

Abstract
Dendritic cells (DCs) are professional antigen-presenting cells equipped with MHC-restricted antigens, co-stimulations, and cytokines that effectively prime and differentiate naïve T cells into distinct functional subsets. The immune signals that DCs carry reflect the route of antigen uptake and the innate stimuli they received. In the mucosal tissues, owing to the great variety of foreign antigens and inflammatory cues, DCs are predominantly activated and migratory. In the small intestine, CD4 Th17 cells are abundant and have been shown to be regulated by DCs and macrophages. Using a mouse commensal bacteria experimental model, we identified that the early priming step of commensal-driven Th17 cells is controlled by bona fide Zbtb46-expressing DCs. CCR7-dependent migration of DC2s from the small intestine to the mesenteric lymph nodes is essential for the activation of naïve CD4 T cells. The migratory DC2 population in the MLN are almost exclusively Esam+ cells. Single-cell RNA sequencing highlighted the abundance of co-stimulatory markers (CD40 and OX40) and chemokines (Ccl22 and Cxcl16) on MLN migratory DCs. Further resolution of MLN migratory DC2s revealed that the Th17-polarizing cytokine IL-6 colocalizes with DC2s expressing CD40, Ccl17, and Ccl22. Thus, early Th17 cell differentiation is initiated by a small subset of migratory DC2s in the gut draining lymph nodes.
INTRODUCTION
Dendritic cells are innate immune effectors that regulate many aspects of adaptive T cells responses. Conventional dendritic cells are classified as DC1s or DC2s with tissue-specific distribution and maturation patterns (1, 2). DC1s express Xcr1 and depends on BATF3 and IRF8 for lineage development (3, 4). Insights gained from infectious disease models and tumor models revealed that DC1s are critically important for generating CD8 cytotoxic T cells (CTL), CD4 T helper 1 (Th1), and T regulatory (Treg) cell responses (5, 6). DC2s are CD11b+Sirpα+ cells with a developmental requirement for Notch2 and Irf4 (7, 8). This DC subset shares many features with macrophages and is mainly involved in Th2 and Th17 responses in the skin and mucosal tissues (9–11). In the small intestine, both DC subsets can be detected as CD103+ cells, and each specialized in priming different types of T cells. DC1s activate CD8 T cells and imprint T cell gut homing by inducing the CCR9 receptor through a retinoic acid-dependent mechanism (12). The absence of DC2s reduces homeostatic Th17 cells in the lung, small intestine, and colon (10, 13). Significant decrease of mucosal Th17 cells post infection with Aspergillus fumigatus or vaccination with intraperitoneal ovalbumin indicate that DC2s control the priming of CD4 T cells for these antigenic challenges (10, 13). However, the role of DC2s in the context of commensal bacteria induced Th17 responses remained relatively unexplored.
Th17 cells are induced by many gut commensal bacteria and segmented filamentous bacteria (SFB) represents the single most potent inducer of intestinal Th17 cells identified (14). Similar to the classically defined Th17 cells (15, 16), SFB-induced Th17 cells are characterized by expression of the master transcription factor RORγt and the signature cytokines IL-17A, IL-17F, and IL-22. As a non-pathogenic inhabitant of the small intestine, SFB does not breach the epithelium, and hence the exact mechanism of bacterial antigen uptake by host DCs remained enigmatic. Adhesion of bacterium to the host epithelial cells and a CDC42-dependent endocytosis process by epithelial cells are necessary for the induction of Th17 cells (17, 18). Downstream of antigen acquisition, it has been demonstrated that small intestinal lamina propria (SILP) CCR2-expressing, CX3CR1+ macrophages are indispensable for intestinal Th17 response to SFB (19). However, macrophages are unlikely to migrate to draining lymph nodes, and naïve CD4 T cells do not enter SILP due to the lack of gut homing receptors. It is unclear how Th17 cell differentiation is accomplished in mesenteric lymph nodes (MLN). One possibility is that the CCR2-expressing phagocytic cells contain a minor subset of emigrating dendritic cells (20) that enter MLN to prime naïve CD4 T cells. Alternatively, antigen transfer could take place between CX3CR1+ macrophages to migratory dendritic cells via gap junction (21).
The availability of an SFB peptide-specific transgenic CD4 T cell model has made studying Th17 differentiation at the clonal level possible (22). In addition, advanced classification of DC subsets is made possible by the recent development in single-cell RNA sequencing (scRNAseq) technology. In the present study, we focused on the priming stage of naïve CD4 T cells in MLN to identify the migratory DC subset that activates and drives Th17 differentiation. Results from genetic models targeting different myeloid cell populations indicated that the Zbtb46-expressing DCs, but not macrophages present SFB antigens in the MLN. CCR7-dependent homing of DCs to the MLN is essential for naïve T cell activation, and IL-6 accounts for a significant fraction of the RORγt expression in T cells. scRNAseq of MLN migratory DCs revealed an increase in 2 transcriptionally distinct DC2 populations post SFB colonization: Cd40/Ccl22- and Cd1d1/Tnfrsf4-expressing subsets. IL-6 can be detected in the Cd40/Ccl22 DC2 cell cluster. Our findings provide insights on the DC2s responsible for mucosal Th17 cell response.
MATERIALS AND METHODS
Mice
C57BL/6, 7B8Tg, zDC-DTR, CCR7−/−, CCR2−/−, BATF3−/−, CX3CR1gfp/gfp, IL-6−/−, MHCIIfl/fl, STAT3fl/fl, IL-6Rαfl/fl, CD4Cre, LysmCre, CD11cCre mice were purchased from The Jackson Laboratory. Mice were bred and maintained at the specific pathogen free animal facilities of the Medical University of South Carolina and later at the Ohio State University. All experiments were performed under protocols approved by Institutional Animal Care and Use Committee at both Universities. Mouse colonies were screened regularly for the presence of fecal SFB. Mice contaminated with SFB was treated with ampicillin in drinking water (1g/ml) for 2 weeks and let recovered before being used for experiments. SFB donor mice include C57BL/6 mice sourced from Taconic and in house SFB+ mice.
SFB screening by quantitative PCR
Fecal pellet (approximately 12mg) was crushed in 2ml PBS, filtered through 40μm cell strainer and homogenized using 0.1mm Zirconia/Silica beads (BioSpec Products) in FastPrep-24 instrument (MP Biomedicals). Bacterial DNA was isolated from cell lysate supernatant using DNA Clean & Concentrator kit (Zymo Research) according to manufacturer’s protocol. qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (Biorad) with published primer sequence (SFB736F – 5’GACGCTGAGGCATGAGAGCAT, SFB844R – 5’GACGGCACGGATTGTTATTCA, UniF340 – 5’ACTCCTACGGGAGGCAGCAGT, UniR514 – 5’ATTACCGCGGCTGCTGGC). Cq value of SFB-specific 16s rRNA was normalized against Cq value of eubacteria 16s rRNA of the same sample.
T cell adoptive transfer
Single fecal pellet from SFB+ donor mice was crushed in 2ml PBS, filtered through 40μm cell strainer and used for 10 recipient mice. One week post SFB oral gavage, MACS beads isolated naïve 7B8Tg cells were transferred via tail vein injection at concentration of 2×105 cells in 200μl PBS per mouse. 7B8Tg cells were labeled with cell trace violet (ThermoFisher Scientific) according to manufacturer’s protocol. Three days post transfer, recipient mice were sacrificed and MLNs harvested for flow cytometry staining. Donor 7B8Tg cells were detected by the expression of congenic marker CD45.1.
Generation of bone marrow chimera mice
Bone marrow cells were collected from Zbtb46-DTR donor mice, and red blood cell lysis was carried out with ACK lysis buffer. Two million cells were injected into lethally irradiated C57BL/6 recipient mice. Two months later, mice were bled to check for donor cell engraftment. Chimeras were orally gavaged with SFB-positive fecal material, and 1 week later, adoptively transferred with 2×105 naïve 7B8Tg cells. A day before and after 7B8Tg cell transfer, diphtheria toxin was administered via intraperitoneal injection to deplete DCs. Three days post 7B8Tg transfer, mice were sacrificed and MLNs were harvested for analysis.
MHCII-Tetramer staining
MHCII tetramer detecting SFB3340-specific endogenous CD4 T cells (I-A(b) SFB SFB3340 200–210 QFSGAVPNKTD, PE- and APC-labeled) were generated by NIH tetramer core facility. Briefly, single cell suspension was incubated with tetramer (1:100 dilution) at 37°C for 30min. Cells were then stained with surface antibodies and viability dye. Samples were recorded at the BDFortessa immediately without fixation.
Cell isolation
Spleens were crushed, filtered through 100μm cell strainer and subjected to red blood cell lysis using ACK buffer. SILP cells were collected as follow: the distal half of small intestine was harvested, Peyer’s patches were removed, and the small intestine was cut open longitudinally before excising into ~1cm segments. Tissue segments were washed in PBS and incubated with DTT/EDTA for 20min in 37°C shaker to disrupt the epithelium. After 3 washes, tissue segments were digested with collagenase D/Dnase1/dispase for 30min in 37°C shaker. Immediately after digestion step, enzymatic reaction was stopped by filling the reaction tube with cold RPMI complete media. Tissue segments were then crushed and filtered through 100μm cell strainers. Single cell suspension was partitioned on a 40%/80% percoll gradient. Lymphocytes at the interphase was collected and washed prior to flow cytometry staining. For the processing of lung lymphocytes, mice were perfused with PBS before harvesting lungs. Lung tissues were excised into smaller fragments and digested with EDTA for 30min in 37°C shaker. After 2 washes, tissue fragments were digested with collagenase D/Dnase1/dispase for 45min in 37°C shaker. Immediately after digestion step, enzymatic reaction was stopped by filling the reaction tube with cold RPMI complete media. Single cell suspension was obtained by following the same procedure as SILP cells.
