

Abstract
The structure of wild-type pentameric C-reactive protein (CRP) is stabilized by two calcium ions and which are required for the binding of CRP to its ligand phosphocholine. CRP in its structurally altered pentameric conformations also binds to proteins that are denatured and aggregated by immobilization on microtiter plates; however, the identity of the ligand on immobilized proteins remains unknown. We tested the hypotheses that immobilization of proteins generated an amyloid-like structure and that amyloid-like structure was the ligand for structurally altered pentameric CRP. We found that the antibodies to amyloid-β peptide 1–42 (Aβ) reacted with immobilized proteins indicating that some immobilized proteins express an Aβ epitope. Accordingly, four different CRP mutants capable of binding to immobilized proteins were constructed and their binding to fluid-phase Aβ was determined. All CRP mutants bound to fluid-phase Aβ suggesting that Aβ is a ligand for structurally altered pentameric CRP. In addition, the interaction between CRP mutants and Aβ prevented the formation of Aβ fibrils. The growth of Aβ fibrils was also halted when CRP mutants were added to growing fibrils. Biochemical analyses of CRP mutants revealed altered topology of the Ca2+-binding site, suggesting a role of this region of CRP in binding to Aβ. Combined with previous reports that structurally altered pentameric CRP is generated in vivo, we conclude that CRP is a dual pattern recognition molecule and an anti-amyloidogenic protein. These findings have implications for Alzheimer’s and other neurodegenerative diseases caused by amyloidosis and for the diseases caused by the deposition of otherwise fluid-phase proteins.
Keywords: Acute phase reactant, Amyloid-β, Amyloidosis, C-reactive protein, Pattern recognition receptor
Introduction
C-reactive protein (CRP) is composed of five identical subunits held together by noncovalent bonds and arranged as a cyclic pentamer (1, 2). Each subunit possesses two Ca2+ ions which are necessary for the structural integrity of the native pentamer and for binding of CRP to its primary ligand phosphocholine (PCh) (2–4). Since CRP recognizes PCh, a pathogen-associated molecular pattern, CRP is regarded as a pattern recognition molecule of the innate immune system (5, 6). Crystallography of CRP-Ca2+-PCh complexes has demonstrated that two Ca2+ ions in CRP are coordinated by Asp60, Asn61, and by residues Glu138, Gln139, Asp140, Glu147 and Gln150 present in a loop (7). In the absence of Ca2+, the Ca2+-coordinating loop moves away from the main body of the CRP molecule exposing an otherwise hidden site of proteolysis (8–10). Thus, bound Ca2+ protects CRP from proteolytic cleavage.
Amyloid-β (Aβ) peptides, 36–43 amino acid residues long, are normal components of plasma and cerebrospinal fluid. Two main variants of this peptide exist in humans and include Aβ40 and Aβ42 which are 40 and 42 amino acid residues long, respectively. The most prevalent circulating Aβ variant is Aβ40; however, Aβ42 has been shown to form amyloid fibrils more rapidly (11–14). The aggregation of Aβ peptides into oligomers and fibrils has been implicated in the development and progression of Alzheimer’s disease (AD) (15–18). CRP, remnants of CRP post-ligand binding, and CRP-like immunoreactivity have all been seen at sites of inflammation including in amyloid plaques of AD (19–25). The functions of CRP, however, at the amyloid plaques are not known.
At physiological pH, the binding of CRP to PCh-containing substances is the primary ligand recognition function of CRP, although the binding of CRP to several protein-ligands have also been reported (26–29). At acidic pH, the pentameric structure of CRP is altered, and such acidic pH-treated CRP has been shown to bind to a variety of proteins that are denatured and aggregated by immobilization, irrespective of the identity of the immobilized protein (10, 30–35). Results similar to that of acidic pH-treated CRP were also observed with H2O2-treated CRP (36). CRP has been structurally altered in vitro by employing site-directed mutagenesis to generate CRP mutants which, like acidic pH-treated and H2O2-treated CRP, bound to immobilized proteins including oxidized low-density lipoprotein (ox-LDL) and complement inhibitor factor H (FH) (37–39). Four such CRP mutants capable of binding to immobilized and aggregated proteins at physiological pH have been reported previously: E42Q, Y40F/E42Q, F66A/T76Y/E81A and E42Q/F66A/T76Y/E81A (37–40). CRP mutants E42Q and Y40F/E42Q retain their PCh-binding activity while CRP mutants F66A/T76Y/E81A and E42Q/F66A/T76Y/E81A do not bind to PCh since in these two mutants the PCh-binding site was mutated (38–42).
The ligand displayed by proteins immobilized on microtiter plates which is recognized by acidic pH-treated, H2O2-treated, and CRP mutants mentioned above has not been defined yet. We hypothesized that the dense immobilization of proteins onto microtiter plates caused aggregation and denaturation of proteins resulting in the generation and exposure of a common structure such as an amyloid-like structure which was being recognized by these modified CRP species. We tested this hypothesis by employing anti-Aβ antibodies to detect proteins immobilized on microtiter plates. We surmised that if our hypothesis turns out to be correct, then the modified CRP species should also be able to bind to purified Aβ peptides in the fluid-phase. Accordingly, we employed CRP mutants E42Q, Y40F/E42Q, F66A/T76Y/E81A and E42Q/F66A/T76Y/E81A (collectively referred to as “structurally altered pentameric CRP” in this study) and tested their binding to both immobilized and fluid-phase Aβ. The consequences of structurally altered pentameric CRP-Aβ interactions on the formation of Aβ fibrils were also evaluated.
Materials and Methods
Assay for the detection of amyloid-like structures on immobilized proteins
For immobilization, five different randomly selected proteins were utilized: Aβ peptide 1–42 (Bachem), ox-LDL (Biomedical Technologies), FH (Complement Technology), fibronectin (Fn) (Sigma) and BSA (Sigma). Microtiter wells (Corning, 9018) were coated with 100 μl of 10 μg/ml of each protein in TBS, pH 7.2, in duplicate, and incubated overnight at 4°C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin for 45 min at room temperature. Both, polyclonal anti-Aβ antibodies (Novus, NBP2-25093) and monoclonal anti-Aβ antibodies (Novus, NBP2-13075) were used to detect the amyloid-like structures formed on the proteins following their immobilization. Normal rabbit IgG and normal mouse IgG were use as controls for the antibodies. The anti-Aβ antibodies (10 μg/ml) diluted in TBS containing 0.1% gelatin and 0.02% Tween 20 were added to the wells and incubated for 1 h at 37°C. After washing the wells, bound polyclonal anti-Aβ antibodies were detected by using HRP-conjugated donkey anti-rabbit IgG (GE Healthcare) and bound monoclonal anti-Aβ antibodies were detected by using HRP-conjugated goat anti-mouse IgG (Thermo Fisher Scientific). Color was developed, and the OD was read at 405 nm.
Preparation of Aβ monomers and Aβ fibrils
Aβ monomers and Aβ fibrils were prepared according to a published method (43). Lyophilized Aβ peptides 1–42 (Bachem, H-1368; m.w. 4514) were dissolved in hexafluoroisopropanol (Sigma) to obtain a 1 mM (4.5 mg/ml) Aβ solution which was then incubated for 2 h at 37°C for monomerization of the oligomers present in the Aβ peptide solution. Aβ monomers were aliquoted (112.8 μg/25 μl/vial) in glass vials. After removing hexafluoroisopropanol by evaporation overnight, the vials containing the film of Aβ monomers were stored at −20°C. When needed, 564 μl ice-cold TBS was added to an aliquot to obtain a 200 μg/ml solution of Aβ monomers. To prepare Aβ fibrils, the 200 μg/ml solution of Aβ monomers was vortexed and transferred to a 96-well microtiter plate (Immunochemistry Technologies, Costar 266). The plate was incubated for 3 h at 37°C with shaking at 300 rpm. The resulting Aβ fibrils were stored at −20°C until needed.
The Aβ monomers and Aβ fibrils were visualized by transmission electron microscopy (TEM). For TEM, Aβ monomers or Aβ fibrils (10 μl) were applied to carbon-coated copper grids followed by counterstaining with 2% uranyl acetate and visualized using a Tecnai 120 kV F20 electron microscope at the Molecular Electron Microscopy Core facility, University of Virginia. The results of TEM (Fig. 1) verified the preparations of both Aβ monomers and Aβ fibrils. In addition to TEM, the Aβ monomers and fibrils were also verified by thioflavin T (ThT) assay for Aβ fibrillation, as described below.
FIGURE 1.
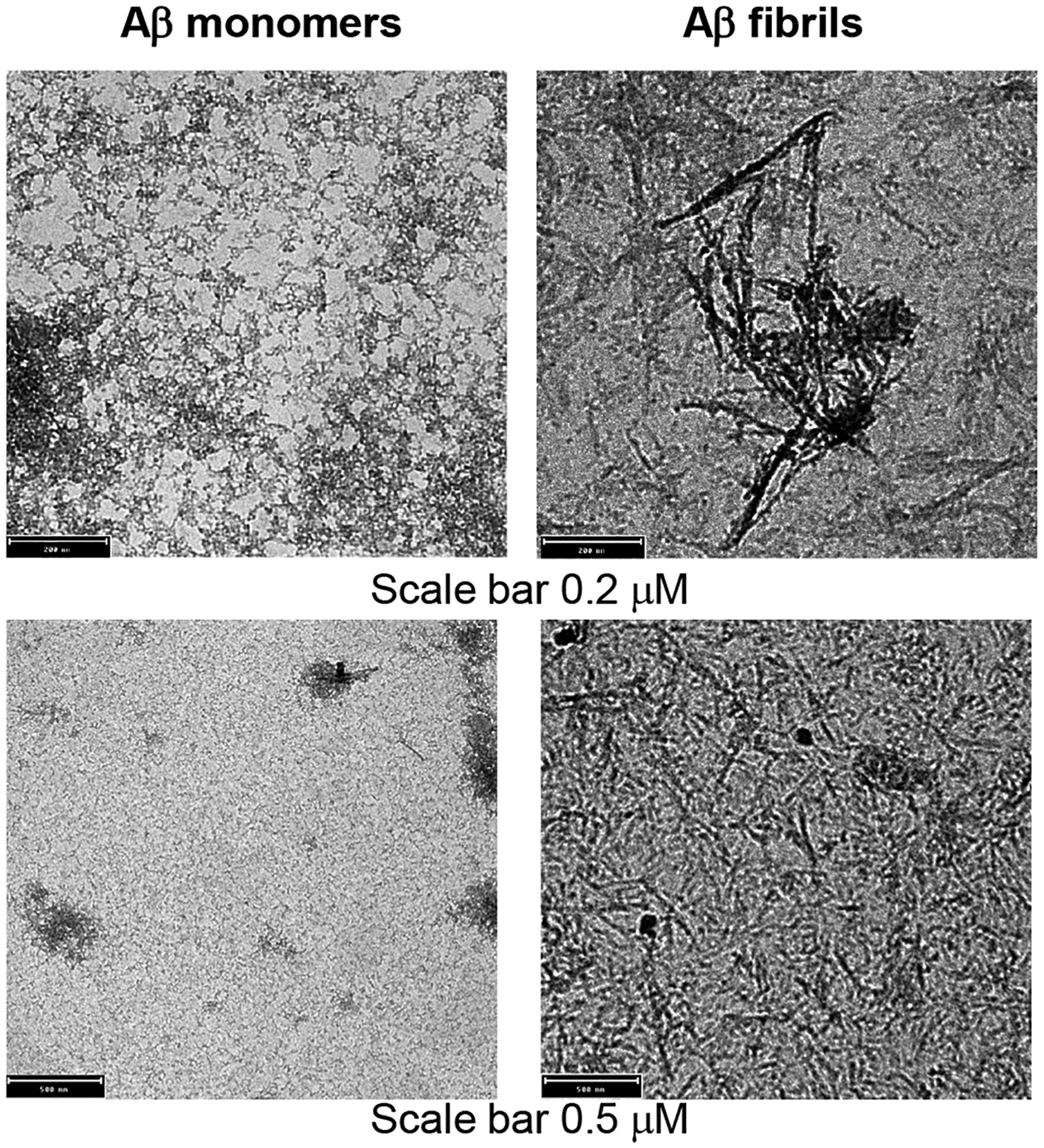
Transmission electron micrographs of Aβ monomers and Aβ fibrils, shown at two different resolutions.
