

Summary
Plant diseases caused by viruses limit crop production and quality, resulting in significant losses. However, options for managing viruses are limited; for example, as systemic obligate parasites, they cannot be killed by chemicals. Sensitive, robust, affordable diagnostic assays are needed to detect the presence of viruses in plant materials such as seeds, vegetative parts, insect vectors, or alternative hosts and then prevent or limit their introduction into the field by destroying infected plant materials or controlling insect hosts. Diagnostics based on biological and physical properties are not very sensitive and are time‐consuming, but assays based on viral proteins and nucleic acids are more specific, sensitive, and rapid. However, most such assays require laboratories with sophisticated equipment and technical skills. By contrast, isothermal‐based assays such as loop‐mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA) are simple, easy to perform, reliable, specific, and rapid and do not require specialized equipment or skills. Isothermal amplification assays can be performed using lateral flow devices, making them suitable for onsite detection or testing in the field. To overcome non‐specific amplification and cross‐contamination issues, isothermal amplification assays can be coupled with CRISPR/Cas technology. Indeed, the collateral activity associated with some CRISPR/Cas systems has been successfully harnessed for visual detection of plant viruses. Here, we briefly describe traditional methods for detecting viruses and then examine the various isothermal assays that are being harnessed to detect viruses.
Keywords: isothermal amplification, nucleic acid detection, LAMP, RPA, CRISPR/Cas‐based diagnosis, onsite diagnosis
Introduction
Crops are susceptible to multiple viral pathogens that cause severe economic losses. Limiting these losses requires producers to identify the causal viruses. The oldest approach for detecting and diagnosing viruses that infect crops is examining their biological properties such as symptoms, host range, and transmission. In the early days of plant virology, symptoms were the primary means by which a viral disease was diagnosed and named, as evidenced by the names of many well‐known plant viruses (Walkey, 1985). The symptoms produced in a range of test plants provide the first clues about the identity of a virus in the field or laboratory. For the grower, the nature and severity of disease symptoms determine the economic importance of a particular virus in terms of yield loss and reduced quality. When considering virus symptoms, it should be remembered that a virus is unlikely to cause just one symptom in an infected plant. Infection usually results in a range of symptoms, and a sequence of symptoms is frequently observed as the disease progresses.
Host range, where a virus can replicate in more than host, also provides key information about viruses, particularly emerging races of known pathogens. The sap transmission test, in which sap from an infected plant is used to inoculate a panel of indicator plants, is still the best means for routine diagnosis of many viruses, as this sensitive test provides essential information about the virus, including whether it is susceptible to specific host resistance genes. However, these tests may take several weeks and require panels of specific test plants. Moreover, not all plant viruses are sap transmissible, and it is often necessary to use natural virus vectors (insect vectors; aphids, beetles, thrips, whiteflies, leafhoppers; mites, nematode vectors, and fungal vectors). In addition, for viruses that cannot be transmitted by sap and vectors, the only means of confirmation may be grafting (Camarço et al., 1998; Hill, 1984).
Biological assays are very important for detecting and diagnosing plant viruses and are the only means to grow viruses. Such assays rely on indicator plants that show specific symptoms when challenged with a specific virus. However, these assays are more time‐consuming and labour‐intensive than laboratory assays. Consistent, reproducible biological assay results require suitable vector‐proof growth facilities and trained workers to produce ideal indicator plants. Also, essential is familiarity with symptomatology. Infectivity assays are often needed to verify the results of laboratory tests, and inoculation of differential hosts is an essential tool for identifying viruses at the species and strain levels (Chin et al., 2007; Davis et al., 2005; González et al., 2002; Roy et al., 1999). For example, biological assays are the only means to differentiate between papaya ringspot virus strains W and P. Viruses are also identified based on biophysical characteristics such as nucleic acid size, type, and sequence, protein composition and size, and particle shape (Stephanidis et al., 2007), as revealed by electron microscopy. However, electron microscopy is expensive and less sensitive and cannot identify viruses to the species level.
The advent of ELISA in the 1970s improved virus detection, as this method was more sensitive than other methods performed at that time. With ELISA, results can be obtained within a few hours, and it can be used to test many samples. However, ELISA requires the production of virus‐specific antisera. Attaching the antibody to an enzyme increases the sensitivity of detection of the antibody–antigen reaction, and the resulting colour reaction may be quantitatively measured. The sensitivity of ELISA is approximately 1–10 ng of virus per ml test sample. Several ELISA procedures have been developed (Clark and Adams, 1977; Clark et al., 1986; Clark and Bar‐Joseph, 1984; Hampton et al., 1990; Mowat and Dawson, 1987). The major advantage of ELISA or other serological assays is that crude samples can be directly used in these assays, as no special procedure or equipment is required for test sample preparation.
Onsite detection of viruses using a serological assay is possible via lateral flow immunoassay (LFIA). Lateral flow or ‘dipstick’ methods are suitable for field use without the need for special equipment or technical skills. A lateral flow test typically incorporates a sample pad in close contact with a conjugate pad (Figure 1). This, in turn, is in contact with a membrane onto which test and control reagents have been immobilized. An absorbent pad wicks fluid away from the membrane. In these kits, virus coat protein‐specific antibodies (capture antibodies) and species‐specific antibodies are pre‐immobilized onto membranes at the test and control lines, respectively. To perform the test, the sample is ground, and a few droplets of the extracted sap are loaded into the sample port of the device (conjugate pad), where it combines with virus‐specific antibodies conjugated with coloured latex beads or gold particles, forming a complex with the tagged antibodies. The bound antigen/antibody complex then moves by lateral flow through the membrane until it reaches the test line (where the virus‐specific capture antibody is immobilized). At the test line, the movement of the virus‐specific antigen–antibody complex is arrested as it binds to the virus‐specific capture antibody. As increasing amounts of antigen–antibody complex become captured at the test line, the line becomes visible as the coloured latex or gold particles accumulate. The second capture line is the control line, comprising species‐specific antibodies intended to arrest all unbound labelled antibodies that would also become visible to confirm that the assay has run correctly. LFIA is only suitable for detecting viruses that occur in high titre in the test samples. It may give false‐negative results for test samples with low concentrations of virus. To overcome this issue, some new lateral flow assays (LFA) involve an initial amplification of the viral nucleic acid using labelled primers/probes, followed by endpoint detection using lateral flow devices (Cassedy et al., 2021).
Figure 1.
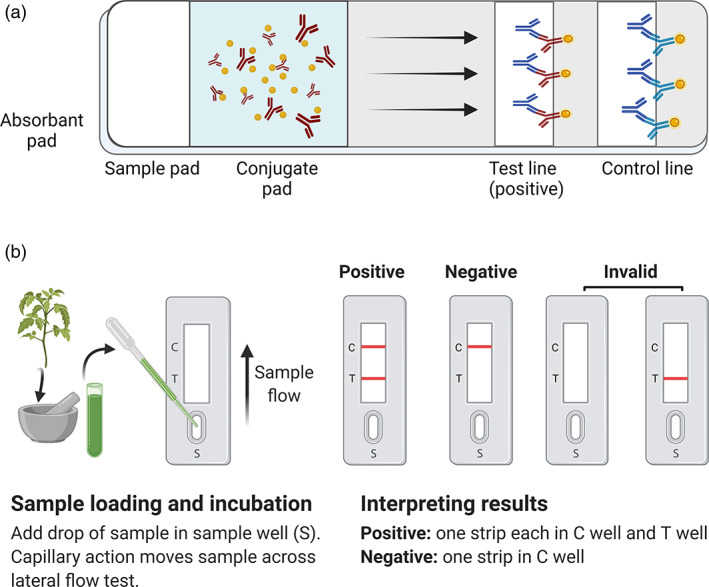
Schematic diagram of the lateral flow immune assay (LFIA). (a) Illustration of a lateral flow strip. The sample is applied to the sample pad and transported to the conjugate pad via an absorbent pad to form an antigen–antibody complex. The complex moves via lateral flow, where virus‐specific antibodies capture it at the test line. The band's intensity at the test line indicates the amount of test antigen. The band at the control line confirms that the assay was done correctly. (b) Representative lateral flow immune assay. The sample sap is added to the sample well (S) and moves along the lateral flow strip via capillary action. Two bands indicate a positive test, while a single control band indicates a negative test. No band or only a band at the test line indicates an invalid result. [Colour figure can be viewed at wileyonlinelibrary.com]
The next breakthrough in virus detection came with the invention of PCR in 1984. The method involves amplifying a specific region of the virus prior to its detection, thus increasing the assay's sensitivity many fold compared with ELISA or other serological assays. The important steps in PCR include extracting nucleic acids from the test plant, synthesizing two virus‐specific primers, and setting up the PCR in a vial by adding extracted nucleic acids, primers, nucleotides, magnesium chloride, and Taq polymerase. The vial is then placed in the thermal cycler for denaturation, primer annealing, and extension using pre‐determined settings. After the run, a positive reaction is identified by running the contents of the vial on an agarose gel. The presence of bands at the expected position indicates that the sample is positive for the virus (Henson and French, 1993).
Different variants of PCR, such as reverse transcription PCR (RT‐PCR), immunocapture PCR, nested PCR, multiplex PCR, real‐time PCR, and so on, have been developed (Bhat and Rao, 2020). PCR can also be used to detect multiple viruses in a sample using multiplex PCR. PCR is more sensitive than ELISA, as even a few copies of the viral nucleic acid present in the test sample can be amplified and detected. However, PCR requires sophisticated laboratory equipment and skilled personnel. PCR‐based methods are cumbersome because they require the initial isolation of nucleic acids, followed by amplification of the target sequence and analysis of the products on an agarose gel.
By contrast, isothermal amplification can be performed at a single temperature without the need for thermal cyclers, and the results can be visualized as colour changes without the need for agarose gel electrophoresis. These methods can be performed in a resource‐poor laboratory, require less time, and produce comparable results to PCR‐based methods. Of the several isothermal amplification assay methods available, loop‐mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA) are commonly used for the detection and diagnosis of infectious agents, including viruses.
Loop‐mediated isothermal amplification
Loop‐mediated isothermal amplification (LAMP) is an isothermal amplification assay that exploits the strand displacement activity of Bst DNA polymerase from Bacillus stearothermophilus for the efficient, robust amplification of any target nucleic acid using a minimum of four primers (F3, B3, FIP, and BIP) (Notomi et al., 2000). The sensitivity of the assay can be further increased by adding one or two additional primers (BL and FL) (Figure 2a) (Nagamine et al., 2002). Designing LAMP primers manually is possible but a bit complicated. Primers can be designed using the free online software package Primer Explorer version 5 (https://primerexplorer.jp/e/) or paid software (www.optigene.co.uk/lamp‐designer/). Unlike PCR, the target nucleic acid is initially converted to a dumbbell‐shaped structure that serves as a starting point for the cyclic phase of LAMP amplification. To convert the nucleic acid into a dumbbell‐shaped structure, outward forward (F3), outward backward (B3), forward inner (FIP), and backward inner (BIP) primers are needed. FIP and BIP are long primers (~45–55 nucleotides) that contain both sense and antisense sequences. Readers can visit http://loopamp.eiken.co.jp/e/lamp/ for detailed guidelines for primer design.
Figure 2.