Flow cytometry
The staining of ex vivo T cells began with 30min of incubation with surface antibodies and viability dye on ice. Transcription factors were stained using the Foxp3 Transcription Factor Staining Buffer Set (eBioscience, ThermoFisher Scientific). Briefly, fixation was done on ice for 30min. RORγt, Foxp3, T-bet, Batf, Irf4, cMaf antibodies were added to the perm buffer and cells were stained at room temperature for 30min. For intracellular cytokine staining, in vitro differentiated Th17 cells were stimulated with PMA and ionomycin in the presence of brefeldin A for 4hrs. Cells were stained with surface antibodies and viability dye as described above before fixation with 4% PFA for 5min at room temperature. Antibodies for IL-17a and TNFα were added to saponin perm buffer and cells were stained for 30min on ice. Flow staining for dendritic cells started with a FcR-blocking step for 10min at room temperature, followed by surface staining for 20min at room temperature. Live cells were recorded immediately or fixed with 4% PFA until acquisition. Flow cytometer instruments used for this study include BDFortessa and Cytek Aurora. Analyses were done using Flowjo v10 software (TreeStar).
Single-cell RNA sequencing and data analysis
C57BL/6 mice were orally gavaged with SFB-positive fecal material. Five days later, 15 naïve mice and 15 SFB-positive mice were sacrificed and MLNs collected. T cells and B cells were depleted using biotinylated anti-mouse CD3e and CD19 followed by anti-biotin MACS beads (Miltenyi Biotec). Negatively selected cells were FACS-sorted for live migratory (CD11c+MHCIIhi) and resident (CD11chiMHCII+) cells. cDNA library generation followed established techniques using the Chromium Single Cell 3’ Library v2 Kit (10x Genomics;https://assets.ctfassets.net/an68im79xiti/RT8DYoZzhDJRBMrJCmVxl/6a0ed8015d89bf9602128a4c9f8962c8/CG00052_SingleCell3_ReagentKitv2UserGuide_RevF.pdf). Briefly, cells in single-cell suspension were loaded onto a 10X Genomics Chip A and emulsified with 3’ Single Cell GEM beads using a Chromium™ Controller (10x Genomics) and libraries are constructed from the barcoded cDNAs (Translational Science Laboratory at the Medical University of South Carolina). RNA sequencing was performed on each sample (approximately 50,000 reads/cell) using a NovaSeq S4 flow cell (Illumina) at the VANTAGE facility (Vanderbilt University Medical Center). Resulting data were processed using Cell Ranger software (10X Genomics) at the sequencing core, followed by analysis in our lab using the R-based package Seurat according to the standard workflow laid out in scRNA-seq integration vignette (Tools for Single Cell Genomics • Seurat (satijalab.org)).
Statistical analysis
Unpaired, equal variance Student’s T test was used to calculate statistical significance in Excel and Prism softwares. * P<0.05, ** P<0.01.
DATA AVAILABILITY
The data discussed in this publication have been deposited in NCMI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE184423 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi).
RESULTS
Gut commensal bacteria induced CD4 Th17 cells undergo distinct clonal expansion and contraction in response to bacterial load
Host immune responses to segmented filamentous bacteria (SFB) are well-studied. Specifically, the induction of gut CD4 Th17 cells and the availability of transgenic CD4 T cells recognizing known bacterial epitope (22) through natural oral infection route made this model an excellent system to identify important factors in homeostatic mucosal T cell responses. In mice, fecal and epithelial SFB can be detected at approximately 1 week post oral gavage, peaked at 2 weeks and maintained a low but detectable level 1 month later (Fig. 1A). The growth and contraction of SFB suggest that the host immune system is capable of controlling the bacteria at a non-pathogenic level. The host T cell receptor (TCR) affinity for the defined SFB3340568–580 epitope was predicted to have a KD value of 89nM (22), which falls within the KD range of anti-pathogen TCR-pMHC (23). This made reliable detection of host SFB-specific CD4 T cells with MHCII tetramer feasible. In the small intestine lamina propria (SILP), antigen (Ag)-specific CD4 T cell expansion reached a maximal level at two weeks post SFB oral gavage (Fig. 1B). We next examined the kinetics of adoptively transferred 7B8Tg cell activation, which recognize the same epitope that was used for generating the MHCII tetramer. Similar to endogenous T cells, expansion of 7B8Tg cells peaked at approximately two weeks (Fig. 1C). Expression of RORγt transcription factor spiked transiently in MLN 7B8Tg cells within the first week of T cell activation but continued to maintain high expression in SILP 7B8Tg cells up until 1 month later (Fig. 1D). The expansion curve of T cell activation suggests that the initial differentiation of Th17 cells takes place in the draining lymph nodes, but complete differentiation requires microbial elements and microenvironment factors in the small intestine. Concluding from these analyses, it is evident that the kinetic of host primary CD4 Th17 cell response to gut commensal bacteria is consistent with pathogenic bacteria-induced CD4 T cells (24, 25).
Figure 1.
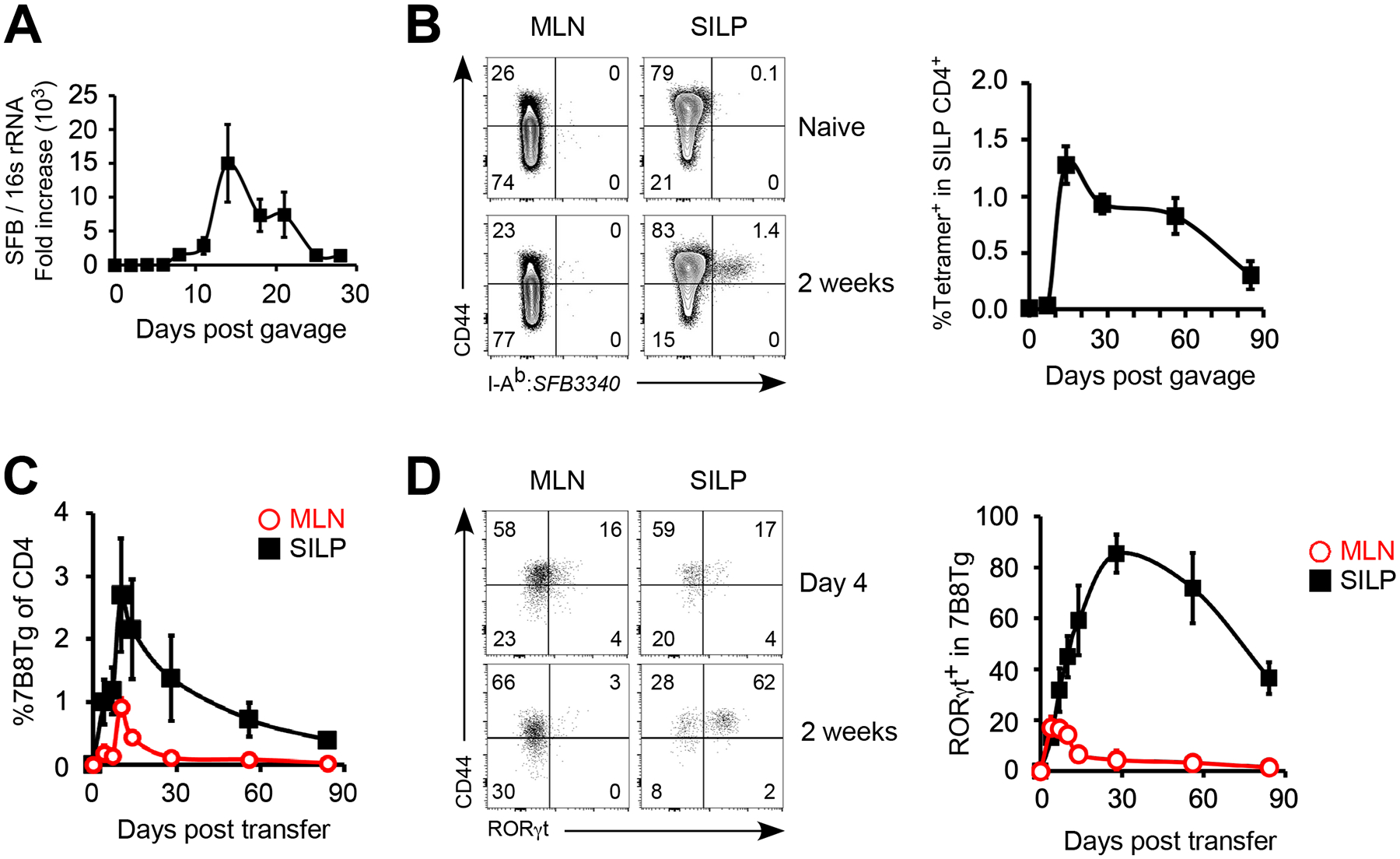
Commensal bacteria induced CD4 Th17 cell response undergoes distinct expansion and contraction in response to bacterial load. (A) qPCR quantitation of recipient fecal SFB post oral gavage with SFB-positive fecal material. (B) Expansion of host SFB antigen-specific CD4 T cells detected by SFB3340568–580:I-Ab tetramer. (C, D) C57BL/6 mice were orally gavaged with SFB-positive fecal material and 2×105 naïve 7B8Tg cells were transferred intravenously 1 week later. Expansion (C) and Th17 specification (D) of adoptively transferred 7B8Tg CD4 T cells were examined. Data shown are representative of 2 independent experiments with 3–4 mice in each group, error bars indicate SEM.