Construction and expression of CRP mutants
The construction of E42Q, Y40F/E42Q, F66A/T76Y/E81A and E42Q/F66A/T76Y/E81A CRP mutants employed in this study has been reported previously (37–41). CRP mutants were expressed in CHO cells using the ExpiCHO Expression System (Thermo Fisher Scientific) according to manufacturer’s instructions and as described previously (44).
Purification of CRP
WT CRP was purified from discarded human pleural fluid as described previously (45). E42Q and Y40F/E42Q CRP mutants were purified from CHO cell culture supernatants by Ca2+-dependent affinity chromatography on a PCh-conjugated Sepharose column (Pierce), followed by ion-exchange chromatography on a MonoQ column and gel filtration on a Superose12 column, as described earlier (37, 38). The E42Q/F66A/T76Y/E81A and F66A/T76Y/E81A CRP mutants were purified from CHO cell culture supernatants by Ca2+-dependent affinity chromatography on a phosphoethanolamine-conjugated Sepharose column, followed by ion-exchange chromatography on a MonoQ column and gel filtration on a Superose12 column, as described earlier (38, 39). Purified CRP in TBS, pH 7.2, containing 2 mm CaCl2, was stored at 4°C, and was used within a week.
Solid-phase Aβ-binding assay
The Aβ-binding assays were performed as described previously (32, 39). In brief, lyophilized Aβ peptides were reconstituted in TBS. Microtiter wells (Corning, 9018) were coated with 10 μg/ml of Aβ, in duplicate, overnight at 4°C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin. CRP, diluted in TBS containing 0.1% gelatin, 0.02% Tween 20, and 2 mm CaCl2 (TBS-Ca), was added and then incubated for 2 h at 37°C. After washing the wells, rabbit polyclonal anti-CRP antibody (Sigma) was used to detect bound CRP. HRP-conjugated donkey anti-rabbit IgG was used as the secondary antibody. Color was developed, and the OD was read at 405 nm. The assays were performed using TBS-Ca, pH 7.2, unless otherwise mentioned.
Aβ-binding inhibition assay
For inhibition assays, Aβ-binding assays were performed as described above, except that CRP was added along with the inhibitors (Aβ monomers and Aβ fibrils) to Aβ-coated wells. Before adding CRP to the wells, CRP was mixed with Aβ monomers, or with Aβ fibrils, in equal volumes to give a final concentration of 100 μg/ml of the inhibitor.
ThT assay for Aβ fibrillation
The fibrillation of Aβ monomers was monitored by employing ThT assay according to a published method (46). In one assay, the effects of CRP on Aβ fibrillation were determined by adding CRP to Aβ monomers at time zero, that is, CRP and Aβ monomers were mixed together before the beginning of the fibrillation reaction. For this assay, the fibrillation reaction mix was prepared, with and without CRP, in which the final concentrations of Aβ and ThT were 150 μg/ml and 10 μM, respectively. In CRP-containing mixture, the final concentrations of CRP were 1, 10 and 100 μg/ml. After vortexing, 240 μl of each mixture was transferred in triplicate wells in a 96-well microtiter plate (Immunochemistry Technologies, Costar 266). Fluorescence was measured by using the Synergy H1 microplate reader (BioTek) with excitation at 440 nm and emission at 480 nm. After the first measurement at 5 min, the plate was incubated for 3 h at 37°C with shaking at 300 rpm; fluorescence was measured every 15 min for 3 h.
In another assay, the effects of CRP on Aβ fibrillation was determined by adding CRP to Aβ 45 min after the fibrillation began. For this assay, the fibrillation reaction mix was prepared without CRP, in which the final concentrations of Aβ and ThT were 167 μg/ml and 10 μM, respectively. After vortexing, 216 μl of each mixture was transferred in triplicate wells in the microtiter plate. After the first measurement at 5 min, the plate was incubated at 37°C with shaking at 300 rpm. The fluorescence was measured every 15 min. After 45 min, either TBS (24 μl) or CRP (24 μl) was added to the wells to obtain a final concentration of 100 μg/ml CRP. More ThT was added to maintain the ThT concentration at 10 μM. The measurement of fluorescence continued every 15 min for another 2.25 h.
Ca2+-requirement assay
The concentration of Ca2+ required for the binding of CRP to PCh was assessed using pneumococcal C-polysaccharide (PnC) (Statens Serum Institut) as the PCh-ligand, as described previously (37, 47). In brief, microtiter plates (Corning, 9018) were coated with 10 μg/ml of PnC in TBS, in duplicate wells, overnight at 4°C. CRP diluted in TBS-Ca (with various concentrations of Ca2+) was added in duplicate wells. After incubating the plates for 2 h at 37°C, the wells were washed with appropriate TBS-Ca. Purified anti-CRP mAb HD2.4 (0.5 μg/ml), diluted in TBS-Ca, was used to detect bound CRP. HRP-conjugated goat anti-mouse IgG, diluted in TBS-Ca, was used as the secondary antibody. Color was developed and the OD was read at 405 nm.
Ca2+-site proteolytic cleavage assay
The Ca2+-binding site-dependent proteolytic cleavage assay of CRP was conducted as described previously (8, 10) with modifications. CRP stored in TBS, pH 7.2, containing 2 mm CaCl2, was first dialyzed against TBS to remove Ca2+. Dialyzed CRP (10 μg; final concentration 2.87 μM) in TBS was incubated with 3 μg protease (Sigma, P8811), with and without 0.5 mM or 5 mM CaCl2, for 2 h at 37°C, and subjected to SDS-PAGE on a 4%−20% polyacrylamide gradient gel under reducing conditions. The gels were stained with Coomassie Brilliant Blue. BioRad’s broad-range marker was used as the m.w. standard.
Results
Immobilized proteins express amyloid-like structures
The results of the experiments in which anti-Aβ antibodies were used to identify the expression of amyloid-like structures on immobilized proteins are shown in Fig. 2. Purified Aβ peptides were immobilized to verify the specificity of the monoclonal (Fig. 2A) and polyclonal anti-Aβ antibodies (Fig. 2B). Among the other four randomly selected proteins for immobilization, ox-LDL was most reactive with anti-Aβ antibodies. Immobilized FH and Fn also reacted with anti-Aβ antibodies. These immobilized proteins were recognized by both polyclonal and monoclonal anti-Aβ antibodies although the reactivities of the two antibodies with the immobilized proteins were different. Both antibodies failed to detect an amyloid-like structure on immobilized BSA. Overall, these data suggest that immobilized proteins express Aβ epitopes or amyloid-like structures and that each protein has different amyloidogenicity, that is, the extent of amyloid-like structures or the number of Aβ epitopes formed on different proteins varies.
FIGURE 2.
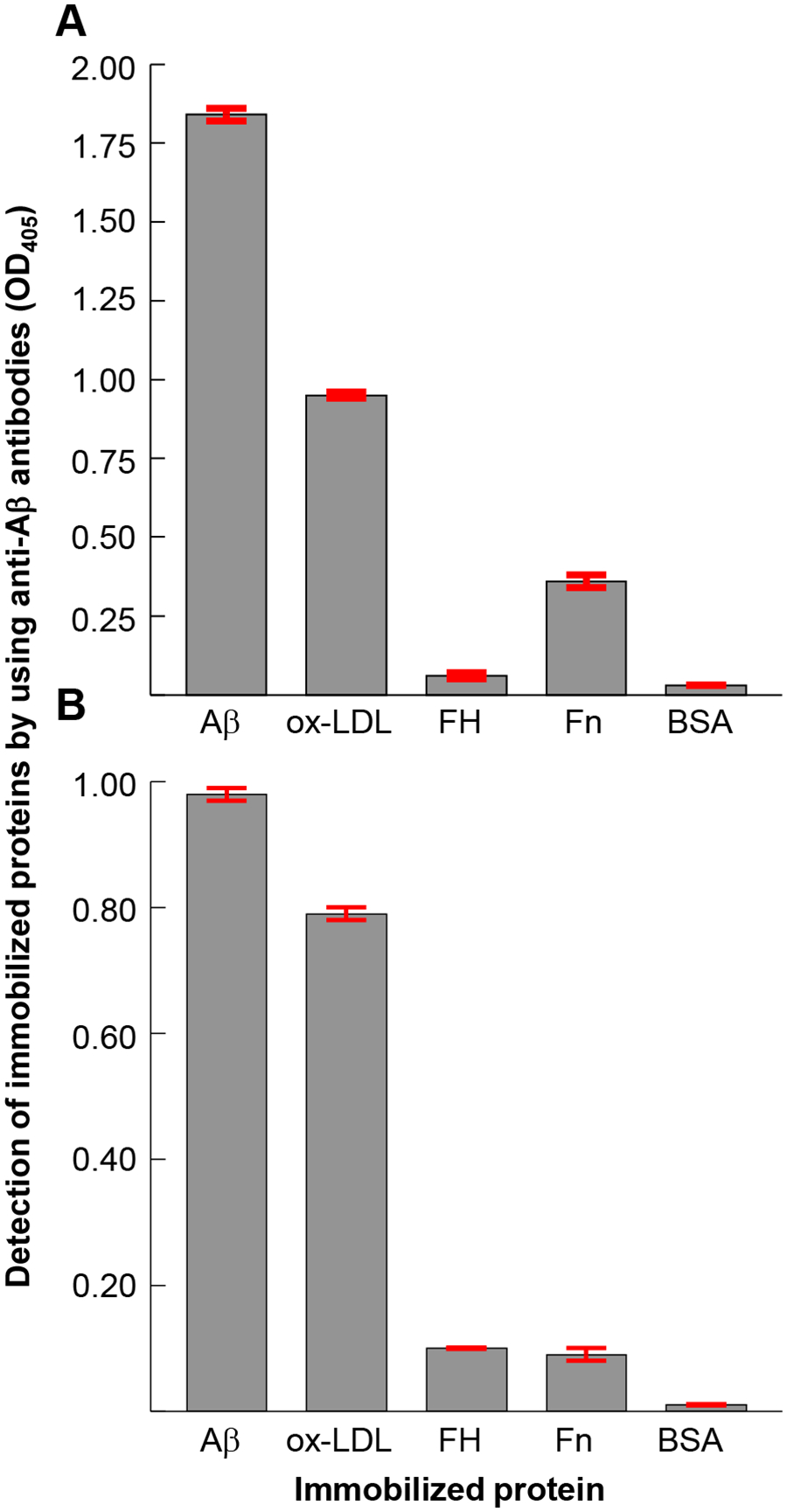
Expression of an Aβ epitope on immobilized proteins. Microtiter wells were coated with Aβ, oxidized LDL (ox-LDL), factor H (FH), fibronectin (Fn) and BSA, in duplicate. The unreacted sites in the wells were blocked with gelatin. Anti-Aβ antibodies were then added to the wells to detect the amyloid-like structures formed on the proteins following their immobilization. (A) Monoclonal anti-Aβ antibodies were used. Bound anti-Aβ antibodies were detected by using HRP-conjugated goat anti-mouse IgG. (B) Polyclonal anti-Aβ antibodies were used. Bound anti-Aβ antibodies were detected by using HRP-conjugated donkey anti-rabbit IgG. The OD of the developed color was read at 405 nm. Data shown are mean ± SEM of three experiments.