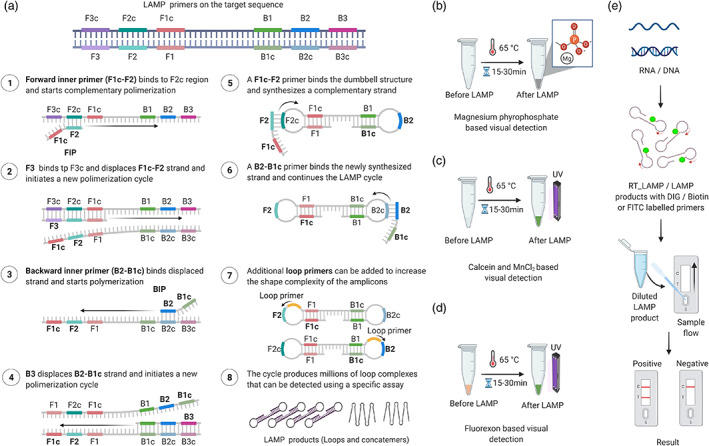
Amplification via loop‐mediated isothermal amplification (LAMP) and various detection methods. (a) Diagram of LAMP primers located on the target sequence and the amplification process. The FIP primer binds to its complementary sequence and begins amplifying the first strand, followed by binding and amplification of the second strand by the F3 primer. The following steps involve the amplification of the FIP‐amplified strand by the B3 primer. Finally, FIP and BIP continue the LAMP cycle, resulting in dumbbell‐shaped amplicons. A full stepwise description of the individual steps is provided in the figure. (b) Magnesium pyrophosphate‐based visual detection of the LAMP product. During LAMP, a large amount of pyrophosphate ion is produced as a by‐product, which reacts with magnesium provided in the reaction mixture. The resulting product, magnesium pyrophosphate, forms a white precipitate that allows easy visual detection. The turbidity of the final LAMP reaction confirms the presence or absence of the targeted nucleic acid. (c) Calcein‐based fluorescence detection of the LAMP product. The reaction of the by‐product pyrophosphate with magnesium or calcein‐manganese aids in the visualization of the LAMP product by producing a precipitate or emitting bright green fluorescence under UV light. Calcein‐based fluorescence is enhanced by the presence of magnesium in the reaction mixture. (d) Fluorexon‐based visual detection of the LAMP product. Upon completion of the LAMP reaction, fluorexon‐MnCl turns from orange to green. The resulting green fluorescence is visualized with the naked eye under UV light. (e) Lateral flow‐based detection of the LAMP product. To visualize LAMP with LFA, the target nucleic acid (RNA/DNA) is amplified via RT‐LAMP or LAMP with DIG, biotin, or FITC‐labelled primers. The labelled LAMP product is diluted and applied to the lateral flow strip. After 5 to 15 min of incubation, the appearance of bands on the test and control lines indicates the presence of the target nucleic acid. [Colour figure can be viewed at wileyonlinelibrary.com]
The first step in the LAMP reaction is the annealing of a portion of the FIP to its complementary sequences (F2 region) in the target nucleic acid, resulting in the synthesis of a complementary strand. This is followed by the binding of primer F3 to its complementary region (F3c), leading to the displacement of the FIP‐linked strand and producing a loop‐structured end (Figure 2a). BIP then initiates the synthesis of another strand by binding to the complementary region (B2) on the FIP‐linked strand. The synthesized strand is displaced upon B3‐primed synthesis, leading to a dumbbell‐shaped structure, which converts into a stem‐loop structure by self‐priming. This stem‐loop structure acts as the template for the cyclic amplification step of LAMP. The conversion of the target nucleic acid into a stem‐loop structure is crucial for the success of LAMP. F3 and B3 are absent in the stem‐loop structure, and hence, these primers are no longer needed for the cyclic phase of LAMP amplification. The cyclic phase utilizes the stem‐loop structure for repeated amplification cycles to produce many amplified strands of different lengths (Figure 2a). The sensitivity of the amplification can be increased using loop primers (LF and LB). LAMP reactions have been successfully coupled with a suitable reverse transcriptase (RT‐LAMP) to transcribe a specific viral RNA sequence to DNA, which serves as a template for subsequent reactions. However, unlike standard RT, it is important to use a thermostable reverse transcriptase enzyme in RT‐LAMP that can withstand temperatures up to 65 °C.
The standard LAMP reaction mix consists of isothermal buffer, MgSO4, betaine, dNTPs, LAMP primers (F3, B3, BIP, FIP, LF, and LB), Bst polymerase, thermostable reverse transcriptase (if performing RT‐LAMP), and template nucleic acid. The LAMP assay does not require a highly purified nucleic acid template; in most cases, it works well with crude extract isolated from an infected plant. The LAMP reaction is incubated at 65 °C (may be optimized between 58 and 70 °C) for 30 to 60 min. The reaction can be stopped by placing the tube at 80 °C for 10 min to inactivate the enzyme (Bhat and Rao, 2020).
The results of LAMP can be visualized in many ways. The pyrophosphate molecule released during nucleotide synthesis in the LAMP reaction combines with magnesium ions to form magnesium pyrophosphate. Magnesium pyrophosphate is insoluble, and its accumulation turns the contents of the tube turbid (Figure 2b). Thus, the most straightforward way to visualize LAMP results is to observe the tube after the reaction with the naked eye for the presence of turbidity (Bhat et al., 2013; Fukuta et al., 2003a,b; Mori et al., 2001; Nagamine et al., 2002; Nie, 2005; Notomi et al., 2000; Tomlinson et al., 2010). The addition of intercalating fluorescent dyes such as SYBR Green, ethidium bromide, or PicoGreen makes positive samples appear fluorescent under UV light (Almasi et al., 2013; Nie et al., 2012; Tomlinson et al., 2007). Similarly, the addition of calcein and MnCl2 in the LAMP reaction results in green fluorescence under UV light (Figure 2c; Mansour et al., 2015). Visual detection is also possible following the addition of fluorexon dye, where a positive reaction is identified by a colour change from light orange to green (Figure 2d). Finally, LAMP results can be visualized by agarose gel electrophoresis, which produces ladder‐like bands when observed under a UV transilluminator. The turbidity of magnesium pyrophosphate, a by‐product of the reaction, can be detected in real‐time with a real‐time turbidimeter (Mori et al., 2001). Real‐time detection is also possible by running the LAMP reaction on a real‐time LAMP instrument and measuring the fluorescence emitted by SYBR Green or other dyes on a real‐time basis). LAMP has been successfully used to detect many infectious agents of humans, animals, and plants. This technique has also been used to detect numerous plant viruses, as listed in Table S1.
LAMP combined with a lateral flow device for onsite detection
Loop‐mediated isothermal amplification results can be visualized with a lateral flow device (LFD) when using labelled loop (FP and BP) or inner primers (FIP and BIP) at the 5′ end with biotin/digoxigenin (DIG)/Texas red or fluorescein isothiocyanate (FITC) (Figure 2e; Peng et al., 2021; Tomlinson et al., 2010). The principle of immunochromatography using LFDs is the same as that described for LFIA. To detect LAMP products using an LFD, the labelled (biotin/DIG/FITC) LAMP product is diluted at the end of the reaction and applied to the sample port of the LFD. The LFD contains coloured latex beads or gold nanoparticles conjugated with the appropriate antibodies produced in rabbit, such as anti‐biotin/anti‐DIG/anti‐FITC, which bind to the LAMP product. This bound complex moves laterally through the membrane until it reaches the test line, where it binds to the label incorporated in the LAMP product, resulting in the formation of a visible coloured line. No coloured line will be visible for negative reactions in which only unincorporated primers are present. The control line will be visible when the coloured latex combines with anti‐rabbit antibody, which takes place within 15 min after the addition of the LAMP product. The presence of two coloured lines indicates a positive reaction, while the presence of only a single control line indicates that the test sample is negative, and the absence of both coloured lines indicates that the test failed. LFDs are also suitable for multiplex detection of different viruses simultaneously (Tomlinson et al., 2010, 2013). Custom‐made or commercial LFDs can be used to detect LAMP products. LAMP, followed by detection using LFDs, has been successfully used to detect many pathogens, including cassava brown streak virus, citrus leaf blotch virus, tomato brown rugose fruit virus, and Ugandan cassava brown streak virus (Peng et al., 2021; Tomlinson et al., 2013; Table S1).
Recombinase polymerase amplification
Recombinase polymerase amplification (RPA) technology was developed by Piepenburg et al. (2006) and commercialized by TwistDx (www.twistdx.co.uk). This technique has been used to detect various pathogens, including fungi, bacteria, and viruses that infect animals, humans, and plants. RPA is an isothermal procedure that requires a single temperature for target amplification and an enzyme to separate the strands of double‐stranded (ds)DNA to achieve primer binding at the target region on the template (Figure 3a). At the beginning of the RPA reaction, in the presence of ATP and a crowding agent (high molecular weight polyethylene glycol), the recombinase protein integrates with the primers to form a recombinase‐primer complex (Zhang et al., 2014). This complex identifies the complementary sequence on the template and allows the primer to anneal after the separation of the dsDNA strands by the recombinase. After the primer annealing, the recombinase separates from the complex, leaving the 3′ end available to the DNA polymerase to extend the chain. The separated DNA strands are then stabilized by single‐stranded binding protein (SSB) as the DNA polymerase extends the chain, forming a new dsDNA that acts as a template for further amplification.
Figure 3.