Early Th17 cell specification takes place in the mesenteric lymph nodes
To visualize the early phase of Th17 differentiation, naïve CD4 7B8Tg T cells were labeled with cell trace dye and adoptively transferred into SFB-colonized C57BL/6 mice. Three days later, T cell proliferation versus expression of lineage determining transcription factors and homing receptors were examined. Although SFB is localized to the gut, 7B8Tg cell activation and proliferation can be observed in distal organs such as the spleen and lung (Fig. 2A), suggesting that SFB antigens are disseminated systemically either in free form or carried by antigen-presenting cells. However, only in the MLN that activated but undivided 7B8Tg cells and their early progeny upregulated RORγt (Fig. 2B), indicating that cellular machinery that supports Th17 differentiation is restricted to the draining lymph nodes. Activated 7B8Tg cells leaving MLN may then accumulate at spleen and lung via systemic circulation. Antigen-presenting cells driving Th17 differentiation are likely limited in numbers since increasing 7B8Tg donor cell numbers resulted in the slower proliferation and reduced RORγt expression (Fig. 2C). As these early Th17 cells progressively strengthen their identity, the expression level of core transcription factors such as IRF4, BATF, and cMaf (26) are amplified, and they displayed no sign of plasticity towards Treg or Th1 lineage (Fig. 2D). Lymphoid organ homing receptor CD62L was downregulated on activated Th17 cells, peripheral tissue homing receptor CCR4 is upregulated (Fig. 2D). Surprisingly, gut homing receptors CCR6 and CCR9 (27, 28) were not significantly upregulated at this early time point (Fig. 2D). Gut homing of these early Th17 cells are likely to dependent solely on α4β7 integrin (29). PD1 was upregulated on Th17 cells (Fig. 2D) but did not seem to have inhibitory effects on either Th17 cell proliferation or differentiation at this early stage (data not shown). Taken together, the limited number of Th17-promoting antigen-presenting cells that prime the first wave of Th17 cells are localized to MLN.
Figure 2.
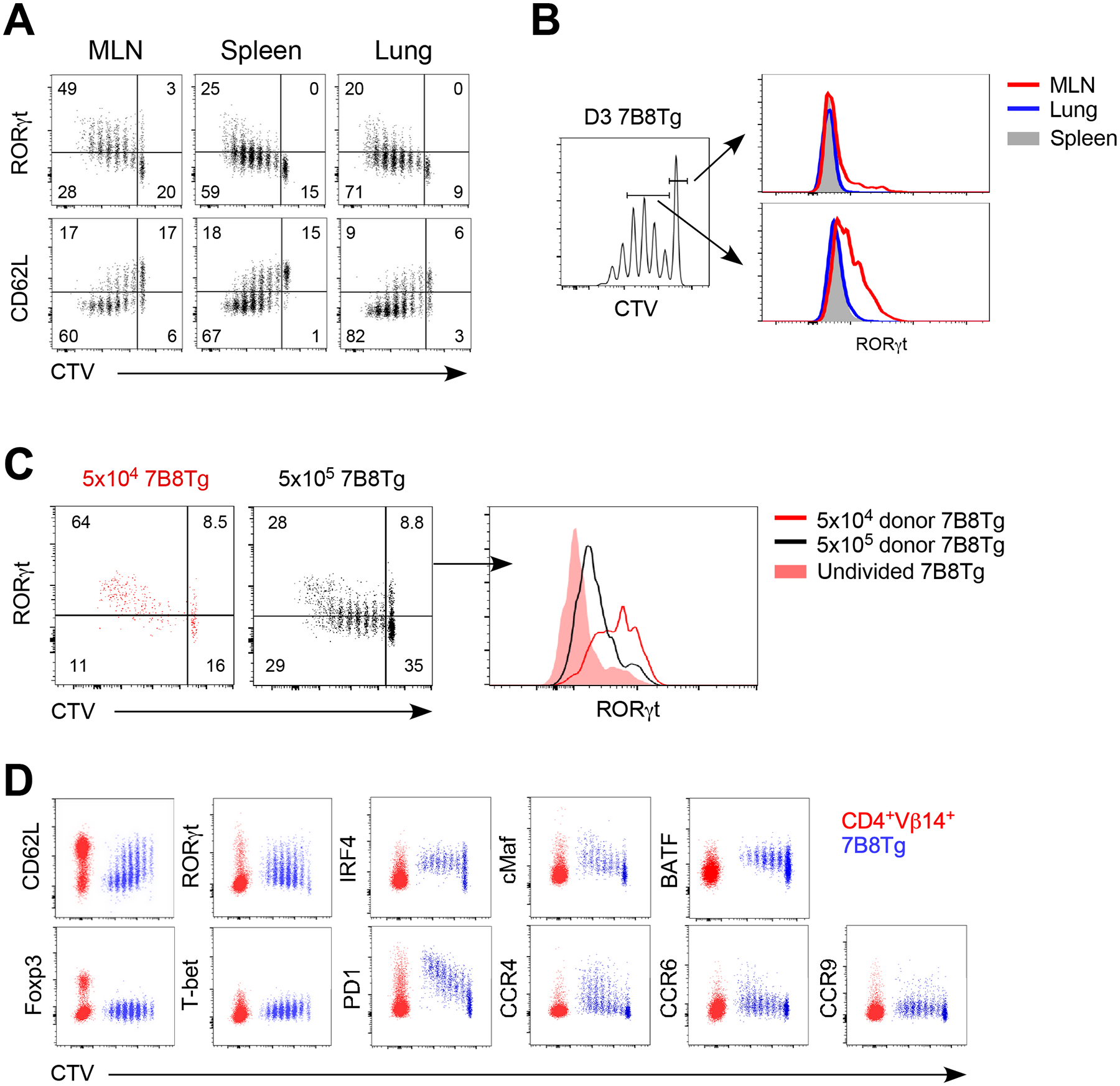
Early Th17 cell specification takes place in the mesenteric lymph nodes. C57BL/6 mice were orally gavaged with SFB-positive fecal material and 2×105 naïve 7B8Tg cells (CD45.1+) were adoptively transferred 1 week later. (A) Donor 7B8Tg cell proliferation and RORγt expression in different tissues on day 3 post adoptive transfer. (B) RORγt expression in undivided 7B8Tg cells and early Th17 progeny (defined as 7B8Tg cells undergone 1–4 cell division post activation) on day 3 post adoptive transfer. (C) Effects of precursor frequency on Th17 cell specification on day 4 post adoptive transfer in MLN. (D) Expression of lineage specifying transcription factors and tissue homing receptors in activated MLN 7B8Tg cells on day 3 post adoptive transfer. Data shown are representative of 2 independent experiments with 3–4 mice in each group, error bars indicate SEM.
Migratory DC2s prime Th17 cells in mesenteric lymph nodes
Myeloid cells at the gut mucosal system are phenotypically and functionally heterogeneous. In tissue draining lymph nodes, dendritic cells are generally divided into migratory (activated and mature) versus resident (steady-state). Both populations contain DC1s (Xcr1+) and DC2s (Sirpα+CD11b+) that are thought to preferentially home to a specific region of the lymph node and induces selected subsets of T cells (9). Both CD103+ DC2s and macrophages have been reported in the induction of gut Th17 cells. Specifically for SFB, the CCR2-expressing CX3CR1+ macrophages but not CD103+ DCs that support the induction of SILP Th17 cells (19). However, a small subset of CCR2+ cells derived from the DC precursors and are thought to be bona fide DCs involved in Th17 cell activation (20). It remains unclear whether Th17 cell-inducing antigen-presenting cells are of macrophage or DC lineage. We took advantage of the SFB-specific 7B8Tg model to address this question during T cell priming. The Zbtb46 transcription factor distinguishes classical dendritic cells from macrophages, monocytes, and plasmacytoid DCs (30). We generated Zbtb46-DTR bone marrow chimera, orally gavaged them with SFB, and administered diphtheria toxin (DT) to the mice before 7B8Tg cell adoptive transfer. A single dose of DT effectively depleted both the migratory (MHCIIhiCD11c+) and resident (MHCII+CD11chi) DCs in the MLN but SILP DCs were not significantly impacted (Fig. 3A). Naïve 7B8Tg cells failed to activate in these hosts (Fig. 3B), suggesting that classical DCs are indispensable for the priming step of Th17 cells. In MHCIIfl/flLysMCre (Fig. S1A and B), CX3CR1gfp/gfp (Fig. S1C) and CCR2−/− (Fig. S1D and E) recipients however, where antigen-presenting cells of the macrophage or monocyte lineages were targeted, Th17 differentiation was not affected. Next, in CCR7−/− mice, where the majority of the migratory DCs are absent (Fig. 3C), 7B8Tg cells failed to be activated (Fig. 3D), confirming that the sources of SFB antigens are gut-derived activated, migratory DCs. CCR7 deficiency impairs migration of all classes of gut derived DCs and is insufficient for specific DC-T cell axis. To further examine the role of DC subtypes in Th17 induction, we made use of DC lineage-specific model. The MLN migratory DC population consists of DC1s (Xcr1+CD103+CD11b−) and DC2s (Sirpα+CD11b+). In Batf3−/− mice, Xcr1+ DC1s are absent, but the DC2 compartment remained intact (Fig. 3E). CD11b+CD103+DC2s are present but with reduced CD103 expression (Fig. 3E, lower panel). We found that DC1s are not involved in SFB-induced Th17 cells (Fig. 3F), consistent with the previous report (19). Altogether, these findings suggest that migratory DC2s are the antigen-presenting cells that prime CD4 Th17 cells in the intestinal draining lymph nodes.
Figure 3.