Aβ and amyloid-like structures are ligands of structurally altered pentameric CRP
We first determined the binding of various structurally altered pentameric CRP species to immobilized Aβ. The results of the solid phase Aβ-binding assays are shown in Fig. 3. WT CRP at pH 5.0 and pH 7.2 were included as positive and negative controls for the binding of CRP to immobilized Aβ. As shown, and as has been reported previously (31, 32), WT CRP did not bind to Aβ at pH 7.2, but, at pH 5.0, WT CRP bound to Aβ in a CRP concentration-dependent manner. Similar to the binding of WT CRP to Aβ at acidic pH, all four CRP mutants bound to Aβ at physiological pH in a CRP concentration-dependent manner, irrespective of whether the PCh-binding site in the CRP mutants was mutated or not. As shown, the avidity of the binding of E42Q/F66A/T76Y/E81A CRP to Aβ was highest while the avidity of the binding of E42Q CRP to Aβ was lowest of all other mutants.
FIGURE 3.
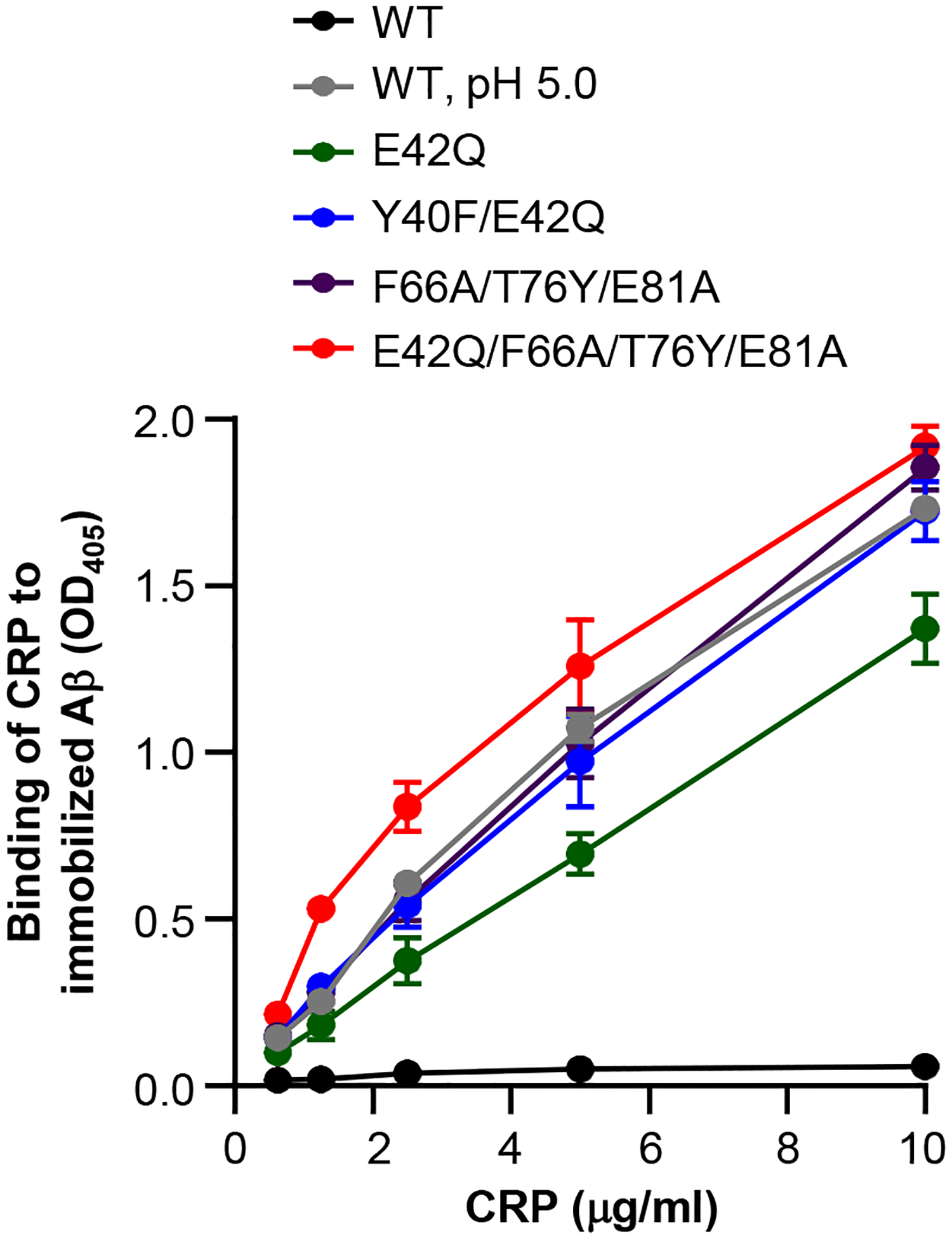
Binding of CRP to immobilized Aβ. Microtiter wells were coated with Aβ. The unreacted sites in the wells were blocked with gelatin. WT CRP, diluted in TBS-Ca, pH 7.2, and diluted in TBS-Ca, pH 5.0, was added in duplicate wells. Mutant CRP was diluted in TBS-Ca, pH 7.2, only and was added in duplicate wells. Bound CRP was detected by using a rabbit polyclonal anti-CRP antibody as the primary antibody and HRP-conjugated donkey anti-rabbit IgG as the secondary antibody. The OD of the developed color was read at 405 nm. Data shown are mean ± SEM of three experiments.
Previously, we reported that structurally altered pentameric CRP binds to immobilized proteins regardless of the identity of the protein (10, 29, 31, 32, 36–39). If the binding of structurally altered pentameric CRP to immobilized proteins was through the ligand Aβ, then CRP mutants employed in this study should be able to bind to purified Aβ in the fluid phase. Accordingly, Aβ-binding inhibition assays were performed in which the inhibition of binding of CRP mutants to immobilized Aβ by fluid-phase Aβ monomers and Aβ fibrils were determined. Two of the four CRP mutants were tested; one that retains the PCh-binding site (Y40F/E42Q) and one with the mutated PCh-binding site (E42Q/F66A/T76Y/E81A). Both Aβ monomers and Aβ fibrils inhibited the binding of Y40F/E42Q CRP to immobilized Aβ, indicating interactions between Y40F/E42Q CRP and Aβ, either monomers or fibrils, in the fluid phase (Fig. 4, left). Greater inhibition by Aβ fibrils than by Aβ monomers suggested that the avidity of binding of Y40F/E42Q CRP to Aβ fibrils was higher than that of Aβ monomers. Similar results were obtained with the CRP mutant E42Q/F66A/T76Y/E81A (Fig. 4, right). These data suggest that Aβ is a ligand of structurally altered pentameric CRP. Inhibition assays for the binding of CRP mutants to immobilized proteins other than Aβ could not be performed since fluid-phase Aβ interacted directly with immobilized proteins (data not shown).
FIGURE 4.
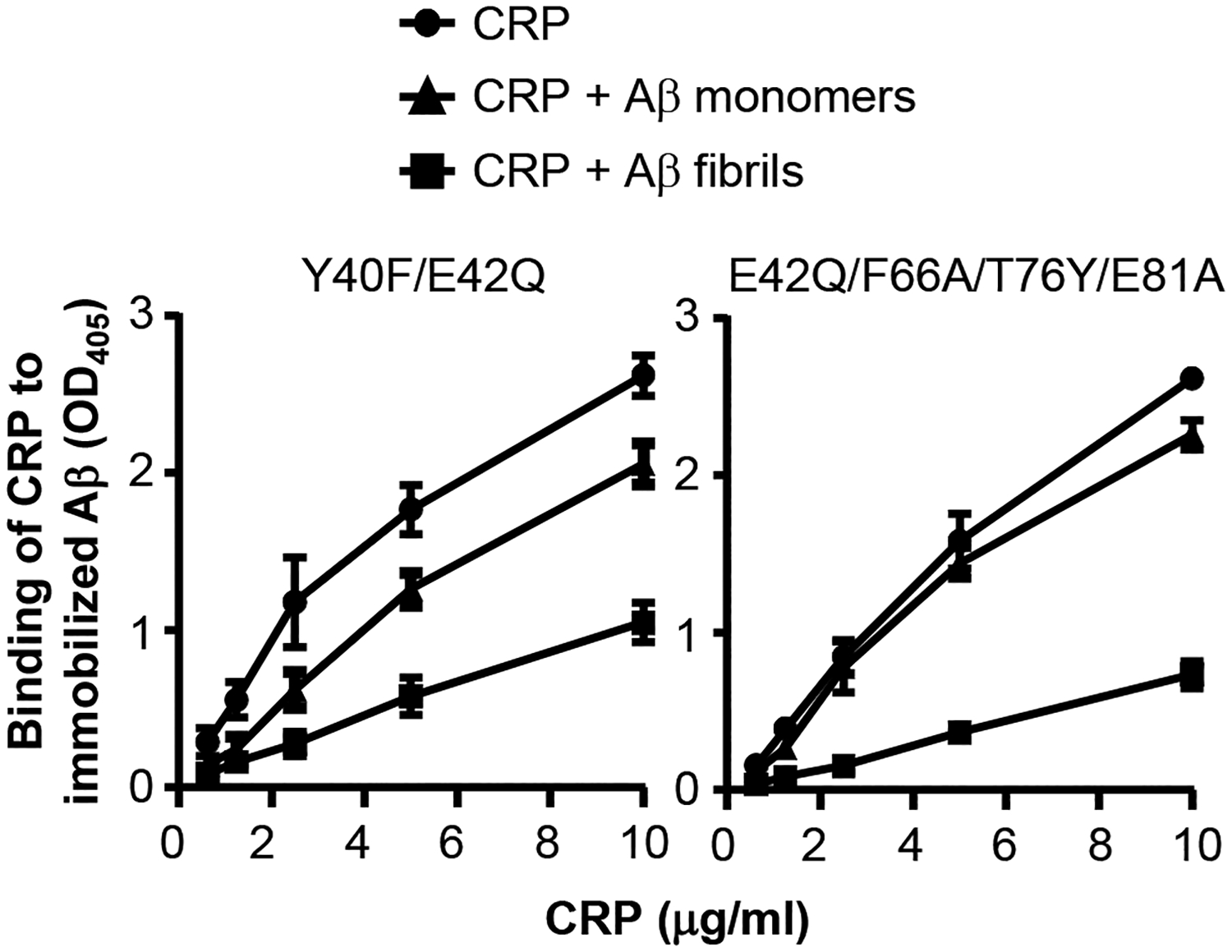
Inhibition of binding of CRP mutants to immobilized Aβ by fluid-phase Aβ. Aβ monomers (100 μg/ml) or Aβ fibrils (100 μg/ml) were mixed with CRP. The mixture of CRP and Aβ was then added to the wells coated with Aβ. Bound CRP was detected by using a rabbit polyclonal anti-CRP antibody and HRP-conjugated donkey anti-rabbit IgG. The OD of the developed color was read at 405 nm. Data shown are mean ± SEM of three experiments. The p values were determined by employing linear regression analysis of the slopes. For both CRP mutants, the differences between all the curves when compared to each other were statistically significant (p<0.05; not shown).
