

Abstract
Protein kinase 2 (CK2) is a serine/threonine kinase composed of two catalytic subunits (CK2α and/or CK2α’) and two regulatory subunits (CK2β). CK2 promotes cancer progression by activating the NF-κB, PI3K/AKT/mTOR and JAK/STAT pathways, and also is critical for immune cell development and function. The potential involvement of CK2 in CD8+ T-cell function has not been explored. We demonstrate that CK2 protein levels and kinase activity are enhanced upon mouse CD8+ T-cell activation. CK2α deficiency results in impaired CD8+ T-cell activation and proliferation upon T-cell receptor stimulation. Furthermore, CK2α is involved in CD8+ T-cell metabolic reprogramming through regulating the AKT/mTOR pathway. Lastly, using a mouse Listeria monocytogenes infection model, we demonstrate that CK2α is required for CD8+ T-cell expansion, maintenance and effector function in both primary and memory immune responses. Collectively, our study implicates CK2α as an important regulator of mouse CD8+ T-cell activation, metabolic reprogramming and differentiation both in vitro and in vivo.
Keywords: Protein kinase 2, CD8+ T-cell, mTOR, Metabolic reprogramming, Infection
INTRODUCTION
CD8+ T-cells are critical in controlling infection caused by intracellular pathogens including viruses and intracellular bacteria, and controlling cancer progression by cytolysis and recruitment and activation of other immune cells (1). In response to infection, naive CD8+ T-cells differentiate into heterogeneous populations of pathogen-specific effector CD8+ T-cells. Most effector cells undergo apoptosis and contract after resolution of the infection, while a small percentage of effector CD8+ T-cells differentiate into memory cells and provide lasting protection against reinfection (2–4). Heterogeneous populations of effector CD8+ T-cells can be distinguished by the expression of several markers, including KLRG1, IL-7Rα, CD27, CXCR3 and CD62L. For instance, in acute infection, effector CD8+ T-cell pools can be distinguished as short-lived effector T-cells (SLECs, KLRG1hiIL-7Rαlow), which have a shorter lifespan and reduced proliferative capacity in response to secondary antigenic challenge. Conversely, memory precursor effector cells (MPECs, KLRG1lowIL-7Rαhi) differentiate into long-lived memory cells, and proliferate vigorously in response to secondary challenge (5, 6). Current evidence suggests that multiple signals orchestrate CD8+ T-cell fate decisions, including T-cell receptor (TCR), co-stimulatory signaling, cytokines, transcription factors and changes in metabolism (3, 7).
Protein Kinase CK2 is a highly conserved serine-threonine kinase that is expressed in all eukaryotic organisms. CK2 is often present as a tetrameric complex of two catalytic subunits (CK2α and/or CK2α’) and two regulatory subunits (CK2β) which phosphorylate serine and threonine as well as tyrosine residues on hundreds of substrates (8, 9). CK2 is involved in a wide range of biological processes and cellular functions, including cell growth, proliferation, differentiation, transcription and translation (10, 11). Aberrant expression and high CK2 kinase activity are characteristic of many cancers, promoting tumor survival and growth, and CK2 is a promising therapeutic target for malignant diseases (12–14). CK2 enhances the activity of several signaling pathways that are essential for cancer progression, including the NF-κB, PI3K/AKT/mTOR, JAK/STAT and HIF-1α pathways, which are also critical for immune cell development and function (15).
Emerging evidence further suggests that CK2 plays important roles in inflammatory responses and pathologies associated with inflammation (16–20). Both Ulges et al. (16), and our group (17, 21) demonstrated that deletion of CK2 subunits, either CK2α or CK2β, resulted in significant protection in a model of Multiple Sclerosis, Experimental Autoimmune Encephalomyelitis (EAE), by promoting differentiation of T regulatory cells (Treg) and inhibiting Th17 cell differentiation. CK2α contributes to the pathogenesis of colitis by promoting CD4+ T-cell proliferation and Th1 and Th17 responses (18). CK2 is also involved in the suppressive function of CD4+ Foxp3+ Tregs against allergy-promoting Th2 cells (22). In addition, CK2 is critical for monocyte-derived dendritic cells to mature and produce cytokines that polarize effector T-cells in response to chemicals related to allergic contact dermatitis (23). CK2α deficiency in myeloid cells increases inflammatory myeloid cell recruitment, activation, and resistance following systemic Listeria monocytogenes infection (19). However, the function of CK2 in CD8+ T-cells is completely unknown.
Herein, we demonstrate that CK2 protein levels and kinase activity are enhanced upon CD8+ T-cell activation and that CK2α is required for CD8+ T-cell activation and proliferation upon TCR stimulation. Importantly, CK2α is involved in CD8+ T-cell metabolic reprogramming during activation through regulating the AKT/mTOR signaling pathway. The function of CK2α was further explored in a Listeria monocytogenes infection model, and our findings indicate that CK2α is required for CD8+ T-cell expansion, maintenance and effector function in both primary and memory stages during Listeria monocytogenes infection. Taken together, our study demonstrates that CK2α regulates CD8+ T-cell activation and differentiation both in vitro and in vivo.
MATERIALS AND METHODS
Mice.
CK2αfl/fl, CK2αfl/fldLck-Cre (CK2α−/−) and CD45.1 C57BL/6 mice were previously described (21). All mice were already backcrossed to C57BL/6 for at least 12 generations. CK2αfl/fl or CK2αfl/fldLck-Cre mice were further bred with OT-I mice to obtain OVA antigen-specific CD8+ T-cells. All mice were maintained under specific pathogen free conditions in the animal facility at the University of Alabama at Birmingham (UAB). Male and female mice between 8 and 12-weeks old were used for all experiments. All experimental procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of UAB.
Listeria monocytogenes Infection.
For the study of primary immune responses, CD45.1 C57BL/6 host mice were first intravenously (i.v.) transferred with 1 × 105 OT-I cells from either OT-I CK2αfl/fl or OT-I CK2αfl/fldLck-Cre (OT-I CK2α−/−) mice. One day post-transfer, recipient mice were infected with 1 × 103 colony-forming units (cfu) of Listeria monocytogenes expressing OVA (LM-OVA) (24) by i.v. injection. CD8+ T-cell responses were determined at day 5 (early effector phase), day 7 (effector phase) and day 42 (early memory phase) post infection (25). For analysis of recall immune responses, donor derived memory CD8+ T-cells were first sorted from CD45.1 host mice at early memory phase (day 42 post primary infection). Then, OT-I CK2αfl/fl and OT-I CK2α−/− memory CD8+ T-cells (5 × 103) were separately transferred into naive CD45.1 congenic recipient mice, which were then infected with 1 × 105 cfu of LM-OVA. Seven days post-infection, CD8+ T-cell responses in the spleen were analyzed.
Lymphocyte Preparation.
Single-cell suspensions of spleen and lymph nodes were prepared as previously described (18) and resuspended in R10 medium (RPMI 1640 with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 50 μM β-mercaptoethanol).
CD8+ T-cell Purification and Activation.
For in vivo transfer, CD8+ T-cells were first purified from the spleen using the EasySep™ Mouse CD8α Positive Selection Kit II (STEMCELL Technologies Inc, Vancouver, BC), then naïve OT-I cells were further sorted as CD8+Vβ5.1+CD44−CD62L+ using FACS Aria II (BD Bioscience, San Jose, CA). For memory CD8+ T-cell transfer, memory OT-I cells were sorted as CD8+CD45.2+Vα2+CD44+CD62L+ on an Aria II, routinely to higher than 98% purity. For in vitro activation and stimulation, naïve CD8+ T-cells were enriched from the spleen and peripheral lymph nodes using the EasySep™ Mouse Naïve CD8+ T Cell Isolation Kit (STEMCELL Technologies Inc, Vancouver, BC), routinely to 90– 95% purity.
For polyclonal activation, naïve CD8+ T-cells were stimulated with plate-bound anti-CD3 (1 μg/ml) (Clone 145-2C11, BioX Cell, West Lebanon, NH) and soluble anti-CD28 (1 μg/ml) (Clone 37.51, BioX Cell, West Lebanon, NH) Abs for the indicated times. For antigen-specific T-cell activation, splenocytes (1 × 106/ml) from OT-I CK2αfl/fl or OT-I CK2α−/− mice were cultured in R10 medium, incubated with OVA257–264 peptide (0.1 ng/ml) (InvivoGene, San Diego, CA), and then cells were harvested at the indicated time points.
Proliferation and Apoptosis Assays.
For the polyclonal proliferation assay, naïve CD8+ T-cells were labelled with 5 μM CellTrace Violet (CTV), washed, and activated with plate-bound anti-CD3 (1 μg/ml) and soluble CD28 (1 μg/ml) Abs for 48 and 72 h. Cell proliferation indicated by CTV dilution was detected by flow cytometry. For the cell apoptosis assay, cells were stained with Annexin V APC (BioLegend, San Diego, CA) according to the manufacturer’s protocol and analyzed by flow cytometry, as previously described (18). For the antigen-specific T-cell proliferation assay, 5 μM CTV labeled splenocytes (1 × 106/ml) from OT-I CK2αfl/fl or OT-I CK2α−/− mice were cultured in R10 medium and incubated with OVA257–264 peptide (0.1 ng/ml) for 48 h. CD8+ T-cell proliferation was detected by CTV dilution.
Flow Cytometry.