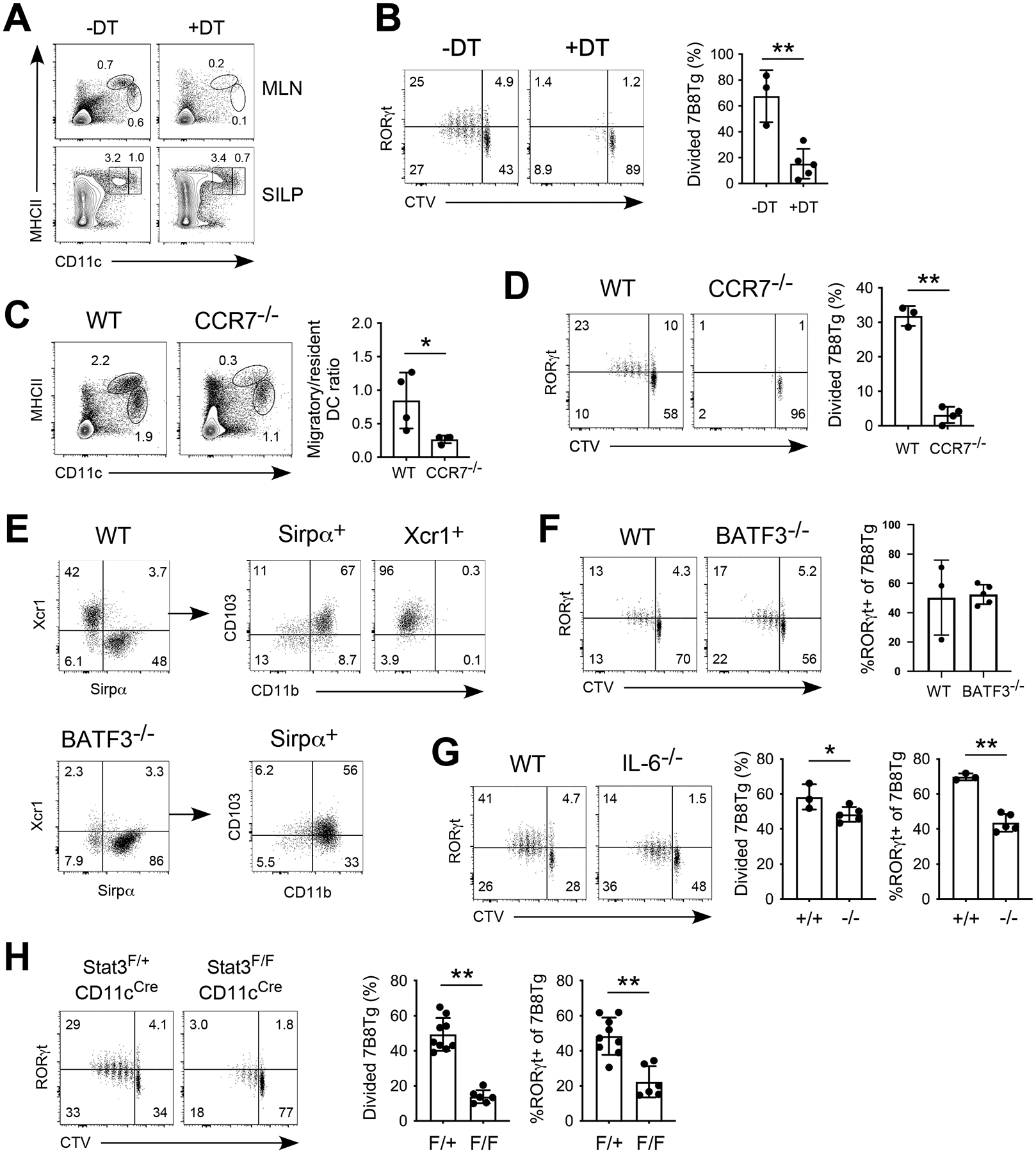
Migratory DC2s prime Th17 cells in the MLN in an IL-6-dependent manner. (A) Depletion of Zbtb46-DTR expressing DCs in bone marrow chimeras after diphtheria toxin administration. (B) 7B8Tg activation in the MLN of DCs-depleted bone marrow chimeras on day 3 post adoptive transfer. (C) Loss of migratory DCs (MHCIIhiCD11c+) in CCR7−/− mice. (D) 7B8Tg activation in the MLN of CCR7−/− mice on day 3 post adoptive transfer. (E) Loss of Xcr1+ DC1s but not Sirpα+ DC2s in the MLN of BATF3−/− mice. (F) 7B8Tg activation in the MLN of BATF3−/− mice on day 3 post adoptive transfer. (G) 7B8Tg activation in the MLN of IL-6−/− mice on day 3 post adoptive transfer. (H) 7B8Tg activation in the MLN of Stat3fl/flCD11cCre mice on day 3 post adoptive transfer. Data shown are representative of 2 independent experiments with 3–5 mice in each group, error bars indicate SEM. * p<0.05, ** p<0.01.
Gut commensal driven Th17 differentiation requires IL-6 and Stat3 signaling in antigen presenting cells
Cytokines are critical drivers of CD4 T helper differentiation. Ample evidence established that IL-1β, IL-6, IL-21, IL-23, and TGFβ are all implicated in Th17 cell differentiation and pathogenicity (31, 32). Temporally speaking, IL-6 acts at an earlier window since IL-6Rα expression is detected on naïve CD4 T cells (Fig. S1F). IL-1β, IL-21, and IL-23 are likely to act on differentiated Th17 cells because IL-1R, IL-21, and IL-23R expression are sequential to IL-6 stimulation and RORγt expression (33–35). We transferred 7B8Tg cells into SFB-colonized IL-6−/− recipients and found that indeed, IL-6 is important for early Th17 differentiation (Fig. 3G). Despite normal proliferation, activated 7B8Tg cells showed suboptimal RORγt upregulation in IL-6−/− mice (Fig. 3G). IL-6 sensing by CD4 T cells is generally thought to be achieved through classical signaling, acting on the IL-6Rα-gp130 complex on T cells. We generated IL-6RαflfCD4Cre 7B8Tg mice and confirmed that IL-6Rα expression on naïve 7B8Tg CD4 T cells was absent (Fig. S1G). IL-6R-null 7B8Tg cells showed no deficiency in early Th17 differentiation (Fig. S1H). On the contrary, T cell-intrinsic IL-6R signaling is indispensable for in vitro Th17 differentiation with direct IL-6 and TGFβ stimulation (Fig. S1I, upper panel). The lack of IL-6R signaling selectively affects IL-17A production but not a non-Th17 cytokine TNFα (Fig. S1I, lower panel). Interestingly, deletion of a IL-6 downstream signaling molecule Stat3 in antigen-presenting cells significantly decreased 7B8Tg cell proliferation and Th17 differentiation (Fig. 3H). Stat3 deficiency did not impact DC1 or DC2 development and ratio in the MLN migratory DC compartment (Fig. S1J). Our findings indicate that in vivo, commensal bacteria-induced Th17 cells required IL-6, which indirectly promotes Th17 differentiation through Stat3 signaling in antigen-presenting cells.
MLN migratory DC2s are exclusively Esam+ with costimulation capacity
Characterization of splenic DCs by single-cell sequencing and flow cytometry revealed considerable heterogeneity within DC2s (36). T-bet+Esam+ DC2s have an anti-inflammatory signature, whereas RORγt+Clec12a+ DC2s are pro-inflammatory and support Th17 cell differentiation in vitro (36). Using Esam and Clec12a markers, we refined DC2s in the spleen, SILP, and MLN (Fig. 4A and B). To distinguish bona fide DCs from macrophages, we used CD24 marker (Fig. S2A and B), a surface receptor that distinguishes DCs from macrophages and has co-stimulation function on T cells (13, 37, 38). In the SFB-colonized mice, MLN migratory population is dominated by Sirpα+ DC2s, mirroring the small intestine, whereas MLN resident DCs are mostly Xcr1+ DC1s, similar to the spleen (Fig. 4A and B). Secondarily, unlike splenic and resident DCs that contain both Esam+ and Clec12a+ cells, migratory DCs are largely Esam+ and small intestinal DC2s are partially Clec12a+ (Fig. 4A and C). Hence, MLN migratory DC2s cannot be further subclassified using Esam and Clec12a. Overall, both migratory DC1s and DC2s express high level of co-stimulation receptors (CD86, CD40, and CD70) and migration marker (CCR7) compared to MLN resident DCs, splenic DCs, or F4/80+CD24lo macrophages (Fig. 4D and S2C), highlighting their maturation status and T cell priming capacity. Altogether, currently known co-stimulatory receptors (Fig. 4D) could not adequately distinguish migratory DC2s from DC1s functionally. Additionally, we were unable to detect the essential Th17-polarizing cytokine IL-6 in the MLN DCs by flow cytometry. To identify the source of IL-6 and further interrogate DC2 heterogeneity in MLN, we subjected both migratory and resident DCs in SFB newly-colonized mice (5 days post oral gavage when fecal SFB was beginning to be detectable) to single-cell RNA sequencing.
Figure 4.