Structurally altered pentameric CRP prevents fibrillation of Aβ
Under the conditions of the fibrillation assay, the fibrillation of Aβ monomers began within 5 min and continued till ~2 h when further fibrillation stopped (Fig. 5, black curves in all panels). WT CRP, even at the highest concentration of 100 μg/ml did not affect the fibrillation significantly. These results supported our findings shown in Figs. 3–4 that WT CRP is incapable of recognizing and binding to Aβ. In contrast to WT CRP, all four CRP mutants inhibited the fibrillation in a CRP concentration-dependent manner. There was no statistically significant difference in fibrillation without or with 1 μg/ml of CRP, except for the Y40F/E42Q CRP mutant. There were statistically significant differences in the fibrillation when CRP mutants were used at 10 μg/ml and 100 μg/ml. As shown in Fig. 5, CRP mutants significantly inhibited the rate of fibrillation over time and also significantly inhibited the total amount of the fibrils formed by the end of the fibrillation reaction. Fibrillation assays employing acidic pH-treated WT CRP was not performed since the modified structure of WT CRP at pH 5.0 was reversible at pH 7.0 and the fibrillation assay failed to work at pH 5.0.
FIGURE 5.
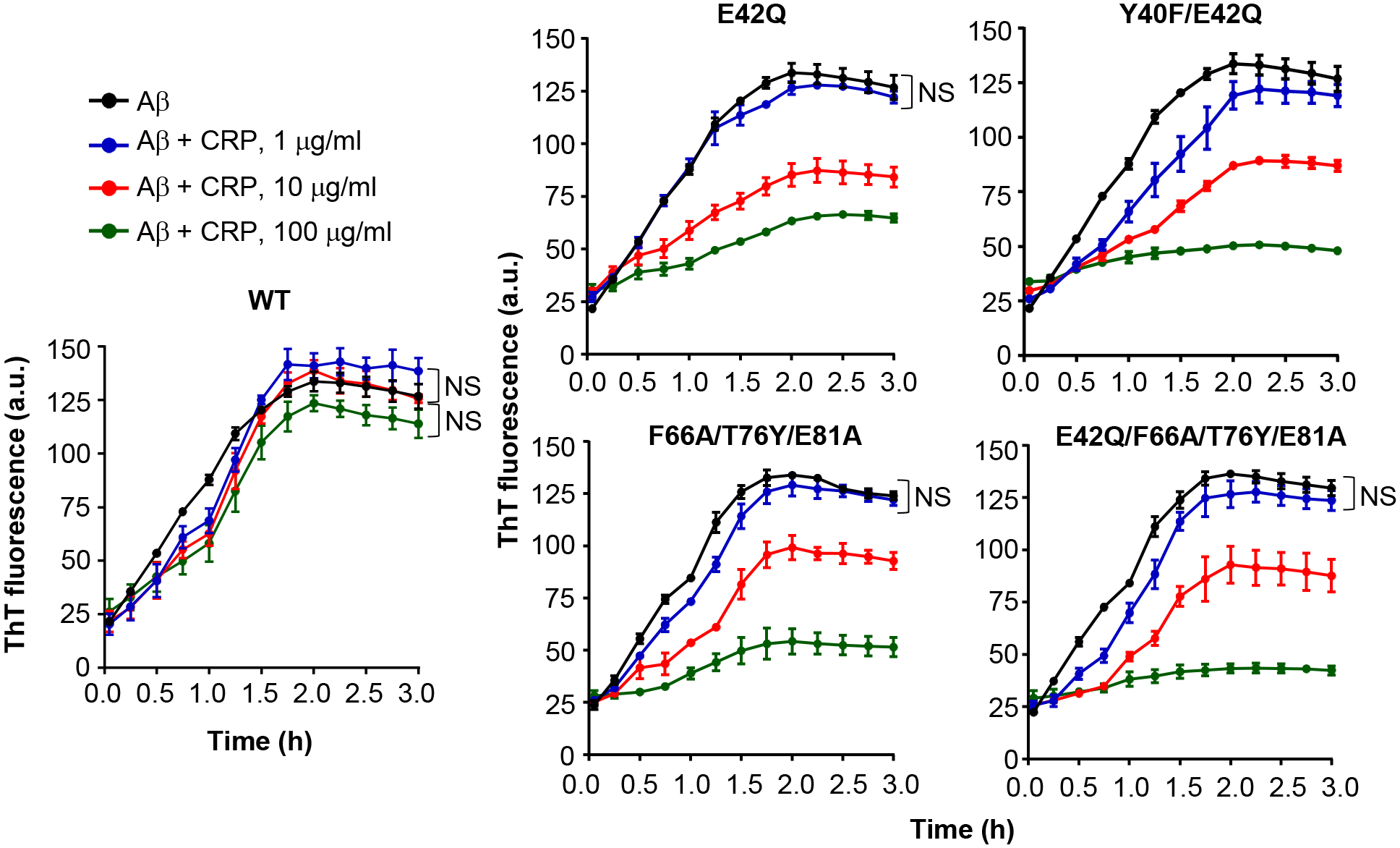
Effects of CRP on the formation of Aβ fibrils. The fibrillation of Aβ peptides was monitored by ThT fluorescence in the absence (black) or presence of 1 μg/ml CRP (blue), 10 μg/ml CRP (red) and 100 μg/ml CRP (green). CRP and Aβ monomers were mixed at time zero and added to microtiter wells. After the first measurement at 5 min, the plate was incubated at 37°C with shaking; fluorescence was measured every 15 min. Results are plotted as mean arbitrary units (a.u.) ± SEM of three experiments. For the time period of 5 min to 2 h, p values were determined by employing linear regression analysis of the slopes. For the time period of 2.25 h to 3 h, p values were determined by taking the mean of all points and employing student unpaired t test. For clarity, only those differences are indicated (NS) where the difference was statistically not significant (p>0.05). The difference between all other curves when compared to each other was statistically significant (p<0.001; not shown).
Next, we performed fibrillation assays in which CRP was added to Aβ 45 min after the fibrillation began (Fig. 6). WT CRP did not significantly affect the fibrillation of Aβ. Two of the four mutants were tested in this assay: Y40F/E42Q which binds to PCh and E42Q/F66A/T76Y/E81A which does not bind to PCh. Both CRP mutants, as soon as they were added to growing fibrils, completely prevented further fibrillation. The total amount of the fibrils formed at the end of 3 h was significantly less than the total amount of the fibrils formed without CRP. There was no statistically significant difference between the inhibitory effects of the two mutants suggesting that the effects of CRP mutants on Aβ fibrillation was independent of the PCh-binding site of CRP. Taken together, these data indicate that structurally altered pentameric CRP binds to Aβ monomers and prevents fibrillation. Structurally altered pentameric CRP can also bind to Aβ fibrils to prevent further fibrillation.
FIGURE 6.
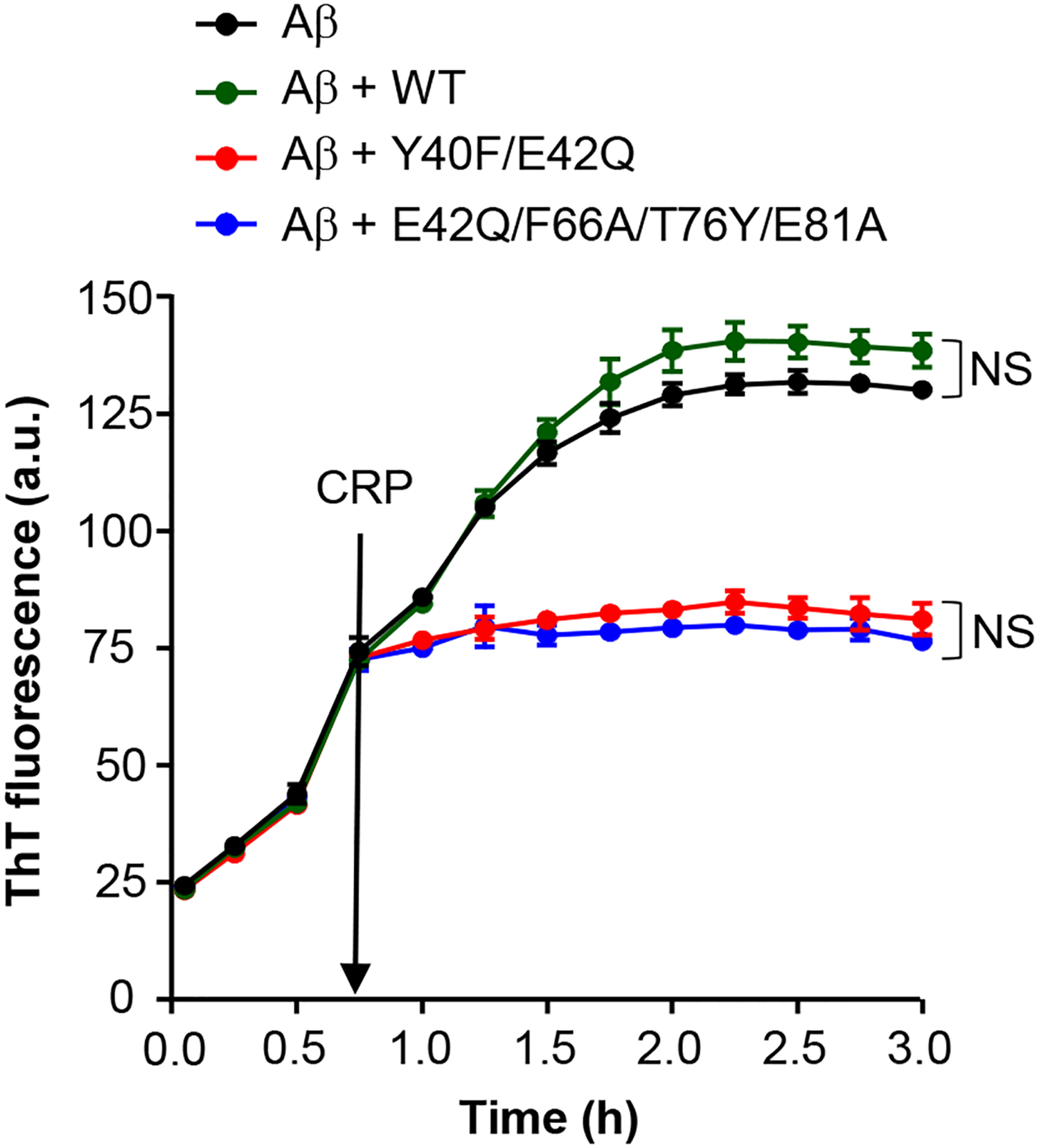
Effects of CRP on the growth of pre-formed Aβ fibrils. The time course of fibrillation of Aβ peptides was monitored by ThT fluorescence in the absence or presence of 100 μg/ml CRP. CRP was added to Aβ 45 min after the fibrillation began in the microtiter plate which was incubated at 37°C throughout with shaking. Fluorescence was measured every 15 min. Results are plotted as mean a.u. ± SEM of three experiments. Statistical analysis was performed for the time period of 2 h to 3 h; p values were determined by taking the mean of all points and employing student unpaired t test. For clarity, only those differences are indicated (NS) where the difference was statistically not significant (p>0.05). The difference between all other curves when compared to each other was statistically significant (p<0.001; not shown).