Cell surface staining and intracellular staining was performed as previously described (18). For cytokine production analysis, cells were stimulated with 50 ng/ml of PMA (Sigma-Aldrich, St. Louis, MO) and 750 ng/ml of ionomycin (Sigma-Aldrich, St. Louis, MO), or 50 ng/ml of OVA257–264 peptide, in the presence of GolgiStop (BD Biosciences, San Jose, CA) for 4 h. After surface staining, cells were fixed and permeabilized using Cytofix/Cytoperm Fixation/Permeabilization Solution Kits (BD Biosciences). For intracellular staining of transcription factors or CK2α, cells were fixed and permeabilized using the Foxp3 Staining Buffer Set (eBioscience, Grand Island, NY), as previously described (21). For phosphorylated protein detection, cells were fixed and permeabilized using Cytofix/Cytoperm Fixation/Permeabilization Solution Kits. For glucose uptake, cells were incubated with 2-NBDG (50 μg/ml) for 30 min at 37°C, washed, and stained for viability and surface markers (17). For MitoTracker Green (MTG) staining, cells were stained for viability and surface markers first, then washed and incubated with MTG (50 nM) for 30 min at 37°C. Samples were acquired on an LSRII flow cytometer using FACSDiva (BD Biosciences, San Jose, CA), and data were analyzed using FlowJo software (Tree Star, Inc, Ashland, OR). The following antibodies were used in this study (all Biolegend except where noted otherwise): anti-CD3ε PerCP-Cy5.5/PE-Cy7/APC (clone 145-2C11); anti-CD8 Pacific Blue/Alexa Fluor 488/PE-Cy7/BUV395 (clone 53–6.7); anti-TCR Vβ5.1/5.2 PerCP/Cyanine5.5 (clone MR9-4); anti-CD45.1 Alexa Fluor 488/PerCP-Cy5.5 (clone A20); anti-CD45.2 Alexa 647/APC-Cy7 (clone 104); anti-CD44 Alexa Fluor 488/APC/BV786 (clone IM7); anti-CD25 Alexa Fluor 647/PE-Cy7 (clone PC61.5); anti-CD69 PE (clone H1.2F3); anti-CD62L PE/Brilliant Violet 650 (clone MEL-14); anti-CD127 (IL-7Rα) Brilliant Violet 605 (clone A7R34); anti-KLRG1 Brilliant Violet 421 (clone 2F1/KLRG1); anti-CD98 Alexa Fluor 647 (clone RL388); anti-CD71 Brilliant Violet 421(clone C2); anti-IFN-γ Pacific Blue/PE-Cy7 (clone XMG1.2); anti-Granzyme B Pacific Blue (clone GB11); anti-IL-2 Brilliant Violet 421 (clone JES6-5H4); anti-Bcl2 PE (clone 3F11, BD Bioscience); anti-Tcf7 Brilliant Violet 421 (clone S33-966, BD Bioscience), anti-Bcl6 Alexa Fluor 647 (clone K112-91, BD Bioscience); Phospho-ERK1/2 (pT202/pY204) Alexa 488 (clone 20A; BD Bioscience); Phospho-Akt (Ser473) APC (clone D9E, Cell Signaling Technology); Phospho-S6 Ribosomal Protein (Ser235/236) Pacific Blue (clone D57.2.2E, Cell Signaling Technology); c-Myc Rabbit mAb (D84C12, CST); HIF-1α Rabbit mAb (CST); anti-CSNK2A1 (CK2α) (abcam); and Alexa Fluor 488 Anti-Rabbit IgG (H+L) (Jackson Immuno Research Labs).
Immunoblotting.
CD8+ T-cells were lysed in buffer containing 1% Triton X-100 (Sigma- Aldrich), protein lysates separated by electrophoresis, transferred to a nitrocellulose membrane, and then blotted with CK2α (ab76040), CK2β (ab76025) (Abcam, Cambridge, MA), CK2α’ (sc-514403) (Santa Cruz Biotechnology, Dallas, TX) and β-Actin (Sigma-Aldrich, St. Louis, MO) Abs, as previously described (26).
CK2 Kinase Activity.
The Casein Kinase 2 Assay Kit (Millipore) was used to assess CK2 kinase activity. Cells were lysed, and both catalytic subunits (CK2α and CK2α’) were immunoprecipitated. The lysates were assayed for CK2 kinase activity as previously described (17).
Extracellular Flux Assay.
Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) measurements were performed using the Seahorse XF96 analyzer (Agilent Technologies, Santa Clara, CA). Naive CK2αfl/fl or CK2α−/− CD8+ T-cells were activated with plate-bound anti-CD3 (1 μg/ml) and soluble CD28 (1 μg/ml) Abs for 24, 48 and 72 h. For the glycolytic stress test (ECAR), naïve and activated cells were plated at 2 × 105 cells/well into a XF96 plate coated with Cell-Tak (Corning) and resuspended in assay medium (Seahorse basic DMEM supplemented with 1 mM Pyruvate, 2 mM Glutamine, and 5 mM HEPES, pH 7.4). The plated cells were maintained in a non-CO2 incubator at 37°C for 1 h prior to the assay. The glycolysis stress test was conducted by subsequent injections of glucose (10 mM), oligomycin (1 μg/ml), antimycin A (10 μM) and 2-deoxyglucose (2-DG; 50 mM) as described in (27). The values of the indicated parameters were calculated as: glycolysis = ECAR postglucose − ECAR basal and glycolytic capacity = ECAR post-AA − ECAR post-2-DG). For the mitochondrial stress test (OCR), cells were plated at 2 × 105/well into a XF96 plate coated with cell-tak, and switched to XF media (DMEM supplemented with 5.5 mM Glucose, 1 mM Pyruvate, 2 mM Glutamine, and 5 mM HEPES, pH 7.35). OCR was measured under basal conditions and upon the sequential addition of oligomycin (1 μg/ml), fluorocarbonyl cyanide phenylhydrazone (FCCP; 2 μM), and antimycin A (10 μM), to determine the mitochondrial respiration of cells. The value of the indicated parameters were calculated as: basal OCR = OCR initial − OCR post–AA; maximal respiration = OCR post-FCCP − OCR post–AA; ATP-linked respiration = OCR basal − OCR post-oligomycin; Proton leak = OCR post-oligomycin − OCR post-AA; and Reserve capacity = OCR max − OCR basal (28). For the ex vivo assay, OT-I CK2αfl/fl and OT-I CK2α−/− CD8+ T-cells were sorted from the spleen at day 7 post infection, then the mitochondrial stress test was performed as described above.
Quantification of Mitochondrial DNA (mtDNA).
Total DNA was extracted from CD8+ T-cells. The mitochondrial small fragment (mtDNA) and nuclear 18S DNA were amplified using RT Real-Time SYBR Green PCR master mix (Fisher Scientific) in an ABI 7500. The primer sequences used for mtDNA were mtF (5′-CCCCAGCCATAACACAGTATCAAAC-3′) and mtR (5′- GCCCAAAGAATCAGAACAGATGC-3′). The primer sequences for nuclear DNA were 18SF (5′-AAACGGCTACCACATCCAAG-3′) and 18SR (5′-CAATTACAGGGCCTCGAAAG-3′). mtDNA copy number was normalized to amplification of an 18S nuclear amplicon (29).
RNA Isolation, RNA Sequencing, and Quantitative RT-PCR.
RNA sequencing was performed as previously described (17). Briefly, OT-I CK2αfl/fl and OT-I CK2α−/− CD8+ T-cells were sorted from the spleen at day 7 post infection. Total RNA was extracted from FACS-sorted CD8+ T-cells using the miRNeasy Mini Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s protocols and submitted to GENEWIZ (South Plainfield, NJ) for RNA sequencing (RNA-seq) and bioinformatics analysis. RNA sequencing data was submitted to the Gene Expression Omnibus (GEO) Repository under accession number GSE175941 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175941). Genes with an FDR less than 0.05 with a fold change more than 1.5 were considered as differentially expressed genes (DEG). Further pathway analysis was performed by Gene Set Enrichment Analysis (GSEA) available through the Broad Institute.
For quantitative RT-PCR (qRT-PCR) analysis, 500–1000 ng of RNA was reverse transcribed into cDNA using M-MLV Reverse Transcriptase (Promega) as previously described (17, 18). cDNA was subjected to qRT-PCR using TaqMan primers (Thermo Fisher Scientific). Relative gene expression was calculated according to the ΔΔ threshold cycle (Ct) method.
Statistics.
Levels of significance for comparison between two groups was determined by Student’s t test distribution. Multiple comparisons were performed by ordinary one-way analysis of variance. The p values are indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All error bars represent mean ± SD. All statistical analyses (excluding RNA-seq, described above) were performed using Prism software (GraphPad).
RESULTS
CK2 Subunit Protein Expression and Kinase Activity are Induced upon CD8+ T-cell Activation.
We first determined the expression patterns of CK2α and CK2α’, the catalytic subunits of CK2, and CK2β, the regulatory subunit of CK2, in naïve and activated primary murine CD8+ T-cells. Expression of CK2α, CK2α’ and CK2β was induced post activation with anti-CD3 and anti-CD28 Abs, and increased with time (Fig. 1A). The expression of CK2α was confirmed by flow cytometry at 24 h (Fig. 1B). Most importantly, CK2 kinase activity was enhanced upon activation with anti-CD3 and anti-CD28 Abs (Fig. 1C). These results demonstrate the induction of CK2 expression and kinase activity upon CD8+ T-cell activation, which indicates a potential role of CK2 in CD8+ T-cell responses.
Figure 1. CK2 Subunit Protein Expression and Kinase Activity are Induced in CD8+ T-cells upon Activation, and CK2α is Required for CD8+ T-cell Activation and Proliferation.
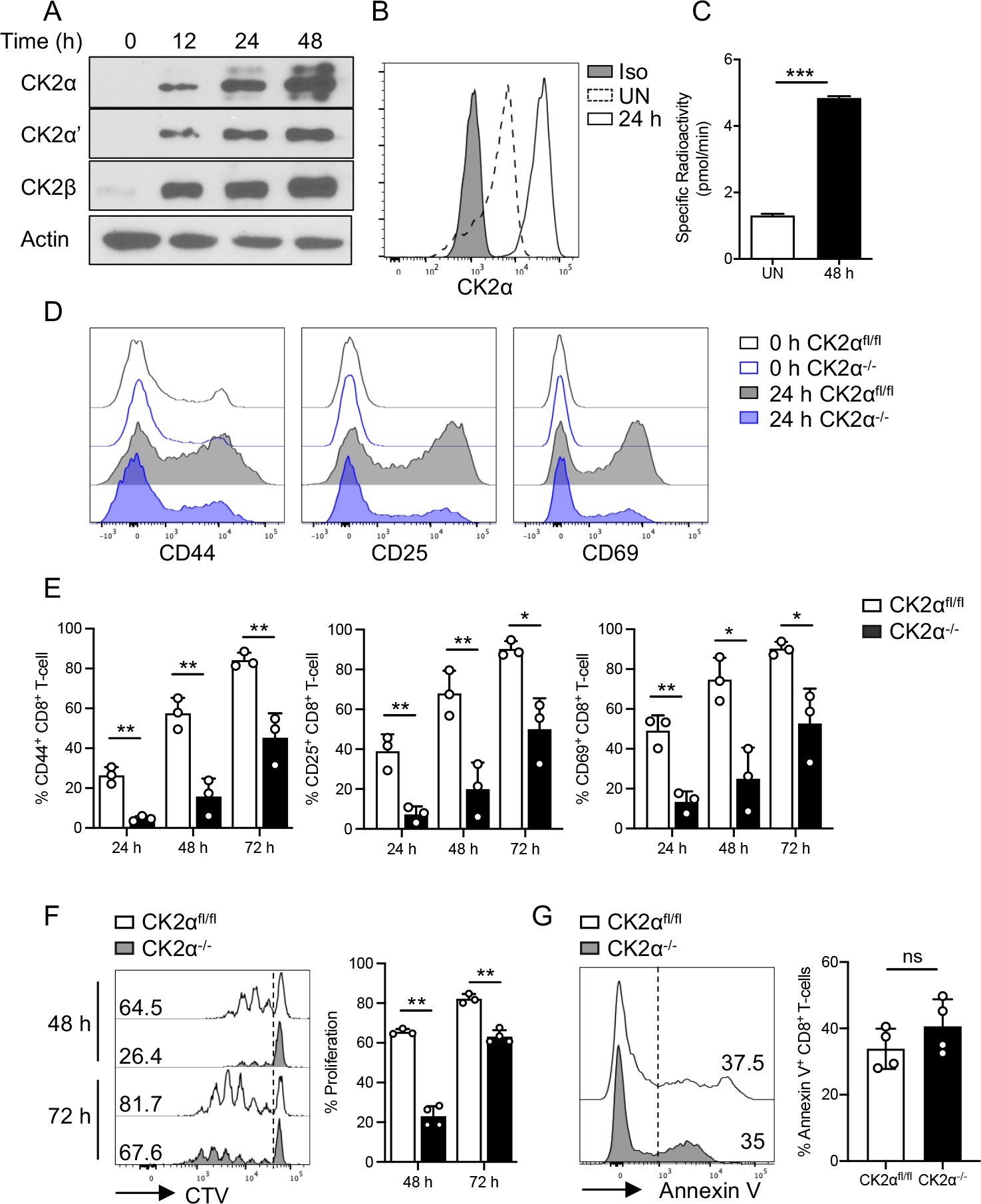
Naïve CD8+ T-cells were enriched from the spleen of C57BL/6 mice and activated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (1 μg/ml) Abs for the indicated time points. (A) Expression of CK2 subunits CK2α, CK2α’ and CK2β was detected by immunoblotting at the indicated time points. Actin serves as a loading control. (B) CK2α expression was detected by flow cytometry at 24 h under unstimulated conditions (UN) and with anti-CD3 and anti-CD28 Abs treatment. (C) CK2 kinase activity was measured at 48 h by the Casein Kinase 2 Assay Kit. (D) Naïve CD8+ T-cells from CK2αfl/fl and CK2α−/− mice were activated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (1 μg/ml) Abs for 24, 48 and 72 h. Expression of CD44, CD25 and CD69 was detected by flow cytometry. Representative line graphs of 24 h are shown. (E) Percentages of CD44, CD25 and CD69 positive cells at the indicated time points are shown. (F) Naïve CD8+ T-cells were labeled with 5 μM CTV, then activated with anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml) Abs for 48 and 72 h, and proliferation assessed by CTV dilution. Representative line graphs are shown. (G) Dead and apoptotic cells were assessed by Annexin V staining at 48 h. Representative line graphs are shown. n = 3 in each group. Each experiment was performed two times with three biological replicates per experiment. Bars represent mean ± SD. * p< 0.05, ** p< 0.01, *** p< 0.001. ns = not significant.
CK2α Kinase Activity is Required for CD8+ T-cell Activation and Proliferation.
To study the function of CK2α in T-cells, we generated transgenic mice in which CK2α was specifically deleted in both CD4+ and CD8+ T-cells, as described previously (21). CK2α protein was undetectable in CK2α−/− CD8+ T-cells upon TCR stimulation (Sup. Fig. 1A). Although CK2α−/− mice have normal immune phenotypes with no signs of systemic or organ-specific inflammation (21), there was a decrease in the percentage of total leukocytes and absolute number of CD8+ T-cells in the spleen but not lymph nodes (LNs) compared with CK2αfl/fl mice (Sup. Fig. 1B). In addition, CK2α−/− mice had an increase in naïve CD8+ T-cells (CD44−CD62L+) in the spleen, no change in the LNs, and a decrease in central memory CD8+ T-cells (Tcm, CD44+CD62L+), but no change of effector memory CD8+ T-cells (Tem, CD44+CD62L−) in the spleen and LNs compared with CK2αfl/fl mice (Sup. Figs. 1C and 1D).
Next, we sought to determine the contribution of CK2α to CD8+ T-cell activation. Upon anti-CD3 and anti-CD28 stimulation, CK2α−/− CD8+ T-cells exhibited significantly lower expression levels of CD44, CD25 and CD69 compared with CK2αfl/fl CD8+ T-cells (Figs. 1D and 1E). We next examined the role of CK2 in proliferation and cell survival in vitro. CD8+ T-cells from CK2αfl/fl and CK2α−/− mice were labeled with CellTrace Violet (CTV) dye followed by anti-CD3 and anti-CD28 activation. Interestingly, CK2α−/− CD8+ T-cells exhibited a significant decrease in proliferation, as determined by the frequency of cells undergoing more than two divisions at both 48 and 72 h (Fig. 1F). When we analyzed the survival of CK2αfl/fl and CK2α−/− CD8+ T-cells by Annexin V staining, no difference was observed (Fig. 1G). These findings indicate that CK2α regulates CD8+ T-cell activation and proliferation but does not affect cell survival.
CK2α is Required for CD8+ T-cell Glycolysis and Mitochondrial Respiration Upon Activation In Vitro.
Following TCR activation, T-cells undergo substantial metabolic reprogramming, specifically increases in glucose uptake and metabolism and mitochondrial function (30). To determine whether CK2α is involved in CD8+ T-cell glycolytic and mitochondrial metabolic switch during activation, we first examined the function of CK2α on glycolysis. mRNA levels of glucose transporter 1 (Scl2a1), glucose transporter 3 (Scl2a3) and hexokinase 2 (Hk2) were upregulated in CK2αfl/fl CD8+ T-cells upon TCR stimulation, while expression levels of these genes were significantly lower in CK2α−/− CD8+ T-cells (Fig. 2A). Consistent with these findings, CK2α−/− CD8+ T-cells had a decreased ability to uptake glucose as measured by 2-NBDG (Fig. 2B). Furthermore, we measured the glycolytic metabolism of naïve and activated CD8+ T-cells via extracellular flux analysis. After glucose supplementation, activated CK2αfl/fl CD8+ T-cells promptly engaged glycolysis, as manifested by a sharp increase in the extracellular acidification rate (ECAR), which was further enhanced by adding oligomycin and antimycin A. Activated CK2α−/− CD8+ T-cells had significantly lower responses than CK2αfl/fl CD8+ T-cells (Figs. 2C and 2D, Sup. Figs. 2A and 2B). These findings indicate that activated CK2α−/− CD8+ T-cells show less glycolytic capacity than activated CK2αfl/fl CD8+ T-cells, while there was no difference between naïve CK2αfl/fl and CK2α−/− CD8+ T-cells.
Figure 2. CK2α is Required for CD8+ T-cell Glycolysis and Mitochondrial Respiration In Vitro.