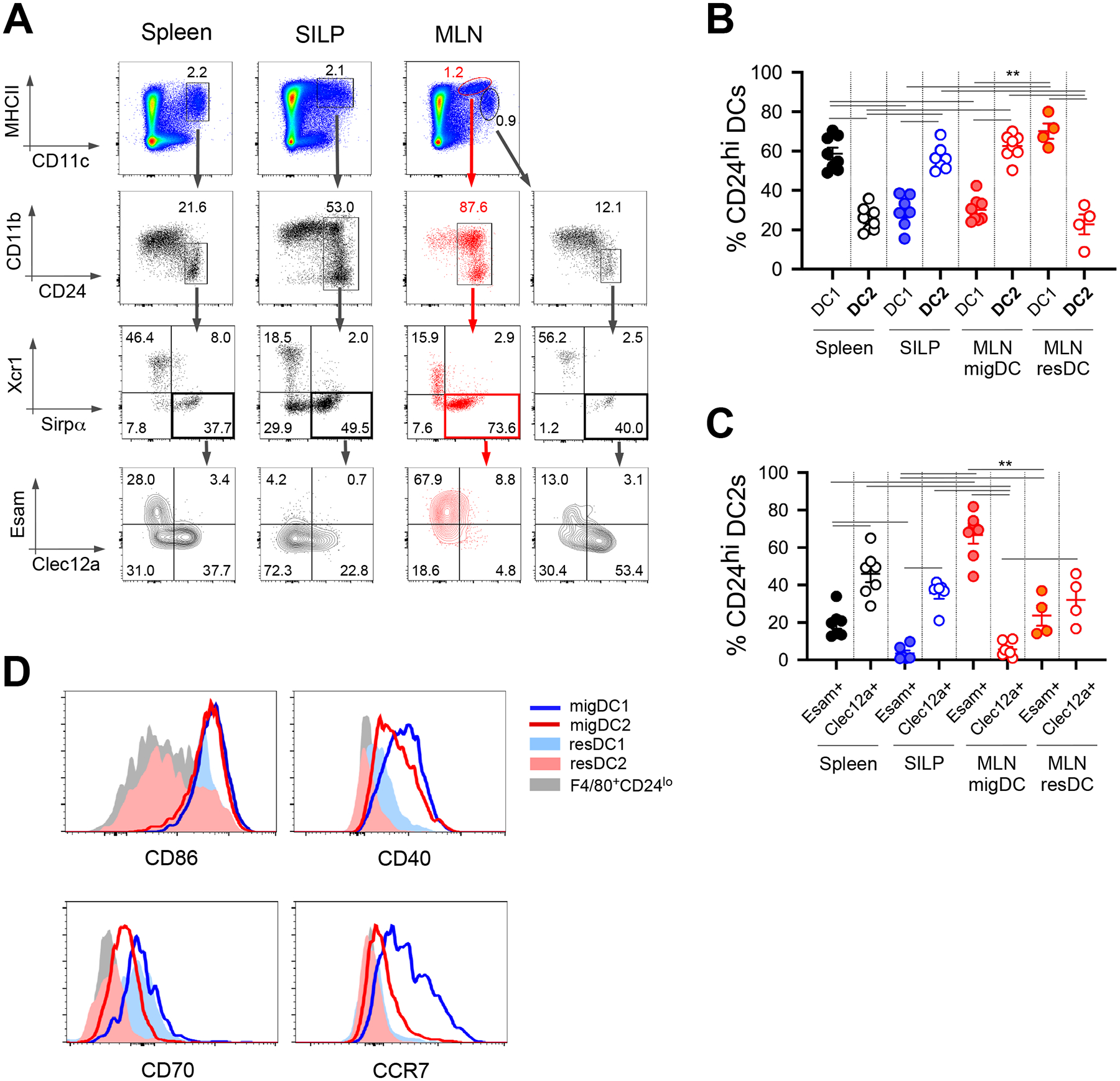
Migratory DC2s are predominantly Esam+ and express high level of surface costimulation receptors. (A) Flow cytometry gating scheme for splenic, small intestinal and MLN DCs. (B) Frequency of DC1s and DC2s in SFB-colonized C57BL/6 mice. (C) Frequency of Esam+ and Clec12a+ cells within DC2s. (D) Expression of costimulatory and migration markers on DC subsets and macrophages. Data shown consists of 2 independent experiments with 2–4 mice in each group, error bars indicate SEM. ** p<0.01.
Single-cell RNA sequencing revealed novel costimulatory marker and chemokines on MLN migratory DCs
From SFB-negative and SFB-positive mice (15 mice in each group), MLNs were harvested, DCs were FACS-sorted based on live Lineage (CD3e, CD19, NK1.1)−CD11c+MHCII+ gating scheme. Migratory (MHCIIhiCD11c+) and resident (MHCII+CD11chi) DCs were partitioned, and single-cell cDNA libraries were generated using the 10xGenomics Chromium system. Sequencing was done on Illumina NovaSeq 6000, and downstream analyses were done using Seurat (39). Integrated analysis performed on the migratory DCs (4335 cells, 14023 genes) and resident DCs (4604 cells, 13850 genes) from SFB-positive mice resulted in 14 DC clusters (Fig. 5A and S3A). Migratory DCs formed 2 transcriptionally distinct cell clusters that are non-overlapping with all resident DC clusters (Fig. 5B). Based on known DC genes (Flt3, Ly86) and DC1/DC2 (Xcr1, Clec9a, Sirpa, Irf4) gene signatures, lineage identity is assigned to each cluster (Fig. 5C). Consistent with flow cytometry data (Fig. 4D), migratory DC1s (C1) and DC2 (C0) are enriched in Ccr7 and CD40. Interestingly, OX40 (Tnfrsf4), typically detected on CD4 T cells, is highly expressed by MLN migratory DCs (Fig. 5D). In addition, selected chemokines (Ccl22, Ccl5, and Cxcl16) are abundant in migratory DCs (Fig. 5D). These and other genes (Apol7c, Serpinb6b, AW112010,) that distinguish migratory DC2s from resident DC2s are mostly shared by migratory DC1s (Fig. S3B). Nevertheless, we identified genes that are unique to migratory DC2s (Cd1d1, Car2, Gbp2) versus resident DC2s (Ltb, Il1b, Fcer1g) that are minimally expressed by DC1s (Fig. 5E). Altogether, results and analyses from single-cell sequencing data reinforce the general observations that migratory DCs are activated with elevated expression of costimulatory receptors and chemokines.
Figure 5.
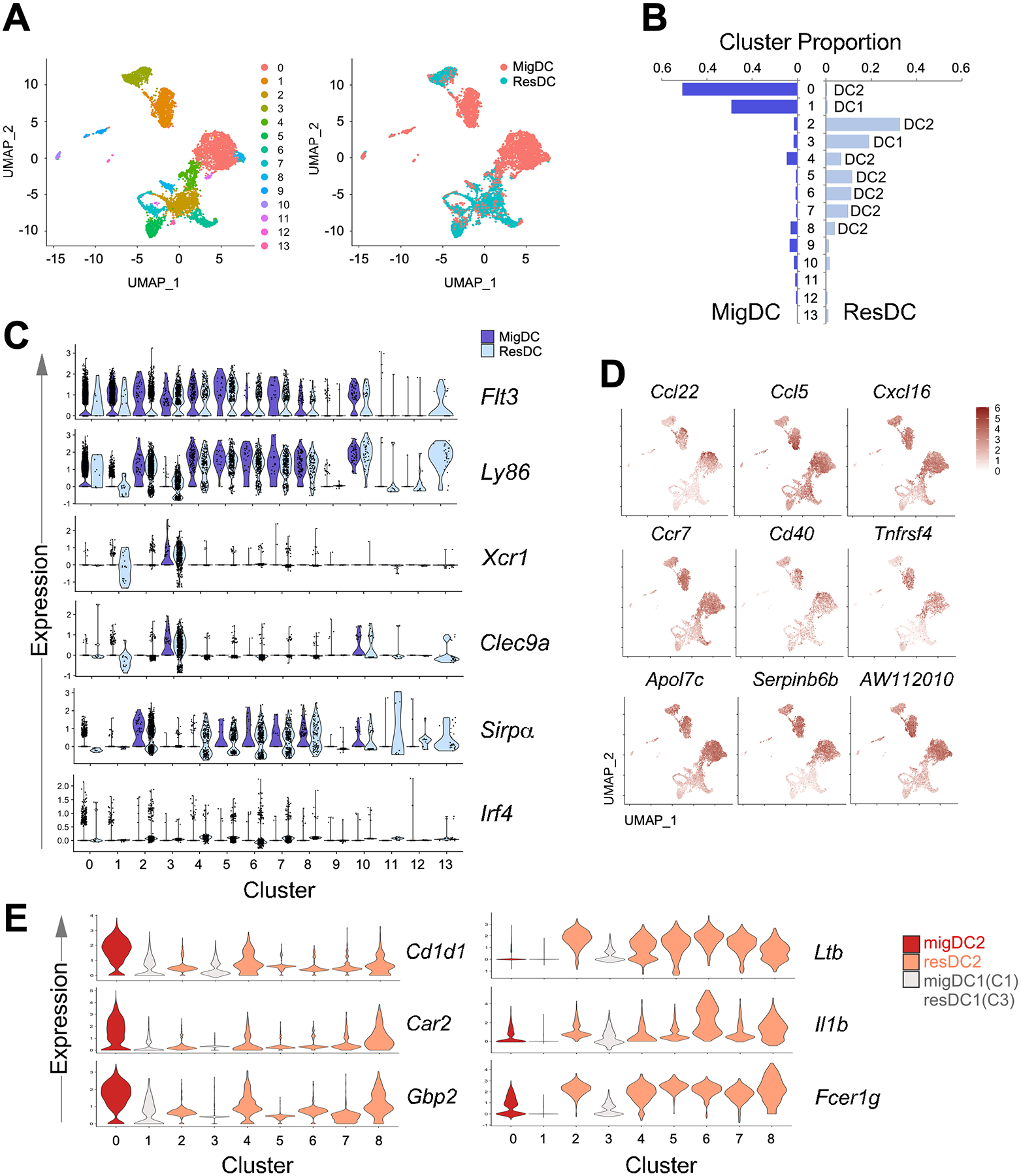
Single cell RNA sequencing of MLN DCs from SFB acutely colonized C57BL/6 mice showed enrichment of costimulation markers and chemokines on migratory DCs. (A) UMAP visualization of transcriptionally distinct cells clusters from integrated analysis of MLN migratory DC and resident DC datasets. (B) Proportion of clusters within migratory or resident DC. DC identity assignment was based on expression of lineage markers in (C): DC1 (Flt3, Ly86, Xcr1, Clec9a), DC2 (Flt3, Ly86, Sirpa, Irf4). (C) Expression of DC lineage markers in cell clusters. (D) Selected transcripts that are preferentially expressed in migratory DC2s compared to resident DC2s. (E) Top transcripts that are expressed by migratory DC2s and resident DC2s.