Ca2+-binding site is perturbed in structurally altered pentameric CRP
We hypothesized previously that the creation of the Aβ-binding site in acidic pH-treated WT CRP was probably due to the loss of one of the two Ca2+ from each CRP subunit (32). This hypothesis was based on the findings that acidic pH-treated WT CRP had reduced avidity for Ca2+. Since all four CRP mutants bound to Aβ as well as acidic pH-treated WT CRP did, it was possible that the avidity of CRP mutants for Ca2+ was also reduced similar to the reduction in the avidity of acidic pH-treated WT CRP for Ca2+. This possibility was explored by employing two different approaches.
In the first approach, we performed Ca2+-requirement assays to determine the requirement of Ca2+ for the binding of CRP mutants to PCh. Only two of the four CRP mutants, E42Q and Y40F/E42Q, which retained the intact PCh-binding site could be tested employing this assay. The concentration of Ca2+ required for 50% binding of each CRP species to PCh was determined; a change in the Ca2+-requirement for comparable PCh-binding activities of CRP would reflect a change in the avidity of CRP for Ca2+. As shown in Fig. 7, approximately five-fold more Ca2+ was required for equivalent binding of CRP mutants E42Q and Y40F/E42Q to PCh compared to WT CRP. These data suggested that the Ca2+-binding site was perturbed in the E42Q and Y40F/E42Q CRP mutants.
FIGURE 7.
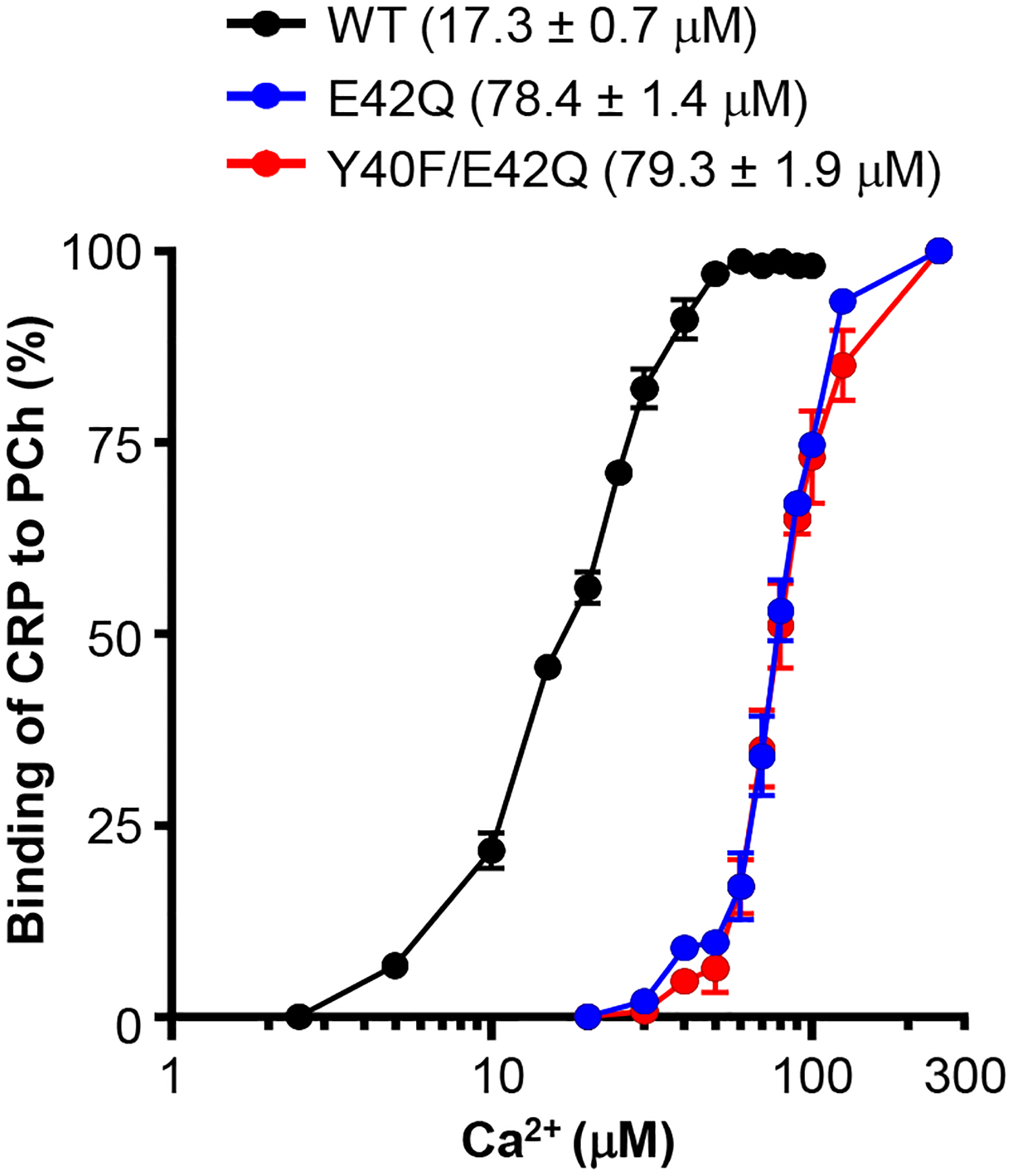
Effects of mutations on the Ca2+-dependent binding of CRP to PCh. The PCh-binding activity of CRP as a function of Ca2+ concentration is shown. Microtiter wells were coated with PnC. The unreacted sites in the wells were blocked with gelatin. CRP (50 ng/ml), diluted in TBS-Ca (various concentrations of Ca2+), pH 7.2, was added in duplicate wells. Bound CRP was detected by using the anti-CRP mAb HD2.4 as the primary antibody and HRP-conjugated goat anti-mouse IgG as the secondary antibody. The OD of the developed color was read at 405 nm. The highest OD obtained was taken as 100%. The values on the y-axis represent the percentage of binding of CRP to PCh. The concentration of Ca2+ for 50% of maximal binding of each CRP species is shown. Results were plotted as mean ± SEM of three experiments. Statistical differences were calculated using the unpaired student t-test.
In the second approach, we performed protease cleavage assays to assess the Ca2+-dependent protection of the protease sensitive sites in CRP. All four CRP mutants were tested employing this assay. First, we performed a titration assay to determine the concentration of Ca2+ needed to protect WT CRP from protease cleavage. As shown (Supplemental Fig. 1), WT CRP was protected from cleavage in a Ca2+ concentration-dependent manner. There was no protection of CRP from cleavage at 0.05 mM Ca2+, the protection began with a concentration of Ca2+ ≥ 0.5 mM, and 5 mM Ca2+ was fully protective. From this data, 5 mM and 0.5 mM Ca2+ were chosen to evaluate the Ca2+-binding site of structurally altered pentameric CRP. A change in the Ca2+-requirement for protecting CRP mutants from cleavage would reflect a change in the avidity of CRP for Ca2+.
The results of the cleavage assay performed in the presence of 5 mM Ca2+ are shown in Fig. 8. Protease treatment of WT CRP in the absence of Ca2+ generated 3 fragments (lane 3). The m.w. of the fragments designated A1 and A2 were 17 and 16 kD, respectively. The 16 kD A2 fragment is generated due to a secondary cleavage site in the larger fragment A1. The m.w. of fragment B was estimated to be of ~6 kD, consistent with previously published reports (8, 10). WT CRP was completely protected from this digestion in the presence of 5 mM Ca2+ (lane 4). Results of the protease treatment of E42Q and Y40F/E42Q CRP mutants either in the absence of (lanes 6 and 9) or presence of (lanes 7 and 10) 5 mM Ca2+ were similar to that of WT CRP. Protease treatment of F66A/T76Y/E81A and E42Q/F66A/T76Y/E81A CRP mutants in the absence of Ca2+ generated 3 fragments (lanes 12 and 15); however, the intensity of the cleaved products was stronger than that of WT CRP, indicating that the digestion of CRP mutants was faster than that of WT CRP. In the presence of 5 mM Ca2+, F66A/T76Y/E81A and E42Q/F66A/T76Y/E81A CRP mutants were not fully protected from protease cleavage. These data suggested that the accessibility of the proteolytic site in WT CRP to the protease was different from the accessibility of the proteolytic site in CRP mutants F66A/T76Y/E81A and E42Q/F66A/T76Y/E81A, but not in E42Q and Y40F/E42Q, to the protease.
FIGURE 8.
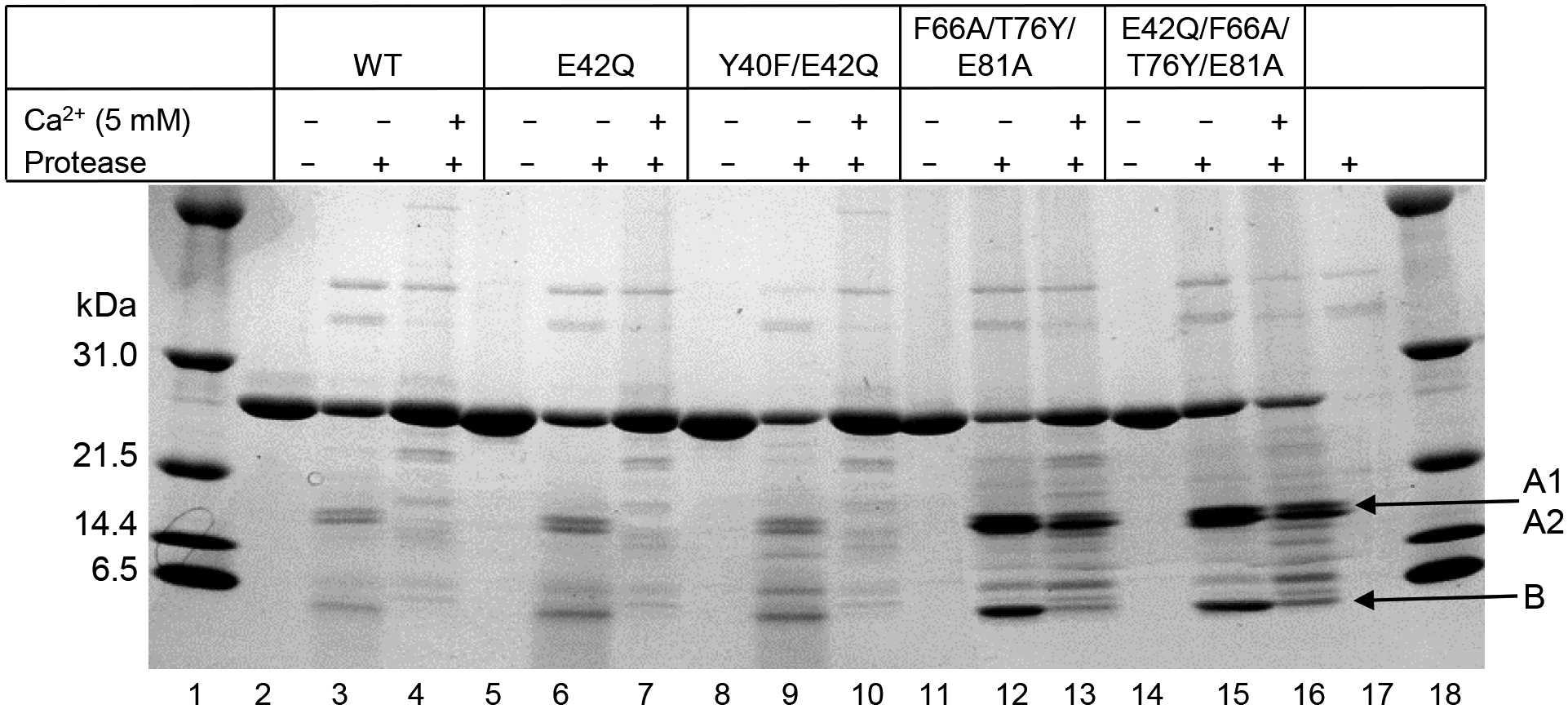
Effects of mutations on the Ca2+-site-dependent proteolytic cleavage of CRP. Reducing SDS-PAGE of CRP treated with protease in the absence and presence of 5 mM Ca2+ was run on a 4–20% polyacrylamide gradient gel. Bands were visualized by Coomassie brilliant blue staining. Protease alone is shown in lane 17. The m.w. markers are shown in lanes 1 and 18.