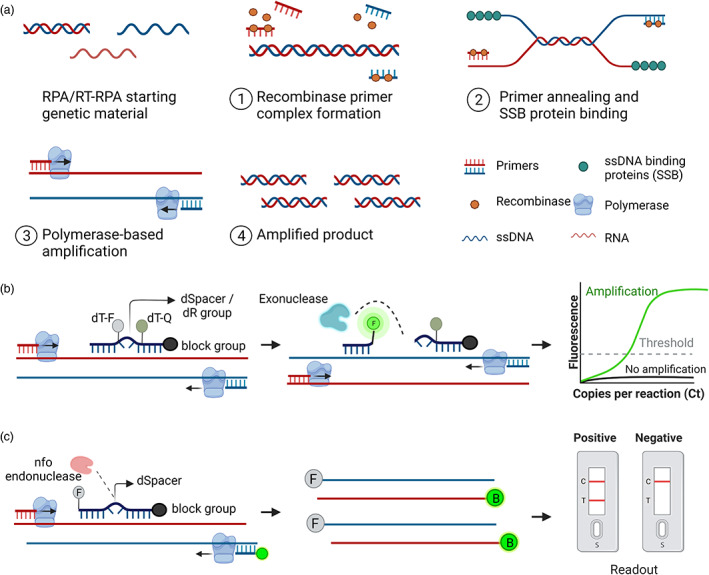
Recombinase polymerase amplification (RPA)‐based amplification and its detection methods. (a) Schematic diagram of the RPA assay. An ideal RPA assay consists of forward and reverse primers, a recombinase protein that helps bind primers to the target nucleic acid, ssDNA‐binding proteins to stabilize the ssDNA, and a polymerase to amplify the primer‐bound strands. The stepwise amplification process is shown in the figure. (b) Exonuclease‐based detection of the RPA product. A 46–52 bp long Exo probe flanked by a quencher and fluorophore binds to the amplified product. Exo probe contains a THF (tetrahydrofuran) residue known as dSpacer, which is cleaved by the exonuclease, thus releasing the fluorophore from the quencher. The observed fluorescent signal indicates the presence of the target nucleic acid. (c) Endonuclease‐based detection of the RPA product. A 46–52 bp long oligonucleotide probe labelled with FAM or Alexa fluor binds to the target strand. The annealed probe is cleaved by the nfo enzyme, freeing the 3′‐OH group of the probe and is used as a primer in the subsequent reactions. The resulting amplicons are produced with FAM and biotin using a biotin‐labelled reverse primer. The final RPA product is applied to the LFA strip, and the results are visualized after the appearance of matching lanes. [Colour figure can be viewed at wileyonlinelibrary.com]
Although the manufacturer recommends using primers 30–35 nucleotides long, some recent reports describe successful RPA using standard PCR primers (Lobato and O'Sullivan, 2018). Primers with long tracts of guanines and cytidines at the 3′ end and guanines at the 5′ end are not recommended (www.twistdx.uk.com). Similarly, primers with a GC content of >70% or <30% are not recommended. The ideal amplicon length is ~100–200 bases. RPA is generally carried out at 37–42 °C for 15–30 min (Piepenburg et al., 2006). The template for RPA can be dsDNA, single‐stranded ssDNA, or cDNA. In addition, primer design is simple, and there is no need to consider the annealing temperature (Boyle et al., 2014). RPA assays can be performed on crude samples (does not require a highly purified template) and are less prone to contamination than conventional PCR assays (Rojas et al., 2017). RPA can be performed in a simple incubator, dry bath, or at room temperature without the need for a thermocycler. Finally, RPA reagents are supplied in lyophilized form without the need for a freezer for transport and storage. These features make RPA suitable for use onsite or in a laboratory with minimum facilities. RPA can be combined with a lateral flow assay, making it ideally suited for the onsite detection of viruses in only 15–30 min. Another advantage of RPA is its suitability for multiplexing, depending on the sequence of the target pathogen, target amplicon size, and primer design (Kersting et al., 2014). As primers can compete for recombinase proteins, the ratios and concentrations of primers must be optimized for multiplex RPA assays. In most cases, RPA is highly specific, with 100% specificity for the target sequence.
Recombinase polymerase amplification is performed using the basic kit available from TwistDx. Primers (forward and reverse), template, and rehydration buffer are added to the freeze‐dried reaction pellets, followed by mixing. The addition of magnesium acetate will immediately initiate the RPA reaction. The tube is placed in an incubator at a suitable temperature (optimum 37–42 °C). The tube is removed from the incubator after 4 min, inverted vigorously, and placed back in the incubator for 20–40 min. The results of RPA can be visualized by agarose gel electrophoresis. To avoid smeared bands, the RPA product can be purified to remove proteins and crowding agent prior to gel electrophoresis. Colorimetric detection of the RPA product is possible if either primers or dNTPs labelled with biotin are used in the reaction (Lobato and O'Sullivan, 2018). After the reaction, the addition of streptavidin‐HRP conjugate, followed by the substrate (tetramethylbenzidine and hydrogen peroxide), results in a change in colour whose intensity depends on the amplicon concentration.
Recombinase polymerase amplification can also be performed using an RNA template (RNA viruses) by adding a compatible reverse transcriptase enzyme directly into the RPA reaction mix in a one‐step format called RT‐RPA. Alternatively, the RNA can be converted into cDNA separately and a portion of the cDNA can be used as a template for RPA (Aman et al., 2020a,b, 2022). RPA has been successfully used to detect several infectious agents, including viruses. Plant viruses that have been detected using the RPA assay are listed in Table 1. The sensitivity of detection by RPA can be enhanced by including probes in the reaction for real‐time detection via fluorescence or endpoint detection in lateral flow assays (Daher et al., 2016).
Table 1.
Recombinase polymerase amplification (RPA) and reverse transcription (RT)‐RPA‐based assays used for the detection of different plant viruses
| Virus name (Genus; Family) | Target gene | Host | Detection method | Time (min) | Temperature (°C) | Sensitivity | Reference |
|---|---|---|---|---|---|---|---|
| DNA viruses | |||||||
| Banana bunchy top virus (Babuvirus; Nanoviridae) | replicase initiator protein | Banana | AGE | 30 | 37 | 10 times more than PCR | Kapoor et al. (2017) |
| Bean golden yellow mosaic virus (BGYMV) (Begomoviurs; Geminiviridae) | C1 region | Bean | AGE | 30 | 37 | Equal to ELISA | Londoño et al. (2016) |
| Citrus yellow mosaic virus (CYMV) (Badnavirus; Caulimoviridae) | ORF3 | Citrus | AGE | 30 | 37 | 10 times less than PCR | Kumar et al. (2018) |
| Milk vetch dwarf virus (MDV) (Nanovirus; Nanoviridae) | CP | Cowpea | AGE, LFD | 30 | 37 | 101 copies of MDV | Cao et al. (2020) |
| Piper yellow mottle virus (PYMoV) (Badnavirus; Caulimoviridae) | ORF 2 | Black pepper | AGE | 40 | 37 | 10 times more than PCR | Mohandas and Bhat (2020) |
| Tomato mottle virus (ToMoV) (Begomoviurs; Geminiviridae) | C1 region | Tomato | AGE | 30 | 37 | Equal to ELISA | Londoño et al. (2016) |
| Tomato yellow leaf curl virus (TYLCV) (Begomoviurs; Geminiviridae) | C1 region | Tomato, tobacco, bean |
AGE |
30 | 37 | Equal to ELISA | Londoño et al. (2016) |
| RNA viruses | |||||||
| Apple chlorotic leaf spot virus (ACLSV) (Trichovirus; Betaflexiviridae) | – | Apple | CRISPR/Cas12a with oligonucleotide‐conjugated gold nanoparticle | 20 | 37 | RNA transcripts of 0.01–1 fM (25 viral copies) sensitivity equal to RT‐qPCR | Jiao et al. (2021) |
| Apple necrotic mosaic virus (ApNMV) (Ilarvirus; Bromoviridae) | – | Apple | CRISPR/Cas12a with oligonucleotide‐conjugated gold nanoparticle | 20 | 37 | RNA transcripts of 0.01–1 fM (25 viral copies) sensitivity equal to RT‐qPCR | Jiao et al. (2021) |
| Apple stem grooving virus (ASGV) (Capillovirus; Betaflexiviridae) | CP | Apple and pear | AGE | 1 | 42 | 10 times less than RT‐PCR; Total RNA diluted up to 4.7 ng/μl | Kim et al. (2018) |
| – | Apple | CRISPR/Cas12a with oligonucleotide‐conjugated gold nanoparticle | 20 | 37 | RNA transcripts of 0.01–1 fM (25 viral copies) sensitivity equal to RT‐qPCR | Jiao et al. (2021) | |
| Apple stem pitting virus (ASPV) (Foveavirus; Betaflexiviridae) | CP | Pear | Capillary gel electrophoresis | 4 | 42 | 1000‐fold higher than RT‐PCR; 1 fg/μL of RNA | Kim et al. (2019) |
| Apple | CRISPR/Cas12a with oligonucleotide‐conjugated gold nanoparticle | 20 | 37 | RNA transcripts of 0.01–1 fM (25 viral copies) sensitivity equal to RT‐qPCR | Jiao et al. (2021) | ||
| Barley yellow dwarf virus (BYDV) (Luteovirus; Luteoviridae) | CP | Oat | AGE | 5 | 42 | 100 times more than RT‐PCR; 50 fg/μl RNA | Kim et al. (2020) |
| Beet necrotic yellow vein virus (BNYVV) (Benyvirus; Benyviridae) | RNA‐1 | Sugarbeet | CRISPR‐Cas12a Reporter Assay with fluorescence signal | 60 | 42 | 0.1 pM of Target DNA | Ramachandran et al. (2021) |
| Chilli veinal mottle virus (ChiVMV) (Potyvirus; Potyviridae) | CP | Tobacco | AGE | 20 | 38 | 10‐fold more sensitive than RT‐PCR; 10 fg RNA | Jiao et al. (2020) |
| Citrus concave gum‐associated virus (CCGaV) (Coguvirus; Phenuiviridae) | RNA‐1 | Apple | AGE | 30 | 38 | 10‐fold more sensitive than RT‐PCR | Liu et al. (2021) |
| Cucumber green mottle mosaic virus (CGMMV) (Tobamovirus; Virgaviridae) | CP | Watermelon | AGE | 30 | 38 | 10‐fold more sensitive than RT‐PCR; 1.0 × 10−6 μg RNA | Jiao et al. (2019b) |
| Cucumber mosaic virus (CMV) (Cucumovirus; Bromoviridae) | CP | Banana | Real‐time visual fluorescence (Exo probe) | 25 | 40 | 3 pg/μl of RNA; up to 10−5 dilution of the crude leaf extract | Srivastava et al. (2019) |
| Cucurbit yellow stunting disorder virus (CYSDV) (Crinivirus; Closteroviridae) | CP | Watermelon, squash | Real‐time visual fluorescence (Exo probe) | 30 | 40 | 2.5 pg purified total RNA | Kalischuk et al. (2020) |
| Ginger chlorotic fleck‐associated virus 1 (GCFaV‐1) (Tombusviridae) | CP | Ginger | AGE | 50 | 39 | 100‐fold more sensitive than RT‐PCR | Naveen and Bhat (2020) |
| ginger chlorotic fleck‐associated virus 2 (GCFaV‐2) (Ampleovirus; Closteroviridae) | CP | Ginger | AGE | 30 | 39 | 1000‐fold more sensitive than RT‐PCR | Naveen and Bhat (2020) |
| Little cherry virus 2 (LChV2) (Ampelovirus; Closterovridae) | CP | Sweet cherry, mealybug | LFD | 15 | 39 | More than RT‐PCR | Mekuria et al. (2014) |
| Maize chlorotic mottle virus (MCMV) (Machlomovirus; Tombusviridae) | CP | Maize | AGE | 30 | 38 | 10‐fold more than or less than RT‐PCR | Gao et al. (2021); Jiao et al. (2019a) |
| Onion yellow dwarf virus (OYDV) (Potyvirus; Potyviridae) | CP | Onion | AGE | 25 | 42 | 10‐fold more than RT‐PCR | Kumar et al. (2021b) |
| Plum pox virus (PPV) (Potyvirus; Potyviridae) | CP | Apricot, cherry, peach, plum | LFD, fluorescence (AmplifyRP) | 15 | 39 | More sensitive than Immunostrip | Zhang et al. (2014) |
| Potato virus X (PVX) (Potexvirus; Alphaflexiviridae) | CP | Potato | AGE | 30 | 39 | 100 times more than RT‐PCR | Kumar et al. (2021a) |
| CP | Nicotiana benthamiana | CRISPR/Cas12a‐induced fluorescence | 30 | 37 | Picomolar range (femtomolar range with longer reaction times) | Aman et al. (2020b) | |
| Potato virus Y (PVY) (Potyvirus; Potyviridae) | CP | Potato | AGE, LFD, fluorescence | 20 | 25–40 | Equal to RT‐PCR | Babujee et al. (2019); Cassedy et al. (2022); Wang et al. (2020) |
| CP | Nicotiana benthamiana | CRISPR/Cas12a‐induced fluorescence | 30 | 37 | Picomolar range (femtomolar range with longer reaction times) | Aman et al. (2020b) | |
| Rice black‐streaked dwarf virus (RBSDV) (Fijivirus; Reoviridae) | P10 | Rice | LFD | 20 | 37 | Equal to RT‐PCR | Zhao et al. (2019) |