SFB colonization increases CD40- and CCL22-expressing migratory DC2 subsets
To gain insight into the cellular source of IL-6, we performed an integrated analysis on SFB-negative migratory DCs (4220 cells, 13767 genes) and SFB-positive migratory DCs (4335 cells, 14023 genes) datasets. Eighteen cell clusters were identified (Fig. 6A), with 3 major DC1 and 2 major DC2 clusters (Fig. 6B and C). While the cluster-specific markers of the 3 DC1 clusters are largely overlapping, the major DC2 clusters can be distinguished by the higher level of Csrp1, Ccl22, and Isg15 in C4 (Fig. S4A). In addition to classical DC1/DC2 lineage markers (Irf8 and Fcer1g), these DC subsets can be identified using additional markers as revealed in this analysis, such as Cd81 and Glipr2 versus Cd1d1 and Fabp5 (Fig. 6D and S4B). SFB colonization increased the proportion of DC2s (C0 and C4, Fig. 6B) that are characterized by Ccl22, Cd40, and Cd1d1 compared to the unchanged DC2 clusters (Ltb, S100a4, and S100a6) (Fig. 6E). IL-6 was detected in both of these DC2 clusters but much less or nearly absent in DC1 clusters (Fig. 6F). In contrast, the transcripts of TGFβ, another essential cytokine for in vivo Th17 activation (32), is widely distributed across DC1s and DC2s (Fig. 6F, lower panel). We identified several transcripts that are enriched in migratory DC2s and examined their relationship to IL-6 expression. Il6, Cd40, Ccl17, and Ccl22 are mostly detected on the C4 DC2 cluster, C0 DC2 cluster on the other hand, expressed a higher level of Cd1d1, and Tnfrsf4 (Fig. 7A). However, the transcriptional differences between these DC2 clusters were not resolvable at the protein level using CD1d (Cd1d1) and OX40 (Tnfrsf4), which present an expression spectrum instead of distinct cell populations (Fig. 7B). Nonetheless, we validated that there is an increased expression of these markers compared to lymph node resident and splenic DCs (Fig. 7B). To identify gene signature that is unique to IL-6-producing DCs, we performed differential expression analysis using migratory DCs from SFB-positive mice. Dll4, a Notch ligand was found to be enriched in this DC subset (Fig. 7C), in agreement with the role of this ligand for Th17 cell differentiation (40). additionally, novel transcripts associated with Il6 expression such as Ifitm1, Htra1, Mylk1 and Ccnd2 were revealed. Altogether, our single-cell sequencing data revealed that IL-6-producing, Th17-polarizing DC2s constitute a small fraction of migratory DC2s that co-express Cd40, Ccl17, and Ccl22.
Figure 6.
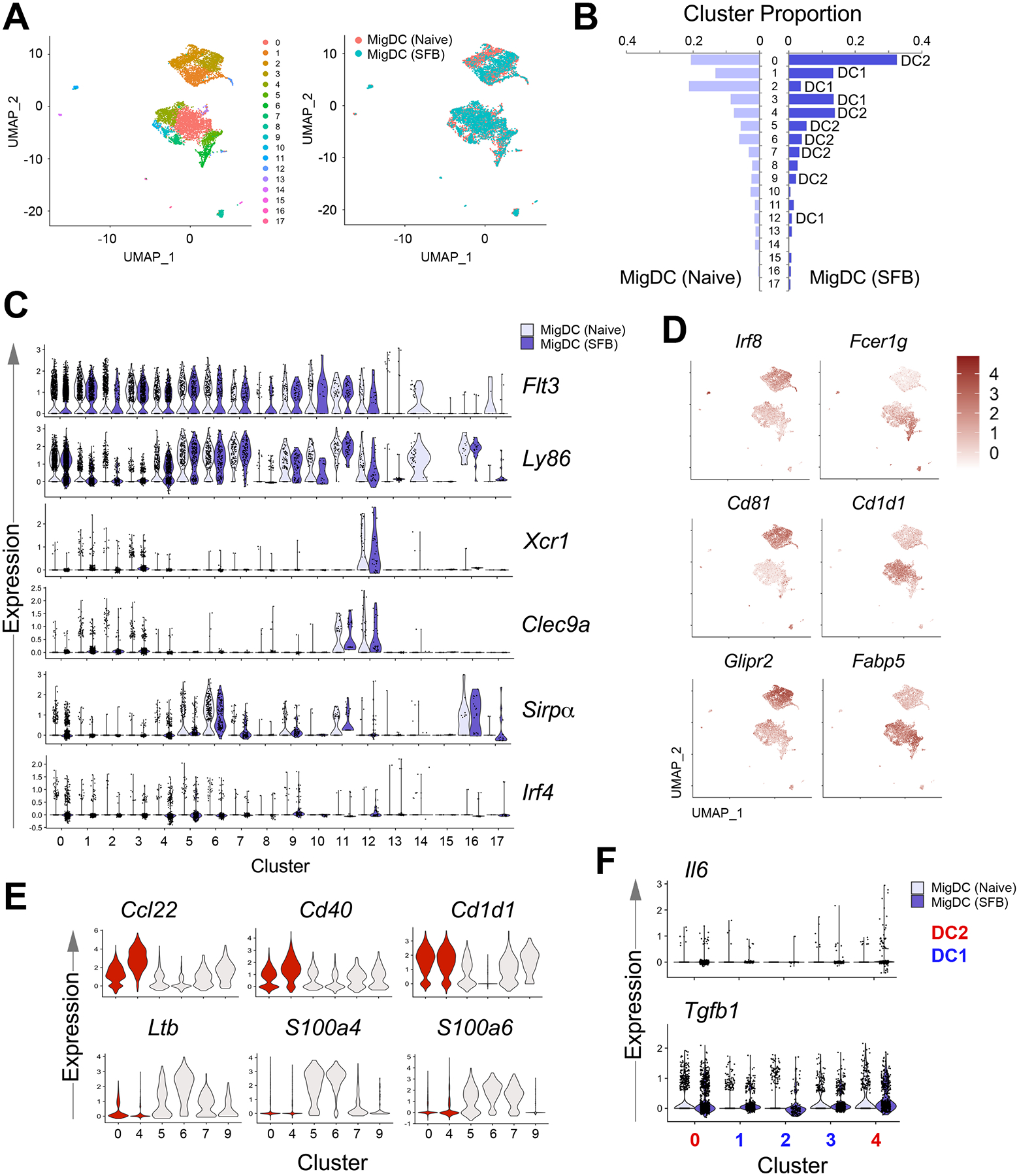
Single cell RNA sequencing of MLN migratory DCs from SFB acutely colonized C57BL/6 mice revealed increase in 2 major DC2 clusters. (A) UMAP visualization of transcriptionally distinct cells clusters from integrated analysis of MLN migratory DC datasets from naive and SFB-colonized mice. (B) Proportion of migratory DC clusters within naive or SFB-colonized mice. DC identity assignment was based on expression of lineage markers in (C): DC1 (Flt3, Ly86, Xcr1, Clec9a), DC2 (Flt3, Ly86, Sirpa, Irf4). (C) Expression of DC lineage markers in cell clusters. (D) Selected transcripts that are preferentially expressed in migratory DC1s compared to DC2s. (E) Top transcripts that are expressed by DC2 clusters that were increased in proportion (red) compared to unchanged DC2 clusters (light grey) in SFB-colonized mice. (F) Detection of the Th17-polarizing cytokines in major DC clusters.
Figure 7.
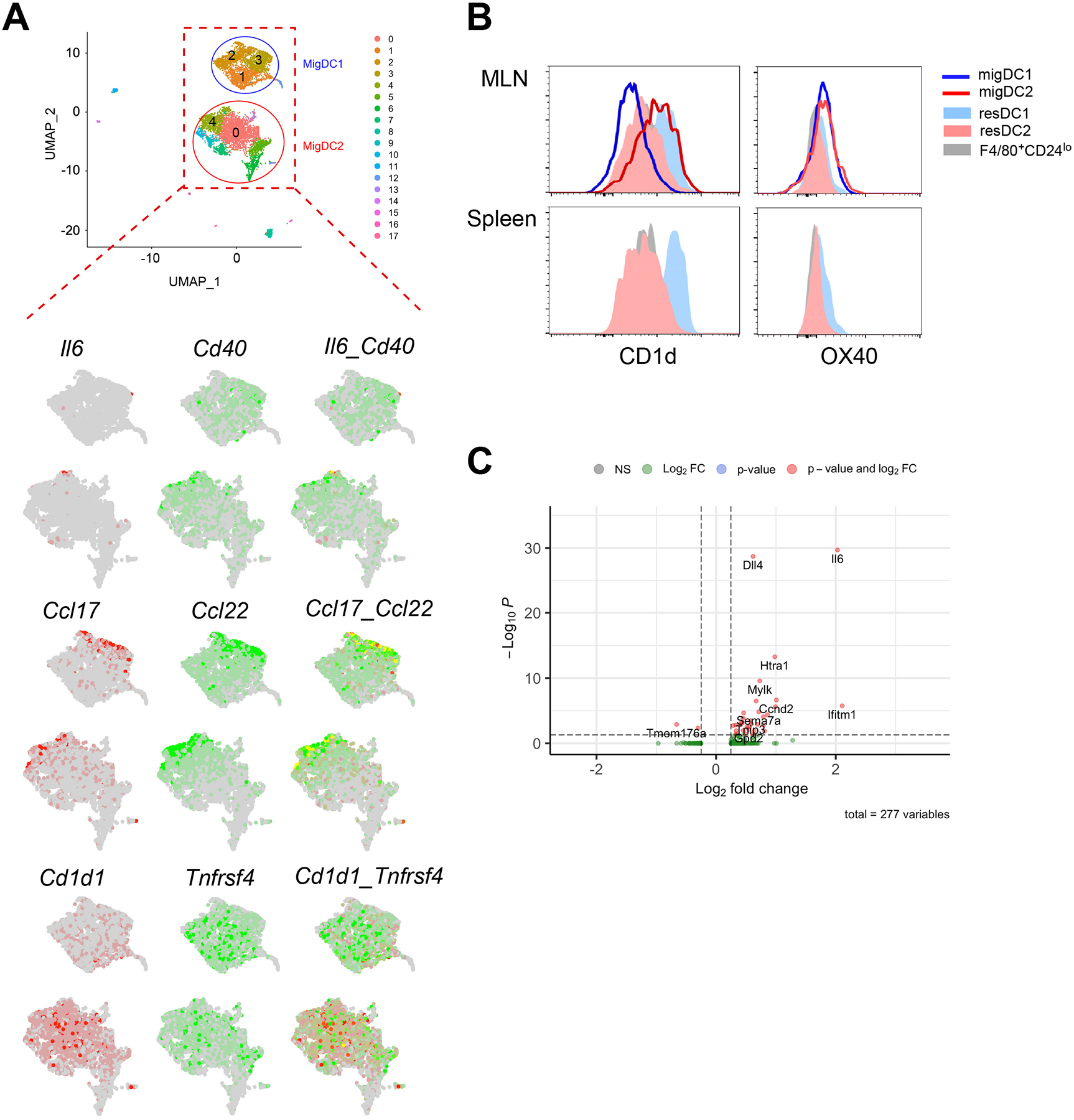
IL-6-producing DCs are a subset of Cd40/Ccl22-expressing DC2s. (A) Merged feature plots of transcripts of interest that are highly expressed by migratory DC2s (Il6-Cd40, Ccl17-Ccl22 and Cd1d1-Tnfrsf4). (B) Flow cytometry validation CD1d and OX40 expression on DC subsets. Data shown are representative of 2 independent experiments with 2–3 mice. (C) Volcano plot displaying differentially expressed genes in IL-6+ versus IL-6− MLN migratory DCs from SFB-colonized mice.