Since the data obtained from the Ca2+-requirement assay (Fig. 7) suggested that E42Q and Y40F/E42Q CRP mutants were different from WT CRP and the data obtained from the protease cleavage assay (Fig. 8) suggested that E42Q and Y40F/E42Q CRP mutants were not different from WT CRP, we performed another protease cleavage assay with 0.5 mM Ca2+ instead of 5 mM Ca2+. As shown (Fig. 9), 0.5 mM Ca2+ protected WT CRP from cleavage (lane 4) but did not fully protect E42Q and Y40F/E42Q from cleavage (lanes 7 and 10). Since 5 mM Ca2+ did not protect the other mutants from cleavage (Fig. 8), it was expected that 0.5 mM Ca2+ would also not protect from cleavage (lanes 13 and 16).
FIGURE 9.
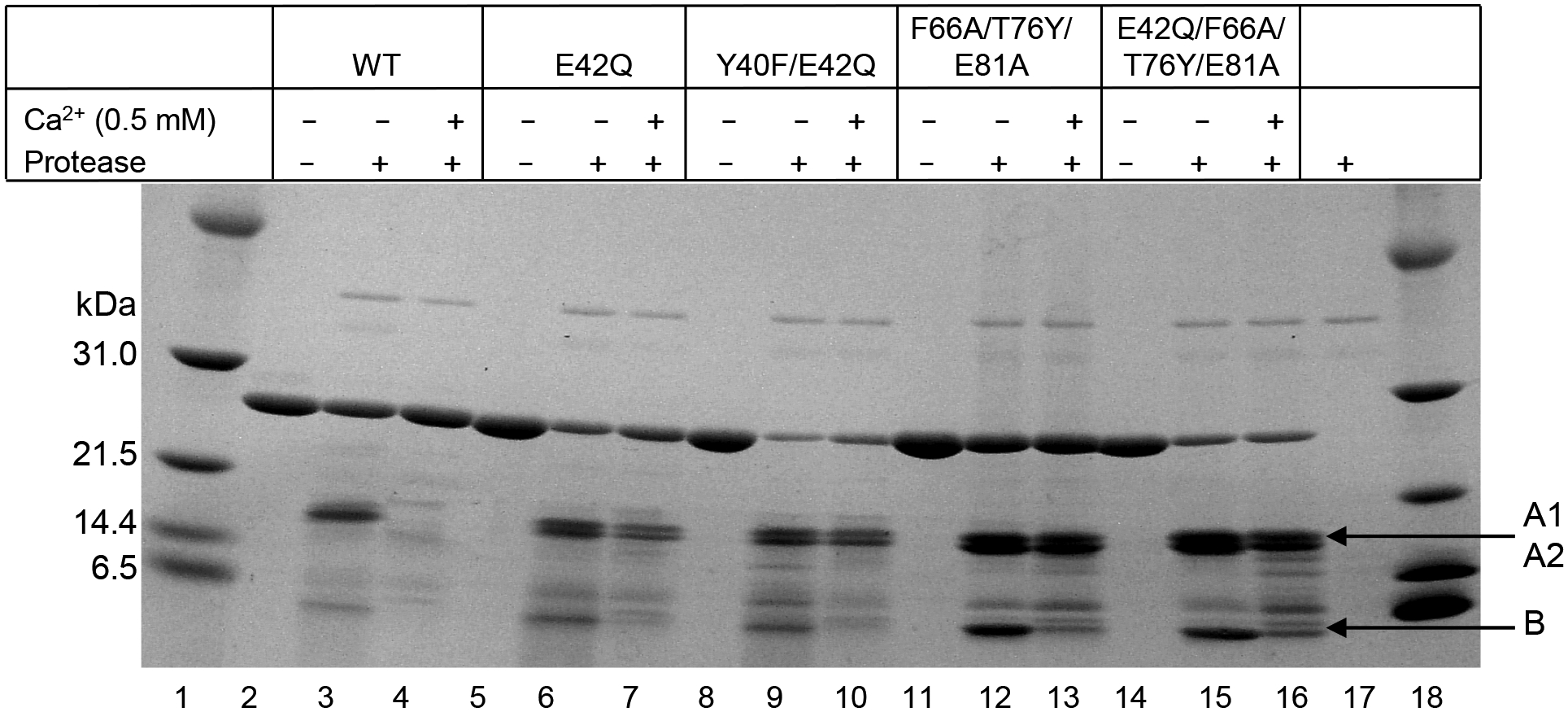
Effects of mutations on the Ca2+-site-dependent proteolytic cleavage of CRP. Reducing SDS-PAGE of CRP treated with protease in the absence and presence of 0.5 mM Ca2+ was run on a 4–20% polyacrylamide gradient gel. Bands were visualized by Coomassie brilliant blue staining. Protease alone is shown in lane 17. The m.w. markers are shown in lanes 1 and 18.
Combined data obtained from the Ca2+-requirement and protease cleavage assays indicated that the topology of the Ca2+-binding site in structurally altered pentameric CRP was perturbed and different from that of WT CRP, despite the fact that the amino acid residues coordinating the two Ca2+ were not mutated in any of the CRP mutants.
Discussion
Our major findings in this study were: 1. Proteins immobilized on microtiter wells expressed amyloid-like structures. 2. Structurally altered pentameric CRP (four different CRP mutants) bound to immobilized Aβ at physiological pH similar to the binding of WT CRP to immobilized Aβ at acidic pH. 3. Structurally altered pentameric CRP also bound to Aβ in the fluid phase. 4. Structurally altered pentameric CRP, not WT CRP, inhibited the fibrillation of Aβ. 5. Structurally altered pentameric CRP also halted the progression of fibril formation when added to growing Aβ fibrils. 6. There was one commonality in all four CRP mutants: the topology of the Ca2+-binding site of all four CRP mutants was different from that of WT CRP.
In a previously reported study (48, 49), at physiological pH, WT CRP was also found to bind to Aβ and inhibit fibrillation, which is in contrast to our findings. However, there were methodological differences between the two studies. First, Aβ42 was used in this study while Aβ40 was used in the previous study. These two types of Aβ peptides have different rates of fibrillation; the rate of fibrillation of Aβ42 is faster than that of Aβ40 (50–52). Second, the fibrillation assays were performed while shaking the assay plate in this study while there was no shaking in the previous study. Shaking enhances the rate of fibrillation (50–54). Third, perhaps due to the two differences mentioned above, fibrillation began within 5 min under the conditions of our assay while it began after 30 h in the previous study. Based on a previous publication on the stability of purified CRP (55), it was likely that when WT CRP was incubated at 37°C for 30 h (48), a fraction of CRP pentamers might have monomerized. If this happened, then it could be monomeric CRP (mCRP) which bound to Aβ and inhibited fibrillation. Indeed, WT CRP used in the previous study did contain mCRP as determined by gel filtration analysis of the protein (48). Fourth, CRP was used at a concentration of 100 μg/ml and less in this study while it was 500 μg/ml in the previous study. Higher concentration of purified and buffered CRP could have generated enough mCRP and their aggregates, when incubated at 37°C for 30 h, for detectable inhibition of fibrillation. We suspect that in the previous study, if CRP was added to Aβ after 30 h, that is, at the time of initiation of fibrillation, and if used at a lower concentration, the results could have been similar to ours. Indeed, in another study on CRP-Aβ interactions (56), biotinylated WT CRP was not found to bind to Aβ.
It has been reported that the binding of WT CRP to certain ligands, including Aβ plaques in vivo, leads to dissociation of pentamers into monomers (23, 24, 35, 57). Such monomerization of pentameric CRP into mCRP involves an intermediate or transitional or non-native stage when the conformation of the ligand-bound CRP pentamer is different from that of native pentamer. Several monoclonal anti-CRP antibodies (3H12, 6B7, 8C10 and 9C9) are available that can differentiate between native and non-native pentameric CRP; however, these antibodies do not differentiate between non-native pentameric CRP and mCRP (23–25, 35, 57–61). By employing these antibodies in immunohistochemistry, it has been concluded that mCRP was deposited at the amyloid plaques of AD (19–25). Because the sites of inflammation including the site of Aβ deposition are characterized by acidic and redox conditions (62), we propose that structurally altered pentameric CRP is generated at the site of Aβ deposition first, followed by the binding of structurally altered pentameric CRP to deposited Aβ which can then lead to monomerization of CRP. It is unclear though if CRP, in any structural conformation, before or after monomerization, leaves the surface of the molecule to which CRP was initially bound.
All amyloid deposits contain serum amyloid P component (SAP) which is a protein homologous to CRP (63). SAP binds to Aβ fibrils, prevents further fibrillation, and stabilizes the fibrils by protecting against proteolysis (64–68). Since structurally altered pentameric CRP is generated at the site of Aβ deposition, our data raise the possibility of competition between SAP and structurally altered pentameric CRP for binding to Aβ fibrils, and, therefore, the two proteins may be regulating the extent of incorporation of SAP into Aβ fibrils. It remains to be investigated whether the binding of structurally altered pentameric CRP to Aβ fibrils facilitates proteolysis of the fibrils.