| Rice yellow mottle virus (RYMV) (Sobemovirus; Solemoviridae) | ORF 2 | Rice | AGE | 05 | 41 | Equal to RT‐PCR | Juma et al. (2021) |
| Rose rosette virus (RRV) (Emaravirus; Fimoviridae) | RNA 3 | Rose | AGE, fluorescence | 20 | 42 | 1 fg/μl of viral transcript | Babu et al. (2017a,b) |
| Sugarcane mosaic virus (SCMV) (Potyvirus; Potyviridae) | CP | Maize | AGE | 30 | 38 | 10‐fold more than or less than RT‐PCR | Gao et al. (2021) |
| Sugarcane streak mosaic virus (SCSMV) (Poacevirus; Potyviridae) | CP | Sugarcane | AGE | 05 | 38 | 100‐fold higher than RT‐PCR | Feng et al. (2018b) |
| Sugarcane yellow leaf virus (SCYLV) (Polerovirus; Solemoviridae) | CP | Sugarcane | AGE | 10–20 | 27–39 | 10 times lower than RT‐PCR | Feng et al. (2018a) |
| Tobacco mosaic virus (TMV) (Tobamovirus; Virgaviridae) | CP | Nicotiana benthamiana | CRISPR/Cas12a‐induced fluorescence | 30 | 37 | Picomolar range (femtomolar range with longer reaction times) | Aman et al. (2020b) |
| Tomato spotted wilt virus (TSWV) (Orthotospovirus; Tospoviridae) | CP | Pepper | LFD | 10 | 38 | Equal to RT‐PCR | Lee et al. (2021) |
| Yam mild mosaic virus (YMMV) (Potyvirus; Potyviridae) | CP | Yam | Fluorescence | 30 | 37 | 1 × 10−3 dilution of crude extract | Silva et al. (2018) |
| Yam mosaic virus (YMV) (Potyvirus; Potyviridae) | CP | Yam | Fluorescence | 30 | 37 | 1 × 10−3 dilution of crude extract | Silva et al. (2018) |
| Viroids | |||||||
| Apple scar skin viroid (ASSVd) (Apscaviroid; (Pospiviroidae) | Full | Apple | AGE | 10 | 42 | 10 times more than RT‐PCR; Total RNA diluted up to 20 pg/μl | Kim et al. (2021a) |
| Full | Apple | CRISPR/Cas12a with oligonucleotide‐conjugated gold nanoparticle | 20 | 37 | RNA transcripts of 0.01–1 fM (25 viral copies) sensitivity equal to RT‐qPCR | Jiao et al. (2021) | |
| Hop stunt viroid (HSVd) (Hostuviroid; Pospiviroidae) | Full | Hops | LFD | 20 | 39 | 2 × 109 copies of HSVd trimeric transcript (less than RT‐PCR) | Kappagantu et al. (2017) |
| Peach latent mosaic viroid (PLMVd) (Pelamoviroid; Avsunviroidae) | Full | Peach | AGE | 05 | 42 | 1000‐fold more than RT‐PCR. | Lee et al. (2020) |
| Potato spindle tuber viroid (PSTVd) (Pospiviroid; Pospiviroidae) | Full | Potato | LFD | 30 | 39 | 106 copies of in vitro transcribed PSTVd RNA | Ivanov et al. (2020) |
| Tomato apical stunt viroid (TASVd) (Pospiviroid; Pospiviroidae) | Full | Tomato | Fluorometer | 20 | 39 | 27 to 81‐ fold dilution of crude extract | Kovalskaya and Hammond (2022) |
| Tomato chlorotic dwarf viroid (TCDVd) (Pospiviroid; Pospiviroidae) | Full | Tomato | LFD | 19 | 39 | 1 pg pure RNA. Sensitivity equal to RT‐PCR | Hammond and Zhang (2016) |
Real‐time detection of RPA products based on fluorescence
Recombinase polymerase amplification products can also be viewed and quantified in real‐time with a fluorimeter using fluorescent probes. This assay is performed using a TwistAmp Exo kit or TwistAmp fpg kit (Daher et al., 2016; Powell et al., 2018; Stringer et al., 2018). The TwistAmp Exo kit uses a specially designed probe called the Exo probe, which consists of a complementary 46–52 nucleotide sequence internal to both primers. The Exo probe contains a polymerase extension blocking group at the 3′ end and an internal abasic nucleotide analogue (the tetrahydrofuran residue THF, which replaces a conventional nucleotide, also called dSpacer) flanked by a dT‐fluorophore and a matching dT‐quencher group (Figure 3b) (Lobato and O'Sullivan, 2018). During the RPA reaction, primers generate targets for annealing of the probe. When the probe is annealed to the template strand, the THF residue is cleaved by the exonuclease, separating the fluorophore and quencher, thus generating a fluorescent signal that can be monitored with a fluorimeter. The amount of signal produced is directly proportional to the concentration of amplicons.
Like the Exo probe, the fpg probe in the fpg kit also contains a complementary sequence internal to both primers that is approximately 35 nucleotides long (Powell et al., 2018). This probe is modified at the 5′ end with a quencher group and contains a fluorophore on an abasic nucleotide analogue 4–5 nucleotides downstream of the quencher. The fluorophore is attached to the ribose group of the abasic nucleotide through a C‐O‐C linker termed the dR group. During the RPA reaction, primers generate targets for annealing of the probe. When the probe is annealed to the template strand, the fpg cleaves the probe at the dR position, thus separating the fluorophore and quencher, thereby generating a fluorescent signal that can be monitored with a fluorimeter. The amount of signal produced is directly proportional to the concentration of the amplicon. These approaches were used to develop detection assays for cucumber mosaic virus infecting banana (Srivastava et al., 2019), cucurbit yellow stunting disorder virus infecting watermelon and squash (Kalischuk et al., 2020), plum pox virus infecting stone fruits (Zhang et al., 2014), potato virus Y (Cassedy et al., 2022), rose rosette virus (Babu et al., 2017b), yam mosaic virus, and yam mild mosaic virus (Silva et al., 2018; Table 1).
Instead of a specific probe, real‐time detection is also possible using intercalating dyes in the RPA reaction, such as SYBR Green or Eva Green (Lobato and O'Sullivan, 2018). However, these dyes cannot discriminate RPA amplicons from non‐specific amplified dsDNA products or primer‐dimers.
Detection of RPA products via lateral flow assay
To increase the sensitivity and facilitate the instrument‐free detection of RPA products, a lateral flow assay (LFA) can be used, allowing the results to be visualized extremely rapidly. This assay is performed using a TwistAmp nfo kit containing two primers (a conventional forward primer and a reverse primer labelled at its 5′ end, usually with biotin), one probe, and the enzyme nfo (endonuclease IV). The probe should have a complementary sequence internal to both primers with lengths varying from 46 to 52 nucleotides. The probe is modified at the 5′ end with an antigenic label (usually a carboxyfluorescein group (FAM), or Alexa Fluor 488 or digoxigenin), a polymerase extension blocking group at the 3′ end, and an internal abasic nucleotide analogue (the tetrahydrofuran residue THF, which replaces the conventional nucleotide, also called dSpacer) placed at least 30 nucleotides from the 5′ end and 15 nucleotides from the 3′ end (Figure 3c) (Lobato and O'Sullivan, 2018).
During the RPA reaction, primers generate targets for annealing of the probe. When the probe is annealed to the template strand, the THF residue is cleaved by the nfo enzyme, leaving a 3′ hydroxyl group at the 3′ end of the probe, thus converting the probe into a primer that can extend the chain. RPA performed in this manner produces amplicons with two antigenic labels (FAM and biotin) on either side and can be processed via a lateral flow assay to visualize the results. The diluted (1:10 or 1:100) products of RPA are loaded onto the sample pad of the lateral flow strip, and the sample pad region of the strip is placed into PBST buffer. The labelled amplicons bind to gold‐labelled FITC‐specific antibodies and move through the capillarity. The amplicons bound with gold particles become immobilized when they combine with biotin ligand molecules at the test line and generate a red or blue line. Gold particles that have not been captured move further to reach the control lines, where they become immobilized by species‐specific antibodies to produce a red or blue line. Thus, the development of coloured test and control lines indicates a positive reaction, while the development of only the control line indicates that the sample is negative for the test virus.
By combining RPA with a lateral flow assay, results can be obtained in less than 1 h with a sensitivity of detection ranging from 1 to 10 DNA copies (Lobato and O'Sullivan, 2018). The method is also suitable for detecting multiple viruses if different antigenic labels are used for different viruses. RPA‐LFD assays have been developed to detect milk vetch dwarf virus, little cherry virus 2, plum pox virus, potato virus Y, rice black‐streaked dwarf virus, and tomato spotted wilt virus (Cao et al., 2020; Cassedy et al., 2022; Lee et al., 2021; Mekuria et al., 2014; Zhang et al., 2014). Similar assays have also been developed for the detection of viroids such as hop stunt viroid, potato spindle tuber viroid, and tomato chlorotic dwarf viroid (Hammond and Zhang, 2016; Ivanov et al., 2020; Kappagantu et al., 2017; Table 1). Further improvements of LFA were reported by several researchers (Cordray and Richards‐Kortum, 2015; Crannell et al., 2014; Jauset‐Rubio et al., 2016; Rohrman et al., 2012).
CRISPR/Cas for onsite detection
CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR‐associated) is a naturally occurring adaptive immune system used by prokaryotes such as bacteria and archaea to avoid invading viruses and plasmids (Doudna and Charpentier, 2014; Jinek et al., 2012). When a virus infects a bacterium for the first time, it incorporates a small portion of the viral sequence into the host's genome. Later, when the same bacterium is infected by the same virus for the second time, it transcribes the incorporated viral sequences into CRISPR RNA. CRISPR RNA forms a complex with tracrRNA and CRISPR‐associated (Cas) protein, which binds to the complementary sequence in the invading viral DNA/RNA and generates double‐strand breaks (DSBs), thereby destroying the virus. The CRISPR/Cas system can be introduced into a eukaryotic cell to cut its DNA at the desired site using single‐guide RNA (sgRNA) containing a complementary sequence to the targeted genomic region. Upon introduction of sgRNA and Cas9 in the nucleus of the target organism, the sgRNA binds to and guides Cas9 to the target sequence to generate a site‐specific DSB. The DSB is then repaired by non‐homologous end joining (NHEJ) or homology‐directed DNA repair (HDR) if a donor DNA molecule is provided (Jinek et al., 2012).
CRISPR/Cas is widely used as a genome‐editing tool due to its target specificity and the ease of targeting any gene simply by varying the gRNA sequence. Due to their ease of target‐dependent programmability, CRISPR/Cas systems have also been used for virus interference in various organisms including plants (Ali et al., 2015a,b, 2018; Aman et al., 2018a,b, 2020a; Mahas et al., 2018, 2019; Mahas and Mahfouz, 2018). Following the discovery of increasing numbers of Cas proteins with different activities, CRISPR/Cas systems have been used to diagnose infectious organisms, including viruses. CRISPR/Cas is also employed for the detection of microRNAs (miRNAs), single‐nucleotide polymorphisms (SNPs), and DNA methylation (Kim et al., 2021b).
Among the various Cas proteins, Cas3 and Cas9 cleave dsDNA; Cas12 and Cas14 cut either dsDNA or ssDNA; and Cas13 cleaves ssRNA (Abudayyeh et al., 2016; Aman et al., 2018a,b, 2020a; Harrington et al., 2018; Kim et al., 2021b; Zetsche et al., 2015). To enable the specific binding of gRNA‐Cas protein complexes and cleavage, most Cas proteins (Cas3, Cas9, and Cas12) require a tri/tetranucleotide sequence next to the gRNA binding region known as the protospacer adjacent motif (PAM). The Cas13 recognition site next to the crRNA consists of a single nucleotide and is called the protospacer recognition site (PFS). By contrast, Cas14 cleaves ssDNA in a PAM‐independent manner and dsDNA in a PAM‐dependent manner (Kim et al., 2021b). Upon binding and cleaving of the specific target nucleic acid, certain Cas proteins (Cas3, Cas12, Cas13, and Cas14) show collateral, non‐specific activities against ssRNA (Cas13a) or ssDNA (Cas 12a and Cas14a) (Abudayyeh et al., 2016; Aman et al., 2020a; Chen et al., 2018; Harrington et al., 2018). This collateral (trans‐cleavage) activity occurs multiple times for a single target bound by the CRISPR‐gRNA complex.
This non‐specific cleavage has been exploited for diagnostic purpose (Abudayyeh et al., 2016; Aman et al., 2020a,b, 2022; Chen et al., 2018, 2021), leading to the development of a Cas13‐based detection assay for RNA viruses known as SHERLOCK (specific high‐sensitivity enzymatic reporter unlocking) (Gootenberg et al., 2017, 2018). In this method, the RNA template is converted into cDNA via reverse transcription. The cDNA is then subjected to RPA, and the RPA‐amplified DNA molecules are subjected to in vitro transcription to obtain RNA templates that serve as the substrate for specific cleavage by the Cas13a‐gRNA complex. Following target recognition and cleavage, the non‐specific trans‐cleavage nuclease activity of Cas13a becomes activated, and this enzyme cleaves the labelled ssRNA probe (labelled with a quencher and reporter). Once cleaved, the released reporter generates a fluorescent signal that can easily be quantified or detected under UV light. Different reporters that are compatible with LFA devices have also been used to facilitate signal detection at point‐of‐care (Figure 4; Gootenberg et al., 2018).
Figure 4.
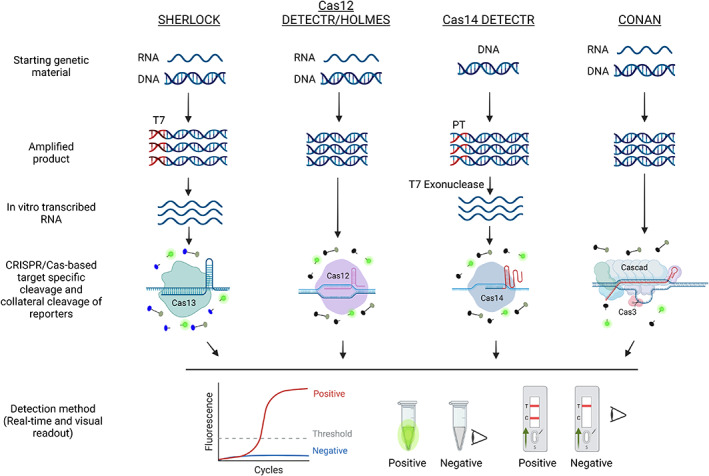
CRISPR‐based diagnostic platforms. A schematic diagram of the different CRISPR‐based diagnostic platforms for nucleic acid detection. SHERLOCK, Cas12‐DETECTR, Cas12‐HOLMES, and Cas14‐DETECTR make use of a single effector, while CONAN uses a multi‐component Cas3 system for nucleic acid detection with their respective gRNAs. In the SHERLOCK system, the target is amplified with a forward primer appended with a T7 promoter sequence. The amplified product is reverse transcribed and used as a template for Cas13. In the Cas14‐DETECTR system, the template is amplified with a phosphorothioate (PT)‐containing primer to protect one strand from T7 exonuclease activity. The PT‐protected strand is then recognized by Cas14. CONAN is based on a multi‐component system. After target recognition, the highly specific collateral activity of these proteins is activated, causing them to cleave the corresponding reporter. Due to this activity, all of these systems have been effectively harnessed for efficient, robust, precise CRISPR‐based nucleic acid platforms. [Colour figure can be viewed at wileyonlinelibrary.com]
Another detection assay called DETECTR (DNA endonuclease‐targeted CRISPR trans‐reporter) was subsequently developed for the detection of human papillomavirus (Chen et al., 2018). In this method, the target region is initially amplified using RPA to increase the assay's sensitivity. The resulting dsDNA is used as the target for Cas12a‐gRNA‐mediated cleavage. A ssDNA probe labelled with a reporter and quencher molecules is used to monitor the collateral (trans‐cleavage) activity of Cas12a. The other Cas protein exploited for diagnosis is Cas14, which does not require a PAM sequence for specific cleavage of the target ssDNA and is small (nearly half the size of Cas12a). Cas14‐DETECTR is useful for detecting ssDNA pathogens and SNPs (Aquino‐Jarquin, 2019; Harrington et al., 2018). Subsequently, Cas12a‐based HOLMES (one‐hour low‐cost multipurpose highly efficient system) and a Cas3‐based assay named CONAN (Cas3 operated nucleic acid detection) were developed. CONAN is similar to DETECTR, except that it uses multiple Cas proteins to achieve higher sensitivity (Figure 4; Li et al., 2018; Yoshimi et al., 2020). The sensitivity of CRISPR‐based diagnostic assays is comparable to that of PCR‐based assays. However, the current CRISPR‐based diagnostic assays require prior amplification of the target nucleic acid molecule by RPA, LAMP, or PCR, adding to the time and cost of the assays (Dong et al., 2018; Gootenberg et al., 2017). To overcome this problem, non‐amplification‐based CRISPR/Cas assays are currently being developed (Aman et al., 2020a; Bruch et al., 2019; Hajian et al., 2019; Kim et al., 2021b).
CRISPR/Cas‐based onsite detection of plant DNA viruses
Loop‐mediated isothermal amplification followed by CRISPR–Cas12a‐based detection using a real‐time fluorescence assay was used to detect tomato yellow leaf curl virus and tomato leaf curl New Delhi virus (Mahas et al., 2021) (Table S1). This assay takes less than an hour and successfully detects both viruses in plants with high sensitivity and specificity. The assay provides easy‐to‐interpret visual readouts using a simple, low‐cost fluorescence visualizer, making it suitable for point‐of‐use applications. Similarly, Bernabé‐Orts et al., successfully detected tomato brown rugose fruit virus using RT‐LAMP followed by CRISPR–Cas12a‐based detection using both real‐time fluorescence detection and calorimetrically using LFD (Figure 5) (Bernabé‐Orts et al.). Furthermore, Alon et al. (2021) performed differential detection of tomato brown rugose fruit virus and tomato mosaic virus infecting tomatoes using a similar approach, except that they used RT‐PCR instead of RT‐LAMP or RT‐RPA for the initial conversion of viral RNA into dsDNA to act as a substrate for Cas12a.
Figure 5.
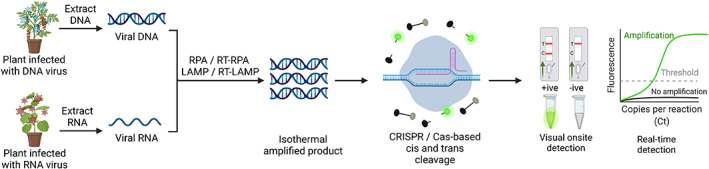
Onsite CRISPR‐based detection of plant viruses. A schematic flow diagram of nucleic acid detection using CRISPR/Cas‐based biosensing technologies. The extracted nucleic acid from plants infected with either RNA or DNA viruses is amplified via RT‐RPA/RT‐LAMP (for an RNA template) or RPA/LAMP (for a DNA template) using target‐specific primers. The amplified product is subjected to CRISPR/Cas‐based detection, and the signal readout can be detected via real‐time PCR or visually using different reporter systems. FAM/biotin and FAM or HEX reporters are primarily used for LFA and visual detection (under UV or LED light), respectively. All figures were created with BioRender. [Colour figure can be viewed at wileyonlinelibrary.com]
CRISPR/Cas‐based onsite detection of plant RNA viruses
Recently, Aman et al. (2020b) developed a one‐pot detection assay named iSCAN one‐pot (iSCAN‐OP), a simple, quick, efficient RT‐RPA method coupled with a CRISPR/Cas12a‐based one‐step detection assay to detect potato virus X, potato virus Y, and tobacco mosaic virus (Table 1). In this method, viral RNA is initially converted into dsDNA via RT‐RPA, allowing it to act as a substrate for Cas12a cis activity (Figure 5). The targeting of the dsDNA by Cas12a elicits its collateral action, which in turn cuts the ssDNA reporter molecules and releases the signal combined with the iSCAN‐OP assay with a commercially available P51 fluorescence viewer device to enable rapid, inexpensive, in‐field diagnosis of viruses (Aman et al., 2020b; Chen et al., 2018). This assay can be performed at a single temperature within 30 min and provides a robust system for plant virus detection.
RT‐RPA followed by CRISPR/Cas12a‐based detection using real‐time fluorescence was also used to detect beet necrotic yellow vein virus (BNYVV) (Ramachandran et al., 2021). Jiao et al. (2021) performed CRISPR/Cas12a‐based detection of viruses infecting apples, such as apple necrotic mosaic virus, apple stem pitting virus, apple stem grooving virus, apple chlorotic leaf spot virus, and apple scar skin viroid (Table 1). Each virus/viroid was detected directly from crude leaf extracts after simultaneous multiplex RT‐RPA with high specificity. The results could be identified with the naked eye using oligonucleotide‐conjugated gold nanoparticles and LFDs. The sensitivity of the CRISPR/Cas12a‐RT‐RPA platform is equivalent to that of RT‐qPCR, with detection limits ranging from 250 to 2500 copies of virus. This method is simple and quick, requiring less than an hour from sample collection to the visualization of results.
Concluding remarks
Plant diseases impose serious limitations on the cultivation of many crops in all growing regions due to a lack of rapid and sensitive methods for proper diagnosis. The key factors in any efficient disease management programme are the reliable identification of pathogens and an understanding of their natural dissemination mechanisms. Immunoassay and nucleic acid‐based techniques are currently the most used diagnostic methods in plant pathology. Among these techniques, PCR and its modifications as well as various ELISA formats are the most popular. With the development of isothermal assays such as LAMP and RPA and their coupling with CRISPR/Cas systems, the detection of viruses and other pathogens has become far easier, as these assays do not require laboratory setup or specific technical skills. These tests can be performed onsite using crude extracts from plants as a template. The assays are more sensitive and require less time compared with conventional PCR assays, and the results can easily be interpreted visually.