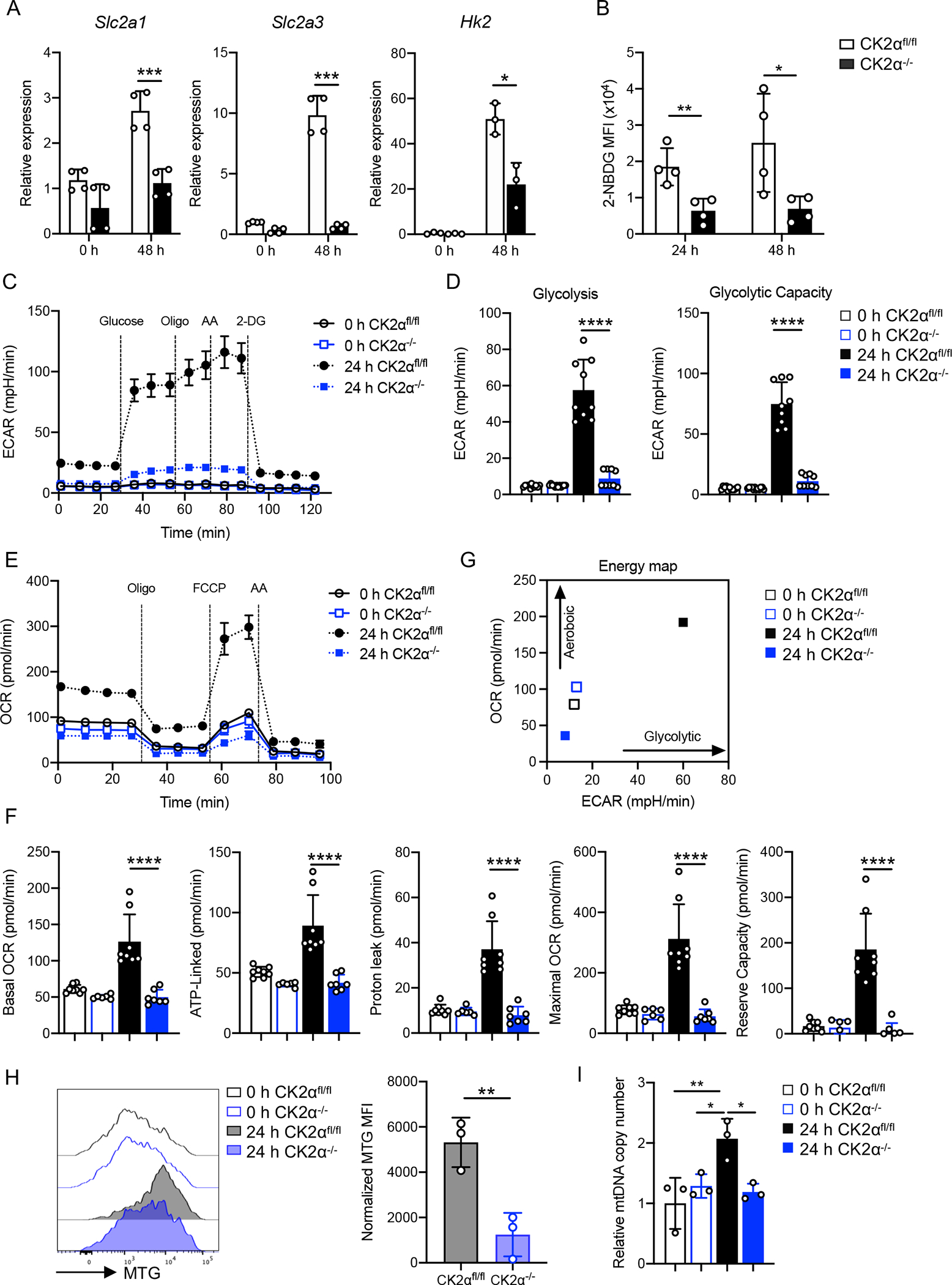
Naïve CD8+ T-cells from CK2αfl/fl and CK2α−/− mice were activated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (1 μg/ml) Abs for 24 or 48 h. (A) Gene expression levels of Glut 1 (Slc2a1), Glut 3 (Slc2a3) and Hk2 were detected by qPCR at 48 h. (B) Glucose update was measured by 2-NBDG uptake. n = 3–4 in each group. (C) Glycolysis was measured using extracellular acidification rate (ECAR) with injections of glucose (10 mM), oligomycin (1 μg/ml), antimycin A (10 μM) and 2-DG (50 mM). (D) ECAR values were pooled from two independent glycolysis stress tests (glycolysis =ECARpostglucose− ECARbasal; glycolytic capacity = ECARpost-AA−ECARpost-2-DG). (E) Oxidative phosphorylation profiles were measured by the oxygen consumption rate (OCR) with injections of oligomycin (1 μg/ml), FCCP (2 μM) and antimycin A/rotenone (AA) (10 μM/1 μM). (F) Calculation of basal OCR, ATP-linked, proton-leak, maximal OCR and reserve respiration capacity. (G) Energy map of CD8+ T-cells at basal OCR and ECAR condition. Data pooled from three independent mitochondrial stress tests. (H) Mitochondrial content of CD8+ T-cells was assessed by flow cytometry with MitoTracker Green (MTG); n = 3 in each group. MFI was normalized to corresponding untreated controls. (I) Mitochondrial DNA copy numbers relative to genomic DNA measured using qPCR; n = 3 in each group. Each experiment was performed two times with three to four technical replicates per experiment. Bars represent the mean ± SD. * p< 0.05, ** p< 0.01, *** p< 0.001 **** p< 0.0001.
Activated T-cells show a considerable increase in mitochondrial respiration (31). Next, we sought to test whether CK2α was involved in mitochondrial respiration in CD8+ T-cells by analyzing the oxygen consumption rate (OCR). Activated CK2α−/− CD8+ T-cells showed significantly lower levels of basal OCR, ATP-linked, proton leak, maximal and reserve capacity OCR compared to activated CK2αfl/fl CD8+ T-cells, while no significant difference was observed between naïve CK2αfl/fl and CK2α−/− CD8+ T-cells (Figs. 2E and 2F, Sup. Figs. 2C and 2D). CK2α−/− CD8+ T-cells failed to complete metabolic reprogramming to achieve the aerobic glycolytic phenotype characteristic of T-cell activation, as shown by the energy map (Fig. 2G and Sup. Fig. 2E). Mitochondrial mass and mtDNA also increase during T-cell activation (32). We next sought to define whether CK2α regulated mitochondrial mass and mtDNA during activation. Our data demonstrated that activated CK2α−/− CD8+ T-cells had significantly less mitochondrial mass as indicated by staining with Mitotracker Green (MTG) compared to activated CK2αfl/fl CD8+ T-cells (Fig. 2H). Also, CK2α−/− CD8+ T-cells had less mitochondrial DNA compared to activated CK2αfl/fl CD8+ T-cells (Fig. 2I). These findings support the involvement of CK2α in CD8+ T-cell metabolic reprogramming during TCR activation.
CK2α Controls CD8+ T-cell Metabolic Reprogramming by Regulating the AKT/mTOR Pathway.
Our previous studies showed that CK2α regulated AKT/mTOR activity in CD4+ T-cells (17, 21). mTOR is a protein kinase that acts as a central integrator of various environmental cues and is able to regulate multiple cellular processes accordingly (33). As the mTOR pathway is a major pathway regulating T-cell metabolism (33), we sought to test whether CK2α regulated CD8+ T-cell metabolism through mTOR signaling. Expression levels of mTORC1-dependent cell surface markers CD71 and CD98 were significantly decreased in CK2α−/− CD8+ T-cells compared to CK2αfl/fl CD8+ T-cells upon activation (Figs. 3A and 3B). In addition, phosphorylation of the downstream target of mTORC1, ribosomal protein S6, was decreased in CK2α−/− CD8+ T-cells compared with CK2αfl/fl CD8+ T-cells (Fig. 3C). CK2α−/−CD8+ T-cells also exhibited a significant decrease in phosphorylation of Akt at S473, which is essential for Akt activity (34) (Fig. 3D). It is well known that mTOR signaling pathways modulate T-cell metabolism through controlling hypoxia inducible factor-1 alpha (HIF-1α) and c-Myc (35, 36). HIF-1α and c-Myc promote and regulate glycolysis and mitochondrial biogenesis (35). CK2α−/− CD8+ T-cells showed significant decreases in HIF-1α and c-Myc compared with CK2αfl/fl CD8+ T-cells upon activation (Figs. 3E and 3F). Furthermore, phosphorylation levels of ERK1/2 in CK2α−/− CD8+ T-cells were significantly lower compared with CK2αfl/fl CD8+ T-cells (Fig. 3G), which indicates that CK2 may control CD8+ T-cell activation by regulating ERK1/2 activation. Altogether, these findings suggest that CK2α plays an important role in regulating CD8+ T-cell metabolism reprogramming through promoting AKT/mTOR and ERK1/2 signaling and regulating HIF-1α and c-Myc expression in CD8+ T-cells upon activation.
Figure 3. CK2α Controls CD8+ T-cell Metabolic Reprogramming by Regulating the AKT/mTOR Pathway.
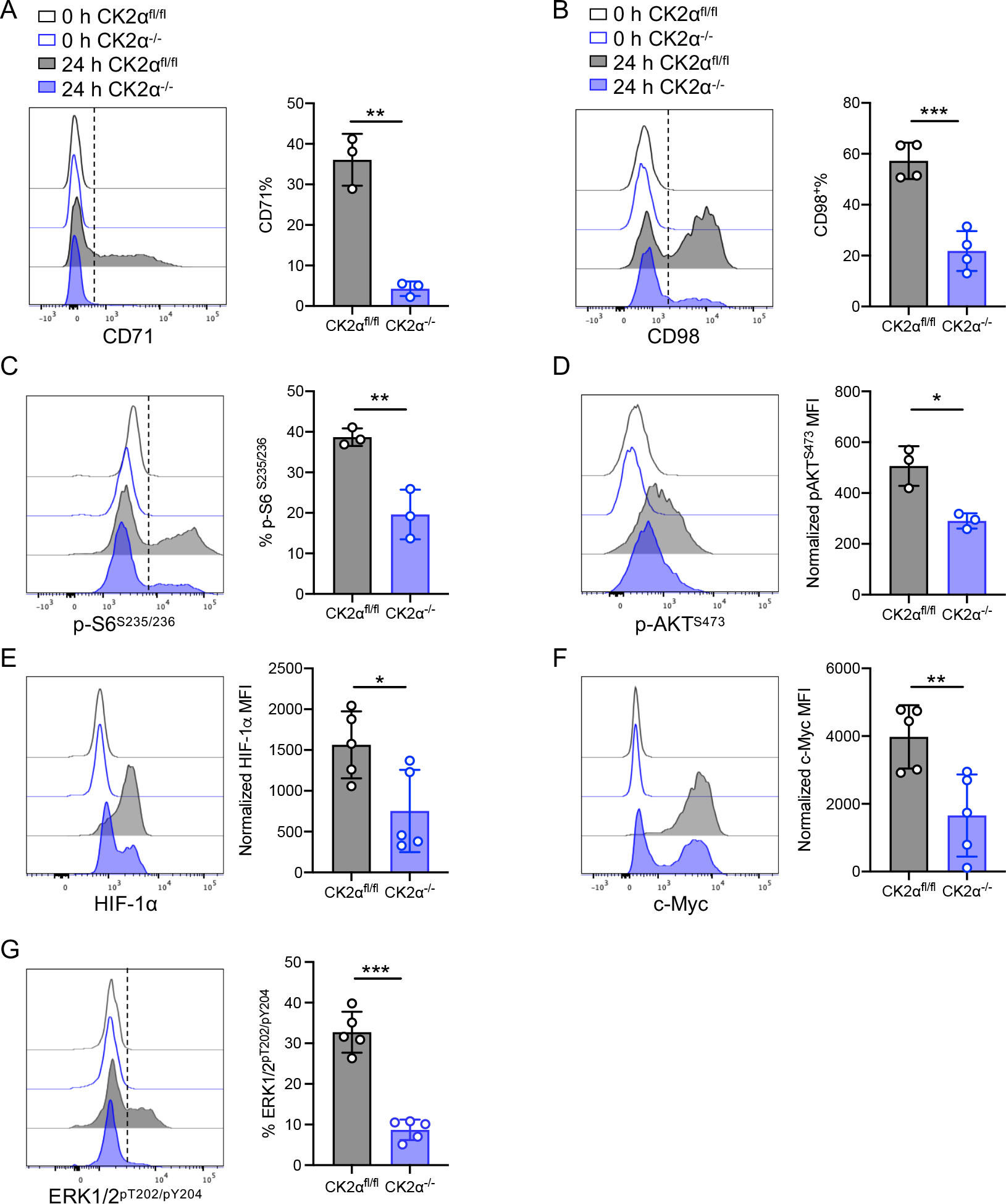
Naïve CD8+ T-cells from CK2αfl/fl and CK2α−/− mice were activated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (1 μg/ml) Abs for 24 h. (A) CD71 and (B) CD98 expression as detected by flow cytometry is shown. (C) Phosphorylated S6 ribosomal protein S235/236 and (D) AKT S473 were detected by flow cytometry. (E) HIF-1α, (F) c-Myc, and (G) ERK1/2pT202/pY204 expression was detected by flow cytometry. n = 3–5 in each group. Each experiment was performed two times with two to three biological replicates per experiment, each dot indicates one mouse. Bars represent the mean ± SD. * p< 0.05, ** p< 0.01, *** p< 0.001.
CK2α is Vital for CD8+ T-cell Primary Responses in LM-OVA Infection.
To determine the function of CK2α in regulating CD8+ T-cell responses to infection, we crossed CK2α−/− mice to OT-I TCR transgenic mice, whose TCR recognizes the SIINFEKL epitope from chicken ovalbumin (OVA) (37). Similar to the phenotype observed in polyclonal CD8+ T-cells, CK2α−/− OT-I cells exhibited significantly lower expression levels of CD44, CD25 and CD69 compared with CK2αfl/fl OT-I cells upon OVA257–263 peptide stimulation (Sup. Figs. 3A and 3B). In addition, CK2α deficiency impaired OT-I cell proliferation (Sup. Fig. 3C).
We transferred CD45.2 CK2αfl/fl or CK2α−/− OT-I CD8+ T-cells into CD45.1 congenic C57BL/6 recipients, followed by infection with Listeria monocytogenes expressing OVA (LM-OVA), and analyzed CD8+ T-cell responses at day 7 post-infection (Fig. 4A). The frequencies of CK2α−/− OT-I cells in the peripheral blood and spleen were significantly lower than CK2αfl/fl OT-I cells, as were the absolute number of CK2α−/− OT-I cells in the spleen compared with CK2αfl/fl OT-I cells (Figs. 4B and 4C). We next examined whether CK2α deficiency affected CD8+ T-cell differentiation. CD62L and KLRG1 expression was used to classify different subsets of CD8+ T-cells into CD62L+ central memory cells (Tcm), CD62L−KLRG1− effector cells (Eff) and KLRG1+ terminally differentiated effector (TD) cells (3). We observed a decrease in TD KLRG1+ cells and an increase in CD62L+ Tcm cells in CK2α−/− OT-I cells compared to CK2αfl/fl OT-I cells at day 7 after infection (Fig. 4D). No change was observed in Eff cells (Fig. 4D). Expression of the cytotoxic related molecule granzyme B was significantly reduced in CK2α−/− OT-I cells (Fig. 4E). Furthermore, upon restimulation with OVA peptide, CK2α−/− OT-I cells produced similar levels of IFN-γ, but increased IL-2 production compared to CK2αfl/fl OT-I cells (Fig. 4F). In particular, there was an increase in IFN-γ+IL-2+ producing cells in CK2α−/− OT-I cells, which suggests that CK2α−/− OT-I cells have more potential to develop into memory cells (38).
Figure 4. CK2α Promotes CD8+ T-cell Effector Responses during LM-OVA Infection.
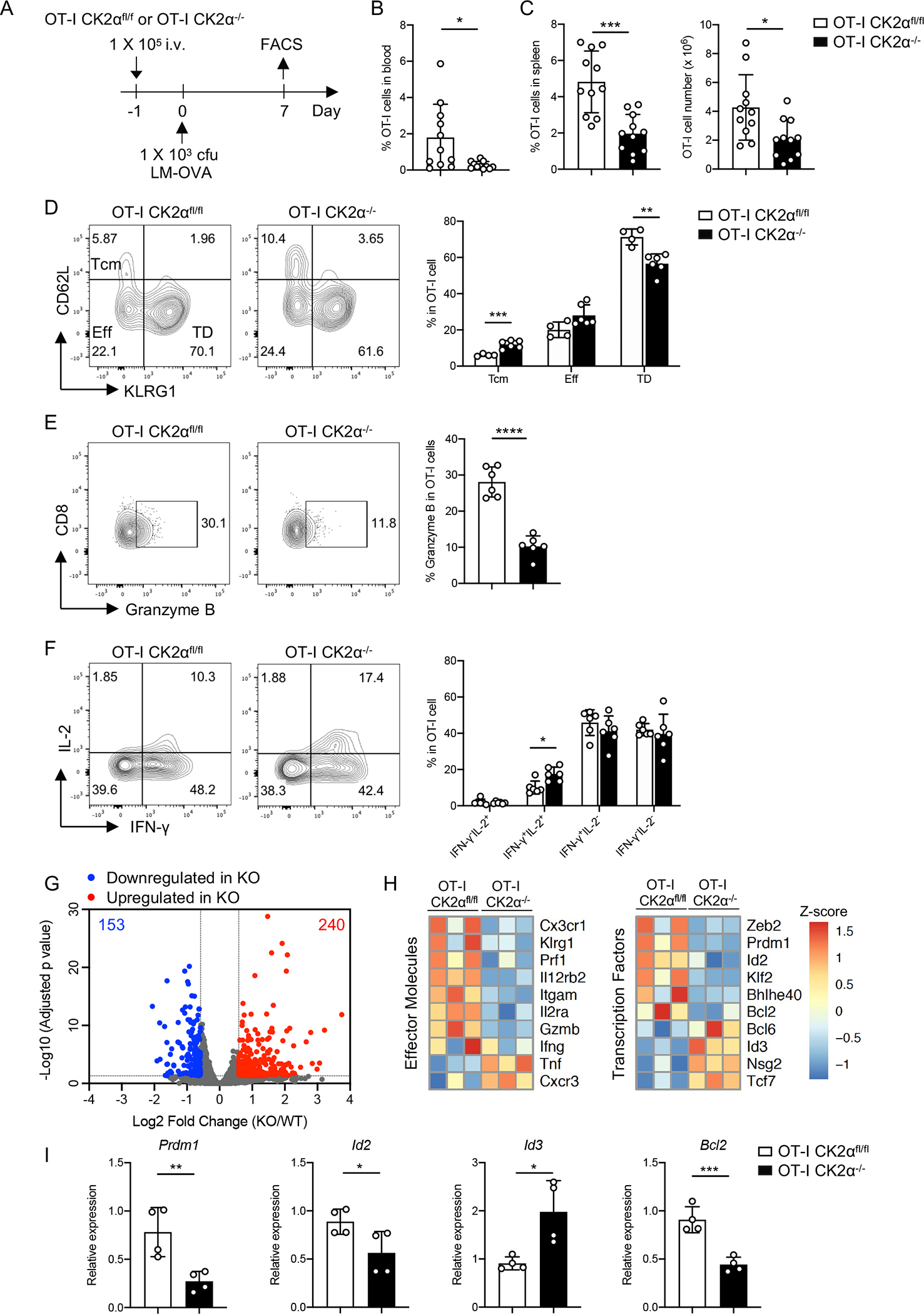
(A) One × 105 naïve CD8+ T-cells (CD45.2+) from OT-I CK2αfl/fl or OT-I CK2α−/− mice were adoptively transferred into age- and sex-matched CD45.1 C57BL/6 mice. Twenty-four hour later, recipients were infected with 1 × 103 cfu of LM-OVA by i.v. injection. Donor CD8+ T-cells from the blood and spleen were analyzed by flow cytometry at day 7. (B) The percentages of OT-I CK2αfl/fl or OT-I CK2α−/− derived CD8+ T-cells in total leukocytes in the blood of recipients at day 7 post-infection are shown. (C) The percentages and numbers of donor CD8+ T-cells in the spleen at day 7 post infection are shown. n=12 in each group. (D) Central memory cells (Tcm), effector cells (Eff) and terminally differentiated effector cells (TD) were detected by CD62L and KLRG1 staining of donor CD8+ T-cells. (E) Granzyme B expression was detected in donor CD8+ T-cells by intracellular staining. (F) IFN-γ and IL-2 expression was detected in donor CD8+ T-cells by intracellular staining. Quantitation of IFN-γ−IL-2+, IFN-γ+IL-2+, IFN-γ+IL-2− and IFN-γ−IL-2− is shown. n = 4–6 in each group. (G) RNA sequencing of OT-I CK2αfl/fl and OT-I CK2α−/− CD8+ T-cells from the spleen at day 7 post infection. Summary of genes differentially regulated by CK2α using the following cutoffs are shown: p < 0.05, fold change >1.5. (H) Heat map shows relative gene expression. (I) Real-time PCR analysis of the indicated genes. n=4 in each group. Each experiment was performed at least two times with two to four biological replicates per experiment, each dot indicates one mouse. Bars represent mean ± SD. * p< 0.05, ** p< 0.01, *** p< 0.001, **** p< 0.0001.
To identify CK2α-dependent transcriptional programs in CD8+ T-cells, CK2αfl/fl and CK2α−/− OT-I cells from recipient mice were sorted at day 7 after LM-OVA infection and subjected to RNA sequencing. Using a setting of p < 0.05 and fold change > 1.5 to define significant differences in DEGs, 153 genes were upregulated and 240 genes downregulated as a result of CK2α deficiency (Fig. 4G). CK2α−/− OT-I cells showed decreased expression of effector function related molecules, including Klgr1, Prf1, Gzmb and IL12rb2 (Fig. 4H). These data are consistent with the flow staining analyses (Figs. 4D and 4E). Furthermore, CK2α−/− OT-I cells showed a memory-like transcriptional signature (39), as evidenced by the downregulation of genes that regulate effector cell differentiation, including Zeb2, Prdm1, Id2 and Bcl2, and upregulation of genes that control memory cell development, including Id3, Nsg2 and Tcf7 (Fig. 4H). The expression of select genes was validated by qPCR (Fig. 4I).
We noticed that CK2α−/− OT-I cells contain twice as many CD62L+ (TCM) and fewer KLGR1+ cells (TD) compared with CK2αfl/fl OT-I cells (Fig. 4D). To rule out the possibility that the RNA-Seq data was only reflective of population differences, we verified certain key genes identified in Fig. 4H at day 5 post infection (Sup. Fig. 4), which is early effector phase with fewer KLRG1+ cells. Consistent with day 7, the frequencies and absolute number of CK2α−/− OT-I cells in the spleen were significantly lower than CK2αfl/fl OT-I cells at day 5 post-infection (Sup. Fig. 4B). Importantly, expression of the activation marker CD25 and effector marker KLRG1 was significantly lower on CK2α−/− OT-I cells compared with CK2αfl/fl OT-I cells (Sup. Figs. 4C and 4D). Expression of granzyme B was significantly reduced in CK2α−/− OT-I cells (Sup. Fig. 4E), while IFN-γ producing cells were comparable between CK2α−/− OT-I and CK2αfl/fl OT-I cells (Sup. Fig. 4F). Moreover, transcription factors Tcf7 and Bcl6, which controls memory cell development, were significantly increased in CK2α−/− OT-I cells (Sup. Figs. 4G and 4H). Taken together, our data verified that CK2α is also critical for the CD8+ T-cell response in the early effector phase, and it is more likely our RNA-Seq data reflects the key gene differences derived from T-cell programing rather than population changes.
Furthermore, expression of the pro-survival protein Bcl2 in CK2αfl/fl OT-I and CK2α−/− OT-I cells was detected by intracellular staining at day 7 post-infection, showing that the expression level of Bcl2 in CK2α−/− OT-I cells was significantly lower compared to CK2αfl/fl OT-I cells (Sup. Fig. 4I). We used Annexin V and Live/Dead co-staining to detect live, early apoptotic and late apoptotic cells. The percentage of late apoptotic cells among CK2α−/− OT-I cells was significantly higher compared to CK2αfl/fl OT-I cells (Sup. Fig. 4J). These data indicate CK2 controls CD8+ T-cell cellularity by regulating cell survival. Furthermore, gene set enrichment analysis (GSEA) based on the RNA sequencing data revealed that differentially expressed genes are associated with mitochondrial proteins, including components of the respiratory chain and oxidoreductase complexes (Sup. Fig. 4K). It is known that memory CD8+ T-cells exhibit enhanced oxidative phosphorylation and fatty acid oxidation compared to effector CD8+ T-cells (40). Our extracellular flux assays show that CK2α−/− OT-I cells have a higher basal and ATP-linked OCR compared to CK2αfl/fl OT-I cells (Sup. Fig. 4L), which suggest that CK2α regulates CD8+ T-cell function in a manner that engages mitochondrial metabolism during infection. Taken together, our findings demonstrate that deletion of CK2α in CD8+ T-cells affects cell expansion, differentiation and effector functions in response to Listeria monocytogenes infection.
CK2α Promotes Memory CD8+ T-cell Maintenance and Function during LM-OVA Infection.
To investigate the role of CK2α in the control of memory T-cell differentiation, we assessed the CD8+ T-cell response in the spleen at day 42 post-infection (Fig. 5A). The percentage and absolute number of CK2α−/− OT-I cells were significantly lower compared to CK2αfl/fl OT-I cells (Fig. 5B). Interestingly, no difference was detected in the differentiation of CD8+ T-cell subsets (Fig. 5C). As for cytokine production capacity, IFN-γ production by CK2α−/− OT-I cells was significantly lower than CK2αfl/fl OT-I cells upon restimulation with OVA peptide (Fig. 5D), but no difference was observed in IL-2 production (data not shown). Expression of Bcl2 was significantly decreased in CK2α−/− OT-I compared to CK2αfl/fl OT-I cells (Fig. 5E). These findings suggest that CK2α is required for maintenance of the memory CD8+ T-cell pool.
Figure 5. CK2α is Required for CD8+ T-cell Memory Maintenance and Function.
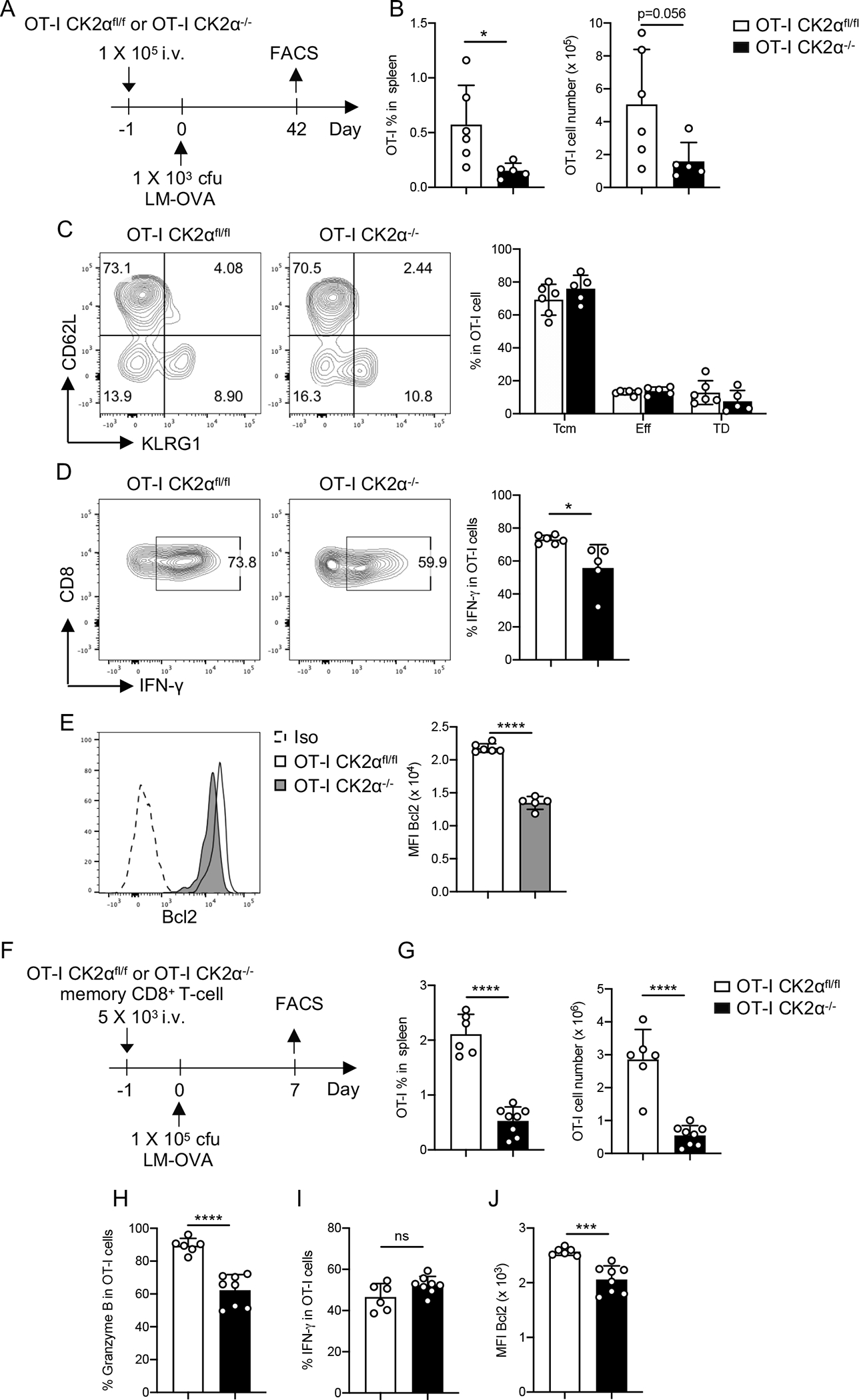
(A) One × 105 naïve CD8+ T-cells (CD45.2+) from OT-I CK2αfl/fl or OT-I CK2α−/− mice were adoptively transferred into age- and sex-matched CD45.1 C57BL/6 mice. Twenty-four hour later, recipients were infected with 1 × 103 cfu of LM-OVA by i.v. injection. Donor CD8+ T-cells from the spleen were analyzed by flow cytometry at day 42. (B) The percentages and numbers of donor CD8+ T-cells in the spleen at day 42 post-infection are shown. (C) Central memory cells (Tcm), effector cells (Eff) and terminally differentiated effector cells (TD) were detected by CD62L and KLRG1 staining of donor CD8+ T-cells. (D) IFN-γ production was detected in donor CD8+ T-cells by intracellular staining. (E) Bcl2 expression was detected in donor CD8+ T-cells by intracellular staining. n = 5–6 in each group (F) Five × 103 memory CD8+ T-cells (CD8+ CD45.2+ Vα2+ CD44+ CD62L+) from OT-I CK2αfl/fl or OT-I CK2α−/− mice were adoptively transferred into CD45.1 C57BL/6 mice. One day later, recipients were infected with 1 × 105 cfu of LM-OVA by i.v. injection. Secondary expansion of memory CD8+ T-cells in the spleens was detected at day 7. (G) Percentages and absolute numbers of OT-I CK2αfl/fl or OT-I CK2α−/− CD8+ T-cells in the spleen were analyzed by flow cytometry at day 7. (H) Granzyme B, (I) IFN-γ and (J) Bcl2 expression in OT-I CK2αfl/fl or OT-I CK2α−/− CD8+ T-cells was detected by intracellular staining. n = 6–8 in each group. Each experiment was performed at least two times with three or four biological replicates per experiment, each dot indicates one mouse. Bars represent mean ± SD. * p< 0.05, *** p< 0.001, **** p< 0.0001. ns = not significant.
We next sought to assess the function of CK2α in the quality of CD8+ T-cell memory in secondary responses. We transferred the same number of sorted CK2αfl/fl OT-I or CK2α−/− OT-I memory cells at day 42 post-infection into naïve CD45.1 congenic recipients (Fig. 5F). After infection with 1 × 105 cfu LM-OVA, secondary proliferation of CK2α−/− OT-I cells was impaired in the spleen (Fig. 5G). Expression of granzyme B was decreased in CK2α−/− OT-I cells compared to CK2αfl/fl OT-I cells (Fig. 5H), while no difference was observed in IFN-γ production (Fig. 5I). Bcl2 expression in CK2α−/− OT-I cells was significantly lower compared to CK2αfl/fl OT-I cells (Fig. 5J). These data suggest that CK2α is critical for the fitness of memory CD8+ T-cells, not only in the maintenance of cell number but also for their expansion to secondary challenge.
DISCUSSION
Herein, we identify a critical role for CK2α in regulating CD8+ T-cell activation, proliferation, differentiation and function both in vitro and in vivo. CK2 kinase activity is induced in CD8+ T-cells upon TCR stimulation and is required for CD8+ T-cell activation and proliferation in vitro. To explore the function of CK2 in CD8+ T-cell responses in vivo, we introduced a mouse model to abrogate CK2α expression in antigen-specific CD8+ T-cells in response to LM-OVA infection. Our findings demonstrate that CK2α−/− mice develop significantly less effector and memory T-cells, and have impaired memory responses against bacterial rechallenge. This study is the first to examine and define the function of CK2α in CD8+ T-cell primary responses and memory formation.
Our previous findings identified that treatment with CX-4945, a small molecule inhibitor of CK2, results in a shift from a CD4+ Th17-associated glycolytic metabolic gene expression profile to expression of genes associated with oxidative metabolism, preferred by Tregs in vitro (17). These findings suggest that CK2α controls CD4+ T-cell fate by regulating the metabolic switch enabling aerobic glycolysis (17). In this study, we demonstrate that CK2α is involved in CD8+ T-cell metabolic reprogramming upon TCR stimulation by regulating both glycolysis and mitochondrial respiration. CK2α regulates CD8+ T-cell glycolysis by controlling the expression of glucose transporters and key enzymes of the glycolytic pathway, which are important for glucose uptake. Mechanistically, CK2α controls metabolic reprogramming in CD8+ T-cells through regulating the activity of AKT/mTOR, ERK1/2 and c-Myc, which are the primary regulators of early metabolic changes associated with T-cell activation and differentiation (35, 36, 41). However, the ex vivo flux assay showed that CK2α-deficient CD8+ T-cell exhibited enhanced mitochondrial function. The explanation for this inconsistency in CK2 function on CD8+ T-cell metabolism between in vitro and ex vivo is not clear. One possibility is the fact that the population of cells generated in vivo were exposed to asynchronous stimulation over a different time course compared to in vitro activation. Although the exact role of CK2α in CD8+ T-cell metabolism needs to be explored further, our findings suggest that CK2α is involved in T-cell metabolic reprogramming, which is important for CD8+ T-cell function and differentiation.
Previous studies identified a transcriptional network regulating effector to memory CD8+ T-cell development. Expression or activation of T-bet, Blimp-1, Id2 and STAT4 drives CD8+ T-cell terminal differentiation, while the expression of Eomes, Bcl-6, Id3 and STAT3 promotes memory CD8+ T-cell formation (3, 42–46). In our study, we found that CD8+ T-cells developed into more central memory CD8+ T-cells in the absence of CK2α, accompanied by a decrease in granzyme B expression and an increase in IL-2 production. These findings indicate that CK2α regulates CD8+ T-cell effector and memory differentiation. To characterize transcriptional features in CD8+ T-cells in the presence and absence of CK2α, we performed gene expression profiling in CK2αfl/fl and CK2α−/− CD8+ T-cells at day 7 post-infection with Listeria monocytogenes. Consistent with previous studies (43–45), expression levels of Id2 and Blimp1 decreased and levels of Id3 increased in CK2α−/− CD8+ T-cells. Based upon gene expression profiling, CK2α−/− CD8+ T-cells exhibited a memory signature. These findings suggest that CK2α may regulate CD8+ T-cell effector and memory differentiation through controlling these key transcription factors. However, the precise mechanism of how CK2α regulates these transcription factors and their activity will be further studied.
Our study has demonstrated an essential role for CK2α in maintaining the CD8+ T-cell memory pool and regulating the recall response, which is vital in the immune response against infection. Although CK2α−/− CD8+ T-cells at day 7 post-infection exhibited more potential to develop into memory cells, CK2α−/− CD8+ T-cells had compromised memory cell numbers at day 42 post-infection compared with CK2αfl/fl CD8+ T-cells. These findings do not necessarily contradict each other. One explanation is the initial total number of memory precursor cells is still lower in CK2α−/− mice compared to CK2αfl/fl mice at day 7 post-infection, which eventually results in less memory CK2α−/− CD8+ T-cells. Another explanation is linked with cell survival. Our data demonstrated that the expression of the pro-survival gene Bcl2 was significant lower in CK2α−/− memory CD8+ T-cells, which indicates that CK2α may control CD8+ T-cell responses by regulating cell survival. Although our in vitro data suggested that CK2α is not critical for CD8+ T-cell survival upon TCR stimulation, for the CD8+ T-cell response against infection in vivo, this was not the case. A possible explanation is additional CK2-dependent survival signals are required for CD8+ T-cell survival in vivo but not in vitro. Moreover, our data are consistent with findings that using a CK2 inhibitor downregulates Bcl2 expression in a leukemia cell line, while overexpression of CK2 induces Bcl2 expression (47). However, whether CK2 directly regulates Bcl2 expression is still unclear.
As noted above, CK2α−/− naïve and memory CD8+ T-cells exhibited compromised expansion capacity and expressed less granzyme B during primary and secondary challenge, respectively. However, the biological function of CK2α in CD8+ T-cells, particularly CD8+ T-cell cytotoxic function, remains to be explored. A recent study showed that CK2 is enhanced post SARS-CoV-2 infection (48), and CX-4945, an inhibitor of CK2, is being tested in a phase II clinical trial for COVID-19 patients, potentially targeting infected cells. Our data suggest that CK2 is vital for CD8+ T-cell function, which may affect bacterial and viral clearance. Conversely, LysMcreCK2αfl/fl mice, in which CK2α is specifically deleted in myeloid cells, are more resistant to systemic LM infection (19), which indicates that CK2α has a divergent function in myeloid cells compared to CD8+ T-cells related to pathogen infection. Therefore, systematic CK2 inhibition may compromise the function of CD8+ T-cells, while enhancing the function of myeloid cells, with the ultimate biological consequence uncertain. Our study highlights the complexity of CK2 biology with respect to CD8+ T-cell function and underlines caution for therapeutic use of CK2 inhibitors.
Supplementary Material
KEY POINTS.
TCR engagement induces CK2 expression and kinase activity in CD8+ T-cells
CK2α impacts CD8+ T-cell activation, proliferation and metabolic reprogramming
CK2α regulates CD8+ T-cell effector and memory differentiation in infection.
ACKNOWLEDGMENTS
We thank the Comprehensive Flow Cytometry Core at the University of Alabama at Birmingham for their assistance with flow cytometry.
This work was supported by National Institutes of Health Grants R01NS057563 and R01CA194414 (to E.N.B.), National Multiple Sclerosis Society Grant RG-1606-24794 (to H.Q.), R21AI142641 and R01DK125870 (to L.E.H.) and R01AG050886R01 (to J.Z. and V.D-U.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Zhang N, and Bevan MJ. 2011. CD8(+) T cells: foot soldiers of the immune system. Immunity 35: 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed R, and Gray D. 1996. Immunological memory and protective immunity: understanding their relation. Science 272: 54–60. [DOI] [PubMed] [Google Scholar]
- 3.Kaech SM, and Cui W. 2012. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams MA, and Bevan MJ. 2007. Effector and memory CTL differentiation. Annu Rev Immunol 25: 171–192. [DOI] [PubMed] [Google Scholar]
- 5.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, and Ahmed R. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4: 1191–1198. [DOI] [PubMed] [Google Scholar]
- 6.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM. 2007. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27: 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L, and Romero P. 2018. Metabolic control of CD8(+) T cell fate fecisions and antitumor immunity. Trends Mol Med 24: 30–48. [DOI] [PubMed] [Google Scholar]
- 8.Litchfield DW 2003. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J 369: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominguez I, Sonenshein GE, and Seldin DC. 2009. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol Life Sci 66: 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan JS, and Litchfield DW. 2008. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim Biophys Acta 1784: 33–47. [DOI] [PubMed] [Google Scholar]
- 11.Singh NN, and Ramji DP. 2008. Protein kinase CK2, an important regulator of the inflammatory response? J. Mol. Med 86: 887–897. [DOI] [PubMed] [Google Scholar]
- 12.Rabalski AJ, Gyenis L, and Litchfield DW. 2016. Molecular pathways: Emergence of Protein Kinase CK2 (CSNK2) as a potential target to inhibit survival and DNA damage response and repair pathways in cancer cells. Clin Cancer Res 22: 2840–2847. [DOI] [PubMed] [Google Scholar]
- 13.Borgo C, and Ruzzene M. 2021. Protein kinase CK2 inhibition as a pharmacological strategy. Adv Protein Chem Struct Biol 124: 23–46. [DOI] [PubMed] [Google Scholar]
- 14.Silva-Pavez E, and Tapia JC. 2020. Protein Kinase CK2 in Cancer Energetics. Front Oncol 10: 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Husain K, Williamson TT, Nelson N, and Ghansah T. 2020. Protein kinase 2 (CK2): a potential regulator of immune cell development and function in cancer. Immunol Med: 1–16. [DOI] [PubMed] [Google Scholar]
- 16.Ulges A, Witsch EJ, Pramanik G, Klein M, Birkner K, Buhler U, Wasser B, Luessi F, Stergiou N, Dietzen S, Bruhl TJ, Bohn T, Bundgen G, Kunz H, Waisman A, Schild H, Schmitt E, Zipp F, and Bopp T. 2016. Protein kinase CK2 governs the molecular decision between encephalitogenic TH17 cell and Treg cell development. Proc Natl Acad Sci U S A 113: 10145–10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson SA, Yang W, Yan Z, Liu Y, Rowse AL, Weinmann AS, Qin H, and Benveniste EN. 2017. Protein kinase CK2 controls the fate between Th17 cell and regulatory T cell differentiation. J Immunol 198: 4244–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang W, Gibson SA, Yan Z, Wei H, Tao J, Sha B, Qin H, and Benveniste EN. 2020. Protein kinase 2 (CK2) controls CD4(+) T cell effector function in the pathogenesis of colitis. Mucosal Immunol 13: 788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larson SR, Bortell N, Illies A, Crisler WJ, Matsuda JL, and Lenz LL. 2020. Myeloid cell CK2 regulates inflammation and resistance to bacterial infection. Front Immunol 11: 590266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson SA, and Benveniste EN. 2018. Protein Kinase CK2: An emerging regulator of immunity. Trends Immunol 39: 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibson SA, Yang W, Yan Z, Qin H, and Benveniste EN. 2018. CK2 controls Th17 and regulatory T cell differentiation through inhibition of FoxO1. J Immunol 201: 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ulges A, Klein M, Reuter S, Gerlitzki B, Hoffmann M, Grebe N, Staudt V, Stergiou N, Bohn T, Bruhl TJ, Muth S, Yurugi H, Rajalingam K, Bellinghausen I, Tuettenberg A, Hahn S, Reissig S, Haben I, Zipp F, Waisman A, Probst HC, Beilhack A, Buchou T, Filhol-Cochet O, Boldyreff B, Breloer M, Jonuleit H, Schild H, Schmitt E, and Bopp T. 2015. Protein kinase CK2 enables regulatory T cells to suppress excessive T2 responses in vivo. Nat Immunol 16: 267–275. [DOI] [PubMed] [Google Scholar]
- 23.de Bourayne M, Gallais Y, El Ali Z, Rousseau P, Damiens MH, Cochet C, Filhol O, Chollet-Martin S, Pallardy M, and Kerdine-Romer S. 2016. Protein kinase CK2 controls T-cell polarization through dendritic cell activation in response to contact sensitizers. J Leukoc Biol 101: 703–715. [DOI] [PubMed] [Google Scholar]
- 24.Harrington LE, Janowski KM, Oliver JR, Zajac AJ, and Weaver CT. 2008. Memory CD4 T cells emerge from effector T-cell progenitors. Nature 452: 356–360. [DOI] [PubMed] [Google Scholar]
- 25.Kim MV, Ouyang W, Liao W, Zhang MQ, and Li MO. 2013. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity 39: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng Y, McFarland BC, Drygin D, Yu H, Bellis SL, Kim H, Bredel M, and Benveniste EN. 2013. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin Cancer Res 19: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hill BG, Benavides GA, Lancaster JR Jr., Ballinger S, Dell’Italia L, Jianhua Z, and Darley-Usmar VM. 2012. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem 393: 1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, Landar A, and Darley-Usmar VM. 2011. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med 51: 1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quiros PM, Goyal A, Jha P, and Auwerx J. 2017. Analysis of mtDNA/nDNA Ratio in Mice. Curr Protoc Mouse Biol 7: 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, and Pearce EL. 2013. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153: 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Almeida L, Lochner M, Berod L, and Sparwasser T. 2016. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol 28: 514–524. [DOI] [PubMed] [Google Scholar]
- 32.Desdin-Mico G, Soto-Heredero G, and Mittelbrunn M. 2018. Mitochondrial activity in T cells. Mitochondrion 41: 51–57. [DOI] [PubMed] [Google Scholar]
- 33.Chi H 2012. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 12: 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manning BD, and Toker A. 2017. AKT/PKB signaling: Navigating the network. Cell 169: 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollizzi KN, and Powell JD. 2014. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol 14: 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, and Green DR. 2011. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76: 17–27. [DOI] [PubMed] [Google Scholar]
- 38.Kahan SM, Bakshi RK, Luther R, Harrington LE, Hendrickson RC, Lefkowitz EJ, Weaver CT, and Zajac AJ. 2017. IL-2 producing and non-producing effector CD8 T cells phenotypically and transcriptionally coalesce to form memory subsets with similar protective properties. J Immunol 198: 212.216–212.216. [Google Scholar]
- 39.Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW, and Immunological Genome Project C. 2013. Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat Immunol 14: 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buck MD, O’Sullivan D, and Pearce EL. 2015. T cell metabolism drives immunity. J Exp Med 212: 1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, and Thompson CB. 2003. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol 23: 7315–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, and Reiner SL. 2005. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol 6: 1236–1244. [DOI] [PubMed] [Google Scholar]
- 43.Cannarile MA, Lind NA, Rivera R, Sheridan AD, Camfield KA, Wu BB, Cheung KP, Ding Z, and Goldrath AW. 2006. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat Immunol 7: 1317–1325. [DOI] [PubMed] [Google Scholar]
- 44.Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, Watowich SS, Murre C, and Goldrath AW. 2011. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol 12: 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kallies A, Xin A, Belz GT, and Nutt SL. 2009. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity 31: 283–295. [DOI] [PubMed] [Google Scholar]
- 46.Ichii H, Sakamoto A, Kuroda Y, and Tokuhisa T. 2004. Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J Immunol 173: 883–891. [DOI] [PubMed] [Google Scholar]
- 47.Kim JS, Eom JI, Cheong JW, Choi AJ, Lee JK, Yang WI, and Min YH. 2007. Protein kinase CK2alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin Cancer Res 13: 1019–1028. [DOI] [PubMed] [Google Scholar]
- 48.Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Marrero MC, Polacco BJ, Melnyk JE, Ulferts S, Kaake RM, Batra J, Richards AL, Stevenson E, Gordon DE, Rojc A, Obernier K, Fabius JM, Soucheray M, Miorin L, Moreno E, Koh C, Tran QD, Hardy A, Robinot R, Vallet T, Nilsson-Payant BE, Hernandez-Armenta C, Dunham A, Weigang S, Knerr J, Modak M, Quintero D, Zhou Y, Dugourd A, Valdeolivas A, Patil T, Li Q, Hüttenhain R, Cakir M, Muralidharan M, Kim M, Jang G, Tutuncuoglu B, Hiatt J, Guo JZ, Xu J, Bouhaddou S, Mathy CJP, Gaulton A, Manners EJ, Félix E, Shi Y, Goff M, Lim JK, McBride T, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, De wit E, Leach AR, Kortemme T, Shoichet B, Ott M, Saez-Rodriguez J, tenOever BR, Mullins D, Fischer ER, Kochs G, Grosse R, García-Sastre A, Vignuzzi M, Johnson JR, Shokat KM, Swaney DL, Beltrao P, and Krogan NJ. 2020. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182: 685–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.