Since SAP binds to and prevents proteolysis of Aβ fibrils, both SAP and Aβ fibrils have been therapeutic targets for treating amyloidosis (67). In SAP-deficient mice, in which the gene coding for SAP was inactivated, the deposition of Aβ was found to be delayed (69). Accordingly, treatment with anti-SAP antibodies and blocking SAP with small chemical compounds have been shown to remove Aβ deposits in vivo (70). Strategies to prevent the aggregation of Aβ into fibrils and to enable the dissociation of Aβ fibril plaques have also been explored as possible treatments of amyloidosis. Some Aβ-binding synthetic peptides, including one that was derived from CRP, have been found to block the toxic effects of Aβ on neuronal cells in culture (71). Whether exogenously prepared structurally altered pentameric CRP can be delivered to the site of Aβ deposition and whether there would be a protective effect in established animal models and in ex vivo cell culture model systems remain to be explored (69–72).
The amino acid residues which participate in the formation of the Aβ-binding site on structurally altered pentameric CRP have not been identified yet. These amino acid residues must be hidden in WT CRP. Our data suggest that a perturbation of the topology of the Ca2+-binding sites is necessary to form the Aβ-binding site. However, there were no mutations of the amino acid residues coordinating Ca2+ in any of the four mutants used in this study. Since EDTA-treated, Ca2+-free WT CRP does not bind to Aβ (32), it is likely that in all four CRP mutants, there was loss of binding of one Ca2+ per CRP subunit, leaving half of the Ca2+-site vacant generating Aβ-binding activity and sensitivity to protease cleavage. Cryo-electron microscopy of all four CRP mutants, as performed for WT CRP at acidic pH (73), would reveal the topology of the Aβ-binding site on structurally altered pentameric CRP.
There are dozens of known amyloidogenic proteins, including ox-LDL employed in this study, that are responsible for clinically significant amyloidoses (74–76). Our hypothesis that dense immobilization of some proteins onto microtiter plates may cause aggregation and denaturation of proteins resulting in the generation of amyloid-like structures creating Aβ epitopes was also found to be correct. Considering that immobilized proteins have a common amyloid-like structure on their surface, we conclude that Aβ peptides and amyloid-like structures expressed on immobilized proteins are ligands of structurally altered pentameric CRP. Based on the Aβ recognition function of structurally altered pentameric CRP, we propose that CRP should be considered as a dual pattern recognition molecule of the immune system. One, CRP recognizes conserved patterns such as PCh present on the surface of pathogens. Two, CRP recognizes non-conserved, newly created, molecular patterns such as amyloid-like structures formed by amyloidogenic and pathogenic proteins. This recognition function is exhibited by structurally altered pentameric CRP.
CRP is a phylogenetically conserved protein (77, 78). Also, a CRP-deficient human has not been reported so far. Therefore, it has been suggested that CRP must have an important host defense function that applies across the animal kingdom. We conclude that CRP is an anti-amyloidogenic protein. CRP functions as a scavenger to get rid of misfolded, denatured, deposited and non-functional proteins to prevent the toxicity they generate. This function of CRP would favor the conservation of CRP throughout evolution. The anti-amyloidogenic function of CRP also has implications for many human inflammatory and neurodegenerative diseases such as AD, Parkinson’s disease, the prion-related diseases and also non-neurodegenerative diseases such as type II diabetes which are characterized by abnormal amyloid fibril aggregates (74, 75). The anti-amyloidogenic function of CRP also has implications for diseases caused by the deposition of otherwise fluid-phase proteins.
Supplementary Material
KEY POINTS.
Some proteins, when immobilized, express an Aβ epitope.
Aβ and amyloid-like structures are the ligands of structurally altered pentameric CRP.
Structurally altered pentameric CRP prevents formation of Aβ fibrils.
Acknowledgements
We thank Sanjay Singh, PhD, for his assistance with the preparation of CRP mutants.
This work was supported by National Institutes of Health Grants AR068787 and AI151561.
Abbreviations used in this article:
- AD
Alzheimer’s disease
- Aβ
amyloid-β
- CRP
C-reactive protein
- FH
Factor H
- Fn
Fibronectin
- mCRP
monomeric CRP
- ox-LDL
Oxidized low density lipoprotein
- PCh
phosphocholine
- PnC
pneumococcal C-polysaccharide
- SAP
serum amyloid P component
- TBS-Ca
TBS, pH 7.2, containing 0.1% gelatin, 0.02% Tween-20 and 2 mM CaCl2
- TEM
Transmission electron microscopy
- ThT
Thioflavin T
- WT
wild-type
References
- 1.Srinivasan N, White HE, Emsley J, Wood SP, Pepys MB, and Blundell TL. 1994. Comparative analyses of pentraxins: Implications for protomer assembly and ligand binding. Structure 2: 1017–1027. [DOI] [PubMed] [Google Scholar]
- 2.Shrive AK, Cheetham GM, Holden D, Myles DA, Turnell WG, Volanakis JE, Pepys MB, Bloomer AC, and Greenhough TJ. 1996. Three-dimensional structure of human C-reactive protein. Nat. Struct. Biol 3: 346–354. [DOI] [PubMed] [Google Scholar]
- 3.Abernethy TJ, and Avery OT. 1941. The occurrence during acute infections of a protein not normally present in the blood. I. Distribution of the reactive protein in patients’ sera and the effect of calcium on the flocculation reaction with C polysaccharide of pneumococcus. J. Exp. Med 73: 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volanakis JE, and Kaplan MH. 1971. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc. Soc. Exp.Biol. Med 136: 612–614. [DOI] [PubMed] [Google Scholar]
- 5.Janeway CA Jr. 1989. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol 54: 1–13. [DOI] [PubMed] [Google Scholar]
- 6.Labarrere CA, and Kassab GS. 2021. Pattern recognition proteins: First Line of defense against coronaviruses. Front. Immunol 12: 652252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson D, Pepys MB, and Wood SP. 1999. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure 7: 169–177. [DOI] [PubMed] [Google Scholar]
- 8.Kinoshita CM, Ying S-C, Hugli TE, Siegel JN, Potempa LA, Jiang H, Houghten RA, and Gewurz H. 1989. Elucidation of a protease-sensitive site involved in the binding of calcium to C-reactive protein. Biochemistry 28: 9840–9848. [DOI] [PubMed] [Google Scholar]
- 9.Ramadan MA, Shrive AK, Holden D, Myles DA, Volanakis JE, DeLucas LJ, and Greenhough TJ. 2002. The three-dimensional structure of calcium-depleted human C-reactive protein from perfectly twinned crystals. Acta Crystallogr. D Biol. Crystallogr 58: 992–1001. [DOI] [PubMed] [Google Scholar]
- 10.Suresh MV, Singh SK, and Agrawal A. 2004. Interaction of calcium-bound C-reactive protein with fibronectin is controlled by pH: In vivo implications. J. Biol. Chem 279:52552–52557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarrett JT, Berger EP, and Lansbury PT. 1993. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32: 4693–4697. [DOI] [PubMed] [Google Scholar]
- 12.Harper JD, Wong SS, Lieber CM, and Lansbury PT. 1997. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol 4: 119–125. [DOI] [PubMed] [Google Scholar]
- 13.Sgourakis NG, Yan Y, McCallum SA, Wang C, and Garcia AE. 2007. The Alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: A combined MD / NMR study. J. Mol. Biol 368: 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawaya MR, Hughes MP, Rodriguez JA, Riek R, and Eisenberg DS. 2021. The expanding amyloid family: Structure, stability, function, and pathogenesis. Cell 184: 4857–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardy J, and Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: 353–356. [DOI] [PubMed] [Google Scholar]
- 16.Lansbury PT, and Lashuel HA. 2006. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443: 774–779. [DOI] [PubMed] [Google Scholar]
- 17.Hamley IW 2012. The amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev 112: 5147–5192. [DOI] [PubMed] [Google Scholar]
- 18.Long JM, and Holtzman DM. 2019. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179: 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwamoto N, Nishiyama E, Ohwada J, and Arai H. 1994. Demonstration of CRP immunoreactivity in brains of Alzheimer’s disease: Immunohistochemical study using formic acid pretreatment of tissue sections. Neurosci. Lett 177: 23–26. [DOI] [PubMed] [Google Scholar]
- 20.Duong T, Nikolaeva M, and Acton PJ. 1997. C-reactive protein-like immunoreactivity in the neurofibrillary tangles of Alzheimer’s disease. Brain Res 749: 152–156. [DOI] [PubMed] [Google Scholar]
- 21.Yasojima K, Schwab C, McGeer EG, and McGeer PL. 2000. Human neurons generate C-reactive protein and amyloid P: Upregulation in Alzheimer’s disease. Brain Res. 887: 80–89. [DOI] [PubMed] [Google Scholar]
- 22.McGeer EG, Yasojima K, Schwab C, and McGeer PL. 2001. The pentraxins: Possible role in Alzheimer’s disease and other innate inflammatory diseases. Neurobiol. Aging 22: 843–848. [DOI] [PubMed] [Google Scholar]
- 23.Strang F, Scheichl A, Chen Y-C, Wang X, Htun N-M, Bassler N, Eisenhardt SU, Habersberger J, and Peter K. 2012. Amyloid plaques dissociate pentameric to monomeric C-reactive protein: A novel pathomechanism driving cortical inflammation in Alzheimer’s disease? Brain Pathol. 22: 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiele JR, Habersberger J, Braig D, Schmidt Y, Goerendt K, Maurer V, Bannasch H, Scheichl A, Woollard KJ, von Dobschütz E, Kolodgie F, Virmani R, Stark GB, Peter K, and Eisenhardt SU. 2014. Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation: In vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy. Circulation 130: 35–50. [DOI] [PubMed] [Google Scholar]
- 25.Slevin M, Matou S, Zeinolabediny Y, Corpas R, Weston R, Liu D, Boras E, Di Napoli M, Petcu E, Sarroca S, Popa-Wagner A, Love S, Font MA, Potempa LA, Al-Baradie R, Sanfeliu C, Revilla S, Badimon L, and Krupinski J. 2015. Monomeric C-reactive protein: A key molecule driving development of Alzheimer’s disease associated with brain ischaemia? Sci. Rep 5: 13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, and Sun PD. 2008. Structural recognition and functional activation of FcγR by innate pentraxins. Nature 456: 989–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mihlan M, Hebecker M, Dahse H-M, Hälbich S, Huber-Lang M, Dahse R, Zipfel PF, and Józsi M. 2009. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol. Immunol 46: 335–344. [DOI] [PubMed] [Google Scholar]
- 28.Hebecker M, Okemefuna AI, Perkins SJ, Mihlan M, Huber-Lang M, and Józsi M. 2010. Molecular basis of C-reactive protein binding and modulation of complement activation by factor H-related protein 4. Mol. Immunol 47: 1347–1355. [DOI] [PubMed] [Google Scholar]
- 29.Agrawal A, Gang TB, and Rusinol AE. 2014. Recognition functions of pentameric C-reactive protein in cardiovascular disease. Mediators Inflamm. 2014: 319215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salonen E-M, Vartio T, Hedman K, and Vaheri A. 1984. Binding of fibronectin by the acute phase reactant C-reactive protein. J. Biol. Chem 259: 1496–1501. [PubMed] [Google Scholar]
- 31.Singh SK, Hammond DJ Jr., Beeler BW, and Agrawal A. 2009. The binding of C-reactive protein, in the presence of phosphoethanolamine, to low-density lipoproteins is due to phosphoethanolamine-generated acidic pH. Clin. Chim. Acta 409: 143–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammond DJ Jr., Singh SK, Thompson JA, Beeler BW, Rusinol AE, Pangburn MK, Potempa LA, and Agrawal A. 2010. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J. Biol. Chem 285: 36235–36244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li SL, Feng JR, Zhou HH, Zhang CM, Lv GB, Tan YB, Ge ZB, and Wang MY. 2018. Acidic pH promotes oxidation-induced dissociation of C-reactive protein. Mol. Immunol 104: 47–53. [DOI] [PubMed] [Google Scholar]
- 34.Ullah N, Ma F-R, Han J, Liu X-L, Fu Y, Liu Y-T, Liang Y-L, Ouyang H, and Li H-Y. 2020. Monomeric C-reactive protein regulates fibronectin mediated monocyte adhesion. Mol. Immunol 117: 122–30. [DOI] [PubMed] [Google Scholar]
- 35.Rajab IM, Hart PC, and Potempa LA. 2020. How C-reactive protein structural isoforms with distinctive bioactivities affect disease progression. Front. Immunol 11: 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh SK, Thirumalai A, Pathak A, Ngwa DN, and Agrawal A. 2017. Functional transformation of C-reactive protein by hydrogen peroxide. J. Biol. Chem 292: 3129–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh SK, Thirumalai A, Hammond DJ Jr., Pangburn MK, Mishra VK, Johnson DA, Rusiñol AE, and Agrawal A. 2012. Exposing a hidden functional site of C-reactive protein by site-directed mutagenesis. J. Biol. Chem 287: 3550–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ngwa DN, Singh SK, Gang TB, and Agrawal A. 2020. Treatment of pneumococcal infection by using engineered human C-reactive protein in a mouse model. Front. Immunol 11: 586669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pathak A, Singh SK, Thewke DP, and Agrawal A. 2020. Conformationally altered C-reactive protein capable of binding to atherogenic lipoproteins reduces atherosclerosis. Front. Immunol 11: 1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agrawal A, Xu Y, Ansardi D, Macon KJ, and Volanakis JE. 1992. Probing the phosphocholine-binding site of human C-reactive protein by site-directed mutagenesis. J. Biol. Chem 267: 25352–25358. [PMC free article] [PubMed] [Google Scholar]
- 41.Gang TB, Hammond DJ Jr., Singh SK, Ferguson DA Jr., Mishra VK, and Agrawal A. 2012. The phosphocholine-binding pocket on C-reactive protein is necessary for initial protection of mice against pneumococcal infection. J. Biol. Chem 287: 43116–43125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gang TB, Hanley GA, and Agrawal A. 2015. C-reactive protein protects mice against pneumococcal infection via both phosphocholine-dependent and phosphocholine-independent mechanisms. Infect. Immun 83: 1845–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stine WB, Jungbauer L, Yu C, LaDu MJ. 2011. Preparing synthetic Aβ in different aggregation states. Methods Mol. Biol 670: 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh SK, Ngwa DN, and Agrawal A. 2020. Complement activation by C-reactive protein is critical for protection of mice against pneumococcal infection. Front. Immunol 11: 1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thirumalai A, Singh SK, Hammond DJ Jr., Gang TB, Ngwa DN, Pathak A, and Agrawal A. 2017. Purification of recombinant C-reactive protein mutants. J. Immunol. Methods 443: 26–32. [DOI] [PubMed] [Google Scholar]
- 46.Blancas-Mejia LM, Misra P, Dick CJ, Marin-Argany M, Redhage KR, Cooper SA, and Ramirez-Alvarado M. 2019. Assays for light chain amyloidosis formation and cytotoxicity. Methods Mol. Biol 1873: 123–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agrawal A, Lee S, Carson M, Narayana SVL, Greenhough TJ, and Volanakis JE. 1997. Site-directed mutagenesis of the phosphocholine-binding site of human C-reactive protein: Role of Thr76 and Trp67. J. Immunol 158: 345–350. [PubMed] [Google Scholar]
- 48.Ozawa D, Nomura R, Mangione PP, Hasegawa K, Okoshi T, Porcari R, Bellotti V, and Naiki H. 2016. Multifaceted anti-amyloidogenic and pro-amyloidogenic effects of C-reactive protein and serum amyloid P component in vitro. Sci. Rep 6: 29077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozawa D, Nomura R, Mangione PP, Hasegawa K, Okoshi T, Porcari R, Bellotti V, and Naiki H. 2017. Antiamyloidogenic and proamyloidogenic chaperone effects of C-reactive protein and serum amyloid P component. Amyloid 24: 28–29. [DOI] [PubMed] [Google Scholar]
- 50.Xue C, Lin TY, Chang D, and Guo Z. 2017. Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci 4: 160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nirmalraj PN, List J, Battacharya S, Howe G, Xu L, Thompson D, and Mayer M. 2020. Complete aggregation pathway of amyloid β (1–40) and (1–42) resolved on an atomically clean interface. Sci. Adv 6: 6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L, Eom K, and Kwon T. 2021. Different aggregation pathways and structures for Aβ40 and Aβ42 peptides. Biomolecules 11: 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu J, Cao Q, Wang C, Zheng J, Luo F, Xie J, Li Y, Ma X, He L, Eisenberg D, Nowick J, Jiang L, and Li D. 2019. Structure-based peptide inhibitor design of amyloid-β aggregation. Front. Mol. Neurosci 12: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee CF, Bird S, Shaw M, Jean L, and Vaux DJ. 2012. Combined effects of agitation, macromolecular crowding, and interfaces on amyloidogenesis. J. Biol. Chem 287: 38006–38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh SK, Suresh MV, Hammond DJ Jr, Rusinol AE, Potempa LA, and Agrawal A. 2009. Binding of the monomeric form of C-reactive protein to enzymatically-modified low-density lipoprotein: effects of phosphoethanolamine. Clin. Chim. Acta 406: 151–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dorta-Estremera SM, and Cao W. 2015. Human pentraxins bind to misfolded proteins and inhibit production of type I interferon induced by nucleic acid-containing amyloid. J. Clin. Cell Immunol 6:1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ji SR, Wu Y, Zhu L, Potempa LA, Sheng FL, Lu W, and Zhao J. 2007. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRP(m). FASEB J 21: 284–294. [DOI] [PubMed] [Google Scholar]
- 58.Schwedler SB, Guderian F, Dämmrich J, Potempa LA, and Wanner C. 2003. Tubular staining of structurally altered C-reactive protein in diabetic chronic kidney disease. Nephrol. Dial. Transplant 18: 2300–2307. [DOI] [PubMed] [Google Scholar]
- 59.Al-Baradie RS, Pu S, Liu D, Zeinolabediny Y, Ferris G, Sanfeli C, Corpas R, Garcia-Lara E, Alsagaby SA, Alshehri BM, Abdel-Hadi AM, Ahmad F, Moatari P, Heidari N, and Slevin M. 2021. Monomeric C-reactive protein localized in the cerebral tissue of damaged vascular brain regions is associated with neuro-inflammation and neurodegeneration-An immunohistochemical study. Front. Immunol 12: 644213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McFadyen JD, Kiefer J, Braig D, Loseff-Silver J, Potempa LA, Eisenhardt SU, and Peter K. 2018. Dissociation of C-reactive protein localizes and amplifies inflammation: Evidence for a direct biological role of C-reactive protein and its conformational changes. Front. Immunol 9: 1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braig D, Nero TL, Koch H-G, Kaiser B, Wang X, Thiele JR, Morton CJ, Zeller J, Kiefer J, Potempa LA, Mellett NA, Miles LA, Du X-J, Meikle PJ, Huber-Long M, Stark GB, Parker MW, Peter K, and Eisenhardt SU. 2017. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat. Commun 8: 14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tönnies E, and Trushina E. 2017. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis 57: 1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hawkins PN, Myers MJ, Epenetos AA, Caspi D, and Pepys MB. 1988. Specific localization and imaging of amyloid deposits in vivo using 123I-labeled serum amyloid P component. J. Exp. Med 167: 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pepys MB, Dyck RF, de Beer FC, Skinner M, and Cohen AS. 1979. Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clin. Exp. Immunol 38: 284–293. [PMC free article] [PubMed] [Google Scholar]
- 65.Hamazaki H 1995. Ca2+-dependent binding of human serum amyloid P component to Alzheimer’s β-amyloid peptide. J. Biol. Chem 270: 10392–10394. [DOI] [PubMed] [Google Scholar]
- 66.Janciauskiene S, de Frutos PG, Carlemalm E, Dahlbäck B, and Eriksson S. 1995. Inhibition of Alzheimer β-peptide fibril formation by serum amyloid P component. J. Biol. Chem 270: 26041–26044. [DOI] [PubMed] [Google Scholar]
- 67.Tennent GA, Lovat LB, and Pepys MB. 1995. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc. Natl. Acad. Sci. USA 92: 4299–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Danielsen B, Sørensen IJ, Nybo M, Nielsen EH, Kaplan B, and Svehag S-E. 1997. Calcium-dependent and -independent binding of the pentraxin serum amyloid P component to glycosaminoglycans and amyloid proteins: enhanced binding at slightly acid pH. Biochim. Biophys. Acta 1339: 73–78. [DOI] [PubMed] [Google Scholar]
- 69.Botto M, Hawkins PM, Bickerstaff MCM, Herbert J, Bygrave AE, McBride A, Hutchinson WL, Tennent GA, Walport MJ, and Pepys MB. 1997. Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nature Med 3: 855–859. [DOI] [PubMed] [Google Scholar]
- 70.Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, Hutchinson WL, Mangione PP, Gallimore JR, Millar DJ, Minogue S, Dhillon AP, Taylor GW, Bradwell AR, Petrie A, Gillmore JD, Bellotti V, Botto M, Hawkins PN, and Pepys MB. 2010. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 468: 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelson TJ, and Alkon DL. 2007. Protection against β-amyloid-induced apoptosis by peptides interacting with β-amyloid. J. Biol. Chem 282: 31238–31249. [DOI] [PubMed] [Google Scholar]
- 72.Hirko AC, Meyer EM, King MA, and Hughes JA. 2007. Peripheral transgene expression of plasma gelsolin reduces amyloid in transgenic mouse models of Alzheimer’s disease. Mol. Ther 15: 1623–1629. [DOI] [PubMed] [Google Scholar]
- 73.Noone DP, van der Velden TT, and Sharp TH. 2021. Cryo-electron microscopy and biochemical analysis offer insights into the effects of acidic pH, such as occur during acidosis, on the complement binding properties of C-reactive protein. Front. Immunol 12: 757633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dobson CM 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci 24: 329–332. [DOI] [PubMed] [Google Scholar]
- 75.Chiti F, and Dobson CM. 2017. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem 86: 27–68. [DOI] [PubMed] [Google Scholar]
- 76.Stewart CR, Tseng AA, Mok YF, Staples MK, Schiesser CH, Lawrence LJ, Varghese JN, Moore KJ, and Howlett GJ. 2005. Oxidation of low-density lipoproteins induces amyloid-like structures that are recognized by macrophages. Biochemistry 44: 9108–9116. [DOI] [PubMed] [Google Scholar]
- 77.Torzewski M 2022. C-reactive protein: Friend or foe? Phylogeny from heavy metals to modified lipoproteins and SARS-CoV-2. Front. Cardiovasc. Med 9: 797116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pathak A, and Agrawal A. 2019. Evolution of C-reactive protein. Front. Immunol 10: 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.