These assays are useful for detecting viruses in seeds, vegetative propagules, weed hosts, and vectors that harbour viruses so that appropriate measures can be taken to contain the spread of the viruses within the field/locality. These assays can be used for screening germplasm accessions of crops for resistance against viruses and would also serve as a tool to study epidemiology and predict the outbreak of the disease.
Both isothermal assays discussed here are suitable for onsite detection of plant viruses and other infectious agents in resource‐poor settings without the need for any special technical skills. Results of both assays can be visualized in real‐time or endpoint detection using a lateral flow device. Further, among the two assays, the RPA assay is more suited for onsite detection as it can be performed using a pair of primers, lyophilized reagents (that can be stored at room temperature) and crude extracts from the test plant at low temperature (37–42 °C), in a short time (5–20 min) and amenable for multiplexing. However, RPA is affected by the initial concentration of the target template, concentration of magnesium acetate, mixing step, and the need for a purification step if results are viewed through agarose gel electrophoresis. On the contrary, LAMP is performed using 4–6 primers at higher temperatures (58–68 °C) for a duration of up to 1 h and is not easily amenable for multiplexing. Further, like RPA, LAMP is also affected by the initial concentration of the template, concentrations of magnesium sulphate, betaine, temperature, and time. CRISPR‐based diagnostic assays are very specific, sensitive, and suitable for onsite detection either by real‐time through fluorescence or endpoint through a lateral flow device. Besides its utility in the detection of infectious agents, it is used in the detection of miRNA, DNA methylation, and non‐nucleic acids such as proteins. The current CRISPR‐based diagnostic assays require prior amplification of the target nucleic acid molecule by RPA, LAMP, or PCR, adding to the time and cost of the assays. A small beginning has been made on the development of non‐amplification‐based CRISPR/Cas assay but more research efforts are needed in this direction.
Future efforts on the onsite detection should focus on the development of affordable, cost‐effective, and robust assays. RPA‐ and LAMP‐based technologies coupled with CRISPR enzymes are revolutionizing pathogen diagnostics. Future improvements should focus on (i) building an inexpensive and portable device for in‐field deployment (ii) quick extraction methods to bypass the need for nucleic acid purification (iii) multiplexing, to establish and optimize the detection of multiple pathogens simultaneously (iv) to develop an artificial intelligence (AI) application to be able to share the data locally and globally which would aid in the better decision making process to manage the outbreak of diseases. With improvements and technology transfer to a simple yet affordable device, CRISPR‐based diagnostic technologies have the ability to revolutionize onsite pathogen diagnostics.
Conflict of interest
All authors declare that they have no conflict of interest.
Author contributions
MM conceived the idea. A.I Bhat, RA, and MM wrote the paper.
Funding
This publication was supported by the King Abdullah University of Science and Technology (KAUST) baseline fund (BAS/1/1035‐01‐01) to Professor Magdy Mahfouz.
Supporting information
Table S1 Loop‐mediated isothermal amplification (LAMP) and reverse transcription (RT)‐LAMP‐based assays used for the detection of different plant viruses.
Acknowledgements
We would like to thank members of genome engineering and synthetic biology laboratory for thoughtful discussions. AIB is thankful to the Science and Engineering Research Board (SERB), Government of India for supporting the project (CRG/2021/000292).
References
- Abudayyeh, O.O. , Gootenberg, J.S. , Konermann, S. , Joung, J. , Slaymaker, I.M. , Cox, D.B. , Shmakov, S. et al. (2016) C2c2 is a single‐component programmable RNA‐guided RNA‐targeting CRISPR effector. Science, 353, aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali, Z. , Abul‐faraj, A. , Li, L. , Ghosh, N. , Piatek, M. , Mahjoub, A. , Aouida, M. et al. (2015a) Efficient virus‐mediated genome editing in plants using the CRISPR/Cas9 system. Mol Plant, 8, 1288–1291. [DOI] [PubMed] [Google Scholar]
- Ali, Z. , Abulfaraj, A. , Idris, A. , Ali, S. , Tashkandi, M. and Mahfouz, M.M. (2015b) CRISPR/Cas9‐mediated viral interference in plants. Genome Biol. 16, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali, Z. , Mahas, A. and Mahfouz, M. (2018) CRISPR/Cas13 as a tool for RNA interference. Trends Plant Sci. 23, 374–378. [DOI] [PubMed] [Google Scholar]
- Almasi, M.A. , Ojaghkandi, M. and Aghaei, S. (2013) Visual detection of curly top virus by the colorimetric loop‐mediated isothermal amplification. J. Plant Pathol. Microbiol. 4, 9. [Google Scholar]
- Alon, D.M. , Hak, H. , Bornstein, M. , Pines, G. and Spiegelman, Z. (2021) Differential detection of the Tobamoviruses tomato mosaic virus (ToMV) and tomato Brown rugose fruit virus (ToBRFV) using CRISPR‐Cas12a. Plants, 10, 1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman, R. , Ali, Z. , Butt, H. , Mahas, A. , Aljedaani, F. , Khan, M.Z. , Ding, S. et al. (2018b) RNA virus interference via CRISPR/Cas13a system in plants. Genome Biol. 19, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman, R. , Mahas, A. , Butt, H. , Aljedaani, F. and Mahfouz, M. (2018a) Engineering RNA virus interference via the CRISPR/Cas13 machinery in Arabidopsis. Viruses, 10, 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman, R. , Mahas, A. and Mahfouz, M. (2020b) Nucleic acid detection using CRISPR/Cas biosensing technologies. ACS Synth. Biol. 9, 1226–1233. [DOI] [PubMed] [Google Scholar]
- Aman, R. , Mahas, A. , Marsic, T. , Hassan, N. and Mahfouz, M.M. (2020a) Efficient, rapid, and sensitive detection of plant RNA viruses with one‐pot RT‐RPA‐CRISPR/Cas12a assay. Front. Microbiol. 11, 610872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman, R. , Marsic, T. , Sivakrishna Rao, G. , Mahas, A. , Ali, Z. , Alsanea, M. , Al‐Qahtani, A. et al. (2022) iSCAN‐V2: a one‐pot RT‐RPA–CRISPR/Cas12b assay for point‐of‐care SARS‐CoV‐2 detection. Front. Bioeng. Biotechnol. 9, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquino‐Jarquin, G. (2019) CRISPR‐Cas14 is now part of the artillery for gene editing and molecular diagnostic. Nanomedicine, 18, 428–431. [DOI] [PubMed] [Google Scholar]
- Babu, B. , Washburn, B.K. , Ertek, T.S. , Miller, S.H. , Riddle, C.B. , Knox, G.W. , Ochoa‐Corona, F.M. et al. (2017b) A field based detection method for rose rosette virus using isothermal probe‐based reverse transcription‐recombinase polymerase amplification assay. J. Virol. Methods, 247, 81–90. [DOI] [PubMed] [Google Scholar]
- Babu, B. , Washburn, B.K. , Miller, S.H. , Poduch, K. , Sarigul, T. , Knox, G.W. , Ochoa‐Corona, F.M. et al. (2017a) A rapid assay for detection of rose rosette virus using reverse transcription‐recombinase polymerase amplification using multiple gene targets. J. Virol. Methods, 240, 78–84. [DOI] [PubMed] [Google Scholar]
- Babujee, L. , Witherell, R.A. , Mikami, K. , Aiuchi, D. , Charkowski, A.O. and Rakotondrafara, A.M. (2019) Optimization of an isothermal recombinase polymerase amplification method for real‐time detection of potato virus Y O and N types in potato. J. Virol. Methods, 267, 16–21. [DOI] [PubMed] [Google Scholar]
- Bernabé‐Orts, J.M. , Hernando, Y. and Aranda, M.A. (2021) Toward a CRISPR‐based point‐of‐care test for tomato brown rugose fruit virus detection. PhytoFrontiers™. 10.1094/PHYTOFR-08-21-0053-TA [DOI] [Google Scholar]
- Bhat, A.I. and Rao, G.P. (2020) Characterization of Plant Viruses. New York, NY: Springer. [Google Scholar]
- Bhat, A.I. , Siljo, A. and Deeshma, K.P. (2013) Rapid detection of piper yellow mottle virus and cucumber mosaic virus infecting black pepper (Piper nigrum) by loop‐mediated isothermal amplification (LAMP). J. Virol. Methods, 193, 190–196. [DOI] [PubMed] [Google Scholar]
- Boyle, D. , McNerney, R. , Low, H. , Leader, B. , Pérez‐Osorio, A. , Meyer, J. , O'Sullivan, D. et al. (2014) Rapid detection of Mycobacterium tuberculosis by recombinase polymerase amplification. PLoS ONE, 9, e103091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruch, R. , Baaske, J. , Chatelle, C. , Meirich, M. , Madlener, S. , Weber, W. , Dincer, C. et al. (2019) CRISPR/Cas13a‐powered electrochemical microfluidic biosensor for nucleic acid amplification‐free miRNA diagnostics. Adv. Mater. 31, 1905311. [DOI] [PubMed] [Google Scholar]
- Camarço, R.F.E.A. , Lima, J.A.A. and Pio‐Ribeiro, G. (1998) Transmission and presence in the soil of papaya lethal yellowing virus. Fitopatologia Brasileira (Brazil).