DISCUSSION
Immune cells at the mucosal tissues are charged with the responsibility of host protection and consequently, have devised a plethora of cellular and molecular mechanisms to balance immunity and tolerance at the barrier tissues. Dendritic cells and macrophages are abundant and phenotypically diverse at the intestinal lamina propria (41), making the examination of gut T cell responses challenging.
Historically, CD11b+CD103+ DC2s were shown to be essential for inducing homeostatic CD4 Th17 cells in the gut (10, 13). More recently, along with the emerging interests in the gut microbiome and the identification of specific Th17-inducing commensal bacteria species (SFB), intestinal macrophages were found to control Th17 and Treg responses in the context of microbiota (19, 42). Using the SFB antigen-specific T cell model, our results indicated that dendritic cells control the priming of naive CD4 T cells and the early differentiation of Th17 response (Fig. 1–3). At this early stage of Th17 differentiation, RORγt expression is relatively low, and cell frequency is rare in the small intestine (Fig. 1D). Notably, activated Th17 cells do not persist in the MLN but instead, they home to the small intestine and continue to reinforce RORγt expression and expand (Fig. 1C and D). The kinetics of the Th17 response suggest that early priming steps are initiated by MLN dendritic cells and later, full differentiation and maintenance of the Th17 cell population are likely supported by intestinal macrophages.
Effective T cell differentiation requires proper co-stimulation and cytokine signals delivered by antigen-presenting cells. Ample evidence suggested that IL-6, IL-1β, IL-21, and IL-23 are all implicated in Th17 responses both in physiological and inflammatory settings (32–35, 43, 44). We found that IL-6 is important for Th17 polarization in CD4 T cells, however, IL-6Rα expression is not essential for the optimal expression of RORγt (Fig. 3G and S1H). Examination of the IL-6R signaling pathway revealed that the expression of downstream signaling molecule Stat3 in T cells is required for overall gut Th17 response (29). It is possible that deficiency in T cell-intrinsic IL-6R signaling could be compensated by dendritic cells trans-presenting IL-6 to naïve T cells, which has been shown to support pathogenic Th17 priming (45). Additionally, other Th17-related cytokines could also compensate for the loss of IL-6 (29). Overall, IL-6 may play an important role in early Th17 differentiation or when it is the sole source of polarizing factor (Fig. S1I) but is not absolutely required in vivo.
Despite the redundant role of IL-6, we found it to be informative when probing the DC2 subsets that prime Th17 cells. Concluding from the results obtained with CCR7−/− and BATF3−/− mice (Fig. 3D and F), migratory DC2s are the most likely source of Th17 antigens. DC2s are considerably heterogeneous and can be further subset into Esam+ and Clec12a+ cells in the spleen, both of which are capable of supporting Th17 differentiation (7, 36). Our characterization of MLN migratory DC2s revealed them to be mostly Esam+ and CD24hi and have increased level of costimulatory markers compared to splenic or resident DC2s (Fig. 4 and S2). On the contrary, small intestine DC2s are partially Clec12a+ and devoid of Esam+ cells (Fig. 4). To further understand the composition and function of DC2s, we took an unbiased approach and subjected MLN DCs to single-cell sequencing. IL-6 transcripts were found in a minor subset of DC2s, mostly colocalized with Cd40-high DC2s (Fig. 7A). CD40-CD40L crosstalk has been shown to promote Th17 response (46). However, in the MLN CD40 is expressed by both migratory DC1s and DC2s (Fig. 4D), it is unlikely to be the determining element for CD4 Th17 cell activation in this context. In the lymph nodes, migratory DC1s (with high CCR7 expression) are observed to travel deep into the T cell zone, attracted by the gradient of CCL19/CCL21 (9). This was observed with skin draining lymph node informed using Xcr1 marker (47). The T-B border localization of DC2s due to their lower expression of CCR7 (Fig. 4D, and reviewed in (9)) could be responsible for their preferential interaction with naïve CD4 T cells. Interestingly, we found that CD1d effectively distinguished migratory DC2s from DC1s at the transcript and protein level, displaying a complete reverse trend compared to the resident DCs (Fig. 6D and 7B). CD1d is known to present lipid antigens and activate natural killer T cells (48), but early reports suggest that it can interact with CD4 co-receptor, contributing to CD4+ iNKT cell activation (49). It will be interesting to investigate its role in CD4 T cell activation.
In the MLN, chemokines are constitutively expressed by migratory DCs and are generally more abundant than cytokines. Among the chemokines identified, Ccl17 and Ccl22 overlapped with Il6 in the DC2 cluster that was increased by SFB colonization (Fig. 7A). Conversely, CCR4, the receptor for these 2 chemokines, was detected on newly activated Th17 cells in MLN (Fig. 2D). CCR4/CCR6-deficient mice showed suppressed Th17 cell response in experimental autoimmune encephalomyelitis (50). We speculate that the CCL17/CCL22 produced by DC2s may act as a chemoattractant for Th17 cells for sustained interaction in the MLN, reinforcing Th17 cell differentiation.
Our study identified the pivotal role of migratory DC2s in early Th17 programming at the small intestine. However, it is important to note that DC2s play functionally diverse roles in adaptive T cell responses. They have been shown to induce CD4 Th1, Th2, Th17, TFH and Treg cells (10, 21, 51, 52) when responding to different immunizations. In the MLN, DC1s and DC2s play redundant roles in Th1 response (52, 53), but DC2s are uniquely required for Th2 and Th17 responses. This was clearly demonstrated by subjecting IRF4fl/flCD11cCre mice to S. Mansoni infection or when examined under steady state, whereby Th2/Th17 cytokines were greatly diminished but IFNg production remained unaffected (10, 13, 52). On closer examination using Ag-specific transgenic T cells, it was found that the absence of DC2s did not impair CD4 OTII cells activation and proliferation but specifically blocked IL-17 production after ovalbumin immunization (10). These results suggest that for Th17 differentiation, DC2s deliver mucosal tissues or pathogen-imprinted molecular factors that are not found on DC1s. IL-6, the key cytokine that directs early stage of Th2 and Th17 differentiation (35, 54), represent an important clue for identifying DC2 subset that is specific for this role. Moving forward, the novel markers identified on migratory DC2s could be potential targets for therapeutic intervention in Th17-related inflammatory diseases.
Supplementary Material
Key points:
Gut Th17 response is type 2 DC-driven.
IL-6 is required for Th17 differentiation but do not act directly on T cells.
IL-6 is derived from a type 2 DCs enriched with CD40 and CCL17/CCL21.
ACKNOWLEDGEMENTS
We would like to acknowledge Cynthia Timmers and Marty Romeo of the Translational Science Core, Hollings Cancer Center, Medical University of South Carolina for processing the samples for single cell RNA sequencing. We are grateful to the Flow Cytometry & Cell Sorting Unit of the Medical University of South Carolina, and the Immune Monitoring and Discovery Platform, Pelotonia Institute of Immuno Oncology of the Ohio State University for flow cytometry services. We thank Dr. Zihai Li and Tong Xiao for their advice and help with the bioinformatic analysis.
This work was supported by the NIAID U01Al125859 grant.
