

Abstract
Inflammatory processes induced by brain injury are important for recovery; however, when uncontrolled, inflammation can be deleterious, likely explaining why most anti-inflammatory treatments have failed to improve neurological outcomes after brain injury in clinical trials. In the thalamus, chronic activation of glial cells, a proxy of inflammation, has been suggested as indicator of increased seizure risk and cognitive deficits that develop after cortical injury. Furthermore, lesions in the thalamus, more than other brain regions, have been reported in patients with viral infections associated with neurological deficits, such as SARS-CoV-2. However, the extent to which thalamic inflammation is a driver or byproduct of neurological deficits remains unknown. Here, we found that thalamic inflammation in mice was sufficient to phenocopy the cellular and circuit hyperexcitability, enhanced seizure risk, and disruptions in cortical rhythms that develop after cortical injury. In our model, downregulation of the GABA transporter GAT-3 in thalamic astrocytes mediated this neurological dysfunction. In addition, GAT-3 was decreased in regions of thalamic reactive astrocytes in mouse models of cortical injury. Enhancing GAT-3 in thalamic astrocytes prevented seizure risk, restored cortical states, and was protective against severe chemoconvulsant-induced seizures and mortality in a mouse model of traumatic brain injury, emphasizing the potential of therapeutically targeting this pathway. Altogether, our results identified a potential therapeutic target for reducing negative outcomes after brain injury.
One Sentence Summary:
Thalamic inflammation mediates enhanced seizure risk and altered cognition-related cortical states through astrocytic GAT-3 in mice.
INTRODUCTION
Neuroinflammation that occurs after brain lesions such as traumatic brain injury (TBI) and stroke is a double-edged sword, contributing to both recovery and pathogenesis (1–5). Accordingly, most anti-inflammatory treatments have failed to prevent neurological deficits after brain injuries in clinical trials (1, 2). Untangling the adaptive and maladaptive aspects of neuroinflammation is a crucial step in developing targeted treatments that could promote brain recovery without jeopardizing the adaptive aspects of the neuroinflammatory process.
Accumulating evidence suggests that the thalamus is particularly vulnerable to secondary damage, even when the initial injury occurred at a remote location. In both humans and rodent models, chronic thalamic inflammation is observed after TBI (6–11) and cortical stroke (12–14). The development of secondary and persistent thalamic inflammation—particularly gliosis—has been suggested to be an indicator of neurological dysfunction after injury resulting in cognitive impairment (8) and seizure risk (12). In addition, thalamic lesions and/or thrombotic strokes have been reported among patients with viral infections caused by West Nile Virus (15, 16), and those with altered mental status following SARS coronavirus-2 (SARS-CoV-2) infection (17, 18). Mice intranasally infected with SARS-CoV-2 have high viral antigen presence in the thalamus compared to other brain regions (19). Since the thalamus plays a central role in cognition, sleep and seizures in humans and rodents (20–25), it is well-positioned to be involved in diverse neurological deficits observed across insults, such as cognitive impairments, sleep disruption and epileptogenesis (6, 26, 27). Thalamic astrogliosis—in which astrocytes become reactive and undergo changes that affect neuronal function (5, 28–33)—has been observed in both humans and rodents after cortical injuries (9–13).
Here, we sought to determine the extent to which thalamic astrogliosis is a driver or bystander of neurological dysfunction, and whether this process could be therapeutically targeted. To this end, we developed a mouse model of thalamic astrogliosis and investigated its effects on local neurons, micro-circuits, and on cortical states associated with cognition.
RESULTS
Thalamic astrogliosis is observed in post-mortem human tissue with a history of cortical injury and in mouse models of cortical injury
Human imaging studies in patients with mild TBI have suggested that the chronic thalamic gliosis that develops secondarily after cortical lesions is an indicator of cognitive dysfunction (6). To investigate the role of astrogliosis in cortical stroke and TBI, we first performed immunostaining for the astrocytic marker glial fibrillary acidic protein (GFAP)—a molecular marker of many types of reactive astrocytes (28)—in post-mortem thalamic and peri-lesional cortical tissue obtained from human brains with a history of ischemic stroke or TBI; a subset of subjects also had a history of epilepsy (2 of 7 subjects had epilepsy prior to injury, and 3 of 7 subjects developed epilepsy after injury) (Fig. 1, and tables S1 and S2). The thalamus and peri-lesional cortex of subjects with stroke or TBI exhibited discrete GFAP-positive cells with astrocyte-like morphology, compared to the dim, diffuse GFAP labeling in control subjects (Fig. 1A). Putative astrocyte morphology ranged from resting (Fig. 1A; cortex, control, inset) to reactive, featuring somatic hypertrophy and thick processes (Fig. 1A; thalamus and cortex, stroke, inset). These changes were evident both acutely and years after stroke or TBI, and were supported by a semi-quantitative analysis of GFAP immunoreactivity (table S2).
Fig. 1. Thalamic astrogliosis is a consistent feature of cortical lesions, recapitulated by viral-mediated activation.
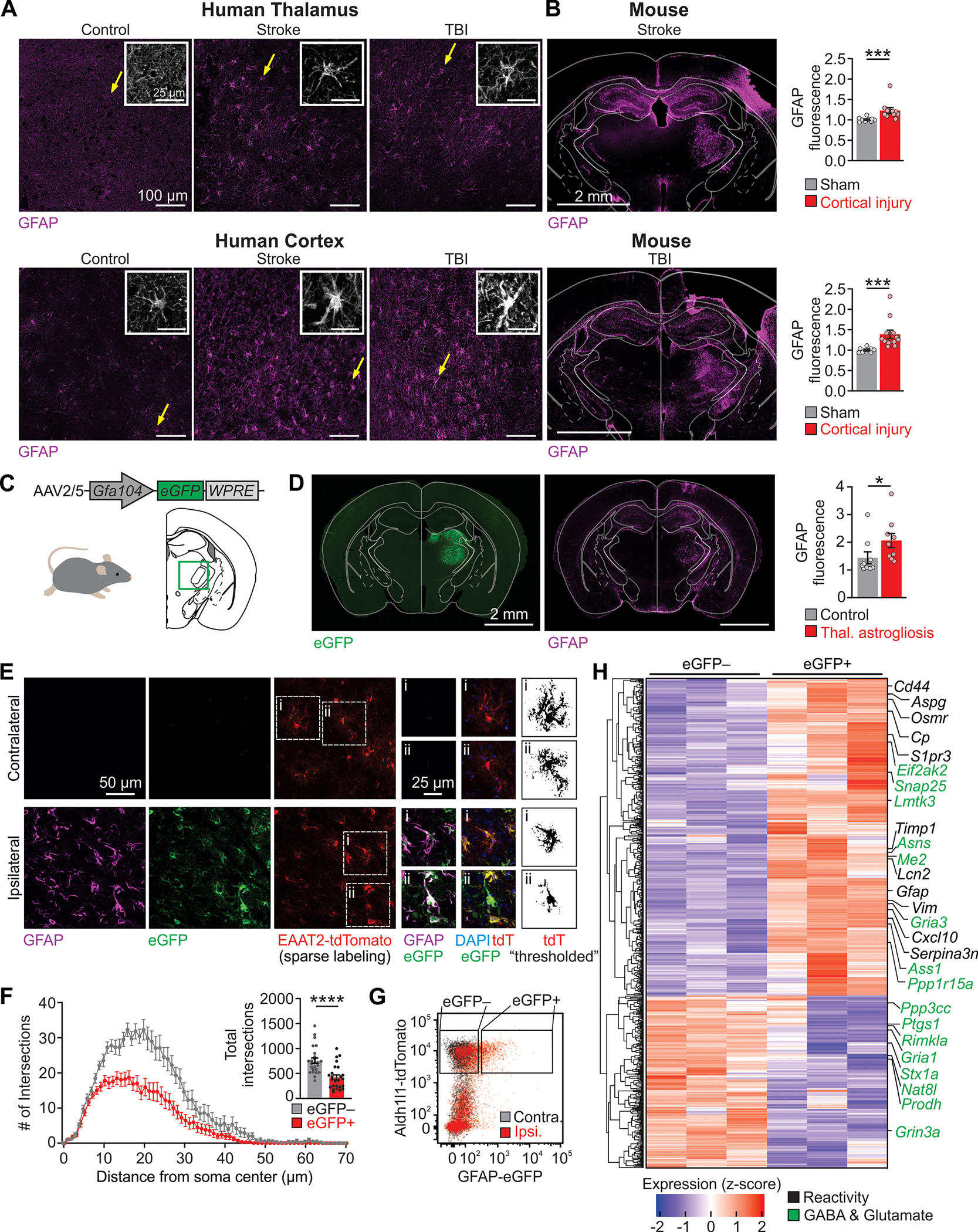
(A) GFAP immunofluorescence in post-mortem thalamus and peri-lesional cortex from human subjects with history of ischemic stroke or TBI, and age-matched controls, representative of 3 control subjects, 3 subjects with stroke, and 4 subjects with TBI. Insets: magnification of GFAP+ cells with astrocyte-like morphology (putative astrocytes, yellow arrow). (B) GFAP immunofluorescence in mouse models of photothrombotic stroke and controlled cortical impact. Right: thalamic GFAP fluorescence ratios between ipsilateral and contralateral regions. Stroke: Mann-Whitney U test, ***P=0.0002. Sham: n=9 mice; Stroke: n=10 mice. TBI: Mann-Whitney U-test, ***P=0.0002. Sham: n=7 mice; TBI: n=12 mice. (C) Viral induction of unilateral thalamic astrogliosis (brain schematic adapted from (76)). (D) Immunofluorescence three weeks after astrogliosis induction. GFAP immunofluorescence quantified as in (B). Mann-Whitney U test, *P=0.019. n=9 mice per group. Quantification in other brain regions shown in fig. S1. (E) Representative images of GFAP, eGFP, tdTomato, and DAPI fluorescence in thalamic astrocytes contralateral and ipsilateral to viral transduction of astrocytes in EAAT2-tdTomato reporter mice. (F) Sholl analysis of thalamic astrocytes. Inset: Total number of intersections. Mann-Whitney U test, ****P<0.0001. n=25 astrocytes per condition. (G) Flow plot of thalamic astrocytes, ipsilateral and contralateral to site of viral transduction in Aldh1l1-tdTomato reporter mice. Gates indicate sorting strategy detailed in fig. S2. (H) Differentially expressed genes in ipsilateral eGFP+ versus contralateral eGFP− thalamic astrocytes (Padjusted<0.05), highlighting genes related to astrocyte reactivity (31) and astrocyte modulation of glutamate and GABA (54, 77) on the right. Heat map including ipsilateral eGFP− in fig. S3.
Similarly, in two mouse models of injury—unilateral photothrombotic stroke or controlled cortical impact in the somatosensory cortex (a model of TBI)—GFAP was elevated in the thalamus, without gross hippocampal damage (Fig. 1B), in agreement with previous findings (10–13, 34).
Viral transduction of thalamic astrocytes induces persistent reactive astrocytes
To generate reactive astrocytes in the thalamus, we injected an AAV construct expressing enhanced green fluorescent protein (eGFP) from a truncated portion of the GFAP promoter (AAV2/5-Gfa104-eGFP) (35) unilaterally in the ventrobasal (VB) somatosensory thalamus (Fig. 1C and fig. S1, A and B). This paradigm was originally developed to selectively transduce hippocampal astrocytes and render them reactive, as determined by hypertrophy and increased GFAP expression (35), although the molecular mechanisms of virus-induced reactive astrogliosis remain unknown.
To characterize the effects of the viral transduction on thalamic astrocytes, we assessed immunohistochemical, morphological and transcriptomic features of astrocytes three weeks following viral transduction. GFAP was increased in the thalamus ipsilateral to injection (Fig. 1D), consistent with the work in the hippocampus (35). The location of thalamic astrogliosis in our model was similar to that observed in rodent models of cortical injuries (Fig. 1B). The increase in GFAP in the viral transduction model persisted for at least nine months specifically in the ipsilateral thalamus, but not in other brain regions (fig. S1C). Thus, the viral transduction model recapitulated two key features of astrogliosis following cortical injury—the specific location within the dorsal thalamus, and the chronic presence of reactive astrocytes in the thalamus (10).
To evaluate astrocyte morphology, we used an EAAT2-tdTomato transgenic reporter mouse line (36), which enabled sparse labeling and tracing of individual thalamic astrocytes (Fig. 1E). Using Sholl analysis to assess arborization of astrocytic processes (37), we observed somatic hypertrophy and a reduction in process branching in eGFP-positive astrocytes ipsilateral to viral injection (eGFP-positive/tdT-positive) relative to eGFP-negative/tdT-positive astrocytes (Fig. 1F). Thus, viral transduction of thalamic astrocytes recapitulates morphological features of reactive astrogliosis (32, 35, 38).
We next performed transcriptomic profiling of virally-transduced thalamic astrocytes acutely isolated by fluorescence-activated cell sorting (FACS; Fig. 1, G and H, and fig. S2). Hierarchical clustering of FACS-isolated samples revealed that eGFP-positive astrocytes, which represented ~18% of astrocytes isolated from the thalamus, were distinct from their eGFP-negative counterparts (fig. S3, A to C). We found robust upregulation of canonical reactivity genes Gfap, Vim, CD44, and Serpina3n (31, 33) in eGFP-positive astrocytes (Fig. 1H). Comparing this transcriptomic profile with a published analysis of reactive astrocytes revealed upregulation of “pan-reactive” astrocyte genes (31), although there was no bias towards the subsets of reactive astrocyte genes that were selectively induced by lipopolysaccharide injection or ischemic stroke (middle cerebral artery occlusion model) in that study (31) (fig. S3D). Finally, we found alterations in multiple genes related to astrocytic modulation of glutamate and GABA (Fig. 1H), pinpointing molecular features of reactive astrocytes which might impact neural activity. Given that viral transduction of thalamic astrocytes reproducibly induces immunohistochemical, morphological and transcriptomic features of astrocyte reactivity observed across models (28, 32, 35, 38), hereon we refer to virally transduced astrocytes as reactive astrocytes.
Viral-induced thalamic astrogliosis did not lead to abnormalities in standard assays of spontaneous locomotion, anxiety, olfaction, nociception, context-dependent learning, and grooming three weeks post-injection (fig. S4).
Reactive astrocytes enhance intra-thalamic microcircuit excitability
The effects of reactive astrocytes on local microcircuit excitability were assessed in acute brain slices that preserve the connectivity between VB thalamus and the reticular thalamic nucleus (nRT). We focused on the VB-nRT microcircuit because: 1) VB thalamus—the center of viral transduction—showed the strongest and most persistent astrogliosis overtime (fig. S1); 2) the VB-nRT connections can be maintained in slices and reliably generate circuit oscillations, unlike connections between nRT and other thalamic nuclei; 3) Multi-unit activity in VB thalamus is a proxy for the strength of the oscillatory activity in the VB-nRT microcircuit (39–43).
Electrical stimulation was delivered in the internal capsule, which contains corticothalamic and thalamocortical axons, and the resulting multi-unit activity was recorded three weeks following induction of reactive astrocytes (Fig. 2). Stimulation of the internal capsule produced robust multi-unit activity, characterized by an immediate “direct response,” due to direct activation of corticothalamic axons and/or retrograde activation of thalamocortical neurons, followed by a “delayed response,” characterized by repeated action potential bursts (Fig. 2, A to C), characteristic of the reciprocal VB-nRT oscillatory circuit activity (39–43). Thalamic slices with astrogliosis exhibited an augmented delayed response characterized by more bursts and longer circuit oscillations (Fig. 2D). Beyond the site of maximal responsiveness in each slice, we also examined the spatial and temporal synchrony of neural activity across all recording sites. Evoked oscillatory activity in thalamic slices with astrogliosis had higher oscillation indices (39, 44) (Fig. 2, B and E), although the spatial extent of the direct response was not altered, suggesting that astrogliosis enhanced the rhythmogenic properties of the intra-thalamic microcircuit across a larger region.
Fig. 2. Viral-mediated thalamic astrogliosis enhances intra-thalamic circuit rhythmogenesis.

(A) Representative intra-thalamic multi-unit activity evoked by stimulation of the internal capsule (lightning bolt) in thalamic slices, as schematized in (C). Only nine out of 16 channels are shown for clarity. Shaded boxes highlight bursts (clusters of ≥3 spikes), horizontal arrow indicates a sustained circuit oscillation (clusters of ≥2 bursts). (B) Peri-stimulus time histograms of recordings shown in (A). Color represents multi-unit activity normalized to maximal firing in each slice. White traces represent the spatial summation of firing activity across all recording sites and all trials within each slice during the delayed response (excluding the first 150 ms of direct response). (C) Schematic of multi-unit activity recordings in thalamic slices following electrical stimulation of the internal capsule (yellow). Probe schematic adapted from NeuroNexus, brain schematic adapted from (76). (D) Quantification of bursts and circuit oscillations, as shown in (A). Each dot represents the site with maximal multi-unit activity in each slice from n=13 Control and n=16 Thalamic Astrogliosis mice (1 slice/mouse; 20 evoked responses averaged per slice). Mann-Whitney U test; **P=0.007 (bursts) and **P=0.003 (oscillations). (E) Left: auto-correlation analyses of multi-unit activity in the delayed response from (B). Multi-unit activity is summed across all recording sites and all trials within a slice. Right: oscillation index calculated for the first peak of the auto-correlated signal. Mann-Whitney U test, *P<0.05. n=13 Control, n=16 Thalamic Astrogliosis mice.
Reactive astrocytes facilitate low-threshold-calcium spikes in thalamocortical neurons by increasing extrasynaptic tonic inhibition
To determine how reactive astrocytes cause hyperexcitability in the VB-nRT microcircuit, we investigated cellular excitability and synaptic function in excitatory and inhibitory neurons in thalamic brain slices (12, 45, 46) (tables S3 to S8).
Thalamocortical neurons in VB thalamus from mice with thalamic astrogliosis exhibited hyperpolarized action potential (AP) threshold (Fig. 3, A and B), and no difference in the resting membrane potential (Fig. 3C). The difference in AP threshold was abolished when membrane potential was held at −75 mV in both groups (table S4). Furthermore, the post-inhibitory rebound low-threshold calcium spike (LTS), mediated by T-type Ca2+ currents (47), occurred at more hyperpolarized membrane potentials (Fig. 3D) and was accompanied by enhanced AP firing (fig. S5, A to C). T-type Ca2+ current properties were similar in thalamocortical neurons from both groups (table S5), as was the hyperpolarization-activated depolarizing sag potential (fig. S5, D to G).
Fig. 3. Viral-mediated thalamic astrogliosis boosts cellular excitability via extrasynaptic GABAAR-mediated tonic inhibition.

(A) Representative whole-cell recordings showing response to hyperpolarizing and depolarizing intracellular current injections. (B) Left: Overlay of action potentials (APs) from two representative neurons. Right: AP generation threshold in the presence or absence of Picrotoxin (50 μM). No picrotoxin condition: Mann-Whitney U test, *P=0.02; Control: n=17 cells (from 8 slices, 4 mice); Thalamic Astrogliosis: n=14 cells (from 7 slices, 5 mice). Picrotoxin condition: P=0.28; Control: n=9 cells (from 5 slices, 3 mice). Thalamic Astrogliosis: n=13 cells (from 9 slices, 5 mice). ns: not significant. (C) Resting membrane potentials of thalamocortical neurons. No picrotoxin condition: Mann-Whitney U test, P=0.053. Picrotoxin condition: Mann-Whitney U-test, P=0.52. See (A) for sample size. (D) Left: Overlay of the hyperpolarization-induced rebound burst from two representative neurons. Center: Threshold for the T-type Ca2+-mediated low threshold spike (LTS), plotted as a function of the pre-pulse hyperpolarized membrane potential following current injection. Black and burgundy crosses indicate mean±SEM for membrane potential and LTS threshold. Right: LTS threshold. No picrotoxin condition: Mann-Whitney U test, ***P=0.0005. Control: n=12 cells (from 7 slices, 4 mice). Thalamic Astrogliosis: n=12 cells (from 7 slices, 5 mice). Picrotoxin condition: P=0.36. Control: n=13 cells (from 9 slices, 4 mice). Thalamic Astrogliosis: n=11 cells (7 slices, 5 mice). (E) Representative whole-cell voltage-clamp recordings. Gabazine (GBZ; 50 μM) was bath-applied to isolate the tonic current, measured by the shift in baseline holding current (black arrow). Mann-Whitney U test, ****P<0.0001. Control: n=20 cells (from 20 slices, 10 mice); Thalamic Astrogliosis: n=22 cells (from 22 slices, 11 mice). (F) Representative phasic GABAA currents. Mann-Whitney U test, P=0.88. Control: n=36 cells (from 36 slices, 14 mice); Thalamic Astrogliosis: n=40 cells (from 40 slices, 16 mice).
In mice with thalamic astrogliosis, thalamocortical neurons showed a three-fold increase in extrasynaptic GABAAR-mediated current (hereon referred to as tonic GABA current or ITONIC) (Fig. 3E), but no change in the frequency or biophysical properties of spontaneous inhibitory and excitatory synaptic currents (Fig. 3F and table S6). GABAAR antagonist picrotoxin normalized the AP threshold, LTS threshold, and rebound burst AP firing (Fig. 3, B to D, and fig. S5C).
In contrast with thalamocortical neurons, nRT neurons showed no change in intrinsic membrane excitability (table S7) or in spontaneous synaptic currents (table S8).
Thalamic reactive astrocytes enhance seizure risk in mice via tonic inhibition in thalamocortical neurons
Given that enhanced ITONIC in the rodent thalamus has been implicated in hyperexcitability and seizures in genetic and pharmacological models of epilepsies (46, 48), we asked whether thalamic astrogliosis—which increased ITONIC in adult wildtype mice (Fig. 3E)—led to heightened seizure susceptibility in mice.
Three weeks following induction of thalamic astrogliosis, a low dose of the pro-convulsive agent Pentylenetetrazol (PTZ; 5 mg/kg, intra-peritoneal, i.p.) was sufficient to induce epileptiform discharges associated with behavioral freezing in mice with thalamic astrogliosis but not in control mice (Fig. 4, A to D). Epileptiform discharges were prominent in the primary somatosensory cortex S1, a major recipient of inputs from VB thalamus (49), and generalized to other cortical areas (fig. S6A). These epileptiform discharges were similar to those reported in rodent models of epilepsies in terms of peak frequency and generalization (43, 46, 50). Increased seizure susceptibility persisted for at least seven months (fig. S6B), consistent with the persistent thalamic astrogliosis (fig. S1). Mice in which astrocytes were transduced with a low titer of the same viral construct (previously shown to not induce astrocyte reactivity (35)) did not exhibit enhanced seizure risk (fig. S6C). Mice with thalamic astrogliosis also had a heightened response to Kainic Acid (10 mg/kg i.p.) (Fig. 4, E to G, and fig. S6D). Altogether, these results show that unilateral induction of reactive astrocytes in the thalamus enhances susceptibility to both non-convulsive and generalized tonic-clonic seizures, as assessed by pro-convulsive agents that act through different mechanisms (GABA and glutamate).
Fig. 4. Conditional deletion of the extrasynaptic GABAAR δ-subunit (δ-GABAAR-cKO) in thalamocortical neurons prevents thalamic astrogliosis-induced seizure risk.
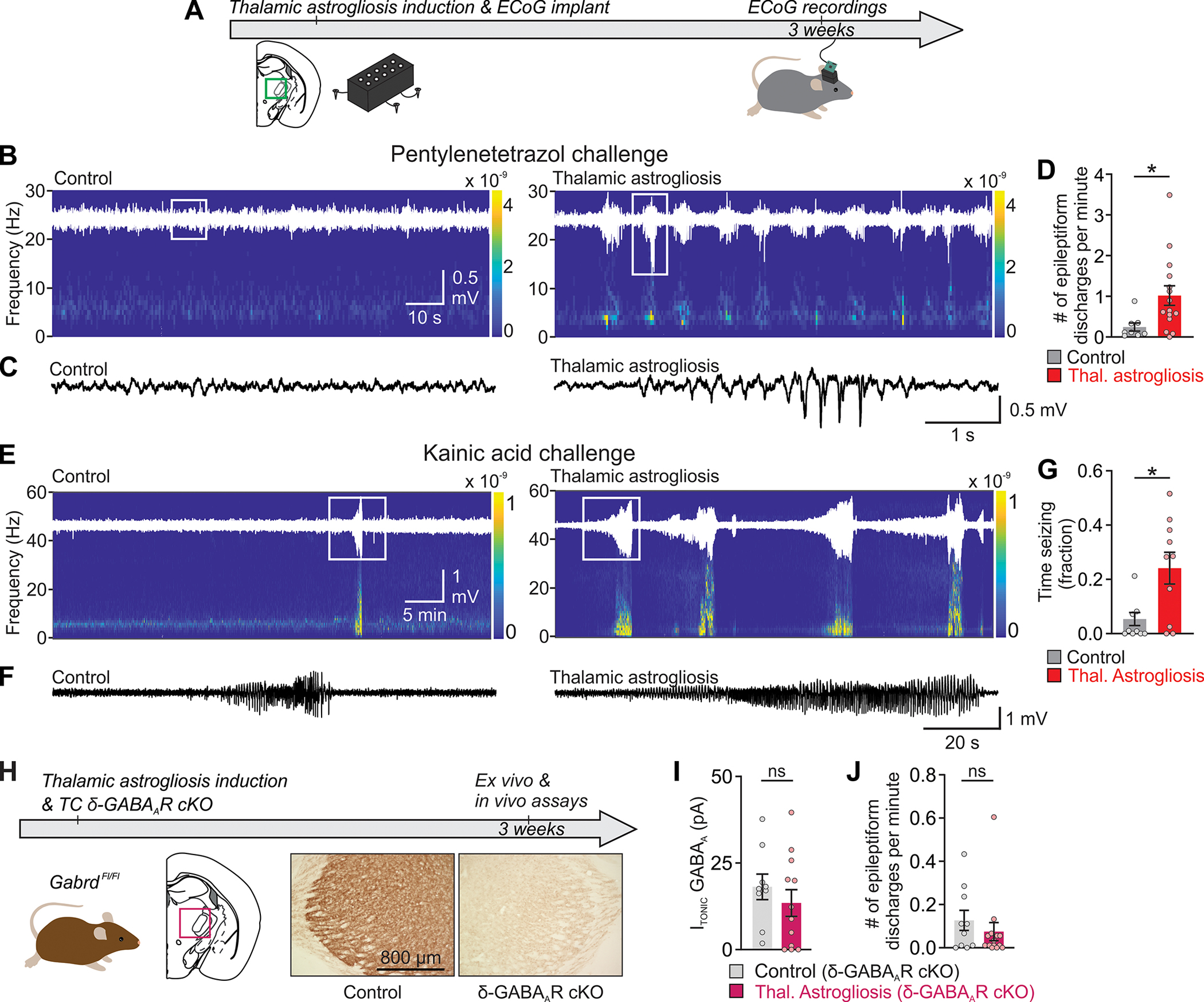
(A) Experimental timeline of ECoG recordings. (B, E) Representative ECoG recordings from ipsilateral primary somatosensory cortex (S1), following i.p. injection of pentylenetetrazol (PTZ, 5 mg/kg; B) or of kainic acid (10 mg/kg; E), with raw ECoG traces (white) overlaid onto corresponding spectrograms. (C, F) Enlargement of ECoG traces in boxed areas in (B) and (E). (D) Frequency of epileptiform discharges in ipsilateral S1 ECoG in the PTZ challenge experiment. Mann Whitney U test: *P=0.013. Control, n=8 mice; Thalamic Astrogliosis, n=15 mice. (G) Time spent seizing in response to Kainic Acid-challenge. Mann-Whitney U test, *P=0.025. Control: n=10 mice; Thalamic Astrogliosis: n=10 mice. (H) Unilateral, conditional deletion of the extrasynaptic GABAAR δ-subunit (δ-GABAAR-cKO) in thalamocortical neurons via viral Cre-mediated (AAV2/5-CaMKIIα-mCherry-Cre) recombination in GabrdFl/Fl mice. Timeline (top) and DAB labeling in thalamic sections (bottom). (I) Quantification of ITONIC in thalamocortical neurons in thalamic ex-vivo slices from GabrdFl/Fl mice. Mann-Whitney U test, P=0.42. ns: not significant. Control δ-GABAAR-cKO (intra-thalamic transduction of thalamocortical neurons with AAV2/5-CaMKIIα-mCherry-Cre in mice without astrogliosis): n=9 cells (from 9 slices, 5 mice); Thalamic Astrogliosis δ-GABAAR-cKO (intra-thalamic transduction of thalamocortical neurons with AAV2/5-CaMKIIα-mCherry-Cre in mice with astrogliosis): n=12 cells (from 12 slices, 5 mice). (J) Epileptiform discharges from ipsilateral S1 in response to PTZ challenge (5mg/kg) in GabrdFl/Fl mice with and without thalamic astrogliosis. Mann-Whitney U Test, P=0.37. Control δ-GABAAR-cKO: n=10 mice; Thalamic astrogliosis δ-cKO: n=14 mice.
In the thalamus, δ-subunit-containing extrasynaptic GABAA receptors generate ITONIC (45, 46, 48). Thus, to investigate whether preventing the increase in ITONIC could prevent astrogliosis-induced seizure risk, we conditionally deleted the GABAA receptor δ-subunit (δ-GABAAR) unilaterally in thalamocortical neurons of adult GabrdFl/Fl mice (51) (Fig. 4H). Thalamic astrogliosis did not increase ITONIC in thalamocortical neurons in these mice (Fig. 4I), and did not increase seizure risk (Fig. 4J), indicating that astrogliosis-induced increase in ITONIC in thalamocortical neurons is necessary for the development of hyperexcitability.
Enhancing GAT-3 in thalamic astrocytes prevents astrogliosis-induced seizure risk and cortical rhythm perturbations in mice
Astrocytes take up GABA from the extracellular space, mainly via the GABA transporters GAT-1 and GAT-3 (46, 52, 53). Given the alterations in multiple genes linked to GAT regulation (54) in our transcriptomic study (Fig. 1H), we investigated whether the expression of these transporters was altered in reactive astrocytes. Three weeks following viral induction of astrogliosis, GAT-3 expression was reduced in the ipsilateral thalamus, and Slc6a11 transcripts (encoding GAT-3) were reduced in reactive astrocytes (Fig. 5, A to C). The reduction of GAT-3 mRNA was specific to eGFP-transfected reactive astrocytes (Fig. 5C, and fig. S7A and B), and there was no evidence of compensatory changes in GAT-1 mRNA (fig. S7C and D).
Fig. 5. Enhancing GAT-3 in thalamic astrocytes prevents astrogliosis-induced tonic inhibition in neurons and seizure risk.

(A) GAT-3 immunofluorescence in mouse coronal brain sections (left) and confocal images of GAT-3 and GFAP co-localization in VB thalamus (right), three weeks after unilateral thalamic astrogliosis induction. (B) GAT-3 immunofluorescence ratio between regions ipsilateral-and contralateral to thalamic astrogliosis, three weeks following induction. Mann-Whitney U test, *P=0.028; n=8 mice per group. (C) Quantitative PCR analysis of Slc6a11 expression (encoding GAT-3), normalized to Aldh1l1 expression in astrocytes FACS-isolated from contralateral and ipsilateral VB thalamus of Aldh1l1-tdTomato mice, three weeks following thalamic astrogliosis induction. Sorting strategy detailed in fig. S2. One-way ANOVA, *P=0.01; Shapiro-Wilk normality test, P>0.05, Tukey’s post-hoc tests, *P=0.012 and *P=0.024; n=3 mice. (D) Increased GAT-3 expression in thalamic astrocytes was achieved by transducing thalamic astrocytes with AAV2/5-GfaABC1D-GAT3-mCherry (54) in adult mice with and without thalamic astrogliosis. Representative confocal images show GAT-3 expression in eGFP+ astrocytes in mice with thalamic astrogliosis, with and without enhanced GAT-3 enhancement. (E) Quantification of ITONIC in thalamocortical neurons from thalamic ex vivo slices, three weeks following GAT-3 enhancement in mice with and without thalamic astrogliosis. Mann-Whitney U test, P=0.60. Control + GAT-3: n=10 cells (from 10 slices, 7 mice); Thalamic Astrogliosis + GAT-3: n=13 cells (from 13 slices, 9 mice). (F) Epileptiform discharges in response to PTZ (15 mg/kg). Mann-Whitney U Test, P=0.42. n=9 mice per group.
Enhancing GAT-3 expression unilaterally in thalamic astrocytes by transducing mice with an astrocyte-specific viral construct (AAV2/5-GfaABC1D-GAT3) (54) at the same time as the induction of thalamic astrogliosis (Fig. 5D) counteracted the enhanced ITONIC in thalamocortical neurons (Fig. 5E) and protected mice from increased seizure risk (Fig. 5F).
We next investigated whether induction of reactive astrocytes in the thalamus could initiate long-term changes in cortical rhythms. Seven weeks post-induction of thalamic astrogliosis, spectral analysis revealed a unilateral reduction in the sigma (12–15 Hz) and gamma (30–75 Hz) power, but not in total power (Fig. 6, A to C and table S9). The reduction of sigma power was specific to the 12-hr light cycle, whereas the reduction of gamma power was specific to the 12-hr dark cycle (table S9).
Fig. 6. Enhancing GAT-3 in thalamic astrocytes prevents astrogliosis-induced reduction in cortical sigma and gamma frequency power.

(A) Schematic of experimental timeline. Chronic wireless ECoG from S1 was obtained bilaterally in freely-behaving mice in their home cage for continuous one-week periods, up to seven weeks following induction of thalamic astrogliosis. (B) Representative plots of delta (1–4 Hz), sigma (12–15 Hz), gamma (30–75 Hz), and total band power (1–75 Hz), across weeks 1, 3, 5, and 7, obtained from a mouse with thalamic astrogliosis. Spectral analysis was performed on S1 ECoG in 1-hour bins. Horizontal lines indicate the average power of the frequency band during week one. (C) Average ECoG power for each mouse, seven weeks after thalamic astrogliosis induction, during the light (top) and dark cycle (bottom). Control: n=6 mice; Thalamic Astrogliosis: n=6 mice. (D) Average ECoG power for each mouse, seven weeks after thalamic astrogliosis induction and astrocytic GAT-3 enhancement. Control + GAT-3: n=3 mice; Thalamic Astrogliosis + GAT-3: n=4 mice. See table S9 for comparison of all frequency bands. Wilcoxon matched-pairs signed rank tests, *P<0.05.
Enhancement of GAT-3 expression in thalamic astrocytes protected from the loss of sigma and gamma power in the ipsilateral cortex (Fig. 6D and table S9).
Enhancing GAT-3 in thalamic astrocytes reduces chemoconvulsant-induced seizure severity and mortality in a mouse model of cortical injury
We investigated whether similar reductions of thalamic GAT-3 transcripts occurred, and whether enhancing thalamic GAT-3 would confer resilience, in a mouse model of cortical injury. First, we performed 10x Visium Spatial Transcriptomics on tissue obtained from mice after TBI, stroke, or a sham procedure (Fig. 7 and fig. S8). Portions of the thalamus exhibited a higher number of Gfap transcripts and other markers of astrogliosis six weeks following stroke or TBI (Fig. 7, A to C, and fig. S9). Elevated Gfap expression was associated with reduced Slc6a11 expression in the thalamus (Fig. 7, D and E), whereas no correlation was found in the cortex (fig. S9).
Fig. 7. Cortical injury-induced thalamic Gfap is negatively correlated with thalamic Slc6a11 expression.
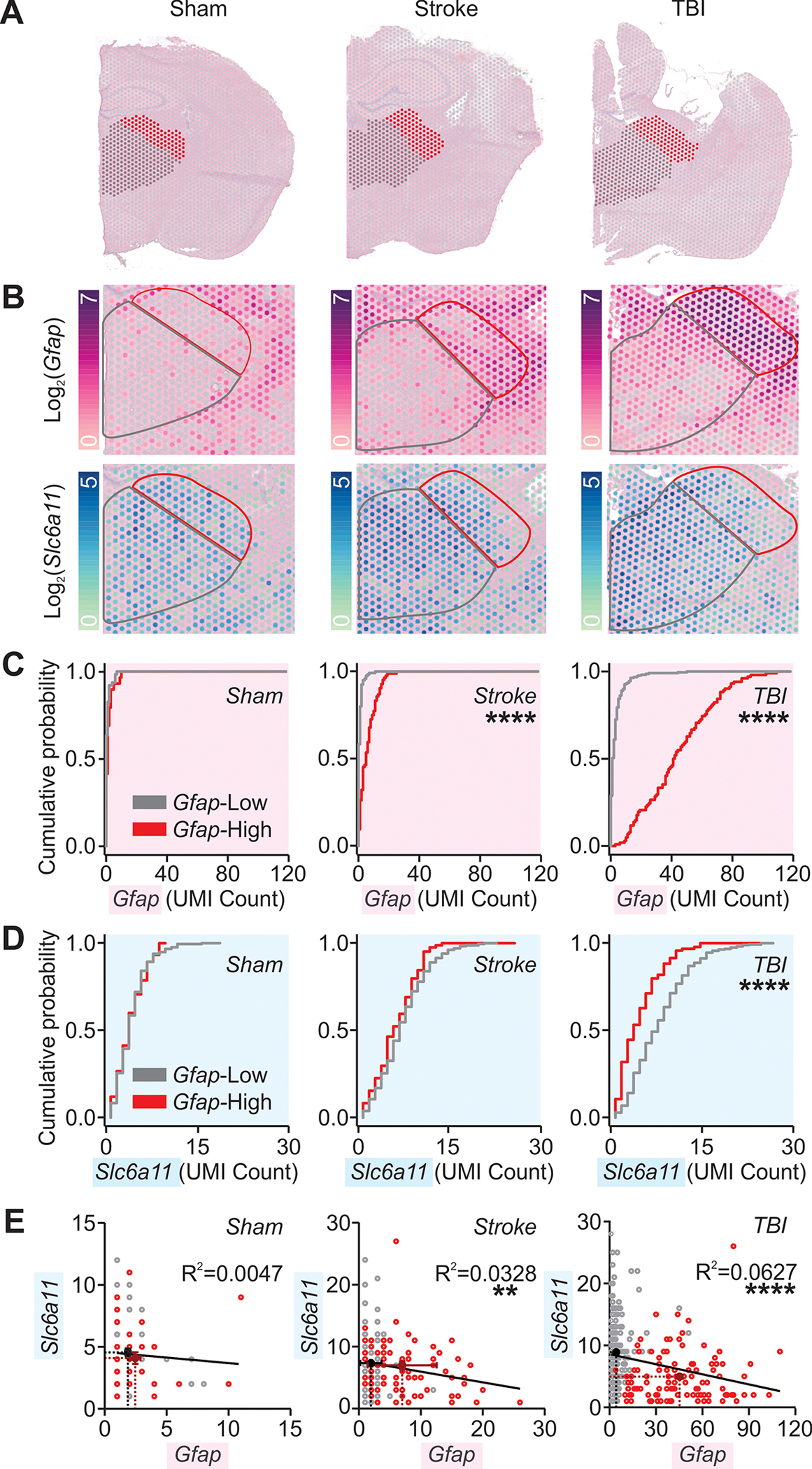
(A) Hemi-brain sections from mice after sham, stroke, and TBI procedures six weeks after surgery overlaid with thalamic regions of interest (grey and red) analyzed with 10x Visium spatial transcriptomics and magnified in (B). (B) Thalamic expression of Gfap and Slc6a11 transcripts. Color map indicates log2 of the detected counts of the gene’s unique molecular identifier (UMI). Red and black outlines correspond to regions of interest shown in (A) and to regions with high and low Gfap expression in cortical injury, as detailed in (C). (C, D) Cumulative probability distribution of Gfap (C) or Slc6a11 (D) expression per spot in regions indicated in (A-B). Kolmogorov-Smirnov test, ****P<0.0001. All spots containing non-zero UMI counts were included. Number of spots from low- and high-Gfap regions, respectively, in (C): sham (n=63 & 29); stroke (n=120 & 84); TBI (n=218 & 102); in (D): sham (n=216 & 76); stroke (n=237 & 85); TBI (n=277 & 95). Adjusted α=0.025 for multiple comparisons. (E) Relationship between Gfap and Slc6a11 expression. Circles represent UMI counts per spot in low- (grey) and high-Gfap (red) areas; black and burgundy crosses, and corresponding dotted lines, mark mean±SEM for low- and high-Gfap areas respectively. Black lines plot the best-fit slopes and intercepts from a simple linear regression. **P=0.0095, ****P<0.0001. Number of spots: sham (n=93); stroke (n=204); TBI (n=322). Similar analysis for cortical regions distal and proximal to injury is described in fig. S9.
Enhancing GAT-3 in thalamic astrocytes ipsilateral to cortical TBI (fig. S10A) increased survival following PTZ challenge; 5/12 TBI mice died following the PTZ challenge, whereas all 10 TBI mice that received astrocytic GAT-3 treatment survived (fig. S10B). Furthermore, GAT-3 treatment resulted in reduced seizure severity following TBI (fig. S10C, D).
We next investigated GAT-3 expression in human brain samples. Using a recently described method to deconvolute cell-type specific and brain-region specific transcriptional signatures from thousands of neurotypical adult human brain samples (55), we found that SLC6A11 (encoding GAT-3) was highly expressed in the thalamus-containing diencephalon relative to all genes detected in this region (Fig. 8A). Comparing the fidelity of SLC6A11 expression to transcriptional signatures of the major brain cell types showed that SLC6A11 expression was highly correlated to an astrocyte transcriptional signature and not to neurons, oligodendrocytes, or microglia (Fig. 8B and fig. S11), consistent with ultrastructural studies suggesting that GAT-3 is predominantly expressed in astrocytes in the rodent thalamus (56, 57). We next performed immunostaining for GAT-3 in post-mortem thalamic and peri-lesional cortical tissue obtained from human brains with a history of ischemic stroke or TBI (Fig. 8, Fig, S11, and tables S1 and S2). The extent of co-localization between GAT-3 and GFAP-positive putative astrocytes varied, prominent in some cases (such as thalamus and cortex of control subject, Fig. 8, C and D) and less prominent in others (such as thalamus of stroke and TBI subject, Fig. 8C). In a blinded, semi-quantitative scoring of GAT-3 intensity, thalamic samples from control subjects displayed a median score of 3 (corresponding to dense GAT-3 immunoreactivity), whereas thalamic samples from subjects with stroke or TBI displayed, respectively, a median score of 2 (moderate GAT-3) and 2.5 (moderate to dense GAT-3) (table S2). Cortical samples from control subjects, as well as peri-lesional cortical samples from subjects with stroke or TBI, displayed a median score of 2 (moderate GAT-3) (table S2).
Fig. 8. Decreased GAT-3 immunoreactivity in human post-mortem thalamic tissue after cortical injuries.
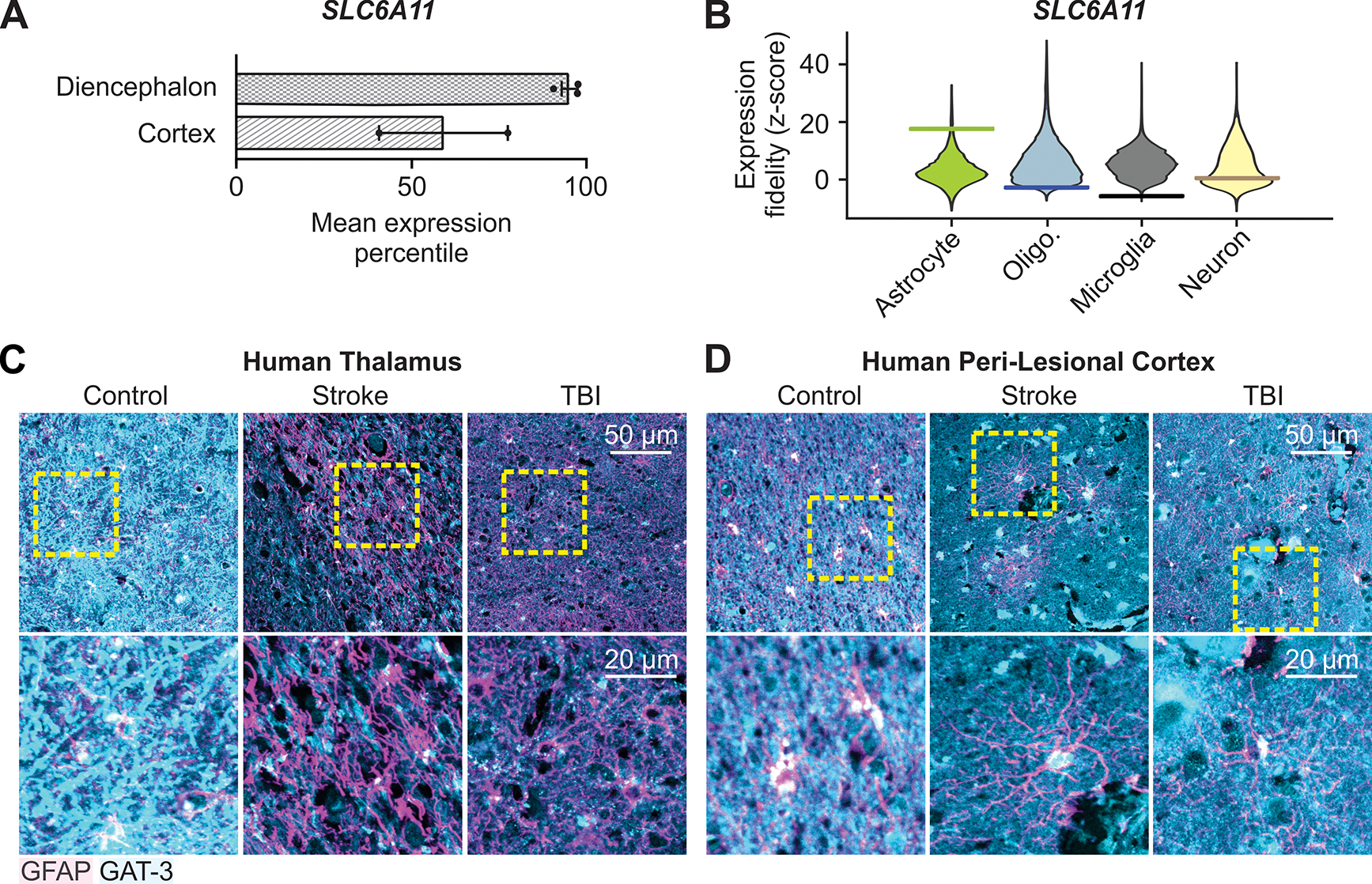
(A) Mean expression percentile of SLC6A11 gene relative to all genes in neurotypical adult human diencephalon and parietal cortex. Data obtained from (55) from 535 human samples. (B) Expression fidelity of SLC6A11 from cell-type specific transcriptomic profiling in the adult human diencephalon compared to transcriptional signatures of major brain cell types, relative to all human genes detected in the database from (55). Higher fidelity indicates higher correlation of SLC6A11 to cell type. Horizontal bar indicates Z-score for SLC6A11 relative to that cell type (17.6, −2.8, −5.8, and 0.4 for astrocytes, oliogodendrocytes, microglia, and neurons respectively), violin plot indicates Z-score distribution for all genes detected in that cell type. (C, D) GFAP and GAT-3 immunofluorescence in post-mortem thalamic tissue (C) and peri-lesional cortex (D) from human subjects with a history of ischemic stroke or TBI, and age-matched control subjects. Images are representative of 3 control subjects, 3 subjects with stroke, and 4 subjects with TBI. Yellow boxes: regions magnified below. Summary of all data shown in Fig. S11 and table S2.
DISCUSSION
Here we demonstrate that neuroinflammation in the mouse thalamus can drive enhanced cellular and microcircuit excitability, seizure risk, and aberrant changes in cortical states in mice. Our findings pinpoint astrocytic GAT-3 as a link between neuroinflammation and long-term network dysfunction, and as a potential therapeutic target for repairing circuit dynamics and promoting resilience following brain insults characterized by secondary and persistent thalamic neuroinflammation. Although astrocyte dysfunction has been widely documented in cellular and circuit hyperexcitability in the context of epilepsy (3, 38, 58–62), untangling whether reactive astrocytes are a cause and/or consequence of changes in neuronal network activity remains challenging (3, 35, 58, 59). In the thalamus, astrogliosis has been suggested to be a predictor of long-term consequences of cortical lesions such as epilepsy and cognitive impairment (6, 8, 12, 27). Given the role of the thalamus as a regulator of thalamocortical rhythms and higher cognitive processes, understanding if and how reactive astrocytes in the thalamus can initiate development of pathological circuit dynamics is of broad fundamental and translational interest. To address this gap, we used a viral construct (35) to selectively transduce astrocytes in the mouse thalamus, recapitulating secondary thalamic astrogliosis, but in the absence of a cortical lesion. The long-term consequences of thalamic astrogliosis included increased seizure risk, and disruption of adaptive cortical rhythms in the sigma and gamma bands. These effects of thalamic astrogliosis are reminiscent of maladaptive outcomes of cortical injuries, such as increased seizure risk in mice and patients (63), and reduced sigma-related sleep spindles in mice (10, 64) linked to sleep-wake disturbances in patients (65). Therefore, rather than being a mere bystander in the pathological sequelae of brain injuries, thalamic astrogliosis in mice can initiate robust, sustained aberrations in thalamocortical rhythms—gain-of-function in pathological rhythms and loss-of-function in adaptive rhythms—as had been suggested by previous studies (10) but not yet demonstrated directly.
How might thalamic reactive astrocytes drive maladaptive circuit function? In the mouse model of thalamic astrogliosis, reactive astrocytes downregulated the GAT-3 GABA transporter, leading to increased ITONIC in thalamocortical neurons, resulting in increased neuronal excitability, and increased intra-thalamic micro-circuit excitability (fig. S12). Enhancing GAT-3 in mouse thalamic astrocytes prevented astrogliosis-induced neuronal hyperexcitability and restored cortical sigma and gamma power, supporting the conclusion that the maladaptive outcomes were driven by reactive astrocytes. As in our mouse model of thalamic astrogliosis, GAT-3 was also decreased in regions of thalamic reactive astrocytes in mouse models of cortical stroke and TBI. The identification of SLC6A11—encoding GAT-3—as a risk gene for epilepsy in humans (66), and the data showing that pharmacological blockade of GAT-3 strengthens epileptiform oscillations in rats ex-vivo (67) suggest a link between GAT-3 and brain hyperexcitability. We found that enhancing astrocytic GAT-3 unilaterally in the thalamus prevented the increased seizure risk in the mouse model of thalamic astrogliosis, and reduced chemoconvulsant-induced seizure severity and mortality in a mouse model of cortical injury.
At the mechanistic level, our study suggests that enhancing GAT-3 could act through reducing excess ITONIC in thalamocortical neurons. In these cells, ITONIC is mediated by the δ-subunit-containing GABAAR (encoded by Gabrd). GABRD is a susceptibility locus for generalized epilepsies, and increased ITONIC has been implicated in epilepsy (46, 48). Astrocytic GAT-3 plays a key role in the regulation of extrasynaptic GABA in the thalamus (52), which contributes to ITONIC in thalamocortical neurons. In support of our hypothesis, preventing the increase in ITONIC via conditional Gabrd deletion in thalamocortical neurons also prevented astrogliosis-driven hyperexcitability.
In the peri-lesional cortex of rodents, phasic and tonic GABA currents may play opposing roles in the aftermath of stroke (68, 69). ITONIC has diverse region- and cell type-specific effects (48). Thus, selectively counteracting the maladaptive components is critical in promoting repair. Although we did not examine changes in ITONIC in thalamocortical neurons in TBI mice, our finding that enhancing thalamic GAT-3 prevented PTZ-induced mortality in TBI mice supports the idea that targeting thalamic astrocytic GAT-3 may be a viable strategy to decrease seizure risk following cortical injury.
Astrocytic GAT-3 activity and modulation of neuronal ITONIC should be evaluated as possible therapeutic targets in the context of neuroinflammation associated with increased seizure risk. Indeed, dysfunction of GATs and ITONIC have been implicated in various disorders associated with both chronic neuroinflammation and increased seizure risk, such as stroke (69) and Alzheimer’s disease (70).Secondary and persistent astrogliosis of the thalamus is a predictor of long-term consequences of cortical lesions such as epilepsy and cognitive impairment (6, 8, 12, 27), and perturbations of cortical rhythms driven by thalamic astrogliosis (discussed above) phenocopy the changes observed in the aftermath of brain injuries. The secondarily damaged thalamus can be targeted to abort post-stroke epileptic seizures in rodents (41) and protect against chemoconvulsant-induced mortality in mice with TBI (discussed above). Based on our findings showing that GAT-3 loss-of-function is a hallmark of thalamic astrogliosis, and that increasing GAT-3 specifically in thalamic astrocytes is beneficial in multiple mouse models, we propose that GAT-3 loss-of-function is a node of failed homeostatic plasticity and a key component of reactive astrocyte dysfunction which enables the far-reaching consequences of thalamic astrogliosis. In the sclerotic hippocampus, loss of GAT-1 is accompanied by an upregulation in GAT-3, suggesting a compensatory homeostatic mechanism (71, 72). However, in the setting of thalamic inflammation, loss of GAT-3 was not associated with transcriptional upregulation of GAT-1. In the thalamus of humans and mice, Slc6a11 (encoding GAT-3) is almost exclusively expressed in astrocytes (56, 57), whereas Slc6a1 (encoding GAT-1) is expressed similarly across astrocytes and neurons. Therefore, we speculate that loss of GAT-3 in thalamic astrocytes might be particularly detrimental because of a homeostatic failure to compensate for lost function. We propose that enhancing GAT-3 in reactive astrocytes might promote brain resilience to injuries.The mouse model of thalamic astrogliosis allows us to dissect the role of thalamic astrogliosis bypassing cortical injuries, but it has several limitations. First, a single experimental manipulation of astrocytes cannot fully phenocopy the molecular and functional features of reactive astrocytes. Nevertheless, our analyses highlight some features of astrogliosis conserved across experimental conditions—including somatic hypertrophy and reduction in process branching (32), and differential expression of canonical reactivity genes (31, 33). Second, we did not investigate other elements of the neuroinflammatory cascade—such as the secondary activation of microglia—and therefore do not exclude their possible influence on circuit dysfunction after cortical lesions. Third, we did not investigate the extent to which astrogliosis-induced changes in cortical sigma and gamma power directly relate to disease-relevant cognitive or behavioral impairments. The absence of major abnormalities in mice with thalamic astrogliosis in standard behavioral assays relying on distributed brain circuits does not preclude other thalamocortical circuit-specific changes such as somatosensation or attention. Fourth, only male mice were included in our study. Given that the link between ITONIC and neuronal excitability is ovarian cycle-dependent (73), further investigations are needed to determine the relevance of our findings in female mice. Another limitation of this study was the limited sample size for post-mortem thalamic human tissue, which excluded reliable statistical analysis of immunofluorescence data. We hope our study will encourage inclusion of the thalamus in standard resection protocols of human post-mortem brain tissue. In addition, because some data included in our study were obtained from patients with a history of epilepsy before and/or after injury, we cannot conclusively demonstrate that the changes in GFAP or GAT-3 were solely due to injury rather than recurrent seizures. Finally, although we present evidence of thalamic GAT-3 perturbation across various model systems and proof-of-concept that thalamic GAT-3 can be targeted to prevent seizure risk in mice, further work in pre-clinical models of cortical injury is needed to understand whether GAT-3 can be safely targeted to ameliorate disease-relevant cellular and circuit pathologies such as spontaneous seizures without compromising the adaptive aspects of neuroinflammation.
MATERIALS AND METHODS
Study Design
The main objective of this study was to investigate whether thalamic astrogliosis acts as a node of vulnerability after brain injuries, and if so, whether it represents a potential disease-modifying target. We developed a mouse model of unilateral thalamic astrogliosis using intra-thalamic delivery of a viral construct previously shown to selectively induce reactive astrocytes in the hippocampus (35); used established mouse models of cortical injury (photothrombotic stroke and controlled cortical impact); and determined the potential restorative effects of counteracting GAT-3 loss using a viral construct to unilaterally enhance GAT-3 expression (54) in thalamic astrocytes. Transduction of reactive astrocytes was validated using established transcriptomic, morphological, and immunohistochemical markers (28), using a combination of transgenic reporter mice, FACS, bulk RNA-Seq, and microscopy. Cellular excitability and synaptic function were assessed ex vivo with whole-cell current- and voltage-clamp recordings in thalamic neurons, and exclusion criteria are detailed below. Microcircuit excitability was assessed with extracellular recordings in ex vivo thalamic slices. In vivo brain excitability and cortical states were assessed with pharmacological seizure assays and electrocorticography recordings. Dysregulation of GAT-3 was assessed using a combination of immunohistochemistry, qPCR, bulk RNASeq, and spatial transcriptomics in mice with thalamic astrogliosis and cortical injury. To assess relevance to human disease, immunohistochemical markers of astrogliosis and GAT-3 were assessed in post-mortem thalamic tissue obtained from human subjects with a history of cortical injuries.
All mouse experiments were performed in adult, male, age-matched mice with similar numbers randomly assigned to different experimental groups. Mice with thalamic astrogliosis were compared to mice injected unilaterally with sterile saline. Mice with cortical injury were compared to data from mice which received identical surgical procedures but no illumination of the light-sensitive dye (stroke) or impact (TBI). All procedures were approved by the ethics committees at the University of California San Francisco, Gladstone Institutes, and Amsterdam UMC. Sample sizes were based on similar previously published work (10, 12, 43, 74) and are provided in figure legends. Data collection and analyses were performed blinded. Raw data are reported as separate Excel document in data file S1.
Mice
All protocols were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco and Gladstone Institutes. Precautions were taken to minimize stress and the number of animals used in each set of experiments. Mice were separately housed after surgical implants.
Viral injections
Stereotaxic viral injections were carried out as described (43, 44). Briefly, mice were anesthetized with 2–5% isoflurane. Adeno-associated viral (AAV) constructs were delivered into the ventrobasal nucleus of the thalamus (VB) using the following coordinates: 1.65 mm posterior to Bregma, 1.5 lateral relative to midline, and 3.3 to 3.5 mm ventral to the cortical surface. For all injections, the MicroSyringe Pump Controller (Micro4, WPI), NanoFil syringes (10μL, WPI), and 33g beveled NanoFil needles (WPI; NF33BV-2) were used. Viral constructs were infused at a rate of 100–200 nL/min, with a 5–10 minute pause before withdrawing the needle. All viral injections were performed unilaterally. Experiments were performed at least 3 weeks following viral injections. Astrogliosis was induced with a high titer injection of AAV2/5-Gfa104-PI.eGFP.WPRE.bGH (AAV2/5-Gfa104-eGFP), which transduces astrocytes with enhanced green fluorescent protein (eGFP) expression driven by a Gfap promoter (35) (Addgene #100896-AAV5; previously available through Penn Vector Core, Catalog #AV-5-PV2380). Mice received unilateral injections of 750 nL of AAV2/5-Gfa104-eGFP, or saline in control conditions (0.9% sodium chloride; Hospira). The titer of the construct, across various lots and experiments, was validated by Penn Vector Core using qPCR and dd-qPCR, and fell into the range of 0.85–1.7e10 g.c. per injection. For low-titer control experiments, we delivered 1.4e7 g.c. per injection. Previous work demonstrated that this titer does not induce astrogliosis (35). In describing the results of this study, we use “Control” to refer to mice in which we performed intra-thalamic injection of saline, and we use “Thalamic Astrogliosis” to refer to mice in which we performed intra-thalamic injections of AAV2/5-Gfa104-eGFP, unless specified otherwise. To selectively delete the delta subunit in GabrdFl/Fl mice, 600 nL of AAV-CaMKIIα-mCherry-Cre (UNC Vector Core) was used, in combination with 750 nL of either AAV2/5-Gfa104-eGFP, or saline. AAV2/5-GfaABC1D-GAT3-HA-mCherry (generated by B.S.K. and X.Y.) was used to selectively enhance expression of GAT-3 in astrocytes. 600 nL of AAV-GAT3 was delivered in combination with 750 nL of either AAV2/5-Gfa104-eGFP, or saline.
Patch-clamp electrophysiology in thalamic slices
Recordings were performed as described (12, 43, 75). Thalamocortical neurons in the VB thalamus, and neurons in the reticular thalamic nucleus (nRT), were visually identified by differential contrast optics with an Olympus microscope (60x objective, NA 1.1, WD 1.5 mm; SKU 1-U2M592). Recording electrodes made of borosilicate glass (Sutter Instruments) had a resistance of 2.5–4 MΩ when filled with intracellular solution. Access resistance was monitored in all the recordings, and cells were included for analysis only if the access resistance was <25 MΩ.
To record intrinsic membrane properties and bursting properties in current-clamp mode, and spontaneous excitatory post-synaptic currents in voltage-clamp mode, an internal solution containing 120 mM potassium gluconate, 11 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, and 1 mM EGTA, pH adjusted to 7.4 with KOH (290 mOsm) was used; these recordings were performed in the presence of picrotoxin (50 μM, Sigma-Aldrich, #P1675) in the artificial cerebrospinal fluid (aCSF). The potentials for were corrected offline for −15 mV liquid junction potential.
To record spontaneous inhibitory post-synaptic currents, an internal solution containing 135 mM CsCl, 10 mM HEPES, 10 mM EGTA, 2 mM MgCl2—6H2O, and 5 mM QX-314, pH adjusted to 7.3 with CsOH (290 mOsm) was used. Recordings were performed in the presence of kynurenic acid (2 mM, Sigma-Aldrich #K3375) in aCSF.
To record tonic GABAA receptor-mediated currents, an internal solution containing 130 mM CsCl, 2 mM MgCl2, 4 mM Mg-ATP, 0.3 mM Na-GTP, 10 mM Na-HEPES, 0.1 mM EGTA, pH adjusted to 7.3 with CsOH (290 mOsm) was used (45).
Recordings were performed in the presence of kynurenic acid (2 mM) in aCSF which was kept at 30 ± 1 °C. To obtain tonic GABA measurements, a bath of 50 μM Gabazine (GBZ; Sigma #SR-95531) prepared in dimethyl sulfoxide (DMSO; Sigma #D8418) was applied. Following GBZ application, slices were recorded for at least 10 minutes. Tonic current was defined as the shift in holding current due to GBZ bath application, and marked by the stopping of phasic IPSCs. Average currents were obtained for at least 2 minutes pre- and post-stabilization of the shift in holding current. Currents were subtracted using Clampfit 10.5 (Molecular Devices, SCR_011323). Currents are presented without normalizing to cell capacitance, as capacitance did not differ between groups (table S3).
In vivo electrocorticography (ECoG) data acquisition
To control for circadian rhythms, animals were housed using a regular light/dark cycle, and recordings performed between 7:00 AM and 7:00 PM. Prior to acquisition of multi-site EcoG recordings (Figs. 4, 5, S6), mice were allowed to recover for at least one week. EcoG signals were recorded using RZ5 via Synapse software (Tucker Davis Technologies) and sampled at 1221 Hz. Animals were continuously monitored during recordings using a video camera that was synchronized to the signal acquisition via RZ5 (43).
Chronic, continuous EcoG recordings (Fig. 6, table S9) were acquired using wireless telemetry devices (PhysioTel HD-X02 implants, Data Sciences International) and sampled at 500 Hz via Ponemah software (DSI). 24/7 recordings were performed for one-week periods (168 hours) at the following timepoints: 1, 3, 5 and 7 weeks after thalamic astrogliosis (Fig. 6C), and 7 weeks after thalamic astrogliosis with GAT-3 enhancement (Fig. 6D). Acquisition started immediately following implantation, as the wireless devices allow mice to remain in their home cages.
Quantification and Statistical Analysis
In vivo electrocorticography
Detection of epileptiform discharges:
To quantify the number of epileptiform discharges, induced by PTZ, we used a custom MATLAB script, which enabled detection of synchronous discharges with amplitude three times baseline signal noise and required user input to reject or accept events. Quantification was performed for the somatosensory cortex ipsilateral to thalamic astrogliosis, and compared with a Mann-Whitney U-test. This detection method was validated against manual quantification of epileptiform discharges from the raw ECoG and simultaneously acquired video recordings of mice. A subset of mice presented in Fig. 4D underwent the PTZ seizure assay again at 4 and/or 7 months to determine the chronic effects of astrogliosis (presented in fig. S6).
Kainic acid seizure detection:
Seizures on EEG traces were identified based on high frequency, synchronous discharges with amplitude three times baseline signal noise, using Spike2 software. Detection was cross-validated with behavioral signs of seizures from simultaneously acquired video recordings of mice.
ECoG spectral analysis:
For chronic, continuous ECoG recordings, spectral analysis was performed in MATLAB. Absolute power of individual frequency bands was calculated using the bandpower function in MATLAB. Total band power was calculated between 1 to 75 Hz, in order to avoid edge effects of the wireless device bandwidth (0.5 to 80 Hz). For Weeks 1, 5, and 7, we obtained 168 1-hr bins; for Week 3, we obtained 152 1-hr bins. Total band power was calculated between 1 to 75 Hz, in order to avoid edge effects of the wireless device bandwidth (0.5 to 80 Hz). To analyze spectral features at week 7 (the most chronic time-point tested), we calculated the absolute power of individual frequency bands for 12-hour bins corresponding to the light and dark cycle. To investigate spectral features of the somatosensory cortex (S1) functionally connected to the site of thalamic astrogliosis, we performed paired t-tests of frequency bands obtained from ipsilateral and contralateral S1. All statistical comparisons are reported in table S9.
Statistical Analysis
All numerical values are given as means and error bars are standard error of the mean (SEM) unless stated otherwise. For comparison between two groups, the non-parametric Mann-Whitney U test was performed. For comparisons between multiple groups, a one- or two-way analysis of variance (ANOVA; ordinary or repeated measures) was performed, in addition to post-hoc comparisons with corrections for multiple comparisons (detailed in figure legends). When parametric tests were used, results of the Shapiro-Wilk normality test are provided. The threshold for statistical significance was set to α=0.05, unless specified otherwise. Each n represents an independent biological sample, unless specified otherwise in figure legends. Data analysis was performed with MATLAB, GraphPad Prism 7, OriginPro, SigmaPlot, and SPSS.
Supplementary Material
Acknowledgments:
We thank Dr. Jamie Maguire, for kindly providing the GabrdFl/Fl mice; Drs. John Huguenard and Jordan Sorokin, for providing the custom MATLAB script for thalamic oscillation analysis; Drs. Kevin Kelley and Michael Oldham, for providing assistance in using their published database of human transcriptomic data; Irene Lew and Hiromi Inoue, for help with animal husbandry; Naznin Jahan, for technical assistance; Drs. Kathryn Claiborn and Francoise Chanut, for providing critical editorial feedback on the manuscript; Dr. Reuben Thomas (Gladstone Bioinformatics Core), for consultation of statistical analyses; Drs. Lennart Mucke and Michael Gill, and Iris Lo and Jeffrey Sims (Gladstone Behavioral Core), for consultation on behavioral experiments; Jim McGuire (Gladstone Genomics Core), for conducting the RNA sequencing library preparation and QCs for sequencing at the Center For Advanced Technology at UCSF; and the Gladstone Histology Core, for technical advice.
Funding:
National Institute of Neurological Disorders and Stroke grant F31 NS111819 (to FSC)
National Institute of Neurological Disorders and Stroke grant R01 NS096369 (to JTP)
National Institute of Neurological Disorders and Stroke grant R01 NS121287 (to JTP)
National Institute of Neurological Disorders and Stroke grant R00 NS078118 (to JTP)
National Institute of Neurological Disorders and Stroke grant R35 NS111583 (to BSK)
National Institute of Mental Health grant DP1 MH104069 (to BSK)
National Institute of Mental Health grant DP2 MH116507 (to AVM)
National Institute of Mental Health grant R01 MH119349 (to AVM)
National Cancer Institute grant P30 CA082103 (to UCSF Laboratory for Cell Analysis)
National Science Foundation grant 1144247 (to FSC)
UCSF Discovery Fellowship (to FSC)
Department of Defense grant EP150038 (to JTP)
Department of Defense grant EP190020 (to JTP)
Gladstone Institutes Animal Facility grant RR18928 (to JTP)
Pew Charitable Trusts (to AVM)
European Union Seventh Framework Programme EPITARGET grant 602102 (to EAV, EA)
European Union Horizon 2020 Research and Innovation Programme Marie Sklodowska-Curie grant 722053 (to EA)
Dutch Epilepsy Foundation project 16–05 (to DWMB, EA)
European Union Horizon 2020 WIDESPREAD-05-2020-Twinning, EpiEpiNet grant agreement 952455 (to EA, EAV)
Footnotes
Competing interests: Authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper and Supplementary Data Files. Bulk transcriptomic data are available through GEO (GSE203111). Correspondence and request for materials should be addressed to J.T.P.
References and Notes
- 1.Russo MV, McGavern DB, Inflammatory neuroprotection following traumatic brain injury, Science 353, 783–785 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon DW, McGeachy MJ, Baylr H, Clark RSB, Loane DJ, Kochanek PM, The far-reaching scope of neuroinflammation after traumatic brain injury, Nat. Rev. Neurol 13, 171–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vezzani A, Balosso S, Ravizza T, Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy, Nat. Rev. Neurol. 15, 459–472 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Shi SX, Shi K, Liu Q, Brain injury instructs bone marrow cellular lineage destination to reduce neuroinflammation, Sci. Transl. Med. 13, eabc7029 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Llorente IL, Xie Y, Mazzitelli JA, Hatanaka EA, Cinkornpumin J, Miller DR, Lin Y, Lowry WE, Carmichael ST, Patient-derived glial enriched progenitors repair functional deficits due to white matter stroke and vascular dementia in rodents., Sci. Transl. Med. 13, 1–18 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Grossman EJ, Inglese M, The role of thalamic damage in mild traumatic brain injury, J. Neurotrauma 33, 163–167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott G, Hellyer PJ, Ramlackhansingh AF, Brooks DJ, Matthews PM, Sharp DJ, Thalamic inflammation after brain trauma is associated with thalamo-cortical white matter damage, J. Neuroinflammation 12, 1–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ, Inflammation after trauma: Microglial activation and traumatic brain injury, Ann. Neurol. 70, 374–383 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI, Thalamic nuclei after human blunt head injury, J. Neuropathol. Exp. Neurol. 65, 478–488 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Holden SS, Grandi FC, Aboubakr O, Higashikubo B, Cho FS, Chang AH, Forero AO, Morningstar AR, Mathur V, Kuhn LJ, Suri P, Sankaranarayanan S, Andrews-Zwilling Y, Tenner AJ, Luthi A, Aronica E, Corces MR, Yednock T, Paz JT, Complement factor C1q mediates sleep spindle loss and epileptic spikes after mild brain injury, Science 373 (2021), doi: 10.1126/science.abj2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hazra A, Macolino C, Elliott MB, Chin J, Delayed thalamic astrocytosis and disrupted sleep-wake patterns in a preclinical model of traumatic brain injury, J. Neurosci. Res. 92, 1434–1445 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Paz JT, Christian CA, Parada I, Prince DA, Huguenard JR, Focal Cortical Infarcts Alter Intrinsic Excitability and Synaptic Excitation in the Reticular Thalamic Nucleus, J. Neurosci. 30, 5465–5479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao Z, Harvey SS, Bliss TM, Cheng MY, Steinberg GK, Inflammatory Responses in the Secondary Thalamic Injury After Cortical Ischemic Stroke, Front. Neurol. 11, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pappata S, Levasseur M, Gunn RN, Myers R, Crouzel C, Syrota A, Jones T, Kreutzberg GW, Banati RB, Thalamic microglial activation in ischemic stroke detected in vivo by PET and [(11)C]PK11195, Neurology 55, 1052–1054 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Beattie GC, Glaser CA, Sheriff H, Messenger S, Preas CP, Shahkarami M, Venkatesan A, Encephalitis with thalamic and basal ganglia abnormalities: Etiologies, neuroimaging, and potential role of respiratory viruses, Clin. Infect. Dis. 56, 825–832 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Guth JC, Futterer SA, Hijaz TA, Liotta EM, Rosenberg NF, Naidech AM, Maas MB, Pearls & Oy-sters: Bilateral thalamic involvement in West Nile virus encephalitis, Neurology 83, 16–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abel D, Shen MY, Abid Z, Hennigan C, Boneparth A, Miller EH, Uhlemann AC, McBrian DK, Thakur K, Silver W, Bain JM, Encephalopathy and bilateral thalamic lesions in a child with MIS-C associated with COVID-19, Neurology 95, 745–748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B, COVID-19-associated acute hemorrhagic necrotizing encephalopathy: Imaging features, Radiology 296, E119–E120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng J, Wong LYR, Li K, Verma AK, Ortiz M, Wohlford-Lenane C, Leidinger MR, Knudson CM, Meyerholz DK, McCray PB, Perlman S, COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice, Nature (2020), doi: 10.1038/s41586-020-2943-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeanmonod D, Magnin M, Morel A, Low-threshold calcium spike bursts in the human thalamus: Common physiopathology for sensory, motor and limbic positive symptoms, Brain 119, 363–375 (1996). [DOI] [PubMed] [Google Scholar]
- 21.Steriade M, McCormick DA, Sejnowski TJ, Thalamocortical oscillations in the sleeping and aroused brain., Science 262, 679–85 (1993). [DOI] [PubMed] [Google Scholar]
- 22.Steriade M, Contreras D, Amzica F, Timofeev I, Synchronization of fast (30–40 Hz) spontaneous oscillations in intrathalamic and thalamocortical networks, J. Neurosci. 16, 2788–2808 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crunelli V, Hughes SW, The slow (1 Hz) rhythm of non-REM sleep: A dialogue between three cardinal oscillators, Nat. Neurosci. 13, 9–17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fogerson PM, Huguenard JR, Tapping the Brakes: Cellular and Synaptic Mechanisms that Regulate Thalamic Oscillations, Neuron 92, 687–704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones EG, The thalamic matrix and thalamocortical synchrony, Trends Neurosci. 24, 595–601 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Paz JT, Huguenard JR, Microcircuits and their interactions in epilepsy: is the focus out of focus?, Nat. Neurosci. 18, 351–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grossman EJ, Ge Y, Jensen JH, Babb JS, Miles L, Reaume J, Silver JM, Grossman RI, Inglese M, Thalamus and Cognitive Impairment in Mild Traumatic Brain Injury: A Diffusional Kurtosis Imaging Study, J. Neurotrauma 29, 2318–2327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen W-T, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Díaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Götz M, Gutiérrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, V Kastanenka K, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, MacVicar BA, Magistretti P, Messing A, Mishra A, V Molofsky A, Murai KK, Norris CM, Okada S, Oliet SHR, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Pérez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner I-B, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, V Sofroniew M, Verkhratsky A, Reactive astrocyte nomenclature, definitions, and future directions., Nat. Neurosci. (2021), doi: 10.1038/s41593-020-00783-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burda JE, V Sofroniew M, Review Reactive Gliosis and the Multicellular Response to CNS Damage and Disease, Neuron 81, 229–248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colombo E, Farina C, Astrocytes: Key Regulators of Neuroinflammation, Trends Immunol. 37, 608–620 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, Neurotoxic reactive astrocytes are induced by activated microglia, Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun D, Jakobs TC, Structural remodeling of astrocytes in the injured CNS, Neuroscientist 18, 567–588 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zamanian J, Xu L, Foo L, Nouri N, Zhou L, Giffard R, Barres B, Genomic Analysis of Reactive Astrogliosis, J. Neurosci. 32, 6391–6410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Necula D, Cho FS, He A, Paz JT, Secondary thalamic neuroinflammation after focal cortical stroke and traumatic injury mirrors corticothalamic functional connectivity, J. Comp. Neurol. (2021), doi: 10.1002/cne.25259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA, Selective induction of astrocytic gliosis generates deficits in neuronal inhibition., Nat. Neurosci. 13, 584–91 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morel L, Higashimori H, Tolman M, Yang Y, VGluT1+ Neuronal Glutamatergic signaling regulates postnatal developmental maturation of cortical protoplasmic astroglia, J. Neurosci. 34, 10950–10962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjöström PJ, Van Meyel DJ, Neuronal morphometry directly from bitmap images, Nat. Methods 11, 982–984 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel DC, Tewari BP, Chaunsali L, Sontheimer H, Neuron–glia interactions in the pathophysiology of epilepsy, Nat. Rev. Neurosci. 20, 282–297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR, Reciprocal Inhibitory Connections and Network Synchrony in the Mammalian Thalamus, Science 283, 541–543 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Lüthi A, T-type Ca 2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites, Nat. Neurosci. 11, 683–692 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng K, Deisseroth K, Huguenard JR, Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury., Nat. Neurosci. 16, 64–70 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho FS, Clemente A, Holden S, Paz JT, in Pitkänen Buckmaster, Galanopoulou Moshé (editors): Models of Seizures and Epilepsy, Second Edition, (Academic Press, 2017), pp. 273–284. [Google Scholar]
- 43.Ritter-Makinson S, Clemente-Perez A, Higashikubo B, Cho FS, Holden SS, Bennett E, Chkaidze A, Rooda OHJE, Cornet MC, Hoebeek FE, Yamakawa K, Cilio MR, Delord B, Paz JT, Augmented Reticular Thalamic Bursting and Seizures in Scn1a-Dravet Syndrome, Cell Rep. 26, 54–64 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorokin JM, Davidson TJ, Frechette E, Abramian AM, Deisseroth K, Huguenard JR, Paz JT, Bidirectional Control of Generalized Epilepsy Networks via Rapid Real-Time Switching of Firing Mode, Neuron 93, 194–210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cope DW, Hughes SW, Crunelli V, GABAA Receptor-Mediated Tonic Inhibition in Thalamic Neurons, J. Neurosci. 25, 11553–11563 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lőrincz ML, Gould TM, Carter DA, Crunelli V, Enhanced tonic GABAA inhibition in typical absence epilepsy, Nat. Med. 15, 1392–1398 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coulter DA, Huguenard JR, Prince DA, Calcium currents in rat thalamocortical relay neurones: kinetic properties of the transient, low-threshold current, J. Physiol. 414, 587–604 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee V, Maguire J, The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type, Front. Neural Circuits 8, 1–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agmon A, Yang LT, O’Dowd DK, Jones EG, Organized growth of thalamocortical axons from the deep tier of terminations into layer IV of developing mouse barrel cortex, J. Neurosci. 13, 5365–5382 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, Murphy GG, Parent JM, Meisler MH, Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy, Hum. Mol. Genet. 24, 506–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee V, Maguire J, Impact of inhibitory constraint of interneurons on neuronal excitability, J. Neurophysiol. 110, 2520–2535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beenhakker MP, Huguenard JR, Astrocytes as gatekeepers of GABAB receptor function., J. Neurosci. 30, 15262–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pirttimaki T, Parri HR, Crunelli V, Astrocytic GABA transporter GAT-1 dysfunction in experimental absence seizures., J. Physiol. 591, 823–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu X, Taylor AMW, Nagai J, Golshani P, Evans CJ, Coppola G, Khakh BS, Reducing Astrocyte Calcium Signaling In Vivo Alters Striatal Microcircuits and Causes Repetitive Behavior, Neuron 99, 1170–1187.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelley KW, Nakao-Inoue H, Molofsky AV, Oldham MC, Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes, Nat. Neurosci. 21, 1171–1184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Biasi S, Vitellaro-Zuccarello L, Brecha NC, Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron- microscopic immunolocalization, Neuroscience 83, 815–828 (1998). [DOI] [PubMed] [Google Scholar]
- 57.Vitellaro-Zuccarello L, Calvaresi N, De Biasi S, Expression of GABA transporters, GAT-1 and GAT-3, in the cerebral cortex and thalamus of the rat during postnatal development, Cell Tissue Res. 313, 245–257 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, Sutor B, Sontheimer H, Reactive Astrogliosis Causes the Development of Spontaneous Seizures, J. Neurosci. 35, 3330–3345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robel S, Sontheimer H, Glia as drivers of abnormal neuronal activity, Nat. Neurosci. 19, 28–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A, Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures, Nat. Med. 16, 413–419 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Senatorov VV, Friedman AR, Milikovsky DZ, Ofer J, Saar-Ashkenazy R, Charbash A, Jahan N, Chin G, Mihaly E, Lin JM, Ramsay HJ, Moghbel A, Preininger MK, Eddings CR, Harrison HV, Patel R, Shen Y, Ghanim H, Sheng H, Veksler R, Sudmant PH, Becker A, Hart B, Rogawski MA, Dillin A, Friedman A, Kaufer D, Blood-brain barrier dysfunction in aging induces hyper-activation of TGF-beta signaling and chronic yet reversible neural dysfunction, Sci. Transl. Med. 11 (2019), doi: 10.1101/537431. [DOI] [PubMed] [Google Scholar]
- 62.Steinhäuser C, Seifert G, in Noebels JL, Avoli M, Rogawski MA, et al. , editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th edition. Bethesda (MD): National Center for Biotechnology Information (US), (2012). [PubMed] [Google Scholar]
- 63.Klein P, Dingledine R, Aronica E, Bernard C, Blümcke I, Boison D, Brodie MJ, Brooks-Kayal AR, Engel J, Forcelli PA, Hirsch LJ, Kaminski RM, Klitgaard H, Kobow K, Lowenstein DH, Pearl PL, Pitkänen A, Puhakka N, Rogawski MA, Schmidt D, Sillanpää M, Sloviter RS, Steinhäuser C, Vezzani A, Walker MC, Löscher W, Commonalities in epileptogenic processes from different acute brain insults: Do they translate?, Epilepsia 59, 37–66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andrade P, Nissinen J, Pitkänen A, Generalized Seizures after Experimental Traumatic Brain Injury Occur at the Transition from Slow-Wave to Rapid Eye Movement Sleep, J. Neurotrauma 34, 1482–1487 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Ouellet MC, Beaulieu-Bonneau S, Morin CM, Sleep-wake disturbances after traumatic brain injury, Lancet Neurol. 14, 746–757 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Schijns OEMG, Bisschop J, Rijkers K, Dings J, Vanherle S, Lindsey P, Smeets HJM, Hoogland G, GAT-1 (rs2697153) and GAT-3 (rs2272400) polymorphisms are associated with febrile seizures and temporal lobe epilepsy, Epileptic Disord. 22, 176–182 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Lu AC, Lee CK, Kleiman-Weiner M, Truong B, Wang M, Huguenard JR, Beenhakker MP, Nonlinearities between inhibition and t-type calcium channel activity bidirectionally regulate thalamic oscillations, Elife 9, 1–36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hiu T, Farzampour Z, Paz JT, Wang EHJ, Badgely C, Olson A, Micheva KD, Wang G, Lemmens R, Tran KV, Nishiyama Y, Liang X, Hamilton SA, O’Rourke N, Smith SJ, Huguenard JR, Bliss TM, Steinberg GK, Enhanced phasic GABA inhibition during the repair phase of stroke: A novel therapeutic target, Brain 139, 468–480 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST, Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke, Nature 468, 305–309 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee DY, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ, GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease, Nat. Med. 20, 886–896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu Z, Guo Z, Gearing M, Chen G, Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzhiemer’ s disease model, Nat. Commun. 5, 1–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee TS, Bjørnsen LP, Paz C, Kim JH, Spencer SS, Spencer DD, Eid T, Lanerolle NC, GAT1 and GAT3 expression are differently localized in the human epileptogenic hippocampus, Acta Neuropathol. 111, 351–363 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Maguire JL, Stell BM, Rafizadeh M, Mody I, Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety, Nat. Neurosci. 8, 797–804 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-inoue H, Dorman LC, Akil O, Joshita S, Barres BA, Paz JT, Molofsky AB, V Molofsky A, Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development, Science 359, 1269–1273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, Huguenard JR, A new mode of corticothalamic transmission revealed in the Gria4(−/−) model of absence epilepsy., Nat. Neurosci. 14, 1167–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Franklin KBJ, Paxinos G, The Mouse Brain in Stereotaxic Coordinates (Elsevier Academic Press, 3rd Editio., 2007). [Google Scholar]
- 77.Schousboe A, Bak LK, Waagepetersen HS, Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA, Front. Endocrinol. (Lausanne). 4, 1–11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, Veras MM, Pereira TF, Leite REP, Möller T, Wes PD, Sogayar MC, Laman JD, den Dunnen W, Pasqualucci CA, Oba-Shinjo SM, Boddeke EWGM, Marie SKN, Eggen BJL, Transcriptomic analysis of purified human cortical microglia reveals age-associated changes, Nat. Neurosci. 20, 1162–1171 (2017). [DOI] [PubMed] [Google Scholar]
- 79.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA, A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function, J. Neurosci. 28, 264–278 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van Vliet EA, Ndode-Ekane XE, Lehto LJ, Gorter JA, Andrade P, Aronica E, Gröhn O, Pitkänen A, Long-lasting blood-brain barrier dysfunction and neuroinflammation after traumatic brain injury, Neurobiol. Dis. 145, 105080 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Pangratz-Fuehrer S, Sieghart W, Rudolph U, Parada I, Huguenard JR, Early postnatal switch in GABAA receptor α-subunits in the reticular thalamic nucleus, J. Neurophysiol. 115, 1183–1195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCormick DA, Pape H-C, Properties of a Hyperpolarization-Activated Cation Current and Its Role in Rhythmic Oscillation in Thalamic Relay Neurones, J. Physiol., 291–318 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mirski MA, Ferrendelli JA, Anterior thalamic mediation of generalized pentylenetetrazol seizures, Brain Res. 399, 212–223 (1986). [DOI] [PubMed] [Google Scholar]
- 84.Bolkvadze T, Pitkänen A, Development of Post-Traumatic Epilepsy after Controlled Cortical Impact and Lateral Fluid-Percussion-Induced Brain Injury in the Mouse, J. Neurotrauma 29, 789–812 (2012). [DOI] [PubMed] [Google Scholar]
- 85.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L, Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model, Science 316, 750–754 (2007). [DOI] [PubMed] [Google Scholar]
- 86.Manninen E, Chary K, Lapinlampi N, Andrade P, Paananen T, Sierra A, Tohka J, Gröhn O, Pitkänen A, Acute thalamic damage as a prognostic biomarker for post-traumatic epileptogenesis, Epilepsia, 1–13 (2021). [DOI] [PubMed] [Google Scholar]
- 87.Hunt RF, Scheff SW, Smith BN, Posttraumatic epilepsy after controlled cortical impact injury in mice, Exp. Neurol. 215, 243–252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper and Supplementary Data Files. Bulk transcriptomic data are available through GEO (GSE203111). Correspondence and request for materials should be addressed to J.T.P.