- Cao, Y. , Yan, D. , Wu, X. , Chen, Z. , Lai, Y. , Lv, L. , Yan, F. et al. (2020) Rapid and visual detection of milk vetch dwarf virus using recombinase polymerase amplification combined with lateral flow strips. Virol. J. 17, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassedy, A. , Della Bartola, M. , Parle‐McDermott, A. , Mullins, E. and O'Kennedy, R. (2022) A one‐step reverse transcription recombinase polymerase amplification assay for lateral flow‐based visual detection of PVY. Anal. Biochem. 642, 114526. [DOI] [PubMed] [Google Scholar]
- Cassedy, A. , Parle‐McDermott, A. and O'Kennedy, R. (2021) Virus detection: a review of the current and emerging molecular and immunological methods. Front. Mol. Biosci. 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.S. , Ma, E. , Harrington, L.B. , Da Costa, M. , Tian, X. , Palefsky, J.M. and Doudna, J.A. (2018) CRISPR‐Cas12a target binding unleashes indiscriminate single‐stranded DNase activity. Science, 360, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Mao, S. , Zhang, W. , Fan, X. , Wu, W. , Liu, C. , Zhao, K. et al. (2021) Rapid visual detection method for barley yellow mosaic virus using reverse transcription loop‐mediated isothermal amplification (RT‐LAMP). Plant Dis. 105, 2658–2663. [DOI] [PubMed] [Google Scholar]
- Chin, M. , Ahmad, M.H. and Tennant, P. (2007) Momordica charantia is a weed host reservoir for papaya ringspot virus type P in Jamaica. Plant Dis. 91, 1518. [DOI] [PubMed] [Google Scholar]
- Clark, M.F. and Adams, A.N. (1977) Characteristics of the microplate method of enzyme‐linked immunosorbent assay for the detection of plant viruses. J. Gen Virol. 34, 475–483. [DOI] [PubMed] [Google Scholar]
- Clark, M. and Bar‐Joseph, M. (1984) Enzyme immunosorbent assays in plant virology. In: Clark, M. & Hilary, K. (Eds.) Methods in Virology, pp. 51–85. Orlando, FL: Academic Press, Inc. [Google Scholar]
- Clark, M.F. , Lister, R.M. and Bar‐Joseph, M. (1986) ELISA techniques. In Colowick, S. P. & Kaplan, N. O. (Eds.) Methods in Enzymology, Vol. 118, pp. 3–829. Academic Press. [Google Scholar]
- Cordray, M.S. and Richards‐Kortum, R.R. (2015) A paper and plastic device for the combined isothermal amplification and lateral flow detection of plasmodium DNA. Malaria J. 14, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crannell, Z.A. , Castellanos‐Gonzalez, A. , Irani, A. , Rohrman, B. , White, A.C. and Richards‐Kortum, R. (2014) Nucleic acid test to diagnose cryptosporidiosis: lab assessment in animal and patient specimens. Anal. Chem. 86, 2565–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher, R.K. , Stewart, G. , Boissinot, M. and Bergeron, M.G. (2016) Recombinase polymerase amplification for diagnostic applications. Clin. Chem. 62, 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, R. , Mu, L. , Maireroa, N. , Wigmore, W. , Grison, M. , Bateson, M. and Thomas, J. (2005) First records of the papaya strain of papaya ringspot virus (PRSV‐P) in French Polynesia and The Cook Islands. Aust. Plant Pathol. 34, 125–126. [Google Scholar]
- Dong, J. , Chen, G. , Wang, W. , Huang, X. , Peng, H. , Pu, Q. , Du, F. et al. (2018) Colorimetric PCR‐based microRNA detection method based on small organic dye and single enzyme. Anal. Chem. 90, 7107–7111. [DOI] [PubMed] [Google Scholar]
- Doudna, J.A. and Charpentier, E. (2014) The new frontier of genome engineering with CRISPR‐Cas9. Science, 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Feng, X.‐Y. , Shen, L.‐B. , Wang, W.‐Z. , Wang, J.‐G. , Cao, Z.‐Y. , Feng, C.‐L. , Zhao, T.‐T. et al. (2018a) Development of a reverse transcription‐recombinase polymerase amplification assay for detection of sugarcane yellow leaf virus. Sugar Tech. 20, 700–707. [Google Scholar]
- Feng, X.‐Y. , Shen, L.‐B. , Wang, W.‐Z. , Wang, J.‐G. , Cao, Z.‐Y. and Zhang, S.‐Z. (2018b) Reverse transcription–recombinase polymerase amplification assay for the detection of sugarcane streak mosaic virus in sugarcane. Sugar Tech. 21, 645–652. [Google Scholar]
- Fukuta, S. , Iida, T. , Mizukami, Y. , Ishida, A. , Ueda, J. , Kanbe, M. and Ishimoto, Y. (2003a) Detection of Japanese yam mosaic virus by RT‐LAMP. Arch. Virol. 148, 1713–1720. [DOI] [PubMed] [Google Scholar]
- Fukuta, S. , Kato, S. , Yoshida, K. , Mizukami, Y. , Ishida, A. , Ueda, J. , Kanbe, M. et al. (2003b) Detection of tomato yellow leaf curl virus by loop‐mediated isothermal amplification reaction. J. Virol. Methods, 112, 35–40. [DOI] [PubMed] [Google Scholar]
- Gao, X. , Chen, Y. , Luo, X. , Du, Z. , Hao, K. , An, M. , Xia, Z. et al. (2021) Recombinase polymerase amplification assay for simultaneous detection of maize chlorotic mottle virus and sugarcane mosaic virus in maize. ACS Omega, 6, 18008–18013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González, A. , Trujillo, G. , Vegas, A. and Garrido, M.J. (2002) Hospedantes de cepas del virus de la mancha anillada de la lechosa en Venezuela. Fitopatol. Venez. 15, 7–12. [Google Scholar]
- Gootenberg, J.S. , Abudayyeh, O.O. , Kellner, M.J. , Joung, J. , Collins, J.J. and Zhang, F. (2018) Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science, 360, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg, J.S. , Abudayyeh, O.O. , Lee, J.W. , Essletzbichler, P. , Dy, A.J. , Joung, J. , Verdine, V. et al. (2017) Nucleic acid detection with CRISPR‐Cas13a/C2c2. Science, 356, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajian, R. , Balderston, S. , Tran, T. , deBoer, T. , Etienne, J. , Sandhu, M. , Wauford, N.A. et al. (2019) Detection of unamplified target genes via CRISPR‐Cas9 immobilized on a graphene field‐effect transistor. Nat. Biomed. Eng. 3, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, R.W. and Zhang, S. (2016) Development of a rapid diagnostic assay for the detection of tomato chlorotic dwarf viroid based on isothermal reverse‐transcription‐recombinase polymerase amplification. J. Virol. Methods, 236, 62–67. [DOI] [PubMed] [Google Scholar]
- Hampton, R.O. , Ball, E.M. and DeBoer, S. (1990) Serological methods for detection and identification of viral and bacterial plant pathogens‐a laboratory manual. Indian Phytopathol. 44, 263–264. [Google Scholar]
- Harrington, L.B. , Burstein, D. , Chen, J.S. , Paez‐Espino, D. , Ma, E. , Witte, I.P. , Cofsky, J.C. et al. (2018) Programmed DNA destruction by miniature CRISPR‐Cas14 enzymes. Science, 362, 839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson, J.M. and French, R. (1993) The polymerase chain reaction and plant disease diagnosis. Ann. Rev. Phytopathol. 31, 81–109. [DOI] [PubMed] [Google Scholar]
- Hill, S.A. (1984) Methods in Plant Virology/Stephen A. Hill. Oxford [Oxfordshire]; Boston: Published on behalf of the British Society for Plant Pathology by Blackwell Scientific publications. [Google Scholar]
- Ivanov, A.V. , Shmyglya, I.V. , Zherdev, A.V. , Dzantiev, B.B. and Safenkova, I.V. (2020) The challenge for rapid detection of high‐structured circular RNA: assay of potato spindle tuber viroid based on recombinase polymerase amplification and lateral flow tests. Plants, 9, 1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauset‐Rubio, M. , Svobodová, M. , Mairal, T. , McNeil, C. , Keegan, N. , El‐Shahawi, M.S. , Bashammakh, A.S. et al. (2016) Aptamer lateral flow assays for ultrasensitive detection of β‐conglutin combining recombinase polymerase amplification and tailed primers. Anal. Chem. 88, 10701–10709. [DOI] [PubMed] [Google Scholar]
- Jiao, Y. , Jiang, J. , An, M. , Xia, Z. and Wu, Y. (2019a) Recombinase polymerase amplification assay for rapid detection of maize chlorotic mottle virus in maize. Arch. Virol. 164, 2581–2584. [DOI] [PubMed] [Google Scholar]
- Jiao, Y. , Jiang, J. , Wu, Y. and Xia, Z. (2019b) Rapid detection of cucumber green mottle mosaic virus in watermelon through a recombinase polymerase amplification assay. J. Viro. Methods, 270, 146–149. [DOI] [PubMed] [Google Scholar]
- Jiao, J. , Kong, K. , Han, J. , Song, S. , Bai, T. , Song, C. , Wang, M. et al. (2021) Field detection of multiple RNA viruses/viroids in apple using a CRISPR/Cas12a‐based visual assay. Plant Biotechnol. J. 19, 394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, Y. , Xu, C. , Li, J. , Gu, Y. , Xia, C. , Xie, Q. , Xie, Y. et al. (2020) Characterization and a RT‐RPA assay for rapid detection of Chilli Veinal mottle virus (ChiVMV) in tobacco. Virol. J. 17, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J.A. and Charpentier, E. (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juma, K.M. , Kojima, K. , Takita, T. , Natsuaki, T. and Yasukawa, K. (2021) Comparison of sensitivity and rapidness of PCR, recombinase polymerase amplification, and RNA‐specific amplification for detection of Rice yellow mottle virus. J. Biol. Macromol. 21, 27. [Google Scholar]
- Kalischuk, M.L. , Roberts, P.D. and Paret, M.L. (2020) A rapid fluorescence‐based real‐time isothermal assay for the detection of cucurbit yellow stunting disorder virus in squash and watermelon plants. Mol. Cell Probes, 53, 101613. [DOI] [PubMed] [Google Scholar]
- Kapoor, R. , Srivastava, N. , Kumar, S. , Saritha, R.K. , Sharma, S.K. , Jain, R.K. and Baranwal, V.K. (2017) Development of a recombinase polymerase amplification assay for the diagnosis of banana bunchy top virus in different banana cultivars. Arch. Virol. 162, 2791–2796. [DOI] [PubMed] [Google Scholar]
- Kappagantu, M. , Villamor, D.E.V. , Bullock, J.M. and Eastwell, K.C. (2017) A rapid isothermal assay for the detection of hop stunt viroid in hop plants (Humulus lupulus), and its application in disease surveys. J. Virol. Methods, 245, 81–85. [DOI] [PubMed] [Google Scholar]
- Kersting, S. , Rausch, V. , Bier, F.F. and von Nickisch‐Rosenegk, M. (2014) Rapid detection of plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malaria J. 13, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, N.‐K. , Lee, H.‐J. , Ryu, T.‐H. , Cho, I.‐S. , Ju, H.‐J. and Jeong, R.‐D. (2021a) Detection of apple scar skin viroid by reverse transcription recombinase polymerase amplification assay. Res. Plant Dis. 27, 79–83. [Google Scholar]
- Kim, N.K. , Kim, S.M. and Jeong, R.D. (2020) Reverse transcription recombinase polymerase amplification assay for rapid and sensitive detection of barley yellow dwarf virus in oat. Plant Pathol. J. 36, 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, N.Y. , Lee, H.J. and Jeong, R.D. (2019) A portable detection assay for apple stem pitting virus using reverse transcription‐recombinase polymerase amplification. J. Virol. Methods, 274, 113747. [DOI] [PubMed] [Google Scholar]
- Kim, N.Y. , Oh, J. , Lee, S.H. , Kim, H. , Moon, J.S. and Jeong, R.D. (2018) Rapid and specific detection of apple stem grooving virus by reverse transcription‐recombinase polymerase amplification. Plant Pathol. J. 34, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. , Ji, S. and Koh, H.R. (2021b) CRISPR as a diagnostic tool. Biomolecules, 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalskaya, N. and Hammond, R.W. (2022) Rapid diagnostic detection of tomato apical stunt viroid based on isothermal reverse transcription‐recombinase polymerase amplification. J. Virol. Methods, 300, 114353. [DOI] [PubMed] [Google Scholar]
- Kumar, R. , Kaundal, P. , Tiwari, R.K. , Siddappa, S. , Kumari, H. , Chandra Naga, K. , Sharma, S. et al. (2021a) Rapid and sensitive detection of potato virus X by one‐step reverse transcription‐recombinase polymerase amplification method in potato leaves and dormant tubers. Mol. Cell Probes, 58, 101743. [DOI] [PubMed] [Google Scholar]
- Kumar, R. , Pant, R.P. , Kapoor, S. , Khar, A. and Baranwal, V.K. (2021b) Development of a reverse transcription‐recombinase polymerase amplification (RT‐RPA) assay for the detection of onion yellow dwarf virus (OYDV) in onion cultivars. Indian Phytopathol. 74, 201–207. [Google Scholar]
- Kumar, P.V. , Sharma, S.K. , Rishi, N. , Ghosh, D.K. and Baranwal, V.K. (2018) An isothermal based recombinase polymerase amplification assay for rapid, sensitive and robust indexing of citrus yellow mosaic virus. Acta Virol. 62, 104–108. [DOI] [PubMed] [Google Scholar]
- Lee, H.J. , Cho, I.S. , Ju, H.J. and Jeong, R.D. (2021) Rapid and visual detection of tomato spotted wilt virus using recombinase polymerase amplification combined with lateral flow strips. Mol. Cell Probes, 57, 101727. [DOI] [PubMed] [Google Scholar]
- Lee, H.J. , Kim, H.J. , Lee, K. and Jeong, R.D. (2020) Rapid detection of peach latent mosaic viroid by reverse transcription recombinase polymerase amplification. Mol. Cell Probes, 53, 101627. [DOI] [PubMed] [Google Scholar]
- Li, S.Y. , Cheng, Q.X. , Wang, J.M. , Li, X.Y. , Zhang, Z.L. , Gao, S. , Cao, R.B. et al. (2018) CRISPR‐Cas12a‐assisted nucleic acid detection. Cell Discov. 4, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Dong, Z. , Zhan, B. and Li, S. (2021) Characterization of an isolate of citrus concave gum‐associated virus from apples in China and development of an RT‐RPA assay for the rapid detection of the virus. Plants, 10, 2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato, I.M. and O'Sullivan, C.K. (2018) Recombinase polymerase amplification: basics, applications and recent advances. Tras. Trends Anal. Chem. 98, 19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londoño, M.A. , Harmon, C.L. and Polston, J.E. (2016) Evaluation of recombinase polymerase amplification for detection of begomoviruses by plant diagnostic clinics. Virol. J. 13, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahas, A. , Aman, R. and Mahfouz, M. (2019) CRISPR‐Cas13d mediates robust RNA virus interference in plants. Genome Biol. 20, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahas, A. , Hassan, N. , Aman, R. , Marsic, T. , Wang, Q. , Ali, Z. and Mahfouz, M.M. (2021) LAMP‐coupled CRISPR‐Cas12a module for rapid and sensitive detection of plant DNA viruses. Viruses, 13, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahas, A. and Mahfouz, M. (2018) Engineering virus resistance via CRISPR‐Cas systems. Curr. Opin. Virol. 32, 1–8. [DOI] [PubMed] [Google Scholar]
- Mahas, A. , Neal Stewart, C., Jr. and Mahfouz, M.M. (2018) Harnessing CRISPR/Cas systems for programmable transcriptional and post‐transcriptional regulation. Biotechnol. Adv. 36, 295–310. [DOI] [PubMed] [Google Scholar]
- Mansour, S. , Ali, H. , Chase, C. and Cepica, A. (2015) Loop‐mediated isothermal amplification for diagnosis of 18 world Organization for Animal Health (OIE) notifiable viral diseases of ruminants, swine and poultry. Anim. Health Res. Rev. 16, 89–106. [DOI] [PubMed] [Google Scholar]
- Mekuria, T.A. , Zhang, S. and Eastwell, K.C. (2014) Rapid and sensitive detection of little cherry virus 2 using isothermal reverse transcription‐recombinase polymerase amplification. J. Virol. Methods, 205, 24–30. [DOI] [PubMed] [Google Scholar]
- Mohandas, A. and Bhat, A.I. (2020) Recombinase polymerase amplification assay for the detection of piper yellow mottle virus infecting black pepper. Virusdisease, 31, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, Y. , Nagamine, K. , Tomita, N. and Notomi, T. (2001) Detection of loop‐mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 289, 150–154. [DOI] [PubMed] [Google Scholar]
- Mowat, W.P. and Dawson, S. (1987) Detection and identification of plant viruses by ELISA using crude sap extracts and unfractionated antisera. J. Virol. Methods, 15, 233–247. [DOI] [PubMed] [Google Scholar]
- Nagamine, K. , Hase, T. and Notomi, T. (2002) Accelerated reaction by loop‐mediated isothermal amplification using loop primers. Mol. Cell. Probes, 16, 223–229. [DOI] [PubMed] [Google Scholar]
- Naveen, K.P. and Bhat, A.I. (2020) Development of reverse transcription loop‐mediated isothermal amplification (RT‐LAMP) and reverse transcription recombinase polymerase amplification (RT‐RPA) assays for the detection of two novel viruses infecting ginger. J. Virol. Methods, 282, 113884. [DOI] [PubMed] [Google Scholar]
- Nie, X. (2005) Reverse transcription loop‐mediated isothermal amplification of DNA for detection of potato virus Y. Plant Dis. 89, 605–610. [DOI] [PubMed] [Google Scholar]
- Nie, K. , Qi, S.‐X. , Zhang, Y. , Luo, L. , Xie, Y. , Yang, M.‐J. , Zhang, Y. et al. (2012) Evaluation of a direct reverse transcription loop‐mediated isothermal amplification method without RNA extraction for the detection of human enterovirus 71 subgenotype C4 in nasopharyngeal swab specimens. PLoS ONE, 7, e52486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi, T. , Okayama, H. , Masubuchi, H. , Yonekawa, T. , Watanabe, K. , Amino, N. and Hase, T. (2000) Loop‐mediated isothermal amplification of DNA. Nucleic Acids Res. 28, E63–E663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Q. , Ning, J. , Xu, Q. , Yang, T. , Wang, Y. , Zheng, T. , Zhuang, Q. et al. (2021) Development and application of a reverse transcription loop‐mediated isothermal amplification combined with lateral flow dipstick for rapid and visual detection of citrus leaf blotch virus in kiwifruit. Crop Prot. 143, 105555. [Google Scholar]
- Piepenburg, O. , Williams, C.H. , Stemple, D.L. and Armes, N.A. (2006) DNA detection using recombination proteins. PLoS Biol. 4, e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, M.L. , Bowler, F.R. , Martinez, A.J. , Greenwood, C.J. , Armes, N. and Piepenburg, O. (2018) New Fpg probe chemistry for direct detection of recombinase polymerase amplification on lateral flow strips. Anal. Biochem. 543, 108–115. [DOI] [PubMed] [Google Scholar]
- Ramachandran, V. , Weiland, J.J. and Bolton, M.D. (2021) CRISPR‐based isothermal next‐generation diagnostic method for virus detection in Sugarbeet. Front. Microbiol. 12, 679994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrman, B.A. , Leautaud, V. , Molyneux, E. and Richards‐Kortum, R.R. (2012) A lateral flow assay for quantitative detection of amplified HIV‐1 RNA. PLoS ONE, 7, e45611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas, J.A. , Miles, T.D. , Coffey, M.D. , Martin, F.N. and Chilvers, M.I. (2017) Development and application of qPCR and RPA genus‐and species‐specific detection of phytophthora sojae and P. sansomeana root rot pathogens of soybean. Plant Dis. 101, 1171–1181. [DOI] [PubMed] [Google Scholar]
- Roy, G. , Jain, R. , Bhat, A. and Varma, A. (1999) Comparative host range and serological studies of papaya ringspot potyvirus isolates. Indian Phytopathol. 52, 14–17. [Google Scholar]
- Silva, G. , Oyekanmi, J. , Nkere, C.K. , Bömer, M. , Kumar, P.L. and Seal, S.E. (2018) Rapid detection of potyviruses from crude plant extracts. Anal. Biochem. 546, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, N. , Kapoor, R. , Kumar, R. , Kumar, S. , Saritha, R. , Kumar, S. and Baranwal, V.K. (2019) Rapid diagnosis of cucumber mosaic virus in banana plants using a fluorescence‐based real‐time isothermal reverse transcription‐recombinase polymerase amplification assay. J. Virol. Methods, 270, 52–58. [DOI] [PubMed] [Google Scholar]
- Stephanidis, B. , Adichtchev, S. , Gouet, P. , McPherson, A. and Mermet, A. (2007) Elastic properties of viruses. Biophys. J. 93, 1354–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer, O.W. , Andrews, J.M. , Greetham, H.L. and Forrest, M.S. (2018) TwistAmp® liquid: a versatile amplification method to replace PCR. Nat. Methods, 15, 395.29855569 [Google Scholar]
- Tomlinson, J. , Barker, I. and Boonham, N. (2007) Faster, simpler, more‐specific methods for improved molecular detection of Phytophthora ramorum in the field. Appl. Environ. Microbiol. 73, 4040–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson, J. , Dickinson, M. and Boonham, N. (2010) Rapid detection of Phytophthora ramorum and P. kernoviae by two‐minute DNA extraction followed by isothermal amplification and amplicon detection by generic lateral flow device. Phytopathology, 100, 143–149. [DOI] [PubMed] [Google Scholar]
- Tomlinson, J. , Ostoja‐Starzewska, S. , Adams, I. , Miano, D. , Abidrabo, P. , Kinyua, Z. , Alicai, T. et al. (2013) Loop‐mediated isothermal amplification for rapid detection of the causal agents of cassava brown streak disease. J. Virol. Methods, 191, 148–154. [DOI] [PubMed] [Google Scholar]
- Walkey, D. (1985) Applied Plant Virology. London, UK: William Heinemann Ltd. [Google Scholar]
- Wang, Y. , Chen, R. , Nie, X. , Zhong, Z. , Li, C. , Li, K. , Huang, W. et al. (2020) Rapid and sensitive detection of potato virus Y by isothermal reverse transcription‐recombinase polymerase amplification assay in potato. Mol. Cell. Probes, 50, 101505. [DOI] [PubMed] [Google Scholar]
- Yoshimi, K. , Takeshita, K. , Yamayoshi, S. , Shibumura, S. , Yamauchi, Y. , Yamamoto, M. , Yotsuyanagi, H. et al. (2020) Rapid and accurate detection of novel coronavirus SARS‐CoV‐2 using CRISPR‐Cas3. Available at SSRN 3640844.
- Zetsche, B. , Gootenberg, J.S. , Abudayyeh, O.O. , Slaymaker, I.M. , Makarova, K.S. , Essletzbichler, P. , Volz, S.E. et al. (2015) Cpf1 is a single RNA‐guided endonuclease of a class 2 CRISPR‐Cas system. Cell, 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Ravelonandro, M. , Russell, P. , McOwen, N. , Briard, P. , Bohannon, S. and Vrient, A. (2014) Rapid diagnostic detection of plum pox virus in prunus plants by isothermal AmplifyRP® using reverse transcription‐recombinase polymerase amplification. J. Virol. Methods, 207, 114–120. [DOI] [PubMed] [Google Scholar]
- Zhao, C. , Sun, F. , Li, X. , Lan, Y. , Du, L. , Zhou, T. and Zhou, Y. (2019) Reverse transcription‐recombinase polymerase amplification combined with lateral flow strip for detection of rice black‐streaked dwarf virus in plants. J. Virol. Methods, 263, 96–100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Loop‐mediated isothermal amplification (LAMP) and reverse transcription (RT)‐LAMP‐based assays used for the detection of different plant viruses.