Footnotes
COMPETING INTEREST STATEMENT
The authors declare no competing interests
REFERENCES
- 1.Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, Miron M, Kumar BV, Griesemer A, Ho SH, Lerner H, Thome JJC, Connors T, Reizis B, and Farber DL. 2017. Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics over Human Life. Immunity 46: 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, Van Gassen S, Chen J, Poidinger M, De Prijck S, Tavernier SJ, Low I, Irac SE, Mattar CN, Sumatoh HR, Low GHL, Chung TJK, Chan DKH, Tan KK, Hon TLK, Fossum E, Bogen B, Choolani M, Chan JKY, Larbi A, Luche H, Henri S, Saeys Y, Newell EW, Lambrecht BN, Malissen B, and Ginhoux F. 2016. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 45: 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Guttler S, Hutloff A, Mages HW, Ranke K, Schaefer M, Jack RS, Henn V, and Kroczek RA. 2009. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 31: 823–833. [DOI] [PubMed] [Google Scholar]
- 4.Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, Kretzer NM, Durai V, and Murphy KM. 2016. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol 34: 93–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnold IC, Zhang X, Artola-Boran M, Fallegger A, Sander P, Johansen P, and Muller A. 2019. BATF3-dependent dendritic cells drive both effector and regulatory T-cell responses in bacterially infected tissues. PLoS Pathog 15: e1007866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, and Murphy KM. 2008. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322: 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, Klinakis A, Charo IF, Jung S, Gommerman JL, Ivanov II, Liu K, Merad M, and Reizis B. 2011. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35: 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, Yamamoto K, Suematsu T, Nakamura M, Yui K, and Kumatori A. 2004. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc Natl Acad Sci U S A 101: 8981–8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisenbarth SC 2019. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol 19: 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hagerbrand K, Marsal J, Gudjonsson S, Hakansson U, Reizis B, Kotarsky K, and Agace WW. 2013. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 38: 958–969. [DOI] [PubMed] [Google Scholar]
- 11.Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, Decker DC, Blaine KM, Fixsen BR, Singh H, Sciammas R, and Sperling AI. 2013. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun 4: 2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luda KM, Joeris T, Persson EK, Rivollier A, Demiri M, Sitnik KM, Pool L, Holm JB, Melo-Gonzalez F, Richter L, Lambrecht BN, Kristiansen K, Travis MA, Svensson-Frej M, Kotarsky K, and Agace WW. 2016. IRF8 Transcription-Factor-Dependent Classical Dendritic Cells Are Essential for Intestinal T Cell Homeostasis. Immunity 44: 860–874. [DOI] [PubMed] [Google Scholar]
- 13.Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AW, See P, Shin A, Wasan PS, Hoeffel G, Malleret B, Heiseke A, Chew S, Jardine L, Purvis HA, Hilkens CM, Tam J, Poidinger M, Stanley ER, Krug AB, Renia L, Sivasankar B, Ng LG, Collin M, Ricciardi-Castagnoli P, Honda K, Haniffa M, and Ginhoux F. 2013. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 38: 970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, and Littman DR. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, and Weaver CT. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6: 1123–1132. [DOI] [PubMed] [Google Scholar]
- 16.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, and Dong C. 2005. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6: 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, Ishikawa E, Shima T, Hara T, Kado S, Jinnohara T, Ohno H, Kondo T, Toyooka K, Watanabe E, Yokoyama S, Tokoro S, Mori H, Noguchi Y, Morita H, Ivanov II, Sugiyama T, Nunez G, Camp JG, Hattori M, Umesaki Y, and Honda K. 2015. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163: 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ladinsky MS, Araujo LP, Zhang X, Veltri J, Galan-Diez M, Soualhi S, Lee C, Irie K, Pinker EY, Narushima S, Bandyopadhyay S, Nagayama M, Elhenawy W, Coombes BK, Ferraris RP, Honda K, Iliev ID, Gao N, Bjorkman PJ, and Ivanov II. 2019. Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panea C, Farkas AM, Goto Y, Abdollahi-Roodsaz S, Lee C, Koscso B, Gowda K, Hohl TM, Bogunovic M, and Ivanov II. 2015. Intestinal Monocyte-Derived Macrophages Control Commensal-Specific Th17 Responses. Cell Rep 12: 1314–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott CL, Bain CC, Wright PB, Sichien D, Kotarsky K, Persson EK, Luda K, Guilliams M, Lambrecht BN, Agace WW, Milling SW, and Mowat AM. 2015. CCR2(+)CD103(−) intestinal dendritic cells develop from DC-committed precursors and induce interleukin-17 production by T cells. Mucosal Immunol 8: 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazzini E, Massimiliano L, Penna G, and Rescigno M. 2014. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1(+) macrophages to CD103(+) dendritic cells. Immunity 40: 248–261. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, Sczesnak A, Liao JJ, Torres VJ, Jenkins MK, Lafaille JJ, and Littman DR. 2014. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510: 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dolton G, Zervoudi E, Rius C, Wall A, Thomas HL, Fuller A, Yeo L, Legut M, Wheeler S, Attaf M, Chudakov DM, Choy E, Peakman M, and Sewell AK. 2018. Optimized Peptide-MHC Multimer Protocols for Detection and Isolation of Autoimmune T-Cells. Front Immunol 9: 1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romagnoli PA, Fu HH, Qiu Z, Khairallah C, Pham QM, Puddington L, Khanna KM, Lefrancois L, and Sheridan BS. 2017. Differentiation of distinct long-lived memory CD4 T cells in intestinal tissues after oral Listeria monocytogenes infection. Mucosal Immunol 10: 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srinivasan A, Foley J, and McSorley SJ. 2004. Massive number of antigen-specific CD4 T cells during vaccination with live attenuated Salmonella causes interclonal competition. J Immunol 172: 6884–6893. [DOI] [PubMed] [Google Scholar]
- 26.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Myers RM, Bonneau R, and Littman DR. 2012. A validated regulatory network for Th17 cell specification. Cell 151: 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, and Song SY. 2004. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21: 527–538. [DOI] [PubMed] [Google Scholar]
- 28.Wang C, Kang SG, Lee J, Sun Z, and Kim CH. 2009. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol 2: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sano T, Kageyama T, Fang V, Kedmi R, Martinez CS, Talbot J, Chen A, Cabrera I, Gorshko O, Kurakake R, Yang Y, Ng C, Schwab SR, and Littman DR. 2021. Redundant cytokine requirement for intestinal microbiota-induced Th17 cell differentiation in draining lymph nodes. Cell Rep 36: 109608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, and Murphy KM. 2012. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med 209: 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, and O’Shea JJ. 2010. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467: 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, and Littman DR. 2008. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4: 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, and Dong C. 2009. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30: 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 35.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, and Littman DR. 2007. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8: 967–974. [DOI] [PubMed] [Google Scholar]
- 36.Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallee VP, Mendoza A, Fromme R, Mazutis L, Ariyan C, Leslie C, Pe’er D, and Rudensky AY. 2019. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 179: 846–863 e824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Askew D, and Harding CV. 2008. Antigen processing and CD24 expression determine antigen presentation by splenic CD4+ and CD8+ dendritic cells. Immunology 123: 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim TS, Gorski SA, Hahn S, Murphy KM, and Braciale TJ. 2014. Distinct dendritic cell subsets dictate the fate decision between effector and memory CD8(+) T cell differentiation by a CD24-dependent mechanism. Immunity 40: 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R. 2019. Comprehensive Integration of Single-Cell Data. Cell 177: 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meng L, Bai Z, He S, Mochizuki K, Liu Y, Purushe J, Sun H, Wang J, Yagita H, Mineishi S, Fung H, Yanik GA, Caricchio R, Fan X, Crisalli LM, Hexner EO, Reshef R, Zhang Y, and Zhang Y. 2016. The Notch Ligand DLL4 Defines a Capability of Human Dendritic Cells in Regulating Th1 and Th17 Differentiation. J Immunol 196: 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gross M, Salame TM, and Jung S. 2015. Guardians of the Gut - Murine Intestinal Macrophages and Dendritic Cells. Front Immunol 6: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, Schady D, Bettini ML, Simpson KW, Longman RS, Littman DR, and Diehl GE. 2018. Critical Role for the Microbiota in CX3CR1(+) Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity 49: 151–163 e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, Blumenschein WM, Sukumar S, Haines CJ, Sadekova S, McClanahan TK, McGeachy MJ, O’Shea JJ, and Cua DJ. 2016. Interleukin-23-Induced Transcription Factor Blimp-1 Promotes Pathogenicity of T Helper 17 Cells. Immunity 44: 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, and Kuchroo VK. 2012. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13: 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, Husterer V, Croxford AL, Moller-Hackbarth K, Bartsch HS, Sotlar K, Krebs S, Regen T, Blum H, Hemmer B, Misgeld T, Wunderlich TF, Hidalgo J, Oukka M, Rose-John S, Schmidt-Supprian M, Waisman A, and Korn T. 2017. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat Immunol 18: 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, and Kopf M. 2009. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci U S A 106: 876–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitano M, Yamazaki C, Takumi A, Ikeno T, Hemmi H, Takahashi N, Shimizu K, Fraser SE, Hoshino K, Kaisho T, and Okada T. 2016. Imaging of the cross-presenting dendritic cell subsets in the skin-draining lymph node. Proc Natl Acad Sci U S A 113: 1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macho-Fernandez E, and Brigl M. 2015. The Extended Family of CD1d-Restricted NKT Cells: Sifting through a Mixed Bag of TCRs, Antigens, and Functions. Front Immunol 6: 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thedrez A, de Lalla C, Allain S, Zaccagnino L, Sidobre S, Garavaglia C, Borsellino G, Dellabona P, Bonneville M, Scotet E, and Casorati G. 2007. CD4 engagement by CD1d potentiates activation of CD4+ invariant NKT cells. Blood 110: 251–258. [DOI] [PubMed] [Google Scholar]
- 50.Scheu S, Ali S, Ruland C, Arolt V, and Alferink J. 2017. The C-C Chemokines CCL17 and CCL22 and Their Receptor CCR4 in CNS Autoimmunity. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Briseno CG, Satpathy AT, Davidson J. T. t., Ferris ST, Durai V, Bagadia P, O’Connor KW, Theisen DJ, Murphy TL, and Murphy KM. 2018. Notch2-dependent DC2s mediate splenic germinal center responses. Proc Natl Acad Sci U S A 115: 10726–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayer JU, Demiri M, Agace WW, MacDonald AS, Svensson-Frej M, and Milling SW. 2017. Different populations of CD11b(+) dendritic cells drive Th2 responses in the small intestine and colon. Nat Commun 8: 15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Everts B, Tussiwand R, Dreesen L, Fairfax KC, Huang SC, Smith AM, O’Neill CM, Lam WY, Edelson BT, Urban JF Jr., Murphy KM, and Pearce EJ. 2016. Migratory CD103+ dendritic cells suppress helminth-driven type 2 immunity through constitutive expression of IL-12. J Exp Med 213: 35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rincon M, Anguita J, Nakamura T, Fikrig E, and Flavell RA. 1997. Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exp Med 185: 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data discussed in this publication have been deposited in NCMI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE184423 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi).