoded by fewer exons than animal proteins. Genom. Proteom. Bioinf. 14, 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes‐Chin‐Wo, S. , Wang, Z. , Yang, X. , Kozik, A. , Arikit, S. , Song, C. , Xia, L. et al. (2017) Genome assembly with in vitro proximity ligation data and whole‐genome triplication in lettuce. Nat. Commun. 8, 14953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.T. , Turner, D. , Durand, N.C. , Thorvaldsdottir, H. , Mesirov, J.P. and Aiden, E.L. (2018) Juicebox.js provides a cloud‐based visualization system for Hi‐C data. Cell Syst. 6, 256–258 e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, K. , Yonekura‐Sakakibara, K. , Nakabayashi, R. , Higashi, Y. , Yamazaki, M. , Tohge, T. and Fernie, A.R. (2013) The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol. Biochem. 72, 21–34. [DOI] [PubMed] [Google Scholar]
- Sanderson, M.J. (2003) r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics, 19, 301–302. [DOI] [PubMed] [Google Scholar]
- Sapir‐Mir, M. , Mett, A. , Belausov, E. , Tal‐Meshulam, S. , Frydman, A. , Gidoni, D. and Eyal, Y. (2008) Peroxisomal localization of Arabidopsis isopentenyl diphosphate isomerases suggests that part of the plant isoprenoid mevalonic acid pathway is compartmentalized to peroxisomes. Plant Physiol. 148, 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione, D. , Reyes‐Chin‐Wo, S. , Acquadro, A. , Froenicke, L. , Portis, E. , Beitel, C. , Tirone, M. et al. (2016) The genome sequence of the outbreeding globe artichoke constructed de novo incorporating a phase‐aware low‐pass sequencing strategy of F1 progeny. Sci. Rep. 6, 19427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, J.M. , Kang, H.M. , Son, K.H. , Kim, J.H. , Lee, C.W. , Kim, H.M. , Chang, S.I. et al. (2003) Antitumor activity of flavones isolated from Artemisia argyi . Planta Med. 69, 218–222. [DOI] [PubMed] [Google Scholar]
- Serafini, M. , Peluso, I. and Raguzzini, A. (2010) Flavonoids as anti‐inflammatory agents. Proc. Nutr. Soc. 69, 273–278. [DOI] [PubMed] [Google Scholar]
- Shen, Q. , Zhang, L.D. , Liao, Z.H. , Wang, S.Y. , Yan, T.X. , Shi, P. , Liu, M. et al. (2018) The genome of Artemisia annua provides insight into the evolution of Asteraceae Family and artemisinin biosynthesis. Mol. Plant, 11, 776–788. [DOI] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Singh, N. , Sahu, D.K. , Chowdhry, R. , Mishra, A. , Goel, M.M. , Faheem, M. , Srivastava, C. et al. (2016) IsoSeq analysis and functional annotation of the infratentorial ependymoma tumor tissue on PacBio RSII platform. Meta. Gene, 7, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, C. , Liu, Y. , Song, A. , Dong, G. , Zhao, H. , Sun, W. , Ramakrishnan, S. et al. (2018) The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant, 11, 1482–1491. [DOI] [PubMed] [Google Scholar]
- Song, X. , Wen, X. , He, J. , Zhao, H. , Li, S. and Wang, M. (2019) Phytochemical components and biological activities of Artemisia argyi . J. Funct. Foods, 52, 648–662. [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, X.Z. and Miller, L.H. (2015) The discovery of artemisinin and the Nobel prize in physiology or medicine. Sci. China Life Sci. 58, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, P., Jiao, B.B. , Yang, Y.Z. , Shan, L.X. , Li, T. , Li, X.N. , Xi, Z.X. , et al. (2021) WGDI: a user‐friendly toolkit for evolutionary analyses of whole‐genome duplications and ancestral karyotypes. bioRxiv . [DOI] [PubMed]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. and Kumar, S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel, S. (2012) Using and understanding RepeatMasker. Methods Mol. Biol. 859, 29–51. [DOI] [PubMed] [Google Scholar]
- Torrell, M. , Cerbah, M. , Siljak‐Yakovlev, S. and Vallès, J. (2003) Molecular cytogenetics of the genus Artemisia (Asteraceae, Anthemideae): fluorochrome banding and fluorescence in situ hybridization. I. Subgenus Seriphidium and related taxa. Plant. Syst. Evol. 239, 13–153. [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D.R. , Pimentel, H. et al. (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkum, N.L. , Lieberman‐Aiden, E. , Williams, L. , Imakaev, M. , Gnirke, A. , Mirny, L.A. , Dekker, J. et al. (2010) Hi‐C: a method to study the three‐dimensional architecture of genomes. J. Vis. Exp. 39, e1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer, Y. , Maere, S. and Meyer, A. (2009) The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10, 725e32. [DOI] [PubMed] [Google Scholar]
- Vranova, E. , Coman, D. and Gruissem, W. (2013) Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 64, 665–700. [DOI] [PubMed] [Google Scholar]
- Wu, Z. , Liu, H. , Zhan, W. , Yu, Z. , Qin, E. , Liu, S. , Yang, T. et al. (2021) The chromosome‐scale reference genome of safflower (Carthamus tinctorius) provides insights into linoleic acid and flavonoid biosynthesis. Plant Biotechnol. J. 19, 1725–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, C. , Mao, X. , Huang, J. , Ding, Y. , Wu, J. , Dong, S. , Kong, L. et al. (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xirau, J.V. and Siljak‐Yakovlev, S. (1997) Cytogenetic studies in the genus Artemisia L.(Asteraceae): fluorochrome‐banded karyotypes of five taxa, including the Iberian endemic species Artemisia barrelieri Besser. Can. J. Bot. 75, 595–606. [Google Scholar]
- Xuan, Y. , Ma, B. , Li, D. , Tian, Y. , Zeng, Q. and He, N. (2022) Chromosome restructuring and number change during the evolution of Morus notabilis and Morus alba . Hortic. Res. 9, uhab030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro, T. , Shiraishi, A. , Satake, H. and Nakayama, K. (2019) Draft genome of Tanacetum cinerariifolium, the natural source of mosquito coil. Sci. Rep. 9, 18249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Q. , Wu, F. , Xu, P. , Sun, Z. , Li, J. , Gao, L. , Lu, L. et al. (2021) The elephant grass (Cenchrus purpureus) genome provides insights into anthocyanidin accumulation and fast growth. Mol. Ecol. Resour. 21, 526–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Zhang, G. , Zhang, J. , Liu, H. , Chen, W. , Wang, X. , Li, Y. et al. (2017) Hybrid de novo genome assembly of the Chinese herbal fleabane Erigeron breviscapus . Gigascience, 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun, C. , Jung, Y. , Chun, W. , Yang, B. , Ryu, J. , Lim, C. , Kim, J.H. et al. (2016) Anti‐inflammatory effects of Artemisia leaf extract in mice with contact dermatitis in vitro and in vivo. Mediators Inflamm. 2016, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.J. (1997) Synopsis of prescriptions of the golden chamber. Beijing: Ancient books of traditional Chinese Medicine Press. [Google Scholar]
- Zhang, Y.Y. , Xu, R.X. , Gao, S. and Cheng Ai, X. (2016) Enzymatic production of oroxylin A and hispidulin using a liverwort flavone 6‐O‐methyltransferase. FEBS Lett. 590, 2619–2628. [DOI] [PubMed] [Google Scholar]
- Zhao, Q. , Yang, J. , Cui, M.Y. , Liu, J. , Fang, Y. , Yan, M. , Qiu, W. et al. (2019) The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of wogonin biosynthesis. Mol. Plant, 12, 935–950. [DOI] [PubMed] [Google Scholar]
- Zimmermann‐Klemd, A.M. , Reinhardt, J.K. , Morath, A. , Schamel, W.W. , Steinberger, P. , Leitner, J. , Huber, R. et al. (2020) Immunosuppressive activity of Artemisia argyi extract and isolated compounds. Front. Pharmacol. 11, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Estimate of the genome size of A. argyi (Xiang ai, 4‐6) by flow cytometry.
Figure S2 Estimate of the genome size and complexity of A. argyi by K‐mers method.
Figure S3 The flowchart of processing pipeline used to assemble the A. argyi genome.
Figure S4 The Hi‐C map of A. argyi genome assembly.
Figure S5 Enriched GO terms for gene families specific to A. argyi.
Figure S6 KEGG enrichment of A. argyi specific genes.
Figure S7 GO enrichment of A. argyi expanded genes.
Figure S8 KEGG enrichment of A. argyi expanded genes.
Figure S9 Syntenic relationships based on pairs of collinear genes of A. argyi.
Figure S10 Clustering of counts of 13‐mers.
Figure S11 The contents of representative flavonoids in seven different tissues of A. argyi. R, root; Rh, rhizome; S, stem; LA, leaf buds, 0 d; LB, young leaves 15 d; LC, mature leaves 30 d; LD, old leaves 45 d.
Figure S12 Expression correlation heat map of pairwise samples.
Figure S13 Expression profiles of TPSs in A. argyi.
Figure S14 Chromosome distribution of TPS family genes in the A. argyi genome.
Table S1 K‐mer analysis of the A. argyi genome using K‐mer = 21.
Table S2 Summary of PacBio sequencing.
Table S3 Statistics of pre‐assembly of the A. argyi genome by using Hifiasm and Hicanu.
Table S4 Summary of Hi‐C sequencing.
Table S5 Summary of chromosome level assembly based on Hi‐C data.
Table S6 Statistics of Illumina and isoform sequencing clean reads mapping rate of A. argyi genome assembly.
Table S7 The assessment of A. argyi genome and annotation completeness with BUSCO.
Table S8 Summary of repeats and transposable elements in the A. argyi genome assembly.
Table S9 Statistics of genes annotated in the A. argyi genome.
Table S10 Functional annotation of predicted protein‐coding genes in the A. argyi genome.
Table S11 Statistics of noncoding RNA genes in the A. argyi genome.
Table S12 Gene family categories.
Table S13 Comparisons of genes and gene families.
Table S14 Significantly expanded genes in the A. argyi genome.
Table 15 Copy numbers of gene families involved in flavonoid biosynthesis among plant species.
Data S1 Summary of RNA sequencing.
Data S2 Enriched GO terms for gene families specific to A. arygi.
Data S3 Enriched KEGG terms for gene families specific to A. arygi.
Data S4 Enriched GO terms for expanded genes in A. arygi.
Data S5 Enriched KEGG terms for expanded genes in A. arygi.
Data S6 Enriched GO terms for lost and novel genes in chromosome 10 by comparision with chromosme 8 and 9.
Data S7 Enriched GO terms for up‐ and down‐regulated genes in chromosome 10 by comparision with chromosme 8 and 9.
Data S8 Location of the flavonoids biosynthesis genes in the A. argyi genome.
Data S9 Genome wide identification, classification of flavonoid o‐methyltransferase (FOMT) gene family in A. argyi genome.
Data S10 Location of the already‐known terpenoids metabolism genes in the A. argyi genome.
Data S11 Genome wide identification, classification of terperne synthase (TPS) gene family in A. argyi genome.
Data Availability Statement
The whole assembled genome data have been submitted in the National Genomics Data Center under the accession number PRJNA804646. The raw data of genome sequencing and RNA sequencing have been deposited at Sequence Read Archive (SRA) under the accession PRJNA804653.